

Visual Abstract

Keywords: glomerular and tubulointerstitial diseases, 16S, bacterial DNA, blood microbiome, gastrointestinal microbiome, gut microbiome, IgA glomerulonephritis, IgA nephropathy, L-form bacteria, microbiota
Key Points
A higher microbiome load, possibly originating from different body sites, may be playing a pathogenic role in IgA nephropathy.
Several microbiome taxonomic differences between patients with IgA nephropathy and healthy controls are observed in blood and stool.
Striking differences between the blood and gut microbiome confirm that the blood microbiome does not directly reflect the gut microbiome.
Abstract
Background
IgA nephropathy (IgAN) has been associated with gut dysbiosis, intestinal membrane disruption, and translocation of bacteria into blood. Our study aimed to understand the association of gut and blood microbiomes in patients with IgAN in relation to healthy controls.
Methods
We conducted a case-control study with 20 patients with progressive IgAN, matched with 20 healthy controls, and analyzed bacterial DNA quantitatively in blood using 16S PCR and qualitatively in blood and stool using 16S metagenomic sequencing. We conducted between-group comparisons as well as comparisons between the blood and gut microbiomes.
Results
Higher median 16S bacterial DNA in blood was found in the IgAN group compared with the healthy controls group (7410 versus 6030 16S rDNA copies/μl blood, P=0.04). α- and β-Diversity in both blood and stool was largely similar between the IgAN and healthy groups. In patients with IgAN, in comparison with healthy controls, we observed higher proportions of the class Coriobacteriia and species of the genera Legionella, Enhydrobacter, and Parabacteroides in blood, and species of the genera Bacteroides, Escherichia-Shigella, and some Ruminococcus in stool. Taxa distribution were markedly different between the blood and stool samples of each subject in both IgAN and healthy groups, without any significant correlation between corresponding gut and blood phyla.
Conclusions
Important bacterial taxonomic differences, quantitatively in blood and qualitatively in both blood and stool samples, that were detected between IgAN and healthy groups warrant further investigation into their roles in the pathogenesis of IgAN. Although gut bacterial translocation into blood may be one of the potential sources of the blood microbiome, marked taxonomic differences between gut and blood samples in each subject in both groups confirms that the blood microbiome does not directly reflect the gut microbiome. Further research is needed into other possible sites of origin and internal regulation of the blood microbiome.
Introduction
IgA nephropathy (IgAN) is the most common primary glomerulopathy and is characterized by deposition of IgA antibodies, usually in the kidney mesangium (1). Although the exact pathogenesis remains unclear, antigens are believed to stimulate the production of poorly galactosylated IgA1 in susceptible hosts, resulting in glomerular mesangium immune complex deposition, thus eliciting inflammation and tissue damage (2). A genome-wide association study showed that genes involved in IgAN were associated with the ability of the gut-associated lymphoid tissue to regulate intestinal pathogens and maintain integrity of the intestinal barrier (3). These results have generated interest in the association and role of gut microbes in IgAN.
Previous gut microbiome studies have shown that the gut microbiome plays a vital role in host nutrition and development of the immune system (4,5). This gut microbiome tends to become imbalanced (dysbiotic) in various disease states, including CKD (6,7). Gut dysbiosis associated with disruption of the intestinal membrane barrier, resulting in translocation of gut bacteria and toxins into blood, has been observed in CKD (8,9). Strong evidence of the gut-renal axis has been recently reported to be associated with the pathogenesis of IgAN (10). As a major Ig of the gut mucosal immune system, IgA in its secretory form plays a crucial role in controlling mucosal inflammation by linking to specific gut microbiota (11). A recent study by De Angelis et al. (12) has shown significant differences in gut microbiota between patients with IgAN and healthy subjects, with a higher proportion of species and genera of the families Ruminococcaceae, Lachnospiraceae, Streptococcaceae, and others identified in patients with IgAN. Subsequent gut microbiome studies in a Chinese population with IgAN have additionally noted a higher prevalence of the genera Escherichia-Shigella and Bacteroides in stool when compared with healthy controls (13,14). We were interested in understanding if such microbiota may be mediating their pathogenic effects by translocating into blood via a disrupted intestinal barrier. A study simultaneously analyzing both blood and gut microbiome in IgAN has not been conducted previously. We hypothesized that the blood microbiome in IgAN will reflect dysbiosis analogous to the gut, and differ from healthy controls. Our study aimed at comparing the blood bacterial quantity of 16S ribosomal DNA (16S rDNA) and blood and stool metagenomic qualitative profiles between patients with IgAN and healthy controls. By analyzing human stool and blood microbiomes simultaneously for the first time in IgAN, we also compared concurrent stool and blood microbiome samples to better understand the relationship of gut microbiota translocating into blood.
