

Abstract
Sickle cell disease (SCD) is caused by a single point mutation in the gene that codes for beta globin synthesis, causing haemoglobin polymerisation, red blood cell stiffening and haemolysis under low oxygen and pH conditions. Downstream effects include widespread vasculopathy due to recurring vaso-occlusive events and haemolytic anaemia, affecting all organ systems. Cardiopulmonary complications are the leading cause of death in patients with SCD, primarily resulting from diastolic heart failure (HF) and/or pulmonary hypertension (PH). HF in SCD often features biventricular cardiac hypertrophy and left ventricular (LV) diastolic dysfunction. Among HF cases in the general population, approximately half occur with preserved ejection fraction (HFpEF). The insidious evolution of HFpEF differs from the relatively acute evolution of HF with reduced ejection fraction. The PH of SCD has diverse origins, which can be pulmonary arterial (precapillary), pulmonary venous (postcapillary) or pulmonary thromboembolic. It is also appreciated that patients with SCD can develop both precapillary and postcapillary PH, with elevations in LV diastolic pressures, as well as elevations in transpulmonary pressure gradient and pulmonary vascular resistance. Regardless of the cause of PH in SCD, its presence significantly reduces functional capacity and increases mortality. PH that occurs in the presence of HFpEF is usually of postcapillary origin. This review aims to assemble what has been learnt from clinical and animal studies about the manifestation of PH-HFpEF in SCD, specifically the contributions of LV diastolic dysfunction and myocardial fibrosis, in an attempt to gain an understanding of its evolution.
CLINICAL HEART FAILURE
Diastolic heart failure (HF) accounts for half of HF cases in the general population, typically manifesting as left ventricular (LV) diastolic dysfunction with normal ejection fraction (EF).1 EF describes systolic function and is blood volume ejected per stroke relative to maximum blood volume from LV filling, represented as per cent. A ≥50%EF is considered normal. HF with reduced EF (HFrEF), defined as <50%EF, typically includes eccentric cardiac remodelling. By contrast, HF with preserved EF (HFpEF), defined as ≥50%EF, occurs with concentric cardiac remodelling. HFpEF often emerges later in life with chronic comorbidities: hypertension, vascular stiffness, diabetes, obesity and anaemia. The molecular evolution of HFpEF relative to HFrEF remains largely unknown. HFpEF does not respond to therapies that are effective for HFrEF. These core differences in development, manifestation and response to treatment have engendered the idea that HFpEF and HFrEF arise from different system aetiologies, with HFrEF starting in the heart and HFpEF starting in the periphery; the cardiocentric lethality of HF is their sole commonality.2
The distribution of HFpEF and HFrEF is approximately 50:50 in the general population.3 The chronic nature of risk factors for HFpEF (hypertension, obesity, hyperlipidaemia, diabetes, insulin resistance, smoking, obstructive sleep apnoea and a sedentary lifestyle)4,5 translates into its slow development relative to HFrEF, allowing pulmonary hypertension (PH) to develop as a comorbidity. Chronic anaemia frequently associates with HFpEF. When that anaemia is haemolytic, as in sickle cell disease (SCD), it predisposes to PH and HFpEF (PH-HFpEF). It is unclear why some patients with chronically elevated LV diastolic pressures in the setting of HFpEF develop adverse pulmonary vascular remodelling with increasing precapillary pressures. These patients present with high transpulmonary pressure and high pulmonary vascular resistance (PVR) and are at the highest risk of death among patients with HFpEF.6-8 This manifestation is referred to as either PH-HFpEF or precapillary and postcapillary PH (CpC-PH). This review will focus on PH-HFpEF in SCD, with attention given to evidence for LV diastolic dysfunction and myocardial fibrosis, as learnt from clinical and animal studies of HF in SCD.
SICKLE CELL DISEASE
Sickle cell anaemia (SCA) results from a single point gene mutation coding for beta globin synthesis. Approximately 100 000 people in the USA have SCA, which means they are homozygous for the HbS mutation.9 When HbS occurs with other beta globin mutations (HbSC and Hb-beta thalassemia), the anaemia and organ pathology is less severe. While HbS mostly affects red blood cells (RBC), causing haemoglobin polymerisation and RBC dysmorphia under low oxygen and pH conditions, its effects are widespread. The current thinking is that SCD pathobiology involves a vicious cycle of HbS polymerisation, endothelial cell dysfunction, vaso-occlusive (VOC) events and sterile inflammation.10 HbS polymerisation causes blood stasis and RBC lysis, promoting adhesion of blood cells to endothelium. Recurring VOC causes ischaemia-reperfusion injury and RBC lysis releases oxygenated haemoglobin that scavenges nitric oxide (NO) and produces methemoglobin. Reactive oxygen species (ROS) result from reactions with degraded methemoglobin and activation of NADPH oxidase, xanthine oxidase and eNOS uncoupling. Heme and danger-associated molecular pattern entities released from cells and tissue cause TLR4 activation and neutrophil extracellular trap and IL1-beta generation. Propagation of this sterile inflammatory cycle underlies much of SCD comorbidity and high mortality rates. Gains have been made in extending lives of patients with SCD in developed countries, primarily due to newborn screening, childhood vaccinations, penicillin and hydroxyurea7 but not without a price. System-wide proliferative vasculopathy, LV diastolic dysfunction, PH, dysrhythmias and chronic kidney disease develop in the ageing SCD population, becoming noticeable in the second decade of life. HFpEF-associated morbidity and mortality in SCD manifests in the second to fourth decades of life.7 Diastolic dysfunction, PH and sudden death are the largest contributors to SCD mortality (figure 1).11
Figure 1.

