

Abstract
BRCA2-mutant cells are defective in homologous recombination, making them vulnerable to the inactivation of other pathways for repairing DNA double-strand breaks (DSBs). This concept can be clinically exploited but is currently limited due to insufficient knowledge about how DSBs are repaired in the absence of BRCA2. We show that DNA polymerase θ (POLθ)-mediated end-joining (TMEJ) repairs DSBs arising during S phase in BRCA2-deficient cells only after the onset of the ensuing mitosis. This process is regulated by RAD52 whose loss causes the premature usage of TMEJ and the formation of chromosomal fusions. Purified RAD52 and BRCA2 proteins both block the DNA polymerase function of POLθ, suggesting a mechanism explaining their synthetic lethal relationships. We propose that the delay of TMEJ until mitosis ensures the conversion of originally one-ended into two-ended DSBs. Mitotic chromatin condensation might further serve to juxtapose correct break ends and limit chromosomal fusions.
Introduction
Mutations in BRCA2 are associated with the development of breast, ovarian, and additional cancers1. BRCA2 functions in homologous recombination (HR) to repair DNA double-strand breaks (DSBs) and other complex DNA damage as well as during DNA replication to protect stalled forks from degradation2. BRCA2-deficient tumours are efficiently treated with PARP inhibitors (PARPi), although the development of tumour resistance represents a limiting factor3. BRCA2-deficient cells rely on other DSB repair pathways to compensate for the loss of HR and allow tumour survival. This makes such tumours selectively sensitive to the inactivation of HR-independent repair pathways, a synthetic lethality concept which can be clinically exploited, e.g., with PARPi4. However, this approach is limited by insufficient knowledge of how HR-deficient cells repair DSBs.
BRCA2 functions during a rate-limiting step of HR by loading RAD51 onto resected DSBs5,6. In the absence of BRCA2, resected DSBs remain unrepaired and must be processed by other pathways. It has been described that the DNA polymerase θ (POLθ)-mediated end-joining (TMEJ) pathway can also repair resected DSBs and that the inactivation of POLθ leads to cell death in BRCA2-mutant but not wild-type (WT) cells7–10. This has motivated the development of chemical inhibitors for POLθ to kill cancer cells and spare healthy tissues in patients with BRCA2 tumours3,4. However, it is unclear how an end-joining pathway can compensate for the loss of HR since most spontaneous and many exogenously induced DSBs arise during replication and have only a single end as opposed to frank DSBs with two ends arising from breaking duplex DNA.
RAD52 is another factor required for the survival of BRCA2-mutant but not WT cells. RAD52 inactivation impairs growth of BRCA2-deficient cells and RAD52 over-expression in BRCA2 mutants with low RAD52 expression substantially enhances cell growth11. In yeast cells, which do not possess BRCA2, Rad52 functions as a Rad51 loader and it has been speculated that human RAD52 may have a similar function which becomes important only when BRCA2 is absent. However, human RAD52 does not mediate RAD51 loading in biochemical assays5 and exerts various RAD51-independent functions in single-strand annealing (SSA), break-induced replication12, DNA-RNA hybrid formation13, and the rescue of under-replicated DNA14,15. Thus, despite major advances in RAD52 inhibitor development16,17, the function of RAD52 in BRCA2-deficient cells remains elusive18.
The gap in knowledge about POLθ and RAD52 function in BRCA2 mutants prevents a rational design of combined inhibition with PARP. Moreover, it is unclear if inhibition of POLθ and/or RAD52 can be applied to other HR-deficient tumours. Here, we delineate the roles and mechanisms of POLθ and RAD52 in the synthetic lethality network of BRCA2-deficient tumours. We uncover an unexpected strict cell cycle regulation of TMEJ and describe new functions of RAD52 and BRCA2 in this process. Our results provide a mechanistic basis for targeting RAD52 and POLθ to improve PARPi treatment and predict which HR-deficient tumours will benefit from such combined treatments.
Results
TMEJ in HR-deficient cells is delayed until mitosis
We first investigated the interplay between BRCA2 and POLθ. We developed an EdU/BrdU labelling approach in which HeLa cells are followed throughout the cell cycle and monitored for the formation and repair of endogenous DSBs by γH2AX foci analysis19 (Fig. 1a,b). DSBs accumulated in S phase and were gradually repaired upon G2 entry (PCNA staining, Fig. 1c). Cells treated with siBRCA2 or siRAD51 showed similar foci numbers as siControl (siCTRL)-treated cells at the end of S phase but elevated foci in G2 (Fig. 2a). Additional depletion of POLθ did not significantly affect foci numbers. We conclude that spontaneous DSBs arise in S phase and repair in G2 depends on HR factors, with no significant contribution of POLθ even in cells deficient for BRCA2 or RAD51.
Figure 1 |. The formation and repair of spontaneous DSBs throughout the cell cycle.
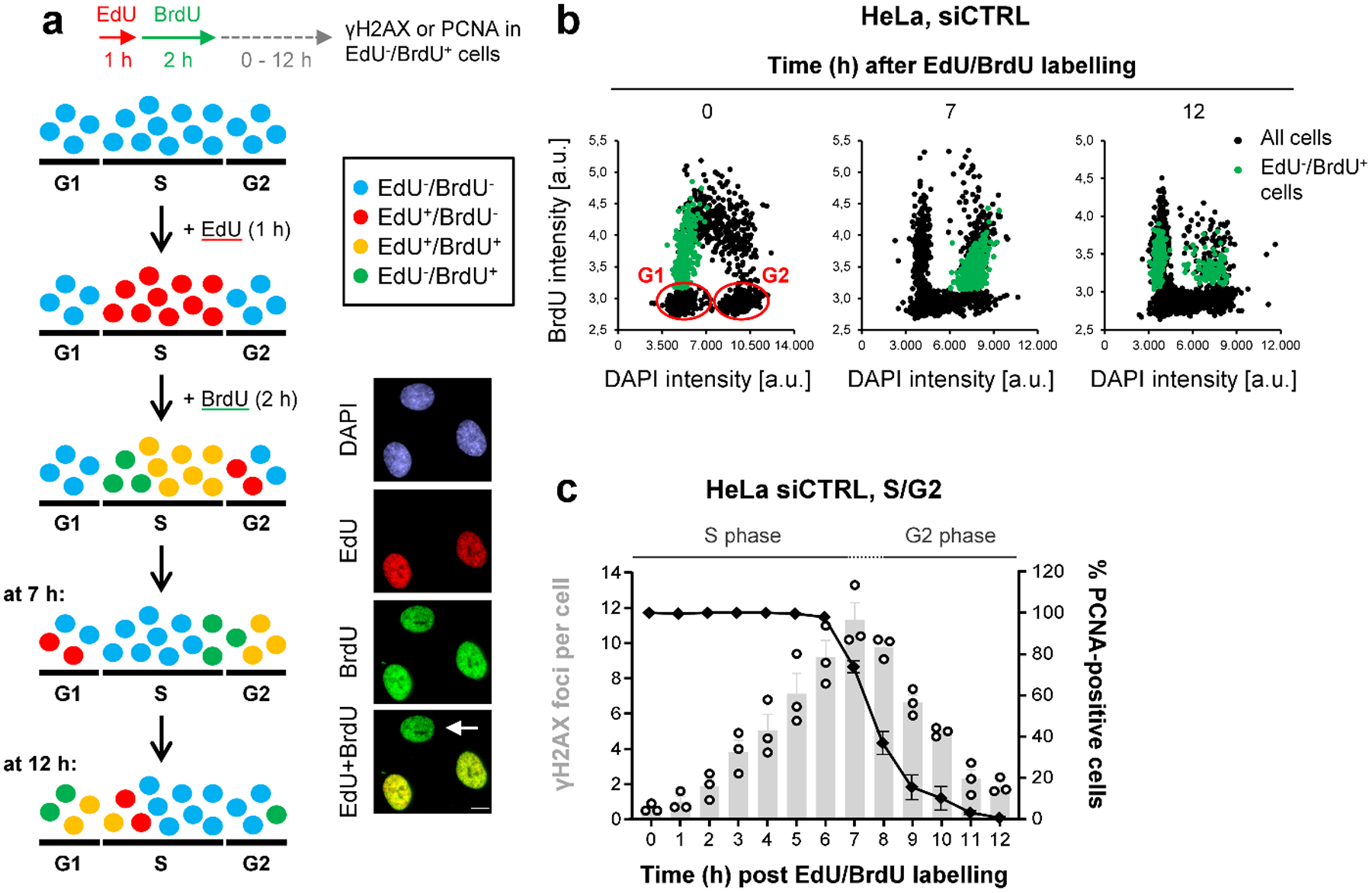
a, Schematic showing the strategy to analyse spontaneous γH2AX foci during the cell cycle. Cells were labelled with EdU for 1 h and then with BrdU for 2 h (see example immunofluorescence (IF) images, scale bar, 10 μm). After BrdU removal, EdU−BrdU+ cells (indicated by the white arrow) are in early S and move to late S and early G2 7 h later. At 12 h after labelling, EdU−BrdU+ cells are in late G2 and early G1. b, Cell cycle progression of HeLa cells. The cells were labelled as in (a) and analysed at the indicated time points. The dot blots, obtained by microscopic scanning, show BrdU versus DAPI intensities, allowing for the discrimination of the different cell cycle phases. At the 0 h time point, S phase cells appear as BrdU+, whilst BrdU− G1 and G2 cells can be distinguished according to their DAPI content (low or high, respectively, indicated in blue). In particular, with this labelling strategy, we could follow the progression of EdU−BrdU+ cells (indicated in green) from early S (0 h) into late S/G2 (7 h) and G2/G1 phase (12 h). c, Spontanous γH2AX and PCNA kinetics in S/G2 HeLa cells. Cells were labelled as in (a) and EdU−BrdU+ cells were analysed at various time points after labelling. Foci numbers (in grey) increase during S and decrease during G2 phase. The transition between S and G2 phase (~7–8 h after labelling) was monitored by chromatin-bound PCNA staining (in black) in parallel experiments (n=3 independent experiments). At later times (>10 h after labelling), cells start entering G1 phaseand these were excluded from the foci analysis. The data show the mean ± SEM. Individual experiments, each derived from 40 cells, are shown as dots. Source data are available online.
Figure 2 |. TMEJ in HR-deficient cells is delayed until mitosis.
