

Abstract
The world is grappling with the COVID-19 pandemic caused by the 2019 novel SARS-CoV-2. To better understand this novel virus and its relationship with other pathogens, new methods for analyzing the genome are required. In this study, intrinsic dinucleotide genomic signatures were analyzed for whole genome sequence data of eight pathogenic species, including SARS-CoV-2. The genome sequences were transformed into dinucleotide relative frequencies and classified using the extreme gradient boosting (XGBoost) model. The classification models were trained to a) distinguish between the sequences of all eight species and b) distinguish between sequences of SARS-CoV-2 that originate from different geographic regions. Our method attained 100% in all performance metrics and for all tasks in the eight-species classification problem. Moreover, the models achieved 67% balanced accuracy for the task of classifying the SARS-CoV-2 sequences into the six continental regions and achieved 86% balanced accuracy for the task of classifying SARS-CoV-2 samples as either originating from Asia or not. Analysis of the dinucleotide genomic profiles of the eight species revealed a similarity between the SARS-CoV-2 and MERS-CoV viral sequences. Further analysis of SARS-CoV-2 viral sequences from the six continents revealed that samples from Oceania had the highest frequency of TT dinucleotides as well as the lowest CG frequency compared to the other continents. The dinucleotide signatures of AC, AG,CA, CT, GA, GT, TC, and TG were well conserved across most genomes, while the frequencies of other dinucleotide signatures varied considerably. Altogether, the results from this study demonstrate the utility of dinucleotide relative frequencies for discriminating and identifying similar species.
Keywords: Alignment-free sequence analysis, COVID-19, dinucleotide frequencies, feature representations, genomic signatures, human pathogens, machine learning, XGBoost
I. Introduction
Coronaviruses (CoVs) are enveloped, linear, positive-sense, single-stranded ribonucleic acid (RNA) viruses approximately 30kb in length [1]. Belonging to the family Coronaviridae and the subfamily Orthocoronavirinae, members of the Betacoronavirus genus have been shown to cause infection in humans [2]. Three coronavirus outbreaks have caused moderate to severe respiratory diseases in the last two decades: the 2002 Severe Acute Respiratory Syndrome (SARS) outbreak [3], the 2012 Middle East Respiratory Syndrome (MERS) outbreak [4], and the current Coronavirus Disease 2019 (COVID-19) pandemic. In December 2019, the first cases of infection with the novel SARS-related CoV-2 (SARS-CoV-2) were reported in Wuhan, China with subsequent spread to more than 180 countries resulting in nearly 15 million COVID-19 cases and more than 600 000 deaths worldwide [5].
During a virus outbreak, the taxonomic classification of a pathogenic species and understanding its relatedness to other pathogens may aid in the development of appropriate mitigation strategies. For example, global efforts to design and develop a vaccine for SARS-CoV-2 and therapeutic drugs may benefit greatly from the early identification of SARS-CoV-2 as a close relative to MERS-CoV and SARS [6], through improved understanding of possible disease progression, host pathogen interactions and potential treatment strategies.
Several alignment-based methods have been used to determine species relatedness [7], [8], but as sequencing technologies improve and available datasets become significantly larger, application of these methods for multiple sequences become computationally inefficient [9], [10]. Alignment-based methods rely on the availability of well-characterized reference sequences, thereby limiting the discovery of novel characteristics embedded within a species’ genome [8]. Thus, several alignment-free methods have been proposed for rapid sequence analyses [11].
This work establishes the usefulness of an alignment-free and machine learning-based taxonomic classification approach using the dinucleotide genomic signatures of several pathogenic species. Specifically, this study examines the relative frequencies of 16 dinucleotide pairs derived from fully assembled whole genome sequences (WGS) of eight human infecting species, including SARS-CoV-2. The same examination was implemented on SARS-CoV-2 sequences only, with an aim to evaluate the usefulness of this approach in an effort to understand a novel pathogen. Understanding the variability of dinucleotide genomic profiles among viruses and other related species is of particular importance during a pandemic. Currently, it is unclear to what extent host species or virus family influence dinucleotide frequencies and whether dinucleotide genomic signatures can be used to accurately predict species using machine learning approaches.
The significance of dinucleotide patterns was first reported in the 1960s when biochemical experiments performed on genomic DNA unraveled remarkable species-specific dinucleotide genomic patterns [12]. However, detailed exploration of dinucleotide patterns was hampered by the limited availability of complete genomes, rendering most earlier inferences speculative [13]. As the field of genomics continues to evolve, more WGS are being made available, providing tremendous opportunities for detailed exploration of dinucleotide genomic signatures. In the aforementioned biochemical experiments, dinucleotide genomic signatures were found to be associated with repair-based enzymes, structural features and replication mechanisms [14], and dinucleotide frequencies were found to be more homogeneous in GC-rich genomes than in AT-rich genomes [15]. Although the main reason for this difference is not clearly understood, genomes with an abundance of the AT genomic signature have often been associated with smaller genomes consisting of fewer genes [16]. In addition, these genomes appear to be prone to mutational bias possibly due to a loss of repair genes [17] or relaxed selective pressures [18]. These early experiments served as motivation for the continued exploration of underlying dinucleotide patterns.
II. Related Work
To date, numerous alignment-free numerical DNA characterization or representation schemes have been proposed [11]. One such widely-used class of representation schemes produces fixed-length numerical representations based on frequency mappings, and these fixed-length vectors are convenient as they facilitate an efficient comparison between DNA sequences. The rationale for using frequency-based mappings is that the occurrence of nucleotides differs in the different regions of the genome both within a species and between species [19]. As a result of these differences, nucleotides can be encoded by their frequency of occurrence. For instance, mononucleotide frequencies are known to differ in the coding and non-coding regions of the genome [20], and have been effectively used in detecting these regions. Besides the use of mononucleotide frequencies, frequencies of dinucleotides, trinucleotides [21]–[25] and tetranucleotides [26], [27] have also been used to numerically represent DNA data. One shortcoming of these methods is the potential loss of valuable genomic information arising when condensing a possibly lengthy DNA sequence into a small fixed set of statistical descriptors [21]. Another popular class of alignment-free representation schemes is one that is based on information theory principles such as entropy, although numerous other representations do not fall in either of these two categories [11]. A comprehensive review of the recent numerical encoding schemes can be found in [28].
The conversion of DNA sequences from nucleotides to numerical representation is a critical component of pipelines in many computational genomics applications. One such application area is taxonomy classification where species are classified into groups based purely on their genomic sequences. For this task, alignment-free approaches have been implemented such as in [29]–[31], where in [29] a deep learning approach was taken to distinguish viral sequences from non-viral sequences in a pool of diverse genomic human samples. Whilst [30] proposed a model to classify subtypes of HIV-1 genomes based on varying sub-sequence lengths (lengths ranging from one through ten), [31] used sub-sequences of length seven in conjunction with chaos game numerical representations to build machine learning models for the purposes of classifying COVID-19 genomic sequences. Specifically, different machine learning models were trained to classify viral genomes at different levels of taxonomy, and these models were utilized to predict the correct classification of the COVID-19 samples within the different taxonomic levels.
