

Abstract
Poly(glycerol sebacate) (PGS) is a new biodegradable polymer with good biocompatibility used in many fields of biomedicine and drug delivery. Sunitinib‐loaded PGS/gelatine nanoparticles were prepared by the de‐solvation method for retinal delivery and treatment of diabetic retinopathy. The nanoparticles were characterised by Fourier‐transform infrared and differential scanning calorimetry. The effects of different formulation variables including drug‐to‐carrier ratio, gelatine‐to‐PGS ratio, and glycerine‐to‐sebacate ratio were assessed on the encapsulation efficiency (EE%), particle size, release efficiency (RE), and zeta potential of the nanoparticles. The in vitro cytotoxicity of PGS/gelatine nanoparticles was studied on L929 cells. Draize test on rabbit eyes was also done to investigate the possible allergic reactions caused by the polymer. Glycerine/sebacic acid was the most effective parameter on the EE and RE. Gelatine‐to‐PGS ratio had the most considerable effect on the particle size while the RE was more affected by the glycerine/sebacic acid ratio. The optimised formulation (S1 G0.7 D21.2) exhibited a particle size of 282 nm, 34.6% EE, zeta potential of −8.9 mV, and RE% of about 27.3% for drug over 228 h. The 3‐(4,5‐dimethylthuazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide assay indicated PGS/gelatine nanoparticles were not cytotoxic and sunitinib‐loaded nanoparticles were not toxic at concentrations <36 nM.
Inspec keywords: polymers, differential scanning calorimetry, toxicology, drug delivery systems, solvation, eye, encapsulation, particle size, drugs, biodegradable materials, nanofabrication, nanomedicine, nanoparticles, gelatin, Fourier transform infrared spectra
Other keywords: gelatine‐to‐PGS ratio; glycerine‐to‐sebacate ratio; particle size; zeta potential; sunitinib‐loaded nanoparticles; biodegradable polymer; retinal delivery; differential scanning calorimetry; drug‐to‐carrier ratio; allergic reactions; physicochemistry; cytotoxicity; poly(glycerol sebacate) nanoparticles; sunitinib ocular delivery; drug delivery; sunitinib‐loaded PGS‐gelatine nanoparticles; Fourier‐transform; in vitro cytotoxicity; biocompatibility; Draize test; rabbit eyes; 3‐(4,5‐dimethylthuazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide assay
1 Introduction
Diabetic retinopathy (DR) is a diabetes complication that affects eyes and damage blood vessels in the retina. If DR is detected, tightening of modifiable risk factors (e.g. blood glucose and blood pressure) can slow the disease progression. When sight‐threatening retinopathy is detected, laser treatment and treatment with vascular endothelial growth factor (VEGF) inhibitors reduce the risk of visual loss [1]. VEGF is very expensive and treatment must be applied frequently, which in turn lowers the patient's compliance. So finding an alternative treatment with more efficacy is essentially felt [2]. Protein kinase C (PKC) inhibitors are new well‐known treatments for DR [3]. PKC has 13 enzymes in their family, of which the β ‐isoform has been assessed more than the other isoforms for diabetic microvascular complications issue. Induction mechanism of PKC is due to an increased diacyl glycerol level followed by increases in retina vascular permeability and neovascularisation in diabetic patients. PKC has a significant role in the signalling of VEGF, which is a major mediator of retinal neovascularisation and permeability in diabetes [4]. So inhibition of this protein could be a helpful way to control DR [5, 6, 7]. For example, ruboxistaurin is a PKC‐β inhibitor that reduced the risk of vision loss and macular oedema up to 40% in DR patients and diminished the need for laser therapy compared to the control group [8, 9]. PKC412 is another PKC inhibitor that is reported to decrease macular oedema significantly in diabetic patients [10]. Sunitinib is also a member of this family widely used for curing some cancers by neovascularisation inhibition, which seems to be a good choice for DR treatment [11].
Poly(glycerol sebacate) (PGS) is a new biodegradable elastic polymer obtained from condensation polymerisation of glycerol and sebacic acid that has tunable mechanical properties to match the requirements of intended applications by controlling curing time, curing temperature, and reactants concentration. In addition, this polymer has good biocompatibility and proper surface degradation profile [12]. Both glycerol and sebacic acid are endogenous components; thus, the degradation products of PGS are often naturally metabolised in the body. In vivo biocompatibility analysis indicates that PGS has a favourable tissue response with little inflammation as a widely utilised biomaterial [13].
Applications of PGS are being expanded to include drug delivery, tissue adhesive, soft and hard tissue (i.e. bone) regeneration. Numerous studies have demonstrated that drug release from PGS carriers is being sustained enough to provide a continuous concentration of cargo drug within the therapeutic window and reduce its toxicity [14]. Some studies have reported that the geometry of PGS carrier is kept constant during the degradation period of 30 days in phosphate‐buffered saline (PBS) and drug release happens within 7 days [15]. Loading the drugs and active molecules into PGS carriers represent some challenges including high temperature needed for drug loading, which is intolerable for many drugs [16], low efficiency of loading and fast release of the hydrophilic drugs due to the hydrophobic nature of this polymer. In order to achieve desirable physicochemical properties of PGS, its combination with an appropriate hydrophilic polymer could be effective.
On the other hand, a cheaper, easier and significantly simpler preparation process for drug loading in PGS polymer is needed [17]. Surface coating of PGS with biocompatible molecules such as laminin, fibronectin, fibrin, and collagen types I/III or elastin is suggested to improve the biocompatibility and physicochemical properties of PGS [18]. These molecules are natural components of the cellular environment and therefore their use as the coating will provide an additional impetus for improving the material–cell interactions and can expand the application potential of PGS.
Over the past few decades, there has been considerable interest in developing protein‐based carriers such as drug delivery devices. By considering medical usage and physicochemical properties of gelatine, this protein could be a good choice for this purpose [19, 20]. Gelatine obtained from partial acid or alkaline hydrolysis of animal collagen is one of the most versatile natural biopolymers widely used in pharmaceutical industries due to its exceptional characteristics [21, 22, 23] including biocompatibility, low‐cost, biodegradability, no antigenicity, abundant renewable sources, extraordinary binding capacity, and active groups for coupling ligands and cross linkers [22, 24]. These advantages have led to its wide applications in drug and gene delivery systems during the last 30 years [25, 26, 27].
Although in some cases, PGS and gelatine were used together in preparation of nanofibres and membranes for tissue engineering applications there is no report on the preparation of their nanoparticles as a drug delivery system [17, 19]. The only report relates to the preparation of PGS nanoparticle by Louage et al. [14], who synthesised PGS nanoparticles in ethanol by the solvent displacement method for encapsulation of hydrophobic anti‐cancer drugs.
In this study, PGS/gelatine nanoparticles were prepared by the desolvation method. A combination of PGS and gelatine could highlight their benefits and overcome the shortcoming of both hydrophilic and hydrophobic systems. Integrating appropriate properties of PGS and gelatine can create a new biocompatible and designable nano‐carrier for delivering water‐soluble and ‐insoluble drugs. PGS/gelatine nanoparticles were synthesised for carrying sunitinib to the retina by a controlled release manner for treatment of DR.
