

Abstract
While significant advancements have been made in the available therapies for metastatic non-small cell lung cancer (NSCLC), acquired resistance remains a major barrier to treatment. We have not yet achieved the ability to cure advanced NSCLC with systemic therapy, despite our growing understanding of many of the oncogenic drivers of this disease. Rather, the emergence of drug-tolerant and drug-resistant cells remains the rule, even in the face of increasingly potent targeted therapies. In this review, we provide a broad overview of the mechanisms of resistance to targeted therapy that have been demonstrated across molecular subtypes of NSCLC, highlighting the dynamic interplay between driver oncogene, bypass signaling pathways, shifting cellular phenotypes, and surrounding tumor microenvironment.
Keywords: Non-small cell lung cancer (NSCLC), Targeted therapies, Acquired Resistance, Tumor microenvironment
Introduction
The clinical landscape for advanced non-small cell lung cancer (NSCLC) has evolved rapidly over the past twenty years. The discovery of activating genomic alterations in EGFR, ALK, ROS1, MET, RET, NTRK, and BRAF and the approval of progressively potent systemic targeted therapies has transformed treatment of these subsets of NSCLC such that the first line(s) of systemic treatment can often involve only single-agent tyrosine kinase inhibitors (TKIs) (Mok et al. 2009; Mitsudomi et al. 2010; Maemondo et al. 2010; Jänne et al. 2012; Soria et al. 2018; Peters et al. 2017). In parallel, immune checkpoint inhibitors have been developed, which have activity in tumors without targetable alterations, in addition to the previous standard platinum doublet mainstay of therapy (Reck et al. 2016; Gandhi et al. 2018; Socinski et al. 2018; Sandler et al. 2006).
Unfortunately, despite these therapeutic advances that have led to significant improvements in progression-free and overall survival, metastatic disease remains incurable. Even with improved response rates in many molecular subsets of NSCLC, our ability to achieve durable remissions remains limited by the inevitable development of resistance to systemic therapy. As the number and range of available therapeutic approaches has expanded, so too has the biological complexity of therapeutic resistance. Ongoing discovery efforts to further define mechanisms of resistance at the level of the genome, epigenome/transcriptome, and tumor microenvironment remain critical to improving the efficacy of our treatments. Herein, we will focus on emerging data and themes of therapeutic resistance to targeted therapies in NSCLC, focusing both on tumor cell-intrinsic and cell-extrinsic mechanisms.
On-target resistance
One of the earliest recognized and genomically simplest mechanisms of resistance to targeted therapy in NSCLC has been ‘on-target’ alterations, or genetic changes in the driver oncogene itself that confer sustained activation of original pro-tumorigenic signaling pathways despite the presence of inhibitor (Figure 1). This is also often described as oncogene-dependent therapeutic resistance. The two most common manifestations of on-target acquired resistance in NSCLC are kinase domain mutations and overexpression of the target oncogene within tumor cells. Examples of on-target resistance mechanisms are seen in response to virtually every targeted therapy in NSCLC, beginning with the earliest generation inhibitors.
Figure 1.
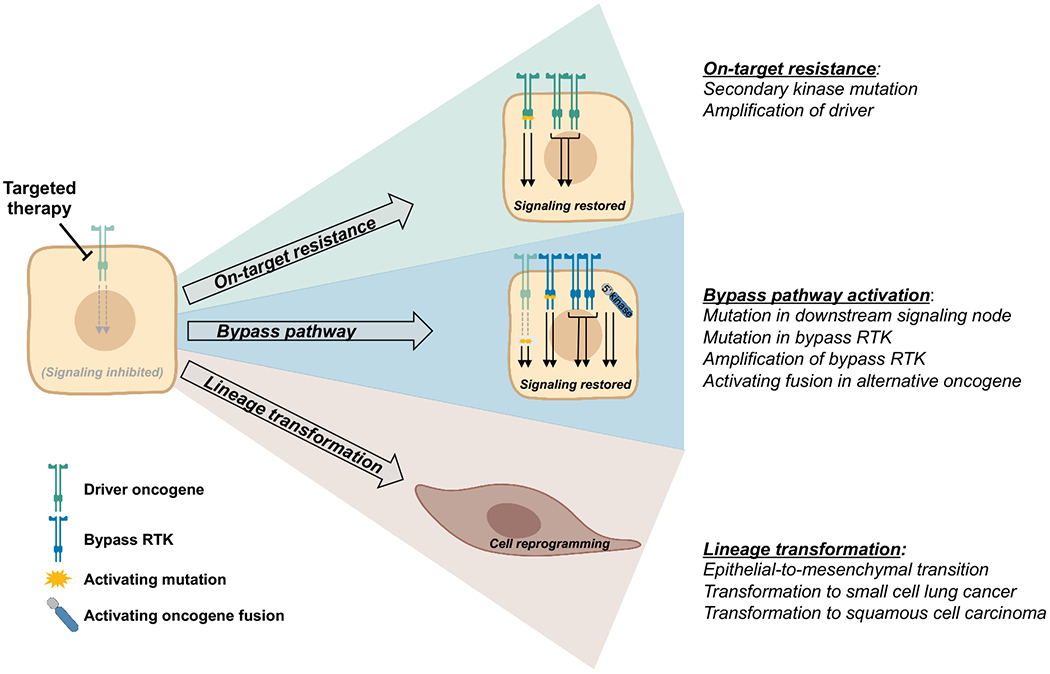
There are three major categories of tumor cell-intrinsic mechanisms of acquired resistance to targeted therapies: on-target resistance, bypass pathway activation, and lineage transformation. On-target resistance and bypass pathway activation maintain reliance on core downstream signaling pathways of the driver oncogene and develop via genetic alterations such as secondary kinase mutations and gene amplification. Lineage transformation is a form of phenotypic plasticity that involves cellular reprogramming and decreased activity of signaling pathways critical to the driver oncogene.
Secondary kinase mutations in EGFR-mutant NSCLC
It has been well established that approximately half of patients with acquired resistance to first-generation EGFR TKIs will develop a secondary mutation in exon 20 of the EGFR kinase domain, leading to a substitution of methionine for threonine at position 790 (T790M) (Kobayashi et al. 2005; Pao et al. 2005). T790M develops in cis with the initial EGFR activating mutation and confers erlotinib and gefitinib resistance primarily by inducing greater ATP affinity of the receptor (Yun et al. 2008). The prevalence of T790M in erlotinib- and geftinib-resistant tumors prompted efforts to develop more selective, irreversible EGFR TKIs with increased affinity for the T790M mutant receptor. Afatinib, dacomitinib and osimertinib are second- and third-generation EGFR TKIs that all act by binding covalently to the cysteine 797 residue in the EGFR kinase domain (Cross et al. 2014; Jänne et al. 2015; Li et al. 2008; Engelman, Zejnullahu, Gale, et al. 2007). Of these three irreversible inhibitors, osimertinib is the only one that was developed as a mutant-selective, wild-type sparing EGFR inhibitor and is capable of inhibiting the growth of T790M-positive tumors in patients. It is now approved for use in both the first-line setting for treatment-naive stage IV EGFR-mutant NSCLC as well as in the setting of T790M-mediated acquired resistance to prior EGFR TKI therapy.
However, despite their significantly more potent inhibition of EGFR signaling, on-target resistance to third-generation EGFR TKIs has also begun to emerge in both the first-line setting (typically in the absence of T790M) and in the second-line setting (in T790M-positive disease). The most well-established EGFR-dependent mechanism of resistance to osimertinib is a C797S substitution mutation in the TKI binding residue, which interferes with covalent bond formation between TKI and EGFR kinase domain. This was first described as a mechanism of resistance in the context of second-line osimertinib treatment for T790M-positive disease (Yu et al. 2015; Thress et al. 2015). It most often occurs as a tertiary EGFR mutation (both T790M and C797S in cis with initial EGFR driver mutation) and thus confers resistance to both first-generation EGFR TKIs and irreversible second- and third-generation EGFR TKIs (Table 1). Case reports have shown that C797S can also rarely emerge in trans to T790M, in which case tumors remain sensitive to the combination of first- and third-generation EGFR TKIs (Z. Wang et al. 2017). In the second-line setting, acquired resistance due to C797S is seen in approximately 10-20% percent of patients (Ramalingam, Yang, et al. 2018; Piotrowska et al. 2018; Papadimitrakopoulou, et al. 2018).
Table 1.
Secondary EGFR kinase mutations acquired in the setting of resistance to third-generation EGFR TKI osimertinib
| Acquired EGFR kinase mutation | Sensitivity to 1st/2nd-gen EGFR TKIs | Sensitivity to 3rd-gen EGFR TKIs | |
|---|---|---|---|
| First-line osimertinib | L718Q | Resistant | Resistant |
| G719X | Sensitive | Resistant | |
| L792X | Sensitive | Resistant | |
| C797S | Sensitive | Resistant | |
| Second-line osimertinib | T790M + L718Q | Resistant | Resistant |
| T790M + G724S | Resistant | Resistant | |
| M766Q | Resistant | Resistant | |
| T790M + L792H | Resistant | Resistant | |
| T790M + G796S | Resistant | Resistant | |
| T790M + C797S (in cis) | Resistant | Resistant | |
| T790M + C797S (in trans) | Sensitive* | Sensitive** |
In combination with a 3rd-generation inhibitor.
In combination with a 1st-generation inhibitor.
Similarly C797S has been shown to emerge in the absence of T790M, in approximately 7% of patients with acquired resistance to osimertinib in the first-line setting (Ramalingam, Cheng, et al. 2018). Although the numbers of patients analyzed are small so far, emergence of C797S appears to be slightly less frequent in the first-line setting than the second-line setting. Preclinical data suggest that these tumor cells should remain sensitive to first-generation EGFR TKIs despite resistance to osimertinib (Thress et al. 2015; Niederst, Hu, et al. 2015). Therefore, patients who are treated in the first-line setting with osimertinib and develop resistance via C797S mutation may benefit from subsequent therapy with first-generation EGFR TKIs following osimertinib resistance. The efficacy of adding first-generation EGFR TKIs at the time of C797S-mediated osimertinib resistance in the first-line setting, as well as up-front combinations of first- and third-generation EGFR TKIs, is currently being tested prospectively in clinical trials.
Other rarer on-target EGFR mutations have been identified that mediate osimertinib resistance and restore EGFR-mediated oncogenic signaling by impairing drug binding, including EGFR L792H, G796R, L718Q, M766Q and G724S mutations (Oztan et al. 2017; Brown et al. 2019; Zhang et al. 2018; Piotrowska et al. 2018; Yang et al. 2018; Table 1). Interestingly, further preclinical investigation has revealed that in some cases, the degree of osimertinib resistance conferred by these compound mutations depends upon the primary EGFR driver mutation present in the cell. For example, Yang and colleagues demonstrate in a Ba/F3 model that, L718Q and L792H mutations confer resistance to osimertinib when present in cis with L858R (L718Q more robust than L792H), but not with Ex19del (Yang et al. 2018). Similarly, Brown and colleagues show that EGFR G724S mutations confer resistance to osimertinib when present in cis with Ex19del driver mutations, but not with L858R driver mutations (Brown et al. 2019). Collectively, these data demonstrate that the combination of mutations, not merely the most recently acquired resistance mutation, may need to be considered when making decisions about potential subsequent targeted therapy options.
