

Abstract
The de novo drug design based on SMILES format is a typical sequence-processing problem. Previous methods based on recurrent neural network (RNN) exhibit limitation in capturing long-range dependency, resulting in a high invalid percentage in generated molecules. Recent studies have shown the potential of Transformer architecture to increase the capacity of handling sequence data. In this work, the encoder module in the Transformer is used to build a generative model. First, we train a Transformer-encoder-based generative model to learn the grammatical rules of known drug molecules and a predictive model to predict the activity of the molecules. Subsequently, transfer learning and reinforcement learning were used to fine-tune and optimize the generative model, respectively, to design new molecules with desirable activity. Compared with previous RNN-based methods, our method has improved the percentage of generating chemically valid molecules (from 95.6 to 98.2%), the structural diversity of the generated molecules, and the feasibility of molecular synthesis. The pipeline is validated by designing inhibitors against the human BRAF protein. Molecular docking and binding mode analysis showed that our method can generate small molecules with higher activity than those carrying ligands in the crystal structure and have similar interaction sites with these ligands, which can provide new ideas and suggestions for pharmaceutical chemists.
1. Introduction
Nowadays, with the dramatic improvement of the computing power of computers and the rapid progress of artificial intelligence and machine learning algorithms, CADD is also developing rapidly due to the integration of new methods.1 Deep-learning-based CADD methods have been widely used in many fields such as performing protein–ligand activity classification, affinity prediction, ADMET prediction, synthesis route design, and protein folding prediction.2−6 Compared with the existing computational methods, this kind of model showed superior performance in terms of calculation speed and precision. In recent years, several deep generative models have been proposed to explore the vast space of drug-like chemistry by encoding molecules into a continuous latent space.7 Segler et al.,8 Popova et al.,9 and Olivecrona et al.10 trained an end-to-end recurrent neural network (RNN) generative model using only SMILES as the input. To improve the efficiency of the model to generate valid molecules, memory-augmented RNN was introduced.9 Similar generative models include variational autoencoders (VAEs)11 and generative adversarial networks (GANs),12 which learn underlying data distribution in an unsupervised setting.
With the excellent performance of reinforcement learning (RL) in solving dynamic decision problems, Popova et al.9 and Olivecrona et al.10 applied it to the problem of designing chemical libraries with required properties.
RL is used to combine the generative model with the predictive model to guide the generator to generate novel chemical entities with desired properties. Similar to it is the particle swarm optimization (PSO) algorithm.13,14 RL guides the generative model through the reward mechanism to find the optimal search space, while the PSO algorithm aims to find the optimal point from the whole chemical space under the guidance of Objective function. Transfer learning (TL) is used to design drugs for specific targets. Several research groups have demonstrated that RNNs can be fine-tuned by TL to generate molecules that are structurally similar to drugs with known activities against particular targets.15,16 However, these generative models are based on RNN, which exhibits limitation in capturing long-range dependency,17 whereas molecules expressed in the SMILES format have longer sequences. The increasingly long sequences significantly affect these models’ prediction capacity.
The more recent Transformer model18 achieved impressive performance in a number of tasks by getting rid of recurrence and introducing a multihead self-attention mechanism, which enabled processing very long sequences. Grechishnikova et al. used a complete Transformer architecture to generate lead compounds for specific proteins with only sequence information, treating molecular generation as a translation task for protein sequences and molecular SMILES.19 Zheng et al. proposed a scaffold hopping model using the Transformer architecture.20 The goal of this model is to predict the “hopped” molecule with improved pharmaceutical activity and a dissimilar two-dimensional (2D) structure but a similar three-dimensional (3D) structure by inputting a reference molecule and a specified protein. All of these works used the complete encoder–decoder architecture of the Transformer.
In this work, we propose a novel ligand-based de novo molecular design approach for targeting specific proteins. First, we trained a Transformer-encoder-based generator on ChEMBL’s 1.6 million data sets to learn the grammatical rules of known drug molecules. Second, TL is used to introduce the prior knowledge of drugs with known activities against particular targets into the generative model to construct new molecules similar to the known ligands. This is then followed by an RL module to combine the generative model and the predictive model to optimize the parameters of the generator to generate small molecules with high scores, that is, molecules with drug-like properties that are expected to bind well with the target. Finally, molecular docking was used to further evaluate the activity of the generated molecules.
The evaluation results showed that the generative model based on the Transformer was superior to the generative model based on RNN (GRU, LSTM) in terms of the stability of the training process, the accuracy of generating valid molecules, the performance of RL, and the feasibility of molecular synthesis. In addition, the samples generated by the generative model almost covered the entire chemical space occupied by BRAF inhibitors, which proves that the Transformer-based de novo drug design method is successful under “low data” conditions and our generative model has the potential to generate BRAF inhibitors with novel structures and higher activity. Finally, molecular docking and binding mode analysis proved that our model could generate quality candidate compounds for BRAF.