III. Materials and Methods
A. Data Collection
Fully assembled, WGS data in FASTA format were retrieved for eight pathogenic species namely, SARS-CoV-2, MERS-CoV, Dengue Virus (DENV), Zaire Ebolavirus (EBOV), Hepatitis B virus (HBV), Hepacivirus C (HCV), Human Immunodeficiency Virus 1 (HIV-1) and Mycobacterium tuberculosis (M. tb). The rationale for including these datasets are firstly because SARS-CoV-2, MERS-CoV, and M. tb all cause diseases affecting the human respiratory system. Secondly, M. tb and HIV-1 are well-established co-infections in low-and middle-income countries such as South Africa [32]. Lastly, EBOV [33], DENV [34], HBV [35], and HCV [36] are responsible for epidemics in the tropical regions, causing similar vascular symptoms to those seen in COVID-19 patients [37].
Complete, high coverage sequences for SARS-CoV-2 from Africa, Asia, Europe, Oceania, North America and South America were downloaded from the GISAID database [38]. In addition, viral and bacterial sequences were sourced from several publicly accessible databases. HCV and HIV-1 sequences were downloaded from the Los Alamos National Laboratory (LANL) database [39], while MERS-CoV, DENV, EBOV, HBV, and M. tb sequences were downloaded from the National Center for Biotechnology Information (NCBI) database [40]. Ethical approval was not required for this study as the samples used were sourced from publicly accessible websites and contain no personally identifiable information. Hereafter, any analyses using the eight species’ sequences are referred to as between species. In addition, any analysis using only the SARS-CoV-2 sequences will be referred to as within species. Analyses were conducted using the Python programming language [41] and R statistical language [42].
B. Data Preprocessing
To ensure that only high quality WGS data was included in this analysis, several preprocessing steps were followed (Fig. 1). To remove duplicate sequences, an in-house Python script was used to identify any sequences that had the same accession number and genomic sequence. Where duplicates were found, only one sequence from each duplicate set was retained for further analysis. An additional in-house Python script was used to detect and identify ambiguous nucleotides. For each species, sequences having any other nucleotides besides A, T, C, and G were excluded, as the presence of ambiguous nucleotides may potentially mask the genomic signature encoded within dinucleotide frequencies. Additionally, samples from Georgia were removed from the dataset due to its transcontinental location between Europe and Asia [43].
FIGURE 1.

Generalized flow diagram showing the methodology.
C. Dinucleotide Frequency Representation
Given the four nucleotides A, T, C, G, there are 42 = 16 unique dinucleotide pairs that can be constructed from them, namely:
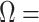 {AT, AA, AC, AG, TT, TA, TC, TG, GT, GA, GC, GG, CT, CA, CC, CG}. If we denote by
{AT, AA, AC, AG, TT, TA, TC, TG, GT, GA, GC, GG, CT, CA, CC, CG}. If we denote by
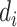 the frequency of the
the frequency of the
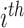 dinucleotide, then a genomic sequence can be represented by a 16-dimensional feature vector:
dinucleotide, then a genomic sequence can be represented by a 16-dimensional feature vector:
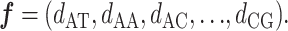 |
Due to the varying sequence lengths of the different species’ genomes, the relative frequencies of the dinucleotides are computed by dividing each frequency by the total number of dinucleotide pairs,
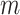 , extracted from the entire genome sequence. Letting
, extracted from the entire genome sequence. Letting
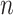 be the length of a genome sequence, and assuming a sliding window of length 1, then there are
be the length of a genome sequence, and assuming a sliding window of length 1, then there are
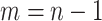 dinucleotide pairs. The refined feature vector is then defined as:
dinucleotide pairs. The refined feature vector is then defined as:
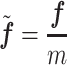 |
where the fraction bar depicts element-wise vector division and each component is the relative frequency of each dinucleotide pair in that sequence.
Relative dinucleotide frequency feature vectors were computed for all genome sequences used in this study and they were used as a numerical representation for all sequence analyses.
D. Dinucleotide Frequency Analysis
1). Exploratory Data Analysis
Two approaches were used to investigate the patterns of the genomic sequences as represented by the dinucleotide features. Firstly, dimensionality reduction techniques were employed to embed the 16-dimensional feature space in two dimensions for visualization. Specifically, principal component analysis (PCA) and t-distributed Stochastic Neighbor Embedding (t-SNE) were used. In the second approach, an unsupervised learning approach using agglomerative hierarchical clustering was utilized to uncover any underlying group structures of genomic sequences. This hierarchical approach was chosen because unlike other clustering algorithms such as
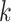 -means [44], this method does not require the number of clusters to be specified. To enable visualization of clustering results, only ten sequences were randomly sampled from each class in both within and between species analysis, resulting in 60 and 80 sequences for the two analyses, respectively. The dinucleotide relative frequency vectors of these sampled sequences were used to construct dendrograms through average linkage of the Euclidean distance matrix of the feature vectors. Validation of the clustering results, which is usually not an easy task, was performed in this study through the known class labels of the samples.
-means [44], this method does not require the number of clusters to be specified. To enable visualization of clustering results, only ten sequences were randomly sampled from each class in both within and between species analysis, resulting in 60 and 80 sequences for the two analyses, respectively. The dinucleotide relative frequency vectors of these sampled sequences were used to construct dendrograms through average linkage of the Euclidean distance matrix of the feature vectors. Validation of the clustering results, which is usually not an easy task, was performed in this study through the known class labels of the samples.
2). Statistical Inference
The Kruskal-Wallis test was used to compare the distribution of the relative frequencies across the different species and continents of SARS-CoV-2 sequences. Pairwise comparisons were conducted using the Wilcoxon Rank Sum test, with adjustment for multiple comparisons using the Bonferroni correction. A p-value of less than 0.05 was considered statistically significant.
3). Classification
For the supervised learning task, two classification problems were investigated for each of the between species and within species analyses. Firstly, the problem was framed directly as a multi-class classification problem where the goal was to classify each sequence into
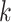 different classes, where
different classes, where
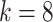 for the different species in the between species analysis and
for the different species in the between species analysis and
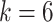 for the different continents in the within species analysis. Secondly, to discriminate SARS-CoV-2 from the other species and to investigate differences between SARS-CoV-2 sampled from different continents, the multi-class classification problem was binarized through the one-vs-all approach. Specifically, one classification model was built to distinguish SARS-CoV-2 from all the other species and another to distinguish SARS-CoV-2 samples originating in Asia from those originating from all the other continents.
for the different continents in the within species analysis. Secondly, to discriminate SARS-CoV-2 from the other species and to investigate differences between SARS-CoV-2 sampled from different continents, the multi-class classification problem was binarized through the one-vs-all approach. Specifically, one classification model was built to distinguish SARS-CoV-2 from all the other species and another to distinguish SARS-CoV-2 samples originating in Asia from those originating from all the other continents.
In the one-vs-all approach, the number of sequences in the class of interest (SARS-CoV-2 and Asia for the two analyses) were greatly outnumbered by the sequences in the respective complement classes (i.e. the complement classes are a combination of all the classes other than the classes of interest). Thus, the complement classes were under sampled to match the classes of interest, thus producing balanced classes.