Secondary kinase mutations in NSCLC harboring ALK rearrangements
Similar patterns of on-target resistance are seen in ALK-positive NSCLC in the face of first- and second-generation ALK TKIs. Approximately one third of ALK-positive tumors treated with the first-generation ALK TKI crizotinib develop some form of on-target genomic alteration, including one of several identified ALK kinase domain mutations (Katayama et al. 2012). Interestingly, despite similar emerging patterns with the development of secondary kinase mutations as a form of on-target resistance in ALK- and EGFR-driven tumors, ALK-positive tumors tend to develop a broader spectrum of secondary kinase mutations (Gainor et al. 2016; Lin, Riely, and Shaw 2017). Mutations frequently differ according to the specific inhibitor used, as opposed to the common T790M ‘gatekeeper’ mutation or C797S covalent site mutation in EGFR described above. The most common ALK kinase domain mutation acquired in the setting of second-generation ALK TKI treatment (now preferred first-line therapy) is the G1202R solvent front mutation, but there are over ten different acquired secondary ALK mutations that have been identified as mechanisms of resistance to first- and second-generation ALK TKIs collectively (Gainor et al. 2016; Lin, Riely, and Shaw 2017). Finally, with the introduction into the clinic of highly potent third-generation ALK TKI lorlatinib, we have seen more frequent emergence of compound ALK kinase domain mutations. In other words, a significant proportion of tumors progressing on lorlatinib preserve their secondary ALK kinase domain mutations, with approximately half acquiring at least one additional ALK mutation at the time of acquired resistance (Dagogo-Jack et al. 2019; Yoda et al. 2018).
Secondary kinase mutations in NSCLC harboring ROS1 and MET rearrangements
Secondary kinase domain mutations in the oncogenic target have also been identified as mechanisms of on-target resistance to therapy in the smaller molecular subsets of NSCLC driven by ROS1 and MET exon 14 rearrangements. In the largest case series yet reported looking at TKI resistance (crizotinib) in ROS1-driven NSCLC, over half of the 16 tumors studied were found to have ROS1 mutations at the time of acquired resistance (Gainor et al. 2017). The most frequently observed ROS1 mutation conferring crizotinib resistance is G2032R, which is structurally analogous to the ALK G1202R mutation, causing steric hindrance that interferes with crizotinib binding. Other ROS1 mutations identified at lower frequencies were D2033N (analogous to ALK D1203N) and S1986F (Gainor et al. 2017). Finally, secondary MET mutations have been identified in preclinical studies and case reports of acquired resistance of tumors driven by MET exon 14 skipping to MET inhibitors crizotinib and capmatinib (D1228 and Y1230 residues) (Engstrom et al. 2017; Heist et al. 2016; Schrock et al. 2017).
Amplification of target oncogene in EGFR-mutant and ALK-rearranged NSCLC
Another common, and often co-existing, mechanism of on-target acquired TKI resistance in oncogene-driven NSCLC is amplification of the driver oncogene. This pattern is seen commonly in EGFR-mutant tumors in response to first-, second- and third-generation EGFR TKIs and most often manifests as amplification of the mutant allele already containing both the initial sensitizing mutation as well as acquired T790M and/or alternative EGFR kinase domain mutation (Ercan et al. 2010; Yang et al. 2018; Piotrowska et al. 2018). This serves to further potentiate the EGFR-dependent, or on-target, resistance afforded to the tumor cell with acquired kinase domain mutations serving to protect against the inhibition of TKIs. Similarly in ALK-positive tumors, amplification of the ALK fusion oncogene has been identified as a mechanism of resistance to ALK TKIs (Gainor et al. 2016; Katayama et al. 2012; Lin, Riely, and Shaw 2017).
Bypass pathway activation
The initial effectiveness of targeted therapies in EGFR-mutant, ALK-positive, and other molecular subsets of NSCLC is due in large part to the degree of oncogene dependence of these tumor cells. This oncogene dependence often persists at the time of acquired resistance to targeted therapy, in the form of secondary kinase mutations and amplification of driver oncogene as outlined above. However, despite the prevalence of on-target resistance in driver-positive NSCLCs, there is a subset of tumors that divert away from dependence on driver oncogenic signaling at the time of acquired resistance and instead develop activation of bypass signaling pathways. Activation of bypass signaling, either in the form of alternative receptor tyrosine kinase (RTK) activation with common downstream signaling pathways, or direct activation and sustained signaling of downstream signaling pathways themselves, is commonly seen in tumors with acquired resistance to targeted therapy (Figure 1). The biological predisposition to activation of bypass pathways is not yet well understood and likely has to do with the genomic makeup of the tumor as well as the potency of TKI-mediated inhibition of driver oncogene.
Upregulation of bypass receptor tyrosine kinase (RTK) signaling
The most common of such bypass signaling pathways activated in EGFR-mutant NSCLC is the MET receptor tyrosine kinase (also known as hepatocyte growth factor receptor; encoded by the gene MET). MET, similar to EGFR, is expressed throughout epithelial tissues and promotes signaling important for cell survival and proliferation, among other functions. The MET gene has been shown to be amplified and mutated in the setting of acquired resistance to both early and later-generation EGFR TKIs, and early phase clinical trials have demonstrated benefit of adding the MET inhibitor capmatinib to the EGFR TKI gefitinib for overcoming MET-mediated acquired resistance (Piotrowska et al. 2018; Ohashi et al. 2013; Engelman, Zejnullahu, Mitsudomi, et al. 2007; Yang et al. 2018; Wu et al. 2018). Mechanistic preclinical studies in the setting of EGFR TKI resistance suggested that MET amplification drives HER3/ERBB3-dependent downstream activation of PI3-kinase signaling (Bean et al. 2007; Engelman, Zejnullahu, Mitsudomi, et al. 2007). MET exon 14 skipping has been identified in one case report of acquired resistance to osimertinib, and rare point mutations in MET have also been identified in clinical specimens with acquired resistance to osimertinib; however, the significance of these mutations in conferring sensitivity to MET inhibition in the setting of osimertinib resistance has not been established (Yang et al. 2018; Piotrowska et al. 2018; Leonetti et al. 2019)
Interestingly, overexpression of MET as well as its ligand, hepatocyte growth factor (HGF), is also seen in EGFR-mutant NSCLC and in some studies has been shown to correlate with EGFR TKI resistance. However, inhibition of MET receptor signaling in the absence of genetic alterations in MET, such as amplification or exon 14 deletion, has not demonstrated impressive clinical benefit in NSCLC (reviewed in(Miranda, Farooqui, and Siegfried 2018)). In addition to MET amplification, activation of bypass signaling pathways by upregulation of other RTKs such as ERBB2 (HER2) and IGF1R is also seen frequently in EGFR-mutant NSCLC in the setting of resistance to early and later-generation EGFR TKIs (Takezawa et al. 2012; Guix et al. 2008; Leonetti et al. 2019). Most recently, a patient with osimertinib resistance in the second-line setting (EGFR L858R + T790M) was found to have a HER2 exon 16 skipping mutation (HER2D16, previously reported in breast cancer), resulting in HER2 splice variant HER2D16 (Hsu et al. 2020). This HER2 variant was originally characterized in breast cancer but has not been previously reported in NSCLC. The authors of this study demonstrate preclinical evidence supporting a unique functional role of the HER2D16 splice variant in mediating osimertinib resistance in NSCLC (Hsu et al. 2020).
Amplification or upregulation of alternative RTKs has also been seen in ALK-positive tumors as a mechanism of resistance to second-generation ALK TKIs. MET amplification has been seen as a mechanism of resistance to later generation ALK inhibitors such as alectinib (Gouji et al. 2014). Notably, MET amplification is not seen in the setting of resistance to first-generation ALK inhibitor crizotinib, as crizotinib also has activity against MET. Preclinical data also suggest that upregulation of IGF-1R, FGFR1, EGFR expression and increased ligand-mediated EGFR and HER2 signaling are potential mediators of resistance to ALK inhibition (Lovly et al. 2014; Miyawaki et al. 2017; Lin, Riely, and Shaw 2017; Katayama et al. 2012; Dardaei et al. 2018). Notably, this upregulation of alternative RTK activity has been seen less commonly in ALK-positive clinical specimens as compared to what has been shown in EGFR-mutant tumors.
One of the challenges in characterizing the full range of RTK-mediated bypass signaling mechanisms of resistance to TKIs is our ability to routinely detect changes in protein expression or signaling activity in clinical samples. While gene amplification can be detected in an unbiased fashion by clinically available next-generation sequencing (NGS) or fluorescence in situ hybridization (FISH) assays, we do not have equivalent assays currently in clinical use to measure levels of mRNA or protein expression of RTKs and their designated ligands. Interestingly, preclinical work in recent years has shown that the SHP2 phosphatase may be a common downstream signaling node responsible for the activation of RAS/ERK signaling downstream by multiple different bypass RTK pathways (Dardaei et al. 2018). In this model, the combination of SHP2 and ALK inhibition in the setting of ALK TKI resistance without a clear genomic mechanism of resistance may prove to be a rational treatment strategy.
Acquired activating fusions in oncogenic signaling pathways
Another mechanism of bypass pathway activation commonly seen in NSCLC is genomic rearrangement resulting in constitutively active fusion proteins. These activating fusions have been seen with highest frequency in alternative RTKs or in BRAF, all of which are known to be important oncogenic drivers in NSCLC. CCDC6-RET, NCOA4-RET, TPM3-NTRK1, FGFR3-TACC3, and multiple unique BRAF fusions have been identified following acquired resistance to first- and second-generation EGFR TKIs (Klempner et al. 2015; Schrock et al. 2018). Acquired fusions are seen even more commonly in the setting of osimertinib resistance; again identified are fusions involving RET, BRAF, NTRK1, and FGFR3, as well as novel GOPC-ROS1 and EML4-ALK fusions (Oxnard et al. 2018; Le et al. 2018; Piotrowska et al. 2018; Zeng, Yang, and Zhang 2018).
Preclinical studies have demonstrated the potential efficacy of combined EGFR/RET inhibition and EGFR/MEK inhibition in the setting of acquired RET and BRAF fusions, respectively (Piotrowska et al. 2018; Vojnic et al. 2018). There are also early clinical data in the form of case reports in which patients with acquired resistance to osimertinib harboring AGK-BRAF fusions demonstrated response to osimertinib plus trametinib, and those harboring CCDC6-RET fusions achieved stable disease in response to combined osimertinib plus cabozantinib (a multi-kinase inhibitor with RET activity) as well as more impressive partial responses to combined osimertinib and BLU-667 (a selective RET inhibitor; Dagogo-Jack et al. 2019; Piotrowska et al. 2018). Similarly, there is one published case report of a patient with acquired GOPC-ROS1 fusion demonstrating a response to osimertinib plus crizotinib at the time of acquired resistance to osimertinib (Zeng et al. 2018). Finally, there are now multiple case reports describing dual EGFR/ALK inhibition in the setting of resistance to EGFR TKIs characterized by an acquired EML4-ALK fusion (Chen et al. 2013; Offin et al. 2018; Tiseo et al. 2011). These data suggest that, similar to the bypass pathway activation achieved by amplification or upregulation of alternative RTKs, acquired fusions are a biologically meaningful mechanism of target-independent resistance to targeted therapy in NSCLC, particularly in EGFR-mutant disease.
Acquired activating mutations in intracellular oncogenic kinases
Bypass signaling, or target-independent resistance, can also be achieved by downstream activation of intracellular pro-proliferative and anti-apoptotic signaling kinases. Although we often differentiate our terminology to describe ‘bypass’ signaling pathways as activation of alternative RTKs -- versus ‘downstream’ activation of intracellular pathways distal to receptor-initiated signaling -- these two mechanisms achieve the same biological endpoint. The clinical relevance of these downstream mutations, as with bypass RTK signaling, lies in the degree to which the tumor cell is dependent upon the particular signaling pathways that the acquired mutations sustain and activate.