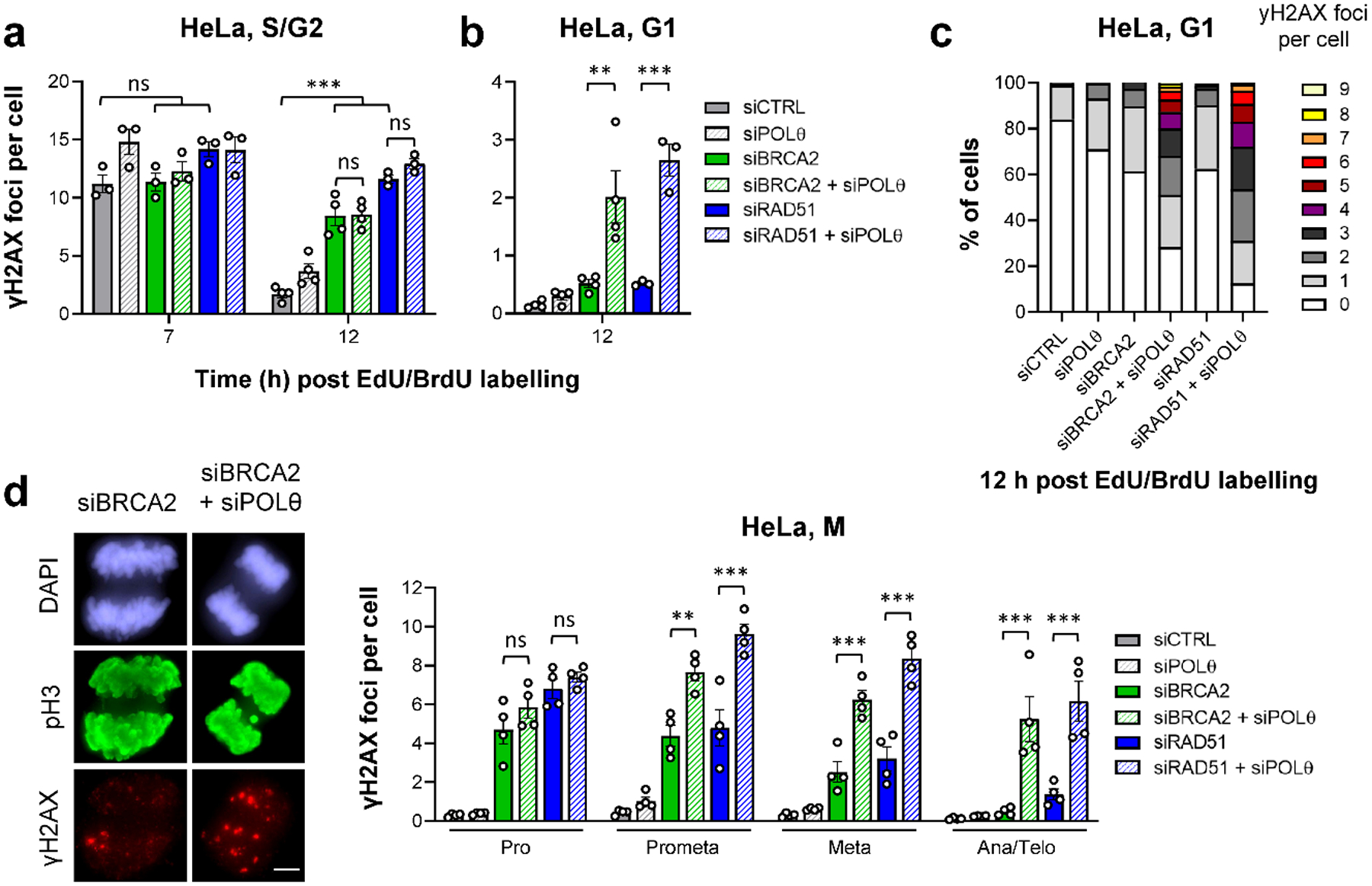
a, Spontaneous γH2AX foci in S/G2 HeLa cells. Cells were transfected with siRNAs, labelled as in Fig. 1a and EdU−BrdU+ cells were analysed in S/G2 (7 h) and G2 (12 h after labelling) (n=3 independent experiments, except siCTRL/siPOLθ/siBRCA2/siBRCA2+siPOLθ at 12 h: n=4 independent experiments). b, Spontaneous γH2AX foci in G1 HeLa cells. Cells were transfected with siRNAs, labelled as in Fig. 1a and EdU−BrdU+ cells were analysed in G1 (12 h after labelling) (n=4 independent experiments, except siRAD51/siRAD51+siPOLθ: n=3 independent experiments). c, Alternative representation of the data in (b). The percentage of G1 cells showing 0–9 spontaneous γH2AX foci is represented. Raw data was combined from the experiments in (b) and in total from ≥ 300 individual cells. d, Spontaneous γH2AX foci in mitotic HeLa cells. Cells were transfected with siRNAs. Mitotic stages were identified by the cell morphology seen in the DAPI and phospho-histone H3(S10) (pH3) staining (see example IF images in the left panel, scale bar, 5 μm) (n=4 independent experiments). All data show the mean ± SEM. Individual experiments, each derived from 40 (S/G2 and M) or 100 cells (G1), are shown as dots. **: P < 0.01; ***: P < 0.001, ns: non-significant (One-way ANOVA). The exact P values are provided as source data. Source data are available online.
We then evaluated cells entering mitosis and G1 phase. Cells depleted for BRCA2 or RAD51 progressively repaired their breaks during mitosis, reaching foci numbers as low as control cells in the next G1 phase. In contrast, depleting POLθ in HR-defective cells caused elevated foci numbers during mitosis and the subsequent G1 phase (Fig. 2b–d). In these and further experiments, we excluded the possibility that the effect was caused by a strong response of a sub-population of cells (Fig. 2c). Similar results were obtained when foci were scored in G1 and G2 HeLa cells without following cell cycle progression (Extended Data Fig. 1a), with a different POLθ siRNA (Extended Data Fig. 1b) and with non-transformed cells (hTERT-immortalised primary human fibroblasts: 82–6 hTERT) (Extended Data Fig. 1c). Finally, the γH2AX foci data were confirmed by the analysis of chromatid breaks in G2 and metaphase (Extended Data Fig. 1d) and by the evaluation of lagging chromosomes in ana/telophase (Extended Data Fig. 1e) and micronuclei in G1 (Extended Data Fig. 1f). Collectively, these results show that POLθ promotes the repair of endogenous DSBs in the absence of HR factors, but its function is delayed until mitosis.
To explore if POLθ also repairs DSBs induced by DNA damaging agents in HR-deficient cells, we employed the topoisomerase 1 inhibitor camptothecin (CPT)20, which induces one-ended DSBs during S phase. HeLa cells showed high foci numbers at 2 h after a CPT pulse treatment for all siRNA conditions (Extended Data Fig. 2a), while 12 h after CPT treatment, when cells had progressed to G2 and G1, siCTRL cells had repaired most of the induced DSBs. In contrast, BRCA2-depleted G2 cells exhibited unrepaired DSBs which were not further elevated after additional depletion of POLθ. BRCA2-depleted cells that had reached G1 showed only a few unrepaired DSBs, while the co-depletion with POLθ strongly increased DSB levels (Extended Data Fig. 2a), confirming previous analyses of endogenous DSBs. We also induced DSBs by ionizing radiation (IR) in G2-phase cells to explore how POLθ repairs two-ended DSBs in the absence of HR factors. Strikingly, even two-ended DSBs were repaired by POLθ only in cells that reached G1 phase (Extended Data Fig. 2b). These results show that the lack of TMEJ of endogenous and CPT-induced DSBs in S/G2 is not simply explained by the absence of a second break end, but instead suggests a regulated process which restricts POLθ function in these cell cycle phases. Finally, we combined the depletion of POLθ with PARPi treatment, which led to very high foci numbers in HR-deficient cells. Of note, the combined treatment had a synergistic effect compared to the individual treatments for cells in late mitosis or G1 (Extended Data Fig. 2c). This suggests that enhancing the spontaneous level of DSBs (due to PARPi) while concomitantly preventing mitotic repair (due to siPOLθ) represents a promising strategy to sensitise HR-deficient cells3.
RAD52 locates to resected DSBs in G2-phase BRCA2 mutants
As BRCA2 deficiency is also synthetically lethal with a RAD52 defect, we next examined the function of RAD52 in BRCA2 mutants. We used a HeLa cell line stably expressing GFP-tagged RAD52 to investigate its recruitment to IR-induced DSBs and compare it to the known HR factors RAD51 and pRPA (a marker for resected DSBs) (Extended Data Fig. 3a). While RAD51 foci numbers in G2 WT cells are maximal at 2 h after IR and decrease as repair proceeds21,22, RAD52 foci numbers increase up to 8 h and reach a value close to that of γH2AX foci (Extended Data Fig. 3b). RAD52 foci co-localise with γH2AX and pRPA but not with RAD51 foci at 8 h post IR (Extended Data Fig. 3c). Depletion of BRCA2 increased RAD52 and pRPA foci levels and, importantly, led to RAD52 foci formation already at 2 h post IR, a time point when RAD52 foci in siCTRL-treated cells are nearly absent (Fig. 3a). These data show that RAD52 and RAD51 display distinct recruitment kinetics to IR-induced DSBs in G2 and that RAD52 is loaded onto resected DSBs if BRCA2 is absent. These findings agree with previous studies suggesting that RAD52 has no major role in RAD51 loading5,6. We also found that RAD52 and pRPA, but not RAD51, foci persist into early M phase and disappear when mitosis proceeds (Fig. 3b).
Figure 3 |. RAD52 locates to resected DSBs to prevent their repair in G2-phase BRCA2 mutants.
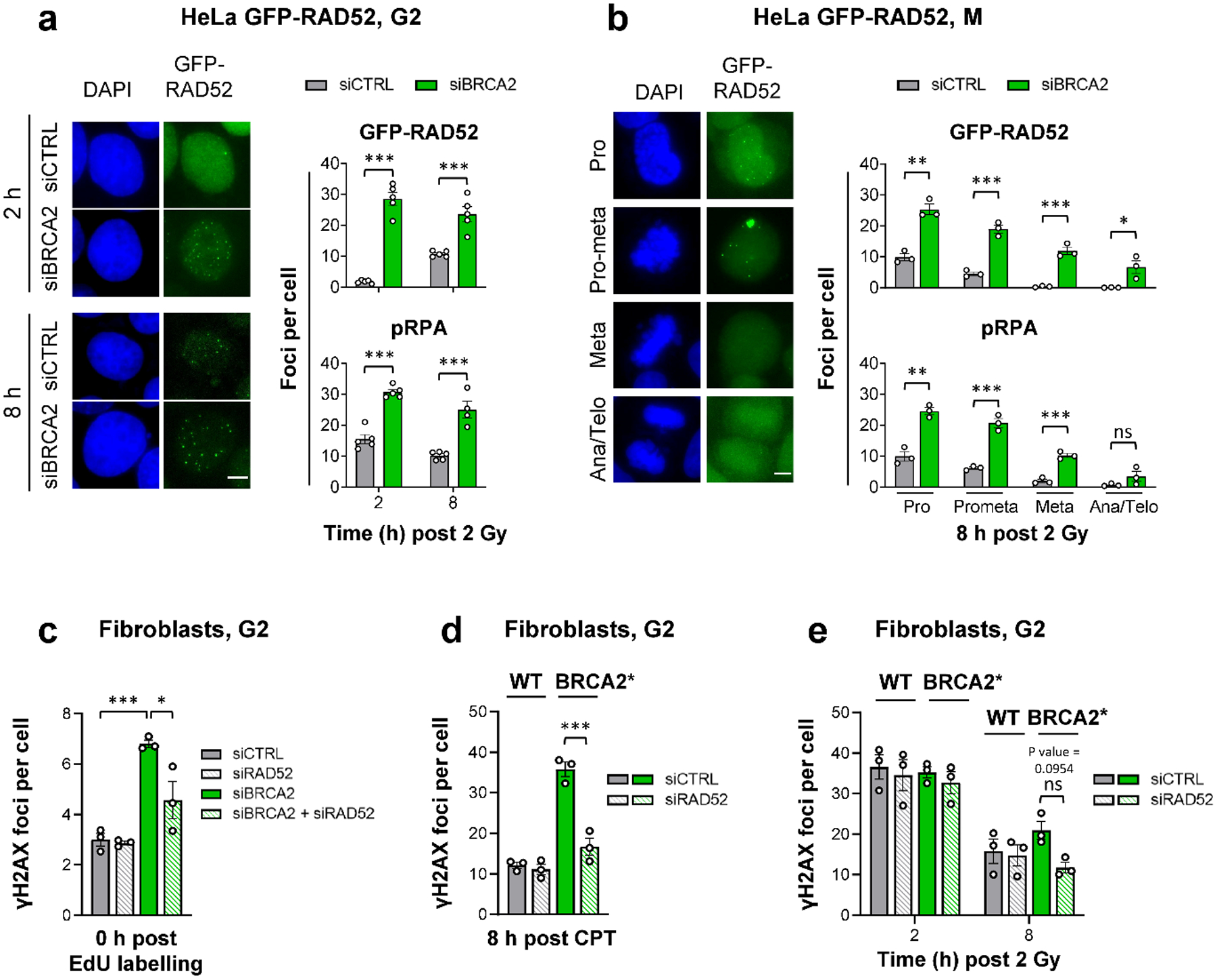
a, GFP-RAD52 and pRPA foci in G2 HeLa GFP-RAD52 cells. Cells stably expressing GFP-RAD52 (see Extended Data Fig. 3a) were transfected with siRNAs, labelled with EdU, irradiated with 2 Gy X-rays and EdU− G2 cells were analysed (see example IF images in the left panel, scale bar, 5 μm) (n=5 independent experiments, except pRPA for siBRCA2 at 8 h: n=4 independent experiments). b, GFP-RAD52 and pRPA foci in mitotic HeLa GFP-RAD52 cells. Experiments were done as in (a). EdU− mitotic cells were identified by their morphology seen in the DAPI staining (see example IF images in the left panel obtained from siCTRL-treated samples, scale bar, 5 μm) (n=3 independent experiments). c, Spontaneous γH2AX foci in G2 fibroblasts. 82–6 hTERT (WT) cells were transfected with siRNAs, labelled with EdU for 1 h and foci were immediately analysed after labelling in EdU− cells in G2. The specificity of the RAD52 siRNA was shown with siRNA resistant GFP-RAD52 HeLa cells (see Fig. 5a and Extended Data Fig. 7c) (n=3 independent experiments). d, γH2AX foci after CPT treatment in G2 fibroblasts. 82–6 hTERT (WT) and HSC-62 hTERT (BRCA2 mutant: BRCA2*) cells were transfected with siRNAs, treated with EdU and 20 nM CPT for 1 h. EdU+ cells were in S during the CPT treatment and progressed to G2 after 8 h. Foci were analysed in this population (n=3 independent experiments). e, γH2AX foci after IR in G2 fibroblasts. WT and BRCA2* cells were transfected with siRNAs, labelled with EdU and irradiated with 2 Gy X-rays. EdU− G2 cells were analysed (n=3 independent experiments). All data show the mean ± SEM. Individual experiments, each derived from 40 (G2) or 20 cells (M), are shown as dots. *: P < 0.05; **: P < 0.01; ***: P < 0.001, ns: non-significant (One-way ANOVA, except for the data in (a,b), where an unpaired two-tailed t-test was used). The exact P values are provided as source data. Source data are available online.