Materials and Methods
Study Design
We conducted a case-control study involving testing of the blood and stool microbiome of 20 patients with IgAN and 20 healthy control subjects. The study was approved by our Partners Institutional Review Board and adhered to the Declaration of Helsinki.
Enrollment of Study Participants
We recruited 20 adult patients in each group, aged 18–65 years, who were enrolled in our hospital electronic medical record system (Figure 1). Patients with IgAN were identified by reviewing kidney biopsy specimen reports and patient charts of individuals followed at Massachusetts General Hospital. Participants with IgAN had biopsy sample–proven IgAN with progressive disease at various stages, had an eGFR of ≥15 ml/min using the Chronic Kidney Disease Epidemiology Collaboration formula (15), were not on any oral or systemic immunosuppressants, and had never received dialysis or a transplant. Healthy controls were frequency matched by age and sex. They were recruited primarily via advertisement of the study using an online platform named “Rally,” which is approved by the institutional review board to foster collaboration between public and the research community. We excluded subjects with diagnosed diabetes, any malignancy, inflammatory bowel disease, history of colon surgery, or intake of antibiotics or probiotics within 30 days of the study visit. Dietary assessment was not performed due to unclear effects of different foods on the microbiome. Notably, previous studies have demonstrated that the overall composition of the gut microbiome at phylum level remains relatively stable despite some diurnal variations (16).
Figure 1.

Enrollment of study subjects. Out of 62 patients with IgA Nephropathy, 20 patients were recruited based on eligibility criteria detailed below. 20 out of 74 healthy subjects met the eligibility criteria for the study after matching for age and sex.
Study Visit and Sample Collection
The study visit involved obtaining written informed consent (per Recommendations for the Conduct, Reporting, Editing, and Publication of Scholarly Work in Medical Journals) and blood and urine samples. Blood was tested for routine chemistries and the microbiome. A preprepared stool kit was given to subjects and all samples were either dropped off personally or mailed to us via overnight shipping within 1–2 days of sample collection. Samples were collected within 2 weeks of signing informed consent. Nine subjects provided a stool specimen on the same day as the study visit, out of which five were provided after using the laboratory restroom within minutes of blood collection. Blood and stool microbiome samples were stored in a −80°C freezer until study completion and then shipped for batch testing.
Microbiome Testing
DNA Extraction and 16S Quantification
After sterilizing skin before venipuncture, 3 ml of whole blood was drawn for microbiome testing in an EDTA tube, midway among other blood draws to eliminate chances of skin contamination. Total DNA was extracted from 100 µl of whole blood using a specific Vaiomer (Toulouse, France) protocol carefully designed to minimize any risk of contamination, as described previously (17–19). To ensure a low background signal from bacterial contamination of reagents and consumables, negative controls, consisting of molecular-grade water, were added separately in an empty tube at the DNA extraction step (extraction negative control) and PCR step (PCR negative control), and then amplified and sequenced at the same time as the extracted DNA of the blood samples. β-Diversity analyses show a clear separation between negative controls and both blood samples (Supplemental Figure 1). These controls confirm that bacterial contamination was well contained in our pipeline and had a negligible effect on the taxonomic profiles of the samples of this study. DNA was extracted from stool samples as described previously (20).
16S rDNA Amplification and Measurement
The quantity of total 16S rDNA extracted from blood samples was measured in triplicate by quantitative PCR using 16S universal primers targeting the V3–V4 region of the bacterial 16S ribosomal gene and normalized using a plasmid-based standard scale (18). The efficiency calculated from the standard curve was 92% (normal, 80%–120%), and the R2 of the standard curve was 0.99 (normal, >0.98). After successful extraction and amplification, 16S rDNA was measured in triplicate as the number of 16S copies per microliter of blood and fell within the standard curve range.