The chronic anaemia of SCD promotes PH-HFpEF development. Chronic anaemia causes a compensatory hyperdynamic state in which both plasma volume and stroke volume increase, while SVR and PVR decrease, leading to enhanced cardiac output (~10.9 L/min) for adequate supply of oxygenated blood to tissues. High LV volumes cause systolic wall stress to increase and the LV to hypertrophy, resulting in LV diastolic dysfunction, SMC and intimal proliferation as well as in situ thrombosis, all of which increases PVR. These effects increase BP and pulmonary artery pressure, leading to PH development. Elevated BP works together with anaemia to keep the cycle of haemodynamic pathology moving forward. If the anaemia is caused by haemolysis, then there is additional direct vascular injury, alterations in NO to ROS ratios and vasoconstriction and proliferation. Importantly, the PH becomes the leading contributor to right heart enlargement, which ultimately ends in RH failure. When PH and LV diastolic dysfunction are both present, then functional capacity of the patient is decreased and mortality increases. BP, blood pressure; HF, heart failure; HFpEF, HF with preserved ejection fraction; LV, left ventricle; NO, nitric oxide; PH, pulmonary hypertension; PVR, pulmonary vascular resistance; ROS, reactive oxygen species; SCD, sickle cell disease; SMC, smooth muscle cell; SVR, systemic vascular resistance.
PULMONARY HYPERTENSION IN SCD
The pathology of PH varies and can involve pulmonary vascular endothelial dysfunction, smooth muscle cell (SMC) proliferation, adventitial fibroblast accumulation and immune system activation. Overall, PH generates right ventricular (RV) pressure overload, causing molecular adaptations that predispose to RV failure. It is important to distinguish between the different haemodynamic types of PH, especially in terms of the primary molecular and cellular basis of the associated pulmonary vascular remodelling; is it precapillary (arterial) or postcapillary (venous) relative to the pulmonary artery (PA)? For almost 45 years, the haemodynamic definition of PH was a mean PA pressure (mPAP) ≥25 mm Hg. The 6th World Symposium on PH recently established a new haemodynamic definition for PH that includes a mPAP >20 mm Hg, which is 2 SD above normal (14.0±3.3 mm Hg). Definitive diagnosis of PH requires accurate measurement of mPAP by RH catheterisation (RHC). Concomitant measurements of PA wedge pressure (>15 mm Hg) and PVR are required to establish involvement of left heart (LH) pathology. When PVR is <3 Wood units (WU), the PH is diagnosed as isolated postcapillary PH. When PVR is ≥3 WU, the PH is diagnosed as CpC-PH. In the setting of postcapillary PH, the risk of death is highest when the transpulmonary pressure gradient (TPG)>12, which is consistent with CpC-PH.12
The prevalence of PH in SCD ranges from 10% to 33%, when measured by RHC or echocardiography, respectively. The distribution of precapillary to postcapillary PH in SCD is roughly 50:50. Spontaneous development of PH from LH pathology in SCD is often found in the setting of biventricular cardiac hypertrophy and myocardial fibrosis.11 Cardiac function in SCD is restrictive but is not ‘restrictive cardiomyopathy’ per se, the latter having idiopathic or infiltrative origins.13 Clinically, the underlying cause of diastolic dysfunction in SCD remains incompletely defined. Epidemiological and clinical data suggest that anaemia stresses the cardiovascular system, impairing filling times; and haemolytic anaemia, in particular, contributes to pulmonary and aortic vascular stiffness that impair afterload.7,11 Recent data suggest microvascular VOC produce myocardial scarring, as detected on CT scan and in mouse models.14,15
While not as definitive as RHC for diagnosing PH, echocardiography for measuring tricuspid regurgitant jet velocity (TRV) has proven useful at estimating mPAP and identifying patients at very high-risk. When TRVs of 2.5–3.0 m/s have been detected in adults, subsequent RHC has shown 25%–39% of those identified had mPAP exceeding 25 mm Hg.16,17 The accuracy of TRV for predicting RHC-confirmed PH climbs to 66%–77% when TRVs>3.0 m/s are detected.16,17 The effectiveness of TRV for predicting PH (mPAP >25 mm Hg) increased to 62% when used with N-terminal pro–B-type natriuretic peptide (NTproBNP >164 pg/mL) and 6 min walk distance (6MWD<333 m). Since the consensus definition of PH has dropped to a mPAP>20 mm Hg, more patients with SCD likely have PH than RHC studies have suggested.18,19
Risk for developing PH in SCD associates with steady state severity of haemolysis and anaemia. Severity of haemolytic anaemia independently associates with TRV values, mPAP measured by RHC and LV diastolic function (LV lateral E/e’ ratio).7,12,20-22 Minitti et al reported an independent association of TRV with haemolysis and oxygen desaturation in paediatric patients with SCD.20 WalkPHaSST found similar associations in adults.23
