

Abstract
Present investigation aimed to prepare, optimise, and characterise lipid nanocapsules (LNCs) for improving the solubility and bioavailability of efavirenz (EFV). EFV‐loaded LNCs were prepared by the phase‐inversion temperature method and the influence of various formulation variables was assessed using Box–Behnken design. The prepared formulations were characterised for particle size, polydispersity index (PdI), zeta potential, encapsulation efficiency (EE), and release efficiency (RE). The biocompatibility of optimised formulation on Caco‐2 cells was determined using 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide assay. Then, it was subjected to ex‐vivo permeation using rat intestine. EFV‐loaded LNCs were found to be spherical shape in the range of 20–100 nm with EE of 82–97%. The best results obtained from LNCs prepared by 17.5% labrafac and 10% solutol HS15 when the volume ratio of the diluting aqueous phase to the initial emulsion was 3.5. The mean particle size, zeta potential, PdI, EE, drug loading%, and RE during 144 h of optimised formulation were confirmed to 60.71 nm, −35.93 mV, 0.09, 92.60, 7.39 and 55.96%, respectively. Optimised LNCs increased the ex vivo intestinal permeation of EFV when compared with drug suspension. Thus, LNCs could be promising for improved oral delivery of EFV.
Inspec keywords: biomedical materials, solubility, drugs, encapsulation, emulsions, nanoparticles, particle size, nanofabrication, suspensions, toxicology, nanomedicine, cellular biophysics, lipid bilayers, electrokinetic effects, drug delivery systems, molecular biophysics
Other keywords: ex‐vivo permeation; diluting aqueous phase; mean particle size; zeta potential; drug loading; optimised formulation; ex vivo intestinal permeation; improved oral delivery; efavirenz oral delivery; optimisation; ex‐vivo gut permeation study; solubility; bioavailability; phase‐inversion temperature method; formulation variables; Box–Behnken design; polydispersity index; encapsulation efficiency; Caco‐2 cells; lipid nanocapsules; 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide assay; EFV‐loaded LNC; drug suspension; size 20.0 nm to 100.0 nm; time 144.0 hour; size 60.71 nm; voltage ‐35.93 mV
1 Introduction
The human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome (AIDS) is a global health problem and one of the most destructive epidemics that caused about 35 million deaths since it was first discovered and continues to grow. At the end of 2016, an estimated 36.7 (30.8–42.9) million people were living with HIV [1]. Highly active antiretroviral therapy (HAART) is the standard treatment of HIV that includes a combination of at least three drugs [2]. HAART has the potential both to decrease mortality and morbidity rates among HIV/AIDS patients but high dose, complex administration schedules, drug side effects, reduced patient compliance, and high cost of treatment have led to a failure of HIV/AIDS therapy [3]. In addition, HIV is known to localise and proliferate in certain unavailable compartments of the body such as lymphatic system and central nervous system (CNS) where the majority of drug cannot be reached in sufficient concentration and retained for the necessary duration to cause therapeutic response [3, 4]. This causes failure to eliminate HIV from these reservoirs and also the development of multi‐drug resistance. Efavirenz (EFV) is non‐nucleoside reverse transcriptase inhibitor and used orally as a part of HAART for the initial treatment of HIV infection. It is also used as the first‐line therapy for children HIV infection. EFV is practically insoluble in water (4 µg/ml) and belongs to class II pharmaceuticals. The low aqueous solubility and extensive first pass metabolism of EFV leads to low (40–45%) oral bioavailability and high inter‐individual (55–58%) and intra‐individual (19–24%) variability in its absorption [5, 6]. High dose and frequent administration of EFV led to several serious side effects including neuropsychiatric disturbances, hepatotoxicity, and metabolic alterations [7]. Furthermore, EFV causes burning mouth syndrome after swallowing which is a limit in development of water‐based liquid formulation [5]. Thus, there is an urgent need to develop an effective drug delivery not only to improve EFV oral bioavailability and reduce needed dose but also to achieve optimum EFV concentration in HIV localised sites in the body. Recent advances in the field of drug delivery demonstrated that nanoparticles (NPs) have a real potential of therapeutic development. Lipid‐based nanocarriers including liposomes, lipid nano‐capsules (LNCs), nano structured lipid carriers (NLCs), and solid lipid NPs (SLNs) are considered as most promising drug delivery systems due to bio‐acceptable, biodegradable nature of these systems, and their ability to enhance solubility of hydrophobic drug, control drug release, increase the stability of the entrapped drug and improve drug bioavailability [8, 9]. Among these, LNCs have been extensively explored as particulate carriers in pharmaceutical and medical sciences. LNCs are spherical vesicular systems with capability for loading of both hydrophobic and hydrophilic drugs. LNCs consists of oily core of labrafac (medium chain triglyceride), coated with a semi‐rigid shell. This shell composed of solutol® HS15 (free polyethylene glycol (PEG) and hydroxy stearate of PEG) in its outer part and lecithin in its inner part. LNCs can be produced on a large industrial scale using solvent free, low energy technique based on the phase inversion process of the emulsion. Their mean size ranging between 20 and 100 nm and can be adjusted by modifying the different parameters involved in the process [10, 11, 12]. Compared with liposomes and/or SLNs, LNCs have higher drug loading (DL) capacity and a longer physical stability (up to 18 months) [13]. LNC formulations need considerably less amount of raw materials than polymeric NPs. In addition, compared with polymeric NPs and liposomes, drug release from the LNCs can be more sustained [14]. LNCs have shown great potential for oral administration drugs with limited oral bioavailability [15, 16, 17, 18]. In studies conducted by Roger et al. [19] and Zhai et al. [20], the stability of LNCs in gastric medium was demonstrated. Groo et al. [21] have also shown stability and diffusion of LNCs in intestinal mucus. In further study, Roger et al. [18] demonstrated the integrity of carrier after crossing intestinal cells using Förster resonance energy transfer technique. Nanoparticulate‐based carriers are also capable of performing specific transport of drugs to intestinal lymphatic through targeting M cells of Peyer patches [22]. Intestinal lymphatic uptake can bypass a hepatic portal route. This can increase the oral bioavailability of drugs that undergo extensive first pass metabolism [4]. In the study conducted by Peltier et al. [15], LNCs enhanced oral bioavailability of paclitaxel (PTX). This finding was explained with different mechanisms such as inhibition of P‐glycoprotein (P‐gp), increased intestinal lymphatic transport, and improved solubilisation. LNCs also have been widely studied for cerebral delivery of different drugs for their ability to enter glioma cells. Solutol HS 15, as one of the constituents of LNCs with P‐gp inhibiting properties can inhibit P‐gp related drug transport [14, 23]. These allow drug penetration more effectively into CNS which acts as a HIV reservoir. In this study, aiming to increase water solubility and oral absorption of EFV, EFV‐loaded LNCs were developed by the phase inversion method. The effects of various parameters on the physicochemical characteristics of LNCs were subsequently evaluated. Finally, ex‐vivo permeation studies of the optimised formulation on rats were carried out using the non‐everted gut sac technique.
2 Materials and methods
2.1 Materials
EFV was kindly provided by Bakhtar Bioshimi Co. and labrafac (capric and caprylic acid triglyceride) was provided by BASF (Ludwigshafen, Germany). Solutol HS 15 and soy lecithin S100 (Lipoid) and dialysis bag (molecular weight cut off 12,000 Da) were provided by Sigma (USA). For a cell culture study, Caco2 cell line was kindly provided by the Iranian Biological Research Center (Iran). 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide (MTT) was purchased from Sigma Company (USA). Trypsin, fetal bovine serum (FBS), phosphate buffer saline (PBS), Dulbecco's modified Eagle medium (DMEM), penicillin, and streptomycin were purchased from Gibco Laboratories (USA).
2.2 Preparation of EFV‐loaded LNCs
The blank and drug‐loaded LNCs were manufactured by following the phase‐inversion temperature method as described previously [10, 11, 12]. To prepare the initial mass of 2 g of LNCs, 50 mg of drug was dissolved in labrafac (10–25%) containing lecithin (1.5%) as a stabilising agent with a magnetic stirrer. Then the aqueous phase containing 1.75% NaCl and different amounts of Solutol HS 15 (10–40%) as the surfactant was added. The amount of each variable is shown in Table 1. The emulsion was subjected to repeated heating and cooling cycles to reach the inversion process (85–60–85–60–85°C). Then at 70°C, an irreversible shock induced by sudden dilution (1.2–3.5 times) with cold deionised water (0°C) was observed. This fast‐cooling dilution process led to the breaking of the microemulsion system and the formation of stable LNCs. Afterwards slow magnetic stirring was applied to cool down the suspension for 5 min.
