

Abstract
This study reports an eco‐friendly‐based method for the preparation of biopolymer Ag–Au nanoparticles (NPs) by using gum kondagogu (GK; Cochlospermum gossypium), as both reducing and protecting agent. The formation of GK‐(Ag–Au) NPs was confirmed by UV‐absorption, fourier transformed infrared (FTIR), atomic force microscopy (AFM), scanning electron microscope (SEM) and transmission electron microscope (TEM). The GK‐(Ag–Au) NPs were of 1–12 nm in size. The anti‐proliferative activity of nanoparticle constructs was assessed by MTT assay, confocal microscopy, flow cytometry and quantitative real‐time polymerase chain reaction (PCR) techniques. Expression studies revealed up‐regulation of p53, caspase‐3, caspase‐9, peroxisome proliferator‐activated receptors (PPAR) PPARa and PPARb, genes and down‐regulation of Bcl‐2 and Bcl‐x(K) genes, in B16F10 cells treated with GK‐(Ag–Au) NPs confirming the anti‐proliferative properties of the nanoparticles.
Inspec keywords: nanomedicine, transmission electron microscopy, genetics, cellular biophysics, molecular biophysics, enzymes, nanofabrication, gold, silver, scanning electron microscopy, nanoparticles, Fourier transform infrared spectra, atomic force microscopy, biomedical materials
Other keywords: size 1.0 nm to 12.0 nm, Ag‐Au, anti‐proliferative assessment, eco‐friendly‐based method, anti‐proliferative activity, anti‐proliferative properties, biopolymer‐based Ag–Au bimetallic nanoparticle, Cochlospermum gossypium, gum kondagogu, biopolymer preparation, biogenic synthesis, UV‐absorption, Fourier transform infrared spectroscopy, scanning electron microscopy, transmission electron microscopy, atomic force microscopy, MTT assay, confocal microscopy, flow cytometry, caspase‐3, caspase‐9, peroxisome proliferator‐activated receptors, Bcl‐2 gene, Bcl‐x(K) gene, B16F10 cells
1 Introduction
The ‘greener’ environmentally friendly processes are becoming increasingly popular due to worldwide problems associated with environmental contamination. Similar to such natural processes, plants [1, 2, 3], fungi [4, 5], and bacteria [6] were widely used for the synthesis of metal nanoparticles. Green synthesis of nanoparticles renders advancement over other methods as it is simple, cost‐effective, and relatively reproducible and often results in more stable materials [7].
The bimetallic nanoparticle synthesis mainly occurs by reduction of two metal components resulting into either core‐shell or alloy arrangement. Very few works on green synthesis of alloy and core‐shell bimetallic nanoparticles are reported in the contemporary literature [8, 9, 10].
Previously, bimetallic nanoparticles have attracted lots of attention because of their importance for magnetic, optical, electrolytic and catalytic applications in a variety of fields, whose properties are distinctly different from monometallic nanoparticles [11, 12, 13, 14]. Hence, a non‐toxic, high yielding, eco‐friendly procedure like green synthesis has attracted a great deal of interest.
Recently, the green synthesis of bimetallic nanostructures has been studied by using biocompatible molecules such as Piper pedicellatum [15], aqueous extract of mahogany leaves [16], extract and dried leaf of Anacardium occidentale [8], Gloriosa superba leaf extract [10], fruit juice of pomegranate [17], dextran [18], glucose and starch [19], fungus, Fusarium semitectum [20] and curdlan [21]. Methods that use polysaccharides to produce bimetallic nanoparticles are gradually becoming more interesting [22].
Earlier studies have demonstrated that combination of both gold and silver in one bimetallic NP confers advantageous biological activities and low toxicity in vivo. Contemporarily, silver and gold nanoparticles encapsulated/immobilised in a polysaccharide are being widely used as antibacterial and anticoagulant. Surface modified nanoparticles have been employed to enhance the total coagulation, thrombin formation and fibrin clotting time [23, 24]. Concurrently biopolymer coatings of the nanoparticles are also being employed [25]; wherein the protein adsorption on NPs plays an important factor in explaining the effect of NPs on cellular behaviour. Previous experimental investigations have shown that hydrophillic surface coatings with embedded biocidal silver nanoparticles and sodium heparin in central venous and skin penetrating catheters uniquely served as both antimicrobial and antithrombogenic, suggesting bifunctional application of surface‐coated nanoparticles [26, 27]. Nanocomposites based on bismuth sulphide‐cellulose have also been synthesised as thin films for applications in optoelectronics [28].
Hence, in this communication, a green approach for the synthesis of Ag–Au bimetallic nanoparticles by reduction of aqueous Ag+ and Au3+ ions using gum kondagogu (GK) biopolymer as both stabilising and reducing agent is reported. Earlier, GK was successfully employed in the synthesis of monometallic nanoparticles [7], while the present investigation deals with green synthesis and characterisation of bimetallic nanoparticles mediated through GK. Further, these bimetallic nanoparticles were evaluated for their anti‐proliferative effect against B16F10 melanoma cells, which was used as a model system.
2 Materials and methods
2.1 Materials
The green synthesis of nanoparticle constructs was carried out by using salts such as AgNO3 (Himedia, Mumbai, India) and HAuCl4 (Sigma‐Aldrich Chemicals, St. Louis, USA). Chemicals such as dNTP's mix, dimethyl sulfoxide, 4′,6‐diamidino‐2‐phenylindole (DAPI), ethidium bromide, fetal calf serum (FCS), fetal bovine serum (FBS), Dulbeccos modified Eagles medium (DMEM), penicillin G and Streptomycin were also procured from Sigma‐Aldrich Chemicals. DNA molecular weight markers and Taq DNA polymerase were from Bangalore Genei Pvt Ltd., India. First‐strand cDNA synthesis kit was procured from Invitrogen‐Life Technologies, Carlsbad, USA. Oligonucleotides were purchased from Bioserve, Hyderabad, India. Real‐time PCR plates and SYBR green were procured from SuperScript® III First‐Strand Synthesis System, Invitrogen‐Life Technologies. The above reagents were of analytical purity and were used without any further purification while other chemicals and reagents were of analytical grade.
2.2 Synthesis of silver–gold bimetallic nanoparticles
The synthesis of bimetallic nanoparticle constructs was carried out with GK. Grade‐1, GK samples were powdered in a high‐speed mechanical blender and were sieved through a test sieve (mesh size – 45 µm). GK powder (1.0 g) was weighed, and dissolved in a plastic beaker containing 100 mL of Milli Q water and was gently stirred overnight at 2000 rpm followed by centrifugation at 5000 rpm for 10 min at 298 K. The supernatant comprising soluble gum solution was freeze dried and was used for the synthesis of nanoparticles [29].