Severity of PH is predictive of death. A large NIH study in 531 patients with SCD found 10.4% had RHC-confirmed PH. Even mildly high TRVs (2.5–2.9) have been found to increase mortality risk.22,24-27 A recent study conducted in France found the >2.5 m/s TRV cut-off predictive for mortality in clinically asymptomatic patients with SCD, despite being only 25% predictive for RHC-confirmed PH.17 A meta-analysis of 45 studies with >6000 patients overall showed a 4.9 HR for mortality with elevated TRV28 Retrospective analysis of a study in patients with SCD (n=34) found the likelihood of death increased 1.7 times for every 10 mm Hg increase in mPAP with RHC-confirmed PH.26 A low 6MWD often accompanies elevated TRV and itself associates with risk of mortality.28
In children with SCD, the low mortality rate in this era lessens TRV’s utility for predicting PH and mortality risk. Two studies, one retrospective29 and the other prospective,30 found that TRV alone insufficiently predicts PH or its associated complications. Children with SCD already have high TRVs due to high cardiac output. Gordeuk et al made this observation and called for setting the paediatric TRV cut-off for predicting PH at >2.7 m/s.31 Coupled with clinical symptoms of vasculopathy and abnormal laboratory findings (haemolysis and proteinuria), TRV becomes more powerful at predicting disease progression in children. Severe haemolysis increases the odds of detecting elevated TRV in children.32 When accompanied by haemolytic anaemia, even a mildly elevated TRV increases predictions of functional deficits (6MWD) by 4.4-fold.31 Hence, clinical experts now call for this combined-risk factor approach to determining need for invasive RHC-confirmed PH.
Guidance on screening for PH in SCD has been overseen by the American Thoracic Society, the American College of Chest Physicians and the Pulmonary Hypertension Association. In adults, screening is recommended every 1–3 years as long as TRV <2.5 m/s. With TRV≥2.5 m/s and <2.9 m/s, frequency of screening should increase and SCD-specific therapy should be intensified. If symptomatic or TRV≥2.9, RHC is recommended for confirming mPAP≥20 mm Hg and determining PH phenotype (precapillary, postcapillary or combined).25 When and how often to screen for PH in children with SCD remains undetermined given the lack of longitudinal data on risk of PH-associated mortality in this population.
LV DIASTOLIC DYSFUNCTION IN SCD
Chronic anaemia in SCD invokes compensatory increases in plasma and stroke volumes and decreases in systemic vascular resistance (SVR) and PVR, which together enhance cardiac output (~10.9 L/min) for tissue oxygenation. Associated high LV volumes increase wall stress during systole, provoking LV hypertrophy, SMC and intima proliferation and in situ thrombosis. These effects increase PVR, blood pressure and pulmonary artery pressure (PAP), promoting PH development, the leading contributor to RH enlargement and failure. When LV diastolic dysfunction also occurs, as with impaired compliance due to progressive myocardial fibrosis, patient functional capacity declines and risk for mortality rises. With haemolytic anaemia, additional direct vascular injury likely occurs as NO-to-ROS ratio changes to favour vasoconstriction and proliferation (figure 1).
Diastolic failure is defined as diastolic pressure that is disproportionate to normal diastolic volume. With LV diastolic dysfunction, adaptation of LV output to RV output via intrinsic myocardial mechanisms (Frank-Starling) becomes impaired. LV diastolic dysfunction is common to HFpEF, often resulting from a stiff LV wall,33 which is thought to result from myocardial fibrosis that often associates with diastolic dysfunction. Hypertrophy or fibrosis of the LV wall impairs compliance, optimal cardiac stretch, active tension development and cardiac contractility. This increases the pressure-to-volume relationship at end of LV diastole (EDPVR). The dilated ventricle may also impair atrioventricular coupling and end-diastolic filling. Patients with HFpEF have abnormally high left atrial (LA) pressures/volumes (E’e), which are secondary to LV diastolic dysfunction. This combination imposes a high afterload on pulmonary venous vasculature, which can also contribute to PH development (figure 2). Not all clinicians agree that diastolic dysfunction is a defining feature of HFpEF, accounting for different European and US guidelines (American Heart Association, American College of Cardiology Foundation) for diagnosis and treatment of HFpEF.2
Figure 2.

