

Abstract
Despite several decades of focused investigation, sepsis remains a major cause of mortality in critically ill patients. Advancements in intensive care have enabled more patients to survive the acute phase of sepsis than previously, but a growing number of them progress to chronic critical illness. The failure of previous randomized clinical trials of anti-inflammatory agents to show any pro-survival benefit in septic patients underscores current thought that simple anti-inflammatory strategies are ineffective because the inhibitory effect of anti-inflammatory agents undermines the immune response to pathogens. New strategies with the dual capability of ameliorating inflammation in organs while stimulating antimicrobial activity are eagerly awaited. On the other hand, the metabolic alterations associated with systemic inflammatory response, including mitochondrial dysfunction and metabolic shift, are closely linked through a nexus of signaling pathways and signaling molecules. Preventing these metabolic derangements may be an alternative way to control excessive inflammation, an intriguing possibility that has not been fully explored. New insight into the molecular pathogenesis of sepsis and sepsis-associated chronic critical illness has led to the recognition of septic cachexia, a life-threatening form of metabolic inflammatory complex associated with multiple organ dysfunction. The potential for septic cachexia to serve as a novel target disease state to improve the clinical outcome of septic patients is discussed in this review.
Keywords: Immune dysfunction, inflammation, metabolism, signaling network, skeletal muscle, therapeutic target
INTRODUCTION
Sepsis was recently redefined as a life-threatening organ dysfunction caused by a dysregulated host response to an infection, as detailed in the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) published in 2016 (1). The diagnosis is made by the concomitant presence of confirmed or suspected infection and multiple organ dysfunction (MOD). Despite advances in critical care involving the management of volume resuscitation and antimicrobial therapy, sepsis remains a leading cause of death in critically ill patients and a major challenge in critical care medicine. Advancements in intensive care have enabled more septic patients to survive the acute phase than previously. Unfortunately, a growing number of those patients, particularly elderly patients, progress to chronic critical illness (CCI) (2). The recently developed definition of CCI, which reflects the current consensus, is one of six eligible clinical conditions (prolonged acute mechanical ventilation, tracheotomy, stroke, traumatic brain injury, sepsis, or severe wounds) plus at least 8 days in the intensive care unit (ICU) (2). New strategies are needed to reduce mortality and improve the long-term clinical outcome of septic patients. This review focuses primarily on septic patients with bacterial infection who develop CCI or are at risk of developing CCI.
The effort to develop new therapeutic strategies for septic patients spans several decades (3). A number of randomized clinical trials (RCTs) have been conducted to test the ability of anti-inflammatory agents, including tumor necrosis factor (TNF)-α antagonist and toll-like receptor (TLR)-4 antagonist, to prolong survival (4). Yet none of these agents has demonstrated any pro-survival effects in sepsis, although some of these trials are still ongoing and their findings are unknown. Consequently, there is no FDA-approved medication today that specifically targets sepsis. To some extent, the failure of previous RCTs may be attributable to the heterogeneity of sepsis which has variable etiology, symptoms, severity, phases of the disease, and so forth. Regardless, there are important lessons to be drawn from previous RCTs.
A fundamental question to consider in the context of previous RCTs is why anti-inflammatory agents have failed to show efficacy in septic patients, particularly when controlling excessive inflammatory response and ameliorating MOD, which is linked to inflammation in organs, is considered essential to improving the clinical course of sepsis. One reason may be that sepsis is often accompanied by a concomitant propagation of hyperinflammatory response and immunosuppressive state (5, 6). The inhibitory actions of anti-inflammatory agents while suppressing inflammation also inhibit immune function, including the bactericidal activity of immune cells, which, in turn, increases susceptibility to secondary infection. Immune suppression and secondary infection are a serious clinical issue in septic patients as previously discussed (7). The failure of the simple anti-inflammatory approach lends support to considering a preventive and/or therapeutic strategy with the dual capability of reducing inflammation in organs (e.g., heart, lung, kidney, liver), while enhancing immune cell function against secondary infection (e.g., bactericidal activity).
This raises the question whether inflammation per se, particularly hyperinflammation in the acute phase, which clearly plays an important role in the pathogenesis of sepsis and in the clinical trajectory of septic patients, is an appropriate therapeutic target. If it is, then the anti-inflammatory agents, TNF-α antagonist and TLR-4 antagonist, would be relevant medications. Yet, their failure to confer a pro-survival benefit in previous RCTs suggests otherwise. It is tempting to speculate that it may be necessary to redefine our pathophysiological target(s) and design a different strategy to prevent and/or treat septic patients based on new understanding of the molecular pathogenesis of sepsis.
The metabolic alterations associated with sepsis include insulin resistance, hyperglycemia, hyperlactatemia, hypoglutaminemia, hypercatabolism, muscle wasting, and lipoatrophy (8–10). These aberrations in metabolism have been linked to inflammatory response and multiple organ dysfunction, although their precise relationship remains unknown. Recent studies have highlighted the biological importance of crosstalk between the complex network of inflammatory signaling pathways and metabolic alterations at multiple levels, including posttranslational protein modifications, transcription, and epigenetics (11–13). This developing knowledge has led to a new concept, termed the “metabolic inflammatory complex, metabolic inflammation, or immunometabolism (14),” which provides new insights into inflammation in sepsis and major trauma (15). In the metabolic inflammatory complex, metabolic dysfunction and inflammatory response are not two separate and distinguishable entities that are simultaneously observed in sepsis. Rather, they are two aspects of the same disease entity (Fig. 1).
Fig. 1. Metabolic inflammatory complex: a new concept.
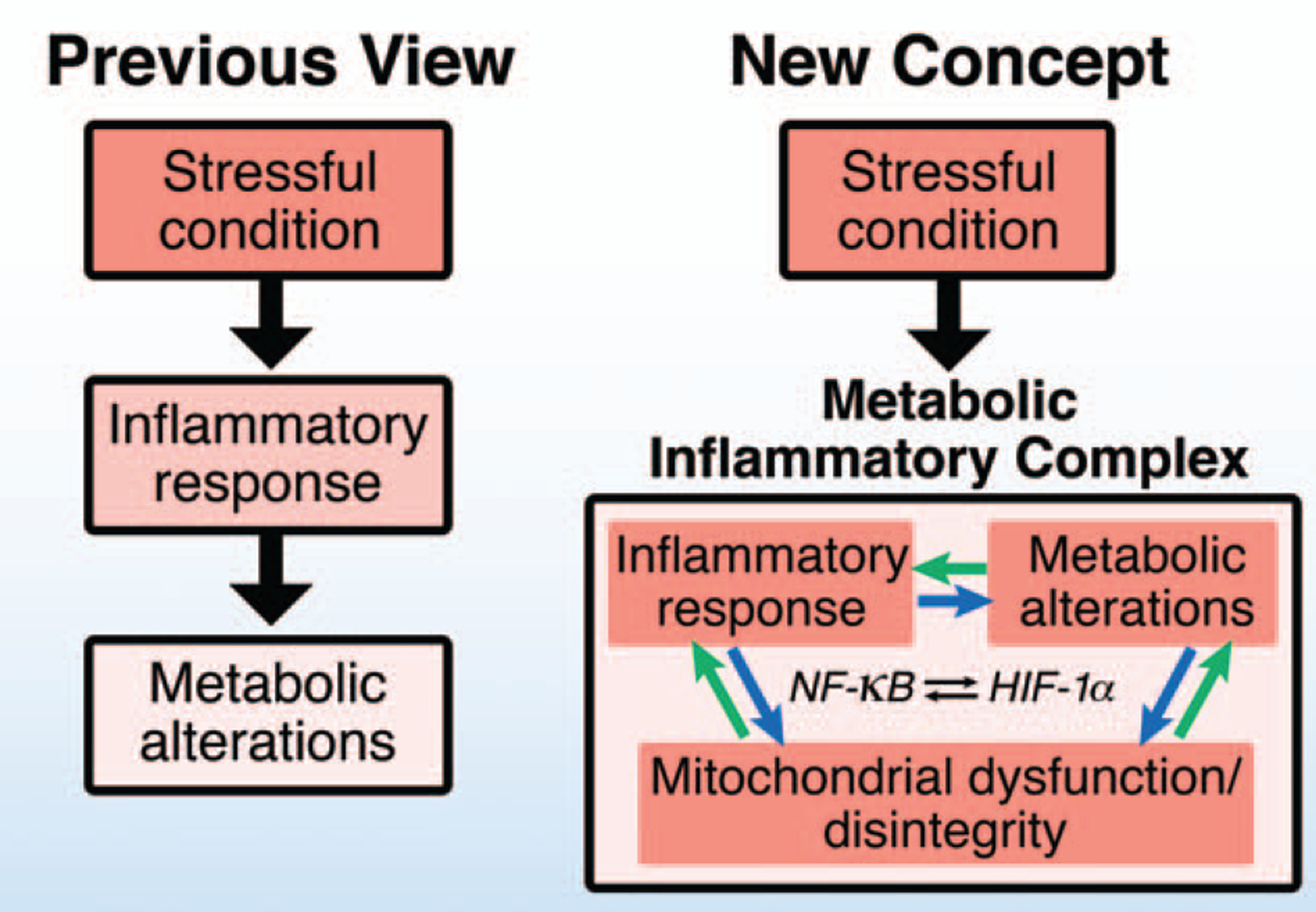
In the previous linear model, inflammatory response induces metabolic alterations under stressful conditions, such as sepsis, and inflammatory response and metabolic alterations are viewed as upstream and downstream events, respectively. In contrast, recently accumulating evidence indicates that metabolic inflammatory complex can be viewed as the nexus of a network consisting of positive feedback and feed-forward mechanisms relating to inflammatory response and metabolic alterations. Crosstalk within the metabolic inflammatory complex takes place at multiple levels, including reciprocal enhancement of the transcriptional activities of NF-κB and HIF-1α. Moreover, mitochondrial dysfunction and disintegrity, which lie at the crossroads of metabolism and inflammatory response, play crucial roles in the development of metabolic inflammatory complex. HIF indicates hypoxia-inducible factor; NF, nuclear factor.