2 Materials and methods
2.1 Materials
Sebacic acid (molecular weight [M w] = 258.35 g/mol), gelatine, and glycerol were purchased from Merck Chemical Company (Germany). Glutaraldehyde was from Sigma (USA) and sunitinib malate was kindly donated by Parsian Pharmaceutical Company (Iran). L929 cell line was from Royan Institute (Iran). Foetal bovine serum (FBS) (GIBCO Laboratories, USA), Dulbecco's PBS (Bioidea, Iran), Roswell Park Memorial Institute medium (RPMI) was from Gibco BRL (Grand Island, NY, USA). 3‐(4,5‐Dimethylthuazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) was purchased from Sigma Chemicals (St. Louis, MO, USA). Tissue culture plates and flasks were from Corning Life Sciences (Corning, NY, USA). Deionised water freshly purged with nitrogen gas was used in all steps for preparing aqueous solutions.
2.2 Synthesis of PGS
PGS was synthesised as previously reported by Wang et al. [28]. Briefly, 2.6, 5.1 and 10.1 g of sebacic acid and 2.85 g of anhydrous glycerol were charged into a 50‐ml three neck round bottom flask to synthesise different types of PGS containing 0.5:1, 1:1 and 2:1 molar ratio of sebacic acid to glycerol ratio, respectively. The reactants were magnetically stirred at 400 rpm and heated under a dry nitrogen blanket to 120°C. After 24 h, the nitrogen line was removed and the flask was placed under vacuum with <50 m Torr pressure for an additional 24 h to yield the viscous PGS. After at least 24 h storing at room temperature, the pre‐polymer was changed from a soft waxy state to a mature solid white texture.
2.3 Box–Behnken experimental design for preparation of nanoparticles
The Box–Behnken design is appropriate for exploring the effect of variables in different levels on desired responses with fitting designs. This experimental design is able to predict the best situation for preparing nanoparticles upon analysing the collected data. Optimisation by the software was done due to the determined desirable responses range [29, 30]. Based on the Box–Behnken design, 17 formulations were suggested by Design Expert Software (version 7.2, Stat‐Ease, USA) in which drug (D), gelatine‐to‐PGS ratio (G) and glycerine‐to‐sebacate ratio (S) were the studied variables. The particle size, zeta potential, encapsulation efficiency (EE%) and RE were assessed as responses. P < 0.05 indicated model terms were significant. The optimum formulation was considered as one with the minimum particle size and drug RE while the maximum EE% and zeta potential being in the range of the obtained values (Table 1). Then the actual and suggested responses of the optimised formulation by the software were compared to assess the precision of the model.
Table 1.
Studied formulations of PGS/gelatine nanoparticles loaded with sunitinib malate
| Code | Sebacic acid/glycerol | Gelatine/PGS | Drug/carrier, % | Particle size ±SD, nm | PDI | Zeta potential, mV | EE% | RE150 % |
|---|---|---|---|---|---|---|---|---|
| S1 G6.5 D20 | 1 | 6.5 | 20 | 484.3 ± 12.8 | 0.3 ± 0.1 | 6.8 | 42.6 ± 0.1 | 22.2 ± 3.9 |
| S1 G6.5 D20 | 1 | 6.5 | 20 | 479.6 ± 54.8 | 0.4 ± 0.3 | 9.8 | 42.2 ± 0.0 | 21.3 ± 2.2 |
| S1 G3 D10 | 1 | 3 | 10 | 446.6 ± 38.8 | 0.5 ± 0.1 | 6.7 | 20.5 ± 0.0 | 35.8 ± 2.5 |
| S1 G6.5 D20 | 1 | 6.5 | 20 | 230.3 ± 3.5 | 0.5 ± 0.1 | 8.1 | 43.8 ± 0.1 | 19.8 ± 2.7 |
| S2 G6.5 D30 | 2 | 6.5 | 30 | 340.7 ± 53.7 | 0.5 ± 0.1 | 20.1 | 21.5 ± 0.2 | 28.9 ± 4.8 |
| S1 G6.5 D20 | 1 | 6.5 | 20 | 231.7 ± 21.2 | 0.5 ± 0.1 | 2.3 | 42.1 ± 1.1 | 18.9 ± 2.5 |
| S1 G10 D10 | 1 | 10 | 10 | 270.3 ± 29.1 | 0.5 ± 0.1 | 7.9 | 20.6 ± 1.6 | 25.1 ± 0.9 |
| S2 G3 D20 | 2 | 3 | 20 | 418.3 ± 0.5 | 0.3 ± 0.2 | 14.7 | 25.3 ± 0.1 | 17.2 ± 1.1 |
| S0.5 G10 D20 | 0.5 | 10 | 20 | 247.7 ± 5.1 | 0.4 ± 0.1 | 9.6 | 17.5 ± 0.1 | 81.2 ± 3.4 |
| S1 G10 D30 | 1 | 10 | 30 | 268.3 ± 17.2 | 0.5 ± 0.2 | 14.9 | 23.8 ± 0.3 | 25.9 ± 1.7 |
| S2 G6.5 D10 | 2 | 6.5 | 10 | 412.6 ± 16.7 | 0.3 ± 0.1 | 14.1 | 19.1 ± 0.3 | 45.7 ± 1.6 |
| S1 G3 D30 | 1 | 3 | 30 | 472.3 ± 6.8 | 0.4 ± 0.1 | 9.6 | 36.3 ± 1.0 | 12.8 ± 1.6 |
| S2 G10 D20 | 2 | 10 | 20 | 319.3 ± 10.9 | 0.4 ± 0.1 | 8.9 | 22.3 ± 1.2 | 55.7 ± 5.9 |
| S0.5 G3 D20 | 0.5 | 3 | 20 | 379.3 ± 18.9 | 0.5 ± 0.1 | 10.3 | 27.8 ± 0.1 | 30.5 ± 2.0 |
| S0.5 G6.5 D30 | 0.5 | 6.5 | 30 | 250.7 ± 40.1 | 0.5 ± 0.1 | 11.3 | 28.1 ± 0.3 | 63.5 ± 3.1 |
| S0.5 G6.5 D10 | 0.5 | 6.5 | 10 | 350.7 ± 3.9 | 0.3 ± 0.2 | 15.3 | 24.9 ± 0.4 | 92.5 ± 2.8 |
| S1 G6.5 D20 | 1 | 6.5 | 20 | 305.3 ± 7.1 | 0.4 ± 0.1 | 5.9 | 41.7 ± 1.8 | 22.5 ± 2.8 |
2.4 Preparation of sunitinib‐loaded PGS/gelatine nanoparticles
Sunitinib‐loaded PGS/gelatine nanoparticles were prepared by desolvation technique. In the first step, sunitinib and gelatine were dissolved in deionised water then PGS solution was prepared separately in EtOH. Three ml of alcoholic solution of PGS was added drop wise to 4 ml of gelatine solution while stirred at 900 rpm. Afterwards, 0.5 ml of glutaraldehyde (25%) was added drop wise to reaction mixture and stirred for 3 h at room temperature to mature cross‐linking reaction in PGS/gelatine nanoparticles.
