

Abstract
A novel three‐dimensional (3D) titanium (Ti)‐doping meso‐macroporous bioactive glasses (BGs)/poly(methyl methacrylate) (PMMA) composite was synthesised using PMMA and EO20 PO70 EO20 (P123) as the macroporous and mesoporous templates, respectively. Unlike the usual calcination method, the acid steam technique was used to improve the polycondensation of Ti‐BGs, and then PMMA was partially extracted via chloroform to induce the macroporous structure. Simultaneously, the residual PMMA which remained in the wall enhanced the compressive strength to 2.4 MPa (0.3 MPa for pure BGs). It is a simple and green method to prepare the macro‐mesoporous Ti‐BGs/PMMA. The materials showed the 3D interconnected hierarchical structure (250 and 3.4 nm), making the fast inducing‐hydroxyapatite growth and the controlled drug release. Besides mentioned above, the good antimicrobial property and biocompatible of the scaffold also ensure it is further of clinical use. Herein, the fabricated materials are expected to have potential application on bone tissue regeneration.
Inspec keywords: titanium, bone, tissue engineering, glass, materials preparation, biomedical materials, polymers, porous materials, drug delivery systems, nanomedicine
Other keywords: poly(methyl methacrylate), PMMA preparation, 3D titanium‐bioactive glass scaffold, bone tissue engineering, titanium‐doping mesomacroporous bioactive glass, bioactive glass‐PMMA composite, macroporous template, mesoporous template, calcination method, acid steam technique, titanium‐bioactive glass polycondensation, macroporous structure, green method, macromesoporous titanium‐bioactive glass‐PMMA, 3D interconnected hierarchical structure, fast inducing‐hydroxyapatite growth, controlled drug release, bone tissue regeneration, Ti
1. Introduction
The development of bone tissue engineering has made it an attractive approach with significant potential for treating bone defects, especially large bone defects caused by trauma, surgical resection and congenital deformity corrections [1]. The design and synthesis of suitable scaffolds (structure and function) as the matrices with suitable physico‐chemical properties play a pivotal role in bone tissue engineering to meet the stringent requirements for tissue regeneration [2, 3]. Bioactive glasses (BGs) in system CaO–P2 O5 –SiO2 are one of the most promising materials for bone regeneration scaffolds [4]. BGs are an inorganic surface‐active biomaterial that can bond with bone tissue via inducing‐hydroxyapatite (HAP) mineralisation in vitro as well as in vivo [5, 6, 7, 8, 9].
Presently, much attention has been paid to the functionalisation of BGs to further satisfy the clinical needs on the structure and property of the implant. First, the abundant porous structure is a key factor for a scaffold to be used as a bone implant. It is known that, the macroporous structure benefits cell migration, delivery of nutrients and gases and eventual vascularisation. A three‐dimensional (3D) interconnecting macroporous structure is vital so as to supply skeletal channels for the diffusion of metabolites and nutrients within a suitable physiological environment for cell proliferation and differentiation culminating in fast bone in‐growth [10]. Furthermore, mesopores (2–50 nm) always reveal high surface areas to enhance bioactivity of the implant due to the accelerated ion release and higher density of exposed Si–OH groups that provide enough nucleation sites for HAP deposition and accelerate the bond with the bone tissue [11]. Besides, reparative process requires the suitable drug impregnation and nutrition supply [12]. Mesoporous structures have been developed to favour the guest molecule storage and the prediction of steady release, leading to a sustained drug release [13]. On the basis of these reasons, the 3D macro‐mesoporous BGs are considered as a potential bone replacement material.
With the development of porous BGs on bone regeneration, the low compressive strength limits their further practical application [14]. Ideal tissue engineering materials should possess the required desirable mechanical property (compressive strength). Therefore, many efforts were made to improve the mechanical properties of these porous BGs. Many polymers, poly(lactic‐co‐glycolic acid), poly(3‐hydroxybutyrate) (PHB) and poly‐DL‐lactide with high mechanical strength were often used as a reinforcing phase to improve the toughness of ceramic (glass)‐based scaffolds [15, 16]. However, their poor hydrophilicity, low porosity and poor bioactivity limit their further application [17]. Thereby, composites composed of organic polymers and porous BGs were expected to combine the advantages of the two components. Bretcanu O synthesised 45S5 Bioglass® ‐derived scaffolds coated with PHB and that composites revealed the considerable increase in the compressive strength to 1.5 MPa at 85% porosity [18]. Furthermore, Wei et al. developed nano‐HAP modified with poly (L‐lactic acid) [19], showing a compressive strength close to that of cancellous bone.
In bone reconstruction surgeries, one influencing factor should be considered: bacterial infection. The doping of antimicrobial agents improves the antibacterial property of BGs is also very important for practical applications [20]. Titanium (Ti) series antimicrobial agent is a typical undissolved antimicrobial agent. TiO2 has received more attention due to its stability, being environmentally benign, safety and broad‐spectrum antibiosis [21].