Two resampling techniques were utilized in this classification system:
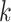 -fold cross-validation for hyper parameter tuning and bootstrap for final model evaluation. For both the within and between species analyses, the data was randomly split, in a stratified manner, into 70% for training and hyper parameter tuning and the remaining 30% was used for final model testing of the fully specified classifier. Randomized hyper parameter search was implemented through stratified 10-fold cross-validation on the 70% of the data reserved for training. Stratification was utilized to maintain the proportion of samples for each class in the train and test sets, as well as in the folds during the 10-fold cross validation procedure. Stratification is especially important when dealing with imbalanced data (such as in the multi-class classification settings). Ten folds were chosen for the cross-validation because they provide a good compromise between model bias and computational efficiency [45]. A smaller number of folds, such as two or three, have a high bias but are computationally efficient. On the other hand, a large number of folds (the extreme case being leave-one-out cross-validation) have a low bias, but are computationally inefficient [45]. Moreover, [46] showed that leave-one-out and 10-fold cross-validation yielded similar results, indicating that using ten folds is more appealing from a computational efficiency perspective.
-fold cross-validation for hyper parameter tuning and bootstrap for final model evaluation. For both the within and between species analyses, the data was randomly split, in a stratified manner, into 70% for training and hyper parameter tuning and the remaining 30% was used for final model testing of the fully specified classifier. Randomized hyper parameter search was implemented through stratified 10-fold cross-validation on the 70% of the data reserved for training. Stratification was utilized to maintain the proportion of samples for each class in the train and test sets, as well as in the folds during the 10-fold cross validation procedure. Stratification is especially important when dealing with imbalanced data (such as in the multi-class classification settings). Ten folds were chosen for the cross-validation because they provide a good compromise between model bias and computational efficiency [45]. A smaller number of folds, such as two or three, have a high bias but are computationally efficient. On the other hand, a large number of folds (the extreme case being leave-one-out cross-validation) have a low bias, but are computationally inefficient [45]. Moreover, [46] showed that leave-one-out and 10-fold cross-validation yielded similar results, indicating that using ten folds is more appealing from a computational efficiency perspective.
The randomized hyper parameter tuning through stratified 10-fold cross validation yielded the best model configuration which was then used to train the model on the entirety of the training data, where “best” was determined through the balanced accuracy metric [47], [48] before being evaluated on the held out 30% testing data. Balanced accuracy is an alternative to the standard accuracy measure that is especially useful when working with imbalanced data. It is defined as the average of recall scores obtained in each class whereas standard accuracy is simply the proportion of all correctly predicted class labels. For evaluation of the final model, 20000 bootstrap resamples from the unseen test data were evaluated on the trained model. In each bootstrap iteration, balanced accuracy, precision, recall and the F1 score performance metrics were computed. The average values of these metrics as well as 95% confidence intervals for uncertainty were computed and reported. The methods performed for hyper parameter optimization and model evaluation are described in greater detail in Fig. S3 and Fig. S4.
IV. Results
A. Data Collection and Preprocessing
A total of 60 063 WGS were downloaded from publicly accessible databases including GISAID, NCBI, and the LANL (Table 1). Of these, 54.8% of the sequences contained only A, T, C, and G nucleotides (Table 1), which were used for the between species dinucleotide frequency analysis. Of the eight species, the SARS-CoV-2 retained the least number of sequences for the between species dinucleotide frequency analysis (Table 1). Most of the SARS-CoV-2 sequences used in this study were sampled in Europe, while South America, Oceania, and Africa had the least number of sequences (Table S1).
TABLE 1. Number of Sequences Downloaded and Selected for Analysis.
| Species | Total number of sequences downloaded | Selected sequences (ATCG only) |
|---|---|---|
| SARS-CoV-2 | 28 067 | 8 252 (29.4%) |
| MERS-CoV | 256 | 198 (77.3%) |
| DENV | 5 448 | 4 749 (87.2%) |
| EBOV | 1 547 | 1 222 (79.0%) |
| HBV | 8 627 | 6 990 (81.0%) |
| HCV | 3 288 | 2 453 (74.6%) |
| HIV-1 | 12 538 | 8 890 (70.9%) |
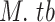 |
292 | 145 (49.7%) |
| Total | 60 063 | 32 899 (54.78%) |
B. Dinucleotide Frequency Analysis
1). Exploratory Data Analysis
Summaries of the relative dinucleotide frequencies for all the species analyzed, and of SARS-CoV-2 samples from the different regions are shown in Fig. S1 and Fig. S2, respectively. The thickness of the lines in each class is a proxy for the number of samples belonging to that class. Substantial diversity is observed in the distribution of the relative frequencies across the dinucleotides of all the species in the study. In contrast, little variation across the dinucleotides was observed among the SARS-CoV-2 samples from the different regions because the lines are superimposed on each other (Fig. S2).
The median relative frequencies of the 16 dinucleotide genomic signatures were calculated for each of the eight pathogens (Table S2) and for the SARS-CoV-2 data sampled in six continental regions (Table S3). Results showed that for SARS-CoV-2 and MERS-CoV, the median relative frequencies for the TT and CG dinucleotides were the most, and least abundant dinucleotides, respectively (Table S2). The median relative frequency for the CG dinucleotide signature was consistently the least abundant across all the species, except for M. tb where it was the most abundant (Table S2). Investigation of the relative dinucleotide frequencies within the SARS-CoV-2 sequences revealed very similar frequencies across the data from the different continents (Table S3). Interestingly, samples from Oceania had the highest frequency of TT dinucleotides as well as the lowest CG frequency across all the continents (Table S3).
Dimensionality Reduction: For the between species analysis, both the PCA and t-SNE visualizations of the relative dinucleotide frequencies revealed a clear separation of the different species (Fig. 2). While the t-SNE is optimized to capture relative distances between the input observations (in this case, genomic sequences that are neighbors in the 16-dimensional space are expected to also be neighbors in the 2-dimensional space), inferences about the observed inter-cluster distances and cluster sizes can seldom be made. In contrast, given that the PCA components are optimized to maximise variability in the original 16-dimensional features, inferences about cluster sizes and inter-cluster distances may be made. In particular, it may be noted that M. tb, the only bacterial genome included in this study, separated the furthest from the other data points. Moreover, SARS-CoV-2 and MERS-CoV clustered together, and this is substantiated by the fact that these two viruses are more closely related to each other than to the other species included in this study.
FIGURE 2.

a) PCA and b) t-SNE visualizations of the eight pathogenic species.
For the within species analysis on the other hand, the sequences collected from the Oceania region were the only group that largely separated from the rest of the sequences, as can be seen in both PCA and t-SNE (Fig. 3). The general lack of separation demonstrated how similar the globally circulating sequences are.
FIGURE 3.

a) PCA and b) t-SNE visualizations of the seven continents of origin for the SARS-CoV-2 dataset.
Hierarchical Clustering: Two dendrograms were constructed for the between and within species analyses. The dendrogram constructed from the subset of 80 species in the between species analysis is characterized by an excellent grouping (Fig. 4), with all species belonging to the same group being placed in the same cluster. For this subset of the data, both intra-cluster and inter-cluster similarities are at their best (Fig. 4). In this analysis, the first branching occurred between M. tb and all the viruses, which likely indicates the biggest dissimilarity in their sequences (Fig. 4). Among the viruses, SARS-CoV-2 and MERS-CoV clustered close together, possibly owing to their relatedness as they are part of the same family (Coronaviridae) and same genus, namely Betacoronavirus. Although HCV and DENV are members of the same Flaviviridae family, their sequences do not form neighbouring clusters. This could mean that at a family level, either the dinucleotide relative frequency feature vectors are not able to capture the similarity well enough or that membership in this family does not guarantee sufficient genome similarity. The dendrogram of the within species analysis was based on 60 sampled SARS-CoV-2 genomes (Fig. 5). In this clustering, the samples were poorly grouped across the continental regions, which may be interpreted as the absence of geography-related markers of the globally circulating SARS-CoV-2 virus genome.
