

Abstract
As in other areas, artificial intelligence (AI) is heavily promoted in different scientific fields, including chemistry. Although chemistry traditionally tends to be a conservative field and slower than others to adapt new concepts, AI is increasingly being investigated across chemical disciplines. In medicinal chemistry, supported by computer-aided drug design and cheminformatics, computational methods have long been employed to aid in the search for and optimization of active compounds. We are currently witnessing a multitude of AI-related publications in the medicinal-chemistry-relevant literature and anticipate that the numbers will further increase. Often, advances through AI promoted in such reports are difficult to reconcile or remain questionable, which hampers the acceptance of computational work in interdisciplinary environments. Herein we attempt to highlight selected investigations in which AI has shown promise to impact medicinal chemistry in areas such as compound design and synthesis.
Introduction
In chemistry, the term artificial intelligence (AI) currently mostly refers to machine learning (ML) and—to a lesser extent—robotics, which only represent a part of the methodological AI spectrum.1 Other AI approaches such as recommendation or expert systems are just beginning to be considered. In medicinal chemistry, supported by cheminformatics, ML has a history of more than two decades, especially for molecular property prediction and virtual compound screening.2 Here AI is by and large associated with deep learning (DL),3 for which a great variety of deep neural network (DNN) architectures and learning strategies have become available.4 Currently many publications are appearing that investigate DL and DNNs for property predictions. In medicinal chemistry, compound property predictions are generally based on predefined molecular representations and data sets that are small in size compared to those used in areas where DL has made a substantial impact, such as image analysis or natural language processing.3 Under these conditions, DNNs have for the most part not yielded significant advances compared to standard ML algorithms.3 More often than not, minor increases in prediction accuracy by DNNs under typical benchmark conditions are promoted as substantial improvements, which is not meaningful, especially if alternative ML approaches already perform at the 90+% accuracy level. Of note, representation learning, a hallmark of DL in other fields, is also being increasingly investigated in cheminformatics and medicinal chemistry. New DNN architectures are frequently proposed to derive (model-internal) molecular representations from graphs or images. Currently available results are heterogeneous. In a number of cases, the performance of learned representations in property predictions was comparable (or inferior) to that of descriptor-based representations. In others, such as molecular-graph-based learning using message-passing DNNs, improvements were observed for predictions on a variety of data sets.5 However, representation learning generally benefits from the availability of large data sets, which limits its general applicability. Moreover, the extent to which relative representation-dependent differences in model performance detected in benchmark settings might affect prospective applications remains largely unknown.
In compound property prediction, DL has not been shown to substantially and consistently outperform standard ML approaches; other applications should better be considered, as discussed herein. In exploring potential advances of DL and DNNs in medicinal chemistry and drug discovery, two important aspects need to be taken into account. First, one is still far away from a situation where algorithms would make decisions beyond human reasoning.6,7 Instead, investigators make the ultimate calls about new or further-improved active compounds with the aid of ML/DL outputs. Second, practical applications using DL beyond benchmarking are still rare in medicinal chemistry,6 and such prospective applications are of critical importance for the further development of the field.7
In the following, we attempt to highlight selected investigations in medicinal chemistry where DL has thus far made a notable impact with promise for the future. By its very nature, our account remains at least in part subjective and, given the scope of a mini-review, cannot possibly be comprehensive. Rather, it aims to point at a number of developments that depend on new learning architectures and have potential for prospective applications.
New Chemical Matter
We begin the discussion by addressing upfront the question that is most interesting and important for the practice of medicinal chemistry: Has AI/DL already impacted compound design and optimization in a measurable way? In other words, have novel molecules been obtained that have advanced—or are expected to advance—drug discovery?