Table 1.
Variables used in Box–Behnken experimental design
| Levels | ||||
|---|---|---|---|---|
| Independent variables | −1 | 0 | 1 | Dependent variables |
| X 1 = oil phase % (O) | 10 | 17.5 | 25 | Y 1 = particle size, nm |
| X 2 = surfactant % (S) | 10 | 25 | 40 | Y 2 = polydispersity index |
| X 3 = volume ratio of the diluting aqueous phase to the initial emulsion, W | 1.2 | 2.35 | 3.5 | Y 3 = zeta potential, mV |
| Y 4 = EE, % | ||||
| Y 5 = DL, % | ||||
| Y 6 = RE, % | ||||
2.3 Experimental design
Our preliminary experiments and other studies showed labrafac (oil) %, solutol HS 15 (surfactant) % and the volume ratio of the diluting aqueous phase to the initial emulsion mostly influenced the preparation of LNCs. To examine these three factors and the relations between the independent factor and their responses as well as interaction, Box–Behnken design as the statistical and mathematical method was used. All studied parameters had three levels and all experiments were performed in triplicate. Studied variables and their levels used in preparation of EFV‐loaded LNCs are shown in Table 1. Box–Behnken designs 17 experimental runs including 12 factorial points with five replicates at the centre point to calculate the pure error sum of squares (Table 2). Dependent parameters including particle size, encapsulation efficiency (EE) %, DL%, zeta potential, polydispersity index (PdI) and release efficiency 144 h (RE144 %) were analysed using design expert software (version 10, USA). To determine each factor was statistically significant, analysis of variance (ANOVA) was performed.
Table 2.
Composition of different studied EFV‐loaded LNCs using Box–Behnken experimental design
| Formulations | Labrafac, % | Solutol HS 15, % | Volume ratio of diluting aqueous phase to initial emulsion |
|---|---|---|---|
| L10 S10 W2.35 | 10 | 10 | 2.35 |
| L25 S10 W2.35 | 25 | 10 | 2.35 |
| L10 S40 W2.35 | 10 | 40 | 2.35 |
| L25 S40 W2.35 | 25 | 40 | 2.35 |
| L10 S25 W1.2 | 10 | 25 | 1.2 |
| L25 S25 W1.2 | 25 | 25 | 1.2 |
| L10 S25 W3.5 | 10 | 25 | 3.5 |
| L25 S25 W3.5 | 25 | 25 | 3.5 |
| L17.5 S10 W1.2 | 17.5 | 10 | 1.2 |
| L17.5 S40 W1.2 | 17.5 | 40 | 1.2 |
| L17.5 S10 W3.5 | 17.5 | 10 | 3.5 |
| L17.5 S40 W3.5 | 17.5 | 40 | 3.5 |
| L17.5 S25 W2.35 | 17.5 | 25 | 2.35 |
| L17.5 S25 W2.35 | 17.5 | 25 | 2.35 |
| L17.5 S25 W2.35 | 17.5 | 25 | 2.35 |
| L17.5 S25 W2.35 | 17.5 | 25 | 2.35 |
| L17.5 S25 W2.35 | 17.5 | 25 | 2.35 |
L, labrafac; S, solutol HS15; W, volume ratio of diluting aqueous phase to initial emulsion.
2.4 Determination of particle size, PdI, and zeta potential of LNCs
Particle size, PdI, and zeta potential of LNCs were determined by using a zeta sizer (PCS, Zeta sizer 3000, Malvern, UK). All formulations were diluted 20 times with deionised water before analysis. Each test was performed in triplicate.
2.5 Determination of EE and DL%
EFV encapsulation was determined by an indirect method. 0.5 ml of each LNC formulation was centrifuged (Sigma 3K30, Germany) using a microcentrifuge filter tube (Amicon Ultra, Ireland, cut off 10 kDa) at 14,000 rpm for 10 min and EFV concentration in the supernatant was determined using the ultraviolet (UV) spectrophotometer method at 257 nm. Plain LNCs without EFV were used as blank samples. The EE and DL % of EFV in LNCs were calculated using the following equations:
| (1) |
| (2) |
2.6 Scanning electron microscopy (SEM)
The morphology of optimised EFV‐loaded LNCs was evaluated using electron microscopy. For performing field emission SEM (HITACHI S‐4160, Japan), freshly prepared dispersion of LNCs deposited on aluminium stubs, sputter‐coated with a thin layer of Au/Pd and characterised by SEM.
2.7 In vitro release of EFV from LNCs
1 ml of each formulation was filled in the dialysis bag (molecular weight cut off 12,000 Da) and then, the bag was immersed in a glass tube containing an appropriate amount of PBS with 0.5% tween 80 at 37°C. At a predetermined time, 1 ml of release medium was removed and refreshed with new medium. Then the content of drug in release medium was determined spectrophotometrically at 257 nm. The parameter of RE144 % was used to compare the release profile and calculated by using the following equation:
| (3) |
where y is the released per cent at time t.
2.8 Study the kinetic of drug release
To study the release kinetics, EFV release data obtained from optimised formulation was studied with different kinetic models including Higuchi (Qt /Q ∞ = kt 1/2), Hixson Crowell (Q 01/3 − Q 1/3 = kt), first order (Ln(1 − Qt /Q ∞) = −kt), zero order (Qt /Q ∞ = kt), Baker–Lonsdale [1 − (1 − Qt /Q ∞)2/3] − Qt /Q ∞ and Korsmeier–Peppas model (Qt /Q ∞ = ktn ), where Q 0 is the initial amount of drug in LNCs, Qt is the amount of drug released at time t, Q is the remaining amount of drug in the system, ∞ is the total amount of drug loaded in LNCs that can be released after infinite time, k is the release constant and n is the release exponent used to describe the drug release mechanism. The model with best fits was selected based on the highest correlation coefficient (R 2). To study the mechanism of drug release, release exponent (n) was determined. For spherical samples, n < 0.43 indicates the drug release controlled by Fickian diffusion. When 0.43<n < 0.85, anomalous transport is the dominant mechanism of drug release. If n value is close to 0.85, the release is described as case II transport.
2.9 Fourier‐transform infrared (FTIR) spectroscopy
FTIR spectroscopy (FTIR‐8400S, Shimadzu, Japan) was performed to understand if there is any possible interaction between EFV and excipients. The FTIR spectra were scanned in the infrared range from 400 to 4000 cm−1 using the KBr Pellet method.
2.10 Storage stability study of EFV‐loaded optimised LNC dispersion
To study the storage stability, the aqueous dispersion of the optimised formulation loaded with EFV was stored at 4°C in a refrigerator for 2 weeks. After this time, the samples were measured to determine the particle size, PdI, and zeta potential as noted above.
2.11 Freeze‐drying of LNCs
Optimised EFV‐loaded LNCs were freeze dried using different kinds of cryoprotectants including PEG 4000, sorbitol, sucrose, aerosil, and lactose in the range of 1–5% concentration. The samples were frozen by placing the glass vials for 24 h in a refrigerator at −20°C, transferred to a freeze‐dryer (Christ, Alpha 2–4 LD plus, Germany). Sublimation lasted 48 h at a temperature of −40°C and pressure of 0.001 bar. After freeze drying, the samples were rehydrated with the same volume of water lost after lyophilisation and evaluated their appearance and reconstitution time. The re‐dispersion of freeze dried products was studied by manual shaking in a small glass vial with deionised water. Then particle size, PdI and zeta potential were assessed as described above.