2. Results and Discussion
2.1. Performance Evaluation of Generative and Predictive Models
Our Transformer-based generative model was trained with ∼1.6 million structures from the ChEMBL database to learn the rules that define SMILES strings corresponding to chemical structures and design new chemical libraries (please see Materials and Methods for technical details). To compare the performance of the Transformer-based generative model with the previous RNN-based one, we trained a stack-augmented gated recurrent unit (GRU) model for molecular generation on the same data set, which is widely used in de novo drug design.9,10,16 We tested the effect of different layers of GRU on generating valid molecular percentages. Unlike Transformer structures, the expressive power of the GRU cannot be increased by deepening the network (Table S1). Therefore, only one layer of GRU was used for molecular generation in our experiment. It can be seen from the training process that our generative model is more stable and has higher accuracy on the validation set (Figure 1). In addition, it was found in the experiment that the bidirectional GRU did not improve the performance of the generative model (Figure 1).
Figure 1.

Training process of three generative models: Transformer-based, bidirectional GRU-based, and GRU-based models. (A) and (B) are the changes of loss on the training set and verification set during the training process, respectively, and (C) and (D) are the accuracy change process on the training set and the validation set during the training process, respectively.
A known drawback of approaches for de novo design is frequent generation of chemical infeasible molecules.7,10 To address this problem, a total of 100 000 compounds were sampled from the pretrained generative model for 10 times, 10 000 compounds at a time. (Among them, the samples of GRU were sampled at the position of iteration = 300, and the samples of the Transformer were sampled at the position of iteration = 500.) As can be seen from the statistical results in Table 1, our generative model reduced the percentage of invalid molecules (which cannot be parsed by the RDKit library) from 4.368 to 1.694%. In addition, we count the lengths of retained and invalid molecules. The average value of the two in the Transformer-based generative model is 46/55, while in the GRU-based generative model it is 44/62, indicating that the RNN model is weaker than the Transformer model in capturing the long-range dependency of small molecules.
Table 1. Sample Statistical Results of Transformer-Based and GRU-Based Generators.
| pretrained model |
optimized model (aver reward = 15) |
|||
|---|---|---|---|---|
| transformer | GRU | transformer | GRU | |
| invalid | 1.694% ± 0.102% | 4.368% ± 0.361% | 1.170% ± 0.052% | 2.690% ± 0.167% |
| redundant | 0.075% ± 0.022% | 0.038% ± 0.015% | 30.273% ± 0.525% | 33.828% ± 0.616% |
| retained | 98.231% ± 0.115% | 95.594% ± 0.351% | 68.557% ± 0.552% | 63.482% ± 0.456% |
In addition to the efficiency of producing valid molecules, another key for de novo-generated molecules is the feasibility of their synthesis. We calculated the synthetic accessibility score (SAS) from the two generator samples, which were in the range of 1–10. The mean SAS of molecules generated by the Transformer was 2.78 and that of GRU was 2.89, compared with 2.84 from the ChEMBL training set. Therefore, our generative model is superior to GRU-based generative model in terms of the accuracy of generating valid molecules and the feasibility of their synthesis.
To introduce the existing structure experience of the specific targets into the generative model, the Transformer-based model and the GRU-based model were, respectively, fine-tuned by TL. A total of 4702 inhibitors (pIC50 ≥ 4) of BRAF and its similar proteins (UniProt ID: P15056, P10398, P04049) were used as a training set of TL. TL was performed for 100 epochs until the distribution of the Tanimoto similarity (TS) between the generated molecules and the training data set showed no further improvement. It can be seen from Figure 2A,B that the TS between the molecules generated after TL and the training set increased, which indicated that the generative model captures the structural features of the existing inhibitors. In addition, the generative model based on GRU generated more structures that were more similar to the training set after TL (44% of the molecules have TS greater than 0.5), while our method enables a bigger structural leap from the existing inhibitors (47% of molecules have TS between 0.3 and 0.5). Therefore, although the similarity between the molecules generated by our generative model after TL and the training set increases, there is no limit to the ability of the generator to produce novel scaffolds.
Figure 2.

Performance of the generative model and the predictive model. (A) Distribution of the maximum Tanimoto similarity between the molecules generated by the model and the BRAF inhibitors. (B) Example of the Tanimoto similarity between the generated molecule and the original structure. (C) Comparison between predicted and observed pIC50 values. Metrics reported are Pearson’s correlation coefficient (R) and root-mean-squared error (RMSE).