The silver–gold bimetallic nanoparticles were synthesised by autoclaving 0.5% soluble gum containing 1 mM AgNO3 and 1 mM HAuCl4 solution at 121°C and 15 psi for 15 min. The effect of different parameters such as (i) reaction time on nanoparticle and (ii) temperature effect on synthesis was studied.
2.3 Characterisation of nanoparticles
Preliminary characterisation for nanoparticles formation was monitored by UV–vis spectroscopy (Agilent Technologies, Cary 60 UV‐vis, Tokyo, Japan). The bioreduction of Ag+ and Au3+ in aqueous solution was monitored by following the UV–vis absorbance of the reaction mixture as a function of time and temperature.
The functional groups involved in the formation of bimetallic nanoparticle constructs (GK‐Ag–Au NP) were identified using FTIR spectrophotometer (TENSOR 27, Bruker Optics, Munich, Germany).
Malvern Zetasizer Nano (Malvern Instruments Ltd., Worcestershire, UK) was used to determine the zeta potential of GK‐(Ag–Au) NP.
Surface morphology of bimetallic nanoparticle constructs was analysed by SEM instrument (Carl Zeiss EVO 18 special edition, 73447 Oberkochen, Germany). The soluble gum and the bimetallic nanoparticle constructs were mounted on a stainless steel stab.
The size of bimetallic nanoparticle constructs was analysed by TEM. The TEM has an accelerated voltage of 120 kV with point resolution of 0.34 nm. A drop of nanoparticle was placed on parafilm (Greenwich, CT), and polystyrene‐coated copper grid. The grid was examined by using FEI‐Phillips Technai‐12 (Eindoven, The Netherlands) at 120 kV filament.
The formation of bimetallic nanoparticle constructs was also examined by AFM (MV 4000, Nanonics, Israel) performed in contact mode using Nanoscope II (Digital Instruments, Santa Barbara, CA, USA). Samples were prepared by placing a drop of solution on freshly cleaved mica followed by drying.
2.4 Quantification of metal by inductively coupled plasma mass spectrometry (ICP‐MS)
The concentration of Ag and Au in their respective ions and nanoparticles was determined by using ICP‐MS (Agilent Technologies‐7700 Series, Tokyo, Japan). The GK‐(Ag–Au) NPs were separated by centrifugation at 13,000 rpm for 30 min. The pellet was redispersed in 1 mL of Milli Q water and was analysed by ICP‐MS [30].
2.5 MTT assay
Melanoma cancer cell line (B16F10) was obtained from National Centre for Cell Sciences, India and was grown in DMEM with 10% FBS, 100 units mL−1 Penicillin G and 1 μg mL−1 Streptomycin, 5 μg mL−1 amphotericin B (Sigma‐Aldrich Chemicals), (2 mM) L‐glutamine and non‐essential amino acids (1×) (Himedia, Mumbai, India) at 37°C in a humidified 5% CO2 incubator (Nectarnova, Symbiogen, Chennai, India). For cytotoxicity studies, after 80% confluence, cells were trypsinised with 0.1% trypsin‐EDTA and were harvested by centrifugation at 500g. Serial dilutions of cells were made from 1 × 106 to 1 × 103 cells/mL. The cells were seeded in triplicates in a 96‐well plate. The cells were treated with 0.25–2.00 µg L−1 of GK‐(Ag–Au) NPs, at two different time points (24 and 48 h). Cells to which no nanoparticles were added served as control. After treatment, the cells were treated with MTT (20 µl, 5 mg mL−1) and the cell viability was determined by measuring the transformation of MTT to a purple coloured formazan dye. The absorbance of samples was measured using an ELISA reader (Micro Scan MS5608A, ECIL, Hyderabad, India) at 570 nm. Per cent viable cells were calculated using the following equation:
| (1) |
where A 570 (sample) corresponds to absorbance obtained from the wells treated with nanoparticles and A 570 (control) represents the absorbance from the wells in which no nanoparticle constructs were added [29].
2.6 Flow cytometry studies
For cell viability studies, after 90% confluence, the cells were treated with 5 µg L−1 of GK‐(Ag–Au) NPs for 48 h. The protocol used for staining was similar to our previous publication [29]. Stained cells were analysed using FACS.
In apoptosis assay Annexin‐V‐FITC staining, the cells were treated with 5 µg L−1 of GK‐(Ag–Au) NPs for 48 h, respectively. 1 × 106 cells were resuspended in 1× binding buffer. To about 500 µl of cell suspension 5 µL of Annexin‐V‐FITC (50 µg mL−1) and 10 µL of PI solution (100 µg mL−1) were added followed by incubation at 28°C for 10 min. Stained cells were analysed using FACS Calibur Flow Cytometer from B.D. Biosceinces, CA, USA. Annexin‐V‐FITC was excited using 488 nm solid state laser and fluorescence emission intensity was captured by 530/30 and 585/42 band pass filters [31].
2.7 Fluorescence microscopy‐based apoptosis assay
In apoptosis assay, B16F10 cells were treated with 5 µg L−1 GK‐(Ag–Au) NPs for 48 h. Then the cells were rinsed with phosphate buffer saline (PBS‐0.01 M pH 7.4) and were incubated with 2.5% formaldehyde for 10 min at 28°C, followed by permeabilisation with 0.5% Triton X‐100 for 5 min. The fixed cells were then incubated with 10% FCS, α tubulin (B‐7) mouse monoclonal IgG (primary antibody) and goat anti‐mouse IgG (secondary antibody) for 1 h each. The cells were then washed with PBS and mounted in mounting medium (Vectashield), containing anti‐fade reagent with DAPI (50 µg mL−1) for 5 min. The cells were rinsed with PBS several times and were viewed using a fluorescence microscope (Axioplan 2, Carl Zeiss, LLC, USA).
2.8 Caspase 8, localisation using confocal microscopy
In caspase‐8 localisation studies, B16F10 cells were treated with 5 µg L−1 GK‐(Ag–Au) NPs for 48 h. Later the cells were rinsed with PBS and were incubated with 2.5% formaldehyde in for 10 min at room temperature, followed by permeabilisation with 0.5% Triton X‐100 for 5 min. The fixed cells were then incubated with 10% FCS, caspase‐8 rabbit polyclonal IgG (primary antibody) and anti‐rabbit IgG (secondary antibody) for 1 h each. The cells were then washed with PBS and mounted in mounting medium (Vectashield), containing anti‐fade reagent with DAPI (50 µg mL−1) for 5 min. The cells were rinsed with PBS several times and were viewed using confocal microscope (Carl Zeiss LSM 880 META, Jena, Germany). Image analysis was done using LSM 880 META software (Carl Zeiss).