Two major downstream pathways of driver RTK signaling in NSCLC are the phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) signaling pathways. Mutations and/or amplifications in each of these pathways are seen commonly in EGFR-mutant and ALK-positive lung cancers with acquired resistance to TKIs. In ALK-positive NSCLC, acquired activating mutations in PIK3CA and MAP2K1 have been identified in the setting of resistance to second-generation ALK TKIs. These mutations have also been shown to confer sensitivity to dual ALK and PI3K/MAPK inhibition, respectively, in preclinical models (Gainor et al. 2016; Lin, Riely, and Shaw 2017; Crystal et al. 2014). Studies have identified acquired novel BRAF G15V and NRAS A155T mutations at the time of resistance to second-generation ALK TKIs, but the functional significance of these mutations, given that they have not been seen in other NSCLC settings and do not occur in ‘hotspot’ regions important for signaling in these proteins, is not well understood.
In primary EGFR-mutant tumors, mutations in BRAF and PIK3CA were identified in early studies of acquired resistance to first- and second-generation EGFR TKIs (Ohashi et al. 2013; Sequist et al. 2011; Ohashi et al. 2012). As described in more detail below, recent studies have revealed an even higher frequency of PI3K and MAPK pathway mutations in the context of acquired resistance to third-generation EGFR TKIs. Importantly, the process of discerning which acquired genomic alterations in downstream signaling pathways are functionally contributing to acquired resistance is complex, as such mutations (commonly PIK3CA mutations) can frequently co-occur with driver mutations, and their impact on response to therapy is not entirely clear (Eng et al. 2015). On the other hand, sequencing of cell-free DNA (cfDNA) and whole-exome sequencing (WES) of longitudinally collected tumor tissue biopsies in a cohort of EGFR-mutant NSCLCs revealed that the frequency of PIK3CA mutations (among others) increases with each line of systemic therapy, supporting their role in acquired resistance (Blakely et al. 2017). Collectively, these data further highlight the need for rigorous preclinical studies proving the functional relevance of these mutations in drug resistance.
Specifically regarding resistance to third-generation EGFR TKIs, preclinical studies identified BRAF G469A and NRAS E63K as novel acquired mutations that can confer resistance to osimertinib but maintain sensitivity to MEK inhibitors selumetinib and trametinib in vitro and in genetically-engineered mouse models (La Monica et al. 2019; Eberlein et al. 2015). Next-generation sequencing analysis of clinical samples with osimertinib resistance across multiple studies has also revealed acquired mutations in MAP3K1, KRAS, NF1, PIK3CA, PIK3R1, and PTEN (Piotrowska et al. 2018; Ortiz-Cuaran et al. 2016; Oxnard et al. 2018; Le et al. 2018; Papadimitrakopoulou et al. 2018; Shi et al. 2018; Yang et al. 2018). While some of these have been validated in preclinical mechanistic studies, the degree to which the full spectrum of these mutations (as well as other downstream point mutations identified in these and other studies), play a role in mediating osimertinib resistance has yet to be fully elucidated in mechanistic studies. Of note, increased RAS/MAPK signaling is also seen as a mechanism of resistance to targeted therapy in primary MET-exon 14 skipping as well as BRAF V600E-driven NSCLC (Rotow et al. 2019; Abravanel et al. 2018).
Lineage transformation
Thus far, we have discussed how oncogene dependence confers exquisite sensitivity to targeted therapies in many molecular subtypes of NSCLC; however, because TKIs cannot eliminate every tumor cell in a heterogeneous mass, resistance occurs. As discussed in previous sections, the mechanism of resistance can involve sustained activation of either the primary oncogene itself or bypass signaling pathways that essentially mimic the original oncogenic driver. A third, biologically distinct category of resistance is that of lineage transformation (Figure 1). This lineage transformation is characterized by a loss of dependence on the original oncogenic signaling pathways and a shift to alternative cellular phenotypes, which exist on a spectrum from subtle transient phenotypic changes to complete, stable transition to new histologic subtypes. The most conceptually well-defined, though mechanistically poorly understood, example of lineage transformation is the histologic transformation from NSCLC to small cell lung cancer (SCLC).
Small-cell lung cancer (SCLC) transformation
SCLC is a less common histologic subtype of lung cancer, characterized by a more aggressive phenotype and fewer targetable oncogenes than NSCLC. Clinically, the majority of SCLC tumors are highly responsive to first-line cytotoxic chemotherapy, though resistance virtually always develops in extensive stage disease (Farago and Keane 2018). Histologically, SCLC is high-grade, with Ki67 staining typically >50%, and appears under the microscope as dense sheets of small cells with scant cytoplasm and finely granular chromatin. It is a tumor of neuroendocrine origin, with the majority of SCLCs expressing neuroendocrine markers such as synaptophysin, chromogranin A, and NCAM1. Genomically, SCLC is characterized by virtually universal loss of function of tumor suppressors p53 and RB (George et al. 2015; Rudin et al. 2012; Peifer et al. 2012). Recent preclinical and translational work has demonstrated a growing appreciation of further molecular subtyping of de novo SCLC, the details of which have been thoroughly reviewed elsewhere (Rudin et al. 2019).
In addition to the proportion of newly diagnosed lung cancers that demonstrate SCLC histology at the time of diagnosis, the phenomenon of histologic transformation from lung adenocarcinoma to SCLC has also been identified as a mechanism of resistance seen in 5-10% of EGFR-mutant NSCLC (Piotrowska et al. 2018; Ohashi et al. 2013; Sequist et al. 2011). The first case of SCLC-associated acquired resistance to EGFR TKI was described in 2006 in a patient who developed acquired resistance to erlotinib (Zakowski et al. 2006). Since this initial case report, multiple case series have shown this as a recurrent mechanism of resistance to both first- and third-generation EGFR TKIs (Piotrowska et al. 2018; Ohashi et al. 2013; Sequist et al. 2011). While this phenomenon appears to be most common in the setting of EGFR-mutant NSCLC, it has also been reported in a series of cases of ALK-positive NSCLC with acquired resistance to TKI (Lin, Riely, and Shaw 2017). The similarities and differences in biology of de novo SCLC versus SCLC histology that evolve as a mechanism of resistance to targeted therapy in NSCLC are an area of active study. The work that has been done to date demonstrates that SCLC tumors emerging in the context of EGFR TKI treatment do appear to universally share classic SCLC characteristics including RB loss, decreased EGFR expression (despite retained EGFR driver mutation), and increased expression of neuroendocrine markers (Niederst, Sequist, et al. 2015).
The molecular predispositions of EGFR-mutant NSCLCs that develop SCLC histologic transformation as a mechanism of resistance to EGFR TKIs are not yet well understood. While theoretically possible, the proposition that tumors acquiring SCLC-mediated resistance to EGFR TKIs possessed a bona fide SCLC subclone at the time of treatment initiation -- one that then grew out under selective inhibition of the EGFR-mutant NSCLC subclone -- does not fit with the natural history of these cases. While we know that SCLC is an aggressive disease capable of high volume growth and metastasis in a matter of weeks to months, many EGFR-mutant NSCLC cases that later evolve to SCLC histology have several years of clinical response to EGFR TKI prior to progression (Oser et al. 2015). If the SCLC clone were present at the time of diagnosis, in the presence of only EGFR TKI, we might expect the outgrowth of that SCLC clone and clinical acquired resistance to occur much earlier in the disease course. Compelling studies employing next-generation sequencing (NGS) analysis to evaluate clonal evolution of NSCLCs that transform into SCLCs with selective pressure of EGFR TKI have shown that the presence of p53 and RB1 loss at the time of NSCLC diagnosis portends a 43x greater likelihood of development of SCLC histology at the time of acquired resistance to TKI (Lee et al. 2017). Based on these and other preclinical data, there is currently a phase I study recruiting patients with EGFR-mutant NSCLC who are found to have p53 and RB1 loss at the time of diagnosis, combining EGFR TKI with SCLC-targeted chemotherapy cisplatin plus etoposide in the first-line setting.
One of the questions raised by the observed prevalence of NSCLC to SCLC histologic transformation is that of tumor cell of origin. Preclinical studies creating genetically engineered mouse models (GEMMs) with cell-type specific promoters have shown that SCLC tumors typically develop from neuroendocrine precursor cells, and adenocarcinoma tumors typically develop from alveolar type II cells. However, surprisingly, SCLC can also develop from alveolar type II cells under the right circumstances with appropriate timing of p53 and RB1 loss (Meuwissen et al. 2003; Sutherland et al. 2011). This suggests the possibility that SCLC and NSCLC share a potential cell of origin, and there may exist plasticity between the two histologic subtypes depending on the genomic and transcriptomic characteristics of the tumor as it develops and evolves. Similar work has been done in prostate cancer, showing that differentially manipulating transcriptional programs governed by p53, RB, AKT, MYC, and BCL2 can drive development of either small cell neuroendocrine versus adenocarcinoma from a common prostate epithelial progenitor (Park et al. 2018).
The degree to which this plasticity depends on the presence of specific types of inhibitors in specific molecular subsets of NSCLC, or the surrounding components of the TME, remains to be definitively shown, although we do rarely find de novo EGFR-mutant SCLC, which suggests lineage transformation can occur in the absence of EGFR TKI (Thai et al. 2017). Interestingly, there have also been several case reports of histologic transformation of EGFR-mutant lung adenocarcinoma to squamous cell carcinoma at the time of acquired resistance (Kuiper et al. 2015; Agita et al. 2016; Levin et al. 2015; Piotrowska et al. 2018). Very little is known about the biology of this type of histologic transformation, but the principles of phenotypic plasticity discussed above are likely to apply, at least conceptually, in these cases as well.
Epithelial-to-mesenchymal transition
Another form of lineage transformation that has been shown to mediate resistance to targeted therapies in NSCLC is epithelial-to-mesenchymal transition (EMT). EMT is a reversible state of phenotypic plasticity characterized by loss of classic features of epithelial cells such as apicobasal polarity, intercellular adhesions and specific cytoskeletal arrangements critical to epithelial cell structure (Tulchinsky et al. 2019). The EMT phenotype is, in reality, a spectrum of phenotypes, and it is the result of a cell differentiation program that is governed by a set of EMT transcription factors, including Snail, Slug, Twist, and Zeb 1 (Lambert, Pattabiraman, and Weinberg 2017; De Craene and Berx 2013). As cells become more mesenchymal in nature, they acquire the migratory and invasive capabilities needed for development of metastases and other features of malignant epithelial tumors. Phenotypic plasticity between epithelial and mesenchymal phenotypes is a critical feature of embryogenesis and organ development, and this phenomenon of EMT in NSCLC and other solid tumors is an example of tumor cell adoption and misappropriation of this central biological pathway (Tulchinsky et al. 2019).
In EGFR-mutant tumors, EMT-like phenotype is seen in significant subset of tumors at the time of acquired resistance to EGFR TKIs (Sequist et al. 2011; Uramoto et al. 2010). Preclinical studies corroborate and further investigate this mechanism, demonstrating correlation between expression of epithelial versus mesenchymal markers and relative resistance to EGFR TKIs, and invoking specific signaling pathways such as AXL, IGF-1R, and SRC/FAK signaling in EMT-characterized resistant tumors (Byers et al. 2013; Yauch et al. 2005; Thomson et al. 2005; C. Wilson et al. 2014; Zhou et al. 2015). The exact frequency of acquired EMT phenotype in the setting of EGFR TKI resistance is difficult to discern, as the definition of EMT transformation encompasses a broad spectrum of potential classifications. Typically, this transformation is defined by loss of expression of epithelial markers such as e-cadherin and gain of expression of mesenchymal markers such as vimentin (Thiery 2002). While there have been studies attempting to more precisely characterize the EMT phenotype via gene expression profiling in both EGFR-mutant and EGFR-wild type NSCLC, these types of molecular characterizations have not been validated in the setting of EGFR TKI resistance and are not routinely performed in the clinic at the time of re-biopsy for treatment decision-making (Byers et al. 2013).