2.12 In vitro cytotoxicity assay
Caco2 cells were originally obtained from the Iranian Biological Research Center and grown in DMEM containing 10% FBS and 1% penicillin–streptomycin at 37°C in a humidified atmosphere containing 5% CO2. The tolerability effects of blank and EFV‐loaded LNCs were evaluated by MTT colorimetric assay. Cells were seeded in 96‐well flat‐bottomed plates at a density of 10 × 103 cells/well for 24 h. Then, the cells were incubated with EFV‐loaded LNCs for 4 and 8 h at 37°C at an equivalent EFV concentration from 10 to 500 μg/ml. Blank LNCs were also tested in equal concentrations as drug‐loaded LNCs on Caco2 cells. At the end of the incubation time, the medium was removed and 20 µl of MTT solution (5 mg/ml in PBS) was added into each well and left to incubate for further 3 h at 37°C. Finally, the media were removed and the intracellular formazon was dissolved in dimethyl sulphoxide. To measure cell viability, the absorbance was determined at 570 nm using a microplate reader. The result is expressed as the cell viability per cent.
2.13 Ex vivo permeation of EFV from the optimised LNCs
A non‐everted rat gut sac method was used to evaluate ex vivo permeation of EFV from its suspension and the optimised LNC formulation containing an equivalent amount of drug [24, 25]. Male Wistar rats weighing 200–250 g were used in this study. All animal procedures were approved by the animal study committee of Isfahan University of Medical Sciences. Animals were fasted for 24 h before test. On the experiment day, rats were anesthetised using excessive ether inhalation. After midline laparotomy, the whole of the small intestine was identified and removed from animal body. A length of 7 cm from jejunum region was separated and was carefully washed with cold Kreb's Ringer phosphate buffer (KRPB) using a syringe equipped with a blunt and then placed inside the KRPB solution oxygenated with O2 /CO2 (95%/5%) at 37°C. An appropriate amount of EFV suspension or the selected EFV LNC suspension (equivalent to 1 mg of EFV) was filled in each sac using a 2 ml syringe. The both ends of the intestine were tied with a cotton tread and immersed in 50 ml of the aerated (15 bubbles/min) KRPB solution containing 0.5% tween 80 and was stirred at 400 rpm at 37 ± 0.5°C. At certain time intervals, the intestinal bags were placed in equal volumes of fresh KRPB. The removed solution was freeze‐dried and then dissolved in ethanol. Then, the amount of EFV transported through the intestinal segments was determined using the UV spectroscopy method at 254 nm. The permeation flux (μg/min cm2), apparent permeability (Papp) and per cent drug permeation in the receptor compartment were calculated. The Papp, expressed in cm/min, was calculated using the following equation:
| (4) |
where is the drug permeation rate, Co is the initial EFV concentration in the mucosal compartment at t = 0 and A is the total cross‐sectional area of tissue (2 ᴨrh). Permeation flux was measured by taking the slope of the linear portion of the graph plot of cumulative amount of EFV permeated through the membrane per unit area (μg/cm2) against time (min).
3 Result and discussion
The nature and ratio of various excipients play a major role in formulation results [11]. LNCs consist of oily core coated with a semi‐rigid shell. Triglycerides of capric and caprylic acids known under the commercial name of labrafac are commonly used in the oily phase for preparation of LNCs. The shells of LNCs are composed of solutol HS15 in its outer part and lecithin in its inner part. Solutol HS15 is a mixture of free PEG 660 and PEG 660 hydroxystearate and was used in the preparation because of its main role in LNC formation and stability. The hydrophilic‐lipophilic balance of solutol HS 15 is altered with temperature, becoming less hydrophilic on heating. As a consequence, the phase inversion method is performed due to the capability of solutol HS 15 to alter its affinity to water and oil as the function of temperature. Lecithin is usually used in small proportion to stabilise the LNCs’ rigid shell and to enhance the biocompatibility with the biological membranes. Sodium chloride in the aqueous phase decreases the phase inversion temperature of solutol HS15 to easily achievable levels and allows the loading of a heat‐sensitive drug. All constituents of LNCs are generally recognised as safe excipients and approved by the Food and Drug Administration for oral, topical, and parenteral administration [10, 11, 12, 13]. By using the phase inversion technique, several formulation variables including labrafac (%), solutol HS15 (%), and the volume ratio of the diluting aqueous phase to the initial emulsion were assessed in order to obtain the optimal preparation technique. According to Table 2, different formulations of EFV‐loaded LNCs were prepared and the basic properties of as‐prepared formulations including particle size, zeta potential, EE%, DL%, PdI, and RE144 % were determined (Table 3). After analysis of results using ANOVA, the best fit model suggested by Design Expert 10.0.1 software for the particle size, zeta potential, DL%, PdI, and RE144 % was a quadratic model. On the other hand, the 2FI model was suggested for EE%. The mathematical relation between responses and independent factors was studied using second‐order polynomial functions:
| (5) |
Table 3.
Physicochemical characteristics of LNCs loaded with EFV
| Formulations | Particle size, nm | PdI | Zeta potential, mV | EE, % | DL, % | RE, % |
|---|---|---|---|---|---|---|
| L10 S10 W2.35 | 43.46 ± 0.65 | 0.14 ± 0.02 | −23.14 ± 2.06 | 97.07 ± 0.77 | 10.14 ± 0.08 | 38.15 ± 1.83 |
| L25 S10 W2.35 | 101.10 ± 1.22 | 0.11 ± 0.00 | −31.46 ± 0.51 | 83.18 ± 0.83 | 5.40 ± 0.05 | 71.54 ± 1.70 |
| L10 S40 W2.35 | 22.13 ± 0.65 | 0.28 ± 0.03 | −21.23 ± 0.80 | 94.29 ± 0.22 | 4.37 ± 0.01 | 56.06 ± 1.26 |
| L25 S40 W2.35 | 30.9 ± 0.71 | 0.10 ± 0.01 | −16.40 ± 3.55 | 91.58 ± 0.03 | 3.30 ± 00 | 37.43 ± 3.01 |
| L10 S25 W1.2 | 34.53 ± 5.28 | 0.21 ± 0.08 | −18.37 ± 2.73 | 82.68 ± 0.20 | 5.36 ± 0.01 | 27.13 ± 2.78 |
| L25 S25 W1.2 | 44.64 ± 0.99 | 0.07 ± 0.00 | −12.86 ± 0.49 | 91.46 ± 0.16 | 4.25 ± 0.00 | 37.99 ± 2.77 |
| L10 S25 W3.5 | 34.00 ± 3.46 | 0.22 ± 0.01 | −15.96 ± 0.87 | 97.72 ± 0.59 | 6.30 ± 0.03 | 44.77 ± 0.43 |
| L25 S25 W3.5 | 45.58 ± 0.60 | 0.08 ± 0.01 | −29.23 ± 2.70 | 93.42 ± 2.00 | 4.34 ± 0.09 | 53.72 ± 0.46 |
| L17.5 S10 W1.2 | 63.72 ± 1.03 | 0.09 ± 0.01 | −21.23 ± 1.56 | 92.99 ± 0.22 | 7.39 ± 0.17 | 22.98 ± 1.84 |
| L17.5 S40 W1.2 | 27.58 ± 0.31 | 0.15 ± 0.03 | −18.13 ± 2.55 | 94.47 ± 0.83 | 3.84 ± 0.03 | 25.85 ± 1.92 |
| L17.5 S10 W3.5 | 60.71 ± 0.58 | 0.09 ± 0.02 | −35.93 ± 1.53 | 92.60 ± 0.70 | 7.39 ± 0.06 | 55.96 ± 3.86 |
| L17.5 S40 W3.5 | 35.46 ± 3.18 | 0.23 ± 0.02 | −22.16 ± 0.70 | 96.97 ± 0.75 | 3.94 ± 0.03 | 37.44 ± 1.61 |
| L17.5 S25 W2.35 | 33.86 ± 0.63 | 0.10 ± 0.00 | −22.1 ± 1.91 | 92.90 ± 1.52 | 5.01 ± 0.08 | 37.17 ± 3.49 |
| L17.5 S25 W2.35 | 37.43 ± 1.64 | 0.12 ± 0.00 | −16.73 ± 1.44 | 93.19 ± 0.01 | 5.03 ± 06 | 44.11 ± 0.96 |
| L17.5 S25 W2.35 | 38.09 ± 0.72 | 0.10 ± 0.00 | −15.13 ± 0.37 | 92.17 ± 0.00 | 4.97 ± 0.02 | 39.37 ± 1.00 |
| L17.5 S25 W2.35 | 37.69 ± 0.65 | 0.19 ± 0.03 | −20.69 ± 1.20 | 93.93 ± 0.01 | 5.07 ± 0.06 | 43.15 ± 0.59 |
| L17.5 S25 W2.35 | 37.42 ± 0.93 | 0.230 ± 0.01 | −24.06 ± 0.91 | 90.35 ± 0.00 | 4.88 ± 0.05 | 45.59 ± 0.20 |
L, labrafac; S, solutol HS15; W, volume ratio of the diluting aqueous phase to the initial emulsion.
where Y is the dependent factor, X 1, X 2, X 3 are independent variables, b 0 is the intercept, b 1, b 2, and b 3 are linear coefficients, b 12, b 13, and b 23 are interaction coefficients of equation, and b 11, b 22, and b 33 are squared coefficients. The one factor and 3‐D surface plot schemed by Design‐Expert software were employed to exhibit the relationship and interaction between the studied variables and responses. In each 3‐D surface plot, the interaction of two independent variables was explored simultaneously and third one was in the middle level value.