2.9 SEM‐energy dispersive X‐ray analysis (EDAX) of B16F10 cells treated with GK‐(Ag–Au) NPs
For SEM‐EDAX studies, B16F10 cells were treated with 5 µg L−1 GK‐(Ag–Au) NPs for 48 h. Later, the cells were rinsed with PBS (0.01 M, pH 7.4) thrice and were heat‐dried before microscopic analysis. The surface structure of the cells before and after nanoparticles treatment was analysed by SEM instrument (Carl Zeiss EVO 18 special edition, 73447 Oberkochen, Germany) equipped with EDAX attachment (INCA Xact, Oxford, UK). The control and nanoparticle (GK‐(Ag–Au) NP) treated B16F10 cells were mounted on a stainless steel stab with double‐sided adhesive tape and were coated with a thin layer of gold metal under high vacuum conditions.
2.10 cDNA synthesis and quantitative real‐time PCR amplification
Gene expression of Bcl‐2, Bcl‐x(K), p53, Caspase‐3, Caspase‐9 and peroxisome proliferator‐activator receptors was determined by qRT‐PCR (ABI Prism 7700 Applied Biosystems) in three independent experiments. B16F10 cells treated with GK‐(Ag–Au) NPs and control group (untreated cells) were homogenised and total RNA was extracted with TRIzol® Reagent (Invitrogen, USA). Total RNA was measured by spectrophotometry (NanoDrop1000, DE, Wilmington, USA) at 260 nm. Purity of RNA was assessed by the quotient of A 260 /A 280 nm. cDNA synthesis was performed using the first‐strand cDNA synthesis kit (Invitrogen‐Life Technologies) primed with oligo (dT)20 in a 20 µl reverse‐transcription reaction on a GeneAmp 2400 thermal cycler system. The cDNA was used as template for qRT‐PCR analysis, with gene‐specific primers, reagents and protocols provided in the SYBR® Select Master Mix kit (Invitrogen‐Life Technologies). All gene‐specific primers were selected using Gene Tools software to amplify a 150–200 bp product with a Tm of 55–60°C. The primers used for the expression studies are shown in Table 1.
Table 1.
Primers designed and used for qRT‐PCR studies
| Name of the gene | Forward primer 5′–3’ | Reverse primer 5′–3′ |
|---|---|---|
| Bcl‐2 | TACCGTCGTGACTTCGCAGAG | GGCAGGCTGAGCAGGGTCTT |
| Bcl‐x(K) | ACAAGGAGATGCAGGTATTG | AGAACTACACCAGCCACAGT |
| p53 | CTTCCCGCCATAAAAAAACA | AGCCCTGAAGTCATAAGACAG |
| caspase‐3 | AAGTGACCATGGACAACAAC | CAGTGCTTTTATGGAAGTTCTTA |
| caspase‐9 | ATCATCAACAACGTGAACTTCT | ATGAGAGAGGATGACCACCAC |
| PPARa | GCAGTGCTGGCTACCTTCAAA | TGGGCTACATCCTCGACTCCT |
| PPARb | TAGAAGCCATCCAGGACACC | CCGTCTTCTTTAGCCACTG |
2.11 Statistical analysis
The results obtained from different experiments were subjected to statistical analysis. Origin (Version 6.1) was used for all the statistical analyses. All the data reported in this paper are represented as mean ± SE value.
3 Result
3.1 Synthesis of GK‐(Ag–Au) nanoparticles
The green synthesis of GK‐(Ag–Au) NPs was done by using soluble gum aqueous solution, silver nitrate and gold chloride solution. The soluble gum (0.5%) (Fig. 1 b (i)) was mixed with silver nitrate (1 mM) and gold chloride (1 mM) for the synthesis of GK‐(Ag–Au) NPs. Conversion of the yellow colour of the HAuCl4 solution into a purple colour [32] indicated the formation of GK‐(Ag–Au) NPs (Fig. 1 b (ii)).
Fig. 1.
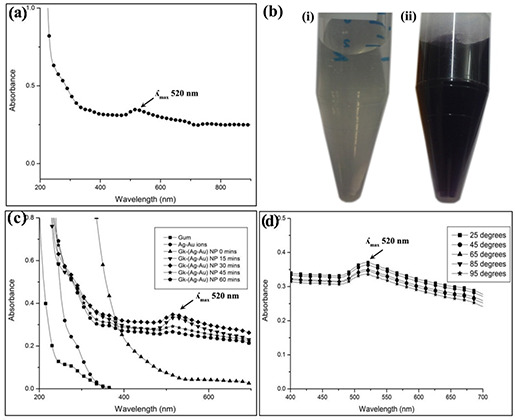
Characterisation of nanoparticles
(a) Synthesis of GK‐(Ag–Au) NP using soluble GK, AgNO3 and HAuCl4. GK‐(Ag–Au) NP shows a characteristic absorbance at 520 nm. (b) (i) Soluble gum (ii) after nanoparticle formation. The soluble gum solution turns to purple colour after the formation of GK‐(Ag–Au) NP (c) . Effect of reaction time (0–60 min) on GK‐(Ag–Au) NP formation. Maximum absorbance was observed at 30 min. (d) . Effect of temperature (25–95°C) on GK‐(Ag–Au) NP. Temperature had no effect on the nanoparticles
3.2 Characterisation of GK‐(Ag–Au) nanoparticles
The confirmation of nanoparticle synthesis was basically done by UV–visible spectroscopy. The GK‐(Ag–Au) NPs showed a characteristic absorption peak at 520 nm in UV spectra (Fig. 1 a). The UV–vis absorption spectra of GK‐(Ag–Au) NPs colloidal solution synthesised at different time points (Fig. 1 c) revealed that by increasing the reaction duration up to 30 min leads to significant increase in absorbance followed by decrease in the absorption intensity was observed from 45 min. The UV–vis spectra of temperature effect (25–95°C) on GK‐(Ag–Au) NPs are depicted in Fig. 1 d. The data indicate that the temperature has no influence in the absorption intensity at 520 nm.
FTIR spectroscopy was carried out to identify the functional groups involved in the formation of nanoparticles. Fig. 2 a shows the presence of six bands at 3442, 2934, 1729, 1621, 1255 and 1046 cm−1 in soluble GK, whereas in case of GK‐(Ag–Au) NPs (Fig. 2 a) the bands were observed at 3356, 1729, 1640, 1262 and 1046 cm−1.
Fig. 2.