While less well characterized, progression to the EMT phenotype has also been shown to be a mechanism of resistance to targeted therapies in ALK-positive NSCLC in preclinical studies. While some studies suggest that ALK-positive NSCLC inherently possesses a more mesenchymal-like phenotype, others report emergence of the EMT phenotype as a byproduct, but not driver of, ALK TKI resistance, and still others report EMT phenotypic transformation as a central mechanism of resistance to ALK TKIs such as crizotinib (Gower et al. 2016; Voena et al. 2016; Kim et al. 2013; Wei et al. 2018; Gao et al. 2016; Lin, Riely, and Shaw 2017). Interestingly, one group showed in preclinical studies that resistance to first- and second-generation ALK TKIs can in some cases be mediated by AXL-expressing drug-tolerant cells that demonstrate an EMT-like phenotype (Nakamichi et al. 2018).
As discussed above, EMT is a widely studied cellular function employed in many physiologic as well as pathologic states. However, the role of EMT in acquired resistance to targeted therapies in NSCLC cannot be simplified to static categorization (via only a handful of cellular markers) of the presence or absence of EMT phenotype on a tumor biopsy at a given moment. Rather, EMT phenotypic transformation is thought to contribute to the complexity of intra-tumoral heterogeneity and is likely to evolve within a cancer over time. Interestingly, there are some biological underpinnings that are shared between the EMT/cancer stem cell state and the SCLC transformed state. For example, cell signaling programs involved in EMT-related phenotypic plasticity, such as SOX2-induced transcription as well as Notch and Hedgehog signaling, are known to be important signaling pathways in SCLC (Vega et al. 2004; Gazdar, Bunn, and Minna 2017; Rudin et al. 2019). In addition, RB1 inactivation, which is required for development of SCLC, results in a loss of regulatory mechanisms for the G1 checkpoint whose activation is a hallmark of EMT (Tulchinsky et al. 2019). These associations suggest, but do not prove, that the phenotypic plasticity seen in development of mesenchymal-like tumor cells could exist on a spectrum with neuroendocrine SCLC histologic transformation.
Drug-tolerant persister cells
In addition to histologic changes identified on re-biopsy at the time of clinical, radiographic acquired resistance to EGFR TKIs, transient phenotypic change as a protective mechanism for cells upon TKI exposure has also been identified in correlative in vitro experiments. Specifically, a subpopulation of initially quiescent drug-tolerant persister cells (DTPs) have been shown to have intrinsic ability to survive initial TKI exposure by reversibly acquiring a stem-cell like phenotype that confers decreased sensitivity to therapy (Sharma et al. 2010). It has been hypothesized that the presence of these DTPs in a reversible phenotypic state explains the phenomenon of successful re-treatment after drug holiday, and DTPs are also suggested to be one mechanism for eventual outgrowth of macroscopic acquired resistance in the setting of persistent TKI exposure (Sharma et al. 2010).
Studies of DTPs in EGFR-mutant NSCLC have shown that these cells develop an EMT-like phenotype under selective TKI pressure, and over time can evolve and develop irreversible genetic resistance to TKIs in the form of secondary EGFR kinase mutation T790M (Hata et al. 2016). Interestingly, this particular subset of T790M+ first-generation EGFR TKI-resistant clones that have transformed via an EMT-like phenotype demonstrate less susceptibility in vitro to third-generation EGFR TKIs and express a gene profile consistent with evolution from cancer stem cell-like DTPs. While this observation has yet to be validated clinically, the presence of two populations of T790M+ resistant clones further demonstrates the dynamic, heterogeneous milieu of TKI-resistant cells present in a given macroscopic tumor and the importance of phenotypic plasticity in the evolution of that tumor. Recent work by our group and others has shown that FGFR signaling plays a role in development of the EGFR TKI-resistant EMT phenotype of DTPs, and that dual EGFR/FGFR inhibition may be a rational clinical approach to preventing or overcome EGFR TKI resistance by re-sensitizing the mesenchymal state of cells to EGFR TKI inhibition (Raoof et al. 2019).
Role of the tumor microenvironment
Up to this point, our discussion has focused primarily on tumor cell-intrinsic mechanisms of resistance to targeted therapy in NSCLC, highlighting data from experiments typically conducted in pure tumor cell populations. However, it has long since been appreciated that the extracellular matrix (ECM), surrounding vasculature, and cancer-associated fibroblasts that make up the tumor microenvironment (TME) play a critical supportive role in the growth of solid tumors (Raghu and Michael 2006; Kalluri 2003; Belli et al. 2018; Quail and Joyce 2013). With technological advancements in single-cell RNA-sequencing techniques, recent studies such as that from Lambrechts and colleagues have further characterized the profound complexity and diversity of the stromal cells in the TME (Lambrechts et al. 2018). Emerging data suggest that the development of acquired resistance to targeted therapies in NSCLC, driven in part by the cell-intrinsic mechanisms, is also inextricably linked to interactions with the surrounding TME. In other words, in addition to tumor cell-intrinsic genomic alterations and phenotypic plasticity described previous sections, the TME is likely playing a role in initiating signaling pathways that enable this transformation.
One of the most prominent elements of the TME, cancer-associated fibroblasts (CAFs) have been described as a subset of activated fibroblasts closely associated with cancer cells that act by synthesis and turnover of ECM, modulation of wound healing and the inflammatory response, and regulation of epithelial cell differentiation and homeostasis via secretion of growth factors and chemokines. In addition to resident fibroblasts that are activated and converted to CAFs, recent literature suggests that other subsets of CAFs originate as bone marrow-derived mesenchymal stem cells or even epithelial tumor cells that have undergone EMT (Davies and Albeck 2018). While a full review of their role in tumor initiation, progression, and invasion is outside the scope of this review, we highlight data supporting their specific role in TKI resistance in NSCLC.
Specifically, signaling by molecules important in the extracellular matrix (ECM) secreted by CAFs, such as collagens and their integrin receptors, has been shown to mediate resistance to EGFR TKIs in preclinical studies. Wang and colleagues recently proposed, based on in vitro experiments in EGFR-mutant TKI-sensitive NSCLC cell lines, that the presence of ECM enhances both gefitinib and osimertinib resistance in a dose-dependent and reversible manner, and this effect is mediated in part by signaling through the collagen receptor Integrin-β1 (Y. Wang et al. 2019). In this study, the authors also demonstrated that inhibition of collagen synthesis potentially has a synergistic effect on osimertinib-mediated inhibition of T790M+ NSCLC cell lines (Y. Wang et al. 2019). Previous work by another group demonstrated that CAF-mediated HGF secretion can confer resistance of EGFR-mutant NSCLC cell lines to gefinitib (W. Wang et al. 2009). In a more recent study, Yi and colleagues showed in preclinical experiments that CAF-mediated secretion of HGF and IGF-1 induced MET/IGF-1R signaling, which resulted in EMT-mediated gefitinib resistance (Yi et al. 2018). In this work, the authors identified annexin A2 (ANXA2), which they found to be expressed at higher levels in CAFs, to be a critical mediator of this process.
While we describe the studies from EGFR-mutant NSCLC here, resistance via influence of the TME on phenotypic plasticity and bypass signaling pathways is relevant across molecular subtypes of NSCLC and in solid tumor oncology more broadly. A recent compelling example of this phenomenon in head and neck cancer was a study in which investigators used single-cell RNA sequencing to identify a mesenchymal sub-population of tumor cells and localized this sub-population (via histological analysis) uniquely to the leading edge of the tumor in close proximity to CAFs, allowing for proposed paracrine signaling (Puram et al. 2017). These types of studies suggest that we still have much to learn about the role of CAFs in facilitating other previously described mechanisms of therapy resistance in solid tumors, and they highlight the blurred lines between ‘intrinsic’ versus ‘extrinsic’ tumor cell adaptation to therapy (Figure 2). Other examples of the ways in which these concepts are likely to be applicable across the spectrum of solid tumors include studies demonstrating CAF-associated EMT transformation and CAF-mediated paracrine signaling via secretion of growth factors such as HGF, EGF, and PDGF to drive therapeutic resistance across biliary tract cancers, breast cancers, melanomas, and others (Yamada et al. 2013; Yu et al. 2013; Davies and Albeck 2018; Junttila and de Sauvage 2013; Straussman et al. 2012; T. R. Wilson et al. 2012).
Figure 2.
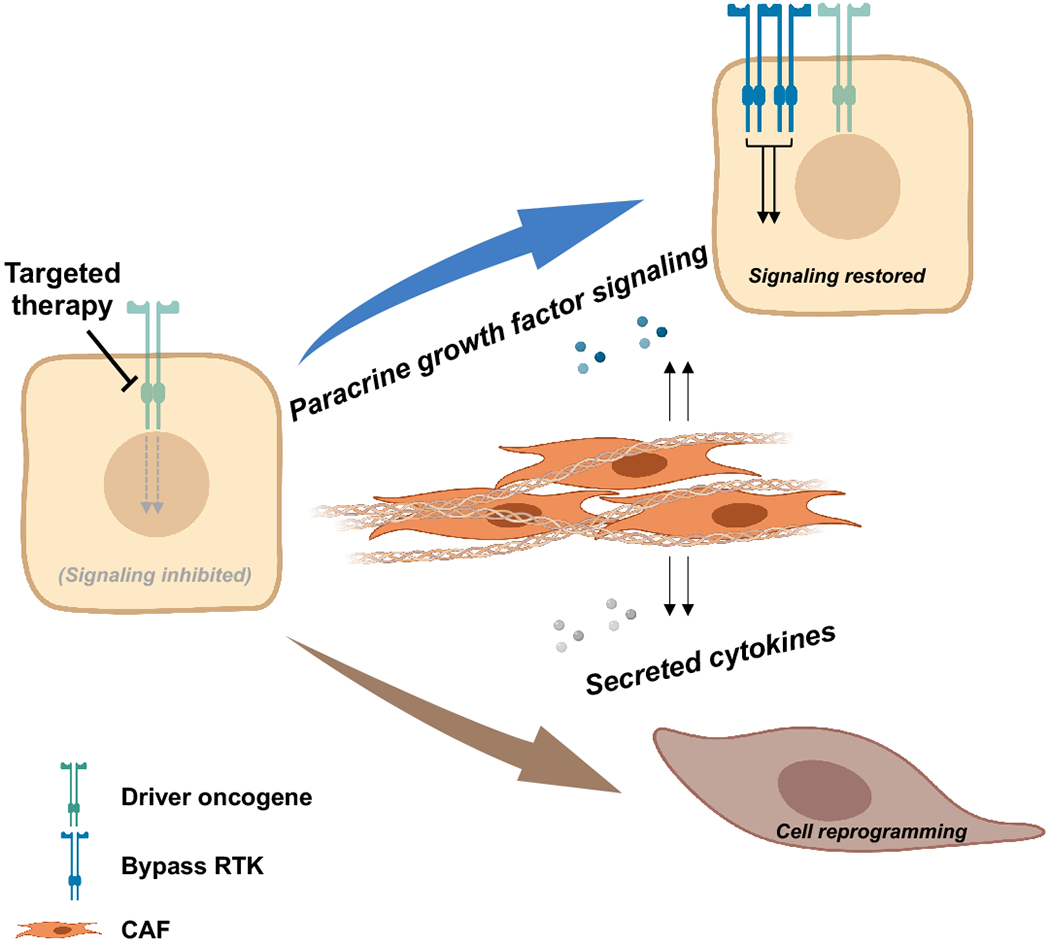
Schema outlining the role of the tumor microenvironment (TME) in mediating acquired resistance to targeted therapies. Proposed mechanisms include paracrine signaling (resulting in activation of bypass RTK signaling) and CAF-mediated collagen production, alteration of the extracellular matrix, and induction of lineage transformation.