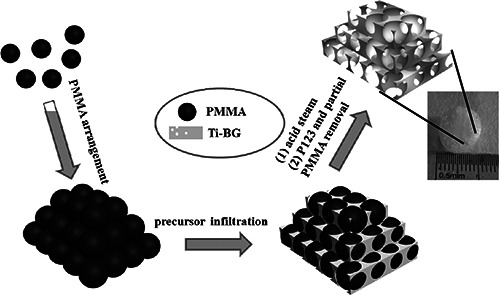
In this paper, a novel 3D macro‐mesoporous BGs doped with Ti has been synthesised using poly(methyl methacrylate) (PMMA) sphere (300 nm) as macroporous template and EO20 PO70 EO20 (P123) as the mesoporous template. It is known that, PMMA possesses non‐toxicity, high modulus, reasonable compressive resistance and versatile processing capabilities. In this paper, to preserve the PMMA component, an acid steam technique was adopted to improve the polycondensation of Ti‐BGs. Then, the partial dissolution of PMMA induces the macroporous structure and the residual enhances the mechanical strength of the composite obviously. Simultaneously, drug delivery and biomineralisation in vitro as well as the biocompatibility assessment were studied in detail.
2. Experimental
2.1 Materials
P123 (average molecular weight: 5800) was supplied by Aldrich. Calcium nitrate (Ca(NO3)2 ·4H2 O), chlorhydric acid (HCl), ethyl silicate (TEOS), tetra‐n ‐butyl titanate (Ti (C4 H9 O)4) and methyl methacrylate (MMA) were purchased from Tianjin Kemiou Chemical Reagent Co., Ltd. Triethylphosphate (TEP) and ethanol were purchased from Harbin Chemical Reagent Factory. Metformin HCl (MH, Shandongkeyuan, Jinan). Chloroform (CHCl3) and potassium persulfate (K2 S2 O8) were obtained from Harbin, China. U2OS cells were applied for subsequent adhesion assays. Dulbecco's modified eagle's medium, penicillin/streptomycin, foetal bovine serum, trypsinase, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) and dimethylsulfoxide (DMSO) were purchased from Sigma–Aldrich.
2.2 Synthesis of PMMA colloidal crystal template
PMMA spheres were synthesised using an optimised version of literature techniques and packed into colloidal crystals. PMMA spheres were synthesised at 70–80 °C from mixtures of typical composition: 200 ml of water, 20 ml of MMA and 0.2 g K2 S2 O8 powder as an azo initiator. Water and MMA were added to a three‐neck round‐bottom flask, to which was attached an electric stirrer driving a glass stirring shaft with a Teflon stirrer blade, a water‐cooled condenser, a pipet connected to a house supply of nitrogen (N2) gas and a thermocouple probe attached to a temperature controller. The mixture was stirred at ∼300 rpm, while being heated to 70–80 °C and purged with N2. After stabilisation of the temperature at an elevated level, the initiator was added. The reaction was stopped after 2 h, producing colloidal PMMA spheres. The colloidal polymer was filtered through glass wool to remove any large agglomerates. PMMA colloidal crystals were formed by centrifuging the colloid at 10,000 rpm for 20 min, decanting the water and allowing the solid to dry for 12 h.
2.3 Synthesis of the 3D‐porous BGs/PMMA/ and Ti‐BGs/PMMA
The colloidal crystals were soaked in a bioglasses precursor ethanol solution for 3 h at 25 °C; herein, a bioglasses precursor was pre‐hydrolysed by mixing 4 g of P123, 0.73 g of TEP, 6.7 g of TEOS, different qualities (0, 0.17, 0.34 g) of [CH3 (CH2)3 O]4 Ti, 1.4 g of Ca(NO3)2 ·4H2 O, 1 g of HCl solution (0.5 M) and 60 g of ethanol. Afterwards, the precursor impregnated colloidal crystals were withdrawn from the solution and allowed to exposure to air for evaporation of ethanol. The above impregnation procedure was repeated for three times to ensure the fully filling of all the interstitial voids in colloid crystals by the bioglasses species. Finally, the composites were placed above HCl solution (5 ml and 0.5 M) in a 50 ml reactor kettle at 40 °C for 24 h to improve the polycondensation of silica and titanic species. After the composite was treated with CHCl3 to dissolve partial PMMA and ethanol to dissolve P123 for 8 h (repeated three times), the sample were labelled as Ti‐0 (0 g Ti (C4 H9 O)4), Ti‐1 (0.17 g Ti (C4 H9 O)4) and Ti‐2 (0.34 g Ti (C4 H9 O)4). Additionally, the precursor impregnated colloidal crystals were calcined at 550 °C to burn out the templates (PMMA and P123), the obtained hierarchical porous materials were labelled as MBG (0 g Ti (C4 H9 O)4).