The concept of metabolic inflammatory complex is based on several lines of evidence. First, the mitochondrion is a hub of inflammatory response as well as a major organelle in metabolism. Alterations in mitochondrial function and integrity play crucial roles in both inflammatory response and metabolic dysfunction. Second, inflammatory response and metabolic alterations induce and/or enhance each other. Therefore, the inhibition of inflammatory response prevents metabolic alterations and vice versa. For example, lipopolysaccharide (LPS) induces inflammatory response and metabolic shift in macrophages, and the metabolic shift is necessary for full activation of inflammatory response by LPS in macrophages (16). Third, alterations in metabolites, such as nicotinamide adenine dinucleotide (NAD+) depletion and increased succinate, which result from metabolic aberration, are an essential component in the activation of the inflammatory signaling pathway (17). Finally, at the transcriptional level, nuclear factor (NF)-κB and hypoxia-inducible factor (HIF)-1α are central transcription factors for inflammation and metabolic alterations, respectively, and they reciprocally activate each other (18–20) (Fig. 1). In many cases, the systemic inflammatory response and metabolic changes are closely connected to each other mainly by positive feed-forward mechanisms as discussed in this review.
In septic patients, multiple organ failure (MOF) often leads to mortality. The molecular etiology of MOF, however, is not fully understood. In the end stage of sepsis, there is exhaustion of the various systems and organs, including central nervous, immune, and cardiovascular systems, which is associated with MOF. Atrophy of skeletal muscle and adipose tissue are prominent as well. Cancer cachexia, which is characterized by a loss of body weight and skeletal muscle, is considered one of the primary causes of mortality in cancer patients. Here, I propose the possibility that septic cachexia may be a novel target disease state, which would help identify new molecular targets to improve outcomes of septic patients, particularly those with CCI. This possibility is discussed in light of new knowledge about the metabolic inflammatory complex.
MITOCHONDRIA AT THE CROSSROADS OF METABOLISM, APOPTOSIS, AND INFLAMMATION
Recent studies have revealed that mitochondria are not simply the power plants of cells, rather they are dynamic organelles that lie at the crossroads of metabolism, apoptosis, and inflammation. Mitochondrial electron transport is responsible for the major portion of adenosine triphosphate (ATP) generation and oxygen consumption in cells. In addition, complex I activity within the mitochondrial electron transport chain is a major source of the oxidized form of NAD+, which is converted from its reduced form, NADH, and thereby regulates the redox balance between NAD+ and NADH. NAD+ is a substrate of many enzymes, including the sirtuin family proteins (Sirt1-7) which play important roles in the regulation of inflammatory signaling pathways, such as NF-κB (21), and mitochondrial function (22). Moreover, the tricarboxylic acid (TCA) cycle (aka the Krebs cycle) provides not only ATP, NADH, and the reduced form of flavin adenine dinucleotide, but also is involved in the flux of numerous substrates for posttranslational protein modifications, such as acetyl-CoA (acetylation) and succinate (succinylation).
The role of the mitochondria in the regulation of apoptosis has been well established. Release of cytochrome C and Smac/DIABLO from the mitochondria to the cytosolic fraction activates caspases, such as caspase-3 and −9, promoting apoptosis. Apoptosis plays a role in sepsis (23–25). Likewise, translocation of mitochondrial DNA from the mitochondria to the cytosolic fraction activates cytosolic DNA sensors, including TLR-9, thereby inducing the activation of nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing-3 (NLRP3) inflammasome (26, 27). NLRP3 inflammasome activation, in turn, activates caspase-1, leading to maturation of interleukin (IL)-1β and IL-18 by cleavage of pro-IL-1β and pro-IL-18 (Fig. 2). Mitochondrial DNA-induced NLRP3 inflammasome activation is a major target of anti-inflammatory action mediated by α7 nicotinic acetylcholine receptor (α7 nAchR) signaling. Acetylcholine attenuates NLRP3 inflammasome activation in parallel with inhibition of mitochondria DNA release in an α7nAchR-dependent manner (26).
Fig. 2. Mitochondrial disintegrity as an inducer of intracellular and systemic inflammatory response.
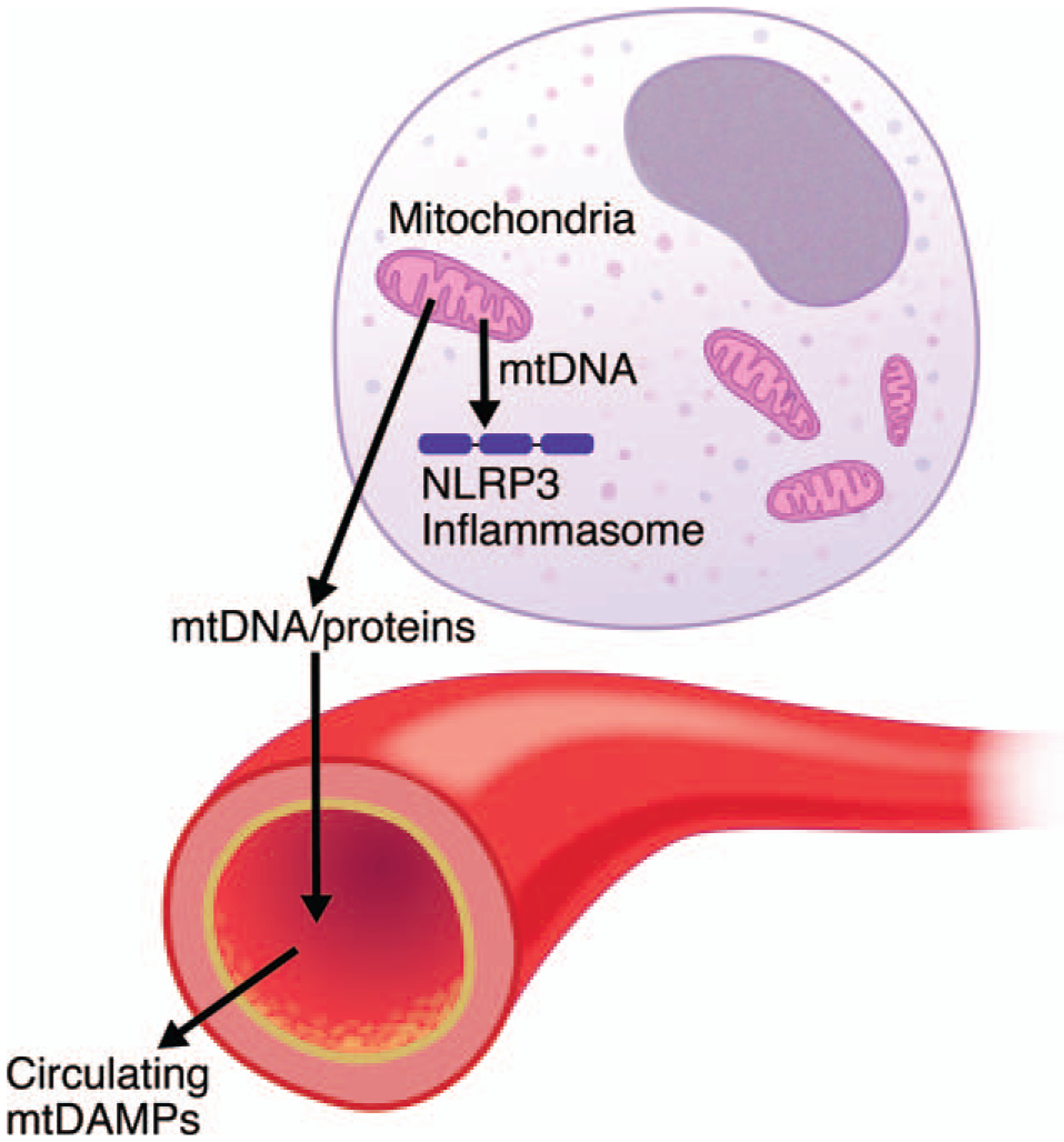
The mitochondria play an essential role in the regulation of inflammatory response as well as metabolism and apoptosis. Disruption of mitochondrial integrity leads to release of mitochondrial DNA (mtDNA) to the cytosolic fraction, which, in turn, induces activation of NLRP3 inflammasome. Moreover, when mtDNA and mitochondrial proteins (e.g., HSP60, formylated peptides) are released to the circulation, they function as mitochondrial DAMPs and thereby cause and/or exacerbate systemic inflammation. DAMPs indicates damage-associated molecular patterns; HSP60, heat shock protein 60; NLRP3, nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing-3.