Another important element of the TME is the immune microenvironment, which we know from the success of immune checkpoint inhibitors in some subsets of NSCLC plays a significant role in growth and survival of some tumors. Currently, our understanding of the role that the immune microenvironment plays in response to TKIs in NSCLC with known driver oncogenes is incomplete. We have seen clinically that EGFR- and ALK-mutant lung cancers are overall less responsive to PD-1/PD-L1 inhibition than other molecular subsets of NSCLC (Gainor et al. 2016). The reasons for this have yet to be fully elucidated, but current hypotheses center around lower PD-L1 expression and CD8+ TIL infiltration, as well as lower overall mutational burden (often considered a surrogate for neoantigen presentation and tumor immunogenicity). While this suggests an overall less active immune microenvironment in EGFR and ALK-mutant NSCLCs, future studies are needed to more fully explore the full role of the immune microenvironment in these tumors.
Conclusion
Targeted systemic therapies for advanced NSCLC have made impressive strides in recent years in improving response rates, progression-free and overall survival. However, surgical intervention or radiation therapy for early stage disease remain the mainstay of curative treatment in lung cancer, as we are unfortunately not yet able to successfully eradicate advanced tumors with systemic therapies. While increasingly effective therapies have been developed, we are limited in our efforts toward cure by acquired resistance to therapy. In this review, we explored the continuum of both tumor cell intrinsic as well as extrinsic (e.g. TME) mechanisms of resistance to targeted therapies.
With our increasingly sophisticated diagnostic capabilities, we will continue to refine our understanding of the biological propensity of a given cell, as defined by its underlying genomics, epigenomics, and surrounding TME, to be sensitive to, or develop resistance to a given therapy by a certain pathway or mechanism. The ultimate goal of understanding these mechanisms is to prevent the emergence of resistance. This will require a deep understanding of not only genetic events causing impaired inhibition of target oncogenic oncogenes, but also knowledge of how drug tolerant persister cells and cancer stem cell-like phenotypes evolve, and what drives transformation of tumor phenotype and surrounding microenvironment to support therapeutic resistance.
Acknowledgments
Figures created in part with BioRender.com.
Conflict of Interest Statement
ANH has received research grants/funding from Amgen, Eli Lilly, Novartis, Pfizer, Relay Therapeutics, and Roche/Genentech.
CBM has no conflicts to disclose.
Abbreviations
- ALK
Anaplastic lymphoma kinase
- AXL
AXL receptor tyrosine kinase
- BRAF
B-Raf proto-oncogene serine/threonine kinase
- EGFR
Epidermal growth factor receptor
- IGF-1
Insulin-like growth factor-1
- MAPK
Mitogen-activated protein kinase
- MET
MET proto-oncogene receptor tyrosine kinase
- NRTK
Neurotrophic tyrosine receptor kinase
- RB1
RB transcriptional corepressor 1
- ROS1
ROS proto-oncogene 1 receptor tyrosine kinase
- SCLC
Small cell lung cancer
- TKI
Tyrosine kinase inhibitor
References
- Abravanel Daniel L., Nishino Mizuki, Sholl Lynette M., Ambrogio Chiara, and Awad Mark M.. 2018. “An Acquired NRAS Q61K Mutation in BRAF V600E-Mutant Lung Adenocarcinoma Resistant to Dabrafenib Plus Trametinib.” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 13 (8): e131–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agita Jukna Cecilia, Gloria Montanari Cecilia, Maria Mengoli Cecilia, Alberto Cavazza Cecilia, Marisa Covi Cecilia, Fausto Barbieri Cecilia, Federica Bertolini Cecilia, and Giulio Rossi Cecilia. 2016. “Squamous Cell Carcinoma ‘Transformation’ Concurrent with Secondary T790M Mutation in Resistant EGFR-Mutated Adenocarcinomas.” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 11 (4): e49–51. [DOI] [PubMed] [Google Scholar]
- Bean James, Brennan Cameron, Shih Jin-Yuan, Riely Gregory, Viale Agnes, Wang Lu, Chitale Dhananjay, et al. 2007. “MET Amplification Occurs with or without T790M Mutations in EGFR Mutant Lung Tumors with Acquired Resistance to Gefitinib or Erlotinib.” Proceedings of the National Academy of Sciences of the United States of America 104 (52): 20932–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belli Carmen, Trapani Dario, Viale Giulia, D’Amico Paolo, Duso Bruno Achutti, Vigna Paolo Della, Orsi Franco, and Curigliano Giuseppe. 2018. “Targeting the Microenvironment in Solid Tumors.” Cancer Treatment Reviews 65 (April): 22–32. [DOI] [PubMed] [Google Scholar]
- Blakely Collin M., Watkins Thomas B. K., Wu Wei, Gini Beatrice, Chabon Jacob J., McCoach Caroline E., Nicholas McGranahan, et al. 2017. “Evolution and Clinical Impact of Co-Occurring Genetic Alterations in Advanced-Stage EGFR-Mutant Lung Cancers.” Nature Genetics 49 (12): 1693–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown Benjamin P., Zhang Yun-Kai, Westover David, Yan Yingjun, Qiao Huan, Huang Vincent, Du Zhenfang, et al. 2019. “On-Target Resistance to the Mutant-Selective EGFR Inhibitor Osimertinib Can Develop in an Allele-Specific Manner Dependent on the Original EGFR-Activating Mutation.” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 25 (11): 3341–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers Lauren Averett, Diao Lixia, Wang Jing, Saintigny Pierre, Girard Luc, Peyton Michael, Shen Li, et al. 2013. “An Epithelial-Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance.” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 19 (1): 279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Xiaoxia, Zhang Jie, Hu Qiong, Li Xuefei, and Zhou Caicun, 2013. “A case of lung adenocarcinoma harboring exon 19 EGFR deletion and EML4-ALK fusion gene.” Lung Cancer 81(2013): 308–310. [DOI] [PubMed] [Google Scholar]
- Cross Darren A. E., Ashton Susan E., Ghiorghiu Serban, Eberlein Cath, Nebhan Caroline A., Spitzler Paula J., Orme Jonathon P., et al. 2014. “AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer.” Cancer Discovery 4 (9): 1046–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal Adam S., Shaw Alice T., Sequist Lecia V., Friboulet Luc, Niederst Matthew J., Lockerman Elizabeth L., Frias Rosa L., et al. 2014. “Patient-Derived Models of Acquired Resistance Can Identify Effective Drug Combinations for Cancer.” Science 346 (6216): 1480–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagogo-Jack Ibiayi, Piotrowska Zofia, Cobb Rosemary, Banwait Mandeep, Lennerz Jochen, Hata Aaron, and Diggumarthy Subba et al. 2019. “Response to the combination of osimertinib and trametinib in a patient with EGFR-mutant NSCLC harboring an acquired BRAF fusion.” J Thorac Oncol 14(10): 226–228. [DOI] [PubMed] [Google Scholar]
- Dagogo-Jack Ibiayi, Rooney Marguerite M., Lin Jessica J., Nagy Rebecca J., Yeap Beow Y., Hubbeling Harper, Chin Emily, et al. 2019. “Treatment with Next-Generation ALK Inhibitors Fuels Plasma ALK Mutation Diversity.” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, July. 10.1158/1078-0432.CCR-19-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardaei Leila, Wang Hui Qin, Singh Manrose, Fordjour Paul, Shaw Katherine X., Yoda Satoshi, Kerr Grainne, et al. 2018. “SHP2 Inhibition Restores Sensitivity in ALK-Rearranged Non-Small-Cell Lung Cancer Resistant to ALK Inhibitors.” Nature Medicine 24 (4): 512–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies Alexander E., and Albeck John G.. 2018. “Microenvironmental Signals and Biochemical Information Processing: Cooperative Determinants of Intratumoral Plasticity and Heterogeneity.” Frontiers in Cell and Developmental Biology 6 (April): 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Craene Bram, and Berx Geert. 2013. “Regulatory Networks Defining EMT during Cancer Initiation and Progression.” Nature Reviews. Cancer 13 (2): 97–110. [DOI] [PubMed] [Google Scholar]
- Eberlein Catherine A., Stetson Daniel, Markovets Aleksandra A., Al-Kadhimi Katherine J., Zhongwu Lai, Fisher Paul R., Meador Catherine B., et al. 2015. “Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models.” Cancer Research 75 (12): 2489–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman Jeffrey A., Zejnullahu Kreshnik, Gale Christopher-Michael, Lifshits Eugene, Gonzales Andrea J., Shimamura Takeshi, Zhao Feng, et al. 2007. “PF00299804, an Irreversible Pan-ERBB Inhibitor, Is Effective in Lung Cancer Models with EGFR and ERBB2 Mutations That Are Resistant to Gefitinib.” Cancer Research 67 (24): 11924–32. [DOI] [PubMed] [Google Scholar]
- Engelman Jeffrey A., Zejnullahu Kreshnik, Mitsudomi Tetsuya, Song Youngchul, Hyland Courtney, Joon Oh Park Neal Lindeman, et al. 2007. “MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling.” Science 316 (5827): 1039–43. [DOI] [PubMed] [Google Scholar]
- Eng Juliana, Woo Kaitlin M., Sima Camelia S., Plodkowski Andrew, Hellmann Matthew D., Chaft Jamie E., Kris Mark G., Arcila Maria E., Ladanyi Marc, and Drilon Alexander. 2015. “Impact of Concurrent PIK3CA Mutations on Response to EGFR Tyrosine Kinase Inhibition in EGFR-Mutant Lung Cancers and on Prognosis in Oncogene-Driven Lung Adenocarcinomas.” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 10 (12): 1713–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrom Lars D., Aranda Ruth, Lee Matthew, Tovar Elizabeth A., Essenburg Curt J., Madaj Zachary, Chiang Harrah, et al. 2017. “Glesatinib Exhibits Antitumor Activity in Lung Cancer Models and Patients Harboring MET Exon 14 Mutations and Overcomes Mutation-Mediated Resistance to Type I MET Inhibitors in Nonclinical Models.” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 23 (21): 6661–72. [DOI] [PubMed] [Google Scholar]