As stated above, reports of prospective AI/DL applications including the generation of new chemical matter are currently rare, and the state of the art is difficult to judge in scientific terms. Currently, news about significant advances predominantly originate from AI companies and startups, claiming the generation of novel chemical matter with attractive activities and/or the achievement of significant shortcuts in bringing new compounds through development pipelines. This may be so, but as long as the results of such efforts are not disclosed and reported in a scientifically rigorous manner, the claims need to be disregarded. Some case studies that have been reported are controversial.8 While virtual compound screening studies using ML including DNNs continue to produce experimentally confirmed hits, as has been the case for many years, these molecules are typically active against well-explored targets for which many potent compounds are already available. We also note that active compounds identified using complex computational methods cannot be claimed to “validate” such approaches as long as it is not conclusively shown that simpler methods would not also identify them. However, there are individual DL studies that have left a mark, such as the discovery of new potential antibiotics, including repurposed kinase inhibitors and other public-domain compounds, using message-passing DNNs.9 In this case, comprehensive control calculations showed that these compounds were not identified using other computational methods, and their antibiotic activities were confirmed in extensive experiments. However, even in this exemplary successful DL application, the potential of the newly identified compounds to ultimately become antibiotics was called into question from a medicinal chemistry perspective, and their chemical novelty was disputed.10 Clearly, the bar for new chemical matter in drug discovery is high, and it is too early to judge the ability of AI to make a substantial difference. Publication of more prospective applications will be required. This also applies to generative de novo design, which is one of the current growth areas of DL in medicinal chemistry, as discussed in the following. Also here, most current work concentrates on what might potentially be accomplished using DL and thus must primarily be considered from a methodological viewpoint, whereas breakthroughs in prospectively generating new chemical matter remain to be reported in a more consistent and rigorous manner.
Synthetic Chemistry
Computer-aided synthesis planning (CASP) aims at accelerating the decision-making process by which medicinal chemists choose the most appropriate routes to synthesize new compounds. In addition to drug exposure, efficacy, and safety, synthetic feasibility represents a key determinant of candidate progression toward the clinic. Accordingly, prediction of chemical reactions and their success rates remains highly relevant for drug discovery, from its early stages focusing on design–make–test–analyze (DMTA) cycles to large-scale manufacturing processes. CASP has its origins in retrosynthetic analysis (decomposing compounds into precursors), which was introduced in the 1960s. ML was first applied for synthesis prediction by Gelernter et al., who developed a production-quality knowledge base named SYNCHEM composed of applications for inductive generalization (ISOLDE), explanation-based learning (TRISTAN), and conceptual clustering (BRANGÄNE).11 Albeit methodologically complex, SYNCHEM established a foundation for a growing number of subsequent ML applications, including both retrosynthetic and forward reaction prediction. CASP via ML is generally complicated because of the lack of “negative” reaction examples, which are typically not reported. Consequently, this precludes supervised learning to distinguish between viable and nonproductive reactions. Accordingly, Coley et al. used data augmentation to supplement negative data samples and predict major reaction products from sets of reactants.12 The strategy was successfully applied to automatically extract reaction templates, requiring no further manual curation and revision of reaction specificity. This was most likely the first large-scale application of generalized reaction templates (rather than individual reactions) combined with harmonization of successful high-yielding reactions and sampled artificial negative representations. Moreover, Coley et al.(12) abandoned the traditional use of reactant and product fingerprints and instead focused on transformations at the reaction sites. The two-step model framework consisting of cross-validated forward enumeration and candidate prioritization resulted in a ranking of final products at the top-1, top-3, and top-5 positions in 72%, 87%, and 91% of the cases, respectively. We note that cross-validation is a widely adopted strategy to evaluate the performance of ML models and is not limited to computational synthesis planning. To evaluate their work in practice, the same group of authors developed an open-source CASP retrosynthetic software that was linked to an experimental robotic platform.13 Once again, generalized reaction templates were applied to suggest appropriate synthetic routes that were sequentially examined in silico, reviewed by expert chemists, and then executed by the robotic platform. The potential of this end-to-end AI setup was demonstrated for 15 drug-like compounds, probed in the order of increasing chemical complexity, setting a milestone for fully autonomous chemical synthesis,13 as schematically illustrated in Figure 1. According to the authors, the growing presence of reaction data should further advance the development AI-mediated robotic synthesis.
Figure 1.

Artificial intelligence in chemical synthesis. Shown is a blueprint for fully automated AI-driven compound generation and synthesis executed by a robotic platform.
Novel methodologies successfully applied in one field often become relevant in another where related yet scientifically different tasks are tackled. An exemplary success story involved the adaptation of DNN architectures originally used in natural language processing for sequence-to-sequence transformation to model retrosynthetic tasks.14 The authors built an encoder-decoder system composed of two recurrent neural networks (RNNs) and trained models on 50 000 atom-mapped U.S. patent reactions from 10 generally defined reaction classes. The work revealed several advantages over rule-based expert systems, including an end-to-end training procedure, better scaling to larger data sets, and global structural environments for reaction species.14 Although retrosynthetic algorithms largely operate on reaction templates, template-free approaches like that of Liu et al. are of interest for several reasons: first, the potential imbalance of specificity versus generality of reaction templates might lead to recommendations that are either of low synthetic quality or incomplete; second, handling subgraph isomorphism in template-based approaches can become computationally unfeasible; and third, template-free approaches have the potential to propose novel synthetic routes.