The mitochondria function as key regulators of NLRP3 inflammasome activation. In addition to mitochondrial DNA, mitochondrial reactive oxygen species (ROS) production and cardiolipin (mitochondrial lipid) play important roles in NLRP3 inflammasome activation (28). Moreover, mitochondrial antiviral signaling protein is also a key trigger in the activation of inflammasomes in response to stressful conditions, including infection, tissue damage, and metabolic dysregulation (29). NLRP3 inflammasome activation also induces pyroptosis, a pro-inflammatory form of cell death that results in the release of cytosolic contents, including high-mobility group protein B1 (HMGB1), mitochondrial damage-associated molecular patterns (DAMPs), and ATP (30).
Furthermore, when mitochondrial DNA and mitochondrial proteins, such as heat shock protein 60 and formylated peptides, are released to the circulation, they function as mitochondrial DAMPs, causing and/or exacerbating systemic inflammation (31) (Fig. 2). Indeed, previous studies have reported that circulating levels of mitochondrial DNA are prognostic of mortality in critically ill patients (32).
Regarding the release of mitochondrial DNA to the cytosol, little is known about active transport machineries or mechanisms. Hence, it is thought that mitochondrial DNA is released passively when mitochondrial integrity is disrupted. Recently, however, endoplasmic reticulum (ER) stress has been shown to be capable of inducing the release of mitochondrial contents, including mitochondrial DNA, by activation of the NLRP3→caspase-2→Bid pathway in cultured macrophages (33). Together, these findings indicate that mitochondrial disintegrity increases inflammatory response by the translocation of mitochondrial DNA to the cytosol where it activates NLRP3 inflammasome, as well as by the release of mitochondrial DNA and proteins to the extracellular space, where they induce a local and systemic inflammatory response, such as the mitochondria-derived DAMPs. In septic rats, mitochondrial ROS induce cardiac inflammation via the pathway that involves mitochondrial DNA, TLR9, and NLRP3 inflammasome (34).
MITOCHONDRIAL DYSFUNCTION/DISINTEGRITY AND MULTIPLE ORGAN DYSFUNCTION (MOD)
Mitochondrial dysfunction has been implicated in the pathogenesis of MOD, including acute kidney injury (AKI), acute respiratory distress syndrome, and cardiovascular and liver dysfunction (35–38). MOD is generally accompanied by mitochondrial dysfunction. Morphological changes of the mitochondria have been shown in dysfunctional organs in rodent models of sepsis and septic patients, including AKI (39). These changes observed in the mitochondria include decreased mitochondrial mass, fragmentation and enlargement of the mitochondria, and loss of cristae structure (40, 41). Conversely, mitochondrial dysfunction can cause organ dysfunction. In rodent models of sepsis and LPS challenge, mitochondria-targeted antioxidants, SS-31, mito-vitamin E, mito-TENPO, and mitoQ, ameliorate organ dysfunction, such as AKI, cardiac dysfunction, and encephalopathy (34, 42, 43). These results clearly implicate dysfunction and/or ROS-mediated damage of the mitochondria in sepsis-associated organ dysfunction. However, the role of mitochondrial dysfunction in the development of MOD remains to be determined (35). Parenthetically, based on the above studies in rodents, the mitochondria-targeted antioxidants appear promising as a new strategy to treat septic patients. Nonetheless, we cannot completely exclude the possibility that a decrease in mitochondrial ROS might reduce the bactericidal activity of immune cells in septic patients because of the important role of mitochondrial ROS in combating pathogens. It has been shown that depletion of mitochondrial ROS by catalase overexpression impairs bacterial clearance by macrophages (44).
Cellular ATP depletion resulting from mitochondrial dysfunction can cause organ dysfunction and a systemic energy crisis which can cause MOF and death in septic patients (45). In rodents, sepsis has been shown to decrease ATP content in organs. There is some question as to whether ATP depletion precedes organ dysfunction during the development of sepsis. A previous study in septic patients with MOD has reported that ATP content in skeletal muscle was significantly lower in septic non-survivors than in septic survivors and healthy controls (46). Of interest, in the latter study there seemed to be a trend toward increased ATP content in septic survivors compared with healthy controls, although there was no statistical significance to this finding (46). It is tempting to speculate that ATP depletion or energy crisis may not be required for the development of organ dysfunction in septic patients, even though ATP depletion can cause organ failure in end-stage sepsis.
The maximum capacity of mitochondrial ATP synthesis is far in excess of demand in the normal state, whereas both ATP consumption and demand are usually increased in sepsis. LPS stimulates mitochondrial oxidative phosphorylation and ATP synthesis. In addition, decreased ATP synthesis caused by mitochondrial oxidative phosphorylation can be compensated for by increased glycolytic ATP synthesis, which is associated with increased lactate production. Consequently, mitochondrial dysfunction does not necessarily result in a significant decrease in ATP content in the early stage during the development of MOD. However, in end-stage sepsis the compensation mechanisms as well as mitochondrial oxidative phosphorylation may not function properly presumably because of general cellular malfunction and/or circulatory failure, which, in turn, leads to a substantial decrease in ATP content and energy crisis, leading to MOF and death.
Even prior to the overt decline in ATP content, mitochondrial dysfunction can contribute to organ dysfunction through multiple mechanisms. Disruption of mitochondrial integrity causes apoptosis and activation of inflammasomes with the latter leading to pyroptosis. Increases in apoptotic and pyroptotic cell death are a feature of sepsis and MOD (47, 48). A decrease in mitochondrial oxidative phosphorylation leads to a decrease not only in mitochondrial ATP synthesis but also in the generation of NAD+ from NADH by complex I. Decreases in NAD+ content or the NAD+/NADH ratio cause redox stress. Decreased intracellular NAD+ concentration impairs NAD+-dependent enzymes, including sirtuins, leading to cellular dysfunction. Recently, protective roles have been shown for Sirt1 and Sirt3, which are NAD+-dependent sirtuin deacetylases, in rodent models of sepsis (49–51). Attenuated complex I activity decreases NAD+ generation and thereby impairs the function of NAD+-dependent enzymes, such as Sirt1, Sirt3, and poly-(ADP-ribose) polymerase (PARP). To compensate for the decrease in NAD+ generation, NAD+ is produced from NADH by lactate dehydrogenase as a by-product of lactate production from pyruvate, leading to increased lactate production and hyperlactatemia. In addition, S-nitrosylation, a covalent attachment of NO to cysteine thiols, of Sirt1 by iNOS inactivates Sirt1 and plays an important role in LPS-induced liver damage in mice (52).
Collectively, over and above ATP depletion, mitochondrial dysfunction/disintegrity may play an important role in the development of MOD in sepsis by promoting the demise and dysfunction of cells, inflammatory responses, and metabolic dysregulation. These possibilities, however, have not been extensively studied in animal models of sepsis or septic patients.
METABOLIC INFLAMMATORY COMPLEX IN SEPSIS
Inflammatory response is associated with metabolic alterations. Metabolic alterations are a major complication of sepsis and associated systemic inflammation, which is linked to the clinical outcome of septic patients (8, 9, 35). It is well recognized that the inflammatory response causes metabolic dysfunction. This is exemplified by the role of inflammatory signaling pathways (e.g., NF-κB, ER stress) in the pathogenesis of obesity-induced insulin resistance (53, 54). Recent studies, however, indicate that metabolic changes modulate the inflammatory response in both immune cells and nonimmune cells. Importantly, the functional roles of metabolites are not limited to being sources of energy or constituents of macromolecules, such as proteins, DNA, RNA, and lipids, but in fact most, if not all, metabolites are important players in intra- and inter-cellular signal transduction. Many metabolites serve as substrates for posttranslational protein and DNA modifications, both of which are major mediators of intracellular signal transduction and regulators of gene expression and epigenetics. In addition, many metabolites function as allosteric inhibitors or activators of enzymes. These metabolites are too numerous to mention, but examples include ATP, acetyl-CoA, and S-adenosylmethio-nine (SAM), the substrates for phosphorylation, acetylation, and methylation, respectively. NAD+ serves as a substrate of sirtuin (e.g., Sirt1, Sirt3)-mediated deacetylation and poly ADP-ribosylation by PARP. Arginine is a substrate for nitric oxide (NO) production, which regulates many pathophysiological processes including inflammation (52, 55). A number of investigators have documented alterations in and the roles of SAM, sirtuins, PARP, and NO in sepsis.
Many observational studies have documented that prior use of statins, inhibitors of hydroxymethylglutamyl coenzyme A reductase, is associated with lower incidence of sepsis in the ICU and improved outcomes in septic patients (56, 57). Similarly, statins improve survival in rodent models of sepsis (25, 58). However, multicenter RCTs have failed to confirm the benefit of statins in septic patients (59). Despite these failures, further studies are beginning to evaluate the preventive effects of statins in the early stage of sepsis development. Statins have been prescribed as cholesterol-lowering drugs and have demonstrated substantial benefit in the prevention of ischemic heart disease. It is important to note that hypercholesterolemia is not a clinical issue in sepsis and that the effects of statins in sepsis are unrelated to cholesterol levels. The cholesterol-unrelated pleiotropic beneficial effects of statins have been highlighted since the 1990s (60). It has been proposed that inhibition of protein isoprenylation, namely farnesylation and geranylgeranylation, mediates the cholesterol-lowering-independent beneficial effects of statins, although direct evidence is lacking. We have previously shown that the amount of farnesylated protein increases in septic mice and that farnesyltransferase inhibitor (FTI) stimulates immune function and improves bacterial clearance and survival in septic mice (61). FTI also reduces morality in parallel with the inhibition of apoptosis and stress signaling in liver in LPS-challenged mice (62). These findings suggest that increases in protein farnesylation, a lipid modification of the cysteine residues, may play a role in immune suppression, inflammation, and mortality of septic mice.