The free un‐entrapped drug was separated by centrifugation at 10,000 rpm for 15 min at room temperature to remove unloaded drug from the surface of nanoparticles and EE% was calculated by measuring the amount of free drug in the centrifuged clear solution by using an ultraviolet spectrophotometer (UV mini 1240, Shimadzu, Japan) at 428 nm using (1). A standard curve of sunitinib in water was used to change the absorbance of the solution to concentration. This standard curve was linear over the concentration range of 1.0–50.0 µg/ml. All measurements were carried out in triplicate
| (1) |
2.5 Particle size, polydispersity index, and zeta potential of PGS/gelatine nanoparticles
The particle size, polydispersity index, and zeta potential of drug‐loaded PGS/gelatine nanoparticles were measured by the dynamic light scattering (DLS) method using the Malvern Nanosizer (ZEN3600, UK). Samples were dispersed in water and sonicated in a bath sonicator for 2 min and measurements were carried out in triplicate at room temperature [30].
2.6 Fourier transform infrared (FTIR) spectroscopy
The FTIR spectra of gelatine, sunitinib, blank nanoparticles, and drug‐loaded nanoparticles were taken by using an FTIR spectrometer at wave numbers of 500–4000 cm−1 with a resolution of 4 cm−1 (FTIR instrument, 6300 Jasco, Japan) using the potassium bromide disc method to explore the possible interactions between sunitinib, gelatine, and PGS during the preparation of nanoparticles.
2.7 Field emission scanning electron microscopy (FE‐SEM)
The morphology and size of PGS/gelatine nanoparticles were assessed using an FE‐SEM microscope (HITACHI S‐4160, Japan). Samples were coated with gold under vacuum before imaging.
2.8 Solid‐state characterisation of PGS/gelatine nanoparticles
The solid state of drug‐loaded nanoparticles, blank nanoparticles, physical mixture of gelatine, PGS and sunitinib, pure PGS, pure gelatine and free sunitinib were evaluated by differential scanning calorimetry (DSC; NETZSCH DSC 200 F3, Japan). For performing this experiment, 6 mg of each sample was heated at the rate of 10°C/min in aluminium pans from room temperature to 400°C under the nitrogen flow and their thermograms were recorded [31].
2.9 In vitro drug release studies from drug‐loaded PGS/gelatine nanoparticles
One ml of each formulation was poured on dialysis membrane with a molecular weight cut off 12000 Da and placed in a beaker containing 50 ml of PBS (pH 7.4) at 37°C. The amount of released drug in PBS media was determined by an ultraviolet spectrophotometer (mini 1240, Shimadzu, Japan) at λ max = 430 nm [30, 32]. The standard curve of sunitinib in PBS was linear over the concentration range of 0.45–20.0 µg/ml. RE per cent (RE%) after 150 h of release test was calculated by :
| (2) |
where y is the per cent of drug released at time t, i.e. 150 h.
2.10 Cell culture and MTT assay
MTT assay was used to ensure that the synthesised nanoparticles were not cytotoxic and to find the highest concentration of the drug‐loaded nanoparticles above its IC50, which could be used with the highest level of cell viability. L929 cell line was selected for this test. The cell cultivation medium was RPMI containing 10% FBS and 1% penicillin–streptomycin. Cells were incubated in an atmosphere containing 5% CO2 at 37°C. Different concentrations of the drug‐loaded nanoparticles ranging from 0.25 to 102 nM of sunitinib [33, 34] were used along with the blank PGS/gelatine nanoparticles in the same concentrations, negative control (cells with no treatment) and the culture medium with no cells and no treatment (blank) in a 96‐well plate. An aqueous solution of free sunitinib was used as a positive control. L929 cells were seeded in a 96‐well plate at a cell density of 13 × 103 and incubated for 24 h to allow cell attachment. Then, the cells were exposed to free sunitinib and different concentrations of blank nanoparticles and drug‐loaded PGS/gelatine nanoparticles as described above for 24 and 72 h. Then, 20 µl of MTT solution (5 mg/ml) was added to each well and incubated for further 3 h. Then all media in each well were removed and 150 µl of dimethylsulphoxide was added to dissolve the formed formazan crystals in wells. An enzyme‐linked immunosorbent assay microplate reader (Biorad, USA) was used to measure the absorbance of formazan. The number of viable cells was determined using the following equation:
| (3) |
2.11 Draize test
For the Draize test, 0.1 ml of the suspension of drug‐loaded nanoparticles was instilled into the right eye of three albino rabbits and the left eye was untreated, which served as the matched control. The test was conducted for 21 days in which the cornea, iris, and conjunctivae were observed for signs of opacity, ulceration, redness, swelling, and discharge. The animals were evaluated before exposure to ensure normal eyes and cages were designed to avoid accidental injury. Instillation was made in the conjunctiva under lower eyelid. Normal blinking was permitted, but the eyelids could be held together for several seconds after instillation. The observation was done after applying the suspension at the moment of instillation, 1, 24, 48, 72 h, 7 and 21 days after exposure. Any ocular alteration was graded by a specific scoring system designed for this test [35].
3 Results and discussion
3.1 Physicochemical properties of sunitinib‐loaded PGS/gelatine nanoparticles
Sunitinib malate is a highly water‐soluble drug. Absolute hydrophobic or hydrophilic carriers are not the best options for delivering water‐soluble drugs. Both have some pros and cons. For example, although the hydrophilic polymers can load these types of drugs efficiently, their release is too fast and uncontrollable. In aqueous medium, these carriers absorb lots of water and swell rapidly so that the drug molecules can diffuse very fast [36]. On the other hand, if the hydrophobic polymers are used to carry water‐soluble drugs they exhibit a sustained release profile but have insufficient capability for drug encapsulation and drug molecules are removed during the drug‐loading process due to the difference in the physicochemical properties of the carrier and the cargo. Therefore, the combination of the hydrophilic and hydrophobic polymers could highlight these benefits and overcome the shortcomings of each system. In the present study, PGS/gelatine nanoparticles were prepared by integrating a hydrophilic polymer (gelatine) and a hydrophobic one (PGS) to produce a novel biocompatible biomaterial with designable capabilities for delivering both water soluble and insoluble drugs [37]. For investigation of this hypothesis, PGS/gelatine nanoparticles were synthesised by desolvation technique. In the first step, PGS was dissolved in ethanol which is an appropriate anti‐solvent for gelatine too. Therefore, when PGS solution was added drop‐wise into gelatine solution, gelatine nanoparticles began to form. On the other hand, water molecules are anti‐solvent of PGS, which is infused in the structure of gelatine. The result of this phenomena is the formation of an interpenetrating polymer network shaped in the PGS/gelatine nanoparticles with PGS being trapped in its structure simultaneously and then glutaraldehyde cross‐links the gelatine segments to stiffen the walls. Fig. 1 shows the schematic representation of the formation and structure of nanoparticles.
Fig. 1.