As discussed above, CASP algorithms mainly operate on known chemical compounds with well-defined synthetic routes that give relatively high yields. In de novo generative design, however, a large fraction of the suggested compounds typically suffers from limited synthesizability, even though currently available tools fairly reproduce external reference drugs. Therefore, synthetic feasibility assessment is highly desirable to support the generation of novel small-molecule therapeutics. To this end, Gao et al. applied a data-driven CASP approach to measure the frequency of de novo-generated compounds with questionable synthetic tractability.15 Their analysis revealed that a significant proportion of de novo-generated molecules contained unrealistic chemical/structural features, despite promising benchmark system performance. Distribution learning methods were frequently found to generate compounds with synthetic potential comparable to that of the corresponding training sets, whereas goal-directed generation methods displayed a higher risk of producing top-ranking compounds with limited or no synthetic feasibility. The authors evaluated several available synthetic accessibility scores, which often improved synthesizability at the cost of detracting from the main objective, i.e., structural novelty. As generative models continue to gather considerable attention in drug discovery, their further refinement was viewed alongside CASP algorithms through either post hoc filtering or implementation of novel accessibility scores.15
The availability of open-source CASP software is of paramount importance not only for quality and reproducibility assessment by the scientific community, but also for medicinal chemistry teams who do not have the capacity to develop a tool of their own. Moreover, intuitive and user-friendly interfaces would further increase their attractiveness and adoption in medicinal chemistry programs. Although a few such software systems currently exist, many others are not publicly available. Recently, Genheden et al. introduced AiZynthFinder, a retrosynthetic planning software that is publicly available for wider use.16 It is based on a Monte Carlo tree search algorithm that recursively decomposes compounds of interest into purchasable educts using a set of template-based reaction rules. The code is highly maintainable, robust, and well-documented for practitioners. Furthermore, the authors encourage users to contribute their ideas to incrementally improve model quality.
Evidently, synthesis prediction is an attractive area with potential to impact and transform medicinal chemistry. Gradual increases in validated learning strategies, reaction data, and user accessibility are some of desirable characteristics for future CASP tools.
Compound Design
Generative de novo design via DNNs aims at proposing structurally novel molecules with desired properties such as potency, suitable drug metabolism and pharmacokinetics (DMPK) profiles, or synthetic accessibility. Earlier computational de novo design employed different methodological frameworks such as incremental compound “growing” or rule-based synthetic schemes. Triggered by the use of DNN architectures, a large number of generative de novo design studies have appeared in recent years (both on preprint servers and in peer-reviewed journals), which are in part controversially viewed.8 However, some studies indicated new potential for medicinal chemistry from a methodological viewpoint (but largely remain to demonstrate impact on practical medicinal chemistry projects). In pioneering work, Segler et al. adopted an RNN architecture trained on large compound data sets to generate novel chemical structures.17 Similar to Liu et al.,14 the authors recognized the potential of RNNs for de novo compound design and hence treated the generation of SMILES representations of molecules as a machine translation problem. The results showed that RNNs based on long-term short memory (LSTM) units were capable of producing chemically reasonable structures. The models successfully captured training set distributions of physicochemical properties and generated novel candidate compounds for library design and virtual screening. Moreover, pretrained RNNs were fine-tuned using a limited sample of biologically active compounds for targets of interest to increase correct predictions of known bioactive molecules.17 While the majority of de novo design approaches construct molecules from scratch (i.e., atom by atom), Arús-Pous et al. followed an alternative approach to grow analogue series from scaffolds;18 which was conceptually reminiscent of earlier compound growing algorithms but methodologically distinct. Initially, a preprocessing algorithm was applied for data augmentation to systematically fragment all acyclic bonds and create a wealth of scaffold-substituent clusters. Then analogue series were generated from diverse chemical scaffolds using an LSTM-based RNN architecture, and synthetically accessible compounds were selected on the basis of retrosynthetic decomposition rules.18
Generative models are typically trained on SMILES notations to infer chemical syntax for compound generation. These simplified textual molecular representations are analogous to words/expressions in natural language processing, where DNNs have yielded considerable success. While one-dimensional representations such as SMILES strings are often assumed to be sufficient for de novo design applications, molecular graph representations are also considered as starting points. Furthermore, among medicinal chemistry practitioners, three-dimensional (3D) structure is predominantly perceived as an information-rich representation. Skalic et al. were among the first to utilize 3D information for generative de novo design.19 Starting with a seed molecule, its shape, and computed pharmacophoric features, the authors aimed at generating lead-like molecules with characteristic shape-based features. 3D structural representations perturbed via a variational autoencoder (VAE) were processed through a combination of convolutional neural network (CNN) and RNN architectures to ultimately extract SMILES token sequences. As expected, a main advantage of this approach is shape-specific compound generation with the potential to propose chemically novel scaffolds.19