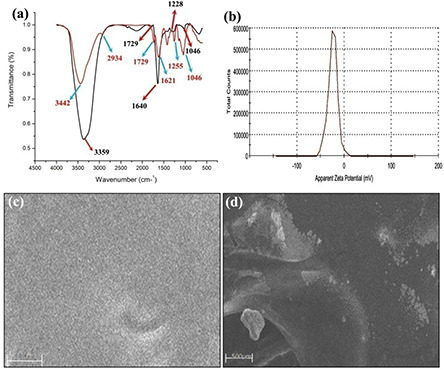
FTIR analysis
(a) Native soluble GK: and after nanoparticle formation indicating the involvement of various functional groups. Functional groups such as hydroxyl, carbonyl, carboxylate, acetyl and C–O were identified to be involved in the nanoparticle formation. (b) Zeta potential of GK‐(Ag–Au) NP, indicating the stability of GK‐(Ag–Au) NP. SEM micrographs of GK (c) soluble gum (d) after nanoparticle formation. GK appeared in the form of a homogenous layer with uniform distribution of the polymer while in the case of GK‐(Ag–Au) NP, it showed uniform distribution of nanoparticles
The zeta potentials of GK‐(Ag–Au) NP composite were −24.3 mV, respectively (Fig. 2 b).
In case of SEM analysis, the GK appeared like a filamentary material in the form of a homogenous layer with uniform distribution of polymer (Fig. 2 c) in SEM, whereas GK‐(Ag–Au) NPs showed the presence of uniformly distributed nanoparticles (Fig. 2 d).
In TEM analysis, gum showed uniform distribution of the polymer without any particles (Fig. 3 a), while GK‐(Ag–Au) NPs showed the presence of tiny particles (Figs. 3 b and c). The shape of the GK‐(Ag–Au) NPs was predominantly spherical with a diameter ranging from 1 to 12 nm ±0.50 S.E.
Fig. 3.
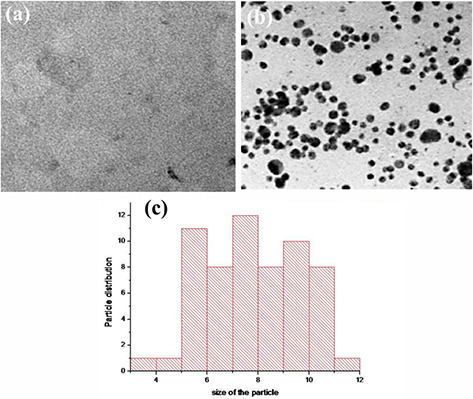
TEM analysis of gum and GK‐ (Ag‐Au) nanoparticles
(a) TEM images of soluble gum, (b) GK‐(Ag–Au) NP, (c) Histogram showing particle size distribution. The shape of GK‐(Ag–Au) NP was predominantly spherical in shape with diameter ranging from 1 to 12 nm ±0.50
The surface morphology of the GK‐(Ag–Au) NPs was studied using AFM. The 3D image (AFM) has been shown in Fig. 4 b displaying spherical‐shaped nanoparticles with smooth surface, without any pin holes or cracks, whereas GK has no major peaks in the 3D image (Fig. 4 a).
Fig. 4.
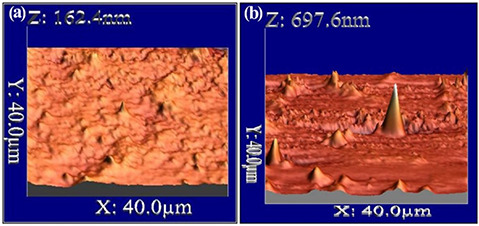
AFM analysis of native soluble GK
(a) Soluble gum, (b) After nanoparticle formation. The 3D images display spherical‐shaped nanoparticles with smooth surface
3.3 Quantification of metal by ICP‐MS
The concentration of Ag and Au in their respective ions (AgNO3 and HAuCl4) and GK‐(Ag–Au) NP was quantified by ICP‐MS. The concentration of Ag–Au in AgNO3 and HAuCl4 was determined to be 154.1 µg mL−1. The concentration of Ag–Au in GK‐(Ag–Au) NP was around 41.66 µg mL−1. The per cent conversion of Ag–Au ions to NPs was 53.88%.
3.4 MTT assay
This assay was used to assess the rate of tumour cell proliferation and the potential of GK‐(Ag–Au) NP in apoptosis induction. IC50 value was calculated to find the effectiveness of nanoparticles inhibiting the cell proliferation up to 50%. MTT assay was performed as per Wilson et al. protocol [33]. The half maximal inhibitory concentration (IC50) for GK‐(Ag–Au) NP with B16F10 cells was around 1.95 µg L−1 for 24 h (Fig. 5 a) and 1 µg L−1 for 48 h, respectively (Fig. 5 b).
Fig. 5.
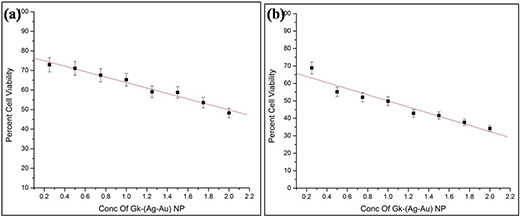
IC50 value of B16F10 cells treated with GK‐(Ag–Au) NP
(a) 24 h, (b) 48 h. At 24 h IC50 value was obtained at 1.95 µg L−1, whereas for 48 h it was achieved at 1 µg L−1
3.5 Flow cytometric studies
Flow cytometry experiments provide insight into the effect of nanoparticles on various phases of cell cycle and their influence on apoptosis. Cell cycle analysis suggests that in control samples, <70% of all cells accumulated in the G1 phase, <10% in the S phase, and <11% in the G2 phase. Only 9% of the cells are in the sub‐G1 phase (Fig. 6 a). B16F10 tumour cell lines on treatment with 5 µg L−1 of GK‐(Ag–Au) NPs, resulted in reduction of percentage of cells in G1, i.e. 62.58%, while S phase showed 14.06% and G2 phase showed 10.14% compared to untreated cells, while the percentage of cells in sub‐G1 phase increased to 10.47%, with cell cycle arrest between G1 and S phase, indicating enhanced DNA fragmentation and induction of apoptosis in tumour cells (Fig. 6 b).
Fig. 6.