ATP is also an important player in inflammatory response. Extracellular ATP activates purinergic receptors, which also contribute to NLRP3 inflammasome activation (63). Circulating levels of ATP are increased in sepsis and play roles in neutrophil activation (64) and sepsis-associated encephalopathy (65).
Collectively, these findings show that inflammatory signaling and metabolic alterations are closely connected to each other through crosstalk conducted by a complex network of signaling pathways and signaling molecules, presumably forming a metabolic inflammatory complex that plays an important role in the development and clinical trajectory of sepsis.
CYTOPATHIC HYPOXIA AND THE WARBURG EFFECT IN SEPSIS
Hyperlactatemia is a hallmark of sepsis and associated with mortality in septic patients (66). Previous studies indicate that increased lactate production rather than decreased lactate clearance has a major role in hyperlactatemia in sepsis. Over and above hypoperfusion and subsequent low tissue oxygen tension, a metabolic shift from mitochondrial oxidative phosphorylation to glycolytic ATP synthesis, even in the presence of sufficient oxygen availability, has been proposed to contribute to hyperlactatemia in sepsis (67). The predominance of glycolytic ATP synthesis over mitochondrial oxidative phosphorylation despite sufficient oxygen availability in sepsis is termed cytopathic hypoxia (68). Essentially the same metabolic shift was discovered in cancer cells by Dr Otto H. Warburg in the 1920s and is referred to as the Warburg effect (aka aerobic glycolysis). Moreover, in diabetes research, essentially the same metabolic shift has been termed pseudohypoxia, which is a complication of hyperglycemia (69). Now, it is recognized that the Warburg effect is not specific to cancer cells but can also be induced by inflammation. Hence, it is reasonable to consider that the Warburg effect, cytopathic hypoxia, and pseudohypoxia are merely different names for the same metabolic reprogramming response to inflammation, despite the differences in etiology and basal disease conditions.
The Warburg effect is characterized by the predominance of aerobic glycolysis over mitochondrial oxidative phosphorylation, increased lactate production, and increased glutaminolysis (70, 71). HIF-1α is a master transcription factor that orchestrates the Warburg effect. HIF-1α upregulates the expression of glycolytic genes, such as glucose transporter-1 and lactate dehydrogenase A, and suppresses mitochondrial function. Inflammation (e.g., NF-κB activation) increases HIF-1α expression and thereby promotes metabolic reprograming at multiple levels including increases in transcription and protein stability of HIF-1α (72), while HIF-1α and metabolic reprogramming modulates inflammatory response (73). LPS causes metabolic shift from mitochondrial oxidative phosphorylation to glycolysis in parallel with HIF-1α induction, both of which are important for activation of macrophages by LPS (16). In LPS-stimulated macrophages, the metabolic shift leads to increased succinate, a TCA cycle intermediate and a substrate of complex II in mitochondrial electron transport, mainly by glutamine-dependent anerplerosis (glutaminolysis). Increased succinate, in turn, enhances HIF-1α expression and IL-1β production by increasing succinylation (74). Conversely, inhibition of HIF-1α activity decreases IL-1β production and HMGB1 release by LPS-activated macrophages (75, 76). HIF-1α induction is required for LPS-induced activation of NLRP3 inflammasome in macrophages. Moreover, the inhibition of aerobic glycolysis (the Warburg effect) blocks LPS-induced activation of NLRP3 inflammasome, although it does not inhibit HIF-1α expression. These results clearly indicate that the metabolic shift plays a crucial role in NLRP3 inflammasome activation by LPS. Thus, metabolic reprogramming functions as the inherent machinery of the inflammatory response in immune cells.
In these studies (74–76), the experiments were performed under normoxic condition. In addition to metabolic shift and HIF-1α induction under normoxic condition (namely the Warburg effect), hypoxia-induced metabolic shift and HIF-1α expression are supposed to have similar or essentially the same effects on inflammatory response based on previous findings that hypoxia induces inflammation via HIF-1α (77).
In contrast to immune cells, limited knowledge is available about the impact of HIF-1α and the Warburg effect on inflammatory response in nonimmune cells in the context of sepsis. Interestingly, estrogen as well as HIF-1α inhibitor suppresses NF-κB activity and IL-6 levels by inhibiting HIF-1α in the heart in a mouse model of trauma-hemorrhage (20). Moreover, TNF-α induces the Warburg effect, including increased aerobic glycolysis and lactate production, through NF-κB-mediated activation of HIF-1α in cultured skeletal muscle cells (78). Collectively, these data raise the possibility that inflammatory response, HIF-1α, and the Warburg effect (metabolic shift) may form a complex by virtue of the nexus of positive feedback and feed-forward mechanisms that occurs not only in immune cells but also in organs such as heart, liver, and skeletal muscle during the development of sepsis.
Based on these data, inhibition of HIF-1α would appear to be a promising strategy for treating sepsis. In fact, inhibition of the pyruvate kinase M2 -HIF-1α pathway improved survival in septic mice (76). HIF-1α in the myeloid lineage is required for LPS-induced septic phenotype, including increased pro-inflammatory cytokine levels and hypotension, and mortality in mice (79). Conversely, increased HIF-1α expression by gene disruption of oxygen sensor, propyl hydroxylase-3, aggravates sepsis via HIF-1α- and NF-κB-mediated enhanced innate immune response in mice (78). However, there is a potential drawback to this strategy. HIF-1α is necessary to adapt to the tissue hypoxia and impaired circulation that frequently occur in sepsis. In addition, HIF-1α induction is necessary for the activation of immune cells (73). In fact, myeloid-specific HIF-1α knockout impairs bactericidal activity in mice (80). It is possible, therefore, that inhibiting HIF-1α in immune cells may increase susceptibility to infection. These considerations suggest that, for this strategy to work, it would be necessary to limit the inhibition of inflammation-induced HIF-1α expression to nonimmune cells, while preserving hypoxia-induced HIF-1α and inflammation-induced HIF-1α expression in immune cells. Otherwise, there may be a risk of worsening the clinical outcome of septic patients.
SEPTIC CACHEXIA
In an attempt to translate the concept of metabolic inflammatory complex to the development of new strategies for septic patients, it is reasonable to look for an unwanted aspect of the metabolic inflammatory complex that may induce or exacerbate immune suppression and can be treated without inhibiting immune function against pathogens. Cachectic condition may be such a candidate disease state (Fig. 3), although it has not attracted much scientific attention in the sepsis research field.
Fig. 3. Septic cachexia: a new potential target to improve the clinical outcome of septic patients.
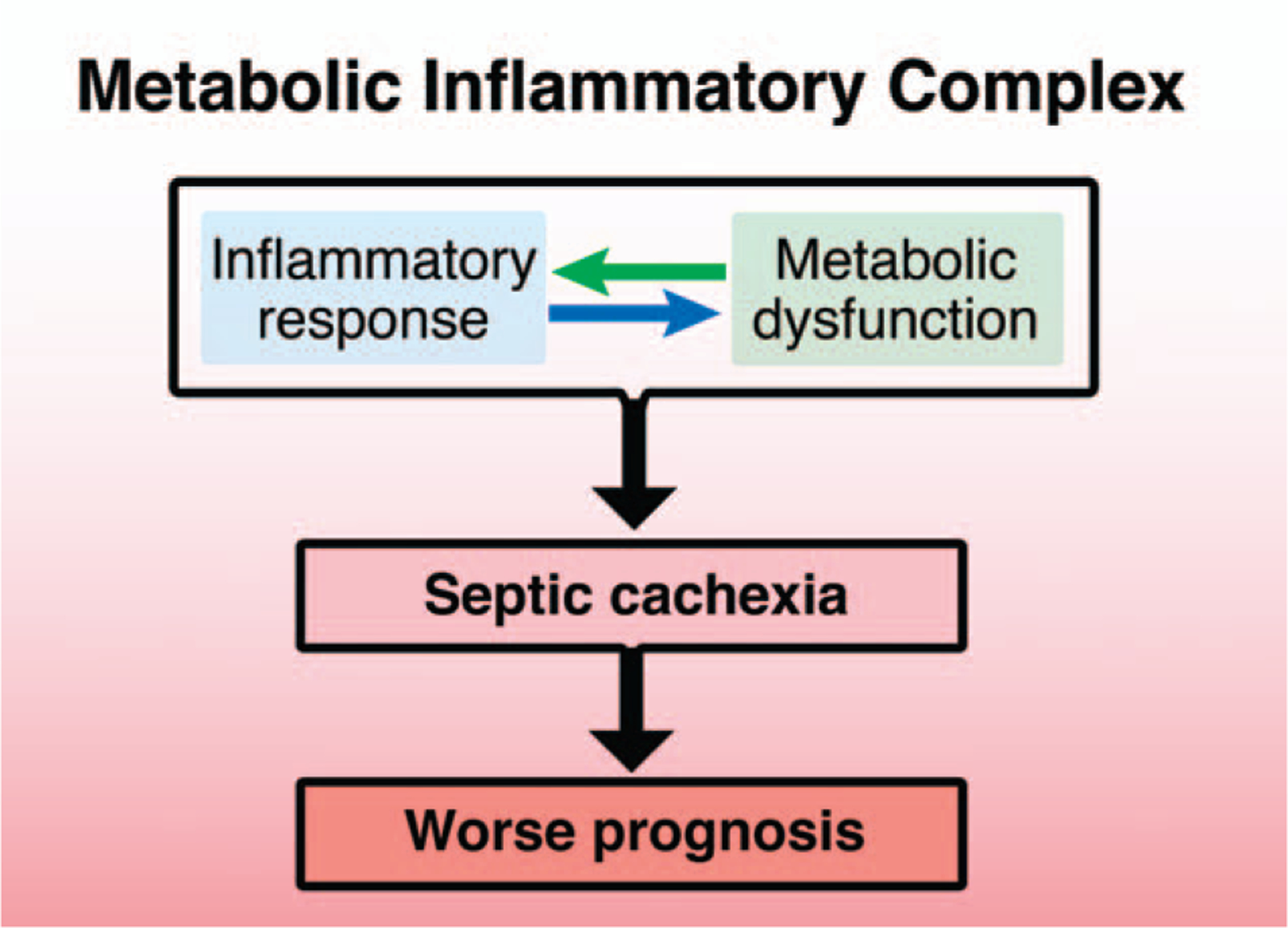
The inflammatory response of immune cells is necessary to combat pathogens in sepsis. On the other hand, excessive inflammation induces or exacerbates not only MOD, but also immune exhaustion. Like cancer cachexia, septic cachexia presumably contributes to worsening prognosis and mortality in sepsis. Preventing the metabolic derangements associated with septic cachexia, while preserving immune function, may be an alternative way to control excessive inflammation and prevent immune exhaustion. Septic cachexia, which is caused by metabolic inflammatory complex, is therefore proposed as a novel target that may potentially improve the clinical outcome of sepsis. MOD indicates multiple organ dysfunction.