LV diastolic dysfunction promotes PH development in SCD. LV diastolic dysfunction, which is increased LV pressure in the presence of normal LV volume (EDPVR), promotes stiffening of the LH and decreased myocardial relaxation (stretch). Less LV relaxation impairs LV filling and LA pressure rises, producing an afterload on the PV circulation. Pulmonary (Pulm) hydrostatic pressure builds, causing oedema to form in the lungs which exerts a back pressure on the PA. High PA pressure develops, giving rise to PH. EDPVR, end diastolic pressure volume relationship; LA, left atrial; LH, left heart; LV, left ventricle; PA, pulmonary artery; PH, pulmonary hypertension; PV, pulmonary venous; TRV, tricuspid regurgitant jet velocity; RVSP, right ventricular systolic pressure; SCD, sickle cell disease.
Whereas LV systolic dysfunction is rare in SCD, LV diastolic dysfunction is common and often occurs with LV enlargement. With severe anaemia, SVR is decreased, resulting in dilated ventricles and increased stroke volumes. Hence, cardiac chamber enlargement and dilation caused by chronic anaemia have limited utility for diagnosing LV diastolic dysfunction in SCD. Large epidemiological studies and clinical trials have identified combined factors that better predict HFpEF and mortality in SCD adults. WalkPHaSST found that LV lateral E/e’ ratio and TRV independently predict 6MWD. The study also found that a low E/A (ratio of early-to-late peak flow velocity during diastole) increases the mortality risk ratio to 3.5.
PH-HFPEF IN SCD
When diastolic dysfunction coexists with high pulmonary pressure, the mortality risk ratio increases to 12.0.7 How LV pathology contributes to PH development and severity in SCD is unknown. Right ventricular diastolic dysfunction also occurs in patients with SCD with HFpEF, frequently with PH, as indicated by high tricuspid inflow pressure and late tricuspid inflow E/A ratio.34 This diastolic dysfunction has been attributed to a stiff RV wall, resulting from high RV afterload caused by PH.34 Epidemiological studies show associations between intravascular haemolysis, PH and mortality in SCD.7 Niss et al screened children with SCD (n=134) using echocardiography and performed a meta-analysis of 68 studies. Both studies revealed enlarged RA dimensions/volumes, an abnormal E/e’ indicating diastolic dysfunction (restrictive cardiac physiology) and mild pulmonary venous hypertension in RHC-confirmed PH,35 all coexisting with normal LV systolic function.35 Damy et al have similarly reported high RA and LV end diastolic dimensions with a high cardiac index, which associated with markers of intravascular haemolysis: lactate dehydrogenase, bilirubin and dense red cells.36 PH in the setting of diastolic dysfunction severely impairs exercise tolerance (6MWD) in patients with SCD. Greater clinical use of non-invasive cardiac magnetic resonance (CMR) technology may help elucidate RH versus LH contributions to PH development in SCD and improve patient profiling for PH therapies. A hypothesis-generating study in patients with SCD, using RHC to measure TPG (n=84) and CMR to detect RV remodelling (n=41), found TPG to be useful in predicting severity of PH-related complications when mPAP in SCD is borderline PH (21–24 mm Hg). High TPG≥12 mm Hg identified patients with low RV EF, indicating RV systolic dysfunction. Low LH versus high RH volumes were found where LV filling volume and cardiac index were reduced, while LV EF was preserved. While HFpEF is widely reported as the predominant form of HF in patients with SCD, HFrEF has also been reported, coinciding with high intravascular haemolysis and TRV >2.5 m/s, as seen in SCD-associated PH. These HFrEF reports likely describe a small, more severe subset of patients with SCD.37
MYOCARDIAL FIBROSIS IN SCD
Myocardial fibrosis is definitively detected in patients either during postmortem autopsy, echocardiography or CMR imaging. While echocardiography is the gold standard for clinical detection of diastolic dysfunction, CMR is superior at detecting myocardial interstitial fibrosis even before onset of diastolic dysfunction.38 Myocardial fibrosis occurs diffusively with ageing. In HFpEF, fibrosis occurs as collagen matrix deposits in the heart, contributing up to 66% of myocardial passive stiffness.39 Functionally, fibrosis restricts diastolic filling and impairs exercise tolerance.39,40 Plasma markers of myocardial fibrosis associate with worse clinical outcomes.39
Ventricular hypertrophy is progressive, compensatory remodelling of the heart with collagen buildup.40,41 Reactive interstitial fibrosis occurs with chronic cardiac stress from elevated pressure, volume, ischaemia or cardiomyopathy and usually does not impair systolic function.42 This may explain why HFpEF predominates over HFrEF in SCD. Some CMR studies have detected myocardial fibrosis in patients with SCD (figure 3). A small study (n=25) in young (23±3 years) non-transfused SCA individuals detected diffuse myocardial fibrosis, measured by extracellular volume fraction (ECV), in all participants with 64% meeting the definition for diastolic dysfunction using adult guidelines.15 Higher ECV correlated with LV diastolic dysfunction, low haemoglobin and high NTproBNP Some atypical findings of vascular remodelling and intimal thickening in young SCA individuals warrant larger, longitudinal studies to mechanistically define their relationship to diastolic dysfunction.15
Figure 3.