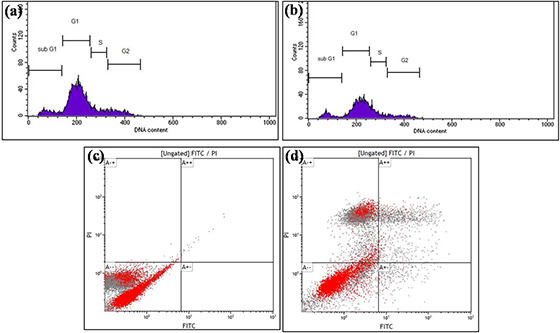
Cell cycle assay obtained with GK‐(Ag–Au)NPs
(a) Untreated, (b) Cells treated with 5 µg L−1 GK‐(Ag–Au)NPs for 48 h. GK‐(Ag–Au)NPs treated B16F10 cell lines were stained with propidium iodide (PI) and cellular DNA content frequency histograms were obtained. The histogram depicts that the subG1 cell population has been enhanced to about 10.47% on treating with GK‐AuNPs. Apoptotic assay with FACS analysis (c) Untreated, (d) Cells treated with 5 µg L−1 GK‐(Ag–Au) NP for 48 h. In case of apoptotic assay, the early apoptotic phase contained 1.77% of cells while in late apoptotic phase there were around 7.11% of cells on treatment with GK‐(Ag–Au) NP
In Annexin staining, the apoptotic effects of GK‐(Ag–Au) NP on cell cycle progression were studied by treating B16F10 cells with 5 µg L−1 of GK‐(Ag–Au) NP for 48 h. As illustrated in Fig. 6 c, apoptotic cells were not found in control cells (unstained). In early apoptotic phase only 0.00% of cell was present while in late apoptotic phase 0.09% of cells were present, whereas 0.18% cells were in death phase. In case of B16F10 cells treated with 5 µg L−1 of GK‐(Ag–Au) NP, the fraction of apoptotic cells has increased substantially. In early apoptotic phase 1.77% of cells were present while in late apoptotic phase there were around 7.11% of cells, while the cells in the death phase were around 36.52% (Fig. 6 d).
3.6 Apoptosis assay with fluorescence microscopy
The flow cytometric analysis clearly revealed that GK‐(Ag–Au) NP effectively induced apoptosis in B16F10 cells. To understand the action of GK‐(Ag–Au) NP, B16F10 cell lines were treated at their respective half maximal inhibitory concentration obtained at 48 h treatment (1 µg L−1) and at higher concentration (5 µg L−1) for 48 h period. GK‐(Ag–Au) NP treated cells viewed under the fluorescence microscope indicated high nuclear condensation and DNA fragmentation than in untreated cells. The cell surface was altered and shrunken, DNA fragmentation and condensation was enhanced, along with the increase in number of apoptotic cells (Fig. 7).
Fig. 7.
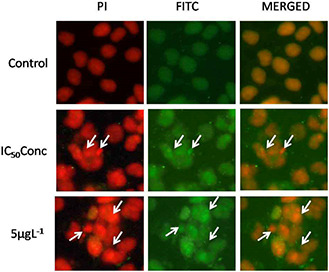
DNA fragmentation in B16F10 cells using fluorescence microscopy upon GK‐(Ag–Au) NP treatment (5 µg L−1) after 48 h. The fluorescence microscopy results confirm higher nuclear condensation and DNA fragmentation leading to apoptosis
3.7 Caspase 8 localisation using confocal microscopy
The sub cellular localisation of Caspase‐8 was carried out by confocal microscopy. In case of control B16F10 cells, Caspase‐8 was distributed throughout the cell in low concentration around the cytoplasm and nucleolus. Whereas in cells treated with GK‐(Ag–Au) NPs, Caspase‐8 was distributed more in the cytoplasmic region. In addition, the localisation of Caspase‐8 in nucleolus was very low when compared to the control cells (Fig. 8).
Fig. 8.
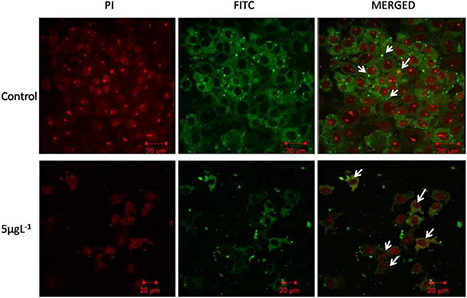
Localisation of caspase‐8 in B16F10 cells using confocal microscopy upon GK‐(Ag–Au) NP treatment (5 µg L−1) after 48 h. Overexpression of caspase‐8 revealed localisation at the cytoplasm in B16f10 cells after GK‐(Ag–Au) NP treatment
3.8 SEM‐EDAX of B16F10 cells treated with GK‐(Ag–Au) NPs
SEM pictures of B16F10 cells showed flaky and porous appearance on the surface. The EDAX spectrum of control cells showed the presence of Na+, O+ and C, whereas the cells treated with GK‐(Ag–Au) NPs showed the presence of both Ag+ and Au+ (Figs. 9 a and b).
Fig. 9.
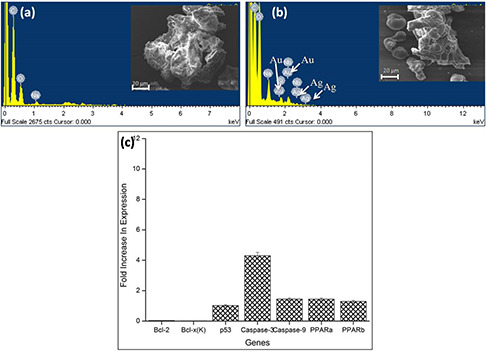
SEM‐EDAX analysis of B16F10 cells and expression status of apoptotic and anti‐apoptotic genes
(a) SEM‐EDAX analysis of control B16F10 cells, (b) B16F10 cells treated with GK‐(Ag–Au) NPs, (c) Expression status of different genes in B16F10 cells treated with 5 µg L−1 GK‐(Ag–Au) NP for 48 h
3.9 Gene expression profile
To study the molecular mechanism of apoptosis, the expression status of apoptotic and anti‐apoptotic genes in B16F10 cell lines at 5 µg L−1 concentration was analysed. In this study, cDNA level of apoptotic genes such as p53, caspase‐3, caspase‐9 was over expressed (Fig. 9 c). p53 was over expressed by 1.02‐fold. Caspase‐3 gene was over expressed by 4.3‐fold, whereas caspase‐9 was over expressed by 1.45‐fold, respectively. In case of anti‐apoptotic genes like Bcl‐2 and Bcl‐x(K) were down regulated by 0.035 and 0.01‐folds, whereas peroxisome proliferator‐activated receptor (PPAR) PPARa was 1.44‐fold over expressed while PPARb was 1.29‐fold over expressed in GK‐(Ag–Au) NP treated B16F10 cells when compared to control.