2.4 Instruments for characterisation
The compressive strength of the specimens was measured by using a universal material testing machine (Instron 5569, Instron, USA) at a cross‐head speed of 1 mm/min. The morphologies of the prepared samples were characterised using a scanning electron microscope (SEM, Hitachi S4800) at an accelerating voltage of 20 kV and sample elements were analysed by an energy dispersive spectrometer (EDS), associated with SEM. Powder X‐ray diffraction (XRD) data were collected on a Bruker D8 ADVANCE diffractometer at 40 kV and 30 mA, using CuK (alpha) radiation. Transmission electron microscopy (TEM) images were recorded on an FEI Tecnai F20 instrument. The specific surface area of samples was determined using Brunauer–Emmett–Teller (BET) (NOVA 4200E Surface Area and Pore Size Analyzer, Quantachrome, USA). The pore size distributions were calculated from the adsorption branches of the N2 adsorption isotherms using the Barrett–Joyner–Halenda (BJH) model. Fourier transform infrared (FTIR) spectra were recorded on a Perkin–Elmer 580B infrared spectrophotometer using the potassium bromide pellet technique. The ultraviolet–visible (UV–vis) absorbance spectra were measured using a Shimadzu UV‐3101PC spectroscope.
2.5 Drug loading and release
In a typical loading procedure, 0.206 g samples (Ti‐0, Ti‐1 and Ti‐2) were added to 10 ml H2 O solution including MH with a concentration of 0.1 M at room temperature under stirring for 4 h. The drug‐loading materials were recovered by vacuum filtration and dried at room temperature. About 10 ml filtrate was extracted and properly diluted. Moreover, then it was analysed by UV–vis spectroscopy at a wavelength of 233 nm to determine the drug loading. The materials are immersed into 300 ml simulated body fluid (SBF, pH = 7.4) at 36.7°C in a thermostat water bath. At predetermined time intervals, 3 ml above solution has been withdrawn and immediately replaced with an equal volume of dissolution medium to keep the volume constant. The withdrawn solution was filtered (0.45 µm), properly diluted and analysed for MH content using UV–vis spectroscopy by monitoring the peak at 233 nm. Calculation of the corrected concentration of the released ibuprofen is based on the following (1):
| (1) |
where C torr is the corrected concentration at time t, Ct is the apparent concentration at time t, v is the volume of sample taken and V is the total volume of the dissolution medium.
2.6 Bioactivity studies
Ti‐2 was immersed in SBF (Na+ 142, K+ 5, Mg2+ 1.5, Ca2+ 2.5, Cl− 147.8, HCO3 − 4.2, HPO4 2− 41 and SO4 2− 0.5 mM) for different times (4, 12, 24, 48 and 96 h) at 37 °C were analysed by SEM to determine their apatite‐formation capability. Furthermore, the scaffolds soaked in SBF for 96 h were further examined by EDS spectra.
2.7 Proliferation of U2OS cells on the Ti‐0, Ti‐1 and Ti‐2 scaffolds
Cell proliferation was measured by MTT method. Briefly, the sterilised scaffolds were placed into 24‐well culture plates and incubated with 1 × 105 U2OS cells in 1.2 ml of proliferation medium for 48 h. After removing the medium, 1.0 ml of 0.5 mg/ml MTT solution was added in each well and incubated for 4 h at 37 °C. After the removal of MTT solution by the adsorption with dry papers, the scaffolds were transferred to 1.5 ml tubes and 500 μl of DMSO was added to each tube. After the formazan was dissolved completely in DMSO, 100 μl of formazan solution was transferred to 96‐well plate and the absorbances were recorded using a microplate reader (SynergyTMHT, BioTek Instruments Inc.) at 570 nm. The results were expressed as the absorbance reading from each well less the optical density value of a blank.
2.8 Antimicrobial test
After comparing tests to choose the best concentration of Escherichia coli, the best quality of the materials and test conditions, the optimal test concentration is 1.6 μl of E. coli was dissolved in 50 ml of physiological saline. Moreover, 0.1 mg of Ti‐0, Ti‐1 and Ti‐2 were added in 2 ml of diluent colibacillus solution, respectively. Then, the samples were cultivated for 1 h in table concentrator at 37 °C. The samples were centrifuged at 1500 r/min for 5 min, and then 20 μl of superstratum clear liquid were homogeneous spread on culture medium, respectively. The culture dishes were cultivated for 24 h in 37 °C constant temperature incubator. After 1 day, the colony amounts were counted out and took a picture, to determine the bactericidal capacity of the materials by the number of bacterial colonies on the petri dish after 24 h. Calculation of kill rate for colibacillus is based on the following (2):
| (2) |
where N 0 is the amount of survived colibacillus of blank sample and N is the amount of survived colibacillus number of the mesoporous material sample.