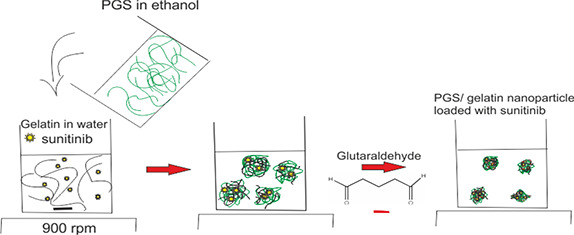
Schematic representation of the structure of glutaraldehyde cross‐linked PGS/gelatine nanoparticles loaded with sunitinib
FTIR spectra of sunitinib‐loaded PGS/gelatine nanoparticles, blank nanoparticles, sunitinib, PGS and gelatine are presented in Fig. 2. A comparison between three spectra of gelatine, PGS, and blank nanoparticles reveals that both PGS and gelatine have contributed to the formation of nanoparticles final structure. A similar conclusion was reported for the formation of chitosan/gelatine/thermoplastic polyurethane blend nanofibres [38]. As can be seen in a drug‐free nanoparticles spectrum, a broad band at 3426 cm−1 and weak multiple peaks at 2920–2850 cm−1 are exhibited, which are allocated to hydroxyl and amine groups of gelatine. These two bands are exhibited in the pure gelatine spectrum too. The characteristic absorption bands of gelatine at 1640 cm−1 (amide I) and 1460 cm−1 (amide II) are observed in pure gelatine spectrum, while in nanoparticles spectrum these peaks are affected by a carbonyl strong peak, which is related to the ester group of PGS. Also, two absorption peaks at 2923 and 2856 cm−1 in PGS, nanoparticles, and drug‐loaded nanoparticles are seen too. Therefore, it may be concluded that gelatine and PGS have been incorporated in the structure of nanoparticles, and their final structure consists of a combination of both polymers. In this study, sunitinib‐loaded PGS/gelatine nanoparticles were prepared by dissolving sunitinib in gelatine solution and the drug was encapsulated in nanoparticles formation. The presence of sunitinib was confirmed by the bands at 1739, 1671, 1473, 1313, and 1260 cm−1, corresponding to C=C–F, C=O stretching of amide, C=C stretching of aromatic ring, and C–N bending and P=O stretching. Also, N–H bending mode of vibration was presented at 1546 cm−1. Except C=C–F, C=O, which are covered by a sharp peak related to PGS ester, other characteristic peaks of sunitinib are presented in drug‐loaded nanoparticles spectra too. These peaks exhibited at the same wave numbers in drug‐loaded nanoparticles spectrum with no shift and changes in shape, which could show process of encapsulation and nanoparticles preparation, caused no changes in sunitinib structure [39].
Fig. 2.
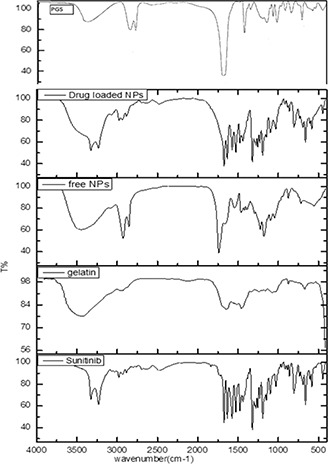
FTIR spectrum of PGS, sunitinib‐loaded PGS/gelatine nanoparticles, drug‐free PGS/gelatine nanoparticles, gelatine, and sunitinib
Based on the Box–Behnken design, 17 formulations were prepared in which drug‐to‐carrier percentage, gelatine‐to‐PGS ratio, and sebacate‐to‐glycerine ratio were studied each in three levels as variables. Drug‐to‐carrier per cent was changed from 10% to 30%, gelatine‐to‐PGS ratio from 3 to 10 and glycerine‐to‐sebacate ratio was 1:1 and 1:2 and 2:1. Details of each formulation are displayed in Table 1.
Table 1 summarises the results of particle size, zeta potential, EE% and sunitinib RE as studied responses of PGS/glycerol nanoparticles.
The hydrodynamic particle size of the nanoparticles and their polydispersity index were 230–484 nm and 0.3–0.5, respectively (Table 1). Statistical analysis of the results in Table 1 by Design Expert Software showed that the most important variable, which affected the particle size of the nanoparticles was gelatine‐to‐PGS ratio (p < 0.05). The particle size reduced as the gelatine‐to‐PGS ratio increased (Table 1). Louage et al. [14] synthesised PGS nanoparticles in different concentrations of PGS in ethanol by the solvent displacement method for encapsulation of hydrophobic anti‐cancer drugs. The sizes of their nanoparticle were 111–208 nm and were dependent on PGS concentration. The particle size increased with increasing PGS concentration [14], which confirms our findings.
In another study, a combination of PGS/gelatine was used to prepare electrospun nanofibres for delivery of an antibiotic in wound‐dressing applications [40]. The authors observed increasing in fibres size after crosslinking the nanofibres, which could be due to more swelling of the nanofibres and increasing their size.
Particle size was fitted to the linear model (p < 0.05) and the software suggested the final equation in terms of coded factors for the particle size of nanoparticles as follows:
| (4) |
One of the most desired qualities of successful nanoparticles is their high drug EE%. Table 1 shows the EE% range for 17 formulations changed between 17.5 and 43.8%. The most effective variable on the EE% of sunitinib in the nanoparticles was the glycerine/sebacic acid ratio although this factor was not statistically significant (p > 0.05). EE% increased with an increase in glycerine/sebacic acid ratio. The results of Table 1 show that in most cases, the ratio of gelatine‐to‐PGS was in the intermediate value; the highest EE% was seen. This could be due to the hydrophilic nature of the drug, which can be better entrapped in the hydrophilic component of the nanoparticles. However, this effect was reversed in higher ratios. The negative sign of G in (5) also confirmed that the variable G decreased the EE% and there was a reverse relationship between these two parameters. As discussed earlier, when the concentration of the hydrophilic polymer (gelatine) was increased, the particle size of nanoparticles decreased (Table 1) and it seems that decreasing in the particle size is the reason for reducing in space for the accommodation of the drug. These findings are in agreement with the study of Meghani et al. [41], who showed with increasing the hydrophobicity of the gelatine–oleic nanoparticles, EE% of doxycycline decreased.
The EE% was fitted to a quadratic model (p < 0.05) and the final equation in terms of coded factors for EE% generated by the Design Expert Software is as follows:
| (5) |
Zeta potential is a key factor for evaluating the stability of colloidal nanoparticles. Surface charge is a very good index for the degree of repulsive and/or interaction between nanoparticles. As zeta potential increases, the particles must overcome a larger energy barrier to aggregate.
Zeta potential of the nanoparticles is shown in Table 1. The zeta potential of different formulations varied between −2.29 and −20.1 mV. The Design Expert analysis results showed none of the studied single factors had a significant effect on the zeta potential of nanoparticles (p > 0.05). The zeta potential of the nanoparticles was fitted to a quadratic model (p < 0.05) and the final equation in terms of coded factors for it was as follows:
| (6) |
Sunitinib release profiles from all studied formulations are presented in Fig. 3 and the calculated release efficiencies over 150 h (RE150 %) are shown in Table 1. Fig. 3 shows that all formulations displayed a biphasic release profile. Burst release at the beginning hours of the release test could be related to the release of the drug located near the surface of the nanoparticles. In further hours, release behaviour followed a sustained pattern by diffusion of the drug from the inner layers of the nanoparticles.
Fig. 3.