3.1 Particle size
Particle size is known as the most important characteristic of NPs that can influence physical stability, the release pattern of the drug, and its absorption from the gastrointestinal tract. Therefore, it is a key parameter to be controlled and assessed [26].
In the present study, the mean particle size of EFV‐loaded LNCs was between 22 and 100 nm and their PdI ranged between 0.08 and 0.28. PdI is an indicator for dispersion homogeneity and is measured from the square root of standard deviation/mean diameter. It ranges from 0 to 1.
Lower PdI values indicate more homogeneity of the formulation [27]. In our study, the PdI varied between 0.08 and 0.28 indicating a mono modal size distribution of LNCs and homogeneity of the formulation. Based on the analysis using Design Expert Software, the size of LNCs has been shown affected by labrafac % (X 1), solutol HS15% (X 2), along with the interaction of labrafac and solutol HS15% (X 1 X 2) (p ‐value <0.05). Equation (6) is used to determine particle size for the given level of each factor
| (6) |
An increase in labrafac % led to an increase in particle size (Fig. 1 a). This finding is in accordance with that reported by Heurtault et al. [11] who observed an increase in particle size with increasing labrafac concentration. This could be due to the enlargement of LNC core with higher concentration proportion of oil in the centre of particles. Particle size significantly decreased with an increase the solutol HS15% (Fig. 1 b). This result is in accordance with the findings of the previous study demonstrating that the higher content of solutol HS15 decreased particle size [28]. The greater stabilisation effect of solutol HS 15 on the oil in water system at higher proportion led to produce smaller particles and protect LNCs from aggregation. Changing the volume ratio of the diluting aqueous phase to the initial emulsion had no significant effect on particle size (Fig. 1 c). 3‐D response surface plots for particle size analyses are shown in Fig. 1. As shown in this figure, the highest particle size was observed when the labrafac at the high level was combined with solutol HS15 at the low level (Fig. 1 d). In addition, when solutol HS15 is at the low level and the high‐volume ratio of the diluting aqueous phase to the initial emulsion of diluting phase was used to increase particle size (Fig. 1 e).
Fig. 1.
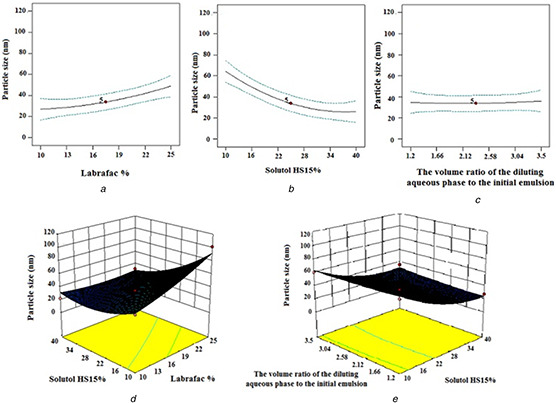
Effect of different levels of
(a) labrafac %,
(b) solutol HS15%,
(c) the volume ratio of the diluting aqueous phase to the initial emulsion and interaction of,
(d) labrafac %‐solutol HS15%,
(e) the volume ratio of the diluting aqueous phase to the initial emulsion ‐ solutol HS15% on the particle size of EFV‐loaded LNCs
3.2 Zeta potential
Zeta potential is electric potential at the surface of NPs in solution and its magnitude could determine the stability of NPs. The zeta potential measured for all formulations ranged from −12.8 to −31.46. The following equation shows the effect of each factor on the zeta potential
| (7) |
Based on the information gained from the ANOVA analysis, solutol HS 15% (X 2) and volume ratio of the diluting aqueous phase to the initial emulsion (X 3), the interaction between labrafac % and solutol HS 15% (X 1 X 2) and the interaction between solutol HS 15% and volume ratio of the diluting aqueous phase to the initial emulsion (X 2 X 3) have significant effects on zeta potential (p ‐value <0.05). As shown in Table 3, all prepared LNCs exhibited negative zeta potential due to the presence of negatively charged phospholipids that are integrated on the surface of LNCs. Increasing the solutol HS15 content led to a decrease in the absolute value of zeta potential (Fig. 2 a). The adsorption of more amount of surfactant on the surface of LNCs and the hiding properties of the PEG steric barrier of the surfactant might decrease these charges [29]. This finding was in agreement with the previous report of Yuan et al. [30] who showed a decrease in the absolute value of zeta potential with the incorporation of PEG‐monostearate in NLCs. Li et al. [31] also showed similar results with the incorporation of cisplatin in NPs of methoxy poly(ethylene glycol)–polycaprolactone. Another factor with a significant effect on zeta potential was the volume ratio of the diluting aqueous phase to the initial emulsion (Fig. 2 b). Negative zeta potential increased with increasing the volume ratio of the diluting aqueous phase to the initial emulsion. Decreasing the final temperature of the system with increasing volume of cold water could be a possible reason for an increase in absolute values of the zeta potential of the LNCs. Phospholipids of lecithin can be considered as electric dipoles in water. Firstly, if phosphoric groups of phospholipidic molecules were present in the outer part of the shell, the presence of phosphoric groups would induce negative zeta potential. On the contrary, if terminal groups of phosphatidylcholine were present in the outer part of the LNC shell, the existence of N1 groups resulted in positive zeta potential [32]. Temperature change can lead to a crystalline reorientation of lipid (lecithin). Different sides of a crystal (lecithin) having a different charge density can cause a change in charges on the particle surface. Freitas et al. [33] also reported that decreasing temperature increased the zeta potential value of poloxamer 188 stabilised SLNs. Increasing labrafac percent results in an increase, though not significant, in the negative zeta potential value of EFV LNCs (Fig. 2 c). The 3‐D response surface plots of zeta potential variations proportional to alterations in independent variables have been shown in Fig. 2. As illustrated in Fig. 2, the maximum absolute value of zeta potential was obtained at the high level of labrafac and the low level of solutol HS15. The highest zeta potential also occurs at the high level of the volume ratio of the diluting aqueous phase to the initial emulsion and labrafac per cent.
Fig. 2.
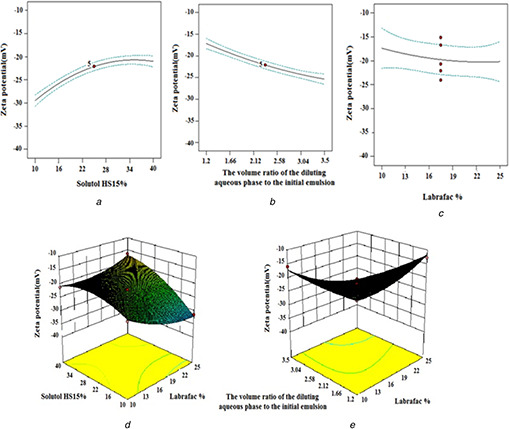
Effect of different levels of
(a) solutol HS15%,
(b) the volume ratio of the diluting aqueous phase to the initial emulsion,
(c) labrafac % and interaction of,
(d) labrafac% ‐solutol HS15%,
(e) the volume ratio of the diluting aqueous phase to the initial emulsion ‐ solutol HS15% on the zeta potential of EFV loaded LNCs
3.3 Determination of EE and DL%
As shown in Table 3, the EE values of EFV‐loaded LNCs were in the range of 83.18 ± 0.83–97.72 ± 0.59%. The effect of formulation parameters on EE% can be comprehended by the following equation:
| (8) |
The amount of drug the can be entrapped in the nano‐carrier is dependent on the physicochemical characteristics of the drug and the preparation process. Owing to the low solubility of EFV in the external aqueous phase, the EE% values for all LNCs prepared were high and did not undergo a significant change from one formulation to another. Overall, within selected ranges of solutol HS 15% and labrafac %, and the volume ratio of the diluting aqueous phase to the initial emulsion, EFV is remarkably encapsulated within LNCs.