Cachexia is a complex metabolic syndrome associated with underlying illness and characterized by loss of skeletal muscle mass with or without loss of fat mass (81–83). Inflammation, insulin resistance, and increased protein breakdown in muscle are frequently associated with cachexia. Anorexia is also commonly associated with cachexia; however, conventional nutritional support cannot fully reverse cachexia. Cachexia is considered one of the primary causes of mortality in cancer patients. Cachexia is associated with many chronic diseases, including cancer, chronic heart failure (CHF), chronic renal failure (CRF), chronic obstructive pulmonary disease (COPD), and human immunodeficiency virus infection/acquired immune deficiency syndrome (HIV/AIDS). Acute disease states can also cause or underlie cachexia. In fact, based on the current definition and criteria, most if not all septic patients, particularly those with CCI, meet criteria for cachexia. Sepsis has been recognized as a major disease state that underlies cachexia.
A number of reports have documented increased protein breakdown and/or decreased protein synthesis in muscle wasting in sepsis. Muscle wasting is a major metabolic complication of sepsis and increased protein breakdown is a feature of muscle wasting in sepsis (84, 85). Protein synthesis has been reported to be decreased, increased, or unaltered in skeletal muscle of septic patients (86). In septic rodents, protein synthesis is decreased in skeletal muscle (87, 88).
Inflammatory response plays a crucial role in muscle wasting as well. NF-κB activation increases the gene expression of ubiquitin ligases, atrogin-1, and Murf-1, thereby increasing protein breakdown (89). One could pose the question, based on the assumption that anti-inflammatory agents are supposed to mitigate cachexia, why have the anti-inflammatory agents tested in RCTs failed to show any pro-survival effects if cachexia is a significant contributor to the mortality of septic patients? Although the assumption seems reasonable, it is not known whether anti-inflammatory agents ameliorate septic cachexia. The effects of anti-inflammatory agents on cachectic changes (e.g., muscle wasting) were not evaluated in those RCTs. Moreover, to the best of my knowledge, the effects of typical anti-inflammatory agents, such as antagonists of pro-inflammatory cytokines and receptors for LPS or DAMPs, have not been studied in patients with or animal models of cachexia of any etiology. It is important to note that the signaling pathways involved in hyperinflammation or “classical” inflammation, which usually takes place in the acute phase of sepsis, and those involved in persistent inflammation, which is associated with cachexia, may differ. The NF-κB pathway is the major downstream signaling cascade of the targets of the anti-inflammatory agents used in the RCTs in septic patients and a key player in “classical” inflammation. In contrast, in addition to the NF-κB pathway, the transforming growth factor [TGF]-β family-Smad pathway plays a critical role in muscle wasting and persistent inflammation in cachexia, as further discussed below. Hence, it is possible that strategies which utilize typical anti-inflammatory agents (e.g., antagonists of TNF-α and TLR-4) and target the NF-κB pathway may not be appropriate for ameliorating cachexia and therefore new targets may need to be explored.
Furthermore, muscle weakness interferes with recovery and weaning from mechanical ventilation, thereby exacerbating ventilator-induced lung injury and prolonging hospital stay and rehabilitation. Obviously, muscle weakness is a clinical issue in critically ill patients. However, the negative impact of muscle wasting may not be limited to muscle weakness. In a mouse model of cancer cachexia, preventing muscle wasting by inhibiting the myostatin (a member of the TGF-β family)—type IIB activin (ActRIIB) receptor-mediated signaling pathway prolongs survival (90, 91). Although little is known about the molecular mechanisms by which prevention of muscle wasting improves survival of cancer-bearing mice, it is reasonable to speculate that some molecular basis beyond muscle weakness per se may contribute to the pro-survival effect of the inhibition of muscle wasting in cancer cachexia. It is an open question whether similar mechanisms may apply in septic cachexia as well.
Cachexia has been known to strongly correlate with poor outcomes, such as mortality, in many chronic diseases, including cancer, CHF, CRF, COPD, and HIV/AIDS. Until the aforementioned landmark study in a mouse model of cancer cachexia (84), however, it was thought that cachexia was a mere epiphenomenon that did not contribute to increased mortality. Likewise, increasing evidence seems to raise a new question, whether septic cachexia, which most but not all septic patients suffer, is a mere bystander or a potential contributor to progression and poor prognosis of sepsis.
To understand the underlying mechanisms, it may be helpful to examine current knowledge about the differences and similarities between cancer and septic cachexia. In addition to the obvious differences in etiology (i.e., cancer vs. sepsis), the time span of cancer cachexia is much longer (i.e., months–years) than septic cachexia (i.e., days–weeks). On the other hand, there are commonalities in the metabolic alterations that affect both conditions. These include muscle wasting, increased negative protein balance, body weight loss, fat mass loss, insulin resistance, hypocholesterolemia, and increased glutaminolysis. Moreover, the molecular signature of immune suppression in cancer cachexia is similar to that of sepsis, particularly sepsis that progresses to CCI. Recently, it has been proposed that PICS (i.e., persistent inflammation, immune suppression, and catabolism syndrome) is the predominant pathophysiology of septic patients with CCI and the principal cause of poor patient outcomes (92). We know that expansion of myeloid-derived suppressor cells (MDSCs) is a feature of immune suppression in both cancer cachexia (93, 94) and sepsis (95). In cachexia associated with cancer and other diseases (e.g., COPD), the TGF-β-Smad pathway has been implicated in the cascade leading to persistent inflammation, immune dysfunction, and muscle wasting (96). However, its potential role in septic cachexia, including that of macrophage inhibitory cytokine-1 (MIC-1)/growth differentiation factor-15 (GDF15), has not been studied. In contrast, the NF-κB pathway is the major signaling pathway downstream of TLR-4 and receptors for the pro-inflammatory cytokines. Therefore, in hyperinflammation, which is associated with acute phase sepsis, the NF-κB pathway is the major player, while TGF-β-Smad has no major role. Other recent studies have indicated the involvement of MIC-1/GDF15, a TGF-β family cytokine, in cachexia of various etiologies, including cancer (96). For example, a recent study demonstrated that MIC-1/GDF15 was required and sufficient to produce cancer cachexia in mice (97). MIC-1/GDF15 causes muscle wasting and fat mass loss, while it induces MDSCs that produce IL-10, an anti-inflammatory cytokine. Moreover, inhibition of MIC-1/GDF15 was found to improve survival in tumor-bearing mice and to prevent muscle atrophy. Plasma levels of MIC-1/GDF15 are increased in cachectic patients with cancer, CHF, CRF, COPD, and HIV/AIDS. Likewise, in a previous report, septic patients had approximately 27-fold greater concentrations of MIC-1/GDF15 compared with healthy controls (94). Another study suggested that MIC-1/GDF15 may mediate ICU-acquired muscle weakness (98). It is possible, therefore, that MIC-1/GDF15 may play a role in the development of septic cachexia and immunosuppression, in parallel with MDSC expansion, as with cachexia of other etiologies. This possibility, however, has not been tested in septic cachexia.
Recently, early mobilization (exercise therapy) has emerged as a promising therapy in critically ill patients. Early mobilization improved the outcome of critically ill patients, including the length of ICU and hospital stay. In a mouse model of acute lung injury, therapeutic exercise inhibited not only muscle atrophy but also neutrophil infiltration to the lung as well as alveolitis (99). These results suggest that physical inactivity (immobilization) (e.g., bed rest) may exacerbate acute lung injury and systemic inflammation in parallel with muscle wasting in critically ill patients.