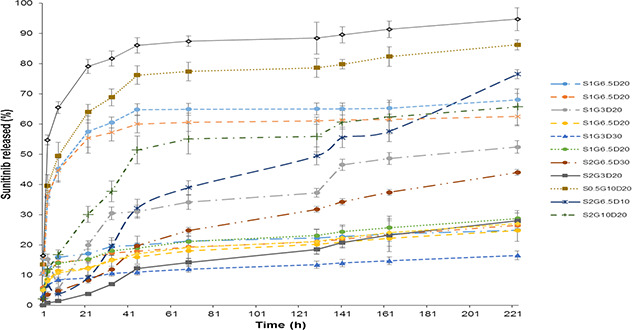
Sunitinib release profiles from PGS/gelatine nanoparticles
The results of Design Expert analysis demonstrated that the most important factor influencing the RE% was the glycerine‐to‐sebacate ratio so that, it increased as glycerine content of the nanoparticles increased. The reason for this finding could be due to hydrophilicity of glycerine, which made the final nanoparticles more hydrophilic compared to the samples with less content of glycerine. Hydrophilic samples absorbed more water and released sunitinib faster. Previous studies also showed that in the nanoparticulate carrier designed by Samimi Gharaie et al. [38], which contained gelatine and poly(lactide‐co‐glycolide) (PLGA), increasing in the hydrophilic polymer concentration (gelatine) caused more swelling of the carrier but when the content of hydrophobic polymer (PLGA) was increased the release of the protein cargo decreased two‐fold. The results of Table 1 also showed that as gelatine‐to‐PGS ratio increased, the RE% increased too. The inherent hydrophilic nature of gelatine describes this phenomenon.
Kumari et al. [42] investigated the in vitro release kinetics of rosiglitazone‐loaded gelatine nanoparticles and observed 80% of the drug released within 32 h. They used glutaraldehyde to cross‐link the nanoparticles, which was hydrophilic by itself and enhanced the swelling ratio of the nanoparticles [43].
The RE% of the nanoparticles was fitted to a quadratic model (p < 0.05) and the final equation in terms of coded factors for RE% is presented in (7):
| (7) |
3.2 Computer optimisation of the nanoparticles formulation
Computer optimisation was done by Design‐Expert Software (version 7.2, Stat‐ Ease, USA) and a desirability function determined the effect of the levels of independent variables on the responses. The optimal conditions for nanoparticles were considered to be the case where the particle size was the lowest and the Polydispersity index (PDI) and zeta were within the range of the results. The goal of EE% was set on maximum and the RE% on a minimum of the obtained data of Table 1. Considering the data of Table 1, optimisation was carried out by the desirability of 81% and the optimised processing situation of the nanoparticles was suggested to be the formulation of S1 G0.7 D21.2. The predicted responses by the Software and the achieved results are displayed in Table 2. The release profile of the optimised drug loaded nanoparticles is shown in Fig. 4. As exhibited in this figure, sunitinib released in a sustained manner with no burst release.
Table 2.
Properties of the optimised sunitinib‐loaded PGS/gelatine nanoparticles (S1 G0.7 D21.2) predicted by the Design Expert Software and the actually obtained data for this formulation (mean ± SD, n = 3)
| Particle size, nm | PDI | Zeta potential, mV | EE% | RE150 % | |
|---|---|---|---|---|---|
| predicted | 342 | — | −6.1 | 41.5 | 22.8 |
| actual | 282 ± 16 | 0.4 ± 0.1 | −8.9 ± 1.4 | 34.6 ± 0.3 | 27.3 ± 3.2 |
| error% | 21.3 | — | 31.5 | 19.9 | −16.5 |
Fig. 4.
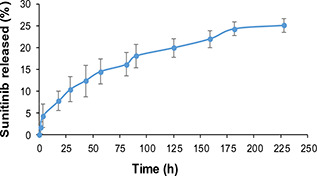
Drug release profile from the optimised formulation of PGS/gelatine nanoparticles (S1 G0.7 D21.2)
3.3 Morphology and particle size of the optimised formulation PGS/gelatin nanoparticles by FE‐SEM
FE‐SEM images of the optimised formulation of PGS/gelatine nanoparticles are shown in Fig. 5. As this figure shows the particles had nearly dimensions of 100 nm in width, 90 nm in length and 130 nm in height. As exhibited in FE‐SEM photographs, the morphology of the nanoparticles was almost mono‐dispersed with a spherical structure and smooth surface. The particle size measured by FE‐SEM was smaller than the results obtained by the DLS method (Table 2). The possible reason may be due to the shrinking of the nanoparticles during the drying process.
Fig. 5.

FE‐SEM images of the optimised PGS/gelatin nanoparticles (S1 G0.7 D21.2)
3.4 DSC analysis
The results of DSC thermograms of free sunitinib, blank PGS/gelatine nanoparticles, drug‐loaded nanoparticles, physical mixture of nanoparticles and sunitinib and PGS are shown in Fig. 6. According to the thermograms, two melting points were seen for PGS at 96 and 105°C, which may be related to two types of polymers present in the sample or two different crystalline forms of the polymer. The pattern of PGS thermogram was quite similar to the previously reported values [44, 45]. Reviewing the literature showed that due to the presence of numerous effective factors in the synthesis of PGS such as the processing parameters and reagents ratio, the melting point of this polymer could be varied. For example, Gaharwar et al. [46] reported the T m of about 12°C for PGS. Aydin et al. [47] synthesised the PGS elastomer via microwave‐assisted pre‐polymerisation in minutes and showed that the PGS synthesised elastomers had a T g around −35.61°C and two melting points; the first at 15.82°C and the second one at 61.70°C corresponding to the glycerol and sebacic acid fragments, respectively. As shown in Fig. 6 the PGS melting endotherms disappeared in the nanoparticles thermogram that could be due to the process, which PGS undergoes during the production of the nanoparticles and becomes amorphous. This result is in agreement with previous studies such as the report of Jiang et al. [45] on the synthesis of electrospun nanofibres of thermoplastic polyurethane/PGS hybrid scaffold prepared for vocal folds tissue engineering. They observed that the pure PGS polymer showed two melting endotherms (the first was sharp and the second was a broad band) at about 12.5 and 42.67°C. Jiang's study showed that the combination of PGS and polyurethane was used in the preparation of nanofibres, the melting endotherms of PGS were eliminated, which is in agreement with our results.
Fig. 6.
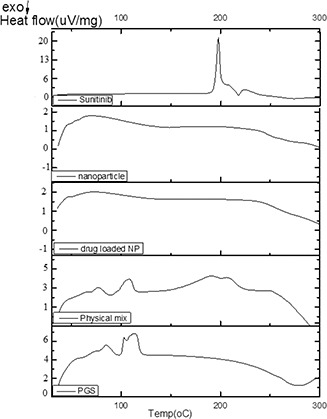
DSC thermograms of free sunitinib, nanoparticles, drug‐loaded PGS/gelatine nanoparticles, physical mixture of sunitinib/PGS and PGS
Gaharwar et al. [46] showed that the melting point of PGS became very weak when it was combined with poly(ɛ‐caprolactone) during the production of the electrospun fibres. In another study, Xu et al. [48] illustrated that the T m of PGS was not detectable in DSC thermogram after preparation of its sheet. Although in some reports after preparation of the formulation and in combination with other polymers, the T m of the PGS was kept intact. For example, Graziano et al. [44] prepared PGS and poly(caprolactone) scaffolds for blood vessel constructs. They showed that after the preparation of the nanofibres, PGS was kept crystalline and in the nanofibres thermogram, the T m of PGS was still recognisable. In addition, it seems that the crystalline structure of sunitinib has altered to the amorphous phase during the drug loading and manufacturing process of nanoparticles as the melting point of the drug at 206°C disappeared, which indicates the complete drug entrapment in the matrix of the nanoparticles. However, the appearance of all endothermic peaks of the drug and polymer in the physical mixture of gelatine/PGS/sunitinib shows the intact crystallinity of the drug and its free presence among the polymeric nanoparticles. Ha et al. [49] also achieved similar results in their study. They prepared sunitinib‐loaded poly(L‐lactide‐co‐caprolactone) films for use in anti‐tumour drug delivery systems against human cholangiocarcinoma cells and observed that the films loaded with sunitinib did not show melting point of the drug even though the melting point of sunitinib itself was detected at about 240°C sharply. This indicates changing the crystalline form of the drug to an amorphous state after encapsulation in the nanoparticles.