We next examined if RAD52 promotes a DSB repair pathway (such as SSA) in BRCA2 mutants. Endogenous γH2AX foci were analysed in HeLa cells and fibroblasts after siRAD52 and/or siBRCA2. While siBRCA2 led to increased foci levels in G2-phase cells, siRAD52 had only a minor or no effect. Strikingly, co-depletion of RAD52 and BRCA2 resulted in fewer unrepaired DSBs compared to siBRCA2 alone (Fig. 3c, Extended Data Fig. 3d). This rescue of the BRCA2 repair defect was also observed for CPT- and IR-induced DSBs in HeLa cells and fibroblasts treated with siBRCA2 or carrying a BRCA2 mutation conferring low residual BRCA2 activity due to an internal deletion of four amino acids23 (HSC-62 hTERT cells, referred to as BRCA2*) (Fig. 3d,e, Extended Data Fig. 3e,f). Thus, RAD52 does not promote but instead prevents repair of resected DSBs in G2-phase BRCA2 mutants.
RAD52 prevents the premature usage of TMEJ in BRCA2 mutants
We next investigated if TMEJ is activated in the absence of BRCA2 and RAD52. POLθ depletion alone or in combination with siRAD52 did not affect γH2AX foci levels in G2-irradiated cells. However, in BRCA2-defective cells, dual siPOLθ/siRAD52 treatment completely reversed the rescue of the BRCA2 repair defect observed after the depletion of RAD52 (Fig. 4a, Extended Data Fig. 4a). In contrast to the absence of POLθ, the lack of canonical non-homologous end-joining factors (LIG4, XLF) did not affect the rescue of the BRCA2 repair defect (Extended Data Fig. 4b,c). To investigate the fidelity of TMEJ in G2, we analysed the formation of chromatid fusions along with unrepaired chromatid breaks (Fig. 4b). BRCA2 mutants with high levels of unrepaired chromatid breaks after IR showed only marginally elevated levels of chromatid fusions compared to WT cells. Depletion of RAD52 in WT cells affected neither chromatid break nor fusion levels. However, RAD52 depletion in BRCA2 mutants, while rescuing the elevated level of chromatid breaks, resulted in a substantial increase in chromatid fusions, both effects dependent on POLθ (Fig. 4c). This dependency on POLθ was confirmed in siBRCA2-treated HeLa cells for endogenous chromatid breaks and fusions (Fig. 4d) and after IR and CPT exposure (Extended Data Fig. 4d).
Figure 4 |. RAD52 prevents the premature usage of TMEJ in BRCA2 mutants.
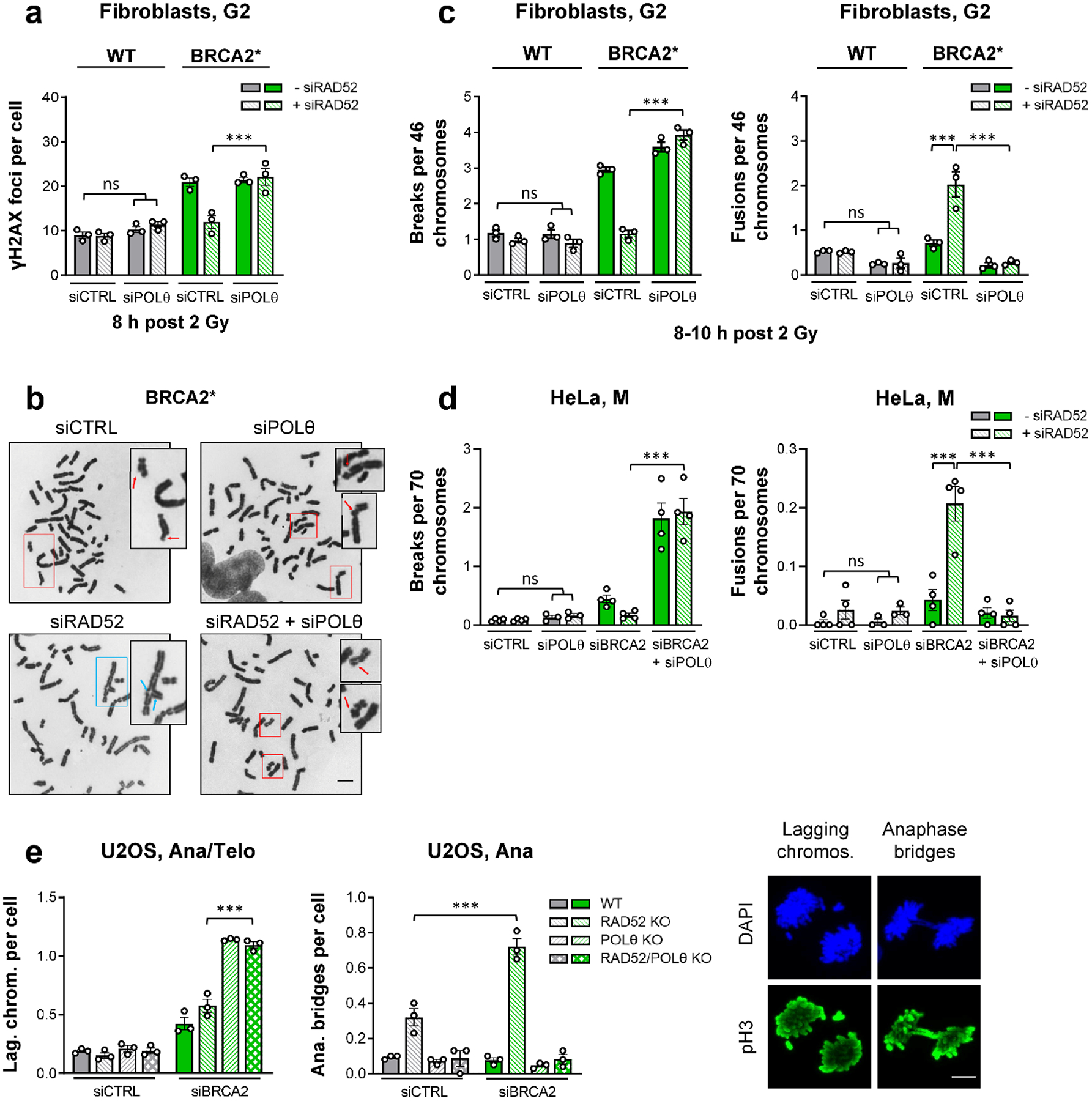
a, γH2AX foci after IR in G2 fibroblasts. WT and BRCA2* cells were transfected with siRNAs, labelled with EdU, irradiated with 2 Gy X-rays and EdU− G2 cells were analysed (n=3 independent experiments). b, Examples of chromatid breaks (red arrows) and fusions (blue arrows) in BRCA2* cells after IR (scale bar, 5 μm). c, Chromatid breaks and fusions after IR in G2 fibroblasts. WT and BRCA2* cells were transfected with siRNAs and irradiated with 2 Gy X-rays. Chromosome spreads were obtained by premature chromosome condensation (PCC) 8–10 h post IR. Chromatid breaks and fusions were quantified per spread and normalised to 46 chromosomes (n=3 independent experiments). d, Spontaneous chromatid breaks and fusions in mitotic HeLa cells. Cells were transfected with siRNAs and mitotic spreads were prepared and analysed for the presence of aberrations. Chromatid breaks and fusions were quantified per spread and normalised to 70 chromosomes (n=4 independent experiments, except siPOLθ/siPOLθ+siRAD52: n=3 independent experiments). e, Spontaneous lagging chromosomes and anaphase bridges in mitotic U2OS cells. U2OS WT and KO cells were transfected with siRNAs and ana/telophase cells were analysed for the presence of lagging chromosomes and anaphase bridges. Cells were identified by their morphology seen in the DAPI and phospho-histone H3(S10) (pH3) stainings. Examples of lagging chromosomes are shown for the POLθ KO, while examples of anaphase bridges are illustrated for the RAD52 KO, both treated with siBRCA2 (scale bar, 5 μm) (n=3 independent experiments). All data show the mean ± SEM. Individual experiments, each derived from 40 (G2 foci and anaphase bridges), 80 (lagging chromosomes), 30 (G2 spreads) or 60 cells (M spreads), are shown as dots. ***: P < 0.001, ns: non-significant (One-way ANOVA). The exact P values are provided as source data. Source data are available online.
To test a link between RAD52 and POLθ with an independent approach, we employed a reporter assay in which a GFP gene is corrupted by an I-SceI recognition site and stop codons flanked by 8 bp of homologous sequences (Extended Data Fig. 5a). End-joning processes utilizing micro-homology of the 8 bp repeats lead to reconstitution and expression of functional GFP24. The frequency of GFP+ cells was significantly increased after siBRCA2, consistent with published data25, but not after siRAD52. However, co-depletion of RAD52 and BRCA2 nearly doubled the GFP+ frequency compared to BRCA2 depletion alone (Extended Data Fig. 5bi). Molecular analysis of the GFP+ cells confirmed micro-homology usage of the 8 bp repeats both in BRCA2-depleted and in BRCA2/RAD52-depleted cells (Extended Data Fig. 5bii,iii). POLθ depletion reduced the elevated GFP+ frequencies in BRCA2- and BRCA2/RAD52-depleted cells to that of siPOLθ alone, consistent with the foci and chromosome studies described above.