- Ercan D, Zejnullahu K, Yonesaka K, Xiao Y, Capelletti M, Rogers A, Lifshits E, et al. 2010. “Amplification of EGFR T790M Causes Resistance to an Irreversible EGFR Inhibitor.” Oncogene 29 (16): 2346–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farago Anna F., and Keane Florence K.. 2018. “Current Standards for Clinical Management of Small Cell Lung Cancer.” Translational Lung Cancer Research 7 (1): 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainor JF, Shaw AT, Sequist LV, Fu X, and Azzoli CG. 2016. “EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non–small Cell Lung Cancer: A Retrospective Analysis.” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. http://clincancerres.aacrjournals.org/content/22/18/4585.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainor JF, Tseng D, Yoda S, Dagogo-Jack I, Friboulet L, Lin JJ, Hubbeling HG, et al. 2017. “Patterns of Metastatic Spread and Mechanisms of Resistance to Crizotinib in ROS1-Positive Non-Small-Cell Lung Cancer. JCO Precis.” Oncologia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainor Justin F., Dardaei Leila, Yoda Satoshi, Friboulet Luc, Leshchiner Ignaty, Katayama Ryohei, Dagogo-Jack Ibiayi, et al. 2016. “Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer.” Cancer Discovery 6 (10): 1118–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi Leena, Rodríguez-Abreu Delvys, Gadgeel Shirish, Esteban Emilio, Felip Enriqueta, Angelis Flávia De, Domine Manuel, et al. 2018. “Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer.” The New England Journal of Medicine 378 (22): 2078–92. [DOI] [PubMed] [Google Scholar]
- Gao Hai-Xiang, Yan Li, Li Chunzhi, Zhao Lian-Mei, and Liu Wei. 2016. “miR-200c Regulates Crizotinib-Resistant ALK-Positive Lung Cancer Cells by Reversing Epithelial-Mesenchymal Transition via Targeting ZEB1.” Molecular Medicine Reports 14 (5): 4135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar Adi F., Bunn Paul A., and Minna John D.. 2017. “Small-Cell Lung Cancer: What We Know, What We Need to Know and the Path Forward.” Nature Reviews. Cancer 17 (12): 765. [DOI] [PubMed] [Google Scholar]
- George Julie, Jing Shan Lim Se Jin Jang, Cun Yupeng, Luka Ozretić Gu Kong, Leenders Frauke, et al. 2015. “Comprehensive Genomic Profiles of Small Cell Lung Cancer.” Nature 524 (7563): 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouji Toyokawa, Takashi Seto, Mitsuhiro Takenoyama, and Yukito Ichinose. 2014. “Crizotinib Can Overcome Acquired Resistance to CH5424802: Is Amplification of the MET Gene a Key Factor?” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 9 (3): e27–28. [DOI] [PubMed] [Google Scholar]
- Gower Arjan, Hsu Wei-Hsun, Hsu Shuo-Tse, Wang Yisong, and Giaccone Giuseppe. 2016. “EMT Is Associated With, but Does Not Drive Resistance to ALK Inhibitors among EML4-ALK Non-Small Cell Lung Cancer.” Molecular Oncology 10 (4): 601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guix Marta, Faber Anthony C., Wang Shizhen Emily, Olivares Maria Graciela, Song Youngchul, Qu Sherman, Rinehart Cammie, et al. 2008. “Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in Cancer Cells Is Mediated by Loss of IGF-Binding Proteins.” The Journal of Clinical Investigation 118 (7): 2609–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata Aaron N., Niederst Matthew J., Archibald Hannah L., Gomez-Caraballo Maria, Siddiqui Faria M., Mulvey Hillary E., Maruvka Yosef E., et al. 2016. “Tumor Cells Can Follow Distinct Evolutionary Paths to Become Resistant to Epidermal Growth Factor Receptor Inhibition.” Nature Medicine 22 (3): 262–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heist Rebecca S., Sequist Lecia V., Borger Darrell, Gainor Justin F., Arellano Ronald S., Le Long P., Dias-Santagata Dora, et al. 2016. “Acquired Resistance to Crizotinib in NSCLC with MET Exon 14 Skipping.” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 11 (8): 1242–45. [DOI] [PubMed] [Google Scholar]
- Hsu Chia-Chi, Liao Bin-Chi, Liao Wei-Yu, Markovets Aleksandra, Stetson Daniel, Thress Kenneth, and Chih-Hsin Yang James. 2020. “Exon 16–Skipping HER2 as a Novel Mechanism of Osimertinib Resistance in EGFR L858R/T790M–Positive Non–Small Cell Lung Cancer.” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 15 (1): 50–61. [DOI] [PubMed] [Google Scholar]
- Jänne Pasi A., Wang Xiaofei, Socinski Mark A., Crawford Jeffrey, Stinchcombe Thomas E., Gu Lin, Capelletti Marzia, et al. 2012. “Randomized Phase II Trial of Erlotinib Alone or With Carboplatin and Paclitaxel in Patients Who Were Never or Light Former Smokers With Advanced Lung Adenocarcinoma: CALGB 30406 Trial.” Journal of Clinical Oncology. 10.1200/jco.2011.40.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jänne Pasi A., Chih-Hsin Yang James, Kim Dong-Wan, Planchard David, Ohe Yuichiro, Ramalingam Suresh S., Ahn Myung-Ju, et al. 2015. “AZD9291 in EGFR Inhibitor-Resistant Non-Small-Cell Lung Cancer.” The New England Journal of Medicine 372 (18): 1689–99. [DOI] [PubMed] [Google Scholar]
- Junttila Melissa R., and de Sauvage Frederic J.. 2013. “Influence of Tumour Micro-Environment Heterogeneity on Therapeutic Response.” Nature 501 (7467): 346–54. [DOI] [PubMed] [Google Scholar]
- Kalluri Raghu. 2003. “Basement Membranes: Structure, Assembly and Role in Tumour Angiogenesis.” Nature Reviews. Cancer 3 (6): 422–33. [DOI] [PubMed] [Google Scholar]
- Katayama Ryohei, Shaw Alice T., Khan Tahsin M., Mino-Kenudson Mari, Solomon Benjamin J., Halmos Balazs, Jessop Nicholas A., et al. 2012. “Mechanisms of Acquired Crizotinib Resistance in ALK-Rearranged Lung Cancers.” Science Translational Medicine 4 (120): 120ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Hyeong Ryul, Kim Woo Sung, Choi Yun Jung, Choi Chang Min, Rho Jin Kyung, and Lee Jae Cheol. 2013. “Epithelial-Mesenchymal Transition Leads to Crizotinib Resistance in H2228 Lung Cancer Cells with EML4-ALK Translocation.” Molecular Oncology. 10.1016/j.molonc.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klempner Samuel J., Bazhenova Lyudmila A., Braiteh Fadi S., Nikolinakos Petros G., Gowen Kyle, Cervantes Claudia M., Chmielecki Juliann, et al. 2015. “Emergence of RET Rearrangement Co-Existing with Activated EGFR Mutation in EGFR-Mutated NSCLC Patients Who Had Progressed on First- or Second-Generation EGFR TKI.” Lung Cancer 89 (3): 357–59. [DOI] [PubMed] [Google Scholar]
- Kobayashi Susumu, Boggon Titus J., Dayaram Tajhal, Jänne Pasi A., Kocher Olivier, Meyerson Matthew, Johnson Bruce E., Eck Michael J., Tenen Daniel G., and Halmos Balázs. 2005. “EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib.” The New England Journal of Medicine 352 (8): 786–92. [DOI] [PubMed] [Google Scholar]
- Kuiper Justine L., Ronden Merle I., Becker Annemarie, Heideman Daniëlle A. M., van Hengel Peter, Ylstra Bauke, Thunnissen Erik, and Smit Egbert F.. 2015. “Transformation to a Squamous Cell Carcinoma Phenotype of an EGFR-Mutated NSCLC Patient after Treatment with an EGFR-Tyrosine Kinase Inhibitor.” Journal of Clinical Pathology 68 (4): 320–21. [DOI] [PubMed] [Google Scholar]
- Lambert Arthur W., Pattabiraman Diwakar R., and Weinberg Robert A.. 2017. “Emerging Biological Principles of Metastasis.” Cell 168 (4): 670–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts Diether, Wauters Els, Boeckx Bram, Aibar Sara, Nittner David, Burton Oliver, and Bassez Ayse. 2018. “Phenotype Molding of Stromal Cells in the Lung Tumor Microenvironment.” Nature Medicine 24 (8): 1277–89. [DOI] [PubMed] [Google Scholar]
- Monica La, Silvia Roberta Minari, Cretella Daniele, Bonelli Mara, Fumarola Claudia, Cavazzoni Andrea, Galetti Maricla, et al. 2019. “Acquired BRAF G469A Mutation as a Resistance Mechanism to First-Line Osimertinib Treatment in NSCLC Cell Lines Harboring an EGFR Exon 19 Deletion.” Targeted Oncology 14 (5): 619–26. [DOI] [PubMed] [Google Scholar]
- Lee June-Koo, Lee Junehawk, Kim Sehui, Kim Soyeon, Youk Jeonghwan, Park Seongyeol, An Yohan, et al. 2017. “Clonal History and Genetic Predictors of Transformation Into Small-Cell Carcinomas From Lung Adenocarcinomas.” Journal of Clinical Oncology. 10.1200/jco.2016.71.9096. [DOI] [PubMed] [Google Scholar]
- Leonetti Alessandro, Sharma Sugandhi, Minari Roberta, Perego Paola, Giovannetti Elisa, and Tiseo Marcello. 2019. “Resistance Mechanisms to Osimertinib in EGFR-Mutated Non-Small Cell Lung Cancer.” British Journal of Cancer, September. 10.1038/s41416-019-0573-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin Pavel A., Mayer Melissa, Hoskin Sharon, Sailors Joseph, Oliver Dwight H., and Gerber David E.. 2015. “Histologic Transformation from Adenocarcinoma to Squamous Cell Carcinoma as a Mechanism of Resistance to EGFR Inhibition.” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 10 (9): e86–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Xiuning, Puri Sonam, Negrao Marcelo V., Nilsson Monique B., Robichaux Jacqulyne, Boyle Theresa, Hicks J. Kevin, et al. 2018. “Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC.” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 24 (24): 6195–6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, et al. 2008. “BIBW2992, an Irreversible EGFR/HER2 Inhibitor Highly Effective in Preclinical Lung Cancer Models.” Oncogene 27 (34): 4702–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Jessica J., Riely Gregory J., and Shaw Alice T.. 2017. “Targeting ALK: Precision Medicine Takes on Drug Resistance.” Cancer Discovery 7 (2): 137–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovly Christine, McDonald Nerina T., Chen Heidi, Ortiz-Cuaran Sandra, Heukamp Lukas, Yan Yingjun, Florin Alexandra, et al. 2014. “Rationale for Co-Targeting IGF-1R and ALK in ALK Fusion–positive Lung Cancer.” Nature Medicine 20 (9): 1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maemondo Makoto, Inoue Akira, Kobayashi Kunihiko, Sugawara Shunichi, Oizumi Satoshi, Isobe Hiroshi, Gemma Akihiko, et al. 2010. “Gefitinib or Chemotherapy for Non–Small-Cell Lung Cancer with Mutated EGFR.” The New England Journal of Medicine 362 (25): 2380–88. [DOI] [PubMed] [Google Scholar]
- Meuwissen Ralph, Linn Sabine C., Linnoila R. Ilona, Zevenhoven John, Mooi Wolter J., and Berns Anton. 2003. “Induction of Small Cell Lung Cancer by Somatic Inactivation of Both Trp53 and Rb1 in a Conditional Mouse Model.” Cancer Cell 4 (3): 181–89. [DOI] [PubMed] [Google Scholar]
- Miranda Oshin, Farooqui Mariya, and Siegfried Jill M.. 2018. “Status of Agents Targeting the HGF/c-Met Axis in Lung Cancer.” Cancers 10 (9). 10.3390/cancers10090280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsudomi Tetsuya, Morita Satoshi, Yatabe Yasushi, Negoro Shunichi, Okamoto Isamu, Tsurutani Junji, Seto Takashi, et al. 2010. “Gefitinib versus Cisplatin plus Docetaxel in Patients with Non-Small-Cell Lung Cancer Harbouring Mutations of the Epidermal Growth Factor Receptor (WJTOG3405): An Open Label, Randomised Phase 3 Trial.” The Lancet Oncology 11 (2): 121–28. [DOI] [PubMed] [Google Scholar]