Predictive models are key to generating molecules with specified desired properties for the de novo drug design. Any QSAR model is generally designed to establish a conversion between the molecular descriptor of a compound and its biological activity.9 In this work, we built a regression model to predict pIC50 values using only molecular SMILES as the input (please see Materials and Methods for technical details). Using 5-fold cross-validation on BRAF and its similar protein inhibitors, we obtained the model accuracy expressed as Pearson’s correlation coefficient (R) = 0.82 and root-mean-squared error (RMSE) = 0.76 for predicting pIC50, indicating a strong positive correlation between the predicted and observed values (Figure 2C). In addition, to confirm the predictive ability of the model, we performed verification on the PDBbind data set. As a ligand-based prediction model, we obtained the model accuracy expressed as Pearson correlation coefficient (R) = 0.68 and RMSE = 1.46 for predicting pIC50 on the PDBbind data set (Table S2). Therefore, our predictive model can establish a good correlation between the molecular structure and the biological activity.
Subsequently, RL is used to guide the generative model (after TL) to generate novel compounds with desired properties (please see Materials and Methods for technical details). The Transformer-based and GRU-based generative models were optimized by the same RL architecture, respectively. To evaluate the generation efficiency of the optimized generative model, a total of 100 000 compounds were sampled from the optimized generative model for 10 times. It can be seen from Table 1 that the optimized model has a higher redundancy percentage, which may be due to the fact that RL has reduced the sampling space of the model to obtain higher rewards. It is worth noting that as the reward increases, the redundancy percentage increases, resulting in a decrease in the proportion of valid and unique molecules (Table S3). Therefore, we stopped optimizing at reward = 15.
Figure 3 shows the optimization results of the molecules generated by the two generative models. It can be seen from Figure 3A that when the two models achieved similar activity distributions, the molecules generated by the Transformer were less similar to the existing BRAF inhibitors (Figure 3B). Therefore, the molecular structures generated by the Transformer were more novel. In addition, the molecules generated by the Transformer performed better in drug-likeness distribution and synthesizable distribution (Figure 3C,D). As a result, our Transformer-based generative model can improve the problems faced by RNN-based generative models and perform better in the property distribution and structural diversity of the generated molecules.
Figure 3.

Property distribution of transformer-based and GRU-based generative models. (A) Distribution of the predicted pIC50 value before and after reinforcement learning. (B) Distribution of the maximum Tanimoto similarity between the molecules generated by the two models and BRAF inhibitors. (C) Distribution of the Quantitative estimate of drug-likeness scores and (D) distribution of the synthetic accessibility scores in the two generative models and existing BRAF inhibitors.
2.2. Discovery of New Candidate Compounds Targeting BRAF
After RL, 50 000 molecules were sampled from the Transformer-based generative model to find candidate compounds targeting BRAF, of which 63.4% of the small molecules were unique and valid (invalid: 1.14%, redundant: 33.21%, training set-identical: 2.17%). Finally, 63.4% (31 714) of the small molecules were retained for further analysis. The Murcko scaffold21 was extracted from the training data set and the retained set of generated molecules to check whether a new structure was generated. The total number of unique Murcko scaffolds were found to be 1404 in existing BRAF inhibitors and 15 058 in generated molecules. A TS cutoff of 0.5 was used to define the dissimilarity between a pair of scaffolds.22 After all pair-wise scaffold similarities were computed, 10 595 scaffolds (70.4%) from the generated molecules were found to be dissimilar to the scaffolds in the training set. Extended Connectivity FingerPrint (ECFP4) was used as the descriptor for embedding, and each small molecule was projected onto a two-dimensional plane using t-SNE. As can be seen from Figure 4, the samples almost covered the entire chemical space occupied by BRAF inhibitors, which indicated that the generative model after RL captures the structural features of the existing inhibitors and has the potential to generate BRAF inhibitors with structural diversity.
Figure 4.
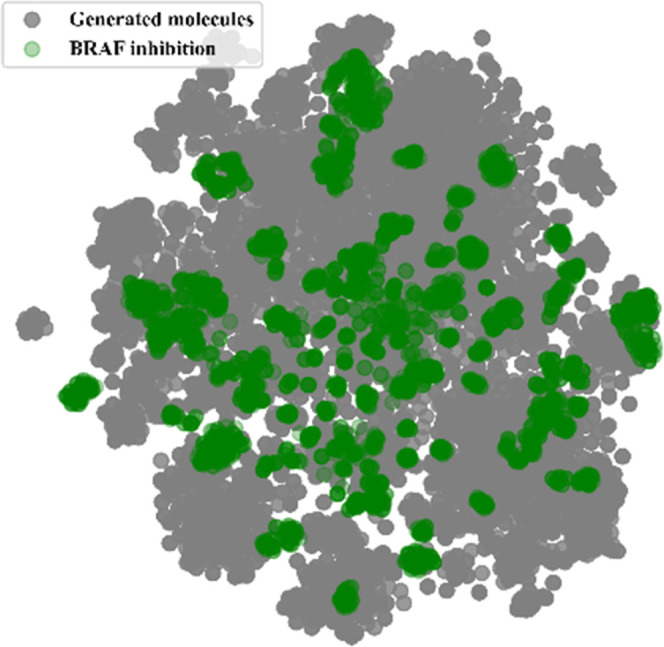
Clustering of the molecules generated by the generative model after reinforcement learning and BRAF inhibitors by t-SNE.