Absent or deregulated TMEJ causes distinct aberrations
We next investigated the cellular consequences of the uncovered roles of RAD52 and POLθ and employed U2OS cells with a disrupted expression of the POLθ polymerase domain (designated henceforth POLθ knock-out (KO) for simplicity), a KO for RAD52, or a combination of both26. We first evaluated the repair of endogenous γH2AX foci in G2 and G1 cells and confirmed similar roles of POLθ and RAD52 as previously observed in other cell systems (Extended Data Fig. 6a).
Since chromatid fusions and unrepaired breaks in G2 are expected to interfere with mitosis, we analysed mitotic aberrations manifesting as lagging chromosomes and anaphase bridges. The level of lagging chromosomes was similar in all cells after siCTRL and only slightly enhanced in siBRCA2-treated WT or RAD52 KO cells. In contrast, BRCA2 depletion in POLθ single or POLθ/RAD52 double KO cells substantially elevated lagging chromosome levels (Fig. 4e), with the majority of cells exhibiting at least one such aberration (Extended Data Fig. 6b). Interestingly, although RAD52 KO cells showed normal foci levels, they exhibited a considerable increase in anaphase bridges which was doubled upon BRCA2 depletion. The presence of anaphase bridges is completely dependent on POLθ function as POLθ/RAD52 double KO cells showed the same bridge level as WT cells (Fig. 4e, Extended Data Fig. 6b). The dependency of anaphase bridges on POLθ was confirmed using HeLa cells (Extended Data Fig. 6c). Collectively, these data suggest that the uncovered functions for RAD52 and POLθ in BRCA2-deficient cells account for the known synthetic lethalities between BRCA2 and RAD5211, as well as BRCA2 and POLθ7,8.
BRCA2 and RAD52 both suppress TMEJ independent of RAD51
To study which of the different RAD52 protein domains are important for preventing TMEJ, we generated siRNA-resistant GFP-tagged deletion mutants, in which the RAD52 self-binding domain (SELF Δ), the RPA-binding domain (RPA Δ) or the RAD51-binding domain (RAD51 Δ) were deleted (Extended Data Fig. 7a). We depleted the endogenous RAD52 and BRCA2 proteins in HeLa cells stably carrying these constructs and tested if expression of the different mutants can restore the siBRCA2 repair defect in G2 (Extended Data Fig. 7b). BRCA2/RAD52-depleted cells expressing the WT or the RAD51 Δ constructs showed similar high γH2AX and pRPA foci levels at 8 h post IR as siBRCA2-treated cells (Fig. 5a, Extended Data Fig. 7c). In contrast, BRCA2/RAD52-depleted cells expressing the SELF Δ or the RPA Δ construct exhibited similar low foci levels as siCTRL-treated cells while the triple depletion of POLθ/BRCA2/RAD52 led to high foci levels in all mutants (Fig. 5a, Extended Data Fig. 7c). In addition, only the WT and RAD51 Δ, but not the SELF Δ or the RPA Δ mutants, were able to form GFP-RAD52 foci, which localised to DSBs (Fig. 5b). These results demonstrate that the RAD51 Δ but not the SELF Δ or the RPA Δ variants of RAD52 exhibit the same functionality as the WT construct, confirming that RAD52 does not suppress TMEJ by loading RAD51. The requirement of the RPA-binding domain of RAD52 is consistent with the localisation of RAD52 at pRPA foci in BRCA2-depleted cells which further requires RAD52’s self-interaction domain, reflecting the heptameric ring formation needed for RAD52 function27.
Figure 5 |. RAD52 and BRCA2 both suppress TMEJ.
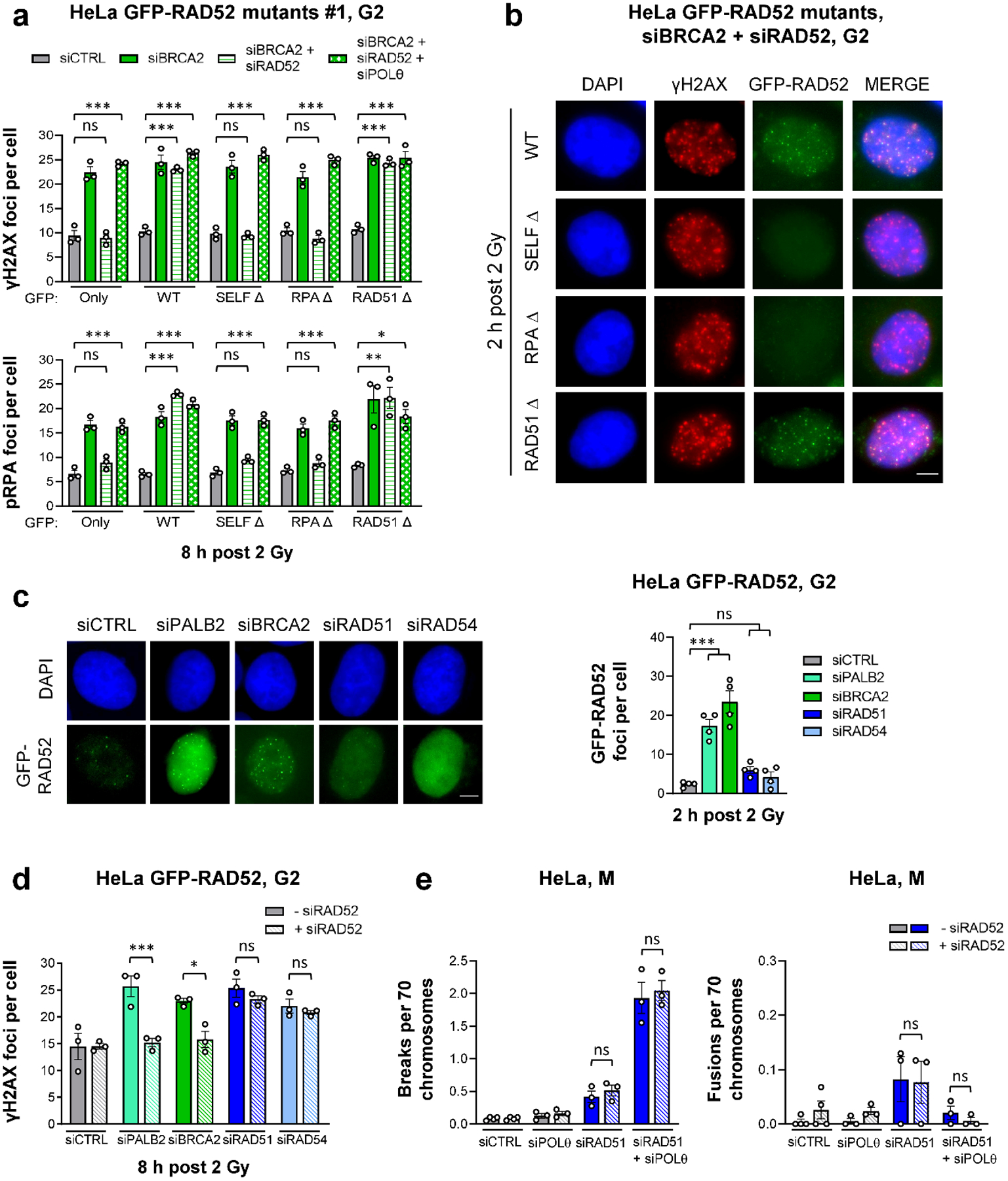
a, γH2AX and pRPA foci in G2 HeLa cells stably expressing different GFP-RAD52 constructs (shown in Extended Data Fig. 7). Two independent clones for each mutant were used: Clone #1 is shown here, while Clone #2 is shown in Extended Data Fig. 7c. Cells were transfected with siRNAs, labelled with EdU, irradiated with 2 Gy X-rays and EdU−GFP+ cells in G2 were analysed (n=3 independent experiments). b, Example IF images of γH2AX and GFP-RAD52 foci for G2 HeLa cells carrying different GFP-RAD52 constructs. Cells were transfected with siRNAs, labelled with EdU, irradiated with 2 Gy X-rays and EdU−GFP+ G2 cells were analysed (scale bar, 5 μM). c, GFP-RAD52 foci in G2 HeLa GFP-RAD52 cells. Cells were transfected with siRNAs, labelled with EdU, irradiated with 2 Gy X-rays and EdU−GFP+ G2 cells were analysed (see example IF images in the left panel, scale bar, 5 μm) (n=4 independent experiments). d, γH2AX foci in G2 HeLa GFP-RAD52 cells. Cells were transfected with siRNAs, labelled with EdU, irradiated with 2 Gy X-rays and EdU− G2 cells were analysed (n=3 independent experiments). e, Spontaneous chromatid breaks and fusions in HeLa cells. Cells were transfected with siRNAs and mitotic spreads were prepared and analysed for the presence of aberrations. Chromatid breaks and fusions were quantified per spread and normalised to 70 chromosomes (n=3 independent experiments, except siCTRL/siRAD52: n=4 independent experiments). The data without siRAD51 are taken from Fig. 4d. All data show the mean ± SEM. Individual experiments, each derived from 40 (G2 foci) and 60 cells (M spreads), are shown as dots. *: P < 0.05; **: P < 0.01; ***: P < 0.001, ns: non-significant (One-way ANOVA). The exact P values are provided as source data. Source data are available online.
We next considered if RAD52 is required to suppress TMEJ specifically when BRCA2 is absent, or in general when HR is inactivated. The depletion of PALB2, which loads BRCA2 onto resected DSBs28, led to RAD52 foci formation in G2 HeLa cells at 2 h post IR similar to the depletion of BRCA2, whilst the depletion of RAD51 or RAD54 caused no significant increase compared to siCTRL cells (Fig. 5c). Moreover, although depletion of BRCA2, PALB2, RAD51 or RAD54 led to a similar repair defect at 8 h post IR in G2, the additional depletion of RAD52 only decreased γH2AX foci numbers in BRCA2- and PALB2-depleted but not in RAD51- or RAD54-depleted cells (Fig. 5d). In addition, while the double depletion of RAD52 and BRCA2 caused a POLθ-dependent reduction in spontaneous chromatid breaks with a concomitant increase in chromatid fusions in comparison to the single depletion of BRCA2 (Fig. 4d), RAD52 depletion did not affect break or fusion numbers in cells depleted for RAD51 (Fig. 5e). This shows that RAD52 regulates TMEJ only if BRCA2 is absent or not loaded onto resected DSBs and that BRCA2 exerts a similar function to RAD52 in suppressing TMEJ. The BRCA2 protection function is independent of its role in loading RAD51, in agreement with a previously suggested RAD51-independent function of BRCA2 in stabilising resected DSBs25.