3.5 Cell viability assay
The in vitro cell viability of free and sunitinib‐loaded PGS/gelatine nanoparticles was assessed using L929 cells by MTT assay. International standard ISO‐10993‐5 suggests that L929 cell line may be used for in vitro cytotoxicity study of biomaterials [50]. MTT assay was used to study the possible cytotoxicity of the synthesised nanoparticles and also to find the highest concentration of the drug‐loaded nanoparticles, which were lower than IC50 of the drug but meanwhile had the highest level of cell viability. To do this, at first the IC50 of the free drug was obtained on the L929 cells. The results indicated that the IC50 of the free sunitinib on L929 cells was around 50 nM (Fig. 7). At 102 nM, the cell viability was about 48% (Fig. 7).
Fig. 7.
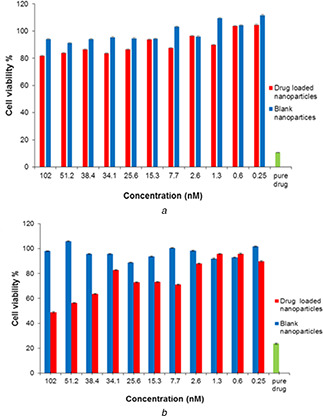
Cell viability of blank nanoparticles and sunitinib‐loaded nanoparticles at various concentrations on L929 cells after
(a) 24 h exposure, (b) 72 h exposure
Different concentrations (0.25–102 nM) of the drug‐loaded nanoparticles (according to the loaded sunitinib concentration) were studied. This range included the higher and lower concentrations than the reported IC50 of free sunitinib [33, 34]. Other studied groups included the blank PGS/gelatine nanoparticles at the same concentrations and negative control groups consisting of the untreated cells. The solution of free sunitinib (5 mg/ml) was used as positive control. L929 cells were seeded in a 96‐well plate at a cell density of 13 × 103 and incubated for 24 h to allow the cells attachment. Then, the cells were exposed with free sunitinib, different concentrations of blank PGS/gelatine nanoparticles and drug‐loaded PGS/gelatine nanoparticles as described above for 24 and 72 h. As shown in Fig. 7 a, blank nanoparticles did not show any cytotoxicity on L929 cells in all concentrations in the first 24 h. However, in the same period of exposure, the drug‐loaded nanoparticles at concentrations >1.3 nM had about 80% cell viability (Fig. 7 a). After 72 h, the viability of the cells of the blank nanoparticles and drug‐loaded nanoparticles changed significantly. After 72 h, none of the concentrations of blank nanoparticles were toxic but cell viability of the drug‐loaded nanoparticles fell to <60% at the same concentration (Fig. 7 b). As indicated in Fig. 7 b at a concentration of 34 nM and lower, the cell viability was still high and could be chosen as an appropriate concentration for further in vivo studies.
3.6 Draize test
The Draize test is an acute ocular toxicity test and provides a method for assessing the irritation potential of materials that might come in contact with human eyes. The test subjects are commonly the albino rabbits, which are observed for up to 14–21 days for signs of redness, swelling, discharge, ulceration, haemorrhaging, cloudiness, or blindness in the tested eye [35]. For Draize test, 0.1 ml of the dispersion of sunitinib‐loaded nanoparticles was instilled into the right eye of three albino rabbits and the left eye was untreated and served as a matched control. The test proceeded for 21 days and the cornea, iris, and conjunctivae were observed for signs of opacity, ulceration, redness, swelling, and discharge. The animals were evaluated before exposure to ensure normal eyes; cages were designed to avoid accidental injury. Instillation was made in the conjunctiva under the lower eyelid. Normal blinking was permitted, but the eyelids could be held together for several seconds after instillation. Observation after applying the dispersion was done at the moment of instillation, 1, 24, 48, 72 h and 7, 21 days after exposure. Applying the dispersion caused tearing in the eye rabbits.
According to the observations at the end of the experiment, this formulation caused just a little redness in the eye at the distillation moment, there was no sign of discharge, and inflammation, which shows the safety of the drug‐loaded nanoparticles after 21 days(Fig. 8).
Fig. 8.
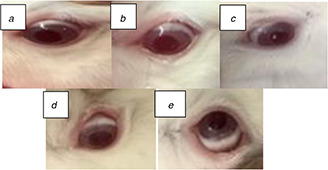
Photographs of rabbits eyes during the experiment
(a) Before distillation, (b) Just at distillation moment, (c) After 7 days, (d) After 21 days, (e) Left eye without treatment
According to Table 3 designed for analysis of this test, any allergic reaction including conjunctiva, corneal, or iris alteration was scored more than 0. As a consequence, the positive response can be defined as the corneal opacity: score >1, iritis: score >1, or conjunctiva redness: score >2 (or an average conjunctiva redness or swelling score >2.5) [35].
Table 3.
Scoring proposed by regulatory agencies for eye irritation tests after 21 days [51]
| Signs | Score | Observation during 21 days |
|---|---|---|
| corneal opacity (the area most dense taken for reading); no ulceration or opacity | 0 | 0 |
| scattered or diffuse areas of opacity (other than slight dulling of normal cluster); details of iris clearly visible | 0–1 | 0 |
| easily discernible translucent area; details of iris slightly obscured | 0–2 | 0 |
| opalescent areas; no details of iris visible, size of pupil barely discernible | 0–3 | 0 |
| opaque cornea; iris not discernible through the opacity | 0–4 | 0 |
| iritis; normal | 0 | 0 |
| deepened iris rugae and/or iris congestion or swelling, with circumcorneal injection | 0–1 | 0 |
| haemorrhage, gross destruction of iris or non‐reactivity to light | 0–2 | 0 |
| conjunctiva redness (palpebral and bulbar conjunctivae); blood vessels normal | 0 | 0 |
| some blood vessels definitely hyperaemia (injected) | 0–1 | 0 |
| diffuse Crimson colour; individual vessels not easily discernible | 0–2 | 0 |
| diffuse beefy red | 0–3 | 0 |
| total score | 0 | |
The results showed no incompatibly with the rabbits’ eyes after 21 days of administration.
4 Conclusion
PGS/gelatine nanoparticles were prepared by integrating a hydrophilic polymer (gelatine) and a hydrophobic one (PGS) for the first time to overcome the shortcoming of both systems and produce a novel biocompatible biomaterial with designable capabilities for delivering of sunitinib. FTIR and DSC analyses confirmed the successful synthesis of the nanoparticles and MTT and Draize test showed acceptable results for biocompatibility and ocular application of nanoparticles. Investigating different variables showed glycerine/sebacic acid was the most effective parameter on the EE% and RE. Gelatine‐to‐PGS ratio had the most considerable effect on the particle size while the RE% was more affected by glycerine/sebacic acid ratio. The optimised formulation (S1 G0.7 D21.2) exhibited a particle size of 282 nm, 34.6% EE, zeta potential of −8.9 mV, and RE% of about 27.3% over 228 h. The nanoparticles were safe with no toxicity at concentrations <36 nM on L929 cells and caused no allergic reaction on the eyes of the rabbits. The sunitinib‐loaded gelatine/PGS nanoparticles may be used for the treatment of DR. Further experiments are needed to show their effectiveness in controlling the angiogenesis in vivo.