Bridging between systems biology and molecular design, Méndez-Lucio et al. used a generative adversarial network (GAN), conditioned with transcriptomics data, to generate compounds with desirable transcriptomic profiles.20De novo-generated molecules shared greater chemical similarity to active compounds than those associated with similar gene expression signatures. Compared with typical de novo methods, no prior knowledge of active compounds, bioactivities, or target annotations was required, which minimized over-representation of chemical entities typically dominating training sets.20
Synthesis planning and de novo compound design, as discussed above, would benefit from well-documented publicly available custom code and computational tools. Such software would lay the groundwork for further development and improvement of compound generation methods and increase their attractiveness for medicinal chemistry applications. One such contribution was made by Blaschke et al., who introduced REINVENT, an intuitive and expandable open-source framework for de novo compound design.21 REINVENT aims to provide both exploitation (users choose a focus area to generate compounds with similar features) and exploration potential (generating compounds with limited structural similarity yet desired features). It is based on a deep reinforcement learning (RL) architecture coupled with a multiparameter optimization scoring scheme and chemical diversity filters.21
Integrating compound design with fully automated synthesis capacity is a particularly attractive task for medicinal chemistry, as exemplified by the work of Coley et al.(13) mentioned above. Recently, Grisoni et al. combined generative design with a microfluidics platform for on-chip chemical synthesis.22 DNN model fine-tuning toward liver X receptor alpha (LXRα) agonists yielded novel bioactive candidates for single-step reactions via the microfluidics-assisted synthesis platform. Twenty-five compounds were successfully synthesized, 17 of which were active, including 12 potent LXRα agonists.22
Uncertainty Estimation and Active Learning
Uncertainty estimation for ML predictions is another important area of research that has the potential to significantly impact ML in medicinal chemistry.23 While model applicability domains have been investigated for years, mostly on the basis of similarity of test instances to training sets (Figure 2),24 state-of-the-art ML models can yield accurate property predictions for test compounds with limited training set resemblance. However, comparably little efforts have been made to quantify the uncertainly associated with given predictions. Different frameworks for uncertainty estimation have been reported, such as frequentist analysis or Bayesian approaches. Modeling on the basis of Bayes’ theorem derives posterior distribution estimates for prediction outcomes, which can be translated into a measure of uncertainty associated with predictions. Despite these efforts, robust and generally applicable approaches for uncertainty quantification are currently not available. In recent years, one of the most popular methodologies has been conformal prediction.25 This framework enables the generation of prediction intervals for test compounds. Depending on the desired confidence level (and nonconformity score), the width of the prediction interval varies.26 Extensions of conformal prediction have also been proposed. However, this methodology also has intrinsic limitations, and the underlying data randomness and exchangeability assumptions are not always met,27,28 hence limiting its general applicability. Ensemble modeling (Figure 2) is another frequentist strategy for probabilistic estimation that relies on the majority or weighted voting of participating predictors. Ensemble models can be generated by the same algorithm, e.g., executed with different random initializations or resampling, but can also integrate distinct ML methods and molecular representations. For example, Cortés-Ciriano and Bender introduced DNN ensemble predictions in which network weights were saved as snapshots at local minima during the optimization phase.27 The authors integrated the conformal prediction framework and obtained results similar to those produced by independently trained DNNs. Of note, Bayesian NNs yield the complete posterior probability distribution for predictions but quickly become impractical for larger data sets.29 In recent work, Hirschfeld et al. have benchmarked different uncertainty quantification approaches for regression modeling.30 None of the evaluated methods succeeded in accurately ranking predictions by absolute error, and the relative performance of the approaches heavily depended on the applied metrics. Nonetheless, the authors recommended a combination of message-passing neural networks (MPNNs) and random forest (RF) as an appropriate approach. Accordingly, an MPNN is trained, and the latent representation is used as input for an RF model to obtain a probabilistic prediction (Figure 2). Other approaches that consistently performed well in different comparisons included the combination of MPNN with Gaussian process modeling as well as a mean-variance-estimation MPNN, which modifies the MPNN output layer to predict both the mean and the variance of a given property (Figure 2). However, a standard RF model trained using molecular fingerprints yielded a strong (baseline) reference.30 In light of these findings, Soleimany et al. proposed a new uncertainty quantification method for property prediction based on evidential DL.31 Following this approach, learning is considered as an evidence acquisition process in which newly selected training instances further support the learned distribution. The loss function and output layer of any NN architecture can be modified to incorporate this uncertainty estimation. The method achieved stronger correlation between estimated uncertainties and model errors than ensemble approaches or dropout sampling methods.31
Figure 2.