BRCA2 and RAD52 inhibit the polymerase function of POLθ
We performed biochemical experiments to characterise the molecular mechanisms by which BRCA2 and RAD52 suppress TMEJ. Since POLθ is a multi-functional enzyme with DNA polymerase and helicase-like domains, the latter suggested to remove RPA prior to DNA strand annealing29, we considered it important to use full-length POLθ for these experiments. We also purified full-length BRCA2, RAD52, RAD51 and RPA (Extended Data Fig. 8a) and studied the DNA polymerase activity of POLθ in a primer extension assay using substrates with an 80 nucleotide 5’-overhang to model the DNA synthesis step of TMEJ (Fig. 6a). Titration experiments determined the minimal POLθ concentration needed for efficient primer extension and 0.2 nM was used throughout (Extended Data Fig. 8b). Titrating RAD52 or BRCA2 (Fig. 6a, Extended Data Fig. 8c,d) showed a concentration-dependent inhibition of POLθ DNA synthesis. The inhibition is evident by the loss of full extension product and accumulation of unused primer on denaturing gels (Fig. 6a) or by the loss of full extended product and accumulation of no extension substrate on non-denaturing gels (Extended Data Fig. 8c,d). Importantly, the DNA motor activity of POLθ did not overcome the inhibitory effects of RAD52 and BRCA2, as RAD52 and BRCA2 inhibited POLθ equally in the presence and absence of ATP (Fig. 6b, Extended Data Fig. 8e,f). The strong inhibitory effect by RAD52 was not species-specific, as also yeast Rad52 inhibited POLθ DNA synthesis to a similar extent as its human counterpart (Extended Data Fig. 8g). This and the given protein:DNA ratios employed in the experiments suggest that RAD52 and BRCA2 inhibit POLθ polymerase activity via binding to single-stranded DNA (ssDNA), although we cannot rule out alternative explanations, such as inhibition due to specific protein contacts with POLθ. To show that this effect was specific for BRCA2 and RAD52 and not for other DNA binding proteins, further studies were performed with RPA and RAD51. Consistent with a previous report29, POLθ was only slightly inhibited by the addition of RPA (Extended Data Fig. 9a), and RPA only slightly, if at all, enhanced the inhibition by RAD52 and BRCA2 (Fig. 6b and Extended Data Fig. 8e–g). Also, the phage ssDNA binding protein T4 gp32 did not inhibit POLθ (Extended Data Fig. 9a). Interestingly, RAD51 which forms filaments on ssDNA did not inhibit DNA polymerase activity of either POLθ and E. coli PolI (Extended Data Fig. 9b). Inhibition of POLθ-mediated DNA synthesis was only observed with higher RAD51 amounts (beyond one RAD51 per three nucleotides), where RAD51 starts saturating the entire substrate, binding to double-stranded DNA and blocking the junction. Importantly, the combination of BRCA2 and RAD52 enhanced the inhibitory effect (Fig. 6c), indicating that they inhibit POLθ DNA synthesis by distinct mechanisms.
Figure 6 |. RAD52 and BRCA2 inhibit the polymerase function of POLθ.
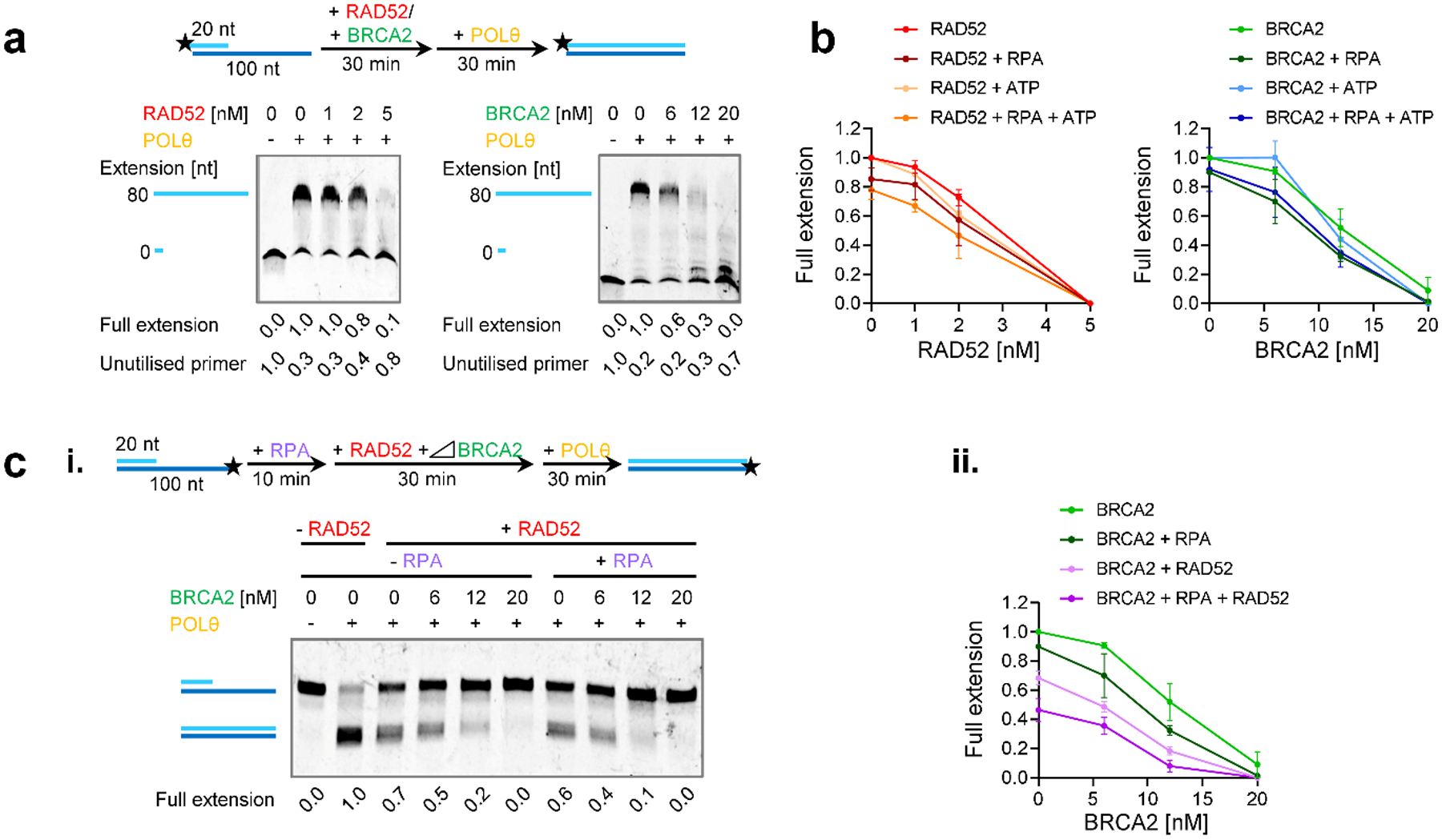
a, Effect of RAD52 and BRCA2 on POLθ-mediated DNA synthesis. Titrations of RAD52 and BRCA2 in the primer extension reaction using the 80 nucleotide 5’-overhang substrate with the 5’-Cy5-labelled 20-mer analysed in denaturing gels. b, Effect of RAD52 and BRCA2 on POLθ-mediated DNA synthesis in the presence or absence of RPA and ATP. The line graphs show quantification of the representative non-denaturing gels shown in Extended Data Fig. 8e,f and additional gels (n=3 independent experiments). c, Effect of the combination of RAD52, BRCA2 and RPA on the polymerase function of POLθ. Titration of BRCA2 in the primer extension assay using the 80 nucleotide 5’-overhang substrate with the Cy5 labelling on 5’-end of the 100-mer ssDNA strand analysed in non-denaturing gels. BRCA2 was added together with 4 nM RPA and 2 nM RAD52 in the reaction. In (i) a representative gel is shown. (ii) The line graph shows quantification of gel in (i) and additional gels (n=3 independent experiments). The quantification is combined with experiments in (b) in the absence of ATP. All data show the mean ± SEM. The schemes of the reactions are shown in the diagrams at the top. Protein concentrations are indicated. Full extension product and unutilised primer values are normalised as described in the Methods section. Source data are available online.
To gain insight into the mechanism of POLθ-inhibition by RAD52 and BRCA2, we carried out primer extension assays using a variety of DNA substrates with various length ssDNA overhangs in the range from 10 to 80 nucleotides (Fig. 7a,b). Both human and yeast full-length RAD52 bind and wrap ssDNA around their homo-heptameric rings and one human RAD52 heptameric ring wraps ~28 nucleotides27,30,31. Consistent with the DNA binding size, RAD52 inhibited POLθ DNA synthesis on the 30, 60 and 80-nucleotide overhang substrates to the same extent but showed almost no inhibition with the 10-nucleotide overhang substrate (Fig. 7a). In addition, the absence of intermediary extension products suggests that RAD52 inhibits POLθ’s ability to bind the primers, as these were fully extended once POLθ started DNA synthesis. We speculate that POLθ primer access is blocked as RAD52 wraps DNA around its ring. Furthermore, this indicates that RAD52 only binds to DNA in its multimeric form and not as a monomer, consistent with our mutant studies showing that the RAD52 self-interaction domain is essential for this function (Fig. 5a, Extended Data Fig. 7c). Surprisingly, BRCA2 inhibited POLθ DNA synthesis similarly on all substrates (Fig. 7b). The appearance of extension intermediates suggests that BRCA2 inhibits both POLθ’s primer binding and DNA extension steps. Given the two DNA binding domains in the N- and C-termini of BRCA232,33, these results suggest that BRCA2 binds to DNA differently and more dynamically than RAD52. Collectively, the in vitro experiments provide independent validation of our in vivo findings and suggest a molecular mechanism of how RAD52 and BRCA2 independently and additively might suppress TMEJ.
Figure 7 |. RAD52 and BRCA2 inhibit POLθ DNA synthesis by distinct mechanisms.