OBESITY PARADOX IN CRITICAL ILLNESS
In contrast to muscle wasting, the role of fat mass loss remains relatively elusive in cachectic states, including cancer cachexia. Interestingly, recent evidence indicates that adipose tissue plays a protective role in critical illness (100, 101). Obesity has been implicated in the pathogenesis of many human diseases, including type 2 diabetes, hypertension, atherosclerosis, ischemic heart disease, and stroke. Intuitively, critically ill obese patients would be anticipated to have worse outcomes compared with their normal-weight counterparts. Contrary to expectations, various observational studies have revealed better outcomes and survival in obese and overweight (but not morbidly obese) patients compared with normal-weight individuals admitted to the ICU, including septic patients (102, 103). This observation is called the obesity paradox (102). Consistent, high-fat diet feeding for 12 weeks improves survival after the induction of sepsis in mice (104), although controversial results have also been reported (105). The reasons for the obesity paradox remain largely unknown. Nonetheless, these findings support the hypothesis that sepsis-related fat mass loss may contribute to worse prognosis and that preservation of fat mass may be protective in septic patients.
Previously, adipose tissue was thought to be a static tissue whose only role was to store excess energy. In contrast, adipose tissue is now recognized as an active endocrine organ that secretes adipokines, including adiponectin. Deficiency of adiponectin reduces survival of septic mice, while adiponectin treatment improves survival (106–108). Hence, it is conceivable that adipose tissue may be an important source of adipokines, such as adiponectin, and possibly exosomes (microvesicles). Alternatively, obesity-induced alterations in metabolism may play a role in the impact of fat mass on outcomes of sepsis. Obesity reduces energy expenditure, whereas sepsis-associated hypermetabolism exacerbates cachectic change. Obesity inhibits fatty acid oxidation capacity (109), while sepsis is associated with increased fat oxidation (8). Increased fatty acid oxidation can accelerate inflammation (110). Thus, obesity and fat deposition may possibly counteract the sepsis-associated alterations in energy and lipid metabolism, while fat mass loss might accelerate them. This possibility is certainly worth consideration.
CONCLUSIONS/FUTURE PERSPECTIVES
Inflammatory response and metabolic derangements are closely connected to each other through a complicated network of positive feedback and feed-forward mechanisms that form a metabolic inflammatory complex in sepsis. Several lines of evidence indicate that metabolic inflammatory complex plays a role in the development of MOD and sepsis.
Recent advancements in intensive care have saved the lives of septic patients in the acute phase, but more and more septic patients are suffering from CCI. The poor outcomes of septic patients with CCI have emerged as a public health issue, which is accelerated by a rapidly increasing aged population. Chronic sepsis or septic CCI represents a new frontier of clinical and basic research. Animal models of chronic sepsis/septic CCI need to be established.
PICS is often associated with chronic sepsis and CCI. Together, the negative results of the previous RCTs of anti-inflammatory agents suggest the importance of developing new strategies with dual capabilities of reducing persistent inflammation in organs while reversing or ameliorating immune dysfunction. In the current predominant paradigm, inflammation, immune dysfunction, and catabolism are thought to be different aspects of sepsis and therefore they are treated individually. In contrast, in the challenging paradigm, cachexia, which substantially overlaps the phenotype of PICS, is a vicious self-reinforcing cycle where these seemingly distinct aspects are closely connected to each other and evolve via the nexus of positive feed-forward mechanisms. Although sepsis progression may not be attributable to any single cause, nodal points of the network involved in this complex syndrome may serve as novel potential targets to improve the clinical outcome of septic patients. In light of the new concept of septic cachexia, recently accumulating evidence raises the possibility that an array of molecules, cellular events and disease states, such as MDSC expansion, MIC-1/GDF15, myostatin, ActRIIB receptor, Smad, and physical inactivity (e.g., bed rest), may serve as new targets to prevent and/or treat chronic septic patients with poor prognosis. From this author’s perspective, the hypothesis that ameliorating cachexia may prevent or mitigate immune dysfunction and persistent inflammation in multiple organs and thereby improve survival in an animal model of chronic sepsis/septic CCI is a concept worth testing.
ACKNOWLEDGMENTS
The author expresses apologies to many authors whose important works are not cited in this review related to the space limitation and thanks Mr Hayato Miki for his technical assistance.
This work was supported by grants from the NIH (R01 GM115552, R01GM117298) and Shriners Hospitals for Children (85600).
Footnotes
The author reports no conflicts of interest.
REFERENCES
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. : The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315(8):801–810, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kahn JM, Le T, Angus DC, Cox CE, Hough CL, White DB, Yende S, Carson SS: The epidemiology of chronic critical illness in the United States. Crit Care Med 43(2):282–287, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marshall JC: Why have clinical trials in sepsis failed? Trends Mol Med 20(4):195–203, 2014. [DOI] [PubMed] [Google Scholar]
- 4.Bernard GR, Francois B, Mira JP, Vincent JL, Dellinger RP, Russell JA, Larosa SP, Laterre PF, Levy MM, Dankner W, et al. : Evaluating the efficacy and safety of two doses of the polyclonal anti-tumor necrosis factor-alpha fragment antibody AZD9773 in adult patients with severe sepsis and/or septic shock. randomized, double-blind, placebo-controlled phase IIb study. Crit Care Med 42:504–511, 2014. [DOI] [PubMed] [Google Scholar]
- 5.Leentjens J, Kox M, van der Hoeven JG, Netea MG, Pickkers P: Immunotherapy for the adjunctive treatment of sepsis: from immunosuppression to immunostimulation. Time for a paradigm change? Am J Respir Crit Care Med 187(12):1287–1293, 2013. [DOI] [PubMed] [Google Scholar]
- 6.Rimmele T, Payen D, Cantaluppi V, Marshall J, Gomez H, Gomez A, Murray P, Kellum JA, Workgroup AX: Immune cell phenotype and function in sepsis. Shock 45(3):282–291, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koch RM, Kox M, de Jonge MI, van der Hoeven JG, Ferwerda G, Pickkers P: Patterns in bacterial- and viral-induced immunosuppression and secondary infections in the ICU. Shock 47(1):5–12, 2017. [DOI] [PubMed] [Google Scholar]
- 8.Tappy L, Chiolero R: Substrate utilization in sepsis and multiple organ failure. Crit Care Med 35(9 suppl):S531–534, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Levy RJ: Mitochondrial dysfunction, bioenergetic impairment, and metabolic down-regulation in sepsis. Shock 28(1):24–28, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Cooney RN, Kimball SR, Vary TC: Regulation of skeletal muscle protein turnover during sepsis: mechanisms and mediators. Shock 7(1):1–16, 1997. [DOI] [PubMed] [Google Scholar]
- 11.Mills E, O’Neill LA: Succinate: a metabolic signal in inflammation. Trends Cell Biol 24(5):313–320, 2014. [DOI] [PubMed] [Google Scholar]
- 12.Moschen AR, Adolph TE, Gerner RR, Wieser V, Tilg H: Lipocalin-2: a master mediator of intestinal and metabolic inflammation. Trends Endocrinol Metab 28(5):388–397, 2017. [DOI] [PubMed] [Google Scholar]
- 13.Raghuraman S, Donkin I, Versteyhe S, Barres R, Simar D: The emerging role of epigenetics in inflammation and immunometabolism. Trends Endocrinol Metab 27(11):782–795, 2016. [DOI] [PubMed] [Google Scholar]
- 14.O’Neill LA, Pearce EJ: Immunometabolism governs dendritic cell and macrophage function. J Exp Med 213(1):15–23, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaplan J: Is leptin a key to metabolic inflammation in trauma and sepsis? Shock 48(1):138, 2017. [DOI] [PubMed] [Google Scholar]
- 16.Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, et al. : Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab 21(1):65–80, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A: Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal 25(10):1939–1948, 2013. [DOI] [PubMed] [Google Scholar]
- 18.van Uden P, Kenneth NS, Rocha S: Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J 412(3):477–484, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Ignazio L, Bandarra D, Rocha S: NF-kappaB and HIF crosstalk in immune responses. FEBS J 283(3):413–424, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nickel EA, Hsieh CH, Chen JG, Schwacha MG, Chaudry IH: Estrogen suppresses cardiac IL-6 after trauma-hemorrhage via a hypoxia-inducible factor 1 alpha-mediated pathway. Shock 31(4):354–358, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW: Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23(12):2369–2380, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, et al. : SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464(7285):121–125, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen G, Li X, Huang M, Zhou X, Li Y, Mao X, Bai J: The role of thioredoxin-1 in suppression sepsis through inhibiting mitochonrial- induced apoptosis in spleen. Shock 46(7):753–758, 2017. [DOI] [PubMed] [Google Scholar]