FIGURE 4.

Dendrogram created from 10 randomly sampled sequences from all classes in the between species analysis.
FIGURE 5.

Dendrogram created from 10 randomly sampled sequences from all classes in the within species analysis.
Although the results of the hierarchical clustering are only based on a small subset of the complete dataset, they are consistent with the visualizations of the relative dinucleotide frequencies of the full dataset (Fig. S1. and Fig. S2.) as well as the results obtained through PCA and t-SNE (Fig. 2 and Fig. 3).
2). Statistical Inference
To statistically compare the relative frequency distributions of the 16 dinucleotides across the different continents, pairwise comparisons were performed using the Wilcoxon Rank Sum test. A subset of the results, which focuses on dinucleotide TT, is presented in Table 2 and the full set of results are given in Table S4. Dinucleotide TT was chosen for discussion as it is one of the pairs that exhibited noticeable patterns in the initial exploratory analysis (Table S2).
TABLE 2. P-Values for the Dinucleotide “TT” Compared Across the Different Continents.
| Continent | Africa | Asia | Europe | North America | Oceania |
|---|---|---|---|---|---|
| Asia |
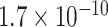 |
||||
| Europe | 0.0033 | <
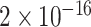
|
|||
| North America | 0.1301 | <
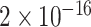
|
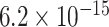 |
||
| Oceania |
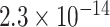 |
<
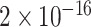
|
<
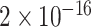
|
<
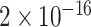
|
|
| South America | 1.0000 | <
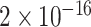
|
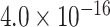 |
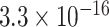 |
<
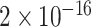
|
Most of the continental pairwise comparisons for the TT dinucleotides were statistically significant (p-value < 0.05) which meant that the relative frequency of TT across these continents differed (Table 2). However, pairwise relative frequency comparison of the TT dinucleotide was not statistically significant for Africa and North America (p-value 0.1301), and Africa and South America (p-value 1.000) (Table 2). This means that the relative frequency distributions of the TT dinucleotide across these continents do not differ.
The statistical significance of the TT dinucleotide in most of the continents provides support that this dinucleotide feature is capable of stratifying the SARS-CoV-2 genome sequences according to geographic region of origin.
3). Classification
All classification models in this work were fitted on the dinucleotide relative frequencies using the extreme gradient boosting (XGBoost) [49] model through its Python implementation. The dinucleotide feature vectors were concatenated to form data frames with
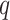 rows and 16 columns in the order {AA, AT, AC, AG, TA, TT, TC, TG, CA, CT, CC, CG, GA, GT, GC, GG} where
rows and 16 columns in the order {AA, AT, AC, AG, TA, TT, TC, TG, CA, CT, CC, CG, GA, GT, GC, GG} where
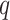 is the number of samples in the different partitions of the data used for training and testing (Table S1 and Table S5).
is the number of samples in the different partitions of the data used for training and testing (Table S1 and Table S5).
The XGBoost model belongs to the decision tree-based family of models. Tree-based models are rule-based systems that are built upon a hierarchy of branching Boolean statements, rendering this class of models highly interpretable [50] and making them suitable in many fields of application. The first tree that was constructed during the training of the XGBoost model for the multi-class classification in the within species analysis is shown in Fig. S5. Default hyper parameter values for the XGBoost models were used except for the parameters noted in Table S6, which were selected through randomized parameter search via 10-fold cross-validation. The final data set sizes are shown in Table S1 and Table S5 for within and between species analysis, respectively.
The performance metrics of the classification models on the test set are shown in Table 3, where the average balanced accuracy, micro-averaged F1, and macro-averaged F1 scores of the learned model are given. These metrics include a 95% confidence interval derived through 20000 iterations of bootstrapping. The distributions of the bootstrapped F-score metrics are shown in Fig. S6 and Fig. S7 and the full performance metrics results are noted in Table S7.
TABLE 3. Balanced Accuracy, Micro F1 and Macro F1 Scores of the Various XGBoost Models.
| Model |
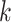 |
Bal. Acc. | Micro F1 | Macro F1 |
|---|---|---|---|---|
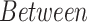 |
2 | 1.00 (1.00,1.00) | 1.00 (1.00,1.00) | 1.00 (1.00,1.00) |
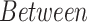 |
8 | 1.00 (1.00,1.00) | 1.00 (1.00,1.00) | 1.00 (1.00,1.00) |
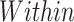 |
2 | 0.869 (0.846, 0.892) | 0.869 (0.845, 0.892) | 0.869 (0.845, 0.892) |
 |
6 | 0.675 (0.641, 0.713) | 0.824 (0.809, 0.838) | 0.686 (0.643, 0.733) |
The results indicate that the between species classification task was a simple one (Table 3 and Fig. S8), both for the multi-class classification and the binary classification, despite facing class imbalances in the multi-class classification task. These results are corroborated by the clear separation of classes that were observed in both the PCA and t-SNE (Fig. 2), and the perfect clustering results from the random subset of the data that was analyzed (Fig. 4).
Feature importance plots for both multi-class and binary classification models are shown in Fig. S9. Dinucleotide pair TC led to the largest average gain in performance in the multi-class model whilst it ranked amongst the lowest in importance in the binary classifier. Similarly, the TG dinucleotide dominated the ranks in the binary model while ranking poorly in the multi-class model. Both GG and CC pairs appeared in the top three for both models. This observation warrants further research and is beyond the scope of this work. The importance of the features diminished exponentially, with some features not contributing to the models at all.
For the within species classification, the models classified SARS-CoV-2 sequences into six continents using a multi-class system and into two classes using a binary approach. The binary classifier produced a 0.869 score across all metrics with very similar confidence (Table 3). Classification of SARS-CoV-2 cases by continent recorded a decrease in performance. Scores of 0.675, 0.824 and 0.686 were achieved for balanced accuracy, micro-averaged F1 and macro-averaged F1 scores, respectively.
With respect to feature importance (Fig. S10), all the dinucleotides contributed to the models, contrary to the observation made in the between species models. Moreover, the reduction in the importance of the dinucleotides was steady, even more so in the multi-class classification setting. This was likely due to the level of difficulty of the problem where the choice of which dinucleotide to base the next split on was not trivial. In decreasing order of importance, dinucleotide pairs TG, CC and GG were the most important for the binary model whilst GT, TG and CC were the most important for the multi-class classification model.
Confusion matrices for the within species binary and multi-class classification tasks are shown in Fig. 6. For the binary classification task, the “Not Asia” class had a slightly higher rate of misclassification (14.32%) compared to the Asia class (11.81%) (Fig. 6). This may be due to the compositionality of the “Not Asia” class where all the diverse samples from the other continents were grouped into one class. For the multi-class classification task, the largest rate of misclassifications (87.5%) was related to the “Africa” class and this was to be expected as this class had the lowest number samples. The “South America” class had the second highest misclassification rate at 45.21%, noting that its sample size was also very low. Asia, Europe, North America, and Oceania recorded misclassification rates of 12.81%, 12.94%, 23.10% and 13.54%, respectively. The highest F1 scores were related to the Oceania and Europe classes with 92.22% and 86.90%, respectively. The strong individual performance of the Oceania class despite a small sample size could also be predicted from the exploratory data analyses.