3. Results and discussion
The fabrication of 3D‐macro‐mesoporous Ti‐BGs/PMMA is illustrated in Scheme 1. First, the macroporous template was obtained through the centrifugation of a homogeneous and stable aqueous dispersion containing monodisperse PMMA colloids. Then, the obtained PMMA scaffold was immersed into the Ti‐mBGs precursor, after the voids were filled completely it was taken out of the precursor solution and dried in vacuum. Afterwards, the acid steam technique was adopted to improve the polycondensation of Ti‐BGs and to preserve PMMA component. After extraction in ethanol, P123 was removed to form the mesoporous structure. Followed by dipping in CHCl3, the partial dissolution of PMMA induced the 3D‐macroporous structure of the sample. Moreover, the 3D‐macro‐mesoporous structure was obtained and hoped to improve the cell migration, delivery of nutrients and gases and eventual vascularisation. In this procedure, PMMA was not only used as the macroporous template but also the components of the composites, which were expected to possess the enhanced mechanical strength of the composite.
Scheme 1.
Synthesis procedure for the antimicrobial 3D ordered macro‐mesoporous Ti‐BGs/PMMA
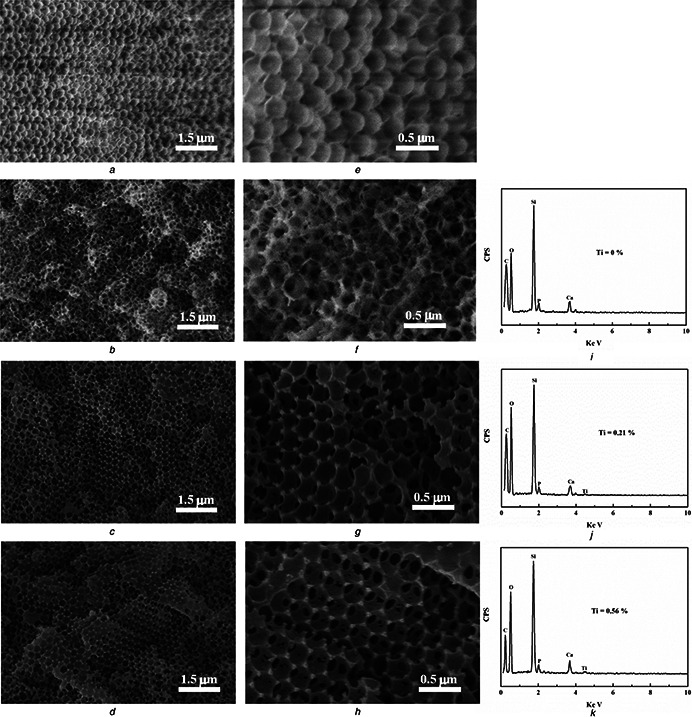
Fig. 1 a shows the SEM image of the monodisperse PMMA colloids synthesised through a soap‐free emulsion polymerisation method. The high‐resolution SEM image shows that the PMMA colloids possess a uniform size of 300 nm (Fig. 1 e). It can be seen that Ti‐0, Ti‐1 and Ti‐2 retain the highly ordered 3D macropore with the interconnected structure (Figs. 1 b –d), testifying the replication of the structure from PMMA scaffold. Furthermore, all samples reveal the decreased macroporous diameters (200–250 nm) in contrast with PMMA template (300 nm) that is ascribed to the partial dissolution of PMMA and the residual is left on the porous walls. The EDS analyses of all samples were recorded as shown in Figs. 1 i –k. It indicates that Ti‐0 was only composed of Si, Ca, P, O and C elements derived from BGs and PMMA. Besides these elements, Ti is found in Ti‐1 and Ti‐2 with the atom per cent about 0.21 and 0.56%, respectively. As displaced in Figs. 1 i –k, it is clearly revealed that Ti has been doped in Ti‐1 and Ti‐2 successfully.
Fig. 1.

SEM images of PMMA, Ti‐0, Ti‐1 and Ti‐2:low‐resolution images (a, b, c, d) and high‐resolution images (e, f, g, h) and EDS spectrum of Ti‐0 (i), Ti‐1 (j) and Ti‐2 (k)
Moreover, TEM analysis was used to further investigate the porous structures of Ti‐0, Ti‐1 and Ti‐2. From Fig. 2, Ti‐0 exhibits the 3D ordered macroporous arrays copying from PMMA template. In the wall, the mesopore, derived from the micellisation of P123, with straight channels can be found, obviously. After the addition of Ti precursor, Ti‐1 and Ti‐2 also reveal the similar mesoporous structure as Ti‐0. This further proved that the Ti (C4 H9 O)4 did not destroy the mesoporous structures in Ti‐1 and Ti‐2.
Fig. 2.
