

Abstract
Clindamycin hydrochloride (CLH) is a clinically important oral antibiotic with wide spectrum of antimicrobial activity that includes gram‐positive aerobes (staphylococci, streptococci etc.), most anaerobic bacteria, Chlamydia and certain protozoa. The current study was focused to develop a stabilised clindamycin encapsulated poly lactic acid (PLA)/poly (D,L‐lactide‐co‐glycolide) (PLGA) nano‐formulation with better drug bioavailability at molecular level. Various nanoparticle (NPs) formulations of PLA and PLGA loaded with CLH were prepared by solvent evaporation method varying drug: polymer concentration (1:20, 1:10 and 1:5) and characterised (size, encapsulation efficiency, drug loading, scanning electron microscope, differential scanning calorimetry [DSC] and Fourier transform infrared [FTIR] studies). The ratio 1:10 was found to be optimal for a monodispersed and stable nano formulation for both the polymers. NP formulations demonstrated a significant controlled release profile extended up to 144 h (both CLH‐PLA and CLH‐PLGA). The thermal behaviour (DSC) studies confirmed the molecular dispersion of the drug within the system. The FTIR studies revealed the intactness as well as unaltered structure of drug. The CLH‐PLA NPs showed enhanced antimicrobial activity against two pathogenic bacteria Streptococcus faecalis and Bacillus cereus. The results notably suggest that encapsulation of CLH into PLA/PLGA significantly increases the bioavailability of the drug and due to this enhanced drug activity; it can be widely applied for number of therapies.
Inspec keywords: drug delivery systems, biomedical materials, antibacterial activity, nanoparticles, nanomedicine, microorganisms, polymers, nanofabrication, differential scanning calorimetry, encapsulation, drugs, scanning electron microscopy, Fourier transform infrared spectra
Other keywords: Streptococcus faecalis; Bacillus cereus; DSC; stable nanoformulation; monodispersed nanoformulation; pathogenic bacteria; FTIR spectra; molecular dispersion; thermal behaviour; controlled release profile; Fourier transform infrared spectra; differential scanning calorimetry; scanning electron microscopy; drug loading; encapsulation efficiency; polymer concentration; solvent evaporation method; molecular level; drug bioavailability; stabilised clindamycin encapsulated poly lactic acid‐poly (D,L‐lactide‐co‐glycolide) nanoformulation; protozoa; Chlamydia; anaerobic bacteria; gram‐positive aerobes; antimicrobial activity; oral antibiotics; oral delivery; PLA‐PLGA based nanoparticle system; clindamycin hydrochloride
1 Introduction
Clindamycin hydrochloride (CLH) (7‐chloro‐7‐deoxylincomycin hydrochloride) is a semi‐synthetic analogue of a natural antibiotic lincomycin. It is available as white crystalline powder and administered orally. It is commonly used in topical treatment for acne and infections of the skin, soft tissue and infections and peritonitis [1]. It can be useful against some methicillin‐resistant Staphylococcus aureus infections [2]. In patients with hypersensitivity to penicillin, clindamycin is used to treat infections caused by many susceptible pathogenic aerobic bacteria. It is also widely used for treatment of many anaerobic infections caused by susceptible anaerobic bacteria, including dental infections [3]. Clindamycin is also known to be useful in reducing the risk of premature births in women diagnosed with bacterial vaginosis [4] and treating toxic shock syndrome in combination with vancomycin [5]. Antimicrobial mechanisms of clindamycin include breakdown of bacterial cell membrane and inhibition of toxin synthesis [6]. The bacteriostatic effect of clindamycin is due to inhibition of bacterial protein synthesis by binding to the 50S rRNA of the large bacterial ribosome subunit that inhibits the ribosomal translocation [6].
Clindamycin is a time‐dependent antibiotic; exerts its best bactericidal effect when drug is maintained above the MIC value in formulation or alone. Hence, the time at which the therapeutic drug concentration is above the MIC (T>MIC) value is considered as the primary parameter and should be kept as minimum standard to achieve the desired clinical outcomes [7]. Therefore, sustained‐release preparation has a primary important role in the clindamycin delivery.
Currently, there is an urgent need for the development of novel carriers which could be target specific to achieve therapeutic drug concentration. Recently nanoparticles (NPs) (diameter: 10–1000 nm) delivery system has been proposed as colloidal drug carriers [8, 9], which serve as excellent carriers by enhancing aqueous solubility, increasing resistance time in the body (increasing half‐life for clearance/specificity for its associated receptors) and targeting drug to specific location in the body. Moreover, polymeric NPs have been proved to enhance the oral bioavailability of orally inactive antibiotics [10]. Various polymers have been employed in the formulation of NPs for drug delivery research to increase therapeutic benefits. Among them, poly (D,L‐lactide‐co‐glycolide) (PLGA), poly lactic acid (PLA), are considered to be highly beneficial because of their biodegradable and biocompatible nature [11]. They are widely used for human research due to their FDA approval (The Food and Drug Administration, USA).
There are several problems associated with conventional methods of clindamycin use. The drug could not reach into appropriate amount to the site of infection due to low bioavailability and drug loss. There are also incidences of low efficacy due to degradability. We hypothesise that if the drug can be encapsulated within a biodegradable system, it would enhance the drug efficiency to a greater extent by conserving its natural therapeutic property. It will not only protect the drug in its native structure but also it will be helpful in delivering the required amount of drug at the target site in an efficient manner. This can be monitored and compared by analysing the changes in structural and physiochemical properties of the free drug and the encapsulated drug. In the current study, an attempt has been made to formulate CLH‐PLA and CLH‐PLGA NPs by double emulsion solvent evaporation method with an aim to overcome the above mentioned demerits. Here, we propose for a better delivery model to offer several advantages over conventional administration and delivery methods, including target specific delivery (e.g. in intracellular infection) [12, 13], reducing systemic toxicity [14] and also to facilitate sustained release of drug (antibiotic), minimising dosing regimens [7]. The characterisation and comparison study of the above formulated NPs were performed in order to foresee possibilities to further design an endocytable controlled drug release system which would provide less alteration to the general structure of the drug and can be available in intact form at molecular level. It might be useful in treatment of various bacterial infections as well as it will reduce other related side effects.