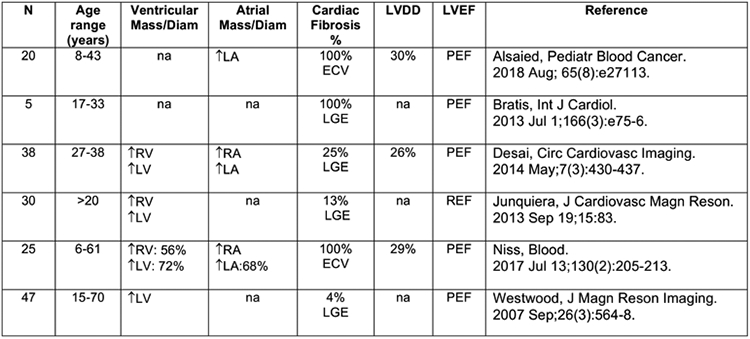
Clinical studies with CMR-assessed cardiac fibrosis in SCD (SCA, HbSC, HbS-thalassemia). CMR, cardiac magnetic resonance; SCD, sickle cell disease.
Definitions: N, number of study participants; %, percentage of study participants with the feature; Mass/Diam, mass or diameter; LVDD, left ventricular diastolic dysfunction; LVEF, left ventricular ejection fraction (PEF, preserved ejection fraction; REF, reduced ejection fraction); LGE, late gadolinium enhancement; ECV, extracellular volume; T, increased versus normal; 4-, decreased versus normal; RA, right atrium; RV, right ventricle; LA, left atrium; LV, left ventricle; na, no available data.
ANIMAL STUDIES OF PH-HFPEF IN SCD
Recent studies in transgenic SCD mice suggest that PH-HFpEF spontaneously develops with age. Bakeer et al longitudinally studied cardiac volume and ECV using serial echo and CMR to determine the relationship of myocardial fibrosis to LV diastolic dysfunction.14 They found gradual development of restrictive physiology, characterised by biventricular and LA hypertrophy at 3 months age, increased thickness of LV end diastolic interventricular septum and posterior wall at 7–8 months age and chaotic sarcomere remodelling at 8 months age. Systolic function was preserved in the presence of diastolic dysfunction caused by diffuse myocardial fibrosis (11%). Prolonged QT intervals, arrhythmias and ischaemic changes preceded sudden death. More groups have detected spontaneous development of PH and RV hypertrophy in SS mice using RHC.43-45 Doppler echo has been used to characterise LH morphology and function in many mouse models of PH and HFpEF; increased RV:LV mass suggests specific pulmonary vascular disease, whereas equal biventricular increases suggest HFpEF. Echo, however, does not provide information on cardiac fibrosis.
Given successes in extending longevity of patients with SCD (42 for men and 48 for women with SCA and approximately 20 more years for the sickle cell variants Hb SC and HbS-beta thal), many cardiovascular and metabolic risk factors for HFpEF development in the general population likely also apply to the SCD population. This suggests the possibility of using bone marrow transplant to create novel SCD animal models of PH-HFpEF with underlying metabolic/cardiovascular disease. A recent, small (n=50) US cross-sectional study in young adults (mid to late 30s) with SCD found risk factors for cardiovascular disease: 14.3% metabolic syndrome, 20% overweight (body mass index 25–30 kg/m2), 26% obesity (>30 kg/m2), 34.7% history of smoking, 18% currently smoking, 69.4% low high density lipoprotein (HDL) and diets exceeding American Heart Association (AHA)-recommended fat and sugar intake.46 When compared with Nigerians, individuals with SCD in the USA are 10-fold more obese by WHO criteria. Others have similarly reported increasing obesity in the SCD population.47 Type 2 diabetes also occurs in patients with SCD at a rate similar to the general African American population.48
Several rodent strains develop features associated with HFpEF, facilitating preclinical study of the disease. These include the C57BL, ApoE-deficient, NON/shiLtJ and AKR/J mouse strains.8 Among 36 inbred mouse strains studied, the AKR/J strain reproducibly developed high fat diet-induced PH with diastolic dysfunction and biventricular hypertrophy.8 Leptin-deficient (ob/ob) mice and high fat diet enable studying how obesity and diabetes contribute to HFpEF development.49 Chronic low dose angiotensin II infusion is another model for stimulating cardiomyocyte hypertrophy and interstitial fibrosis. The vascular endothelial growth factor (VEGF) receptor blocker SU5416 induces features of metabolic syndrome-associated PH-HFpEF in ZSF1 obese rats.50 These strategies may be translatable to studying metabolic syndrome-associated PH-HFpEF development in SCD mice.