At present, the reported BRAF protein has two inhibitor binding sites, as shown in Figure S1. The first binding site is competitive occupation of adenosine triphosphate (ATP) binding sites and these inhibitors are called kinase I inhibitors (Figure S1A); PLX-4032 is a typical kinase I inhibitor. Another is the allosteric binding site, and these inhibitors are called kinase II inhibitors (Figure S1B); belvarafenib belongs to kinase II inhibitors. Due to the binding of allosteric sites, kinase I inhibitors have better selectivity than kinase II inhibitors. Therefore, the reserved 31 714 molecules were virtually screened for the two crystal structures (6XFP, 3OG7) of BRAF, respectively, and conformations with a score less than −8 kcal/mol were retained for study, among which 3533 compounds were reserved for 6XFP and 3519 for 3OG7. Figure 5 shows the representative compounds with different scaffolds generated by the generative model that had a higher docking score (DS) than the ligands from BRAF crystal structures. Some of these molecules differ greatly in structure from existing inhibitors (lower TS) but have high docking scores, which has the potential to become potent BRAF inhibitors with novel structures. More structures with their docking scores are provided in Supporting Information 2.
Figure 5.

Clustering of generated molecules filtered by virtual screening. Molecules are colored by the absolute value of the docking score (DS). Each molecule is expressed by its DS and the maximum Tanimoto similarity (TS) between the training set. Some molecules have a lower TS but a higher DS. (A) and (B) represent BRAF’s two pockets, 6XFP and 3OG7, respectively.
2.3. Comparing the Binding Mode of Representative Compounds with Belvarafenib and PLX-4032
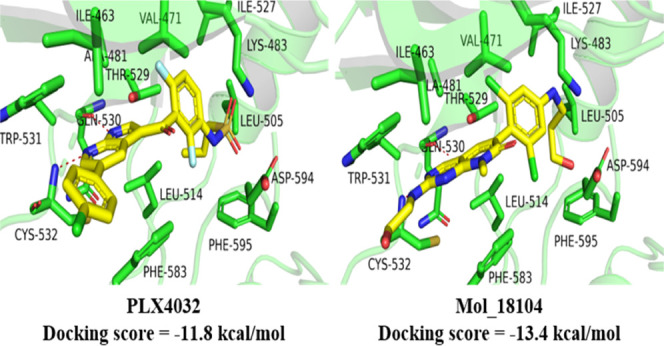
First, the ligand PLX-4032 in 3OG7 and belvarafenib in 6XFP were re-docked in their binding pockets to verify the accuracy of Glide, and the docking scores are −11.8 kcal/mol and −14.3 kcal/mol for PLX-4032/BRAF and belvarafenib/BRAF complexes, respectively. Then, we selected five compounds with different frames for the two BRAF binding pockets and compared the binding modes, as shown in Figures 6 and 7. Figure 6 shows the binding mode of BRAF to the representative compounds with different frames with higher scores than that of PLX-4032, and these compounds also have low similarity (compared with already reported BRAF inhibitors), which illustrates that these compounds have better affinity with BRAF than that with PLX-4032. The important interaction of BRAF and inhibitors is to form hydrogen bonds with the residues in the hinge region of BRAF (GLN530, TRP531 and/or CYS532) and hydrophobic interaction at the ATP binding sites. Obviously, the five compounds form hydrogen bonds and hydrophobic interactions with the residues GLN530, TRP531, and/or CYS532. However, when these compounds bind to ATP binding sites, some groups are exposed to solvents, and these structures will have greater flexibility, such as Mol_00147 and Mol_09590. These two compounds may need to be structurally modified in the future.
Figure 6.

Comparing the bound mode of PLX-4032 and the representative compounds. (A) is the carrying ligand in the crystal structure of 3OG7, while (B), (C), (D), (E), and (F) are representative compounds with high docking scores and different scaffolds.
Figure 7.

Comparing the bound mode of belvarafenib and the representative compounds. (A) is the carrying ligand in the crystal structure of 6XFP, while (B), (C), (D), (E), and (F) are representative compounds with high docking scores and different scaffolds.