2 Materials and methods
2.1 Materials
The drug, CLH, polymer;polylactic acid (PLA; molecular weight: 85,000–160,000 Da), poly (D,L‐lactic‐co‐glycolic) acid (PLGA 50: 50; molecular weight: 40,000–75,000 Da) and poly vinyl alcohol (PVA) were procured from Sigma Aldrich, USA. Dichloromethane and acetone (analytical grade) were purchased from Merck India Pvt Ltd. Ultrapure water from Milli‐Q water system (Millipore, USA) was used throughout the study.
2.2 Preparation of CLH‐PLA and CLH‐PLGA NPs
The CLH‐PLA and CLH‐PLGA NPs were prepared by following double emulsion solvent evaporation method according to Machado and Evangelista [15] with little modifications (Fig. 1). For organic phase (OP), 0.4 gm of PLGA/PLA polymers were dissolved in 8 ml organic mixture (dichloromethane: acetone; 85:15, v/v). For internal aqueous phase (IAP), CLH at varied concentration, that is, 20 mg (drug: polymer; 1:20), 40 mg (drug: polymer; 1:10). 80 mg (drug: polymer; 1:5) were dissolved in phosphate buffered saline (PBS) (67 mM, pH 6.0). The two solutions (OP and IAP) were mixed by ultrasonication (LABSONIC® M, B. Braun Biotech) for 30 s under cooling (output 4, 40% duty cycle) to form W1 /O emulsion. The W1 /O emulsion mixture was slowly added to 100 ml of 1% (w/v) aqueous PVA solution with a high speed homogenisation at 8500 rpm for 8 min. The resulting w1 /o/w2 emulsion was stirred at 300 rpm over night for the maximum evaporation of organic solvent. Then the precipitated NPs were washed three times with ultrapure water at 12,000 rpm for 15 min and the supernatants were stored for estimation of free drug. Finally the pellet samples (NPs) were lyophilised and then used for further characterisation. The prepared CLH‐PLA NPs were named as CLH‐PLA 1(drug: polymer;1:20), CLH‐PLA 2 (drug: polymer;1: 10), CLH‐PLA 3 (drug: polymer;1:5). Similarly CLH‐PLGA NPs were named as CLH‐ PLGA 1(drug: polymer;1:20), CLH‐PLGA 2 (drug: polymer;1: 10), CLH‐ PLGA 3 (drug: polymer;1:5).
Fig. 1.
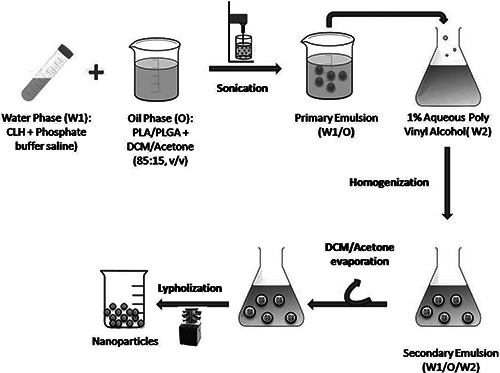
Preparation of CLH‐PLA/PLGA NPs by double emulsion solvent evaporation method
2.3 Characterisation of NPs
2.3.1 Particle size, zeta potential and polydispersity index measurements
The size (hydrodynamic diameter), size distribution (poly dispersity index) and zeta potential (surface charge) of the NPs were analysed by Zeta sizer (Zs 90, Malvern Instruments Ltd, Malvern, UK). The lyophilised samples were made an appropriate dilution with PBS (67 mm, pH 6.0). Aliquots from each preparation batch were sampled in dynamic light scattering (DLS) cuvettes and NPs were then examined for equivalent diameters, size distribution, zeta potential and poly dispersity index (PDI). Particles diameters were assessed at scattering angle of 90° and at a temperature of 25°C. Then determinations for diameter, zeta potential and PDI were measured for each preparation (in triplicate) and the standard deviations were calculated.
2.3.2 Determination of drug loading efficiency
The loading efficiencies of CLH‐PLA and CLH‐PLGANPs were calculated spectrophotometrically using standard curve method prepared by varying concentration of CLH [16]. For standard curve, different dilutions of drug in PBS (3 ml) were taken and heated with 3 ml of 1% KIO4 solution, 2 ml of 30% H2 SO4 and 10 ml of cyclohexane on a water bath at 60° for 45 min then cooled and the cyclohexane layer was removed. The extraction was repeated (×2) with 5 ml of cyclohexane and the combined extracts were diluted to 25 ml before the absorbance was measured at 520 nm. Similar procedure was followed for supernatants (containing free drugs) reserved for determination of drug. The OD value was put in the standard curve equation to find out the total drug content.
The amount of drug loaded in the NPs was calculated by subtracting the free drug present in the supernatant from the total drug added during the preparation of the NPs using the following formula
2.3.3 In vitro drug release study
The rate of CLH released from NPs was measured as a function of time during incubation in 1X PBS. Triplicate samples of 5 mg NPs were suspended in 1 ml PBS in a micro centrifuge tube and sonicated briefly in an ultrasonic water bath. The samples were then incubated on an orbital shaker (200 rpm) at 37°C. At defined time points, the samples were centrifuged. The supernatant was collected and the pellet (NPs) was reconstituted by adding fresh PBS. The drug content in the supernatant was estimated [16] as discussed above. The percentage of drug release was plotted against time and the cumulative release was calculated.
2.3.4 Scanning electron microscope (SEM) study
The morphology of the NPs was investigated by scanning electron microscopy (Jeol 6480LV jsm microscope). The NPs were fixed on adequate support and coated with platinum using platinum sputter module in a higher vacuum evaporator. Observations under different magnifications were performed at 20 kv.