SUMMARY AND FUTURE PERSPECTIVES
SCD is no longer a disease of the young in the developed world. Medical and public health advancements allow individuals with SCD to survive childhood and experience many cardiovascular complications associated with ageing and unhealthy lifestyles. Hence, understanding the evolution of these cardiovascular complications in the setting of SCD is becoming important. HFpEF is reaching similar proportions in the SCD population as in the general population, increasing the mortality risk when coincident with PH. Early diagnosis of diastolic dysfunction and myocardial fibrosis, which increasingly signify diastolic HF in patients with SCD, will require standardised clinical application of high resolution technologies like CMR. Development of more animal models for researching how PH and HFpEF evolve and manifest in SCD, especially when complicated by ageing and cardiovascular disease, are also needed to build a strong therapeutic armamentarium for persons living with the disease.
Current guidelines for clinical management of PH and HF in SCD adults call for intensification of SCD-specific therapy: hydroxyurea, chronic transfusion therapy (CTT), fetal haemoglobin inducers, gene therapy and/or bone marrow transplant.7 Therapies specific for managing PH and HF in SCD are non-existent because the necessary clinical trials have not been conducted. Physiologically, CTT is expected to be effective therapy for PH and HF in SCD since it increases haemoglobin, which improves oxygen delivery per stroke volume and lessens the heart’s work.7 Towering stroke volume increases cardiopulmonary reserve and decreases cardiac wall stress and PAP, reducing RH failure (figure 4). Quality data are needed to overcome the equipoise over CTT’s specific use for PH and HF therapy.
Figure 4.
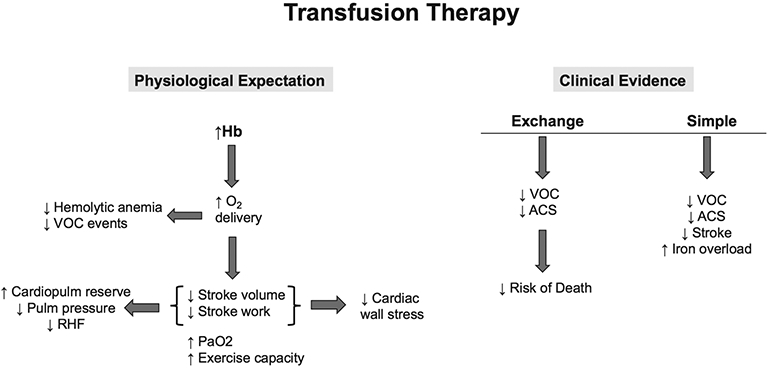
Transfusion therapy for cardiopulmonary disease in adults with SCD. The physiological expectation for transfusion therapy in SCD is that increasing Hb levels will improve oxygen (O2) delivery to tissues, lowering intravascular haemolytic and VOC events. Where the heart is concerned, stroke volume and stroke work are reduced, which decreases cardiac wall stress, increases cardiopulmonary (cardiopulm) reserve, lowers pulmonary (pulm) pressures and reduces risk for RHF. Arterial oxygen tension (PaO2) also increases along with improvement in exercise capacity. The clinical evidence for efficacy of transfusion therapy, however, is limited to observations of decreased VOC and ACS events and subsequently lowered risk of death with exchange transfusion therapy. Simple transfusion therapy, when used prophylactically, appears to decrease VOC, ACS and stroke but may also contribute to iron overload. ACS, acute chest syndrome; Hb, haemoglobin; RHF, right heart failure; VOC, vaso-occlusive; SCD, sickle cell disease.
Funding
This work was supported, in whole or in part, by the Institute for Transfusion Medicine (Vitalant) and National Institutes of Health Grants: R25 HL128640-03 (KW); R01 HL098032, R01 HL125886-01 and P01HL103455, T32 HL007563 (MG) and R01 HL133864, R01 HL128304 and AHA Established Investigator Award (AS).
Footnotes
Competing interests AS receives research support from Bayer Pharmaceuticals. MG is listed as a coinventor on an NIH government patent for the use of nitrite salts in cardiovascular diseases and on provisional patents for the use of recombinant neuroglobin and heme-based molecules as antidotes for CO poisoning; the former has been licensed by United Therapeutics and the latter by Globin Solutions, Inc. MG is a coinvestigator in a research collaboration with Bayer Pharmaceuticals to evaluate riociguat as a treatment for patients with SCD.