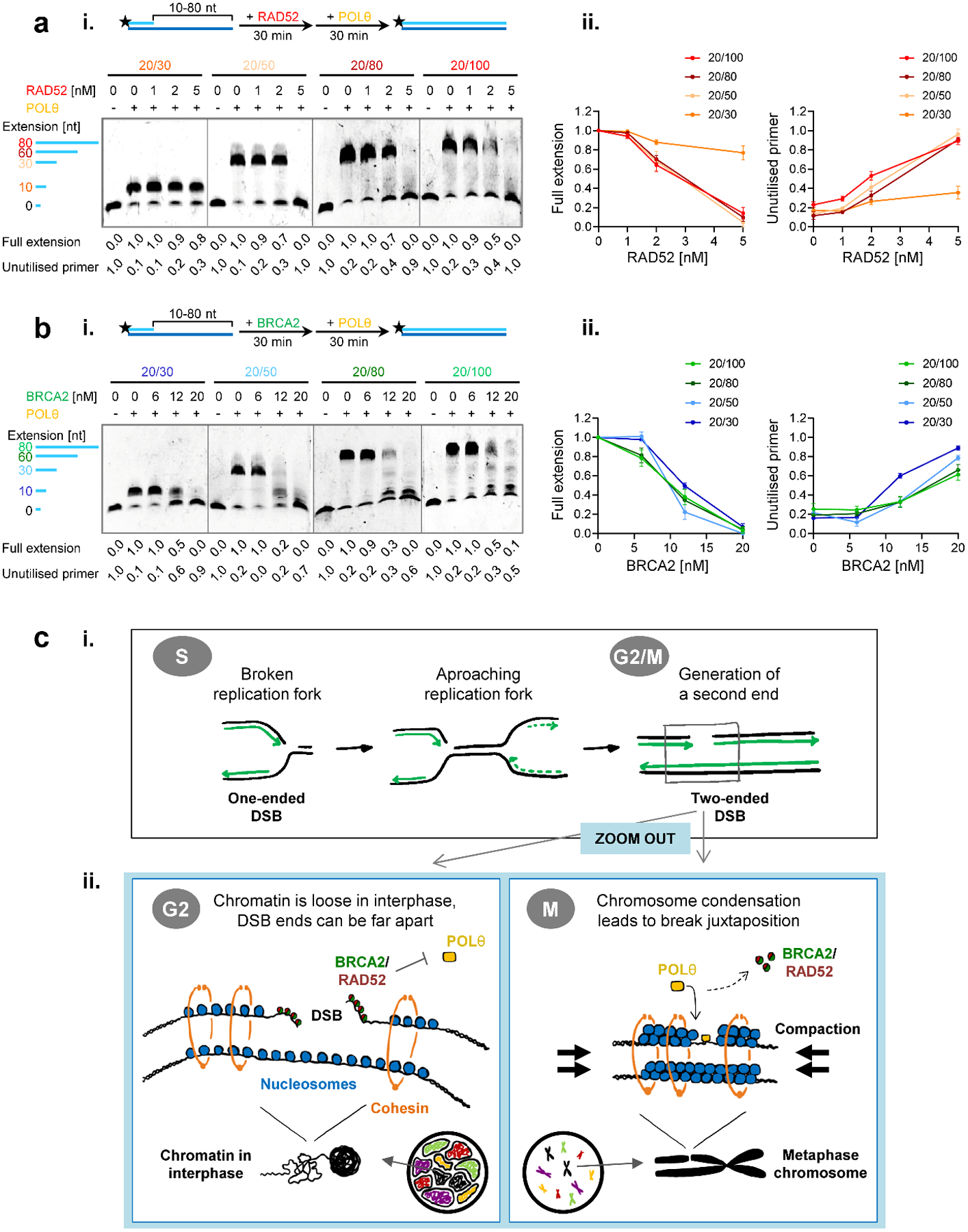
a, Effect of RAD52 on POLθ-mediated DNA synthesis with varying length DNA substrates. (i) Titration of RAD52 in the primer extension reaction using the Cy5-labelled 5’-overhang substrates analised on denaturing gels. (ii) The line graphs show quantification of gels in (i) and additional gels (n=3 independent experiments). b, Effect of BRCA2 on POLθ-mediated DNA synthesis with varying length DNA substrates. (i) Titration of BRCA2 in the primer extension reaction using the Cy5-labelled 5’-overhang substrates analised on denaturing gels. (ii) The line graphs show quantification of gels in (i) and additional gels (n=3 independent experiments). All data show the mean ± SEM. The schemes of the reactions are shown in the diagrams at the top. Protein concentrations are indicated. Full extension product and unutilised primer values are normalised as described in the Methods section. Source data are available online. c, Model for the timely regulation of TMEJ by RAD52 and BRCA2. (i) In the absence of functional HR, a one-ended DSB arising during replication stays unrepaired and can be converted into a two-ended DSB by an approaching replication fork. Such DSBs can be repaired by an end-joining mechanism although the state of chromatin condensation is likely to influence the fidelity of the rejoining process. (ii) During G2, when chromatin consists of intertwined fibres organised in topologically associated domains, the two resected break ends of a two-ended DSB can be far apart, bearing the risk of chromatid fusion formation by connecting ends from different breaks. At this stage, breaks are protected by BRCA2 and/or RAD52. These proteins block access of POLθ to break ends and, thereby, prevent the toxic premature usage of TMEJ, which would result in cell death. In contrast, in M phase, chromosome condensation juxtaposes the correct DSB ends, preventing fusion formation. At this cell cycle stage, BRCA2 and RAD52 vacate the breaks and POLθ becomes essential to repair these lesions via TMEJ and, therefore, cell survival.
Discussion
Here, we show that TMEJ repairs DSBs in BRCA2-defective cells only after the initiation of mitosis. This repair pathway is important for endogenous DSBs arising in unperturbed cells, for the additional breaks arising due to PARPi and for DSBs induced by exogenous agents such as IR and CPT. Delaying TMEJ until the onset of mitosis has the advantage that the second break end has been generated from an approaching replication fork, creating the possibility for TMEJ to rejoin correct break ends (Fig. 7ci). Moreover, we propose that the process of chromatin condensation juxtaposes correct break ends which is particularly important for unrepaired resected DSBs where the presence of long ssDNA regions might favour the joining of incorrect break ends (Fig. 7cii). We further show that RAD52 prevents the premature usage of TMEJ before the onset of mitosis and thereby limits the formation of chromosomal fusions in BRCA2-deficient cells. However, some TMEJ might already occur in the G2 phase, especially in cells where RAD52 insufficiently protects single-stranded overhangs. Although TMEJ of resected DSBs during mitosis leads to deletions, as evidenced by our molecular analysis and the study of BRCA2 tumours34, such events are less likely to compromise cell viability than the formation of chromosomal rearrangements. Thus, two factors whose losses are known to be synthetically lethal with the loss of BRCA2 and who were believed to act in different processes, in fact either regulate (RAD52) or execute (POLθ) the same repair pathway needed for the survival of BRCA2-deficient cells (Fig. 7c).
Intriguingly, RAD52 prevents the premature usage of TMEJ only in HR mutants where BRCA2 is absent or not loaded onto resected DSBs, suggesting that BRCA2 has a similar protective function as RAD52 at resected DSBs. Indeed, we show with in vitro experiments that both human and yeast RAD52 as well as BRCA2, but not ssDNA binding by RPA or RAD51, inhibit the DNA polymerase function of POLθ. Moreover, in contrast to RPA which antagonises annealing of resected DSBs35 but is overcome by the ATPase activity of POLθ29, RAD52 and BRCA2 represent an insurmountable block to POLθ function. However, we would like to note that our in vitro assay models only a specific step of TMEJ and the overall implications for the process remain to be shown.
Our discoveries have several clinical implications. First, the finding that PARPi synergises with the loss of POLθ advises that inhibitors of PARP and POLθ should be combined instead of delivered consecutively. Second, the discovery of a POLθ-blocking function of BRCA2 indicates that RAD52 inhibitors will be effective for tumours with defects in BRCA2 function (e.g., PALB2 mutants) but not for deficiencies downstream of BRCA2 (e.g., RAD51 or RAD54 mutants). Finally, the identification of TMEJ as a mitotic process suggests that even HR-proficient tumours might be treatable with a POLθ inhibitor if combined with a checkpoint inhibitor to drive cells into mitosis.
In summary, we have uncovered novel RAD51-independent roles for RAD52 and BRCA2 in restricting TMEJ until the onset of mitosis. We have shown that they both block the DNA polymerase function of POLθ at resected DSBs and thereby limit the generation of toxic chromatid fusions that arise if TMEJ is employed prematurely. Our mechanistic findings are important for the understanding of the synthetic lethality network of BRCA2-defective cells and will help to stratify tumours that best respond to the inhibition of RAD52, POLθ and/or PARP.
Methods
Cell culture.
HeLa-S3 (ATCC, purchased and authenticated by STR analysis in 2016) and HeLa GFP-RAD52 (this study) were cultured in DMEM (Sigma-Aldrich) supplemented with 10% FCS (Sigma-Aldrich or BioSell) and 1% NEAA (Merck Millipore). Immortalised human primary fibroblast 82–6 hTERT (WT)36, HSC-62 hTERT (BRCA2*)21 and 2BN hTERT (XLF*)37 cells were cultured in MEM (Sigma-Aldrich) with 20% FCS and 1% NEAA. U2OS EJ2-GFP reporter cells24 were cultured in 2 μg/ml puromycin DMEM, supplemented with 10% FCS and 1% NEAA. WT and KO U2OS cell lines (RAD52, POLθ, RAD52/POLθ)26 were cultured in DMEM (Sigma-Aldrich) supplemented with 10% FCS (Sigma-Aldrich or BioSell), 1% Glutamine (Gibco). Cells were routinely tested for mycoplasma contamination by PCR (Minerva Biolabs). All cell lines were maintained at 37 °C in a 5% CO2 incubator.
Generation of RAD52 constructs.
The hRAD52 cDNA was amplified from fibroblast (82–6 hTERT) cells with the P044 and P057 primers, which contained unique restriction sites (SalI/BamHI) used for further cloning steps. Following amplification, the cDNA was cloned into the pEGFP-C1 vector (Clontech) using the SalI and BamHI sites, generating a GFP-RAD52 expressing vector.
To generate an siRNA-resistant construct, this GFP-RAD52 vector was subjected to site-directed mutagenic PCR using RAD52_siRNAresistant_F and RAD52_siRNAresistant_R primers. Q5 polymerase (NEB) was used to amplify the plasmid, which was then digested with the restriction enzyme DpnI to degrade the methylated parent plasmid. The resulting product was then transformed into competent Dh5α E. coli cells, followed by selection and screening of single colonies. The final vector was resistant to siRAD52_3 (see RNA interference section) and was used as WT construct in the RAD52 mutant studies.
Generation of RAD52 internal deletion mutants was achieved by fusion PCR. External primers used in the PCR reactions also contained unique restriction sites (SalI/BamHI), which were used to ligate the resulting GFP-RAD52 mutant constructs (SELF Δ, RPA Δ and RAD51 Δ) into the pEGFP-C1 plasmid.
Plasmid DNA constructs were purified using ZR plasmid miniprep kit (Zymo Research) or for higher yield, peqGOLDXchange plasmid maxi-EF kit (Peqlab). All plasmid constructs were also sequenced (MWG Eurofins) to verify the presence of targeted mutations/deletions, as well as confirming the absence of undesired changes. All mutagenic and sequencing primers are listed in Supplementary Table 1.
Cell line generation
HeLa GFP-RAD52.
The different GFP-RAD52 constructs were transfected into HeLa S3 cells, followed by G418 selection (800 μg/ml) and single clone isolation. An individual clone for the GFP-RAD52 cells and two independent clones for each of the siRNA-resistant WT and deletion mutants (#1 and 2) were used.
U2OS EJ2-GFP cell line with inducible I-SceI.
U2OS EJ2-GFP cells, containing stably integrated alternative non-homologous end-joining (alt-NHEJ) substrates24, were stably transfected with inducible I-SceI-GR-RFP plasmid (gift from Tom Misteli, Addgene plasmid #17654) using FuGENEHD (Promega). The surviving population following G418 selection (800 μg/ml) was used for this study (see Reporter assay section).
Cell irradiation and drug treatment.
X-ray irradiation was performed at 90 kV and 19 mA for all experiments, with a 1 mm aluminium plate serving as a sample holder and a low-energy X-ray filter. The machine used a Philips X-ray tube equipped with a tungsten anode and a thin beryllium window. Irradiation was carried out with consideration of the dose-doubling effect for cells on glass coverslips38, thereby adjusted irradiation durations were used accordingly. The topoisomerase 1 inhibitor camptothecin (CPT, 20 nM, Sigma-Aldrich) was added to cells for 1 h, these were then washed twice with PBS and fresh media was added back during repair incubation. Olaparib was used to inhibit PARP (PARPi, 1 μM, Selleckchem) and was added to cells for 24 h.
RNA interference and plasmid transfection.
Transfection of HeLa, fibroblast and U2OS cells with specific siRNAs (sequences listed in Supplementary Table 2) was carried out using HiPerFect (QIAGEN) or Lipofectamine RNAiMAX (Thermo Fisher Scientific) transfection reagents according to the manufacturer’s instructions. Experiments were performed 48 h after transfection, unless otherwise stated. For HiPerFect transfections, a second round of transfection was necessary to obtain efficient knockdowns, while with Lipofectamine RNAiMAX, one round was sufficient. For RAD52 depletion, a combination of two siRNAs was used in a 1:1 ratio (RAD52_1 and RAD52_2), except for the study of the GFP-RAD52 mutants, where a single siRNA was used (RAD52_3). For plasmid transfection, Lipofectamine LTX (Thermo Fisher Scientific) was used according to the manufacturer’s instructions.
Cell cycle-specific DSB repair analyses
Analysis of total G1 or G2 cells.
To analyse DSB repair in a cell cycle-specific manner, the thymidine analogue EdU (10 μM, Invitrogen) was added to the cells for 1 h. Spontaneous and PARPi-induced foci were analysed immediately after, whilst for IR-induced breaks, EdU was kept throughout the experiment. After fixation, DAPI and EdU intensities were measured and plotted in a diagram using a Zeiss microscope and Metafer4 software (Metasystems). EdU− cells were categorised as either G1 or G2 according to their DNA content (see Extended Data Fig. 1a).