16S Metagenomic Sequencing
The sequencing was performed using Illumina MiSeq technology after a two-step PCR library preparation, as described previously (17,20). The V3–V4 16S region from both blood and stool microbiota were analyzed using the bioinformatics pipeline established by Vaiomer from the FROGS (Find, Rapidly, OTUs with Galaxy Solution) guidelines (21). The taxonomic assignment was performed against the Silva version 132 database to determine community profiles. A total of 4,651,231 raw read pairs were generated; 3,304,534 were kept after quality filters, and 2,447,442 were clustered in operational taxonomic units (OTUs). The following specific filters were applied for this analysis to obtain the best results: (1) the last 10 bases of reads R1 were removed, (2) the last 40 bases of reads R2 were removed, (3) amplicons with a length of <350 or >500 nucleotides were removed, and (4) OTUs with abundance <0.005% of the whole dataset abundance were removed. To increase the specificity of bacterial taxa truly different between the IgAN and healthy groups, we lowered the sensitivity by eliminating taxa with proportions of <0.005% in more than half subjects in both groups and restricting statistical analysis to the genus level, because approximately >70% of taxa at the species level were either unknown or had multiple affiliations.
The α- and β-diversities were compared between the two groups (22). For α-diversity (measuring the richness and evenness of distribution of taxa), we used the Shannon index within each sample (23). β-Diversity (comparing differences in the microbial community between groups) was measured using the weighted UniFrac technique, which calculates the distance between pairs of samples based on the abundance and phylogenetic relatedness of observed taxa (24). Individual bacterial taxonomic differences between groups were determined by comparing OTUs generated using the linear discriminant analysis effect size (LEfSe) algorithm (25).
Statistical Analyses
Demographic characteristics between IgAN and healthy groups were compared using the t test, Mann–Whitney U test, and chi-squared test, as appropriate. Between-group differences in the levels of total 16S rDNA from blood were compared using the Mann–Whitney U test. We performed adjusted analysis using multivariable regression modeling, adjusting for age, albumin, body mass index, and white blood cell (WBC) count. Differences in α-diversity and individual taxonomic differences between groups were compared between groups using the Mann–Whitney U test. The Spearman test was used for correlation analysis. Patients with IgAN were also stratified by eGFR levels ≤60 ml/min (n=11) and eGFR levels >60 ml/min (n=9). Bacterial 16S rDNA quantity and individual taxa in both eGFR groups were compared with the healthy control groups separately to limit confounding by eGFR. Separate analysis was done for blood and stool samples when comparing the IgAN and healthy groups. For all analysis, we used SAS version 9.4, and two-tailed P values of <0.05 were deemed statistically significant. For microbiome differences conducted using LEfSe, significance was also determined on the basis of effect size of microbiota.
Results
Baseline Characteristics
Baseline characteristics of the 40 subjects are shown in Table 1. Demographic characteristics were similar between groups. The IgAN group had a statistically significant higher WBC count (P=0.04) and lower serum albumin levels (P=0.03) compared with healthy controls. As expected, proteinuria and eGFR was significantly worse in the IgAN group. The median eGFR was similar between the healthy and IgAN group with eGFR >60 ml/min (100 versus 91 ml/min).
Table 1.