REFERENCES
- 1.Andersson C, Vasan RS. Epidemiology of heart failure with preserved ejection fraction. Heart Fail Clin 2014;10:377–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.CSP L, Voors AA, de Boer RA. Solomon SD and van Veldhuisen DJ. heart failure with preserved ejection fraction: from mechanisms to therapies. Eur Heart J 2018;39:2780–92. [DOI] [PubMed] [Google Scholar]
- 3.Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation 2015; 131:e29–322. [DOI] [PubMed] [Google Scholar]
- 4.Oktay AA, Rich JD, Shah SJ. The emerging epidemic of heart failure with preserved ejection fraction. Curr Heart Fail Rep 2013;10:401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farr G, Shah K, Markley R, et al. Development of pulmonary hypertension in heart failure with preserved ejection fraction. Prog Cardiovasc Dis 2016;59:52–8. [DOI] [PubMed] [Google Scholar]
- 6.Levine AR, Simon MA, Gladwin MT. Pulmonary vascular disease in the setting of heart failure with preserved ejection fraction. Trends Cardiovasc Med 2019;29:207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gladwin MT. Cardiovascular complications and risk of death in sickle-cell disease. Lancet 2016;387:2565–74. [DOI] [PubMed] [Google Scholar]
- 8.Lai Y-C, Wang L, Gladwin MT. Insights into the pulmonary vascular complications of heart failure with preserved ejection fraction. J Physiol 2019;597:1143–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.CDC. Data & Statistics on Sickle Cell Disease 2019. [Google Scholar]
- 10.Sundd P, Gladwin MT, Novelli EM. Pathophysiology of sickle cell disease. Annu Rev Pathol 2019;14:263–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gladwin MT, Sachdev V. Cardiovascular abnormalities in sickle cell disease. J Am Coll Cardiol 2012;59:1123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kato GJ, Gladwin MT. Evolution of novel small-molecule therapeutics targeting sickle cell vasculopathy JAMA 2008;300:2638–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pereira NL, Grogan M, Dec GW. Spectrum of Restrictive and Infiltrative Cardiomyopathies: Part 2 of a 2-Part Series. J Am Coll Cardiol 2018;71:1149–66. [DOI] [PubMed] [Google Scholar]
- 14.Bakeer N, James J, Roy S, et al. Sickle cell anemia mice develop a unique cardiomyopathy with restrictive physiology. Proc Natl Acad Sci U S A 2016;113:E5182–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niss O, Fleck R, Makue F, et al. Association between diffuse myocardial fibrosis and diastolic dysfunction in sickle cell anemia. Blood 2017;130:205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehari A, Alam S, Tian X, et al. Hemodynamic predictors of mortality in adults with sickle cell disease. Am J Respir Crit Care Med 2013;187:840–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parent F, Bachir D, Inamo J, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med 2011;365:44–53. [DOI] [PubMed] [Google Scholar]
- 18.Maron BA, Brittain EL, Choudhary G, et al. Redefining pulmonary hypertension. Lancet Respir Med 2018;6:168–70. [DOI] [PubMed] [Google Scholar]
- 19.Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019;53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minniti CP, Sable C, Campbell A, et al. Elevated tricuspid regurgitant jet velocity in children and adolescents with sickle cell disease: association with hemolysis and hemoglobin oxygen desaturation. Haematologica 2009;94:340–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Machado RF, Barst RJ, Yovetich NA, et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood 2011;118:855–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med 2008;359:2254–65. [DOI] [PubMed] [Google Scholar]
- 23.Nouraie M, Lee JS, Zhang Y, et al. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica 2013;98:464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sachdev V, Kato GJ, Gibbs JSR, et al. Echocardiographic markers of elevated pulmonary pressure and left ventricular diastolic dysfunction are associated with exercise intolerance in adults and adolescents with homozygous sickle cell anemia in the United States and United Kingdom. Circulation 2011;124:1452–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klings ES, Machado RF, Barst RJ, et al. Gladwin MT and American thoracic Society AD hoc Committee on pulmonary hypertension of sickle cell D. an official American thoracic Society clinical practice guideline: diagnosis, risk stratification, and management of pulmonary hypertension of sickle cell disease. Am J Respir Crit Care Med 2014;189:727–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Castro LM, Jonassaint JC, Graham FL, et al. Pulmonary hypertension associated with sickle cell disease: clinical and laboratory endpoints and disease outcomes. Am J Hematol 2008;83:19–25. [DOI] [PubMed] [Google Scholar]
- 27.Lorch D, Spevack D, Little J. An elevated estimated pulmonary arterial systolic pressure, whenever measured, is associated with excess mortality in adults with sickle cell disease. Acta Haematol 2011;125:225–9. [DOI] [PubMed] [Google Scholar]