Double EdU/BrdU labelling for spontaneous breaks.
To analyse endogenous levels of DSBs in cells progressing from S phase into G2 and G1, a different approach was used. Cells were first incubated with EdU (10 μM). After 1 h, cells were washed with PBS and then incubated with a different thymidine analog, BrdU (10 μM, BD Bioscience). After 2 h, cells were again washed and the EdU−/BrdU+ cell population was followed as it progressed from S phase into the subsequent cell cycle phases (see Fig. 1a,b).
EdU labelling for CPT-induced breaks.
To analyse CPT-treated cells from S phase into G2 and G1, cells were incubated with EdU (10 μM) and CPT for 1 h, washed twice with PBS and, 12 h later, EdU+ cells were analysed (see Extended Data Fig. 2a).
Double EdU/BrdU labelling for IR-induced breaks.
To specifically analyse G2-irradiated cells in the following G1 phase, the strategy above was slightly modified. Cells were first incubated with EdU (10 μM) for 3 h. Next, cells were washed, irradiated and labelled with BrdU (10 μM), which was kept on during repair incubation. We focused on EdU+BrdU− cells, as they were in late S phase during the EdU labelling step, but had already entered G2 phase at the time of irradiation. In particular, EdU+BrdU− cells remaining in G2 or having progressed to G1 were analysed (see Extended Data Fig. 2b).
Analysis of mitotic cells.
Mitotic cells were identified with pH3(S10) staining. Positive cells were then sub-classified into the different mitotic stages according to cell morphology seen in the DAPI/pH3 channel, and γH2AX foci, lagging chromosomes and DNA bridges were scored.
Immunofluorescence.
Cells grown on glass coverslips were fixed with 2% formaldehyde in PBS for 15 min at room temperature (RT), washed three times with PBS and permeabilised with 0.2% Triton X-100 in 1x Roti-ImmunoBlock (Roth) for 10 min at RT. Cells were then washed, blocked for 30–60 min at RT with 1x Roti-ImmunoBlock or with PBS + 5% BSA + 1% FCS and incubated with primary antibodies over night at 4 °C. To stain chromatin-bound PCNA, cells were fixed with methanol for 15 min at −20 °C after a pre-extraction step with 10 μM TRIS-HCl (pH 7.5), 2.5 mM MgCl2, 1 mM PMSF and 0.5% NP-40 Alternative for 8 min at 4 °C. Afterwards cells were washed with PBS and PBS + 0.1% Triton X-100. The following antibodies were used: BrdU (1:500; BD Pharmigen 3D4, #555627), GFP (1:2000; Roche, #11814460001), γH2AX (1:2000; Abcam, #ab81299, or Merck Millipore, #05–636), pH3 (1:1000; Merck Millipore, #06–570), PCNA (1:200; Santa Cruz PC-10, #sc-56), RAD51 (1:10000; Abcam, #ab63801), pRPA (1:10000; Abcam RPA32 pT21, #ab109394). Cells were then washed with PBS and, for cell cycle-specific analysis, EdU was stained using the EdUClick Kit (Baseclick, green or Cy5), according to the manufacturer’s instructions. Cells were then washed with PBS and incubated with fluorescently tagged secondary antibodies (Goat anti-mouse AlexaFluor 488 (Cat#A11001), Goat anti-mouse AlexaFluor 594 (Cat#A11005), Goat anti-rabbit AlexaFluor 488 (Cat#A11008), Goat anti-rabbit AlexaFluor 594 (Cat#A11012), Molecular Probes) for 1 h at RT. Cells were washed in PBS and stained with DAPI (0.4 μg/ml, Sigma-Aldrich) for 5 min at RT then embedded with Vectashield Antifade Mounting Media (Vector Laboratories). Cells were examined with a Zeiss microscope using the Metafer4 or the ZEN 2.6 (Blue Edition) software and, for each experiment, foci were enumerated manually in at least 40 nuclei in G2 and mitosis, and 100 nuclei in G1, unless otherwise stated. Representative pictures were processed with the ImageJ software, adjusting individually the brightness/contrast of every image when necessary.
Western blotting.
Knockdown efficiencies and expression of exogenous plasmids were confirmed by immunoblotting (Extended Data Figs. 7b and 10). Protein extracts were prepared for SDS-PAGE in Laemmli buffer (10% SDS, 300 mMTris-HCL, 10 mM β-mercaptoethanol, 50% glycine, 0.02% bromophenol blue), supplemented with protease inhibitors (Roche, cOmplete inhibitor cocktail). Separated proteins were transferred to a nitrocellulose membrane (Amersham), blocked for 1 h at RT in 1x Roti-Block (Roth) and incubated with the following primary antibodies overnight at 4 °C: BRCA2 (Merck Millipore Ab-1, #OP95), GAPDH (Santa Cruz 6C5, #sc-32233), GFP (Santa Cruz FL, #sc-8334), KU70 (Santa Cruz A-9, #sc-5309), KU80 (Santa Cruz 3241C1a, #sc-81308), LIG4 (Abcam [EPR16531], #ab193353), PALB2 (Bethyl, #A301-246A), RAD51 (Abcam, #ab63801), RAD52 (Santa Cruz F-7, #sc-365341), RAD54 (Santa Cruz A-4, #sc-166370) and Vinculin (Santa Cruz H-10, #sc-25336). All antibodies were used at a dilution of 1:1000. Membranes were then washed and incubated with HRP-conjugated secondary antibodies (Donkey anti-mouse IgG-HRP (1:10000, Cat#715-035-150), Donkey anti-rabbit IgG-HRP (1:30000, Cat#711-035-152), Dianova) for 1 h at RT. Immunoblots were developed using Western Bright Quantum or Sirius Kits (Advansta) and signal detection was performed with a Fusion FX Vilbersystem (Lourmat).
Quantitative Polymerase Chain Reaction (qPCR).
Analysis of POLθ depletion was carried out by RT-qPCR (Extended Data Fig. 10b). RNA was extracted 48 h after siRNA transfection using MasterPure RNA purification kit (Lucigen), according to manufacturer’s instructions. 1μg of RNA was then used for cDNA generation with the RevertAid First Strand cDNA reverse transcription kit (Thermo Fisher Scientific). The obtained cDNA was analysed by qPCR using FastStart Universal SYBR Green Master (Roche) according to manufacturer’s instructions and primers targeting POLθ, and the housekeeping gene GAPDH (primer sequences listed in Supplementary Table 1). Amplification was performed using the Comparative CT settings in a StepOne Real-Time PCR system (Applied Biosystems) with the following program: 95 °C for 10 min, 40 cycles of 95 °C for 15 s, 59 °C for 30 s and 70 °C for 30 s, followed by a melting curve analysis to confirm primer specificity. All samples were run in triplicates (three technical replicates) and Ct values were first normalised to GAPDH, and then compared to siCTRL samples.
Chromosome analysis.
HeLa and fibroblast cells were transfected with siRNAs and treated with 20 nM CPT or irradiated with 2 Gy and harvested at 8 – 10 h post damage induction. For premature chromosome condensation experiments (PCCs) to visualise chromatid breaks in G2, cells were treated with calyculin A (50ng/ml, LC Laboratories) for 30 min prior to fixation. To obtain mitotic spreads, cells were treated with colcemid (10 μg/ml; Thermo Fisher Scientific, KaryoMAX) to arrest/accumulate cells in metaphase. For preparation of chromosome spreads, cells were collected, resuspended in pre-warmed 75 mM KCl for 25 min at 37 °C and centrifuged at 400 × g and 4 °C for 10 min. Cells were then fixed (3:1 methanol:acetic acid) three times and chromosomes were spread onto coverslips, air-dried and stained with Giemsa (Sigma-Aldrich). For each sample, 30–60 chromosome spreads were captured and analysed using a Zeiss microscope with Metafer4 software.
Reporter assay.
U2OS EJ2-GFP cells expressing inducible I-SceI enzyme were transfected with siRNAs and 48 h later treated with the steroid ligand triamcinolone acetonide (TA) (200 nM, Sigma-Aldrich) to induce I-Sce-I nuclear localisation and DSB generation. After 24 h, cells were either fixed or sorted to allow analysis of alt-NHEJ repair events. To score repair efficiencies, fixed cells were stained against GFP and DAPI and at least 10,000 cells were analysed using a Zeiss microscope and Metafer4 software. GFP-positive cells representing alt-NHEJ events were enumerated and normalised to the total cell number. To confirm the usage of alt-NHEJ during repair in this system, at least 20,000 GFP+ cells were recovered using a S3 Bio-Rad cell sorter. Genomic DNA was extracted from sorted GFP− and GFP+ cells using the MasterPure Complete DNA and RNA Purification Kit (Lucigen) and PCR products were then amplified with Q5 polymerase (NEB) using the primers EJ2_FWD and EJ2_REV, as previously described24. The PCR products were digested either with I-SceI or XcmI restriction enzymes (see Extended Data Fig. 5bii,iii).
Oligonucleotide and protein purification
DNA substrates.
All oligonucleotides (sequences listed in Supplementary Table 3) were purified by polyacrylamide gel electrophoresis (PAGE). The different 5’-overhang substrates for analysis on denaturing gels were annealed at a 1:1 molar ratio with the Cy5-labelled 20-mer oligonucleotide ss20-POLQ and the complementary strand in various sizes: 30 nucleotides (ss30-POLQ), 50 nucleotides (ss50-POLQ), 80 nucleotides (ss80-POLQ) and 100 nucleotides (ss100-POLQ) by boiling and then slowly cooling to RT. The 80 nucleotide 5’-overhang substrate for non-denaturing gel analyses was annealed at a 1:1 molar ratio with the ss20-POLQ and 100-mer Cy5-labelled oligonucleotide ss100-POLQ by boiling and then slowly cooling to RT.
Full length human POLθ.
The protein was expressed and purified as described39 with some modifications. The cDNA for POLθ –FL40 was codon-optimised (Genscript, Piscataway NJ) and cloned into phCMV1–2x MBP41. Expi293F cells were transfected with a 1:3 (w/w) mixture of DNA:polyethylenimine per liter. Transfected cells (600 ml, 1 × 106 cells/ml) were incubated in Expi293 Expression Media (Thermo Fisher Scientific) on a shaker platform at 37 °C, 8% CO2 for 48 h. Cells were lysed in 30 ml buffer (50 mM Tris pH 7.5, 500 mM NaCl, 10% Glycerol, 0.01% IGEPAL CA-630, 5 mM 2-mercaptoethanol, 5 mM EDTA, and Roche cOmplete EDTA-free protease inhibitor) for 15 min on ice followed by Dounce homogenization (15 strokes). After centrifugation (20,000 × g, 4 °C, 30 min), the supernatant was diluted to 250 mM NaCl for batch binding with 2.5 ml amylose resin (NEB) for 16 h. The resin was washed with 10 volumes of buffer A (50 mM Tris pH 7.5, 250 mM NaCl, 10% glycerol, 0.01% IGEPAL CA-630, 5 mM 2-mercaptoethanol, 5 mM EDTA, and protease inhibitor tablets) and was eluted with buffer A containing 10 mM maltose. The eluate was loaded onto a 1 ml Hi-Trap HP SP column (GE). The column was washed (AKTA purifier) with 10 column volumes of buffer A and eluted with 10 column volume gradient from buffer A to buffer B (50 mM Tris pH 7.5, 1 M NaCl, 10% glycerol, 0.01% IGEPAL CA-630, 5 mM 2-mercaptoethanol, 5 mM EDTA). Fractions containing POLθ were combined, treated with PreScission Protease and dialyzed with buffer A for 16 h. After incubation with glutathione Sepharose 4B (GE) and amylose resin for 4 h at 4 °C, the flow-through was collected. The POLθ protein was free of unrelated DNA polymerase activities and endo- and exonuclease. The quality of the protein preparation was controlled by mass spectrometry.