Uncertainty estimation methods. Illustrated are approaches for estimating the uncertainty of ML predictions including similarity-based assessment, ensemble models, combined (union-based) models, and mean-variance estimation, as discussed in the text.
The concept of active learning is closely related to uncertainty quantification.32 Active learning refers to the iterative process of retraining a model with increasing numbers of labeled instances, aiming to minimize learning sets by concentrating on the most informative data points (Figure 3). To improve model performance and generalization, the selection strategy typically focuses on uncertainty estimates.32 In addition to uncertainty estimation, greedy selection and other approaches are also applicable to guide active learning. If uncertainty is estimated, compounds predicted with high uncertainty (corresponding to high information entropy) are generally selected for addition to the training set (which is called exploration learning), and selection frequently relies on positive predictions (model exploitation).33 For instance, active learning was applied to predict the outcome of new reactions. In a retrospective analysis, ML models were trained for reaction screening, and the selection of experiments with high information content led to superior models compared with random selection.34 Another recent application of active learning was reported in the context of protein–ligand docking. A DNN was trained to predict docking scores on the basis of chemical structure and applied to a library of 1.36 billion compounds. Predictions were used to update virtual hits and select candidates for docking, which were included in the training set for the next iteration. The approach required 50-times fewer molecules than standard docking for comparably high hit recall.35 Notably, Hie et al. applied an active learning approach based on Gaussian process models in prospective applications leading to the identification of compounds with nanomolar kinase activity as well as inhibitors of Mycobacterium tuberculosis.36 Active learning is of particular interest for medicinal chemistry, as many available data sets are relatively small, and this approach provides a viable complement or alternative to DL. Notably, active learning and DL are not mutually exclusive, as exemplified by the use of multitask and transfer learning strategies that are pursued using DNNs and also benefit from most informative training instances establishing correlations between related yet distinct prediction tasks.
Figure 3.
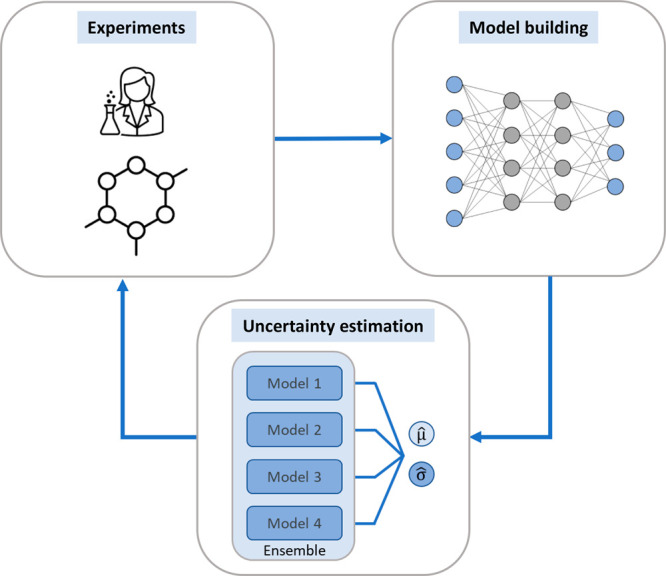
Active learning scheme. Shown is an exemplary iterative active learning cycle. Experimental results are used to train ML models, and uncertainty estimation enables the selection of informative outputs, which are reconfirmed and included in training sets.