TEM image of the porous supported
a Ti‐0 scaffold
b Ti‐1 scaffold
c Ti‐2 scaffold
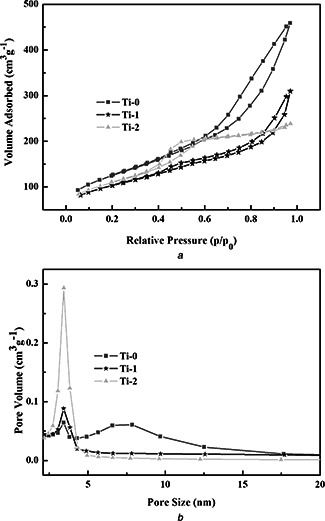
The surface structures of the as‐fabricated materials were analysed by N2 sorption isotherm techniques. The N2 adsorption–desorption isotherms and the BJH pore size distribution curves of Ti‐0, Ti‐1 and Ti‐2 are shown in Fig. 3, respectively. The isotherms show an adsorption hysteresis and narrow peak of the samples in the BJH pore size distribution curves about 3.4 nm for all the samples. That is in agreement with the TEM images. In Fig. 3 b, there is a broad peak of Ti‐0 at 7 nm and the pore size decreased with the increasing of Ti, this is because the introduced Ti joined the formation of the BGs matrix. The corresponding BET surface area and pore volume are listed in Table 1. BET surface areas and pore volumes of Ti‐0, Ti‐1 and Ti‐2 are 442 m2 g−1 and 0.66 cm3 g−1, 357 m2 g−1 and 0.41 cm3 g−1 and 385 m2 g−1 and 0.33 cm3 g−1, respectively.
Fig. 3.

N2 adsorption–desorption isotherms and the BJH pore size distribution curves of Ti‐0, Ti‐1 and Ti‐2
a N2 adsorption–desorption isotherm
b Pore size distribution curve of Ti‐0, Ti‐1 and Ti‐2
Table 1.
Structure parameters and the drug‐loading rate of Ti‐0, Ti‐1 and Ti‐2
| Sample | S BET, m2 /g | Vp , cm3 /g | Dp , nm | Drug loading, % |
|---|---|---|---|---|
| Ti‐0 | 442 | 0.66 | 3.4 | 21.84 |
| Ti‐1 | 357 | 0.41 | 3.4 | 20.06 |
| Ti‐2 | 385 | 0.33 | 3.4 | 20.01 |
FTIR was used to obtain the information of chemical bonds. The FTIR spectra of PMMA, Ti‐0, Ti‐1, Ti‐2, MH‐Ti‐0, MH‐Ti‐1 and MH‐Ti‐2 are presented in Fig. 4. All the samples show adsorption bond at 2929 and 1736 cm−1 indexed as −CH3 and C=O of PMMA. The absorption bands of –OH (3425 cm−1), Si–O–Si antisymmetric stretching vibration (1088 cm−1), Si–O–Si symmetric stretching vibration (810 cm−1) and bending vibration Si–O (469 cm−1) indicate the amorphous silica framework in Ti‐0, Ti‐1 and Ti‐2 (Figs. 4 b –d). Another weak band at 662 cm−1 is corresponding to Si–O–Ti bond in the tetrahedral coordination, testifying that –Ti–O– is incorporated into –Si–O– network in Ti‐1 and Ti‐2. As shown in FTIR spectra of MH‐Ti‐0, MH‐Ti‐1 and MH‐Ti‐2, the absorption peaks located at 3170 and 3360 cm−1 assigned to the stretching vibrations of −NH group that confirms the successful loading of MH molecules onto the scaffolds (Figs. 4 e –g). The MH‐loading capacity is calculated and shown in Table 1. The porous structure causes the high loading amount. With the largest surface area, Ti‐0 possesses 21.84 wt% loading amount, followed with Ti‐1 (20.06 wt%) and Ti‐2 (20.01 wt%), respectively.
Fig. 4.

FTIR spectra of
a PMMA
b Ti‐0
c Ti‐1
d Ti‐2
e MH‐Ti‐0
f MH‐Ti‐1
g MH‐Ti‐2
Fig. 5 shows the release behaviour of MH from the different materials in SBF. In the first hour, MH release about 22.5, 20.6 and 19.1% from Ti‐0, Ti‐1 and Ti‐2, respectively. The initial fast release of HM is attributed to MH molecules adsorbed on the surface of the sample. Moreover, the rapid diffusion of MH molecules from the macroporous structure also leads to the fast initial release. Owing to the drug stored in mesoporous channels diffuses to macropore needs a longer‐time penetration of the medium, so the controlled release sustains to 12 h and the cumulative release reaches 35.1, 27.8 and 27.4% of Ti‐0, Ti‐1 and Ti‐2, respectively. From the drug release curves, it can be seen that Ti‐0 possesses the highest release rate and final release amount than that of Ti‐1 and Ti‐2, ascribing to the largest mesoporous pore of Ti‐0 as revealed in Fig. 3 b.
Fig. 5.

Drug cumulative release profile of Ti‐0, Ti‐1 and Ti‐2
The bioactivity of BGs scaffold, reflected by the ability of inducing mineralisation of HAP, could finally affect the successful transplantation in bone tissue engineering. In this paper, the inducing growth of HAP in vitro was carried out by soaking Ti‐2 in SBF at 37 °C for 4, 12, 24, 48 and 96 h (Figs. 7 a –e). As compared with 3D‐macroporous structure before immersing in SBF (Fig. 1 d), the macroporous walls of Ti‐2 had become thicker gradually with increase of the immersing time. After 96 h, the macropores have been finally blocked completely. From the EDS spectrum of Ti‐2 after 96 h immersion in SBF, it indicates that the precipitated layer is composed of Ca and P with a Ca/P atomic ratio of 1.6 in Fig. 6 f, which is close to the theoretical value 1.67 Ca/P ratio of HAP, implying the precipitated layer on the surface of the sample is HAP.