2.3.5 DSC analysis
The physical states of CLH‐PLA 2, CLH‐PLGA 2 NPs and blank polymeric NPs were characterised by differential scanning calorimetric thermogram analysis (Netzch DSC 200 Fs). The samples (∼12 mg) were sealed in aluminium pans and heated under nitrogen by heating rate of 10°C/min, the heat flow being recorded from 30°C to 200°C. Indium was used as standard reference material to calibrate the temperature and energy scales of the DSC instruments. The data were analysed and DSC thermographs were plotted with help of Microsoft excel.
2.3.6 Fourier transformed infrared spectra
FT‐IR spectra (Shimadzu FTIR spectrophotometer: Model 8400 S) of CLH, CLH‐PLA 2 and CLH‐PLGA 2NPs were recorded in potassium bromide pellets. The spectrum was recorded between 4000 and 400 cm−1 using a high energy ceramic source and DLATGS detectors. Characterisation by Fourier transform infrared (FTIR) was specifically carried out to determine the adsorption of the drug in the prepared NPs by studying the chemical properties of CLH conjugated NPs. After getting the information about the functional groups, its bonding nature with NP was also characterised.
2.3.7 Antibacterial activity
The antibacterial activities of the CLH‐PLA 2, CLH‐PLGA 2 NPs and CLH were studied against Streptococcus faecalis and Bacillus cereus. The minimum inhibitory concentration (MIC) values were determined by following CLSI guidelines for the broth microdilution method [17]. Briefly, bacterial strains were cultured onto Luria Bertani Agar medium (containing 0.5% peptone, 0.5% yeast extract, 1% NaCl and 1.5% Agar, at pH 7.5 ± 0.2). The stock solution was prepared by suspending the NPs in 4 ml of aqueous solution (phosphate buffer 67 mM, pH 7.4, 0.05% Tween 20 and 0.02% sodium azide) contained in borosilicate vials. The NPs were incubated at 37°C under shaking for 24 h. Supernatant samples of 500 µl were collected by centrifugation and stored at 4°C until checked for antimicrobial activities. Chloramphenicol (a common antibiotic) was used as the positive control and PBS was used as the negative control. Bacterial cultures were added to the sterilised Mueller Hinton Broth (MHB) (containing 30% beef extract, 1.75% casein acid hydrolysate and 0.15% starch, pH 7.4 ± 0.2). The MIC was determined by using 2‐fold serial dilutions in the medium (MHB) containing 1.95–1000 µg/ml of the test compounds. To each well of 96 well microtitre plate, 150 µl of medium (MHB) was taken in duplicate, to which 10 µl of 0.5 McFarland standard (1.5 × 108 CFU/ml) culture pathogens from MHB was added. The inoculated plates were incubated at 37°C for 24 h. After incubation, the bacterial growth was monitored by measuring the turbidity of the culture by microtitre plate optical colorimeter (OD600). The MIC was determined as the lowest concentration of compound at which the visible growth of the organisms was completely inhibited.
3 Results and discussion
NPs based drug delivery system is now becoming a useful tool for controlling various acute infectious stages, preventing complications and their relapse. Therefore, it is helpful in overcoming the sensible disadvantages with long‐term therapies [18]. In addition, drugs delivered through nanospheres that target particularly to the infected cells should have reduced cytotoxicity associated with undesired bio distribution of free drug. Depending upon the mode of action, NPs based drug delivery systems could perform better than microparticle based delivery system due to their size and better presentation of antigen [19, 20].
3.1 Physical properties
Various formulation factors were reported to play a key role on the physiochemical properties of polymeric nano/micro particles formed [21]. In this study, CLH loaded in PLA and PLGA NPs were prepared by W1 /O/W2 double emulsion evaporation technique varying the drug to polymer ratio. The effect of the drug concentrations (1:20, 1:10, 1:5) on the size of the obtained NPs were analysed. The physical properties of all the formulations are mentioned in Table 1. The particle size of PLA NPs (blank) was recorded as 42.93 ± 1.77 nm. The particle size increased with the drug encapsulation in to the NPs. The sizes of NPs were 203.35 ± 12.04, 323.5 ± 16.39, 827.4 ± 10.20 nm at different drug to polymer concentration, that is, 1:20, 1:10 and 1:5, respectively. In case of PLGA NPs (blank), the particle size was 178.6 ± 12.11 nm, while for CLH‐PLGA 1 (1:20), CLH‐PLGA 2 (1:10), CLH‐PLGA 3 (1:5), the particle sizes were 196.45 ± 8.78, 258.3 ± 11.23 and 456.5 ± 12.36 nm, respectively. From the particle size analysis report, it can be concluded that the particle sizes were dependent on the initial concentration of the drug used during preparation process. Similar results were also documented earlier [21, 22]. Zeta potential is a scientific term for measurement of electro‐kinetic potential at the surface of the colloidal particles, which has a great significance in comparing the stability of colloidal dispersions [23]. The zeta potential indicates the degree of repulsion between adjacent particles and similarly charged particles in dispersion. For molecules and particles that are small enough, a high zeta potential confers stability (dispersion will resist aggregation). When the zeta potential is low, the attraction between the particles exceeds repulsive force for which leads to flocculate.
Table 1.