FIGURE 6.

Within species XGBoost confusion matrix for a) binary problem b) multi-class classification problem.
V. Discussion
A. Classification Results
For the between species analysis, the classification results show that the XGBoost model is not only able to learn a highly accurate model for distinguishing SARS-CoV-2 samples from non-SARS-CoV-2 samples (based on the species included in this study), but it is also able to learn an accurate model for discriminating between all eight species in a multi-class classification setting.
For the within species analysis, the XGBoost models developed for both the binary and multi-class classification tasks were not as reliable as the classifiers developed in the between species analysis because some incorrect predictions were made, as can be seen from the off-diagonal elements in Fig. 6. These inferior results could be attributed to the identical sequences of the globally circulating SARS-CoV-2 genomes, which a few studies have already confirmed. We hypothesize that this high level of sequence similarity hampers the sensitivity of frequency-based approaches employed here in detecting the very subtle differences in the genomes. Reference [51] analyzed thirteen complete genome sequences of SARS-CoV-2 and found that they had a more than 99% similarity. In addition, [52] analyzed more than 1100 SARS-CoV-2 genome sequences and provided evidence for the absence of distinct evolutionary patterns in the genomes of the currently known major clades of SARS-CoV-2. For the multi-class classification task specifically, these inferior classification results can also be attributed to the poor performance in the classes with small sample sizes, in addition to the limitations brought about by the highly similar genome sequences.
B. Dinucleotide Homogeneity and Conservation
Several dinucleotides are known to be conserved within 50 kb regions across genomes. For instance, the CA dinucleotide is conserved in all retroviruses, including HIV [53]. In a study investigating the frequency conservation or variation of the 16 dinucleotide pairs across bacterial genomes, the frequency of several dinucleotides such as AC, AG, CA, CT, GA, GT, TC, and TG were well-conserved, whereas the frequency of the other dinucleotides varied substantially [54]. Genome inhomogeneity is mostly driven by the AA, TT, AT, TA, GG, CC, GC, and CG dinucleotides (i.e. the combination of two strong (SS) or two weak (WW) nucleotides, where the two strong nucleotides are C and G and the two weak nucleotides are A and T) [55], while homogeneity is driven by the other eight dinucleotides consisting of combinations of the strong and weak nucleotides (SW/WS).
The observation for genome homogeneity held true for most of the SW/WS dinucleotides in our study, namely that of AC, TC, TG, CA, and GT. Deviations from this trend were seen for the AG and GA dinucleotides where HIV-1 had a much higher relative frequency than the other species for AG and DENV had a much higher relative frequency for GA (Table S2). While two of the conserved dinucleotides, AG and CA, were underrepresented in this study’s M. tb sequences, half of the varied dinucleotides, AA, AT, TT, TA, were also underrepresented.
C. CG Signature in RNA Viruses
Several studies exploring the evolution of various human infecting RNA viruses and their interactions with the human host indicate that there is a general strong selection pressure for an underrepresentation of the CG dinucleotides within these viruses [56], [57]. It has been suggested that since human genes eliminate CG dinucleotide motifs, the underrepresentation of this dinucleotide is crucial for the viral genome’s expression and replication, enabling it to shield itself from the host’s immune response [56], [58], [59], [60], [61]. While these studies computed dinucleotide relative abundance values, the present study reports dinucleotide relative frequencies. Here, the CG dinucleotide was largely underrepresented across all the viruses, confirming the existence of this relative frequency conservation.
It is unclear whether taxonomically similar viruses infecting the same host species exhibit similar dinucleotide genomic profiles. Generally, the profiles of dinucleotide genomic signature in viruses are thought to reflect background mutation pressures [62], [63]. Given that all SARS-COV-2 sequences utilized in this study were obtained from human hosts, it may be interesting to investigate how its dinucleotide signature is associated with the human host and if this has any implication on the virus’s pathogenicity and evolution over time. However, this is beyond the scope of this work and is left for future studies.
D. Comparison of M. Tb to Viral Species
Several studies have shown that dinucleotide relative frequencies can be used to adequately discriminate between viral and bacterial species. One study showed that the underrepresentation of the CG dinucleotide may be associated with cytosine methylation, although it may be affected by other aspects of DNA conformation, such as secondary structure and dinucleotide stacking energies, thus demonstrating its potential to differentiate between viral and bacterial species [13], [19]. In our study, when comparing M. tb to the viral species, an underrepresentation of the WW dinucleotide frequencies (AA, AT, TA, TT) and overrepresentation of the SS dinucleotide frequencies (CC, CG, GC, GG) was consistently observed (Table S2). Thus, these dinucleotides may potentially be used to differentiate between M. tb and viral species.
E. Relatedness of SARS-COV-2 and MERS-COV
Dinucleotide biases were observed across most of the WW and SS dinucleotides, alluding to the potential of these dinucleotides to differentiate between species. Even though variation could be observed for the WW/SS relative dinucleotide frequencies, SARS-CoV-2 and MERS-CoV consistently had similar dinucleotide frequencies. Specifically, TT and CG dinucleotides were over and underrepresented, respectively, in both these species, possibly owing to their 50% sequence similarity as indicated by [6], [64], [65]. This demonstrates their phylogenetic relatedness and difference from the other species included in this study. This was further observed in their clustering closer to each other than to other species (Fig. 2 and Fig. 4).
F. Oceania Samples are Distinct
As expected, dinucleotide frequencies within the SARS-CoV-2 were largely conserved, with the TT and CG dinucleotides having the highest, and lowest frequencies, respectively. However, when assessing the SARS-CoV-2 sequences sampled from several continental regions, it was interesting to note that samples from Oceania consistently had an overrepresentation for six of the 16 dinucleotides (AT, AC, TA, TT, TG, and CA) and an underrepresentation for seven of the 16 dinucleotides (AG, TC, CC, CG, GA, GT and GG). This genomic signature for Oceania samples likely led to its distinction from the samples of other continents (Fig. 3b). This further demonstrates the potential use for dinucleotide frequencies to illustrate underlying environmental drivers of evolution within a species where genomes with similar phylogenetic compositions had vastly different GC dinucleotide frequencies [66].
G. Limitations of the Study
Several confounders exist which may have influenced the outcome of the within species classification. Batch effects, or the influence of pooling data sourced from different experiments and locations, is one such confounder [67]. For this study, access to experimental, sequencing, and sample preparation from the source was limited and thus could not be corrected for in the statistical analysis. Furthermore, host pathogen interaction is still largely understudied and investigating its influence on the classification of the SARS-CoV-2 sequences was beyond the scope of this study. It is unclear how the immune response of the host affects the pathogen’s genome. While some of the biological data such as virus clade is available in the GISAID database, inclusion of this information would have further stratified the dataset by seven levels, resulting in a further reduction in the statistical power of analyzing each subgroup against the rest. Furthermore, at the time of this study, no host genotype data was available to enable a paired analysis of the host and pathogen genomes.