Fig. 7.
Wide‐angle XRD patterns of Ti‐2 before (a) and after (b) soaking in SBF for 96 h
Fig. 6.

SEM images of the Ti‐2 after soaking in the SBF for
a 4
b 12
c 24
d 48
e 96 h
f EDS from (e)
To further testify the mineralisation of HAP, the wide‐angle XRD patterns of Ti‐2 before (a) and after (b) soaking in SBF for 96 h are recorded and shown in Fig. 7. The diffraction peaks appearing at 31.7°, 45.3° and 56.1° can be well assigned to correspond to the (211), (203) and (313) crystal planes of HAP (JCPDS no. 09‐0432). From Fig. 6, after immersing in SBF for 4 h, there is HAP mineralisation on the surface that benefits from the various porous structure and large surface area of the sample. It is known that HAP is the main inorganic component of the bone and the spontaneous inorganic mineralisation of HAP makes these materials have potential applications in bone tissue regeneration [22, 23].
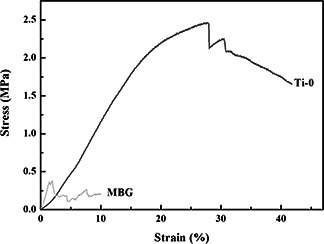
Besides the HAP inducing growth, the suitable mechanical strength is another key factor to the bone tissue regeneration. It is known that the porous materials always possess a low mechanical strength. Here, PMMA was adopted not only as the macroporous template but also as the component of the scaffold in the hope of enhancing mechanical strength. In Fig. 8, stresses and strains were plotted from the compression data (sample volume: about 1 cm3 and test number: three times). The pure MBG reveals a low compressive strength of 0.3 MPa, which then increases to 2.4 MPa for Ti‐0 that is close to the lower values for spongy bone. The improved compressive strength attributes to the residual PMMA, a typical compressive polymer, in the wall.
Fig. 8.
Stress–strain curves of Ti‐0 and MBG
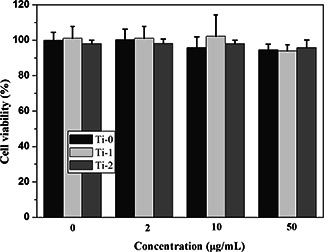
The cell proliferations of U2OS after incubated with Ti‐0, Ti‐1 and Ti‐2 for 48 h were determined quantitatively by MTT assessment (Fig. 9). All of them reveal the negligible cytotoxicity and even when the concentration increases to 50 μg/ml, the cell viability also over 93% for all samples. P values (P Ti‐0 = 0.9999, P Ti‐1 = 0.9838 and P Ti‐2 = 0.9976) were not obvious difference. From Fig. 9, it is confirmed that the as synthesised scaffold does not affect the proliferations of the cells.
Fig. 9.
U2OS cell viabilities after 48 h incubated with Ti‐0, Ti‐1 and Ti‐2 at different concentrations
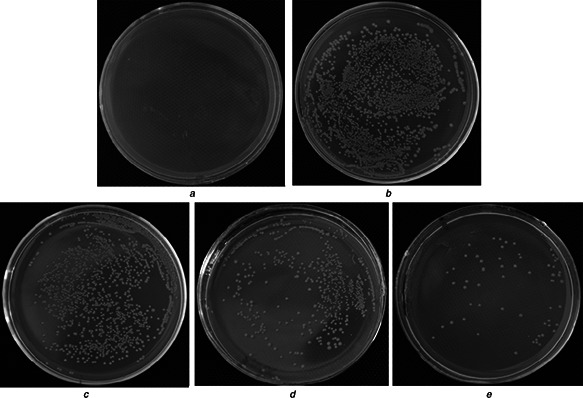
Figs. 10 and 11 exhibit the antimicrobial property of these samples. The colibacillus amounts were counted out and the kill rates were calculated through (2). Each value in the result is the average of the results of three parallel experiments. There are only normal saline solution (Fig. 10 a) and the colibacillus (Fig. 10 b) as the antithesis, which proves the solution is clean and the colibacillus is in good condition. In Figs. 10 c –e, it clearly indicates that these materials display the improved antimicrobial ability with increase of Ti amount. Moreover, the kill rates for colibacillus of Ti‐0, Ti‐1 and Ti‐2 reach 26.48, 76.97 and 94.86%, respectively (Fig. 11 b). The enhanced antimicrobial abilities of Ti‐1 and Ti‐2 depends on the evolved −OH free radicals, which demonstrate stronger oxidisability to organic matters of bacteria. Above all, the Ti‐doped macro‐mesoporous BGs/PMMA displays the excellent antimicrobial property that is significant for the bacterial infection in bone replacement.