Physical properties of CLH loaded PLA and PLGA NPs
| Polymer | Sample (Code) | Drug (mg) | Polymer (mg) | Ratio of drug: polymer | Mean particle size (diameter in nm ± SD) | Zeta potential (mV) ± SD | Poly dispersity index (PDI) ± SD | Encapsulation efficiency, % |
|---|---|---|---|---|---|---|---|---|
| PLA | Blank (PLA) | – | 400 | – | 42.93 ± 1.77 | −24.8 ± 7.67 | 0.454 ± 0.05 | – |
| CLH‐PLA 1 | 20 | 400 | 1:20 | 203.35 ± 12.04 | −19.8 ± 3.88 | 0.332 ± 0.03 | 7.2 ± 2.08 | |
| CLH‐PLA 2 | 40 | 400 | 1:10 | 323.5 ± 16.39 | −30.5 ± 4.95 | 0.219 ± 0.01 | 21.35 ± 3.17 | |
| CLH‐PLA 3 | 80 | 400 | 1:5 | 827.4 ± 10.20 | −17.6 ± 6.55 | 0.423 ± 0.04 | 24.5 ± 4.29 | |
| PLGA | Blank (PLGA) | – | 400 | – | 178.6 ± 12.11 | −32.7 ± 5.01 | 0.524 ± 0.03 | – |
| CLH‐PLGA 1 | 20 | 400 | 1:20 | 196.45 ± 8.78 | −25.5 ± 2.88 | 0.650 ± 0.05 | 45 ± 3.45 | |
| CLH‐PLGA 2 | 40 | 400 | 1:10 | 258.3 ± 11.23 | −33.5 ± 3.0 | 0.176 ± 0.01 | 65.69 ± 2.28 | |
| CLH‐PLGA 3 | 80 | 400 | 1:5 | 456.5 ± 12.36 | −21.7 ± 5.34 | 0.353 ± 0.03 | 72.35 ± 2.31 |
(CLH‐PLA1‐3, CLH‐PLGA 1‐3 were named according to drug: polymer ratio. The drug: polymer ratio of CLH‐PLA 1, CLH‐PLA 2, CLH‐PLA 3 were 1:20, 1:10, and1:5, respectively. Similarly The drug: polymer ratio of CLH‐PLGA 1, CLH‐PLGA 2 and CLH‐PLGA 3 were 1:20, 1:10, and 1:5, respectively). The results were expressed as mean ± standard deviation
The zeta potential values of CLH‐PLA NPs were ranged from −17.6 ± 6.55 mV to −30.5 ± 4.95 mV, while for CLH‐PLGA NPs; it varied from −21.7 ± 5.34 mV to −33.5 ± 3.0 mV. The negative charge confirmed the surface charge of both PLA and PLGA NPs. However, very interestingly, the zeta potential were highest when the drug: polymer concentration was 1:10 (−30.5 ± 4.95 mV for CLH‐PLA and −33.5 ± 3.0 mV for CLH‐PLGA). The zeta potential values of CLH‐PLA NPs and CLH‐PLGA NPs confirmed that the drug: polymer concentration is important for the stability of drug loaded NPs. The size and charge distribution patterns of CLH‐PLA 2, CLH‐PLGA 2 are shown in Figs. 2 and 3, respectively. The PDI is a measurement for distribution of NPs and it gives the distribution range from 0.000 to 0.500. Poly dispersity index >0.5 values indicates the aggregation of particles [24]. The PDI values for all NPs were within good range. In case of CLH‐PLA NPs, the PDI value was lowest for CLH‐PLA 2 (0.219 ± 0.01). Similar results were also found for CLH‐PLGA NPs, where PDI value was found lowest for CLH‐PLGA 2 (0.176 ± 0.01).
Fig. 2.

Distribution pattern of CLH loaded PLA NPs (PLA‐CLH 2). The drug: polymer ratio is 1:10. The average size and zeta potential were 323.5 ± 16.39 nm and −30.5 ± 4.95 mV, respectively
a Size
b Charge
Fig. 3.
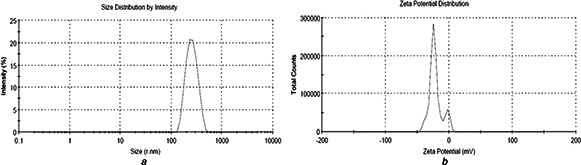
Distribution pattern of CLH loaded PLGA NPs (PLGA‐CLH 2). The drug: polymer ratio is 1:10. The average size and zeta potential were 258.3 ± 11.23 nm and −33.5 ± 3.0 mV, respectively
a Size
b Charge
3.2 Loading efficiency
The loading efficiencies increased with the increase in drug concentration. In case of CLH‐PLA NPs, the loading efficiency (%) increased from 7.2 ± 2.08 to 24.5 ± 4.29, when the drug: polymer concentration increased from 1:20 to 1:5. Similarly in case of CLH‐PLGA NPs, the loading efficiencies (%) increased from 45 ± 3.45 to 72.35 ± 2.31, when the drug: polymer concentration increased from 1:20 to 1:5. This study showed the loading efficiency of CLH to PLGA was higher than CLH to PLA. CLH loading efficiency depended on the polymer type and the physical state of the drug (solid form or in solution form) during processing. The enhanced microencapsulation in case of more hydrophilic polymers might be ascribed to enhanced molecular interactions between the drug and the polymer [25, 26]. This study showed the loading efficiency of CLH to PLGA was higher than CLH to PLA. This might be due to the reason that PLGA is more hydrophilic nature than PLA.
3.3 In vitro drug release
In vitro release of CLH was assessed from PLA and PLGA NPs into PBS (pH 7.4). The release profile of CLH from PLA and PLGA NPs is illustrated in Fig. 4. It is observed that CLH release profile from CLH‐PLGA in a considerably slower than from CLH‐PLA. In case of CLH‐PLA, 50% of the drug was released within 4 h. 75% release of CLH was realised within a period of up more than 48 h. However, in case of CLH‐PLGA, 50% drug was released within 8 h and 75% release of CLH was realised within a period of up more than 72 h. The rapid burst effect is very much delayed in case of CLH‐PLGA. That conferred that binding of drug to PLGA was stronger than to PLA. In vitro release data showed proper drug (CLH) encapsulation that avoided the burst effect and offered an extended release profile. Similar type of drug release studies was observed in etodolac loaded PLGA NPs [27] and minocycline‐loaded PLGA NPs [28]. Thus PLGA‐drug can be a better oral delivery system as it could withstand the drug even after 4 h which is the standard required time for retaining food in stomach.