- 24.Chung CS, Venet F, Chen Y, Jones LN, Wilson DC, Ayala CA, Ayala A: Deficiency of Bid protein reduces sepsis-induced apoptosis and inflammation, while improving septic survival. Shock 34(2):150–161, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buerke U, Carter JM, Schlitt A, Russ M, Schmidt H, Sibelius U, Grandel U, Grimminger F, Seeger W, Mueller-Werdan U, et al. : Apoptosis contributes to septic cardiomyopathy and is improved by simvastatin therapy. Shock 29(4):497–503, 2008. [DOI] [PubMed] [Google Scholar]
- 26.Lu B, Kwan K, Levine YA, Olofsson PS, Yang H, Li J, Joshi S, Wang H, Andersson U, Chavan SS, et al. : alpha7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol Med 20:350–358, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. : Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12(3):222–230, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, et al. : Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39(2):311–323, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN: The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153(2):348–361, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu D, Pan P, Su X, Zhang L, Qin Q, Tan H, Huang L, Li Y: Interferon regulatory factor-1 mediates alveolar macrophage pyroptosis during lps-induced acute lung injury in mice. Shock 46(3):329–338, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Struck J, Uhlein M, Morgenthaler NG, Furst W, Hoflich C, Bahrami S, Bergmann A, Volk HD, Redl H: Release of the mitochondrial enzyme carbamoyl phosphate synthase under septic conditions. Shock 23(6):533–538, 2005. [PubMed] [Google Scholar]
- 32.Nakahira K, Kyung SY, Rogers AJ, Gazourian L, Youn S, Massaro AF, Quintana C, Osorio JC, Wang Z, Zhao Y, et al. : Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med 10(12):e1001577, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bronner DN, Abuaita BH, Chen X, Fitzgerald KA, Nunez G, He Y, Yin XM, O’Riordan MX: Endoplasmic reticulum stress activates the inflammasome via nlrp3- and caspase-2-driven mitochondrial damage. Immunity 43(3):451–462, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao X, Carlson D, Sun Y, Ma L, Wolf SE, Minei JP, Zang QS: Mitochondrial ROS induces cardiac inflammation via a pathway through mtdna damage in a pneumonia-related sepsis model. PLoS One 10(10):e0139416, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arulkumaran N, Deutschman CS, Pinsky MR, Zuckerbraun B, Schumacker PT, Gomez H, Gomez A, Murray P, Kellum JA: Mitochondrial Function in Sepsis. Shock 45(3):271–281, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomez H, Ince C, De Backer D, Pickkers P, Payen D, Hotchkiss J, Kellum JA: A unified theory of sepsis-induced acute kidney injury: inflammation, micro-circulatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock 41(1):3–11, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang LJ, Dong HP, Chuang IC, Liu MS, Yang RC: Attenuation of mitochondrial unfolded protein response is associated with hepatic dysfunction in septic rats. Shock 38(6):642–648, 2012. [DOI] [PubMed] [Google Scholar]
- 38.Karlsson M, Hara N, Morata S, Sjovall F, Kilbaugh T, Hansson MJ, Uchino H, Elmer E: Diverse and tissue-specific mitochondrial respiratory response in a mouse model of sepsis-induced multiple organ failure. Shock 45(4):404–410, 2016. [DOI] [PubMed] [Google Scholar]
- 39.Parikh SM: Therapeutic targeting of the mitochondrial dysfunction in septic acute kidney injury. Curr Opin Crit Care 19(6):554–559, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fredriksson K, Hammarqvist F, Strigard K, Hultenby K, Ljungqvist O, Wernerman J, Rooyackers O: Derangements in mitochondrial metabolism in intercostal and leg muscle of critically ill patients with sepsis-induced multiple organ failure. Am J Physiol Endocrinol Metab 291(5):E1044–1050, 2006. [DOI] [PubMed] [Google Scholar]
- 41.Welty-Wolf KE, Simonson SG, Huang YC, Fracica PJ, Patterson JW, Piantadosi CA: Ultrastructural changes in skeletal muscle mitochondria in gram-negative sepsis. Shock 5(5):378–384, 1996. [DOI] [PubMed] [Google Scholar]
- 42.Patil NK, Parajuli N, MacMillan-Crow LA, Mayeux PR: Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: mitochondria-targeted antioxidant mitigates injury. Am J Physiol Renal Physiol 306(7):F734–743, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zang QS, Sadek H, Maass DL, Martinez B, Ma L, Kilgore JA, Williams NS, Frantz DE, Wigginton JG, Nwariaku FE, et al. : Specific inhibition of mitochondrial oxidative stress suppresses inflammation and improves cardiac function in a rat pneumonia-related sepsis model. Am J Physiol Heart Circ Physiol 302(9):H1847–1859, 2012. [DOI] [PubMed] [Google Scholar]
- 44.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S: TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472(7344): 476–480, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee I, Huttemann M: Energy crisis: the role of oxidative phosphorylation in acute inflammation and sepsis. Biochim Biophys Acta 1842(9):1579–1586, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M: Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 360(9328):219–223, 2002. [DOI] [PubMed] [Google Scholar]
- 47.Aziz M, Jacob A, Wang P: Revisiting caspases in sepsis. Cell Death Dis 5:e1526, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pinheiro da Silva F, Nizet V: Cell death during sepsis: integration of disintegration in the inflammatory response to overwhelming infection. Apoptosis 14(4):509–521, 2009. [DOI] [PubMed] [Google Scholar]
- 49.Liu TF, Vachharajani V, Millet P, Bharadwaj MS, Molina AJ, McCall CE: Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J Biol Chem 290(1):396–408, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Opal S, Ellis JL, Suri V, Freudenberg JM, Vlasuk GP, Li Y, Chahin AB, Palardy JE, Parejo N, Yamamoto M, et al. : Sirt1 activation markedly alters transcription profiles and improves outcome in experimental sepsis. Shock; 2015. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 51.Opal SM, Ellis JL, Suri V, Freudenberg JM, Vlasuk GP, Li Y, Chahin AB, Palardy JE, Parejo N, Yamamoto M, et al. : Pharmacological Sirt1 activation improves mortality and markedly alters transcriptional profiles that accompany experimental sepsis. Shock 45(4):411–418, 2016. [DOI] [PubMed] [Google Scholar]
- 52.Shinozaki S, Chang K, Sakai M, Shimizu N, Yamada M, Tanaka T, Nakazawa H, Ichinose F, Yamada Y, Ishigami A, et al. : Inflammatory stimuli induce inhibitory S-nitrosylation of the deacetylase SIRT1 to increase acetylation and activation of p53 and p65. Sci Signal 7(351):ra106, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaneki M, Shimizu N, Yamada D, Chang K: Nitrosative stress and pathogenesis of insulin resistance. Antioxid Redox Signal 9(3):319–329, 2007. [DOI] [PubMed] [Google Scholar]
- 54.Martyn JA, Kaneki M, Yasuhara S: Obesity-induced insulin resistance and hyperglycemia: etiologic factors and molecular mechanisms. Anesthesiology 109(1):137–148, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu C, Yi C, Wang H, Bruce IC, Xia Q: Mitochondrial nitric oxide synthase participates in septic shock myocardial depression by nitric oxide overproduction and mitochondrial permeability transition pore opening. Shock 37(1):110–115, 2012. [DOI] [PubMed] [Google Scholar]
- 56.Wan YD, Sun TW, Kan QC, Guan FX, Zhang SG: Effect of statin therapy on mortality from infection and sepsis: a meta-analysis of randomized and observational studies. Crit Care 18(2):R71, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schurr JW, Wu W, Smith-Hannah A, Smith CJ, Barrera R: Incidence of sepsis and mortality with prior exposure of HMG-COA reductase inhibitors in a surgical intensive care population. Shock 45(1):10–15, 2016. [DOI] [PubMed] [Google Scholar]
- 58.Zhang S, Luo L, Wang Y, Rahman M, Lepsenyi M, Syk I, Jeppsson B, Thorlacius H: Simvastatin protects against T cell immune dysfunction in abdominal sepsis. Shock 38(5):524–531, 2012. [DOI] [PubMed] [Google Scholar]
- 59.Thomas G, Hraiech S, Loundou A, Truwit J, Kruger P, McAuley DF, Papazian L, Roch A: Statin therapy in critically-ill patients with severe sepsis: a review and meta-analysis of randomized clinical trials. Minerva Anestesiol 81(8):921–930, 2015. [PubMed] [Google Scholar]
- 60.Coldewey SM, Thiemermann C: Pleiotropic effects of atorvastatin in experimental sepsis: preservation of beta1-adrenoreceptor signaling in the heart. Shock 41(5):458–459, 2014. [DOI] [PubMed] [Google Scholar]
- 61.Yang W, Yamada M, Tamura Y, Chang K, Mao J, Zou L, Feng Y, Kida K, Scherrer-Crosbie M, Chao W, et al. : Farnesyltransferase inhibitor FTI-277 reduces mortality of septic mice along with improved bacterial clearance. J Pharmacol Exp Ther 339(3):832–841, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shinozaki S, Inoue Y, Yang W, Fukaya M, Carter EA, Yu YM, Fischman A, Tompkins R, Kaneki M: Farnesyltransferase inhibitor improved survival following endotoxin challenge in mice. Biochem Biophys Res Commun 391(3):1459–1464, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E: Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1beta secretion in response to ATP. Nat Commun 7:10555, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sumi Y, Woehrle T, Chen Y, Bao Y, Li X, Yao Y, Inoue Y, Tanaka H, Junger WG: Plasma ATP is required for neutrophil activation in a mouse sepsis model. Shock 42(2):142–147, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Savio LE, Andrade MG, de Andrade Mello P, Santana PT, Moreira-Souza AC, Kolling J, Longoni A, Feldbrugge L, Wu Y, Wyse AT, et al. : P2X7 receptor signaling contributes to sepsis-associated brain dysfunction. Mol Neurobiol; 2016. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 66.Haas SA, Lange T, Saugel B, Petzoldt M, Fuhrmann V, Metschke M, Kluge S: Severe hyperlactatemia, lactate clearance and mortality in unselected critically ill patients. Intensive Care Med 42(2):202–210, 2016. [DOI] [PubMed] [Google Scholar]