Based on the investigated results, the value of DL% of EFV‐loaded NLCs ranged from 3.3 to 10.14%. The following equation is used to predict the effect of independent variables on DL%:
| (9) |
It is concluded from the results of ANOVA that labrafac % (X 1) and solutol HS15% (X 2) along with their interaction had a significant effect on DL%. The effect of various parameters on DL% is shown in Fig. 3. As shown in Fig. 3, increasing the solutol HS15 and labrafac content (excipients) decreased the DL% of LNCs significantly (p ‐value <0.05) and the lowest DL% was observed when the labrafac and solutol HS15 were combined at the high level. With a constant amount of the drug, when the content of excipients increased, the DL% decreased.
Fig. 3.
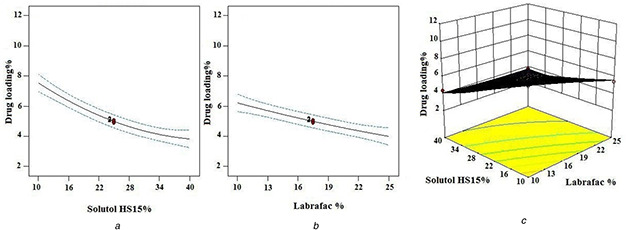
Effect of different levels of
(a) solutol HS15%,
(b) labrafac % and interaction of
(c) labrafac %‐solutol HS15% on DL % of EFV loaded LNCs
3.4 In vitro drug release from LNCs
The in vitro release profiles of EFV from LNCs against time in PBS containing 0.5% Tween 80 are shown in Fig. 4. The LNCs exhibited sustained release properties for EFV. The RE144 % is a parameter employed as an evaluation tool for assessing drug release. The greater RE144 %, the faster the drug release rate will be. The RE144 % for different formulations ranged from 22.98–71.54%. The effect of each factor on the obtained RE144 % can be described by the following quadratic equation:
| (10) |
Fig. 4.
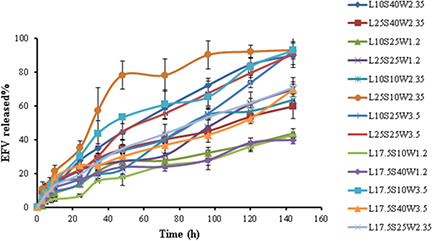
Release profiles of EFV from different studied LNCs
The statistical analysis of RE144% data with Design Expert Software demonstrated that all studied factors and labrafac % in interaction with solutol HS15% (X 1 X 2) and solutol HS15% in interaction with a volume ratio of the diluting aqueous phase to the initial emulsion (X 2 X 3) significantly influenced RE144%. As shown in Fig. 5 a, a decrease in RE144% was attained by an increase in the solutol HS 15 content. Although more amount of solutol HS 15 decreased the size of LNCs, it was expected that the rate of release would increase but increase in solutol HS 15 density at the oil/water interface enhanced the steric constraints and barrier feature of the membrane. This causes slower release from capsules containing a higher content of solutol HS 15. In agreement with our study, Lamprecht et al. [29] and Safvat et al. [12] also demonstrated that higher solutol HS 15 content decreased the release rate. The observation showed that increasing volume ratio of the diluting aqueous phase to the initial emulsion increased RE144 % (Fig. 5 b). Varshosaz et al. [10] also demonstrated that increasing the cold water volume during the dilution step increases the RE. It is possible that decrease in viscosity, on increasing the volume of the diluting phase decreased the resistance to diffusion result in an increase in release rate.
Fig. 5.
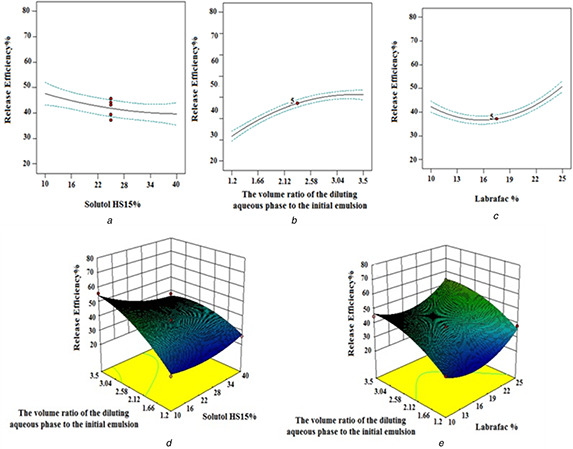
Effect of different levels of
(a) solutol HS15%,
(b) the volume ratio of the diluting aqueous phase to the initial emulsion,
(c) labrafac % and interaction of,
(d) the volume ratio of the diluting aqueous phase to the initial emulsion ‐solutol HS15%,
(e) the volume ratio of the diluting aqueous phase to the initial emulsion ‐ labrafac % on the RE of EFV‐loaded LNCs
The RE144 % increased with an increase in labrafac per cent too (Fig. 5 c). As shown in Figs. 5 d –e, the maximum level of RE144 % occurs at a high volume ratio of the diluting aqueous phase to the initial emulsion along with the low level of solutol HS15% or the high level of labrafac %.
3.5 Optimisation
The computer optimisation process by Design Expert Software (Version 10, USa) and a desirability function determined the effect of the levels of independent variables on the responses. The constraints of particle size and PdI were 22 ≤ Y 1 ≤ 100 nm and 0.08 ≤ Y 2 ≤ 0.28, respectively, with the target set at a minimum the obtained results. The constraints of zeta potential, EE%, DL%, and RE144 % were −12.8 ≤ Y 3 ≤ −31.46 mV, 83.18 ≤ Y 4 ≤ 97.72%, 3.3 ≤ Y 5 ≤ 10.14%, and 22.98 ≤ Y 6 ≤ 71.54% respectively with the goal set at the maximum of the obtained data. The optimised formulation suggested by the desirability of 74% was an L17.5S10W3.5 formulation which was prepared by 17.5% labrafac and 10% solutol HS15 when the volume ratio of the diluting aqueous phase to the initial emulsion was 3.5. The morphology of the selected formulation evaluated by SEM is shown in Fig. 6. As shown in this figure, the LNCs were spherical with the particle size close to that obtained by the dynamic light scattering dynamic light scattering (DLS) method.
Fig. 6.
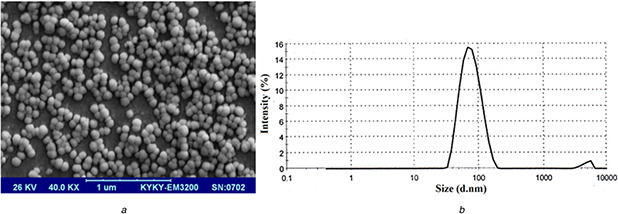
SEM morphology and particle size distribution of EFV loaded optimised LNCs
(a) SEM micrograph,
(b) Size distribution of EFV‐loaded optimised LNCs
The in vitro release data obtained from the optimised LNCs was fitted into different kinetic models and the drug release was found to follow the Higuchi kinetics (Table 4). In this case, the amount of drug released is proportional to the square root of time. To evaluate the mechanism of drug release, Korsemeyer–Peppas equation has been used and good linearity (0.9842) has been observed. The diffusion exponent (n) in Korsmeyer–Peppas equation was about 0.7281 indicating the release was mainly controlled by both diffusion and erosion mechanisms (anomalous).
Table 4.