4 Discussion
In recent years, bionanomaterial has gained the attention of researchers, as they find enormous applications in the field of nanomedicine and tissue engineering. This study deals with the novel method for the synthesis of bimetallic silver–gold nanoparticle construct, employing GK, a natural biopolymer of plant origin. In the recent past, scientists have focused on the preparation of NPs using environmentally benign materials as reducing and stabilising agent such as Neem (Azadirachta indica) leaf broth [3], dextran [18], weed Antigonon leptopus [34], degraded pueraria starch [35], and fruit juice of pomegranate [35]. Hence, in this study, synthesis of silver–gold bimetallic nanoparticles was carried out using GK, a biopolymer which acts as both biological reducing and stabilising agent.
In this investigation, the time period experiments revealed that the absorbance of the bimetallic nanoparticles increased up to 30 min, whereas upon further increase in time point, there was a fall in the absorbance (Fig. 1 c), due to agglomerisation of metal nanoparticles. The physical stability of GK‐(Ag–Au) NP construct was assessed by studying the effect of temperature on the nanoparticles (Fig. 1 d). With increase in temperature from 25 to 95°C, there was no change in absorbance of nanoparticles. This can be attributed to the stability rendered by GK to the nanoparticles.
The FTIR results mainly confirm that hydroxyl, carboxylate, acetyl and C–O functional groups are mainly involved in the formation of the nanoparticles, which clearly indicate the chemical bonding between gum and bimetallic nanoparticles (Fig. 2 a). After autoclaving, a shift in absorbance peak was observed from 3442 to 3356 cm−1 with increased band intensity, signifying the binding of silver and gold ions with hydroxyl groups. Moreover, the band intensities of carboxylate groups were enhanced after autoclaving the gum, indicating oxidised nature of the gum. The band shift in the hydroxyl groups and enhanced band intensities for carbonyl groups in FTIR spectra confirm the oxidation of these functional groups during autoclaving [36]. The bands at 2934 cm−1 in gum disappear as the Au binds to –OH. The band at 1729 cm−1 corresponds to carbonyl group stretching. The stronger bands found at 1640 could be assigned to asymmetrical stretch of carboxylate group. While the bands at 1255 and 1046 cm−1 correspond to acetyl and C–O stretching [29]. FTIR data suggest that the carbonyl groups present in amino acids, peptides and proteins in gum have the ability to bind to metals and facilitate the formation of a coat over the nanoparticles and stabilise them against agglomeration.
The zeta potentials of GK‐(Ag–Au) NP composite were −24.3 mV, respectively, suggesting the stability of metal‐nanocomposite construct (Fig. 2 b). The surface morphology of the nanoparticles was assessed by SEM and AFM analysis which confirmed the presence of spherical‐shaped nanoparticles with smooth surface (Figs. 2c,d and 4a, b). The size of the nanoparticles where assessed by TEM which varied between 1 and 12 nm ±0.50 (Fig. 3 b).
The synthesised nanoparticle was assessed for anti‐proliferative activity on B16F10 cells by MTT assay, confocal microscopy, flow cytometry and real‐time PCR techniques. In MTT assay, the cells were treated with 0.25–2 µg L−1 concentrations of GK‐(Ag–Au) NPs for 24 and 48 h. The IC50 values for 24 h treatment were 1.95 µg L−1, while 48 h treatment showed IC50 concentration of 1 µg L−1 (Figs. 5 a and b). In apoptotic assay, the early apoptotic phase contained 1.77% of cells while in late apoptotic phase there were around 7.11% of cells (Figs. 6 c and d), which clearly indicates that B16F10 cells when treated with GK‐(Ag–Au) NP, inhibits DNA synthesis followed by induction of apoptosis in the tumour cells.
The fluorescence microscopy results revealed high nuclear condensation and DNA fragmentation leading to apoptosis of B16F10 cells treated with GK‐(Ag–Au) NP (Fig. 7), whereas confocal analysis of cells over‐expressing Caspase‐8 revealed localisation at the cytoplasm when the B16f10 cells were treated with GK‐(Ag–Au) NPs (Fig. 8).
qRT‐PCR assay is used to assess gene expression due to its sensitivity. Studies based on qRT‐PCR suggested that expression of apoptotic genes such as p53, caspase3, caspase9 were found to be up‐regulated leading to the apoptosis of the cells (Fig. 9 c) [29]. Up‐regulation of Casp9 transcripts expression was also reported in H9c2 (rat ventricular cell line) and HEK293 (human embryonic kidney cell line) [37], whereas caspase3 up‐regulation was observed in C2C12 (myoblast cell line) [38]. Caspase‐9 forms a multimeric complex with cytochrome c and Apaf‐1 to activate downstream caspases such as caspase‐3 leading to apoptosis mediated by mitochondria [39, 40, 41]. The GK‐(Ag–Au) NP is believed to be involved in cell signalling cascades with the activation of p53 and caspase‐9 which further activates caspase‐3 finally inhibiting cell division through intrinsic pathway. SEM‐EDAX results revealed that the cells treated with GK‐(Ag–Au) NPs showed the presence of both Ag+ and Au+ ions supporting the expression data (Figs. 9 a and b).
The expression of anti‐apoptotic genes such as Bcl‐2 and Bcl‐x(K) was also studied whose expression was down regulated on treatment with GK‐(Ag–Au) NPs [29]. Anti‐apoptotic genes such as Bcl‐2 and Bcl‐x(K) promote cell survival by maintaining the integrity of the external mitochondrial membrane and preventing the release of cytochrome c [42].
In addition, the present investigation also supports that genes such as PPARa and PPARb were up regulated in the cells treated with GK‐(Ag–Au) NPs. PPARs are transcription factors which influence molecular signalling in normal and cancer cells. Activation of PPARα inhibits metastatic potential, whereas activation of PPARγ decreases melanoma cell proliferation while PPARβ/δ activation inhibits melanoma cell proliferation [43].
The outcome of the experimental results entailed in this communication suggests that GK‐(Ag–Au) NP construct induces cytotoxic effects on B16F10 cells, inhibiting their proliferation.
5 Conclusion
This work reveals that the GK has tremendous potential for the synthesis of GK‐(Ag–Au) NP construct. The morphology of the nanoparticles was analysed by SEM, TEM and AFM analysis. The anti‐proliferative efficacy was assessed in B16F10 cell line model by MTT, FACS and confocal microscopy. The qRT‐PCR analysis revealed the up‐regulation of apoptotic genes (p53, caspase‐3 and caspase‐9) followed by the down‐regulation of anti‐apoptotic genes (Bcl‐2, Bcl‐x(K)). The overall results indicated that the GK‐(Ag–Au) NP construct has potential anti‐proliferative activity through induction of apoptosis.