- Miyawaki Masayoshi, Yasuda Hiroyuki, Tani Tetsuo, Hamamoto Junko, Arai Daisuke, Ishioka Kota, Ohgino Keiko, et al. 2017. “Overcoming EGFR Bypass Signal-Induced Acquired Resistance to ALK Tyrosine Kinase Inhibitors in ALK-Translocated Lung Cancer.” Molecular Cancer Research: MCR 15 (1): 106–14. [DOI] [PubMed] [Google Scholar]
- Mok Tony S., Wu Yi-Long, Thongprasert Sumitra, Yang Chih-Hsin, Chu Da-Tong, Saijo Nagahiro, Sunpaweravong Patrapim, et al. 2009. “Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma.” The New England Journal of Medicine 361 (10): 947–57. [DOI] [PubMed] [Google Scholar]
- Nakamichi Shinji, Seike Masahiro, Miyanaga Akihiko, Chiba Mika, Zou Fenfei, Takahashi Akiko, Ishikawa Arimi, et al. 2018. “Overcoming Drug-Tolerant Cancer Cell Subpopulations Showing AXL Activation and Epithelial-Mesenchymal Transition Is Critical in Conquering ALK-Positive Lung Cancer.” Oncotarget 9 (43): 27242–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederst Matthew J., Hu Haichuan, Mulvey Hillary E., Lockerman Elizabeth L., Garcia Angel R., Piotrowska Zofia, Sequist Lecia V., and Engelman Jeffrey A.. 2015. “The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies.” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 21 (17): 3924–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederst Matthew J., Sequist Lecia V., Poirier John T., Mermel Craig H., Lockerman Elizabeth L., Garcia Angel R., Katayama Ryohei, et al. 2015. “RB Loss in Resistant EGFR Mutant Lung Adenocarcinomas That Transform to Small-Cell Lung Cancer.” Nature Communications 6 (March): 6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offin Michael, Somwar Romel, Rekhtman Natasha, Benayed Ryma, Chang Jason C., Plodkowski Andrew, W. Lui Allan J., et al. 2018. “Acquired and Gene Fusions as Mechanisms of Resistance to Osimertinib in -Mutant Lung Cancers.” JCO Precision Oncology 2 (2): 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi Kadoaki, Maruvka Yosef E., Michor Franziska, and Pao William. 2013. “Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor-Resistant Disease.” Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology 31 (8): 1070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi Kadoaki, Sequist Lecia V., Arcila Maria E., Moran Teresa, Chmielecki Juliann, Lin Ya-Lun, Pan Yumei, et al. 2012. “Lung Cancers with Acquired Resistance to EGFR Inhibitors Occasionally Harbor BRAF Gene Mutations but Lack Mutations in KRAS, NRAS, or MEK1.” Proceedings of the National Academy of Sciences of the United States of America 109 (31): E2127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Cuaran Sandra, Scheffler Matthias, Plenker Dennis, Dahmen Llona, Scheel Andreas H., Fernandez-Cuesta Lynnette, Meder Lydia, et al. 2016. “Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors.” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 22 (19): 4837–47. [DOI] [PubMed] [Google Scholar]
- Oser Matthew G., Niederst Matthew J., Sequist Lecia V., and Engelman Jeffrey A.. 2015. “Transformation from Non-Small-Cell Lung Cancer to Small-Cell Lung Cancer: Molecular Drivers and Cells of Origin.” The Lancet Oncology 16 (4): e165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxnard Geoffrey R., Hu Yuebi, Mileham Kathryn F., Husain Hatim, Costa Daniel B., Tracy Philip, Feeney Nora, et al. 2018. “Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib.” JAMA Oncology 4 (11): 1527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oztan A, Fischer S, Schrock AB, Erlich RL, Lovly CM, Stephens PJ, Ross JS, et al. 2017. “Emergence of EGFR G724S Mutation in EGFR-Mutant Lung Adenocarcinoma Post Progression on Osimertinib.” Lung Cancer 111 (September): 84–87. [DOI] [PubMed] [Google Scholar]
- Pao William, Miller Vincent A., Politi Katerina A., Riely Gregory J., Somwar Romel, Zakowski Maureen F., Kris Mark G., and Varmus Harold. 2005. “Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain.” PLoS Medicine 2 (3): e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadimitrakopoulou VA, Wu Yl, Han Jy, Ahn Mj, Ramalingam SS, Johnv T, Okamoto I, Yang Jch, Bulusu Kc, Laus G, Collins B, et al. 2018. “Analysis of Resistance Mechanisms to Osimertinib in Patients with EGFR T790M Advanced NSCLC from the AURA3 Study.” Annals of Oncology: Official Journal of the European Society for Medical Oncology / ESMO 29 (s8): 741–741. [Google Scholar]
- Park Jung Wook, Lee John K., Sheu Katherine M., Wang Liang, Balanis Nikolas G., Nguyen Kim, Smith Bryan A., et al. 2018. “Reprogramming Normal Human Epithelial tissues to a Common, Lethal Neuroendocrine Cancer lineage.” Science 362 (6410): 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer Martin, Fernández-Cuesta Lynnette, Sos Martin L., George Julie, Seidel Danila, Kasper Lawryn H., Plenker Dennis, et al. 2012. “Integrative Genome Analyses Identify Key Somatic Driver Mutations of Small-Cell Lung Cancer.” Nature Genetics 44 (10): 1104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters Solange, Camidge D. Ross, Shaw Alice T., Gadgeel Shirish, Ahn Jin S., Kim Dong-Wan, Ou Sai-Hong I., et al. 2017. “Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer.” The New England Journal of Medicine 377 (9): 829–38. [DOI] [PubMed] [Google Scholar]
- Piotrowska Zofia, Isozaki Hideko, Lennerz Jochen K., Gainor Justin F., Lennes Inga T., Zhu Viola W., Marcoux Nicolas, et al. 2018. “Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion.” Cancer Discovery 8 (12): 1529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram Sidharth V., Tirosh Itay, Parikh Anuraag S., Patel Anoop P., Yizhak Keren, Gillespie Shawn, Rodman Christopher, et al. 2017. “Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer.” Cell 171 (7): 1611–24.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail Daniela F., and Joyce Johanna A.. 2013. “Microenvironmental Regulation of Tumor Progression and Metastasis.” Nature Medicine 19 (11): 1423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu Kalluri, and Michael Zeisberg. 2006. “Fibroblasts in Cancer.” Nature Reviews. Cancer 6 (5): 392–401. [DOI] [PubMed] [Google Scholar]
- Ramalingam SS, Cheng Y, Zhou C, and Ohe Y. 2018. “LBA50 Mechanisms of Acquired Resistance to First-Line Osimertinib: Preliminary Data from the Phase III FLAURA Study.” Annals of. https://academic.oup.com/annonc/article-abstract/29/suppl_8/mdy424.063/5142018. [Google Scholar]
- Ramalingam SS, Yang JC, Lee CK, and Kurata T. 2018. “Osimertinib as First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer.” Jornal Dos Clinicos. https://clf1.medpagetoday.com/content/pdf/reading-room/asco/JCO.LC.Ramalingam031918.pdf. [DOI] [PubMed] [Google Scholar]
- Raoof Sana, Mulford Iain J., Frisco-Cabanos Heidie, Nangia Varuna, Timonina Daria, Labrot Emma, Hafeez Nafeeza, et al. 2019. “Targeting FGFR Overcomes EMT-Mediated Resistance in EGFR Mutant Non-Small Cell Lung Cancer.” Oncogene 38 (37): 6399–6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reck Martin, Rodríguez-Abreu Delvys, Robinson Andrew G., Hui Rina, Tibor Csőszi Andrea Fülöp, Gottfried Maya, et al. 2016. “Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer.” The New England Journal of Medicine 375 (19): 1823–33. [DOI] [PubMed] [Google Scholar]
- Rotow Julia K., Gui Philippe, Wu Wei, Raymond Victoria M., Lanman Richard B., Kaye Frederic J., Peled Nir, et al. 2019. “Co-Occurring Alterations in the RAS-MAPK Pathway Limit Response to MET Inhibitor Treatment in MET Exon 14 Skipping Mutation-Positive Lung Cancer.” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, September. 10.1158/1078-0432.CCR-19-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin Charles M., Durinck Steffen, Stawiski Eric W., Poirier John T., Modrusan Zora, Shames David S., Bergbower Emily A., et al. 2012. “Comprehensive Genomic Analysis Identifies SOX2 as a Frequently Amplified Gene in Small-Cell Lung Cancer.” Nature Genetics 44 (10): 1111–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin Charles M., Poirier John T., Byers Lauren Averett, Dive Caroline, Dowlati Afshin, George Julie, Heymach John V., et al. 2019. “Molecular Subtypes of Small Cell Lung Cancer: A Synthesis of Human and Mouse Model Data.” Nature Reviews. Cancer 19 (5): 289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler Alan, Gray Robert, Perry Michael C., Brahmer Julie, Schiller Joan H., Dowlati Afshin, Lilenbaum Rogerio, and Johnson David H.. 2006. “Paclitaxel-Carboplatin Alone or with Bevacizumab for Non-Small-Cell Lung Cancer.” The New England Journal of Medicine 355 (24): 2542–50. [DOI] [PubMed] [Google Scholar]
- Schrock Alexa B., Lai Andrea, Ali Siraj M., Miller Vincent A., and Raez Luis E.. 2017. “Mutation of MET Y1230 as an Acquired Mechanism of Crizotinib Resistance in NSCLC with MET Exon 14 Skipping.” Journal of Thoracic Oncology. 10.1016/j.jtho.2017.02.017. [DOI] [PubMed] [Google Scholar]
- Schrock Alexa B., Zhu Viola W., Hsieh Wen-Son, Madison Russell, Creelan Benjamin, Silberberg Jeffrey, Costin Dan, et al. 2018. “Receptor Tyrosine Kinase Fusions and BRAF Kinase Fusions Are Rare but Actionable Resistance Mechanisms to EGFR Tyrosine Kinase Inhibitors.” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 13 (9): 1312–23. [DOI] [PubMed] [Google Scholar]
- Sequist Lecia V., Waltman Belinda A., Dias-Santagata Dora, Digumarthy Subba, Turke Alexa B., Fidias Panos, Bergethon Kristin, et al. 2011. “Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors.” Science Translational Medicine 3 (75): 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma Sreenath V., Lee Diana Y., Li Bihua, Quinlan Margaret P., Takahashi Fumiyuki, Maheswaran Shyamala, McDermott Ultan, et al. 2010. “A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations.” Cell 141 (1): 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Xing P, Han X, Wang S, Liu Y, Liu P, Li J, et al. 2018. “Exploring the Resistance Mechanism of Osimertinib and Monitoring the Treatment Response Using Plasma ctDNA in Chinese NSCLC Patients.” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 13 (10): S589–S589. [Google Scholar]
- Socinski Mark A., Jotte Robert M., Cappuzzo Federico, Orlandi Francisco, Stroyakovskiy Daniil, Nogami Naoyuki, Rodríguez-Abreu Delvys, et al. 2018. “Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC.” The New England Journal of Medicine 378 (24): 2288–2301. [DOI] [PubMed] [Google Scholar]