Fig. 10.

Antibacterial results of samples for 24 h
a Blank
b Normal saline solution
c Ti‐0
d Ti‐1
e Ti‐2
Fig. 11.
Bar charts of
a Colonies number
b Sterilisation rate
4. Conclusions
The macro‐mesoporous Ti‐BGs/PMMA scaffolds were synthesised using P123 as the mesoporous template and PMMA as the macroporous template. To obtain the inorganic–organic composites in a one‐pot system, the acid steam technique was adopted to improve the polycondensation of Ti‐BGs. Moreover, PMMA was then partially removed by CHCl3 to produce the macroporous structure, and the residual PMMA remained in the wall to improve the compressive strength to 2.4 MPa (0.3 MPa for pure MBG). It is a simple and green method to synthesize the macro‐mesoporous Ti‐BGs/PMMA. With 3D interconnected hierarchical structure (250 and 3.4 nm), the material demonstrates the fast inducing‐HAP growth (4 h) and the controlled drug release. Furthermore, the scaffold also exhibits good antimicrobial property and biocompatible, associating with the above investigation (bioactivity and mechanical strength) to be expected to the potential application for bone tissue regeneration.
5 Acknowledgments
Financial support for this study was provided by the National Natural Science Foundation of China (21171045 and 21101046), the Natural Science Foundation of Heilongjiang Province of China ZD201214, the Program for Scientific and Technological Innovation team Construction in Universities of Heilongjiang province (2011TD010), and the Technology development pre‐project of Harbin Normal University (12XYG‐11).
6 References
- 1. Xia Y. Mei F. Duan Y.L. et al.: ‘Bone tissue engineering using bone marrow stromal cells and an injectable sodium alginate/gelatin scaffold’, J. Biomed. Mater. Res. A, 2012, 100A, pp. 1044 –1050 (doi: 10.1002/jbm.a.33232) [DOI] [PubMed] [Google Scholar]
- 2. Pulavendran S. Rajam M. Rose C. et al.: ‘Hepatocyte growth factor incorporated chitosan nanoparticles differentiate murine bone marrow mesenchymal stem cell into hepatocytes in vitro’, IET Nanobiotechnol., 2010, 4, pp. 51 –60 (doi: 10.1049/iet-nbt.2009.0014) [DOI] [PubMed] [Google Scholar]
- 3. Suchy T. Ryglova S. Balik K. et al.: ‘Biological evaluation of polydimethylsiloxane modified by calcium phosphate nanoparticles for potential application in spine surgery’, Sci. Adv. Mater., 2013, 5, pp. 484 –493 (doi: 10.1166/sam.2013.1478) [DOI] [Google Scholar]
- 4. Berbecaru C. Alexandru H.V. Ianculescu A. et al.: ‘Bioglass thin films for biomimetic implants’, Appl. Surf. Sci., 2009, 255, pp. 5476 –5479 (doi: 10.1016/j.apsusc.2008.08.020) [DOI] [Google Scholar]
- 5. Hafezi F. Hosseinnejad F. Fooladi A.A.I. Mafi S.M. Amiri A. Nourani M.R.: ‘Transplantation of nano‐bioglass/gelatin scaffold in a non‐autogenous setting for bone regeneration in a rabbit ulna’, J. Mater. Sci., Mater. Med., 2012, 23, pp. 2783 –2792 (doi: 10.1007/s10856-012-4722-3) [DOI] [PubMed] [Google Scholar]
- 6. Meng D.C. Rath S.N. Mordan N. Salih V. Kneser U. Boccaccini A.R.: ‘In vitro evaluation of 45S5 Bioglass® ‐derived glass‐ceramic scaffolds coated with carbon nanotubes’, J. Biomed. Mater. Res. A, 2011, 99A, pp. 435 –444 (doi: 10.1002/jbm.a.33185) [DOI] [PubMed] [Google Scholar]
- 7. Xynos I.D. Edgar A.J. Buttery L.D.K. et al.: ‘Ionic products of bioactive glass dissolution increase proliferation of human osteoblasts and induce insulin‐like growth factor II mRNA expression and protein synthesis’, Biochem. Biophys. Res. Commun., 2000, 276, pp. 461 –465 (doi: 10.1006/bbrc.2000.3503) [DOI] [PubMed] [Google Scholar]
- 8. Mozafari M. Moztarzadeh F. Tahriri M.: ‘Investigation of the physico‐chemical reactivity of a mesoporous bioactive SiO2 –CaO–P2 O5 glass in simulated body fluid’, J. Non‐Cryst. Solids, 2010, 356, pp. 1470 –1478 (doi: 10.1016/j.jnoncrysol.2010.04.040) [DOI] [Google Scholar]