Correlation coefficient (R 2) obtained from curve fitting of EFV release data from optimised LNCs
| Kinetic models (r 2) | Peppas parameters | ||||||
|---|---|---|---|---|---|---|---|
| Formulation | Zero order | Higuchi | Hixson‐Crowell | First order | Baker–Lonsdale | n | r 2 |
| optimised EFV loaded LNCs | 0.9454 | 0.9889 | 0.9454 | 0.9409 | 0.9241 | 0.7281 | 0.9842 |
3.6 FTIR spectroscopy
The FTIR spectra of drug and pure components, blank LNCs, and EFV‐loaded LNCs are shown in Fig. 7. The spectrum of EFV shows characteristic bands at 3318.89 cm−1 (N–H stretching vibration), 2249.56 cm−1 (aromatic C–H stretching vibration), 1746.23 cm−1 (C=O stretching vibration), 1602.56 cm−1 (C=C stretching vibration), 1320 cm−1 (C–O–C stretching vibration), 1240.97 cm−1 (C–F stretching vibration). The FTIR spectrum of solutol HS15 showed the absorption bands of the O–H bond at 3371.71 cm−1 and the ester group at 1733.69 cm−1.
Fig. 7.
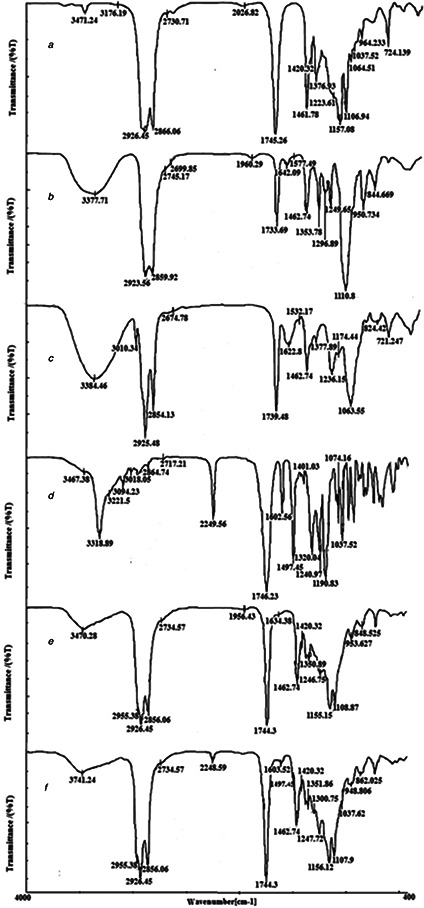
FTIR spectra of
(a) Labrafac,
(b) Solutol HS15,
(c) Lecithin,
(d) EFV,
(e) Blank LNCs,
(f) EFV‐loaded LNCs
Similarly Labrafac revealed the absorption peaks of the O–H stretch band at 3471.24 cm−1, aliphatic C–H bonds at 2865.06 and 2926.55 cm−1, C–O groups at 1106.94 cm−1 and C=O ester groups at 1745.26 cm−1. The spectrum of blank LNCs revealed a C–O band at 1108.87 cm−1, ester groups at 1744.3 cm−1, aliphatic C–H bonds at 2856 and 2926.45 cm−1, and the O–H stretch band at 3470.28 cm−1. All characteristic bands of blank LNCs and EFV were present in the EFV‐loaded LNCs and no significant shift of the existing peaks or the creation of new peaks was observed. This result demonstrated that no physicochemical interaction between EFV and excipients in LNCs’ formulation.
3.7 Storage stability study of EFV‐loaded optimised LNC dispersion
The optimised LNCs loaded with EFV were stored at 4°C for 2 weeks and storage stability was investigated. Table 5 shows the physicochemical properties of the optimised LNCs loaded EFV after 2 weeks storage. As it can be seen in this table, particle size, size distribution, and zeta potential did not show any noticeable change, which supports the high stability of the prepared LNCs [17]. This finding can be explained by electrostatic and steric repulsion between NPs. However, a slight increase (p ‐value >0.05) in particle size during the experimental period due to the formation of aggregates results in a decrease of the absolute value of zeta potential of the EFV dispersions (p ‐value >0.05). This finding was confirmed by other studies such as Taymouri et al. [27] who showed the absolute value of zeta potential decreased with increasing the particle size of folate targeted pluronic F127‐cholesterol polymeric micelles. As the particle size increased, the surface charge density decreased and consequently, the absolute value of zeta potential decreased. Even though the nanodispersion was stable during 2 weeks, we prepared freeze‐dried LNCs to facilitate long‐term storage and shipping.
Table 5.
Physicochemical properties of the optimised LNCs loaded with EFV at an initial time and 2 weeks after preparation
| Formulation | Particle size, nm | PdI | Zeta potential, mV |
|---|---|---|---|
| at initial time | 60.71 ± 0.58 | 0.09 ± 0.02 | −35.93 ± 1.53 |
| after 2 weeks | 65.85 ± 2.07 | 0.10 ± 0.01 | −22.23 ± 4.99 |
3.8 Characterisation of freeze‐dried products
Freeze‐drying is the mostly common used process to ensure high stability, ease of storage, and handling of the formulation. Considering the accelerated degradation of various types of polymers and lipids used in NPs in water, freeze‐drying is a viable option for long‐term stability. In the first set of experiments, EFV‐loaded optimised LNCs were freeze dried without excipients. In the absence of any excipients, the material had turned into sticky form and redispersion of the formulation again into a suspension form was not possible easily. Therefore, different concentrations of the cryoprotectants were added to the formulation to assess the optimum concentration. As shown in Table 6, the particle size of all lyophilised formulations of LNCs after re‐dispersion was larger than that before lyophilisation of 60.0 nm due to the formation of a small population of aggregated particles. Compared with other cryoprotectants, the result of aerosil showed more increase in particle size and PdI with freeze drying. This result is in good agreement with that reported by Varshosaz et al. [34] demonstrating the increasing concentration of aerosil causes the increase in particle size of freeze dried NLCs containing valproate. This finding was explained with the gel forming effect of aerosil in high concentrations. In the case of other excipients, although, freeze drying of formulation led to an increase in particle size, their small particle size distribution (PdI) shows that they are monodispersed.
Table 6.
Effect of different cryoprotectants on the particle size, PdI, zeta potential and dispersion time of freeze dried LNCs
| Cryoprotectants | Concentration %, w/w | Particle size, nm | PdI | Zeta potential, mV | Dispersion time, S |
|---|---|---|---|---|---|
| PEG4000 | 1 | 113.60 ± 1.47 | 0.22 ± 0.01 | −43.23 ± 0.68 | 105 |
| 3 | 135.73 ± 3.15 | 0.31 ± 0.01 | −49.46 ± 0.60 | 75 | |
| 5 | 162.36 ± 3.45 | 0.37 ± 0.01 | −51.10 ± 0.79 | 40 | |
| sorbitol | 1 | 116.00 ± 5.88 | 0.32 ± 0.04 | −48.96 ± 3.76 | 120 |
| 3 | 126.90 ± 0.80 | 0.24 ± 0.02 | −44.53 ± 2.83 | 90 | |
| 5 | 250.86 ± 10.51 | 0.40 ± 0.02 | −50.93 ± 3.08 | 90 | |
| sucrose | 1 | 108.63 ± 1.15 | 0.23 ± 0.01 | −50.23 ± 1.85 | 30 |
| 3 | 115.36 ± 1.42 | 0.21 ± 0.01 | −43.26 ± 0.23 | 20 | |
| 5 | 128.36 ± 4.56 | 0.35 ± 0.00 | −44.56 ± 0.23 | 10 | |
| aerosil | 1 | 177.33 ± 2.17 | 0.40 ± 0.02 | −49.76 ± 3.00 | 45 |
| 3 | 233.76 ± 25.83 | 0.38 ± 0.06 | −49.93 ± 2.05 | 40 | |
| 5 | 436.80 ± 44.38 | 0.52 ± 0.17 | −45.70 ± 1.41 | 40 | |
| lactose | 1 | 121.06 ± 1.68 | 0.28 ± 0.03 | −53.90 ± 2.36 | 60 |
| 3 | 158.13 ± 33.96 | 0.36 ± 0.03 | −54.13 ± 1.09 | 50 | |
| 5 | 149.23 ± 21.01 | 0.28 ± 0.05 | −54.50 ± 1.41 | 30 |
After freeze drying, it was noticed that LNCs freeze dried with 1–5% w/w sorbitol, 1–3% w/w PEG 4000, 1% w/w sucrose and lactose formed a sticky collapsed cake (an adhesive and wet product) while PEG 4000 (5% w/w), lactose (3–5% w/w), and sucrose (3–5% w/w) produced an intact fluffy cake with proper re‐dispersibility characteristics. Table 6 shows the time required for the reconstitution of dry LNC powder from the start of the addition of the solvent. The reconstitution times of dry LNC powders with sucrose as the cryoprotectant were found to be substantially less than those with other cryoprotectants. As shown in Table 6, the freeze dried samples exhibited a higher value of zeta potential which could be due to the presence of cryoprotectants that form a viscous hydration layer on NPs’ surface. A similar behaviour was also observed in Fonte et al. [35] study that demonstrated a similar increase in the zeta potential of insulin‐loaded poly(lactic‐co‐glycolic acid) NPs after freeze drying by adding cryoprotectants. In agreement with the result of freeze dried cake properties, reconstitution behaviour, and DLS data, it can be said that 5% w/w sucrose appeared to be most suitable cryoprotectants for LNCs’ formulation. Additionally, freeze‐dried LNCs with 5% (w/w) of sucrose were stored at 4°C for a period of 6 months to evaluate the long‐term stability. Even the lyophilised powders of LNCs were stored for 6 months, the physicochemical properties including particle size, PdI, and zeta potential did not change significantly (Table 7). These results suggest that the prepared system was highly stable over the observed time span.