Baseline characteristics of the study groups
| Variable | IgAN (n=20) | Healthy (n=20) | P Value |
| Age (yr), median (IQR) | 37 (34–50) | 38 (30–55) | 0.98 |
| Male, n (%) | 9 (45) | 9 (45) | 1.0 |
| White, n (%) | 13 (65) | 13 (65) | 1.0 |
| Body mass index (kg/m2), mean±SD | 29.3±5.8 | 26.2±4.4 | 0.06 |
| White blood cell (per μl), mean±SD | 7.5±2.0 | 6.3±1.5 | 0.04 |
| Serum albumin (g/dl), median (IQR) | 4.2 (3.9–4.4) | 4.4 (4.2–4.7) | 0.03 |
| Urine microalbumin-creatinine ratio (mg/g), median (IQR) | 545.6 (134.8–1168.6) | 2.3 (0.7–5.9) | <0.01 |
| CRP (mg/L), median (IQR) | 2.6 (1.0–6.5) | 1.6 (0.7–3.0) | 0.13 |
| eGFR (ml/min), median (IQR) | 55 (27.5–88) | 100 (81–110) | <0.01 |
| Allergy history, n (%)a | 10 (50) | 6 (30) | 0.19 |
| Perceived stress score, mean±SDb | 15.2±6.2 | 6.7±3.7 | <0.01 |
| 16S DNA (copies/μl blood), median (IQR) | 7410 (6370–8695) | 6030 (4796–7505) | 0.04 |
Data expressed as median (IQR), mean±SD, or n (%) as appropriate. Significant differences between groups detected in white blood cell count, serum albumin, urine microalbumin/creatinine ratio, eGFR, perceived stress score, and 16S DNA copies. eGFR determined using Chronic Kidney Disease Epidemiology Collaboration equation. IgAN, IgA nephropathy; IQR, interquartile range; CRP, C-reactive protein.
Reported or documented allergy to food product or drugs.
Two patients with IgAN and one healthy subject excluded due to language or understanding barrier.
Blood 16S rDNA Quantitative Testing
The median bacterial 16S rDNA concentration in blood was significantly higher in the IgAN group compared with that in the healthy group (7410 versus 6030 16S rDNA copies/μl blood; P=0.04; see Supplemental Figure 2). After stratifying by eGFR, the median blood 16S rDNA concentration remained significantly higher in patients with IgAN who had an eGFR of >60 ml/min when compared with healthy controls (7730 versus 6030 16S rDNA copies/μl blood; P=0.04). This significance was lost when comparing patients with IgAN and an eGFR of ≤60 ml/min with healthy controls (7343 versus 6030 16S rDNA copies/μl blood; p=0.22).
After multivariable adjustment, especially for WBC count, the differences in 16S rDNA between IgAN and healthy groups were no longer significant (P=0.24). There was a strong positive correlation between 16S rDNA copies and WBC count (r=0.7, P<0.001).