The compounds Mol_00605, Mol_22896, Mol_06416, Mol_12256, and Mol_19439 have similar interaction with BRAF at the ATP binding site and with belvarafenib, including the hydrogen bond with the residues GLN530, TRP531, and/or CYS532 and hydrophobic interaction (Figure 7). In addition to these key interactions, the five compounds also have hydrophobic interaction with BRAF at the allosteric binding site. The residues at the allosteric binding site mainly include GLU501, ILE527, ASP594, PHE595, and LEU597, and the interaction of the compounds with these residues improves the selectivity of the compounds to BRAF. In conclusion, the compounds Mol_00147, Mol_05056, Mol_18104, Mol_18796, Mol_09590, Mol_00605, Mol_22896, Mol_06416, Mol_12256, and Mol_19439 exhibited a higher inhibitory activity to BRAF than that of PLX-4032 and belvarafenib.
3. Conclusions
We have proposed a Transformer-encoder-based generative model for a de novo drug design, and sample evaluation of the original pretrained model and the RL-optimized generative model shows that our method is superior to the existing generative models in terms of the percentage of valid compounds generated and the property/activity distribution of generated molecules. It has been observed that our target-specific generative model can capture the features of existing active ligands and has a huge chemical sampling space (the generated molecules almost covered the entire chemical space occupied by BRAF inhibitors). Molecular docking and binding pattern analysis demonstrate that our method can generate small molecules with higher activity than those carrying ligands in the crystal structure and have similar interaction sites with these ligands, which improves the selectivity of these molecules to BRAF. When the three-dimensional structure of the protein and a small number of active ligands are known, the proposed method can be applied to any target protein for the de novo drug design. In addition, the method can also be used for virtual screening of molecular library generation and fragment-based molecular design.
4. Materials and Methods
4.1. Transformer-Based Generator
As with the text generation task, the first step in de novo drug design is to train a generator, which aims to learn the rules of organic chemistry that define SMILES strings corresponding to realistic chemical structures. We collected ∼1.6 million small molecules from the ChEMBL database23 to train the generative model, and each small molecule is represented in SMILES format to train a generator of Seq. 2Seq. The SMILES data set was preprocessed to remove stereochemistry, salts, and molecules with undesirable atoms or groups.9 In the end, ∼1.6 million small molecules with a length of 100 or less were retained for training the generative model.
Since the SMILES string has a natural sequential ordering, it is common to factorize the joint probabilities over symbols as the product of conditional probabilities.24 In recent years, there have been significant improvements in the expressiveness of models capable of calculating these conditional probabilities, such as RNN like GRU9 and self-attention architectures like the Transformer.18 In this work, the encoder module in the Transformer is used to build our generative model (Figure 8A). A similar generative network is GPT-2, which is widely used in commonsense reasoning, question answering, and textual entailment.25 Compared with architectures with similar functions such as RNN architectures, the Transformer framework used by GPT-2 provides a more structured memory for handling long-term dependencies in sequence, resulting in robust transfer performance across diverse tasks.18,26,27 Therefore, in our method, we use a generative model based on the Transformer encoder. The architecture and parameter settings of the generative model are shown in Supporting Information 1—Table S4.
Figure 8.

De novo drug design pipeline: (A) Detailed framework of the generative model, of which N is the number of Transformer blocks. (B) Transfer learning used to fine-tune the generative model to learn the prior structure in small samples. (C) General pipeline of reinforcement learning to design novel molecules with desired properties.
The parameters of the generative model are updated by reducing the cross-entropy loss between the real tokens and the predicted tokens. In the training process, the optimizer we used was AdamW, the learning rate was set as 0.00015, and the model tended to converge after 500 iterations of training. Finally, the trained generator is saved as a pretrained model to be weighted by a task-specific transfer learning model.
4.2. Fine-Tuned by Transfer Learning
In the process of the de novo drug design, random generation without prior experience is too blind to be generated for specific targets. Therefore, how to endow the deep generative model with specific research experience is the key to molecular design for specific tasks. In this work, we introduce the existing leading experience into the pretrained model through transfer learning (TL). We collected the existing BRAF and its similar proteins (UniProt ID: P15056, P10398, P04049) and inhibitors (pIC50 ≥ 4) from the ChEMBL database as small samples (4702 inhibitors) and used TL to guide the generative model to learn the structure feature in those samples.
During TL, all of the layers of the generative model are frozen, which will not be adjusted in the process of gradient calculation and backward transmission, except for the last linear layer (Figure 8B). Only the parameters of the last Linear layer are adjusted, which prevent the model from forgetting the molecular features on small samples, so as to generate compounds that are similar to the training set. The weight of the pretrained model was loaded and trained on small samples during TL.