- 67.Levy B, Perez P, Gibot S, Gerard A: Increased muscle-to-serum lactate gradient predicts progression towards septic shock in septic patients. Intensive Care Med 36(10):1703–1709, 2010. [DOI] [PubMed] [Google Scholar]
- 68.Fink MP: Bench-to-bedside review: cytopathic hypoxia. Crit Care 6(6):491–499, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, Nyengaard JR, van den Enden M, Kilo C, Tilton RG: Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 42(6):801–813, 1993. [DOI] [PubMed] [Google Scholar]
- 70.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. : c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458(7239):762–765, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB: The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7(1):11–20, 2008. [DOI] [PubMed] [Google Scholar]
- 72.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M: NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 453(7196):807–811, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. : HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112(5):645–657, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. : Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496(7444):238–242, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.May CN, Dashwood MR, Whitehead CJ, Mathias CJ: Differential cardiovascular and respiratory responses to central administration of selective opioid agonists in conscious rabbits: correlation with receptor distribution. Br J Pharmacol 98(3):903–913, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, Kang R, Lotze MT, Billiar TR, Wang H, et al. : PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun 5:4436, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fitzpatrick SF, Tambuwala MM, Bruning U, Schaible B, Scholz CC, Byrne A, O’Connor A, Gallagher WM, Lenihan CR, Garvey JF, et al. : An intact canonical NF-kappaB pathway is required for inflammatory gene expression in response to hypoxia. J Immunol 186(2):1091–1096, 2011. [DOI] [PubMed] [Google Scholar]
- 78.Remels AH, Gosker HR, Verhees KJ, Langen RC, Schols AM: TNF-alpha-induced NF-kappaB activation stimulates skeletal muscle glycolytic metabolism through activation of HIF-1alpha. Endocrinology 156(5):1770–1781, 2015. [DOI] [PubMed] [Google Scholar]
- 79.Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V: Cutting edge: essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol 178(12):7516–7519, 2007. [DOI] [PubMed] [Google Scholar]
- 80.Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS: HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest 115(7):1806–1815, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D, Jatoi A, Kalantar-Zadeh K, Lochs H, Mantovani G, et al. : Cachexia: a new definition. Clin Nutr 27(6):793–799, 2008. [DOI] [PubMed] [Google Scholar]
- 82.Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G, et al. : Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 12(5):489–495, 2011. [DOI] [PubMed] [Google Scholar]
- 83.Blum D, Stene GB, Solheim TS, Fayers P, Hjermstad MJ, Baracos VE, Fearon K, Strasser F, Kaasa S: Validation of the Consensus-Definition for Cancer Cachexia and evaluation of a classification model—a study based on data from an international multicentre project (EPCRC-CSA). Ann Oncol 25(8):1635–1642, 2014. [DOI] [PubMed] [Google Scholar]
- 84.Callahan LA, Supinski GS: Sepsis-induced myopathy. Crit Care Med 37(10 suppl):S354–367, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schefold JC, Bierbrauer J, Weber-Carstens S: Intensive care unit-acquired weakness (ICUAW) and muscle wasting in critically ill patients with severe sepsis and septic shock. J Cachexia Sarcopenia Muscle 1(2):147–157, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Klaude M, Mori M, Tjader I, Gustafsson T, Wernerman J, Rooyackers O: Protein metabolism and gene expression in skeletal muscle of critically ill patients with sepsis. Clin Sci (Lond) 122(3):133–142, 2012. [DOI] [PubMed] [Google Scholar]
- 87.Lang CH, Frost RA, Vary TC: Regulation of muscle protein synthesis during sepsis and inflammation. Am J Physiol Endocrinol Metab 293(2):E453–459, 2007. [DOI] [PubMed] [Google Scholar]
- 88.Vary TC, Kimball SR: Sepsis-induced changes in protein synthesis: differential effects on fast- and slow-twitch muscles. Am J Physiol 262(6 pt 1):C1513–1519, 1992. [DOI] [PubMed] [Google Scholar]
- 89.Cai D, Frantz JD, Tawa NE Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, et al. : IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119(2):285–298, 2004. [DOI] [PubMed] [Google Scholar]
- 90.Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, et al. : Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142(4):531–543, 2010. [DOI] [PubMed] [Google Scholar]
- 91.Hatakeyama S, Summermatter S, Jourdain M, Melly S, Minetti GC, Lach-Trifilieff E: ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti-cancer treatments. Skelet Muscle 6:26, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mira JC, Gentile LF, Mathias BJ, Efron PA, Brakenridge SC, Mohr AM, Moore FA, Moldawer LL: Sepsis pathophysiology, chronic critical illness, and persistent inflammation-immunosuppression and catabolism syndrome. Crit Care Med 45(2):253–262, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cuenca AG, Cuenca AL, Winfield RD, Joiner DN, Gentile L, Delano MJ, Kelly-Scumpia KM, Scumpia PO, Matheny MK, Scarpace PJ, et al. : Novel role for tumor-induced expansion of myeloid-derived cells in cancer cachexia. J Immunol 192(12):6111–6119, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mueller T, Leitner I, Egger M, Haltmayer M, Dieplinger B: Association of the biomarkers soluble ST2, galectin-3 and growth-differentiation factor-15 with heart failure and other non-cardiac diseases. Clin Chim Acta 445:155–160, 2015. [DOI] [PubMed] [Google Scholar]
- 95.Mathias B, Delmas AL, Ozrazgat-Baslanti T, Vanzant EL, Szpila BE, Mohr AM, Moore FA, Brakenridge SC, Brumback BA, Moldawer LL, et al. : Human Myeloid-derived suppressor cells are associated with chronic immune suppression after severe sepsis/septic shock. Ann Surg 265(4):827–834, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Johnen H, Lin S, Kuffner T, Brown DA, Tsai VW, Bauskin AR, Wu L, Pankhurst G, Jiang L, Junankar S, et al. : Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat Med 13(11):1333–1340, 2007. [DOI] [PubMed] [Google Scholar]
- 97.Lerner L, Tao J, Liu Q, Nicoletti R, Feng B, Krieger B, Mazsa E, Siddiquee Z, Wang R, Huang L, et al. : MAP3K11/GDF15 axis is a critical driver of cancer cachexia. J Cachexia Sarcopenia Muscle 7(4):467–482, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bloch SA, Lee JY, Syburra T, Rosendahl U, Griffiths MJ, Kemp PR, Polkey MI: Increased expression of GDF-15 may mediate ICU-acquired weakness by down-regulating muscle microRNAs. Thorax 70(3):219–228, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Files DC, Liu C, Pereyra A, Wang ZM, Aggarwal NR, D’Alessio FR, Garibaldi BT, Mock JR, Singer BD, Feng X, et al. : Therapeutic exercise attenuates neutrophilic lung injury and skeletal muscle wasting. Sci Transl Med 7(278). 278ra232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Oliveros H, Villamor E: Obesity and mortality in critically ill adults: a systematic review and meta-analysis. Obesity (Silver Spring) 16(3):515–521, 2008. [DOI] [PubMed] [Google Scholar]
- 101.Robinson MK, Mogensen KM, Casey JD, McKane CK, Moromizato T, Rawn JD, Christopher KB: The relationship among obesity, nutritional status, and mortality in the critically ill. Crit Care Med 43(1):87–100, 2015. [DOI] [PubMed] [Google Scholar]
- 102.Utzolino S, Ditzel CM, Baier PK, Hopt UT, Kaffarnik MF: The obesity paradox in surgical intensive care patients with peritonitis. J Crit Care 29(5):887.e1–887.e5, 2014. [DOI] [PubMed] [Google Scholar]
- 103.Sakr Y, Alhussami I, Nanchal R, Wunderink RG, Pellis T, Wittebole X, Martin-Loeches I, Francois B, Leone M, Vincent JL: Being overweight is associated with greater survival in icu patients: results from the intensive care over nations audit. Crit Care Med 43(12):2623–2632, 2015. [DOI] [PubMed] [Google Scholar]
- 104.Siegl D, Annecke T, Johnson BL 3rd, Schlag C, Martignoni A, Huber N, Conzen P, Caldwell CC, Tschop J: Obesity-induced hyperleptinemia improves survival and immune response in a murine model of sepsis. Anesthesiology 121(1):98–114, 2014. [DOI] [PubMed] [Google Scholar]
- 105.Kaplan JM, Nowell M, Lahni P, O’Connor MP, Hake PW, Zingarelli B: Short-term high fat feeding increases organ injury and mortality after polymicrobial sepsis. Obesity (Silver Spring) 20(10):1995–2002, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.van Meurs M, Castro P, Shapiro NI, Lu S, Yano M, Maeda N, Funahashi T, Shimomura I, Zijlstra JG, Molema G, et al. : Adiponectin diminishes organ-specific microvascular endothelial cell activation associated with sepsis. Shock 37(4):392–398, 2012. [DOI] [PubMed] [Google Scholar]
- 107.Uji Y, Yamamoto H, Tsuchihashi H, Maeda K, Funahashi T, Shimomura I, Shimizu T, Endo Y, Tani T: Adiponectin deficiency is associated with severe polymicrobial sepsis, high inflammatory cytokine levels, and high mortality. Surgery 145(5):550–557, 2009. [DOI] [PubMed] [Google Scholar]
- 108.Teoh H, Quan A, Bang KW, Wang G, Lovren F, Vu V, Haitsma JJ, Szmitko PE, Al-Omran M, Wang CH, et al. : Adiponectin deficiency promotes endothelial activation and profoundly exacerbates sepsis-related mortality. Am J Physiol Endocrinol Metab 295(3):E658–664, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Houmard JA: Intramuscular lipid oxidation and obesity. Am J Physiol Regul Integr Comp Physiol 294(4):R1111–1116, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moon JS, Nakahira K, Chung KP, DeNicola GM, Koo MJ, Pabon MA, Rooney KT, Yoon JH, Ryter SW, Stout-Delgado H, et al. : NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat Med 22(9):1002–1012, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]