5 Acknowledgment
The authors are grateful to Isfahan University of Medical Sciences for their financial support by grant number 196027.
6 References
- 1. Xu X. Weng Y. Xu L. et al.: ‘Sustained release of Avastin® from polysaccharides cross‐linked hydrogels for ocular drug delivery’, Int. J. Biol. Macromol., 2013, 60, pp. 272 –276 [DOI] [PubMed] [Google Scholar]
- 2. Wilkinson C. Ferris F.L. III Klein R.E. et al.: ‘Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales’, Ophthalmology, 2003, 110, (9), pp. 1677 –1682 [DOI] [PubMed] [Google Scholar]
- 3. Raimondi C. Fantin A. Lampropoulou A. et al.: ‘Imatinib inhibits VEGF‐independent angiogenesis by targeting neuropilin 1‐dependent ABL1 activation in endothelial cells’, J. Exp. Med., 2014, 211, (6), pp. 1167 –1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. He Z. King G.L.: ‘Can protein kinase C β‐selective inhibitor, ruboxistaurin, stop vascular complications in diabetic patients?’, Diabetes Care, 2005, 28, (11), pp. 2803 –2805 [DOI] [PubMed] [Google Scholar]
- 5. Aiello L.P. Bursell S.‐E. Clermont A. et al.: ‘Vascular endothelial growth factor‐induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective β‐isoform‐selective inhibitor’, Diabetes, 1997, 46, (9), pp. 1473 –1480 [DOI] [PubMed] [Google Scholar]
- 6. Funatsu H. Yamashita H. Ikeda T. et al.: ‘Angiotensin II and vascular endothelial growth factor in the vitreous fluid of patients with diabetic macular edema and other retinal disorders’, Am. J. Ophthalmol., 2002, 133, (4), pp. 537 –543 [DOI] [PubMed] [Google Scholar]
- 7. Suzuma K. Takahara N. Suzuma I. et al.: ‘Characterization of protein kinase C β isoform's action on retinoblastoma protein phosphorylation, vascular endothelial growth factor‐induced endothelial cell proliferation, and retinal neovascularization’, Proc. Natl. Acad. Sci., 2002, 99, (2), pp. 721 –726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Association Diabetes Association : ‘The effect of ruboxistaurin on visual loss in patients with moderately severe to very severe nonproliferative diabetic retinopathy: initial results of the protein kinase C β inhibitor diabetic retinopathy study (PKC‐DRS) multicenter randomized clinical trial’, Diabetes, 2005, 54, (7), pp. 2188 –2197 [DOI] [PubMed] [Google Scholar]
- 9. Danis R.P. Sheetz M.J.: ‘Ruboxistaurin: PKC‐β inhibition for complications of diabetes’, Expert Opin. Pharmacother., 2009, 10, (17), pp. 2913 –2925 [DOI] [PubMed] [Google Scholar]
- 10. Campochiaro P.A.: ‘Reduction of diabetic macular edema by oral administration of the kinase inhibitor PKC412’, Invest. Ophthalmol. Visual Sci., 2004, 45, (3), pp. 922 –931 [DOI] [PubMed] [Google Scholar]
- 11. Roskoski R. Jr: ‘Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor’, Biochem. Biophys. Res. Commun., 2007, 356, (2), pp. 323 –328 [DOI] [PubMed] [Google Scholar]
- 12. Tamayol A. Najafabadi A.H. Mostafalu P. et al.: ‘Biodegradable elastic nanofibrous platforms with integrated flexible heaters for on‐demand drug delivery’, Sci. Rep., 2017, 7, (1), p. 9220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sundback C.A. Shyu J.Y. Wang Y. et al.: ‘Biocompatibility analysis of poly(glycerol sebacate) as a nerve guide material’, Biomaterials, 2005, 26, (27), pp. 5454 –5464 [DOI] [PubMed] [Google Scholar]
- 14. Louage B. Tack L. Wang Y. et al.: ‘Poly(glycerol sebacate) nanoparticles for encapsulation of hydrophobic anti‐cancer drugs’, Polym. Chem., 2017, 8, (34), pp. 5033 –5038 [Google Scholar]
- 15. Sun Z.‐J. Chen C. Sun M.‐Z. et al.: ‘The application of poly (glycerol‐sebacate) as biodegradable drug carrier’, Biomaterials, 2009, 30, (28), pp. 5209 –5214 [DOI] [PubMed] [Google Scholar]
- 16. Müller K. Bugnicourt E. Latorre M. et al.: ‘Review on the processing and properties of polymer nanocomposites and nanocoatings and their applications in the packaging, automotive and solar energy fields’, Nanomaterials, 2017, 7, (4), p. 74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rai R. Tallawi M. Grigore A. et al.: ‘Synthesis, properties and biomedical applications of poly(glycerol sebacate) (PGS): a review’, Prog. Polym. Sci., 2012, 37, (8), pp. 1051 –1078 [Google Scholar]
- 18. Sales V.L. Engelmayr G.C. Jr Johnson J.A. Jr et al.: ‘Protein precoating of elastomeric tissue‐engineering scaffolds increased cellularity, enhanced extracellular matrix protein production, and differentially regulated the phenotypes of circulating endothelial progenitor cells’, Circulation, 2007, 116, (11_supplement), pp. I‐55 –I‐63 [DOI] [PubMed] [Google Scholar]
- 19. Kharaziha M. Nikkhah M. Shin S.‐R. et al.: ‘PGS: gelatin nanofibrous scaffolds with tunable mechanical and structural properties for engineering cardiac tissues’, Biomaterials, 2013, 34, (27), pp. 6355 –6366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bini R.A. Silva M.F. Varanda L.C. et al.: ‘Soft nanocomposites of gelatin and poly (3‐hydroxybutyrate) nanoparticles for dual drug release’, Colloids Surf. B, Biointerfaces, 2017, 157, pp. 191 –198 [DOI] [PubMed] [Google Scholar]
- 21. Sahoo N. Sahoo R.K. Biswas N. et al.: ‘Recent advancement of gelatin nanoparticles in drug and vaccine delivery’, Int. J. Biol. Macromol., 2015, 81, pp. 317 –331 [DOI] [PubMed] [Google Scholar]
- 22. Elzoghby A.O. Samy W.M. Elgindy N.A.: ‘Protein‐based nanocarriers as promising drug and gene delivery systems’, J. Controlled Release, 2012, 161, (1), pp. 38 –49 [DOI] [PubMed] [Google Scholar]
- 23. Wang H. Boerman O.C. Sariibrahimoglu K. et al.: ‘Comparison of micro‐ vs. nanostructured colloidal gelatin gels for sustained delivery of osteogenic proteins: bone morphogenetic protein‐2 and alkaline phosphatase’, Biomaterials, 2012, 33, (33), pp. 8695 –8703 [DOI] [PubMed] [Google Scholar]
- 24. Zwiorek K. Kloeckner J. Wagner E. et al.: ‘Gelatin nanoparticles as a new and simple gene delivery system’, J. Pharm. Pharm. Sci., 2004, 7, (4), pp. 22 –28 [PubMed] [Google Scholar]