VI. Conclusion
Dinucleotide profiles differ between species, providing a genome signature that is characteristic of the bulk properties of an organism’s DNA. Differences in dinucleotide genomic profiles can be considered as a distinguishing genomic signature for specific taxonomic groups providing important information on molecular evolution mechanisms [68]. This study confirms the utility of alignment-free and machine learning approaches using dinucleotide relative frequencies to discriminate between distantly related species such as viruses and bacteria, closely related species such as SARS-CoV-2 and MERS-CoV, as well as samples of the same species that originate from different regions. This approach may be used for the taxonomic classification of pathogens which are of particular importance during a pandemic such as COVID-19.
Data Availability Statement
The raw whole genome sequencing data is available from GISAID, NCBI, and the LANL (Supplementary file AccessionNumbers.xlsx). The computing code is available upon request from the corresponding author.
Authors’ Contributions
G.D., S.M., R.M. and D.M. designed the experiments and contributed to the interpretation of the results. R.M. and S.M. performed the bioinformatic processing of the raw sequencing data. G.D. performed majority of the statistical analysis and D.M. performed some components of the statistical analysis. R.Y., T.C. and J.M. provided guidance related to the methods. The final manuscript was read and approved by all authors.
Acknowledgment
The authors would like to acknowledge Dr. Sibusisiwe Makhanya for her thoughtful review of this manuscript. The authors would also like to gratefully acknowledge the authors, and the originating and submitting laboratories for their sequence and metadata shared through the GISAID EpiCoV repository, on which this research is based.
Biographies

Gciniwe S. Dlamini received the M.Sc. degree in mathematical statistics from the University of Cape Town, in 2018. She is currently a Research Engineer with IBM Research, Johannesburg, South Africa. Her current research interest includes representation learning.

Stephanie J. Müller received the M.Sc. degree in bioinformatics from the South African National Bioinformatics Institute, University of the Western Cape, in 2016, and the Ph.D. degree in human-genetics from Stellenbosch University, in 2019. She is currently a Research Scientist with IBM Research, Johannesburg, South Africa. Her research interests include analysing big data in human-genetics and human-pathogens.

Rebone L. Meraba received the M.S. degree in bioinformatics from the University of Cape Town, Cape Town, Western Cape, South Africa, in 2017. From 2017 to 2018, she was a DSIDE Participant with the Council for Scientific and Industrial Research. From 2018 to 2019, she was a Bioinformatics Analyst with the Centre for Proteomic and Genomic Research. She is currently a Research Intern with IBM Research, Johannesburg, Gauteng, South Africa. Her research interests include applying artificial intelligence techniques to bioinformatics problems and processing biological/medical data for precision medicine, discovery, diagnosis, and treatment.

Richard A. Young received the B.S. degree (Hons.) in computer science from the University of the Witwatersrand, South Africa, in 2010. He was a Software Developer with the Business Systems Group from 2011 to 2016. He was also a Senior Software Developer with BNRY from 2016 to 2018. He joined IBM Research, South Africa, as a Research Engineer, in 2018. His research interests include building web-based applications and machine learning systems.

James Mashiyane received the Ph.D. degree in physics from the University of the Witwatersrand. He is currently a Research Engineer with IBM Research, Johannesburg, South Africa. His research interest includes applying mathematical algorithms to solving real world problems facing society.

Tapiwa Chiwewe (Senior Member, IEEE) received the B.Eng., M.Eng., and Ph.D. degrees in computer engineering from the University of Pretoria, South Africa, in 2006, 2010, and 2016, respectively. He was a Software Developer with Fifth Dimension Technologies from 2008 to 2011. He was also a Senior Research Engineer with the Council for Scientific and Industrial Research, South Africa, from 2011 to 2015. He is currently a Research Scientist and a Manager with IBM Research, Johannesburg, South Africa. His research interests include cognitive radio networks and machine learning.

Darlington S. Mapiye received the M.Sc. and Ph.D. degrees in bioinformatics from the South African National Bioinformatics Institute, University of the Western Cape, in 2012 and 2016, respectively. He is currently a Research Scientist with IBM Research, Johannesburg, South Africa. His research interests include computational genomics and biostatistics.
References
- [1].Lai M. M. C. and Cavanagh D., “The molecular biology of coronaviruses,” in Advances in Virus Research, vol. 48. Amsterdam, The Netherlands: Elsevier, 1997, pp. 1–100, doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Channappanavar R. and Perlman S., “Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology,” in Semin Immunopathol., vol. 39, no. 5, pp. 529–539, 2017, doi: 10.1007/s00281-017-0629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ksiazek T. G.et al. , “A novel coronavirus associated with severe acute respiratory syndrome,” New England J. Med., vol. 348, no. 20, pp. 1953–1966, May 2003, doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- [4].Zaki A. M., Van Boheemen S., Bestebroer T. M., Osterhaus A. D., and Fouchier R. A., “Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia,” New England J. Med., vol. 367, no. 19, pp. 1814–1820, 2012, doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- [5].WHO. WHO coronavirus Disease (COVID-19) Dashboard [Online Dashboard]. Accessed: Jul. 23, 2020. [Online]. Available: https://covid19.who.int/
- [6].Jin Y., Yang H., Ji W., Wu W., Chen S., Zhang W., and Duan G., “Virology, epidemiology, pathogenesis, and control of COVID-19,” Viruses, vol. 12, no. 4, p. 372, Mar. 2020, doi: 10.3390/v12040372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Eisenhofer R. and Weyrich L. S., “Assessing alignment-based taxonomic classification of ancient microbial DNA,” PeerJ, vol. 7, Mar. 2019, Art. no. e6594, doi: 10.7717/peerj.6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gao X., Lin H., Revanna K., and Dong Q., “A Bayesian taxonomic classification method for 16S rRNA gene sequences with improved species-level accuracy,” BMC Bioinf., vol. 18, no. 1, p. 247, Dec. 2017, doi: 10.1186/s12859-017-1670-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bernard G., Chan C. X., Chan Y.-B., Chua X.-Y., Cong Y., Hogan J. M., Maetschke S. R., and Ragan M. A., “Alignment-free inference of hierarchical and reticulate phylogenomic relationships,” Briefings Bioinf., vol. 20, no. 2, pp. 426–435, Mar. 2019, doi: 10.1093/bib/bbx067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chan C. X. and Ragan M. A., “Next-generation phylogenomics,” Biol. Direct, vol. 8, no. 1, pp. 1–6, Dec. 2013, doi: 10.1186/1745-6150-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zielezinski A., Vinga S., Almeida J., and Karlowski W. M., “Alignment-free sequence comparison: Benefits, applications, and tools,” Genome Biol., vol. 18, no. 1, p. 186, Dec. 2017, doi: 10.1186/s13059-017-1319-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Josse J., Kaiser A. D., and Kornberg A., “Enzymatic synthesis of deoxyribonucleic acid VIII. Frequencies of nearest neighbor base sequences in deoxyribonucleic acid,” J. Biol. Chem., vol. 236, no. 3, pp. 864–875, 1961. [PubMed] [Google Scholar]