4.3. Ligand-Based Score Function
After TL, the small molecules generated by the generator show a high structural similarity to the training set used by TL. Therefore, we designed a QSAR model based on neural network, which is confirmed to be the most potential tool for QSAR analysis and feature extraction,28,29 for predicting IC50 values of small molecules. In this work, we designed a predictive model, RNN taking SMILES as the only input, that contains a GRU for extracting small molecule features and three fully connected layers for reducing features and predicting IC50 values. The data sets used for training the predictive model were the same as those used for TL, and 5-fold cross-validation was used to evaluate the model’s performance. All of the architecture and hyperparameter information for the predictive model are provided in Supporting Information 1—Table S4.
4.4. Reinforcement Learning
The generative model learns a prior policy during training, which is an unbiased probability distribution of different symbols at each position of the SMILES string. In this section, the generative model is combined with the predictive model through reinforcement learning (RL) to generate molecules with specified desirable properties, where the generator acts as the agent to decide what character to choose next given the current state.9,10,16 The set of state S is defined as all possible strings with lengths from zero to value T, and the action space A is defined as all probable characters for the next position. The reward is defined as a function of the predicted value from the predictive model. The goal of the policy gradient method (or RL) is to update the agent to increase the reward for the generated sequences.10
To make the agent inherit the syntax of SMILES and the distribution of the molecular structure in ChEMBL that the prior generator has learned, the agent is anchored to prior policy through an augmented likelihood10
where P(A)Prior denotes the prior likelihood, σ denotes the weight coefficient, and R(A) denotes the reward function calculated as
where x is the predicted value from the predictive model.9
The long-term return G(sT) can be modified by the agreement between the agent likelihood log P(A)A and the augmented likelihood. The goal of the agent is to learn a strategy for maximizing the expected return G(sT), which can be translated into the problem of minimizing the following loss function J(θ)
Therefore, the problem of generating chemical compounds with desired properties can be formulated as a task of finding a vector of parameters θ of the agent generative model. The parameter settings of the model are shown in Supporting Information 1—Table S4.
4.5. Virtual Screening by BRAF Receptor Structure-Based
Molecular docking is one of the most commonly used methods in structure-based drug design, which can predict the affinity between the ligand and the target; so, it can be used to screen bioactive compounds in the early stage of drug development.30,31 Therefore, we chose BRAF inhibitor as an example to verify our mode. First, the crystal structure of the BRAF complex (PDB ID: 3OG7(32) and 6XFP(33)) was downloaded from the PDB database for simple optimization, including adding hydrogen atoms, distributing protonation state of residues, and minimizing the energy with the OPLS-2005 force field.34 The center of mass of the ligand in the complex was defined as the binding site, and the receptor grid box was set to 20 Å × 20 Å × 30 Å. All of the docking procedures were completed in Schrödinger2015.
The reserved compounds produced in the previous training will be docked into the preset BRAF binding pocket through the following three-step protocol: first, all structures were docked and scored by the Glide35 high-throughput virtual screening (HTVS) scoring mode, and the top 10% structures were retained; second, the saved structures from the previous step were re-docked and scored by the Glide standard precision (SP) scoring mode, and the top 10% structures were retained; finally, the saved structures were re-docked and scored by the Glide extra precision (XP) scoring mode.
Acknowledgments
The authors would like to thank the Guangdong Laboratory of Advanced Energy Science and Technology and the Chinese Academy of Sciences for their computing equipment and software support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c05145.
Experimental results of the percentage of small molecules generated when GRUs of different layers are stacked; test results of the predictive model on the PDBbind data set; percentage changes of molecules generated during reinforcement learning; details of the architecture and hyperparameters used for training the generative and predictive models; and two binding pockets of BRAF (PDF)
Structures binded with BRAF and their docking scores (XLSX)
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA21010202).
The authors declare no competing financial interest.
Notes
The source code and data that support the findings of this study are available upon request from the corresponding authors.
Supplementary Material
References
- Chang C.; Hung C.; Tang C. Y. In A Review of Deep Learning in Computer-Aided Drug Design, 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM); IEEE, 2019; pp 1856–1861.