Conclusions
DL and other AI approaches are increasingly being considered across different chemical disciplines, and their popularity is anticipated to further increase. Given the traditionally strong orientation of cheminformatics toward drug discovery, it is not surprising that medicinal chemistry is one of the focal points of DL/AI in chemistry. While most of the current efforts concentrate on the development and calibration of new computational methods, prospective applications demonstrating their impact are still rare. Given strong claims that are occasionally placed concerning the potential of AI to “revolutionize” drug discovery (which no single approach or technology has ever accomplished), it is of utmost importance for the further development and acceptance of new computational concepts to balance the hype associated with AI in this and other fields. In medicinal chemistry, DL using a variety of DNN architectures adapted from other fields and robotics are the prevalent AI approaches. Herein we have attempted to highlight selected studies that have made a demonstrated impact on the field, mostly at the methodological level, given the current sparsity of prospective applications reported in a scientifically rigorous manner. While progress in compound property predictions has been limited, despite heavy use of DNN architectures, for reasons discussed herein, DL has opened the door to exploring tasks in areas such as chemical synthesis or compound design that would be difficult if not impossible to address using shallow ML approaches. Here a methodological framework is being generated with promise to put medicinal chemistry on a new level, provided that it is possible to translate methodological advances into demonstrated success in practical applications, which might eventually circumvent roadblocks on the paths to new small-molecule drugs. Clearly, it is too early to judge, but there is potential in some areas to make a difference if new computational concepts are not oversold, but evaluated in a scientifically rigorous manner.
Biographies
Filip Miljković obtained a Master’s degree in pharmacy from the Medical Faculty, University of Niš, Serbia, in 2014. In 2016 he began doctoral studies in Computational Life Sciences in the Department of Life Science Informatics at the University of Bonn in Germany under the supervision of Prof. Dr. Jürgen Bajorath. He obtained his Ph.D. degree in 2019 and then joined AstraZeneca R&D as a Senior Data Scientist in the Department of Clinical Pharmacology & Safety Sciences in Gothenburg, Sweden. His research interests include cheminformatics and computational methodologies for medicinal chemistry and chemical biology, including machine learning and structure-based approaches. For further details, visit: https://filipm90.github.io/.
Raquel Rodríguez-Pérez studied biomedical engineering at the University of Barcelona and did her Ph.D. work in Computational Life Sciences in Prof. Bajorath’s group at the University of Bonn. She was a Marie Curie Fellow and early-stage researcher at Boehringer Ingelheim in Germany. Currently, she is a Principal Scientist at Novartis Institutes for Biomedical Research and works in the Modeling and Simulation Data Science team in the Translational Medicine Department. Her research interests include bio/cheminformatics, machine learning, and data science for biomedical applications.
Jürgen Bajorath is Professor and Chair of Life Science Informatics and Data Science at the University of Bonn. He is also an Affiliate Professor at the University of Washington in Seattle, WA. His research interests include cheminformatics, machine learning, data science, computational medicinal chemistry, and chemical biology. For further information, see: http://bajorath.bit.uni-bonn.de.
Author Contributions
∥ F.M. and R.R.-P. contributed equally to this work.
The authors declare no competing financial interest.
References
- Pannu A. Artificial Intelligence and its Application in Different Areas. Int. J. Eng. Innovative Technol. 2015, 4, 79–84. [Google Scholar]
- Varnek A.; Baskin I. Machine Learning Methods for Property Prediction in Chemoinformatics: Quo vadis?. J. Chem. Inf. Model. 2012, 52, 1413–1437. 10.1021/ci200409x. [DOI] [PubMed] [Google Scholar]
- Bajorath J. State-of-the-art of Artificial Intelligence in Medicinal Chemistry. Future Sci. OA 2021, 7, FSO702. 10.2144/fsoa-2021-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Wang Z.; Liu X.; Zeng N.; Liu Y.; Alsaadi F. E. A Survey of Deep Neural Network Architectures and Their Applications. Neurocomputing 2017, 234, 11–26. 10.1016/j.neucom.2016.12.038. [DOI] [Google Scholar]
- Yang K.; Swanson K.; Jin W.; Coley C.; Eiden P.; Gao H.; Guzman-Perez A.; Hopper T.; Kelley B.; Mathea M.; Palmer A.; Settels V.; Jaakkola T.; Jensen K.; Barzilay R. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. 10.1021/acs.jcim.9b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearnes S. Pursuing a Prospective Perspective. Trends Chem. 2021, 3, 77–79. 10.1016/j.trechm.2020.10.012. [DOI] [Google Scholar]
- Bajorath J.; Kearnes S.; Walters W. P.; Meanwell N. A.; Georg G. I.; Wang S. Artificial Intelligence in Drug Discovery: Into the Great Wide Open. J. Med. Chem. 2020, 63, 8651–8652. 10.1021/acs.jmedchem.0c01077. [DOI] [PubMed] [Google Scholar]
- Walters W. P.; Murcko M. Assessing the Impact of Generative AI on Medicinal Chemistry. Nat. Biotechnol. 2020, 38, 143–145. 10.1038/s41587-020-0418-2. [DOI] [PubMed] [Google Scholar]
- Stokes J. M.; Yang K.; Swanson K.; Jin W.; Cubillos-Ruiz A.; Donghia N. M.; MacNair C. R.; French S.; Carfrae L. A.; Bloom-Ackermann Z.; Tran V. M.; Chiappino-Pepe A.; Badran A. H.; Andrews I. W.; Chory E. J.; Church G. M.; Brown E. D.; Jaakkola T. S.; Barzilay R.; Collins J. J. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702. 10.1016/j.cell.2020.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemonick S.AI Finds Molecules that Kill Bacteria, but Would They Make Good Antibiotics? Chemistry & Engineering News, February 26, 2020. https://cen.acs.org/physical-chemistry/computational-chemistry/AI-finds-molecules-kill-bacteria/98/web/2020/02 (accessed 2021-10-04).