- [13].Nussinov R., “Some rules in the ordering of nucleotides in the DNA,” Nucleic Acids Res., vol. 8, no. 19, p. 4545, 1980, doi: 10.1093/nar/8.19.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wyrick J. J. and Roberts S. A., “Genomic approaches to DNA repair and mutagenesis,” DNA Repair, vol. 36, pp. 146–155, Dec. 2015, doi: 10.1016/j.dnarep.2015.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bohlin J. and Skjerve E., “Examination of genome homogeneity in prokaryotes using genomic signatures,” PLoS ONE, vol. 4, no. 12, Dec. 2009, Art. no. e8113, doi: 10.1371/journal.pone.0008113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Moran N. A., “Microbial minimalism: Genome reduction in bacterial pathogens,” Cell, vol. 108, no. 5, pp. 583–586, 2002, doi: 10.1016/S0092-8674(02)00665-7. [DOI] [PubMed] [Google Scholar]
- [17].Mann S. and Chen Y.-P.-P., “Bacterial genomic G+C composition-eliciting environmental adaptation,” Genomics, vol. 95, no. 1, pp. 7–15, Jan. 2010, doi: 10.1016/j.ygeno.2009.09.002. [DOI] [PubMed] [Google Scholar]
- [18].Hildebrand F., Meyer A., and Eyre-Walker A., “Evidence of selection upon genomic GC-content in bacteria,” PLoS Genet., vol. 6, no. 9, Sep. 2010, Art. no. e1001107, doi: 10.1371/journal.pgen.1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jabbari K. and Bernardi G., “Cytosine methylation and CpG, TpG (CpA) and TpA frequencies,” Gene, vol. 333, pp. 143–149, May 2004, doi: 10.1016/j.gene.2004.02.043. [DOI] [PubMed] [Google Scholar]
- [20].Cristea P. D., “Conversion of nucleotides sequences into genomic signals,” J. Cellular Mol. Med., vol. 6, no. 2, pp. 279–303, Apr. 2002, doi: 10.1111/j.1582-4934.2002.tb00196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Qi X., Fuller E., Wu Q., and Zhang C.-Q., “Numerical characterization of DNA sequence based on dinucleotides,” Sci. World J., vol. 2012, pp. 1–6, Jan. 2012, doi: 10.1100/2012/104269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nishida H., “Comparative analyses of base compositions, DNA sizes, and dinucleotide frequency profiles in archaeal and bacterial chromosomes and plasmids,” Int. J. Evol. Biol., vol. 2012, pp. 1–5, Jan. 2012, doi: 10.1155/2012/342482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Karlin S., “Global dinucleotide signatures and analysis of genomic heterogeneity,” Current Opinion Microbiol., vol. 1, no. 5, pp. 598–610, 1998, doi: 10.1016/S1369-5274(98)80095-7. [DOI] [PubMed] [Google Scholar]
- [24].Tyagi A., Bag S. K., Shukla V., Roy S., and Tuli R., “Oligonucleotide frequencies of barcoding loci can discriminate species across kingdoms,” PLoS ONE, vol. 5, no. 8, Aug. 2010, Art. no. e12330, doi: 10.1371/journal.pone.0012330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen S., Deng L.-Y., Bowman D., Shiau J.-J.-H., Wong T.-Y., Madahian B., and Lu H. H.-S., “Phylogenetic tree construction using trinucleotide usage profile (TUP),” BMC Bioinf., vol. 17, no. S13, pp. 117–130, Oct. 2016, doi: 10.1186/s12859-016-1222-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pride D. T., “Evolutionary implications of microbial genome tetranucleotide frequency biases,” Genome Res., vol. 13, no. 2, pp. 145–158, Feb. 2003, doi: 10.1101/gr.335003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Noble P. A., Citek R. W., and Ogunseitan O. A., “Tetranucleotide frequencies in microbial genomes,” Electrophoresis, vol. 19, no. 4, pp. 528–535, Apr. 1998, doi: 10.1002/elps.1150190412. [DOI] [PubMed] [Google Scholar]
- [28].Yu N., Li Z., and Yu Z., “Survey on encoding schemes for genomic data representation and feature learning—From signal processing to machine learning,” Big Data Mining Anal., vol. 1, no. 3, pp. 191–210, 2018, doi: 10.26599/BDMA.2018.9020018. [DOI] [Google Scholar]
- [29].Tampuu A., Bzhalava Z., Dillner J., and Vicente R., “ViraMiner: Deep learning on raw DNA sequences for identifying viral genomes in human samples,” PLoS ONE, vol. 14, no. 9, Sep. 2019, Art. no. e0222271, doi: 10.1371/journal.pone.0222271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Solis-Reyes S., Avino M., Poon A., and Kari L., “An open-source k-mer based machine learning tool for fast and accurate subtyping of HIV-1 genomes,” PLoS ONE, vol. 13, no. 11, Nov. 2018, Art. no. e0206409, doi: 10.1371/journal.pone.0206409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Randhawa G. S., Soltysiak M. P. M., El Roz H., de Souza C. P. E., Hill K. A., and Kari L., “Machine learning using intrinsic genomic signatures for rapid classification of novel pathogens: COVID-19 case study,” PLoS ONE, vol. 15, no. 4, Apr. 2020, Art. no. e0232391, doi: 10.1371/journal.pone.0232391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Diedrich C. R., O’Hern J., and Wilkinson R. J., “HIV-1 and the mycobacterium tuberculosis granuloma: A systematic review and meta-analysis,” Tuberculosis, vol. 98, pp. 62–76, May 2016, doi: 10.1016/j.tube.2016.02.010. [DOI] [PubMed] [Google Scholar]
- [33].Baize S.et al. , “Emergence of zaire ebola virus disease in guinea,” New England J. Med., vol. 371, no. 15, pp. 1418–1425, Oct. 2014, doi: 10.1056/NEJMoa1404505. [DOI] [PubMed] [Google Scholar]
- [34].Tuiskunen Bäck A. and Lundkvist Å., “Dengue viruses—An overview,” Infection Ecology Epidemiology, vol. 3, no. 1, p. 19839, Jan. 2013, doi: 10.3402/iee.v3i0.19839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Shepard C. W., “Hepatitis b virus infection: Epidemiology and vaccination,” Epidemiolog. Rev., vol. 28, no. 1, pp. 112–125, Jun. 2006, doi: 10.1093/epirev/mxj009. [DOI] [PubMed] [Google Scholar]
- [36].Bukh J., “The history of hepatitis c virus (HCV): Basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control,” J. Hepatology, vol. 65, no. 1, pp. S2–S21, Oct. 2016, doi: 10.1016/j.jhep.2016.07.035. [DOI] [PubMed] [Google Scholar]
- [37].Ulrich H., Pillat M. M., and Tárnok A., “Dengue fever, COVID-19 (SARS-CoV-2), and Antibody-dependent enhancement (ADE): A perspective,” Cytometry A, vol. 97, no. 7, pp. 662–667, Jul. 2020, doi: 10.1002/cyto.a.24047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].GISAID. GISAID—Initiative. Accessed: Jul. 23, 2020. [Online]. Available: https://www.gisaid.org/