- Yang L.; Yang G.; Chen X.; Yang Q.; Yao X.; Bing Z.; Niu Y.; Huang L.; Yang L. Deep Scoring Neural Network Replacing the Scoring Function Components to Improve the Performance of Structure-Based Molecular Docking. ACS Chem. Neurosci. 2021, 12, 2133–2142. 10.1021/acschemneuro.1c00110. [DOI] [PubMed] [Google Scholar]
- Li S.; Wan F.; Shu H.; Jiang T.; Zeng J.; et al. MONN: A Multi-objective Neural Network for Predicting Compound-Protein Interactions and Affinities. Cell Syst. 2020, 10, 308–322.e11. 10.1016/j.cels.2020.03.002. [DOI] [Google Scholar]
- Kumar A.; Kini S. G.; Rathi E. A recent appraisal of artificial intelligence and in silico ADMET prediction in the early stages of drug discovery. Mini-Rev. Med. Chem. 2021, 21, 2786–2798. 10.2174/1389557521666210401091147. [DOI] [PubMed] [Google Scholar]
- Wang X.; Li Y.; Qiu J.; Chen G.; Liu H.; Liao B.; Hsieh C.-Y.; Yao X. RetroPrime: A Diverse, plausible and Transformer-based method for Single-Step retrosynthesis predictions. Chem. Eng. J. 2021, 420, 129845 10.1016/j.cej.2021.129845. [DOI] [Google Scholar]
- Senior A. W.; Evans R.; Jumper J.; Kirkpatrick J.; Sifre L.; Green T.; Qin C.; Žídek A.; Nelson A. W. R.; Bridgland A.; Penedones H.; Petersen S.; Simonyan K.; Crossan S.; Kohli P.; Jones D. T.; Silver D.; Kavukcuoglu K.; Hassabis D. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- Gómez-Bombarelli R.; Duvenaud D.; Hernández-Lobato J.; Aguilera-Iparraguirre J.; Aspuru-Guzik A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2016, 4, 268–276. 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segler M.; Kogej T.; Tyrchan C.; Waller M. P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 2018, 4, 120–131. 10.1021/acscentsci.7b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova M.; Isayev O.; Tropsha A. Deep Reinforcement Learning for De-Novo Drug Design. Sci. Adv. 2017, 4, eaap7885 10.1126/sciadv.aap7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivecrona M.; Blaschke T.; Engkvist O.; Chen H. Molecular de-novo design through deep reinforcement learning. J. Cheminf. 2017, 9, 48 10.1186/s13321-017-0235-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhavoronkov A.; Ivanenkov Y. A.; Aliper A.; Veselov M. S.; Aladinskiy V. A.; Aladinskaya A. V.; Terentiev V. A.; Polykovskiy D. A.; Kuznetsov M. D.; Asadulaev A.; Volkov Y.; Zholus A.; Shayakhmetov R. R.; Zhebrak A.; Minaeva L. I.; Zagribelnyy B. A.; Lee L. H.; Soll R.; Madge D.; Xing L.; Guo T.; Aspuru-Guzik A. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. 10.1038/s41587-019-0224-x. [DOI] [PubMed] [Google Scholar]
- Prykhodko O.; Johansson S. V.; Kotsias P.-C.; Arús-Pous J.; Bjerrum E. J.; Engkvist O.; Chen H. A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminf. 2019, 11, 74 10.1186/s13321-019-0397-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenfeller M.; Proschak E.; Schüller A.; Schneider G. Concept of combinatorial de novo design of drug-like molecules by particle swarm optimization. Chem. Biol. Drug Des. 2010, 72, 16–26. 10.1111/j.1747-0285.2008.00672.x. [DOI] [PubMed] [Google Scholar]
- Winter R.; Montanari F.; Steffen A.; Briem H.; Clevert D. A.; et al. Efficient Multi-Objective Molecular Optimization in a Continuous Latent Space. Chem. Sci. 2019, 10, 8016–8024. 10.1039/C9SC01928F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C.; Wang S.; Xu Y.; Zhang W.; Tang K.; Ouyang Q.; Lai L.; Pei J. Transfer Learning for Drug Discovery. J. Med. Chem. 2020, 63, 8683–8694. 10.1021/acs.jmedchem.9b02147. [DOI] [PubMed] [Google Scholar]
- Krishnan S. R.; Bung N.; Bulusu G.; Roy A. Accelerating De Novo Drug Design against Novel Proteins Using Deep Learning. J. Chem. Inf. Model. 2021, 61, 621–630. 10.1021/acs.jcim.0c01060. [DOI] [PubMed] [Google Scholar]
- Schmidhuber J.Gradient Flow in Recurrent Nets: the Difficulty of Learning Long-Term Dependencies; IEEE, 2001. [Google Scholar]
- Vaswani A.; Shazeer N.; Parmar N.; Uszkoreit J.; Jones L.; Gomez A. N.; Kaiser L.; Polosukhin I.. Attention Is All You Need. 2017, arXiv:1706.03762. arXiv.org e-Print archive. http://arxiv.org/abs/1706.03762.