Fig. 4.
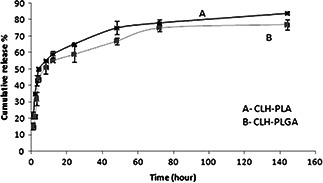
In vitro CLH release from CLH‐PLA 2 and CLH‐PLGA 2 NPs
3.4 SEM study
The morphology of CLH loaded PLA and PLGA NPs were analysed by SEM study. The NPs were spherical structures as confirmed by SEM (Fig. 5). The surface morphologies of the particles were rough and rounded. Particles were found to be regular and isolated in nature. These regular and isolated forms of spherical structures were also observed earlier by Machado and Evangelista [15] in case of cefoxitin loaded D, L‐PLA NPs. The smooth surface can also be correlated to lactide content that adds hydrophobicity to the polymer. Hence, that could prevent the retention of water and the shrinkage could be avoided while drying. Moreover, particle‐particle contact formation was become less due to presence of lactide (which causes hydrophobic) and ultimately prevents agglomeration [29].
Fig. 5.

SEM photograph of
a CLH‐PLA2 (Drug: polymer‐ 1:10)
b CLH‐PLGA2 (Drug: polymer‐ 1:10)
3.5 DSC studies
Thermal analytical studies of polymeric drug delivery systems are vital, since the processes used for their preparation are able to change the organisation of the polymer chains [30]. Fig. 6 shows DSC data of CLH, PLA NPs, PLGA NPs, CLH‐PLA 2 and CLH‐PLGA 2. In the current study, the blank PLA and PLGA polymer showed glass transition temperature (Tg) at 64.24°C and 67.5°C, respectively. Tg (glass transition temperature) represents the measurement of polymer chain flexibility for the lactic acid polymers that indicates the hydrolysis pattern of the ester bonds [31]. The usual Tg values reported for PLA and PLGA systems are more than 37°C (hence, they are glassy in nature) but can be clearly attributed to the presence of crystallites within the samples [32]. Thermal analysis data (Fig. 5) showed that pure CLH possesses an endothermic peak at 150°C related to the melting point. CLH melting peak was depleted in the thermogram for the drug loaded PLA and PLGA NPs, indicating the presence of amorphous CLH in the NPs. Similar results were also observed in minocycline encapsulated PLGA NPs [28] and in cefoxitin loaded DL‐PLA NPs [14]. With this, it can be suggested that the drug was more uniformly dispersed throughout the system in case of PLA and PLGA. The disappearance of the peak referred to the crystalline melting of the drug which indicates that the drug clindamycin is uniformly dispersed throughout the polymer matrix at a molecular level [15, 33].
Fig. 6.
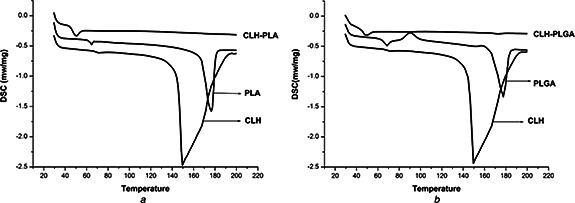
DSC data
a Thermal analysis graph of Clindamycin hydrochloride (CLH), blank PLA, CLH conjugated with PLA (CLH‐PLA2)
b Thermal analysis graph of Clindamycin hydrochloride (CLH), blank PLGA, and CLH conjugated with PLGA (CLH‐PLGA 2)
The results also showed a decrease in glass transition temperature of PLA and PLGA polymer with respect to nanoencapsulation of the drug. This may be due to increase in size of the particles whereas the glass transition temperature is inversely related to size of particles in case of amorphous polymer [34].
3.6 FTIR analysis
FTIR analysis measures the selective absorption of light by the vibration modes of specific chemical bonds in the sample. The observation of vibration spectrum of encapsulated drug permits evaluation of the type of interaction occurring between the drug and polymer. This interaction is due to the vibrations of the atoms involved (in this interaction) can suffer alterations in intensity and frequency [35]. FTIR studies of CLH‐PLA and CLH‐PLGA NPs were performed to characterise the chemical structure of drug conjugated NPs. Fig. 7 shows the FTIR spectra of CLH‐PLA 2 and CLH‐PLGA 2 NPs. Nearly similar peaks were observed in both the prepared NPs some of which occurred at 3648 cm−1 (O‐H stretching), 3505 cm−1 (O‐H stretching), 2946 cm−1 (C‐H stretching), 2996 cm−1 (C‐H stretching), 1761 cm−1 (C=O stretching), 1458 cm−1 (C‐H bending), 1386 cm−1 (N=O bending), 1184 cm−1 (C‐N stretching), 1080 cm−1 (C‐O stretching), 870 cm−1 and 756 cm−1 (N‐H wags) for the CLH‐PLA 2 NPs whereas 3644 cm−1 (O‐H stretching), 2999 cm−1 (C‐H stretching), 2955 cm−1 (C‐H stretching), 1761 cm−1 (C=O stretching), 1458 cm−1 (C‐H bending), 1398 cm−1 (N=O bending), 1183 cm−1, 1086 cm−1 (C‐N stretching), 866 cm−1 and 749 cm−1 (N‐H wags) for the CLH‐PLGA 2 NPs. As drug has a secondary amine group on its general structure whose peak is also detected by FTIR studies. The analysis shows no alteration of secondary amine structure after conjugation with PLA/PLGA NPs. However, changes have been found after conjugation with PLA/PLGA for some functional groups such as C=O, OH those which are abundant in PLGA/pla NPs. These functional groups were found very less in number in case of CLH drug alone. From FTIR studies it can be concluded that there is not much alteration in general structure of CLH drug because of the presence of OH group and the drug is present in its native structure without forming any hydrogen bond with any other functional group. As CH group is also present in CLH drug, there is no alteration of CH group in the conjugated drug. C‐O bond is common to both polymers and CLH drug. Therefore, it has no effect on to the CLH drug's general structure. These observations confirmed that the intact form of CLH drug and it was not involved in any chemical interactions with the polymers which were also observed for nimesulide‐loaded ethylcellulose and methylcellulose NPs and microparticles [21].