Table 7.
Physicochemical properties of freeze dried LNCs with 5% (w/w) of sucrose at an initial time and 6 months after preparation
| Formulation | Particle size, nm | PdI | Zeta potential, mV |
|---|---|---|---|
| at initial time | 128.36 ± 4.56 | 0.35 ± 0.00 | −44.56 ± 0.23 |
| after 6 months | 137.2 ± 1.47 | 0.29 ± 0.01 | −46.96 ± 2.44 |
3.9 In vitro cytotoxicity assay
To predict in vivo toxicity, the cytotoxic activity of EFV‐loaded LNCs and blank LNCs was investigated using Caco‐2 cells. As shown in Fig. 8 A, LNCs entrapping EFV and blank LNCs showed no toxicity to the Caco‐2 cells over a range of concentration from 10 to 500 µg/ml after 4 and 8 h. This result confirmed the prepared LNCs are non‐toxic and are expected to be safe for biomedical application.
Fig. 8.
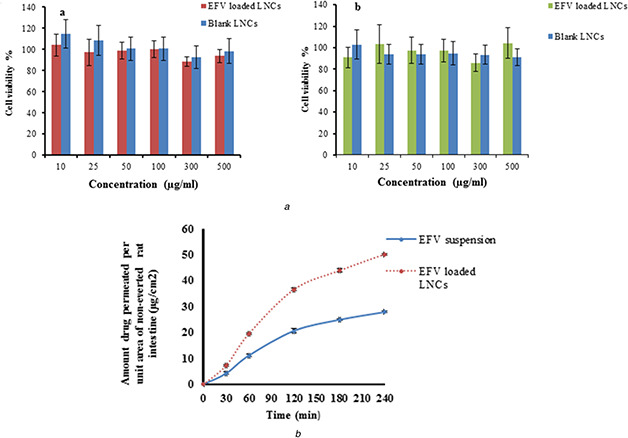
Evaluation of in vitro cytotoxicity and ex vivo permeation of EFV loaded optimised LNCs
(a) In vitro cytotoxicity of blank LNCs and EFV‐loaded LNCs on Caco2 cells (a) 4 h, (b) 8 h,
(b) Permeation profiles of EFV from optimised EFV‐loaded LNCs and drug suspension through rat intestine
3.10 Ex vivo permeation of EFV from the optimised LNCs
The intestinal permeation behaviour of the free EFV and EFV‐loaded LNCs was studied by the non‐everted gut sac model for 4 h. The everted and non‐everted intestinal sac models can provide information on permeability, and drug absorption in humans with lower experimental expenses and labour in comparison with in vivo animal studies [24, 25]. A non‐everted intestinal sac model has many advantages compared with an everted intestinal sac model due to the simple preparation, relatively small amount of drug is required, less intestinal morphological damage, and the ability for successive and frequent sampling from the serosal compartment. The non‐everted intestinal sac model has been used to evaluate the permeation of some drugs from numerous delivery systems [25].
The permeation profiles of EFV from optimised EFV‐loaded LNCs and drug suspension are shown in Fig. 8 B which clearly indicated that LNCs increased the permeation of EFV compared with a drug suspension. As shown in Fig. 8 B after 4 h, a cumulative amount of EFV was permeated to a receiver fluid increased from 26.28 ± 0.27 µg/cm2 for EFV suspension to 47.29 ± 0.27 µg/cm2 for LNCs. It was found for EFV suspension only 27.90% ± 0.29 of EFV permeated after 4 h. On the contrary, the LNCs showed 50.20% ± 0.29 permeation of EFV after 4 h. The Papp obtained for EFV‐loaded LNCs (3.1 × 10−4 cm/min) was also significantly higher than that of EFV suspension (1.8 × 10−4 cm/min). These results could be attributed to enhanced EFV dissolution in addition to enhanced intestinal permeability upon incorporation of the drug within the LNCs. One of the advantages of using NPs is the decrease in particle size and enhancement of the surface area resulting in higher drug dissolution and diffusion as well as greater drug penetration during its stay in the intestine. This result was in agreement with the study of Roger et al. [17, 36]. He showed enhance the intestinal permeability of SN 38 and PTX by incorporation in LNCs.
4 Conclusions
In the present study, LNCs were well fabricated by the phase‐inversion temperature method. The optimised formulation possessed appropriate morphology, small particle size, high EE%. It has been clarified that the incorporation of EFV into LNCs had succeeded to sustain the drug release and increase the ex‐vivo intestinal permeation. These results suggested that LNCs could be promising for improved oral delivery of EFV.
5 Acknowledgments
The authors wish to thank the Research Vice Chancellery of Isfahan University of Medical Sciences for supporting this work.