- Grechishnikova D. Transformer neural network for protein-specific de novo drug generation as a machine translation problem. Sci. Rep. 2021, 11, 321 10.1038/s41598-020-79682-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S.; Lei Z.; Ai H.; Chen H.; Deng D.; Yang Y. Deep Scaffold Hopping with Multi-modal Transformer Neural Networks. ChemRxiv 2020, 11767 10.26434/chemrxiv.13011767.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemis G. W.; Murcko M. A. The Properties of Known Drugs. 1. Molecular Frameworks. J. Med. Chem. 1996, 39, 2887–2893. 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- Shang J.; Sun H.; Liu H.; Chen F.; Tian S.; Pan P.; Li D.; Kong D.; Hou T. Comparative analyses of structural features and scaffold diversity for purchasable compound libraries. J. Cheminf. 2017, 9, 25 10.1186/s13321-017-0212-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anna G.; Anne H.; Michał N.; Patrícia B.; Jon C.; David M.; Prudence M.; Francis A.; Bellis L. J.; Elena C. U. The ChEMBL database in 2017. Nucleic Acids Res. 2017, D1, D945–D954. 10.1093/nar/gkw1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengio Y.; Ducharme R.; Vincent P.; Jauvin C. A Neural Probabilistic Language Model. J. Mach. Learn. Res. 2003, 3, 1137–1155. [Google Scholar]
- Radford A.; Narasimhan K.. Improving Language Understanding by Generative Pre-Training, 2018. https://cdn.openai.com/research-covers/language-unsupervised/language_understanding_paper.pdf.
- Liu P. J.; Saleh M.; Pot E.; Goodrich B.; Sepassi R.; Kaiser L.; Shazeer N.. Generating Wikipedia by Summarizing Long Sequences. 2018, arXiv:1801.10198. arXiv.org e-Print archive. https://arxiv.org/abs/1801.10198v1.
- Kitaev N.; Klein D. In Constituency Parsing with a Self-Attentive Encoder, Proceedings of the 56th Annual Meeting of the Association for Computational Linguistics (Volume 1: Long Papers); Association for Computational Linguistics, 2018; pp 2676–2686.
- Fahimeh G.; Alireza M.; Alfonso P. G.; Horacio P. S. Neural network and deep-learning algorithms used in QSAR studies: merits and drawbacks. Drug Discovery Today 2018, 23, 1784–1790. 10.1016/j.drudis.2018.06.016. [DOI] [PubMed] [Google Scholar]
- Schneider G. Neural networks are useful tools for drug design. Neural Networks 2000, 13, 15–16. 10.1016/S0893-6080(99)00094-5. [DOI] [PubMed] [Google Scholar]
- Fu W.; Wang E.; Ke D.; Yang H.; Chen L.; Shao J.; Hu X.; Xu L.; Liu N.; Hou T. Discovery of a Novel Fusarium Graminearum Mitogen-Activated Protein Kinase (FgGpmk1) Inhibitor for the Treatment of Fusarium Head Blight. J. Med. Chem. 2021, 64, 13841–13852. 10.1021/acs.jmedchem.1c01227. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Xu L.; Zhang Y.; Pan J.; Wang P.-q.; Tian S.; Li H.-t.; Gao B.-w.; Hou T.-j.; Zhen X.-c.; Zheng L.-T. Discovery of novel MIF inhibitors that attenuate microglial inflammatory activation by structures-based virtual screening and in vitro bioassays. Acta Pharmacol. Sin. 2021, 00753 10.1038/s41401-021-00753-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag G.; Hirth P.; Tsai J.; Zhang J.; Ibrahim P. N.; Cho H.; Spevak W.; Zhang C.; Zhang Y.; Habets G.; Burton E. A.; Wong B.; Tsang G.; West B. L.; Powell B.; Shellooe R.; Marimuthu A.; Nguyen H.; Zhang K. Y. J.; Artis D. R.; Schlessinger J.; Su F.; Higgins B.; Iyer R.; D’Andrea K.; Koehler A.; Stumm M.; Lin P. S.; Lee R. J.; Grippo J.; Puzanov I.; Kim K. B.; Ribas A.; McArthur G. A.; Sosman J. A.; Chapman P. B.; Flaherty K. T.; Xu X.; Nathanson K. L.; Nolop K. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen I.; Shanahan F.; Lee J.; Hong Y. S.; Shin S. J.; Moore A. R.; Sudhamsu J.; Chang M. T.; Bae I.; Dela Cruz D.; Hunsaker T.; Klijn C.; Liau N. P. D.; Lin E.; Martin S. E.; Modrusan Z.; Piskol R.; Segal E.; Venkatanarayan A.; Ye X.; Yin J.; Zhang L.; Kim J.-S.; Lim H.-S.; Kim K.-P.; Kim Y. J.; Han H. S.; Lee S. J.; Kim S. T.; Jung M.; Hong Y.-h.; Noh Y. S.; Choi M.; Han O.; Nowicka M.; Srinivasan S.; Yan Y.; Kim T. W.; Malek S. ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma. Nature 2021, 594, 418–423. 10.1038/s41586-021-03515-1. [DOI] [PubMed] [Google Scholar]
- Kaminski G. A.; Friesner R. A.; Tirado-Rives J.; Jorgensen W. L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. 10.1021/jp003919d. [DOI] [Google Scholar]
- LLC, S . Glide; LLC: New York, 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.