- Soria Jean-Charles, Ohe Yuichiro, Vansteenkiste Johan, Reungwetwattana Thanyanan, Chewaskulyong Busyamas, Ki Hyeong Lee Arunee Dechaphunkul, et al. 2018. “Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer.” The New England Journal of Medicine 378 (2): 113–25. [DOI] [PubMed] [Google Scholar]
- Straussman Ravid, Morikawa Teppei, Shee Kevin, Barzily-Rokni Michal, Qian Zhi Rong, Du Jinyan, Davis Ashli, et al. 2012. “Tumour Micro-Environment Elicits Innate Resistance to RAF Inhibitors through HGF Secretion.” Nature 487 (7408): 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland Kate D., Proost Natalie, Brouns Inge, Adriaensen Dirk, Song Ji-Ying, and Berns Anton. 2011. “Cell of Origin of Small Cell Lung Cancer: Inactivation of Trp53 and Rb1 in Distinct Cell Types of Adult Mouse Lung.” Cancer Cell 19 (6): 754–64. [DOI] [PubMed] [Google Scholar]
- Takezawa Ken, Pirazzoli Valentina, Arcila Maria E., Nebhan Caroline A., Song Xiaoling, Stanchina Elisa de, Ohashi Kadoaki, et al. 2012. “HER2 Amplification: A Potential Mechanism of Acquired Resistance to EGFR Inhibition in EGFR-Mutant Lung Cancers That Lack the Second-Site EGFRT790M Mutation.” Cancer Discovery 2 (10): 922–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai Alesha, Chia Puey L., Russell Prudence A., Do Hongdo, Dobrovic Alex, Mitchell Paul, and John Thomas. 2017. “De Novo Activating Epidermal Growth Factor Mutations (EGFR) in Small-Cell Lung Cancer.” Internal Medicine Journal 47 (9): 1071–74. [DOI] [PubMed] [Google Scholar]
- Thiery Jean Paul. 2002. “Epithelial–mesenchymal Transitions in Tumour Progression.” Nature Reviews. Cancer 2 (6): 442–54. [DOI] [PubMed] [Google Scholar]
- Thomson Stuart, Buck Elizabeth, Petti Filippo, Griffin Graeme, Brown Eric, Ramnarine Nishal, Iwata Kenneth K., Gibson Neil, and Haley John D.. 2005. “Epithelial to Mesenchymal Transition Is a Determinant of Sensitivity of Non–Small-Cell Lung Carcinoma Cell Lines and Xenografts to Epidermal Growth Factor Receptor Inhibition.” Cancer Research. 10.1158/0008-5472.can-05-1058. [DOI] [PubMed] [Google Scholar]
- Thress Kenneth S., Paweletz Cloud P., Felip Enriqueta, Cho Byoung Chul, Stetson Daniel, Dougherty Brian, Lai Zhongwu, et al. 2015. “Acquired EGFR C797S Mutation Mediates Resistance to AZD9291 in Non-Small Cell Lung Cancer Harboring EGFR T790M.” Nature Medicine 21 (6): 560–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiseo M, Gelsomino F, Boggiani D, Bortesi B, Bartolotti M, Bozzetti C, Samarelli G, et al. 2011. “EGFR and EML4-ALK gene mutations in NSCLC: A case report of erlotinib-resistant patient with both concomitant mutations.” Lung Cancer 71(2): 241–243. [DOI] [PubMed] [Google Scholar]
- Tulchinsky Eugene, Demidov Oleg, Kriajevska Marina, Barlev Nickolai A., and Imyanitov Evgeny. 2019. “EMT: A Mechanism for Escape from EGFR-Targeted Therapy in Lung Cancer.” Biochimica et Biophysica Acta, Reviews on Cancer 1871 (1): 29–39. [DOI] [PubMed] [Google Scholar]
- Uramoto Hidetaka, Iwata Teruo, Onitsuka Takamitsu, Shimokawa Hidehiko, Hanagiri Takeshi, and Oyama Tsunehiro. 2010. “Epithelial-Mesenchymal Transition in EGFR-TKI Acquired Resistant Lung Adenocarcinoma.” Anticancer Research 30 (7): 2513–17. [PubMed] [Google Scholar]
- Vega Sonia, Morales Aixa V., Ocaña Oscar H., Valdés Francisco, Fabregat Isabel, and Nieto M. Angela. 2004. “Snail Blocks the Cell Cycle and Confers Resistance to Cell Death.” Genes & Development 18 (10): 1131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voena Claudia, Varesio Lydia M., Zhang Liye, Menotti Matteo, Poggio Teresa, Panizza Elena, Wang Qi, et al. 2016. “Oncogenic ALK Regulates EMT in Non-Small Cell Lung Carcinoma through Repression of the Epithelial Splicing Regulatory Protein 1.” Oncotarget 7 (22): 33316–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojnic Morana, Kubota Daesuki, Kurzatkowski Christopher, Offin Michael, Suzawa Ken, Benayed Ryma, and Schoenfeld Adam et al. 2018. J Thorac Oncol 14(5): 802–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Wei, Li Qi, Yamada Tadaaki, Matsumoto Kunio, Matsumoto Isao, Oda Makoto, Watanabe Go, et al. 2009. “Crosstalk to Stromal Fibroblasts Induces Resistance of Lung Cancer to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors.” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 15 (21): 6630–38. [DOI] [PubMed] [Google Scholar]
- Wang Yuanyuan, Zhang Ting, Guo Lixia, Ren Tao, and Yang Yanan. 2019. “Stromal Extracellular Matrix Is a Microenvironmental Cue Promoting Resistance to EGFR Tyrosine Kinase Inhibitors in Lung Cancer Cells.” The International Journal of Biochemistry & Cell Biology 106 (January): 96–106. [DOI] [PubMed] [Google Scholar]
- Wang Zhen, Yang Jin-Ji, Huang Jie, Ye Jun-Yi, Zhang Xu-Chao, Tu Hai-Yan, Han-Zhang Han, and Wu Yi-Long. 2017. “Lung Adenocarcinoma Harboring EGFR T790M and In Trans C797S Responds to Combination Therapy of First- and Third-Generation EGFR TKIs and Shifts Allelic Configuration at Resistance.” Journal of Thoracic Oncology. 10.1016/j.jtho.2017.06.017. [DOI] [PubMed] [Google Scholar]
- Wei Jiacong, van der Wekken Anthonie J., Saber Ali, Terpstra Miente M., Schuuring Ed, Timens Wim, N. Hiltermann T. Jeroen, M. Groen Harry J., van den Berg Anke, and Kok Klaas. 2018. “Mutations in EMT-Related Genes in ALK Positive Crizotinib Resistant Non-Small Cell Lung Cancers.” Cancers 10 (1). 10.3390/cancers10010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson Catherine, Nicholes Katrina, Bustos Daisy, Lin Eva, Song Qinghua, Stephan Jean-Philippe, Kirkpatrick Donald S., and Settleman Jeff. 2014. “Overcoming EMT-Associated Resistance to Anti-Cancer Drugs via Src/FAK Pathway Inhibition.” Oncotarget 5 (17): 7328–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson Timothy R., Fridlyand Jane, Yan Yibing, Penuel Elicia, Burton Luciana, Chan Emily, Peng Jing, et al. 2012. “Widespread Potential for Growth-Factor-Driven Resistance to Anticancer Kinase Inhibitors.” Nature 487 (7408): 505–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Yi Long, Zhang Li, Wan Kim Dong, Liu Xiaoqing, Ho Lee Dae, Hsin Yang James Chih, Ju Ahn Myung, et al. 2018. “Phase Ib/II Study of Capmatinib (INC280) plus Gefitinib after Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients with EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer.” Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology 36 (31): 3101–9. [DOI] [PubMed] [Google Scholar]
- Yamada Daisaku, Kobayashi Shogo, Wada Hiroshi, Kawamoto Koichi, Marubashi Shigeru, Eguchi Hidetoshi, Ishii Hideshi, Nagano Hiroaki, Doki Yuichiro, and Mori Masaki. 2013. “Role of Crosstalk between Interleukin-6 and Transforming Growth Factor-Beta 1 in Epithelial–mesenchymal Transition and Chemoresistance in Biliary Tract Cancer.” European Journal of Cancer 49 (7): 1725–40. [DOI] [PubMed] [Google Scholar]
- Yang Zhe, Yang Nong, Ou Qiuxiang, Xiang Yi, Jiang Tao, Wu Xue, Bao Hua, et al. 2018. “Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients.” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 24 (13): 3097–3107. [DOI] [PubMed] [Google Scholar]
- Yauch Robert L., Januario Thomas, Eberhard David A., Cavet Guy, Zhu Wenjing, Fu Ling, Pham Thinh Q., et al. 2005. “Epithelial versus Mesenchymal Phenotype Determines in Vitro Sensitivity and Predicts Clinical Activity of Erlotinib in Lung Cancer Patients.” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 11 (24 Pt 1): 8686–98. [DOI] [PubMed] [Google Scholar]
- Yi Yanmei, Zeng Shanshan, Wang Zhaotong, Wu Minhua, Ma Yuanhuan, Ye Xiaoxia, Zhang Biao, and Liu Hao. 2018. “Cancer-Associated Fibroblasts Promote Epithelial-Mesenchymal Transition and EGFR-TKI Resistance of Non-Small Cell Lung Cancers via HGF/IGF-1/ANXA2 Signaling.” Biochimica et Biophysica Acta, Molecular Basis of Disease 1864 (3): 793–803. [DOI] [PubMed] [Google Scholar]
- Yoda Satoshi, Lin Jessica J., Lawrence Michael S., Burke Benjamin J., Friboulet Luc, Langenbucher Adam, Dardaei Leila, et al. 2018. “Sequential ALK Inhibitors Can Select for Lorlatinib-Resistant Compound ALK Mutations in ALK-Positive Lung Cancer.” Cancer Discovery 8 (6): 714–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun Cai-Hong, Mengwasser Kristen E., Toms Angela V., Woo Michele S., Greulich Heidi, Wong Kwok-Kin, Meyerson Matthew, and Eck Michael J.. 2008. “The T790M Mutation in EGFR Kinase Causes Drug Resistance by Increasing the Affinity for ATP.” Proceedings of the National Academy of Sciences of the United States of America 105 (6): 2070–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Helena, Tian Shaozhou K., Drilon Alexander E., Borsu Laetitia, Riely Gregory J., Arcila Maria E., and Ladanyi Marc. 2015. “Acquired Resistance of EGFR-Mutant Lung Cancer to a T790M-Specific EGFR Inhibitor: Emergence of a Third Mutation (C797S) in the EGFR Tyrosine Kinase Domain.” JAMA Oncology 1 (7): 982–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, C-H Xiao, L-D Tan, Q-S Wang, X-Q Li, and Y-M Feng. 2013. “Cancer-Associated Fibroblasts Induce Epithelial–mesenchymal Transition of Breast Cancer Cells through Paracrine TGF-β Signalling.” British Journal of Cancer 110 (3): 724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakowski Maureen F., Ladanyi Marc, Kris Mark G., and Memorial Sloan-Kettering Cancer Center Lung Cancer OncoGenome Group. 2006. “EGFR Mutations in Small-Cell Lung Cancers in Patients Who Have Never Smoked.” The New England Journal of Medicine 355 (2): 213–15. [DOI] [PubMed] [Google Scholar]
- Zeng Liang, Yang Nong, and Zhang Yongchang. 2018. “GOPC-ROS1 Rearrangement as an Acquired Resistance Mechanism to Osimertinib and Responding to Crizotinib Combined Treatments in Lung Adenocarcinoma.” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 13 (7): e114–16. [DOI] [PubMed] [Google Scholar]
- Zhang Qi, Zhang Xu-Chao, Yang Jin-Ji, Yang Zhen-Fan, Bai Yu, Su Jian, Wang Zheng, et al. 2018. “EGFR L792H and G796R: Two Novel Mutations Mediating Resistance to the Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib.” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 13 (9): 1415–21. [DOI] [PubMed] [Google Scholar]
- Zhou Juan, Wang Jinjing, Zeng Yunyun, Zhang Xi, Hu Qiaoting, Zheng Jihua, Chen Bei, Xie Bo, and Zhang Wei-Min. 2015. “Implication of Epithelial-Mesenchymal Transition in IGF1R-Induced Resistance to EGFR-TKIs in Advanced Non-Small Cell Lung Cancer.” Oncotarget 6 (42): 44332–45. [DOI] [PMC free article] [PubMed] [Google Scholar]