6 Acknowledgments
The authors acknowledge Department of Science & Technology, New Delhi for providing the infra‐structure facilities required for the research work under DST‐PURSE‐II programme, Osmania University. Ms. S. Kalaignana Selvi also acknowledges University Grants Commission, New Delhi, for the award of BSR Fellowship (370/VRD/HOD/BIOCHEM/OU/2015), at the Department of Biochemistry, UCS, Osmania University. We thank Dr. M.J. Mahesh Kumar, Principal Scientist at Centre for Cellular and Molecular Biology for his constant support for this work.
7 References
- 1. Iravani S.: ‘Green synthesis of metal nanoparticles using plants’, Green Chem., 2011, 13, (10), pp. 2638 –2650 [Google Scholar]
- 2. Mittal A.K. Chisti Y. Banerjee U.C.: ‘Synthesis of metallic nanoparticles using plant extracts’, Biotechnol. Adv., 2013, 31, (2), pp. 346 –356 [DOI] [PubMed] [Google Scholar]
- 3. Shankar S.S. Rai A. Ahmad A. et al.: ‘Rapid synthesis of Au, Ag, and bimetallic Au core–Ag shell nanoparticles using neem (Azadirachta Indica) leaf broth’, J. Colloid Interface Sci., 2004, 275, (2), pp. 496 –502 [DOI] [PubMed] [Google Scholar]
- 4. Sastry M. Ahmad A. Khan M.I. et al.: ‘Biosynthesis of metal nanoparticles using fungi and actinomycete’, Curr. Sci., 2003, 85, (2), pp. 162 –170 [Google Scholar]
- 5. Ahmad A. Mukherjee P. Senapati S. et al.: ‘Extracellular biosynthesis of silver nanoparticles using the fungus Fusarium oxysporum’, Colloids Surf. B Biointerfaces, 2003, 28, (4), pp. 313 –318 [Google Scholar]
- 6. He S. Guo Z. Zhang Y. et al.: ‘Biosynthesis of gold nanoparticles using the bacteria Rhodopseudomonas Capsulata’, Mater. Lett., 2007, 61, (18), pp. 3984 –3987 [Google Scholar]
- 7. Vinod V. Saravanan P. Sreedhar B. et al.: ‘A facile synthesis and characterization of Ag, Au and Pt nanoparticles using a natural hydrocolloid Gum kondagogu (Cochlospermum gossypium)’, Colloids Surf. B Biointerfaces, 2011, 83, (2), pp. 291 –298 [DOI] [PubMed] [Google Scholar]
- 8. Sheny D. Mathew J. Philip D.: ‘Phytosynthesis of Au, Ag and Au–Ag bimetallic nanoparticles using aqueous extract and dried leaf of Anacardium Occidentale’, Spectrochim. Acta A, 2011, 79, (1), pp. 254 –262 [DOI] [PubMed] [Google Scholar]
- 9. AbdelHamid A.A. Al‐Ghobashy M.A. Fawzy M. et al.: ‘Phytosynthesis of Au, Ag, and Au–Ag bimetallic nanoparticles using aqueous extract of sago pondweed (Potamogeton Pectinatus L.)’, ACS Sustain. Chem. Eng., 2013, 1, (12), pp. 1520 –1529 [Google Scholar]
- 10. Gopinath K. Kumaraguru S. Bhakyaraj K. et al.: ‘Green synthesis of silver, gold and silver/gold bimetallic nanoparticles using the Gloriosa Superba leaf extract and their antibacterial and antibiofilm activities’, Microb. Pathog., 2016, 101, pp. 1 –11 [DOI] [PubMed] [Google Scholar]
- 11. Cortie M.B. McDonagh A.M.: ‘Synthesis and optical properties of hybrid and alloy plasmonic nanoparticles’, Chem. Rev., 2011, 111, (6), pp. 3713 –3735 [DOI] [PubMed] [Google Scholar]
- 12. Li Q. Li H. Pol V. et al.: ‘Sonochemical synthesis, structural and magnetic properties of air‐stable Fe/Co alloy nanoparticles’, New J. Chem., 2003, 27, (8), pp. 1194 –1199 [Google Scholar]
- 13. Peng Z. Yang H.: ‘Designer platinum nanoparticles: control of shape, composition in alloy, nanostructure and electrocatalytic property’, Nano Today, 2009, 4, (2), pp. 143 –164 [Google Scholar]
- 14. Link S. Wang Z.L. El‐Sayed M.: ‘Alloy formation of gold‐silver nanoparticles and the dependence of the plasmon absorption on their composition’, J. Phys. Chem. B, 1999, 103, (18), pp. 3529 –3533 [Google Scholar]
- 15. Tamuly C. Hazarika M. Borah S.C. et al.: ‘In situ biosynthesis of Ag, Au and bimetallic nanoparticles using Piper Pedicellatum C. Dc: green chemistry approach’, Colloids Surf. B Biointerfaces, 2013, 102, pp. 627 –634 [DOI] [PubMed] [Google Scholar]
- 16. Mondal S. Roy N. Laskar R.A. et al.: ‘Biogenic synthesis of Ag, Au and bimetallic Au/Ag alloy nanoparticles using aqueous extract of mahogany (Swietenia mahogani Jacq.) leaves’, Colloids Surf. B Biointerfaces, 2011, 82, (2), pp. 497 –504 [DOI] [PubMed] [Google Scholar]
- 17. Kumari M.M. Jacob J. Philip D.: ‘Green synthesis and applications of Au–Ag bimetallic nanoparticles’, Spectrochim. Acta A, 2015, 137, pp. 185 –192 [DOI] [PubMed] [Google Scholar]
- 18. Bankura K. Maity D. Mollick M.M.R. et al.: ‘Antibacterial activity of Ag–Au alloy NPs and chemical sensor property of Au Nps synthesized by dextran’, Carbohydr. Polym., 2014, 107, pp. 151 –157 [DOI] [PubMed] [Google Scholar]
- 19. Raveendran P. Fu J. Wallen S.L.: ‘A simple and ‘green’ method for the synthesis of Au, Ag, and Au–Ag alloy nanoparticles’, Green Chem., 2006, 8, (1), pp. 34 –38 [Google Scholar]
- 20. Sawle B.D. Salimath B. Deshpande R. et al.: ‘Biosynthesis and stabilization of Au and Au–Ag alloy nanoparticles by fungus, Fusarium semitectum’, Sci. Technol. Adv. Mater., 2008, 9, (3), p. 035012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. El‐Naggar M.E. Shaheen T.I. Fouda M.M. et al.: ‘Eco‐friendly microwave‐assisted green and rapid synthesis of well‐stabilized gold and core–shell silver–gold nanoparticles’, Carbohydr. Polym., 2016, 136, pp. 1128 –1136 [DOI] [PubMed] [Google Scholar]