Fig. 7.
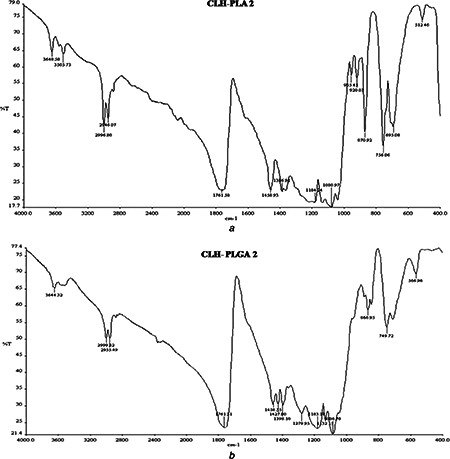
FTIR data analysis of
a CLH‐PLA 2
b CLH‐PLGA‐3
3.7 Antimicrobial activity
The antibacterial activities were evaluated for the selected nano formulations (CLH‐PLA 2, CLH‐PLGA 2) and CLH drug alone by determining their MIC values against pathogenic microorganisms. The MIC values are mentioned in Table 2. In this study, MIC values of CLH were found to be 0.48 ± 0.01 µg/ml for Streptococcus faecalis and 1.95 ± 0.04 µg/ml for Bacillus cereus. However, these MIC values were decreased when the drug was encapsulated into PLA and PLGA NPs. CLH‐PLA 2 showed MIC values of 0.12 ± 0.05 µg/ml and 0.97 ± 0.08 µg/ml, while CLH‐PLGA 2 showed MIC values 0.24 ± 0.05 µg/ml and 0.48 ± 0.06 µg/ml against Streptococcus faecalis and Bacillus cereus, respectively. MIC values of CLH were found to be increased, when the drug was loaded in to PLA and PLGA NPs as proved against Streptococcus faecalis and Bacillus cereus. Therefore, this study confirmed the enhanced antibacterial activity of drug loaded NPs than the standard free drug. Similar enhanced antimicrobial activities of drug loaded NPs were also observed by many researchers [13, 28, 36].
Table 2.
Antibacterial activity of Clindamycin hydrochloride conjugated with PLA (CLH‐PLA 2), Clindamycin hydrochloride conjugated with PLGA (CLH‐PLGA 2) and Clindamycin hydrochloride, in vitro MIC in µg/ml
| MIC (µg/ml) ± Standard deviation | ||
|---|---|---|
| Formulations | Streptococcus faecalis | Bacillus cereus |
| Clindamycin hydrochloride | 0.48 ± 0.01 | 1.95 ± 0.04 |
| PLA‐CLH 2 | 0.12 ± 0.05 | 0.97 ± 0.08 |
| PLGA‐CLH 2 | 0.24 ± 0.05 | 0.48 ± 0.06 |
Hence, proving our hypothesis successful formulations of CLH loaded in PLA and PLGA NPs were carried out. From the DLS analysis, 1:10 dilution (drug: polymer concentration) was proved to be the optimal ratio for the formulation of NPs. From CLH loading efficiency and release profile studies, more hydrophilic PLGA offers better encapsulation as well as extended CLH release profile confirming enhanced molecular interactions. Thermal analysis studies suggested that CLH was more uniformly dispersed throughout PLA and PLGA matrix at a molecular level. FTIR analysis confirmed the unaltered nature of drug CLH (no chemical interaction with polymer matrix), which was very important parameter for better efficacy of CLH‐PLA/PLGA nano complex. Enhanced antimicrobial activities of CLH‐PLA/ CLH‐PLGA NPs were proved against Streptococcus faecalis and Bacillus cereus measured through MIC studies. Hence, the CLH drug was uniformly entrapped in PLA/PLGA NPs and the formulations were proven to exhibit more effective roles used at lower concentration.
4 Conclusions
The therapeutic efficacy of the drug Clindamycin Hydrochloride was significantly enhanced by encapsulating into biodegradable polymer PLA/PLGA based nanoparticulate system when compared with the free drug. The hypothesis with which we initiated the work has been rightfully anticipated and we could successfully overcome the limitations of the drug when used alone. During the current investigation, CLH‐PLA and CLH‐PLGA NPs of various sized particles were formulated by varying the drug to polymer ratio from 1:5 to 1:20. The size, zeta potential and poly dispersity index values of CLH‐PLA NPs and CLH‐PLGA NPs confirmed that the drug: polymer concentration is important for the stability of drug loaded NPs. The drug to polymer ratio 1:10 were found to be optimal ratio for the formulation to be stable and monodispersed CLH loaded PLA and PLGA NPs. NPs formulation (both CLH‐PLA 2 and CLH‐PLGA 2) showed a significantly higher drug loading efficiency and a controlled release profile extended up to 144 h. Higher drug loading and an extended CLH release profile were observed in case of CLH‐PLGA NPs which might be due to higher hydrophilic nature of PLGA that causes enhanced molecular interaction. The spherical and isolated structures of both CLH‐PLA 2 and CLH‐PLGA 2 NPs were confirmed by SEM study. The smooth surface and isolated nature of NPs can also be correlated to presence of lactide that enhances hydrophobicity to the polymer ultimately prevents agglomeration. The thermal behaviour (DSC) studies of CLH‐PLGANPs confirmed the disappearance of CLH endothermic peak (150°C) which indicated the uniform dispersion of drug at a molecular level within the system. From FTIR studies it was demonstrated that there was not much alteration found in general structure of CLH drug (due to conserved OH, CH and C‐O group etc.) after loading into PLA and PLGA NPs. The antimicrobial activities were enhanced in CLH‐PLA NPs and CLH‐PLGANPs than the standard free drug evidenced from decrease in MIC values tested against Streptococcus faecalis and Bacillus cereus. To maintain a sustained release and drug concentration during all the time of medication process, our formulations are shown to be useful and effective. Hence, we can conclude that by encapsulation of CLH into PLA and PLGA NPs, the various physiochemical properties of the drug increased to satisfactory extent. The drug was more effective at lower concentration against pathogenic microorganisms which can be widely applied in number of therapies by further designing successful endocytable controlled drug release system.