- Gelernter H.; Rose J. R.; Chen C. Building and Refining a Knowledge Base for Synthetic Organic Chemistry via The Methodology of Inductive and Deductive Machine Learning. J. Chem. Inf. Comput. Sci. 1990, 30, 492–504. 10.1021/ci00068a023. [DOI] [Google Scholar]
- Coley C. W.; Barzilay R.; Jaakkola T. S.; Green W. H.; Jensen K. F. Prediction of organic reaction outcomes using machine learning. ACS Cent. Sci. 2017, 3, 434–443. 10.1021/acscentsci.7b00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley C. W.; Thomas D. A.; Lummiss J. A. M.; Jaworski J. N.; Breen C. P.; Schultz V.; Hart T.; Fishman J. S.; Rogers L.; Gao H.; Hicklin R. W.; Plehiers P. P.; Byington J.; Piotti J. S.; Green W. H.; Hart A. J.; Jamison T. F.; Jensen K. F. A Robotic Platform for Flow Synthesis of Organic Compounds Informed by AI Planning. Science 2019, 365, eaax1566. 10.1126/science.aax1566. [DOI] [PubMed] [Google Scholar]
- Liu B.; Ramsundar B.; Kawthekar P.; Shi J.; Gomes J.; Nguyen Q. L.; Ho S.; Sloane J.; Wender P.; Pande V. Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models. ACS Cent. Sci. 2017, 3, 1103–1113. 10.1021/acscentsci.7b00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W.; Coley C. W. The Synthesizability of Molecules Proposed by Generative Models. J. Chem. Inf. Model. 2020, 60, 5714–5723. 10.1021/acs.jcim.0c00174. [DOI] [PubMed] [Google Scholar]
- Genheden S.; Thakkar A.; Chadimová V.; Reymond J.; Engkvist O.; Bjerrum E. AiZynthFinder: A Fast, Robust and Flexible Open-Source Software for Retrosynthetic Planning. J. Cheminf. 2020, 12, 70. 10.1186/s13321-020-00472-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segler M. H. S.; Kogej T.; Tyrchan C.; Waller M. P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 2018, 4, 120–131. 10.1021/acscentsci.7b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arús-Pous J.; Patronov A.; Bjerrum E. J.; Tyrchan C.; Reymond J.-L.; Chen H.; Engkvist O. SMILES-based Deep Generative Scaffold Decorator for De-Novo Drug Design. J. Cheminf. 2020, 12, 38. 10.1186/s13321-020-00441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalic M.; Jiménez J.; Sabbadin D.; De Fabritiis G. Shape-based Generative Modeling for De Novo Drug Design. J. Chem. Inf. Model. 2019, 59, 1205–1214. 10.1021/acs.jcim.8b00706. [DOI] [PubMed] [Google Scholar]
- Méndez-Lucio O.; Baillif B.; Clevert D.-A.; Rouquié D.; Wichard J. De Novo Generation of Hit-like Molecules from Gene Expression Signatures Using Artificial Intelligence. Nat. Commun. 2020, 11, 10. 10.1038/s41467-019-13807-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschke T.; Arús-Pous J.; Chen H.; Margreitter C.; Tyrchan C.; Engkvist O.; Papadopoulos K.; Patronov A. REINVENT 2.0: An AI Tool for De Novo Drug Design. J. Chem. Inf. Model. 2020, 60, 5918–5922. 10.1021/acs.jcim.0c00915. [DOI] [PubMed] [Google Scholar]
- Grisoni F.; Huisman B. J. H.; Button A. L.; Moret M.; Atz K.; Merk D.; Schneider G. Combining Generative Artificial Intelligence and On-chip Synthesis for De Novo Drug Design. Sci. Adv. 2021, 7, eabg3338. 10.1126/sciadv.abg3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mervin L. H.; Johansson S.; Semenova E.; Giblin K. A.; Engkvist O. Uncertainty Quantification in Drug Design. Drug Discovery Today 2021, 26, 474–489. 10.1016/j.drudis.2020.11.027. [DOI] [PubMed] [Google Scholar]