- 22. Chtchigrovsky M.L. Lin Y. Ouchaou K. et al.: ‘Dramatic effect of the gelling cation on the catalytic performances of alginate‐supported palladium nanoparticles for the Suzuki–Miyaura reaction’, Chem. Mater., 2012, 24, (8), pp. 1505 –1510 [Google Scholar]
- 23. Breitwieser D. Spirk S. Fasl H. et al.: ‘Design of simultaneous antimicrobial and anticoagulant surfaces based on nanoparticles and polysaccharides’, J. Mater. Chem. B, 2013, 1, (15), pp. 2022 –2030 [DOI] [PubMed] [Google Scholar]
- 24. Ehmann H.M. Breitwieser D. Winter S. et al.: ‘Gold nanoparticles in the engineering of antibacterial and anticoagulant surfaces’, Carbohydr. Polym., 2015, 117, pp. 34 –42 [DOI] [PubMed] [Google Scholar]
- 25. Pasqui D. Golini L. Giovampaola C.D. et al.: ‘Chemical and biological properties of polysaccharide‐coated titania nanoparticles: the key role of proteins’, Biomacromolecules, 2011, 12, (4), pp. 1243 –1249 [DOI] [PubMed] [Google Scholar]
- 26. Stevens K.N. Croes S. Boersma R.S. et al.: ‘Hydrophilic surface coatings with embedded biocidal silver nanoparticles and sodium heparin for central venous catheters’, Biomaterials, 2011, 32, (5), pp. 1264 –1269 [DOI] [PubMed] [Google Scholar]
- 27. Croes S. Stobberingh E.E. Stevens K.N. et al.: ‘Antimicrobial and anti‐thrombogenic features combined in hydrophilic surface coatings for skin‐penetrating catheters. Synergy of Co‐embedded silver particles and heparin’, ACS Appl. Mater. Interfaces, 2011, 3, (7), pp. 2543 –2550 [DOI] [PubMed] [Google Scholar]
- 28. Reishofer D. Ehmann H.M. Amenitsch H. et al.: ‘On the formation of Bi2s3‐cellulose nanocomposite films from bismuth xanthates and trimethylsilyl‐cellulose’, Carbohydr. Polym., 2017, 164, pp. 294 –300 [DOI] [PubMed] [Google Scholar]
- 29. Selvi S.K. Kumar J.M. Sashidhar R.: ‘Anti‐proliferative activity of Gum kondagogu (Cochlospermum gossypium)‐gold nanoparticle constructs on B16f10 melanoma cells: an in vitro model’, Bioactive Carbohydrates Dietary Fibre, 2017, 11, pp. 38 –47 [Google Scholar]
- 30. Sashidhar R. Selvi S.K. Vinod V. et al.: ‘Bioprospecting of Gum kondagogu (Cochlospermum gossypium) for bioremediation of uranium (Vi) from aqueous solution and synthetic nuclear power reactor effluents’, J. Environ. Radioact., 2015, 148, pp. 33 –41 [DOI] [PubMed] [Google Scholar]
- 31. Kumar J.M. Idris M.M. Srinivas G. et al.: ‘Phenyl 1, 2, 3‐triazole‐thymidine ligands stabilize G‐quadruplex DNA, inhibit DNA synthesis and potentially reduce tumor cell proliferation over 3′‐azido deoxythymidine’, PLoS ONE, 2013, 8, (8), p. e70798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nagaonkar D. Rai M.: ‘Sequentially reduced biogenic silver‐gold nanoparticles with enhanced antimicrobial potential over silver and gold monometallic nanoparticles’, Adv. Mater. Lett., 2015, 6, (4), pp. 334 –341 [Google Scholar]
- 33. Wilson J. Sargent J. Elgie A. et al.: ‘A feasibility study of the MTT assay for chemosensitivity testing in ovarian malignancy’, Br. J. Cancer, 1990, 62, (2), p. 189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ganaie S. Abbasi T. Abbasi S.: ‘Rapid and green synthesis of bimetallic Au–Ag nanoparticles using an otherwise worthless weed Antigonon leptopus’, J. Exp. Nanosci., 2016, 11, (6), pp. 395 –417 [Google Scholar]
- 35. Xia B. He F. Li L.: ‘Preparation of bimetallic nanoparticles using a facile green synthesis method and their application’, Langmuir, 2013, 29, (15), pp. 4901 –4907 [DOI] [PubMed] [Google Scholar]
- 36. Kora A.J. Sashidhar R. Arunachalam J.: ‘Gum kondagogu (Cochlospermum gossypium): a template for the green synthesis and stabilization of silver nanoparticles with antibacterial application’, Carbohydr. Polym., 2010, 82, (3), pp. 670 –679 [Google Scholar]
- 37. Huang J. Li Q. Sun D. et al.: ‘Biosynthesis of silver and gold nanoparticles by novel sundried Cinnamomum camphora leaf’, Nanotechnology, 2007, 18, (10), p. 105104 [Google Scholar]
- 38. Wahab R. Dwivedi S. Khan F. et al.: ‘Statistical analysis of gold nanoparticle‐induced oxidative stress and apoptosis in myoblast (C2c12) cells’, Colloids Surf. B, Biointerfaces, 2014, 123, pp. 664 –672 [DOI] [PubMed] [Google Scholar]
- 39. Han Y. Chen Y.‐S. Liu Z. et al.: ‘Overexpression of Hax‐1 protects cardiac myocytes from apoptosis through caspase‐9 inhibition’, Circ. Res., 2006, 99, (4), pp. 415 –423 [DOI] [PubMed] [Google Scholar]
- 40. Bialik S. Cryns V.L. Drincic A. et al.: ‘The mitochondrial apoptotic pathway is activated by serum and glucose deprivation in cardiac myocytes’, Circ. Res., 1999, 85, (5), pp. 403 –414 [DOI] [PubMed] [Google Scholar]
- 41. Kannan K. Jain S.K.: ‘Oxidative stress and apoptosis’, Pathophysiology., 2000, 7, (3), pp. 153 –163 [DOI] [PubMed] [Google Scholar]
- 42. Zhou J.‐Y. Li Y. Zou X. et al.: ‘Hederagenin from the leaves of Ivy (Hedera Helix L.) induces apoptosis in human Lovo colon cells through the mitochondrial pathway’, BMC Complement. Altern. Med., 2014, 14, (1), p. 412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Michiels J.‐F. Perrin C. Leccia N. et al.: ‘Pparβ activation inhibits melanoma cell proliferation involving repression of the Wilms’ tumour suppressor Wt1’, Pflügers Archiv., 2010, 459, (5), pp. 689 –703 [DOI] [PMC free article] [PubMed] [Google Scholar]