5 Acknowledgments
We would like to extend our sincere thanks to Dr Kunal Pal, Dept. of Biotechnology and Medical Engineering; Dr Sasmita Mahapatra, Dept. of Chemistry and Dept. of Metallurgy, NIT Rourkela for carrying out the DSC studies, DLS analysis and FTIR studies, respectively.
6 References
- 1. Darley E.S. MacGowan A.P.: ‘Antibiotic treatment of gram‐positive bone and joint infections’, J. Antimicrob. Chemother., 2004, 53, (6), pp. 928 –935 (doi: 10.1093/jac/dkh191) [DOI] [PubMed] [Google Scholar]
- 2. Daum R.S.: ‘Skin and soft‐tissue infections caused by methicillin‐resistant Staphylococcus aureus ‘, N. Eng. J. Med., 2007, 357, (4), pp. 380 –390 (doi: 10.1056/NEJMcp070747) [DOI] [PubMed] [Google Scholar]
- 3. Brook I. Lewis M.A. Sándor G.K. et al.: ‘Clindamycin in dentistry: more than just effective prophylaxis for endocarditis?’, Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. Endod., 2005, 100, pp. 550 –558 (doi: 10.1016/j.tripleo.2005.02.086) [DOI] [PubMed] [Google Scholar]
- 4. Lell B. Kremsner P.G.: ‘Clindamycin as an antimalarial drug: review of clinical trials’, Antimicrob. Agents. Chemother., 2002, 46, (8), pp. 2315 –2320 (doi: 10.1128/AAC.46.8.2315-2320.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Annane D. Clair B. Salomon J.: ‘Managing toxic shock syndrome with antibiotics’, Expert. Opin. Pharmacother., 2004, 5, (8), pp. 1701 –1710 (doi: 10.1517/14656566.5.8.1701) [DOI] [PubMed] [Google Scholar]
- 6. Coyle E.A.: ‘Targeting bacterial virulence: the role of protein synthesis inhibitors in severe infections’, Soc. Infect. Dis. Pharm. Pharmacother., 2003, 23, (5), pp. 638 –642 [DOI] [PubMed] [Google Scholar]
- 7. Gao P. Nie X. Zou M. et al.: ‘Recent advances in materials for extended‐release antibiotic delivery system’, J. Antibiot. (Tokyo), 2011, 64, pp. 625 –634 (doi: 10.1038/ja.2011.58) [DOI] [PubMed] [Google Scholar]
- 8. Jong W.H.D. Borm P.J.A.: ‘Drug delivery and nanoparticles: applications and hazards’, Int. J. Nanomedicine, 2008, 3, (2), pp. 133 –149 (doi: 10.2147/IJN.S596) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patel A. Patel M. Yang X. et al.: ‘Recent advances in protein and peptide drug delivery: a special emphasis on polymeric nanoparticles’, Protein Pept. Lett., 2014, 21, (11), pp. 1102 –1120 (doi: 10.2174/0929866521666140807114240) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Diab R. Jaafar‐Maalej C. Fessi H. et al.: ‘Engineered nanoparticulate drug delivery systems: the next frontier for oral administration?’, AAPS J., 2012, 14, (4), pp. 688 –702 (doi: 10.1208/s12248-012-9377-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ulery B.D. Nair L.S. Laurencin C.T.: ‘Biomedical applications of biodegradable polymers’, J. Polym. Sci. B: Polym. Phys., 2011, 49, (12), pp. 832 –864 (doi: 10.1002/polb.22259) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pinto‐Alphandary H. Andremont A. Couvreur P.: ‘Targeted delivery of antibiotics using liposomes and nanoparticles: research and applications’, Int. J. Antimicrob. Agents, 2000, 13, pp. 155 –168 (doi: 10.1016/S0924-8579(99)00121-1) [DOI] [PubMed] [Google Scholar]
- 13. Abdelghany S.M. Quinn D.J. Ingram R.J. et al.: ‘Gentamicin‐loaded nanoparticles show improved antimicrobial effects towards Pseudomonas aeruginosa infection’, Int. J. Nanomed., 2012, 7, pp. 4053 –4063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seleem M.N. Munusamy P. Ranjan A. et al.: ‘Silica‐antibiotic hybrid nanoparticles for targeting intracellular pathogens’, Antimicrob. Agents Chemother., 2009, 53, pp. 4270 –4274 (doi: 10.1128/AAC.00815-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Machado S.R.P. Evangelista R.C.: ‘Development and characterization of cefoxitin loaded D, L‐PLA nanoparticles’, J. Basic. Appl. Sci., 2010, 31, (3), pp. 193 –202 [Google Scholar]
- 16. Fawzy A.E. Salah M.B.: ‘Spectrophotometric and titrimetric determination of clindamycin hydrochloride in pharmaceutical preparations’, Analyst., 1993, 118, pp. 577 –579 (doi: 10.1039/an9931800577) [DOI] [Google Scholar]
- 17. CLSI, Wayne : ‘Clinical and laboratory standards institute methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobicall’, Approve Standard M7‐A7, CLSI, seventh ed., PA, USA, 2006.