- Sheridan R. P.; Feuston B. P.; Maiorov V. N.; Kearsley S. K. Similarity to Molecules in the Training Set Is a Good Discriminator for Prediction Accuracy in QSAR. J. Chem. Inf. Model. 2004, 44, 1912–1928. 10.1021/ci049782w. [DOI] [PubMed] [Google Scholar]
- Shafer G.; Vovk V. A Tutorial on Conformal Prediction. J. Mach Learn Res. 2008, 9, 371–421. [Google Scholar]
- Alvarsson J.; Arvidsson McShane S.; Norinder U.; Spjuth O. Predicting with Confidence: Using Conformal Prediction in Drug Discovery. J. Pharm. Sci. 2021, 110, 42–49. 10.1016/j.xphs.2020.09.055. [DOI] [PubMed] [Google Scholar]
- Cortés-Ciriano I.; Bender A. Deep Confidence: A Computationally Efficient Framework for Calculating Reliable Prediction Errors for Deep Neural Networks Using Test-Time Dropout. J. Chem. Inf. Model. 2019, 59, 1269–1281. 10.1021/acs.jcim.8b00542. [DOI] [PubMed] [Google Scholar]
- Krstajic D. Critical Assessment of Conformal Prediction Methods Applied in Binary Classification Settings. J. Chem. Inf. Model. 2021, 61, 4823–4826. 10.1021/acs.jcim.1c00549. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Lee A. A. Bayesian Semi-Supervised Learning for Uncertainty-Calibrated Prediction of Molecular Properties and Active Learning. Chem. 2019, 10, 8154–8163. 10.1039/C9SC00616H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschfeld L.; Swanson K.; Yang K.; Barzilay R.; Coley C. W. Uncertainty Quantification using Neural Networks for Molecular Property Prediction. J. Chem. Inf. Model. 2020, 60, 3770–3780. 10.1021/acs.jcim.0c00502. [DOI] [PubMed] [Google Scholar]
- Soleimany A. P.; Amini A.; Goldman S.; Rus D.; Bhatia S. N.; Coley C. W. Evidential Deep Learning for Guided Molecular Property Prediction and Discovery. ACS Cent. Sci. 2021, 7, 1356–1367. 10.1021/acscentsci.1c00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn D.; Atlas L.; Ladner R. Improving generalization with active learning. Mach. Learn. 1994, 15, 201–221. 10.1007/BF00993277. [DOI] [Google Scholar]
- Reker D. Practical Considerations for Active Machine Learning in Drug Discovery. Drug Discovery Today: Technol. 2019, 32–33, 73–79. 10.1016/j.ddtec.2020.06.001. [DOI] [PubMed] [Google Scholar]
- Eyke N. S.; Green W. H.; Jensen K. F. Iterative Experimental Design Based on Active Machine Learning Reduces the Experimental Burden Associated with Reaction Screening. React. Chem. Eng. 2020, 5, 1963–1972. 10.1039/D0RE00232A. [DOI] [Google Scholar]
- Gentile F.; Agrawal V.; Hsing M.; Ton A. T.; Ban F.; Norinder U.; Gleave M. E.; Cherkasov A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 2020, 6, 939–949. 10.1021/acscentsci.0c00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hie B.; Bryson B. D.; Berger B. Leveraging Uncertainty in Machine Learning Accelerates Biological Discovery and Design. Cell Systems 2020, 11, 461–477. 10.1016/j.cels.2020.09.007. [DOI] [PubMed] [Google Scholar]