Human RAD52.
Plasmid pEG161H1 (pET15b-6HIS-RAD52; pWDH762) was introduced into E. coli Rosetta (DE3) (Novagen). Expression of His6-RAD52 protein was induced on addition of IPTG (0.25 mM) and His6-RAD52 protein was purified essentially as described42 with the following column changes: a HisPur Ni-NTA resin 5 ml column (Thermo Fisher Scientific), a 5 ml Affi-Gel Heparin column (BioRad), and a 1 ml MonoS column (GE Healthcare). Purified His6-RAD52 protein was stored in 50 mM Tris-HCl, pH 7.5, 300 mM KCl, 1 mM EDTA, 1 mM DTT, and 10% glycerol at −80 °C. The RAD52 protein was free of endo- and exonuclease contaminations43. RAD52 concentrations are expressed as nanomolar heptamers, as full-length human RAD52 primarily exists as a heptameric ring27.
Other proteins used.
Human BRCA244, RAD5145, and RPA46 were purified as described. S. cerevisiae RPA47 and Rad5248 were purified as described. PolI (Klenow fragment, 3’→5’ exo) and T4 gp32 were purchased from NEB.
Primer extension assay.
2 nM 5′-Cy5-labelled DNA substrate was pre-incubated with or without RAD52 and/or BRCA2 at the indicated concentrations in the reaction buffer (25 mM potassium phosphate pH 7.0, 5 mM MgCl2, 0.1 mg/ml BSA, 5 mM DTT, 100 μM dNTPs and 100 μM ATP) at 37 °C for 30 min. In the reactions with RPA, 4 nM RPA or the indicated concentrations were pre-incubated with DNA for 10 min prior to adding RAD52 or BRCA2. The polymerase reaction was started by adding 0.2 nM POLθ for 30 min in a total volume of 10 μl. The reactions were terminated by the addition of 2 μl of STOP buffer (3 mg/ml proteinase K, 120 mM EDTA, and 1.2 % SDS) and incubated for another 30 min. The DNA species were resolved on 10% non-denaturing or 15% denaturing polyacrylamide gels and analysed by measuring the Cy5 fluorescence intensity with an Amersham Imager 600 (GE Healthcare). The percentage of extended product was quantified by the ratio between extended product and total DNA substrate. To normalise the percentage of extended product, the level of the POLθ-only reaction was considered as 100% (for ±ATP, respectively). The unutilised primer was quantified by DNA intensity of the remaining substrate and then normalised with total DNA substrate in the reaction without POLθ. Notably, because of the difficulty in purifying high-concentration stocks of full-length BRCA2 and POLθ, we limited the amount of DNA substrate. 2 nM DNA substrate is the lowest detectable amount of fluorescent DNA on the gels, which explains the high background in the gel images.
Statistics & Reproducibility.
Experiments were not randomised and no blinding was used during data analyses. Sample size was determined based on previous experience to obtain statistical significance and reproducibility. Unless indicated differently, all data were derived from at least n=3 biological replicates and for each sample at least 40 nuclei or 30 chromosome spreads were analysed. The particular number of biological replicates for each experiment (n) and the number of samples analysed (cells/spreads, if applicable) is indicated in the figure legends. In general, no data were excluded from the analyses, except in rare cases of contamination of cell culture samples or low staining quality. Background foci were not subtracted from the mean values in experiments using DNA damaging agents. Graphs show the mean value and the error bars show SEM between the experiments, unless otherwise indicated. Images of blots, gels, immunofluorescence stainings or chromosome spreads show representative examples of multiple independent experiments, unless otherwise indicated. Data and images were processed and visualised with Microsoft Office 2010 (Powerpoint and Excel), ImageJ (v 1.53c) and GraphPad Prism 9. All statistical tests were also performed using GraphPad Prism 9. Mean values of each experiment were compared using either an unpaired two-tailed t-test or a one-way ANOVA, depending on the number of samples that were analysed per experiment (two or more, respectively). Time points, cell cycle phases and GFP-RAD52 cell lines were compared independently. If ANOVA showed statistical significance, a Tukey test was subsequently performed to obtain individual P values. *, P < 0.05; **, P < 0.01; ***, P < 0.001. All data points for biological replicates and individual P values are provided as source data.
Reporting summary.
Further information on research design is available in the Nature Reporting Summary attached to this article.
Data availability
Source data are provided with this study. All data supporting the findings of this study are available from the corresponding author on reasonable request.
Supplementary Material
Acknowledgements
We thank Jeremy Stark for providing the U2OS EJ2 reporter and POLθ/RAD52 KO cell lines, as well as Alexander Löwer and Penny Jeggo for helpful discussions. We thank Ratna Weimer, Christel Braun, and Megan P. Lowery for technical assistance and Dorothee Deckbar and Andreas Taubmann for experimental and intellectual contributions during early stages of the project. Work in the ML laboratory is supported by the Deutsche Forschungsgemeinschaft (DFG, project ID 393547839 - SFB 1361) and the Bundesministerium für Bildung und Forschung (02NUK054C, 02NUK050B, 02NUK042D), work in the WDH laboratory is supported by the National Institutes of Health (GM58015, GM137751), and work in the RDW laboratory is supported by the National Institutes of Health (CA247773, CA193124) and by the J. Ralph Meadows Chair in Carcinogenesis Research. This research was also supported by the National Institutes of Health award CA187561 to JL and by core services supported by P30 CA93373.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Roy R, Chun J & Powell SN BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 12, 68–78 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prakash R, Zhang Y, Feng W & Jasin M Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol 7, a01660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Higgins GS & Boulton SJ Beyond PARP-POLθ as an anticancer target. Science 359, 1217–1218 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Ashworth A & Lord CJ Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat. Rev. Clin. Oncol 15, 564–576 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Jensen RB, Carreira A & Kowalczykowski SC Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467, 678–683 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J, Doty T, Gibson B & Heyer WD Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol 17, 1260–1262 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ceccaldi R et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 518, 258–262 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mateos-Gomez PA et al. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wood RD & Doublié S DNA polymerase θ (POLQ), double-strand break repair, and cancer. DNA Repair (Amst) 44, 22–32 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wyatt DW et al. Essential Roles for Polymerase θ-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 63, 662–673 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng Z et al. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. U. S. A 108, 686–691 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sotiriou SK et al. Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol. Cell 64, 1127–1134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mazina OM, Keskin H, Hanamshet K, Storici F & Mazin AV Rad52 Inverse Strand Exchange Drives RNA-Templated DNA Double-Strand Break Repair. Mol. Cell 67, 19–29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhowmick R, Minocherhomji S & Hickson ID RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 64, 1117–1126 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Spies J et al. 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat. Cell Biol 21, 487–497 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Hengel SR et al. Small-molecule inhibitors identify the RAD52-ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. Elife 5, e14740 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang F et al. Targeting BRCA1-and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 44, 4189–4199 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanamshet K, Mazina OM & Mazin AV Reappearance from obscurity: Mammalian Rad52 in homologous recombination. Genes (Basel). 7, 63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Löbrich M et al. γH2AX foci analysis for monitoring DNA double-strand break repair: Strengths, limitations and optimization. Cell Cycle 9, 662–669 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Pommier Y, Sun Y, Huang SYN & Nitiss JL Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol 17, 703–721 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Juhász S, Elbakry A, Mathes A & Löbrich M ATRX Promotes DNA Repair Synthesis and Sister Chromatid Exchange during Homologous Recombination. Mol. Cell 71, 11–24.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Spies J et al. Nek1 Regulates Rad54 to Orchestrate Homologous Recombination and Replication Fork Stability. Mol. Cell 62, 903–917 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howlett NG et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 297, 606–609 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Bennardo N, Cheng A, Huang N & Stark JM Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4, e1000110 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han J et al. BRCA2 antagonizes classical and alternative nonhomologous end-joining to prevent gross genomic instability. Nat. Commun 8, 1470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelso AA, Lopezcolorado FW, Bhargava R & Stark JM Distinct roles of RAD52 and POLQ in chromosomal break repair and replication stress response. PLoS Genet. 15, e1008319 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stasiak AZ et al. The human Rad52 protein exists as a heptameric ring. Curr. Biol 10, 337–340 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Ducy M et al. The Tumor Suppressor PALB2: Inside Out. Trends Biochem. Sci 44, 226–240 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Mateos-Gomez PA et al. The helicase domain of Polθ counteracts RPA to promote alt-NHEJ. Nat. Struct. Mol. Biol 24, 1116–1123 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grimme JM et al. Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes. Nucleic Acids Res. 38, 2917–2930 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shinohara A, Shinohara M, Ohta T, Matsuda S & Ogawa T Rad52 forms ring structures and co-operates with RPA in single-strand DNA annealing. Genes to Cells 3, 145–156 (1998). [DOI] [PubMed] [Google Scholar]
- 32.Von Nicolai C, Ehlén Å, Martin C, Zhang X & Carreira A A second DNA binding site in human BRCA2 promotes homologous recombination. Nat. Commun 7, 1–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang H et al. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science 297, 1837–1848 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Zámborszky J et al. Loss of BRCA1 or BRCA2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene 36, 746–755 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng SK, Gibb B, De Almeida MJ, Greene EC & Symington LS RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol 21, 405–412 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oshima J, Pae C, Huang S, Campisi J, and Schiestl RH Lack of WRN results in extensive deletion at nonhomologous joining ends. Cancer Res. 62, 547–551 (2002). [PubMed] [Google Scholar]
- 37.Ahnesorg P, Smith P & Jackson SP XLF interacts with the XRCC4-DNA Ligase IV complex to promote DNA nonhomologous end-joining. Cell 124, 301–313 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Kegel P, Riballo E, Kühne M, Jeggo PA & Löbrich M X-irradiation of cells on glass slides has a dose doubling impact. DNA Repair (Amst) 6, 1692–1697 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Zahn K, Jensen RB, Wood RD & Doublié S Human DNA polymerase θ harbors a DNA end-trimming activity critical for DNA repair. Mol. Cell, online ahead of print (2021). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Seki M, Marini F & Wood RD POLQ (Pol θ), a DNA polymerase and DNA-dependent ATPase in human cells. Nucleic Acids Res. 31, 6117–6126 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jensen R Purification of Recombinant 2XMBP Tagged Human Proteins from Human Cells. Methods Mol. Biol 1176, 209–217 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Rothenberg E, Grimme JM, Spies M & Ha T Human Rad52-mediated homology search and annealing occurs by continuous interactions between overlapping nucleoprotein complexes. Proc. Natl. Acad. Sci. USA 105, 20274–20279 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang XP & Heyer WD Quality control of purified proteins involved in homologous recombination. Methods Mol. Biol 745, 329–343 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le HP et al. DSS1 and ssDNA regulate oligomerization of BRCA2. Nucleic Acids Res. 48, 7818–7833 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carreira A et al. The BRC Repeats of BRCA2 Modulate the DNA-Binding Selectivity of RAD51. Cell 136, 1032–1043 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sneeden JL, Grossi SM, Tappin I, Hurwitz J & Heyer WD Reconstitution of recombination-associated DNA synthesis with human proteins. Nucleic Acids Res. 41, 4913–4925 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Binz SK, Dickson AM, Haring SJ & Wold MS Functional Assays for Replication Protein A (RPA). Methods in Enzymol. 409, 11–38 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Van Komen S, Macris M, Sehorn MG & Sung P Purification and Assays of Saccharomyces cerevisiae Homologous Recombination Proteins. Methods in Enzymol. 408, 445–463 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data are provided with this study. All data supporting the findings of this study are available from the corresponding author on reasonable request.