- [39].HIV Sequence Compendium. HIV Databases. Accessed: Jul. 23, 2020. [Online]. Available: https://www.hiv.lanl.gov/content/index
- [40].Resource Coordinators N.et al. , “Database resources of the national center for biotechnology information,” Nucleic Acids Res., vol. 46, no. D1, pp. D8–D13, Jan. 2018, doi: 10.1093/nar/gkx1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Van Rossum G. and Drake F. L., Python 3 Reference Manual. Scotts Valley, CA, USA: CreateSpace, 2009. [Google Scholar]
- [42].Team R. C., R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2013. [Online]. Available: http://www.R-project.org/ [Google Scholar]
- [43].Worldview S.. Georgia—Geopolitics, Analysis and News. Accessed: Jul. 19, 2020. [Online]. Available: https://worldview.stratfor.com/region/eurasia/georgia [Google Scholar]
- [44].Lloyd S., “Least squares quantization in PCM,” IEEE Trans. Inf. Theory, vol. IT-28, no. 2, pp. 129–137, Mar. 1982. [Google Scholar]
- [45].Kuhn M. and Johnson K., “Over-fitting and model tuning,” in Applied Predictive Modeling, vol. 26. New York, NY, USA: Springer, 2013, pp. 61–92. [Google Scholar]
- [46].Molinaro A. M., Simon R., and Pfeiffer R. M., “Prediction error estimation: A comparison of resampling methods,” Bioinformatics, vol. 21, no. 15, pp. 3301–3307, Aug. 2005, doi: 10.1093/bioinformatics/bti499. [DOI] [PubMed] [Google Scholar]
- [47].Kelleher J. D., Mac Namee B., and D’arcy A., “Evaluation,” in Fundamentals Mach. Learn. for predictive data analytics: Algorithms, Worked Examples, Case Studies. Cambridge, MA, USA: MIT Press, 2015. [Google Scholar]
- [48].Brodersen K. H., Ong C. S., Stephan K. E., and Buhmann J. M., “The balanced accuracy and its posterior distribution,” in Proc. 20th Int. Conf. Pattern Recognit., Aug. 2010, pp. 3121–3124, doi: 10.1109/ICPR.2010.764. [DOI] [Google Scholar]
- [49].Chen T. and Guestrin C., “XGBoost: A scalable tree boosting system,” in Proc. 22nd ACM SIGKDD Int. Conf. Knowl. Discovery Data Mining, Aug. 2016, pp. 785–794, doi: 10.1145/2939672.2939785. [DOI] [Google Scholar]
- [50].Kingsford C. and Salzberg S. L., “What are decision trees?” Nature Biotechnol., vol. 26, no. 9, pp. 1011–1013, Sep. 2008, doi: 10.1038/nbt0908-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chiara M., Horner D. S., Gissi C., and Pesole G., “Comparative genomics suggests limited variability and similar evolutionary patterns between major clades of SARS-Cov-2,” BioRxiv, to be published, doi: 10.1101/2020.03.30.016790. [DOI]
- [52].Khan M. I., Khan Z. A., Baig M. H., Ahmad I., Farouk A.-E., Song Y. G., and Dong J.-J., “Comparative genome analysis of novel coronavirus (SARS-CoV-2) from different geographical locations and the effect of mutations on major target proteins: An in silico insight,” PLoS ONE, vol. 15, no. 9, Sep. 2020, Art. no. e0238344, doi: 10.1371/journal.pone.0238344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lee I. and Harshey R. M., “Importance of the conserved CA dinucleotide at μtermini 1 1Edited by M. Gottesman,” J. Mol. Biol., vol. 314, no. 3, pp. 433–444, Nov. 2001, doi: 10.1006/jmbi.2001.5177. [DOI] [PubMed] [Google Scholar]
- [54].Zhang H., Li P., Zhong H.-S., and Zhang S.-H., “Conservation vs. Variation of dinucleotide frequencies across bacterial and archaeal genomes: Evolutionary implications,” Frontiers Microbiol., vol. 4, p. 269, Sep. 2013, doi: 10.3389/fmicb.2013.00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kozhukhin C. G. and Pevzner P. A., “Genome inhomogeneity is determined mainly by WW and SS dinucleotides,” Bioinformatics, vol. 7, no. 1, pp. 39–49, 1991, doi: 10.1093/bioinformatics/7.1.39. [DOI] [PubMed] [Google Scholar]
- [56].Cheng X., Virk N., Chen W., Ji S., Ji S., Sun Y., and Wu X., “CpG usage in RNA viruses: Data and hypotheses,” PLoS ONE, vol. 8, no. 9, Sep. 2013, Art. no. e74109, doi: 10.1371/journal.pone.0074109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Auewarakul P., “Composition bias and genome polarity of RNA viruses,” Virus Res., vol. 109, no. 1, pp. 33–37, Apr. 2005, doi: 10.1016/j.virusres.2004.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bahir I., Fromer M., Prat Y., and Linial M., “Viral adaptation to host: A proteome-based analysis of codon usage and amino acid preferences,” Mol. Syst. Biol., vol. 5, no. 1, p. 311, Jan. 2009, doi: 10.1038/msb.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Su M.-W., Lin H.-M., Yuan H. S., and Chu W.-C., “Categorizing host-dependent RNA viruses by principal component analysis of their codon usage preferences,” J. Comput. Biol., vol. 16, no. 11, pp. 1539–1547, Nov. 2009, doi: 10.1089/cmb.2009.0046. [DOI] [PubMed] [Google Scholar]
- [60].Di Giallonardo F., Schlub T. E., Shi M., and Holmes E. C., “Dinucleotide composition in animal RNA viruses is shaped more by virus family than by host species,” J. Virology, vol. 91, no. 8, pp. 1–37, Apr. 2017, doi: 10.1128/JVI.02381-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tulloch F., Atkinson N. J., Evans D. J., Ryan M. D., and Simmonds P., “RNA virus attenuation by codon pair deoptimisation is an artefact of increases in CpG/UpA dinucleotide frequencies,” eLife, vol. 3, Dec. 2014, Art. no. e04531, doi: 10.7554/eLife.04531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jenkins G. M. and Holmes E. C., “The extent of codon usage bias in human RNA viruses and its evolutionary origin,” Virus Res., vol. 92, no. 1, pp. 1–7, 2003, doi: 10.1016/S0168-1702(02)00309-X. [DOI] [PubMed] [Google Scholar]
- [63].Wright F., “The ‘effective number of codons’ used in a gene,” Gene, vol. 87, no. 1, pp. 23–29, 1990, doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- [64].Mousavizadeh L. and Ghasemi S., “Genotype and phenotype of COVID-19: Their roles in pathogenesis,” J. Microbiol., Immunology Infection, to be published, doi: 10.1016/j.jmii.2020.03.022. [DOI] [PMC free article] [PubMed]
- [65].Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., and Bi Y., “Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding,” Lancet, vol. 395, no. 10224, pp. 565–574, 2020, doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Foerstner K. U., Mering C. V., and Bork P., “Comparative analysis of environmental sequences: Potential and challenges,” Phil. Trans. Roy. Soc. B, Biol. Sci., vol. 361, no. 1467, pp. 519–523, Mar. 2006, doi: 10.1098/rstb.2005.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Goh W. W. B., Wang W., and Wong L., “Why batch effects matter in omics data, and how to avoid them,” Trends Biotechnol., vol. 35, no. 6, pp. 498–507, Jun. 2017, doi: 10.1016/j.tibtech.2017.02.012. [DOI] [PubMed] [Google Scholar]
- [68].Kariin S. and Burge C., “Dinucleotide relative abundance extremes: A genomic signature,” Trends Genet., vol. 11, no. 7, pp. 283–290, 1995, doi: 10.1016/s0168-9525(00)89076-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw whole genome sequencing data is available from GISAID, NCBI, and the LANL (Supplementary file AccessionNumbers.xlsx). The computing code is available upon request from the corresponding author.