- 9. Mozafari M. Rabiee M. Azami M. et al.: ‘Biomimetic formation of apatite on the surface of porous gelatin/bioactive glass nanocomposite scaffolds’, Appl. Surf. Sci., 2010, 257, pp. 1740 –1749 (doi: 10.1016/j.apsusc.2010.09.008) [DOI] [Google Scholar]
- 10. Marelli B. Ghezzi C.E. Alessandrino A. Barralet J.E. Freddi G. Nazhat S.N.: ‘Silk fibroin derived polypeptide‐induced biomineralization of collagen’, Biomaterials, 2012, 33, pp. 102 –108 (doi: 10.1016/j.biomaterials.2011.09.039) [DOI] [PubMed] [Google Scholar]
- 11. Yan X.X. Huang X.H. Yu C.Z. et al.: ‘The in‐vitro bioactivity of mesoporous bioactive glasses’, Biomaterials, 2006, 27, pp. 3396 –3403 (doi: 10.1016/j.biomaterials.2006.01.043) [DOI] [PubMed] [Google Scholar]
- 12. Wu F. Liu C.S. O'Neill B. et al.: ‘Fabrication and properties of porous scaffold of magnesium phosphate/polycaprolactone biocomposite for bone tissue engineering’, Appl. Surf. Sci., 2012, 258, pp. 7589 –7595 (doi: 10.1016/j.apsusc.2012.04.094) [DOI] [Google Scholar]
- 13. Liu W.Y. Yeh Y.C. Lipner J. et al.: ‘Enhancing the stiffness of electrospun nanofiber scaffolds with a controlled surface coating and mineralization’, Langmuir, 2011, 27, pp. 9088 –9093 (doi: 10.1021/la2018105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Annabi N. Tamayol A. Uquillas J.A. et al.: ‘25th anniversary article: rational design and applications of hydrogels in regenerative medicine’, Adv. Mater., 2014, 26, pp. 85 –124 (doi: 10.1002/adma.201303233) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ginsac N. Chenal J.M. Meille S. et al.: ‘Crystallization processes at the surface of polylactic acid‐bioactive glass composites during immersion in simulated body fluid’, J. Biomed. Mater. Res. B, 2011, 99B, pp. 412 –419 (doi: 10.1002/jbm.b.31913) [DOI] [PubMed] [Google Scholar]
- 16. Erol M.M. Mournio V. Newby P. et al.: ‘Copper‐releasing, boron‐containing bioactive glass‐based scaffolds coated with alginate for bone tissue engineering’, Acta Biomat., 2012, 8, pp. 792 –801 (doi: 10.1016/j.actbio.2011.10.013) [DOI] [PubMed] [Google Scholar]
- 17. Wei G. Ma Peter X.: ‘Structure and properties of nano‐hydroxyapatite/polymer composite scaffolds for bone tissue engineering’, Biomaterials, 2004, 25, pp. 4749 –4757 (doi: 10.1016/j.biomaterials.2003.12.005) [DOI] [PubMed] [Google Scholar]
- 18. Bretcanu O. Misra S. Roy I. et al.: ‘In vitro biocompatibility of 45S5 Bioglass® ‐derived glass‐ceramic scaffolds coated with poly(3‐hydroxybutyrate)’, J. Tissue Eng. Regen. Med., 2009, 3, pp. 139 –148 (doi: 10.1002/term.150) [DOI] [PubMed] [Google Scholar]
- 19. Stranak V. Wulff H. Rebl H. et al.: ‘Deposition of thin titanium–copper films with antimicrobial effect by advanced magnetron sputtering methods’, Mater. Sci. Eng. C, Mater., 2011, 31, pp. 1512 –1519 (doi: 10.1016/j.msec.2011.06.009) [DOI] [Google Scholar]
- 20. Vallet‐Regi M. Ruiz‐Hernandez E.: ‘Bioceramics: from bone regeneration to cancer nanomedicine’, Adv. Mater., 2011, 23, pp. 5177 –5218 (doi: 10.1002/adma.201101586) [DOI] [PubMed] [Google Scholar]
- 21. Wu C.T. Fan W. Zhu Y.F. et al.: ‘Multifunctional magnetic mesoporous bioactive glass scaffolds with a hierarchical pore structure’, Acta Biomater., 2011, 7, pp. 3563 –3572 (doi: 10.1016/j.actbio.2011.06.028) [DOI] [PubMed] [Google Scholar]
- 22. Mortera R. Fiorilli S. Garrone E. et al.: ‘Pores occlusion in MCM‐41 spheres immersed in SBF and the effect on ibuprofen delivery kinetics: a quantitative model’, Chem. Eng. J., 2010, 156, pp. 184 –192 (doi: 10.1016/j.cej.2009.10.018) [DOI] [Google Scholar]
- 23. Baino F. Fiorilli S. Mortera R. et al.: ‘Mesoporous bioactive glass as a multifunctional system for bone regeneration and controlled drug release’, J. Appl. Biomater. Funct., 2012, 10, pp. 12 –21 [DOI] [PubMed] [Google Scholar]