- 25. Elzoghby A.O.: ‘Gelatin‐based nanoparticles as drug and gene delivery systems: reviewing three decades of research’, J. Controlled Release, 2013, 172, (3), pp. 1075 –1091 [DOI] [PubMed] [Google Scholar]
- 26. Kommareddy S. Shenoy D.B. Amiji M.M.: ‘Gelatin nanoparticles and their biofunctionalization’, Nanotechnol. Life Sci., Online, 2007, 10.1002/9783527610419.ntls0011 [DOI] [Google Scholar]
- 27. Nejat H. Rabiee M. Varshochian R. et al.: ‘Preparation and characterization of cardamom extract‐loaded gelatin nanoparticles as effective targeted drug delivery system to treat glioblastoma’, React. Funct. Polym., 2017, 120, pp. 46 –56 [Google Scholar]
- 28. Wang Y. Ameer G.A. Sheppard B.J. et al.: ‘A tough biodegradable elastomer’, Nat. Biotechnol., 2002, 20, (6), p. 602 [DOI] [PubMed] [Google Scholar]
- 29. Varshosaz J. Eskandari S. Kennedy R. et al.: ‘Factors affecting the production of nanostructure lipid carriers of valproic acid’, J. Biomed. Nanotechnol., 2013, 9, (2), pp. 202 –212 [DOI] [PubMed] [Google Scholar]
- 30. Taymouri S. Varshosaz J. Hassanzadeh F. et al.: ‘Optimisation of processing variables effective on self‐assembly of folate targeted synpronic‐based micelles for docetaxel delivery in melanoma cells’, IET Nanobiotechnol., 2015, 9, (5), pp. 306 –313 [DOI] [PubMed] [Google Scholar]
- 31. Varshosaz J. Ghaffari S. Khoshayand M.R. et al.: ‘Optimization of freeze‐drying condition of amikacin solid lipid nanoparticles using D‐optimal experimental design’, Pharm. Dev. Technol., 2012, 17, (2), pp. 187 –194 [DOI] [PubMed] [Google Scholar]
- 32. Mashak A. Mobedi H. Mahdavi H.: ‘A comparative study of progesterone and lidocaine hydrochloride release from poly(L‐lactide) films’, Pharm. Sci., 2015, 21, (2), p. 77 [Google Scholar]
- 33. Adelaiye‐Ogala R. Damayanti N.P. Orillion A.R. et al.: ‘Therapeutic targeting of sunitinib‐induced Ar phosphorylation in renal cell carcinoma’, Cancer Res., 2018, 78, pp. 2886 –2896. DOI: 10.1177/2515221118769324, canres. 3386.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schenone S. Brullo C. Botta M.: ‘Small molecules ATP‐competitive inhibitors of FLT3: a chemical overview’, Curr. Med. Chem., 2008, 15, (29), pp. 3113 –3132 [DOI] [PubMed] [Google Scholar]
- 35. Wilhelmus K.R.: ‘The draize eye test’, Surv. Ophthalmol., 2001, 45, (6), pp. 493 –515 [DOI] [PubMed] [Google Scholar]
- 36. Jain A.K. Goyal A.K. Gupta P.N. et al.: ‘Synthesis, characterization and evaluation of novel triblock copolymer based nanoparticles for vaccine delivery against hepatitis B’, J. Controlled Release, 2009, 136, (2), pp. 161 –169 [DOI] [PubMed] [Google Scholar]
- 37. Yadav K. Yadav D. Srivastava A.K.: ‘Evaluation of hydrophilic, hydrophobic and waxy matrix excipients for sustained release tablets of venlafaxine hydrochloride’, Drug Dev. Ind. Pharm., 2013, 39, (8), pp. 1197 –1206 [DOI] [PubMed] [Google Scholar]
- 38. Samimi Gharaie S. Habibi S. Nazockdast H.: ‘Fabrication and characterization of chitosan/gelatin/thermoplastic polyurethane blend nanofibers’, J. Text. Fibrous Mater., 2018, 1, 2515221118769324 [Google Scholar]
- 39. Joseph J.J. Sangeetha D. Gomathi T.: ‘Sunitinib loaded chitosan nanoparticles formulation and its evaluation’, Int. J. Biol. Macromol., 2016, 82, pp. 952 –958 [DOI] [PubMed] [Google Scholar]
- 40. Shirazaki P. Varshosaz J. Kharazi A.Z.: ‘Electrospun gelatin/poly(glycerol sebacate) membrane with controlled release of antibiotics for wound dressing’, Adv. Biomed. Res., 2017, 28, (6), 105 –111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meghani N.M. Amin H.H. Park C. et al.: ‘Design and evaluation of clickable gelatin‐oleic nanoparticles using fattigation‐platform for cancer therapy’, Int. J. Pharm., 2018, 545, (1–2), pp. 101 –112 [DOI] [PubMed] [Google Scholar]
- 42. Kumari A. Yadav S.K. Yadav S.C.: ‘Biodegradable polymeric nanoparticles based drug delivery systems’, Colloids Surf. B, Biointerfaces, 2010, 75, (1), pp. 1 –18 [DOI] [PubMed] [Google Scholar]
- 43. Bajpai A. Choubey J.: ‘In vitro release dynamics of an anticancer drug from swellable gelatin nanoparticles’, J. Appl. Polym. Sci., 2006, 101, (4), pp. 2320 –2332 [Google Scholar]
- 44. Graziano R.V. Brera A.A.M. Campos R.M. et al.: ‘Soluble poly(glycerol sebacate) and poly(ɛ‐caprolactone) 3D scaffolds for blood vessel constructs’, MRS Online Proceedings Library Archive, 2016, 1819
- 45. Jiang L. Jiang Y. Stiadle J. et al.: ‘Electrospun nanofibrous thermoplastic polyurethane/poly (glycerol sebacate) hybrid scaffolds for vocal fold tissue engineering applications’, Mater. Sci. Eng. C, 2019, 94, pp. 740 –749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gaharwar A.K. Nikkhah M. Sant S. et al.: ‘Anisotropic poly (glycerol sebacate)‐poly (ɛ‐caprolactone) electrospun fibers promote endothelial cell guidance’, Biofabrication, 2014, 7, (1), p. 015001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aydin H. Salimi K. Rzayev Z. et al.: ‘Microwave‐assisted rapid synthesis of poly(glycerol‐sebacate) elastomers’, Biomater. Sci., 2013, 1, (5), pp. 503 –509 [DOI] [PubMed] [Google Scholar]
- 48. Xu B. Li Y. Zhu C. et al.: ‘Fabrication, mechanical properties and cytocompatibility of elastomeric nanofibrous mats of poly(glycerol sebacate)’, Eur. Polym. J., 2015, 64, pp. 79 –92 [Google Scholar]
- 49. Ha S.H. Hwang J.‐H. Kim D.H. et al.: ‘Sunitinib release from biodegradable films of poly (L‐lactide‐co‐caprolactone)’, Mater. Res. Bull., 2012, 47, (10), pp. 2735 –2738 [Google Scholar]
- 50. Liu L. Koo Y. Collins B. et al.: ‘Biodegradability and platelets adhesion assessment of magnesium‐based alloys using a microfluidic system’, PLOS One, 2017, 12, (8), p. e0182914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chambers W. Green S. Gupta K. et al.: ‘Scoring for eye irritation tests’, Food Chem. Toxicol., 1993, 31, (2), pp. 111 –115 [DOI] [PubMed] [Google Scholar]