- 18. Thomasin C. Corradin G. Men Y. et al.: ‘Tetanus toxoid and synthetic malaria antigen containing poly (lactide)/poly(lactide‐co‐glycolide) microspheres: importance of polymer degradation and antigen release for immune response’, J. Control Release, 1996, 41, pp. 131 –145 (doi: 10.1016/0168-3659(96)01363-6) [DOI] [Google Scholar]
- 19. Rosler A. Vandermeulen G.W.M. Klok H.A.: ‘Advanced drug delivery devices via self‐assembly of amphiphilic block copolymers’, Adv. Drug Deliv. Rev., 2001, 53, pp. 95 –108 (doi: 10.1016/S0169-409X(01)00222-8) [DOI] [PubMed] [Google Scholar]
- 20. Santhi K. Venkatesh D.N. Dhanaraj S.A. et al.: ‘Development and In‐vitro evaluation of a topical drug delivery system containing Betamethazone loaded ethyl cellulose nanospheres’, Trop. J. Pharm. Res., 2005, 4, (20), pp. 495 –500 [Google Scholar]
- 21. Ravikumara N.R. Madhusudhan B. Nagaraj T.S. et al.: ‘Preparation and evaluation of Nimesulide‐loaded ethylcellulose and methylcellulose nanoparticles and microparticles for oral delivery’, J. Biomater. Appl., 2009, 24, pp. 47 –63 (doi: 10.1177/0885328209103406) [DOI] [PubMed] [Google Scholar]
- 22. Nayak B. Panda A.K. Ray P.: ‘Formulation, characterization and evaluation of rotavirus encapsulated PLA and PLGA particles for oral vaccination’, J. Microencapsul., 2009, 26, (2), pp. 154 –165 (doi: 10.1080/02652040802211709) [DOI] [PubMed] [Google Scholar]
- 23. McNaught A.D. Wilkinson A.: Blackwell Scientific Publications, Oxford. Complied version of Nic, M., Jirat, J., Kosata, B. and Jenkins, A. Definition of electro kinetic potential in ‘IUPAC. Compendium of Chemical Terminology’, 2nd ed. (the ‘Gold Book’). XML on‐line corrected version: available at http://goldbook.iupac.org (2006‐) ISBN0‐9678550‐9‐8, 1997 [Google Scholar]
- 24. Tripathi A. Gupta R. Saraf S.A.: ‘PLGA nanoparticles of anti tubercular drug: drug loading and release studies of a water in‐soluble drug’, Int. J. Pharm. Tech. Res., 2010, 2, (3), pp. 2116 –2123 [Google Scholar]
- 25. Gander B. Johansen P. Nam‐trân H. et al.: ‘Thermodynamic approach to protein microencapsulation into poly (D,L‐lactide) by spray drying’, Int. J. Pharm., 1996, 129, pp. 51 –61 (doi: 10.1016/0378-5173(95)04240-7) [DOI] [Google Scholar]
- 26. Prior S. Gamazo C. Irache J.M. et al.: ‘Gentamicin encapsulation in PLA/PLGA microspheres in view of treating Brucella infections’, Int. J.Pharm., 2000, 16, pp. 115 –125 (doi: 10.1016/S0378-5173(99)00448-2) [DOI] [PubMed] [Google Scholar]
- 27. Yasemin C. Robineau C. Çapan Y.: ‘Etodolac loaded Poly (Lactide Co‐Glycolide) nanoparticles: formulation and in vitro characterization’, Hacettepe Univ. J Faculty Pharm., 2009, 29, (2), pp. 105 –114 [Google Scholar]
- 28. Kashi T.S. Eskandarion S. Esfandyari‐Manesh M. et al.: ‘Improved drug loading and antibacterial activity of minocycline‐loaded PLGA nanoparticles prepared by solid/oil/water ion pairing method’, Int. J. Nanomedicine., 2012, 7, pp. 221 –234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rawat A. Majumder Q.H. Ahsan F.: ‘Inhalable large porous microspheres of low molecular weight heparin: in vitro and in vivo evaluation’, J. Control. Release, 2008, 128, pp. 224 –232 (doi: 10.1016/j.jconrel.2008.03.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dubernet C.: ‘Thermoanalysis of microspheres’, Thermo. Chim. Acta., 1995, 248, pp. 259 –269 (doi: 10.1016/0040-6031(94)01947-F) [DOI] [Google Scholar]
- 31. Ford J.L. Timmins P.: ‘Pharmaceutical thermal analysis’ (John Wiley & Sons, Chichester, 1989) [Google Scholar]
- 32. Narladkar A. Balnois E. Vignaud G. et al.: ‘Difference in glass transition behavior between semi crystalline and amorphous poly (lactic acid) thin films’, Macromol. Symp., 2008, 273, pp. 146 –152 (doi: 10.1002/masy.200851321) [DOI] [Google Scholar]
- 33. Hariharan M. Price J.C.: ‘Solvent, emulsifier and drug concentration factors in poly‐(D,L‐Lactic acid) microspheres containing hexamethylmelamine’, J. Microencapsul., 2002, 19, (1), pp. 95 –109 (doi: 10.1080/02652040110081398) [DOI] [PubMed] [Google Scholar]
- 34. Jordan J. Jacob K.I. Tannenbaum R. et al.: ‘Experimental trends in polymer nanocomposites: A review’, Mater. Sci. Eng., 2005, 393, pp. 1 –11 (doi: 10.1016/j.msea.2004.09.044) [DOI] [Google Scholar]
- 35. Devi T.S.R. Gayathri S.: ‘FTIR and FT‐Raman spectral analysis of Paclitaxel drugs’, Int. J. Pharmaceut. Sci. Rev. Res., 2010, 2, (2), pp. 106 –110 [Google Scholar]
- 36. Azhdarzadeh M. Lotfipour F. Zakeri‐Milani P. et al.: ‘Anti‐bacterial performance of azithromycin nanoparticles as colloidal drug delivery system against different gram‐negative and gram positive bacteria’, Adv. Pharmaceut. Bull., 2012, 2, (1), pp. 17 –24 [DOI] [PMC free article] [PubMed] [Google Scholar]