6 References
- 1. Feyera A. Megerssa B. Legesse D. et al.: ‘Prevention of mother to child transmission of HIV/AIDS: service utilization and associated factors among selected public health facilities in Ethiopia’, Med. Pract. Rev., 2017, 8, (1), pp. 1 –3 [Google Scholar]
- 2. Chiappetta D.A. Facorro G. de Celis E.R. et al.: ‘Synergistic encapsulation of the anti‐HIV agent efavirenz within mixed poloxamine/poloxamer polymeric micelles’, Nanomedicine, 2011, 7, (5), pp. 624 –637 [DOI] [PubMed] [Google Scholar]
- 3. Vyas T.K. Shah L. Amiji M.M.: ‘Nanoparticulate drug carriers for delivery of HIV/AIDS therapy to viral reservoir sites’, Expert Opin. Drug Deliv., 2006, 3, (5), pp. 613 –628 [DOI] [PubMed] [Google Scholar]
- 4. Makwana V. Jain R. Patel K. et al.: ‘Solid lipid nanoparticles (SLN) of efavirenz as lymph targeting drug delivery system: elucidation of mechanism of uptake using chylomicron flow blocking approach’, Int. J. Pharm., 2015, 495, (1), pp. 439 –446 [DOI] [PubMed] [Google Scholar]
- 5. Chiappetta D.A. Hocht C. Sosnik A.: ‘A highly concentrated and taste‐improved aqueous formulation of efavirenz for a more appropriate pediatric management of the anti‐HIV therapy’, Curr. HIV Res., 2010, 8, (3), pp. 223 –231 [DOI] [PubMed] [Google Scholar]
- 6. Avachat A.M. Parpani S.S.: ‘Formulation and development of bicontinuous nanostructured liquid crystalline particles of efavirenz’, Colloids Surf. B, 2015, 126, pp. 87 –97 [DOI] [PubMed] [Google Scholar]
- 7. McDonald T.O. Giardiello M. Martin P. et al.: ‘Antiretroviral solid drug nanoparticles with enhanced oral bioavailability: production, characterization, and in vitro–in vivo correlation’, Adv. Healthc. Mater., 2014, 3, (3), pp. 400 –411 [DOI] [PubMed] [Google Scholar]
- 8. Pardeike J. Weber S. Haber T. et al.: ‘Development of an itraconazole‐loaded nanostructured lipid carrier (NLC) formulation for pulmonary application’, Int. J. Pharm., 2011, 419, (1), pp. 329 –338 [DOI] [PubMed] [Google Scholar]
- 9. Jia L. Zhang D. Li Z. et al.: ‘Nanostructured lipid carriers for parenteral delivery of silybin: biodistribution and pharmacokinetic studies’, Colloids Surf. B, 2010, 80, (2), pp. 213 –218 [DOI] [PubMed] [Google Scholar]
- 10. Varshosaz J. Hajhashemi V. Soltanzadeh S.: ‘Lipid nanocapsule‐based gels for enhancement of transdermal delivery of ketorolac tromethamine’, J Drug Deliv., 2011, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heurtault B. Saulnier P. Pech B. et al.: ‘The influence of lipid nanocapsule composition on their size distribution’, Eur. J. Pharm. Sci., 2003, 18, (1), pp. 55 –61 [DOI] [PubMed] [Google Scholar]
- 12. Safwat S. Hathout R.M. Ishak R.A. et al.: ‘Augmented simvastatin cytotoxicity using optimized lipid nanocapsules: a potential for breast cancer treatment’, J. Liposome Res., 2017, 27, (1), pp. 1 –10 [DOI] [PubMed] [Google Scholar]
- 13. Huynh N.T. Passirani C. Saulnier P. et al.: ‘Lipid nanocapsules: a new platform for nanomedicine’, Int. J. Pharm., 2009, 379, (2), pp. 201 –209 [DOI] [PubMed] [Google Scholar]
- 14. Karim R. Palazzo C. Evrard B. et al.: ‘Nanocarriers for the treatment of glioblastoma multiforme: current state‐of‐the‐art’, J. Control. Release, 2016, 227, pp. 23 –37 [DOI] [PubMed] [Google Scholar]
- 15. Peltier S. Oger J.M. Lagarce F. et al.: ‘Enhanced oral paclitaxel bioavailability after administration of paclitaxel‐loaded lipid nanocapsules’, Pharm. Res., 2006, 23, (6), pp. 1243 –1250 [DOI] [PubMed] [Google Scholar]
- 16. Groo A.C. Bossiere M. Trichard L. et al.: ‘In vivo evaluation of paclitaxel‐loaded lipid nanocapsules after intravenous and oral administration on resistant tumor’, Nanomedicine, 2015, 10, (4), pp. 589 –601 [DOI] [PubMed] [Google Scholar]
- 17. Roger E. Lagarce F. Benoit J.P.: ‘Development and characterization of a novel lipid nanocapsule formulation of Sn38 for oral administration’, Eur. J. Pharm. Biopharm., 2011, 79, (1), pp. 181 –188 [DOI] [PubMed] [Google Scholar]
- 18. Roger E. Gimel J.C. Bensley C. et al.: ‘Lipid nanocapsules maintain full integrity after crossing a human intestinal epithelium model’, J. Control. Release, 2017, 253, pp. 11 –18 [DOI] [PubMed] [Google Scholar]
- 19. Roger E. Lagarce F. Benoit J.P.: ‘The gastrointestinal stability of lipid nanocapsules’, Int. J. Pharm., 2009, 379, (2), pp. 260 –265 [DOI] [PubMed] [Google Scholar]
- 20. Zhai Y. Liu M. Wan M. et al.: ‘Preparation and characterization of puerarin‐loaded lipid nanocapsules’, J. Nanosci. Nanotechnol., 2015, 15, (4), pp. 2643 –2649 [DOI] [PubMed] [Google Scholar]
- 21. Groo A.C. Saulnier P. Gimel J.C. et al.: ‘Fate of paclitaxel lipid nanocapsules in intestinal mucus in view of their oral delivery’, Int. J. Nanomed., 2013, 8, pp. 4291 –4302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kreuter J.: ‘Nanoparticles and microparticles for drug and vaccine delivery’, J. Anat., 1996, 189, pp. 503 –505 [PMC free article] [PubMed] [Google Scholar]
- 23. Bastiancich C. Vanvarenberg K. Ucakar B. et al.: ‘Lauroyl‐gemcitabine‐loaded lipid nanocapsule hydrogel for the treatment of glioblastoma’, J. Control. Release, 2016, 225, pp. 283 –293 [DOI] [PubMed] [Google Scholar]
- 24. Radwan S.E. Sokar M.S. Abdelmonsif D.A. et al.: ‘Mucopenetrating nanoparticles for enhancement of oral bioavailability of furosemide: In vitro and in vivo evaluation/sub‐acute toxicity study’, Int. J. Pharm., 2017, 526, (1), pp. 366 –379 [DOI] [PubMed] [Google Scholar]
- 25. Shehata E.M. Elnaggar Y.S. Galal S. et al.: ‘Self‐emulsifying phospholipid pre‐concentrates (SEPPs) for improved oral delivery of the anti‐cancer genistein: development, appraisal and ex‐vivo intestinal permeation’, Int. J. Pharm., 2016, 511, (2), pp. 745 –756 [DOI] [PubMed] [Google Scholar]
- 26. Emami J. Boushehri M.S. Varshosaz J.: ‘Preparation, characterization and optimization of glipizide controlled release nanoparticles’, Res. Pharm. Sci., 2014, 9, (5), pp. 301 –314 [PMC free article] [PubMed] [Google Scholar]
- 27. Taymouri S. Varshosaz J. Hassanzadeh F. et al.: ‘Optimisation of processing variables effective on self‐assembly of folate targeted synpronic‐based micelles for docetaxel delivery in melanoma cells’, IET Nanobiotechnol., 2015, 9, (5), pp. 306 –313 [DOI] [PubMed] [Google Scholar]
- 28. Barras A. Mezzetti A. Richard A. et al.: ‘Formulation and characterization of polyphenol‐loaded lipid nanocapsules’, Int. J. Pharm., 2009, 379, (2), pp. 270 –277 [DOI] [PubMed] [Google Scholar]
- 29. Lamprecht A. Bouligand Y. Benoit J.P.: ‘New lipid nanocapsules exhibit sustained release properties for amiodarone’, J. Control. Release, 2002, 84, (1), pp. 59 –68 [DOI] [PubMed] [Google Scholar]
- 30. Yuan H. Wang L.L. Du Y.Z. et al.: ‘Preparation and characteristics of nanostructured lipid carriers for control‐releasing progesterone by melt‐emulsification’, Colloids Surf. B, 2007, 60, (2), pp. 174 –179 [DOI] [PubMed] [Google Scholar]
- 31. Li X. Li R. Qian X. et al.: ‘Superior antitumor efficiency of cisplatin‐loaded nanoparticles by intratumoral delivery with decreased tumor metabolism rate’, Eur. J. Pharm. Biopharm., 2008, 70, (3), pp. 726 –734 [DOI] [PubMed] [Google Scholar]
- 32. Vonarbourg A. Saulnier P. Passirani C. et al.: ‘Electrokinetic properties of noncharged lipid nanocapsules: influence of the dipolar distribution at the interface’, Electrophoresis, 2005, 26, (11), pp. 2066 –2075 [DOI] [PubMed] [Google Scholar]
- 33. Freitas C. Müller R.H.: ‘Effect of light and temperature on zeta potential and physical stability in solid lipid nanoparticle (SLN™) dispersions’, Int. J. Pharm., 1998, 168, (2), pp. 221 –229 [Google Scholar]
- 34. Varshosaz J. Eskandari S. Tabbakhian M.: ‘Freeze‐drying of nanostructure lipid carriers by different carbohydrate polymers used as cryoprotectants’, Carbohydr. Polym., 2012, 88, (4), pp. 1157 –1163 [Google Scholar]
- 35. Fonte P. Soares S. Costa A. et al.: ‘Effect of cryoprotectants on the porosity and stability of insulin‐loaded PLGA nanoparticles after freeze‐drying’, Biomatter, 2012, 2, (4), pp. 329 –339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roger E. Lagarce F. Garcion E. et al.: ‘Lipid nanocarriers improve paclitaxel transport throughout human intestinal epithelial cells by using vesicle‐mediated transcytosis’, J. Control. Release, 2009, 140, (2), pp. 174 –181 [DOI] [PubMed] [Google Scholar]