- 28.Caughey MC, Poole C, Ataga KI, et al. Estimated pulmonary artery systolic pressure and sickle cell disease: a meta-analysis and systematic review. Br J Haematol 2015;170:416–24. [DOI] [PubMed] [Google Scholar]
- 29.Hebson C, New T, Record E, et al. Elevated tricuspid regurgitant velocity as a marker for pulmonary hypertension in children with sickle cell disease: less prevalent and predictive than previously thought? J Pediatr Hematol Oncol 2015;37:134–9. [DOI] [PubMed] [Google Scholar]
- 30.Liem RI, Nevin MA, Prestridge A, et al. Tricuspid regurgitant jet velocity elevation and its relationship to lung function in pediatric sickle cell disease. Pediatr Pulmonol 2009;44:281–9. [DOI] [PubMed] [Google Scholar]
- 31.Gordeuk VR, Minniti CP, Nouraie M, et al. Elevated tricuspid regurgitation velocity and decline in exercise capacity over 22 months of follow up in children and adolescents with sickle cell anemia. Haematologica 2011;96:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niu X, Nouraie M, Campbell A, et al. Angiogenic and inflammatory markers of cardiopulmonary changes in children and adolescents with sickle cell disease. PLoS One 2009;4:e7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borbély A, van der Velden J, Papp Z, et al. Cardiomyocyte stiffness in diastolic heart failure. Circulation 2005;111:774–81. [DOI] [PubMed] [Google Scholar]
- 34.Akgül F, Yalçin F, Seyfeli E, et al. Pulmonary hypertension in sickle-cell disease: comorbidities and echocardiographic findings. Acta Haematol 2007;118:53–60. [DOI] [PubMed] [Google Scholar]
- 35.Niss O, Quinn CT, Lane A, et al. Cardiomyopathy with restrictive physiology in sickle cell disease. JACC Cardiovasc Imaging 2016;9:243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Damy T, Bodez D, Habibi A, et al. Haematological determinants of cardiac involvement in adults with sickle cell disease. Eur Heart J 2016;37:1158–67. [DOI] [PubMed] [Google Scholar]
- 37.Guedeney P, Lionnet F, Ceccaldi A, et al. Cardiac manifestations in sickle cell disease varies with patient genotype. Br J Haematol 2018;181:664–71. [DOI] [PubMed] [Google Scholar]
- 38.Niss O, Taylor MD. Applications of cardiac magnetic resonance imaging in sickle cell disease. Blood Cells Mol Dis 2017;67:126–34. [DOI] [PubMed] [Google Scholar]
- 39.Zile MR, Baicu CF, Ikonomidis JS, et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation 2015;131:1247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plitt GD, Spring JT, Moulton MJ, et al. Mechanisms, diagnosis, and treatment of heart failure with preserved ejection fraction and diastolic dysfunction. Expert Rev Cardiovasc Ther 2018;16:579–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Travers JG, Kamal FA, Robbins J, et al. Cardiac fibrosis: the fibroblast Awakens. Circ Res 2016;118:1021–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biernacka A, Frangogiannis NG, Aging FNG, et al. Aging and cardiac fibrosis.. Aging Dis 2011;2:158–73. [PMC free article] [PubMed] [Google Scholar]
- 43.Potoka KP, Wood KC, Baust JJ, et al. Nitric oxide-independent soluble guanylate cyclase activation improves vascular function and cardiac remodeling in sickle cell disease. Am J Respir Cell Mol Biol 2018;58:636–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang B-Y, Park K, Kleinhenz JM, et al. Peroxisome proliferator-activated receptor γ regulates the v-ets avian erythroblastosis virus E26 oncogene homolog 1/microRNA-27a axis to reduce endothelin-1 and endothelial dysfunction in the sickle cell mouse lung. Am J Respir Cell Mol Biol 2017;56:131–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsu LL, Champion HC, Campbell-Lee SA, et al. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood 2007;109:3088–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ogunsile FJ, Bediako SM, Nelson J, et al. Metabolic syndrome among adults living with sickle cell disease. Blood Cells Mol Dis 2019;74:25–9. [DOI] [PubMed] [Google Scholar]
- 47.Ballas SK. The sixth vital sign: body mass index in patients with sickle cell disease. J Clin Med Res 2017;9:889–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou J, Han J, Nutescu EA, et al. Similar burden of type 2 diabetes among adult patients with sickle cell disease relative to African Americans in the U.S. population: a six-year population-based cohort analysis. Br J Haematol 2019;185:116–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horgan S, Watson C, Glezeva N, et al. Murine models of diastolic dysfunction and heart failure with preserved ejection fraction. J Card Fail 2014;20:984–95. [DOI] [PubMed] [Google Scholar]
- 50.Lai Y-C, Tabima DM, Dube JJ, et al. SIRT3-AMP-Activated protein kinase activation by nitrite and metformin improves hyperglycemia and normalizes pulmonary hypertension associated with heart failure with preserved ejection fraction. Circulation 2016;133:717–31 [DOI] [PMC free article] [PubMed] [Google Scholar]