16S Metagenomic Sequencing
α-Diversity and β-Diversity
α-Diversity (Figure 2A), by the Shannon index, between the IgAN and healthy groups was not significantly different in blood (2.65 versus 2.68, P=0.79) or stool (3.25 versus 3.31, P=0.82). No significant differences were observed, even after stratification of patients with IgAN by eGFR. The overall β-diversity, measured using the weighted UniFrac technique, was largely similar between IgAN and healthy groups in both blood and stool (indicated by the high degree of overlap in Figure 2B).
Figure 2.
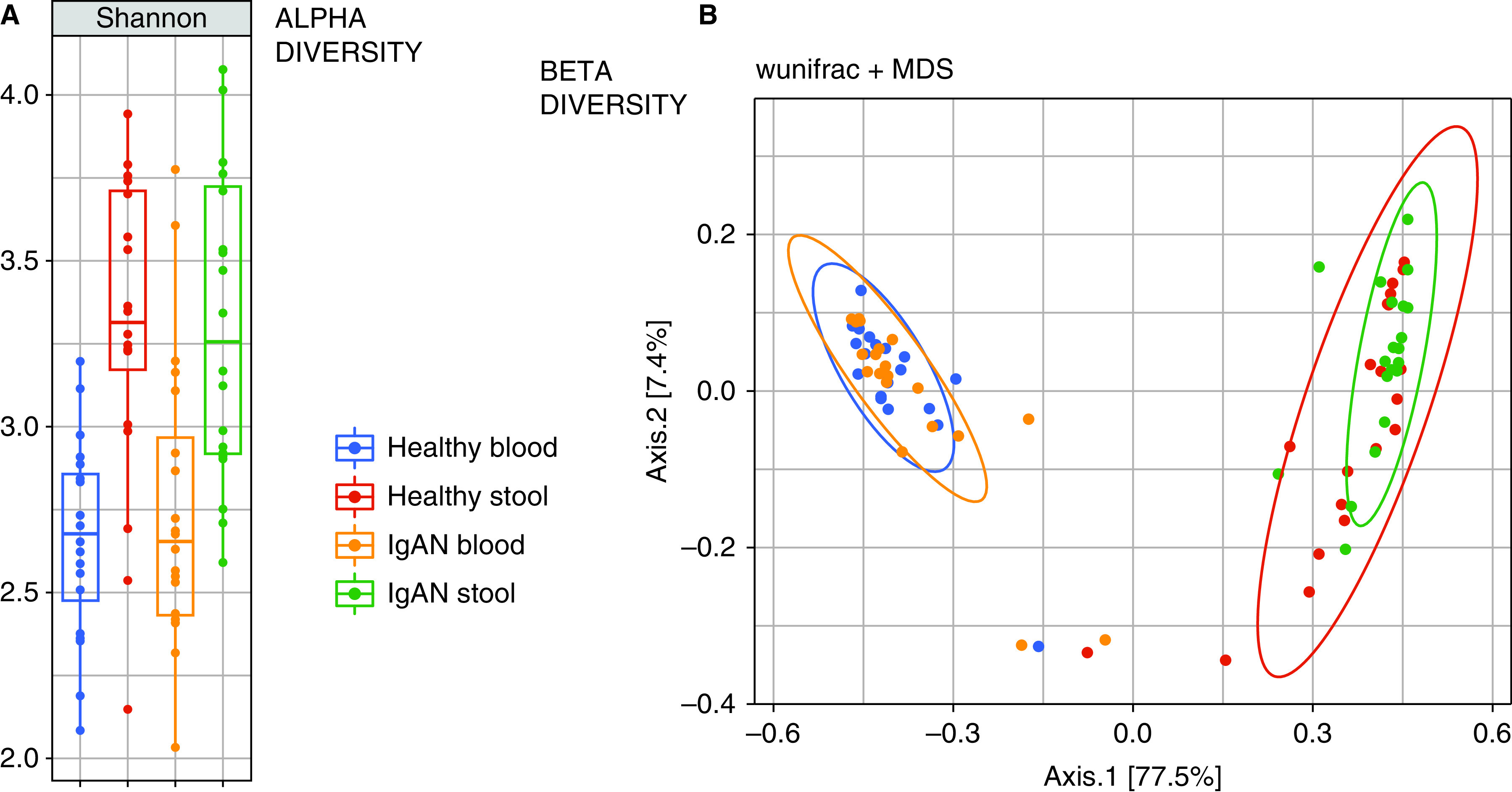
Alpha and beta diversity comparison between IgA Nephropathy and Healthy Control groups. Similar α- (Shannon index) and β-diversities (weighted UniFrac index) between patients with IgA nephropathy and healthy subjects, both in blood and stool samples. Different α- and β-diversities between blood and stool samples in all subjects. IgAN, IgA nephropathy; MDS, multidimensional scaling.
Taxonomic Signature Analysis
Analyses of differences in individual taxa proportions between biologic groups were performed using the LEfSe algorithm, which combines statistical significance with biologic effect size. There were ten OTU differences in blood and 41 OTU differences in the gut between the IgAN and healthy groups (Table 2). After eliminating OTUs with proportions <0.005% and restricting analysis to the genus level to increase specificity, a significantly higher prevalence of the Coriobacteriia class (P=0.01), and the genera Legionella (P=0.05) and Enhydrobacter (P=0.04), were present in the blood of those in the IgAN group (Table 3). After stratifying patients with IgAN by eGFR, such differences were no longer significant, despite having a higher prevalence in both eGFR-stratified IgAN groups when compared with healthy controls. The proportion of the genus Legionella was higher in patients with IgAN and an eGFR of ≤60 ml/min compared with those with an eGFR of >60 ml/min. Additionally, proportions of both Staphylococcus and Streptococcus genera, from the class Bacilli of phylum Firmicutes, were higher in patients with IgAN and an eGFR of ≤60 ml/min in comparison with both the eGFR >60 ml/min group and the healthy control group.
Table 2.
Taxonomic differences between IgAN and healthy groups
| Sample | High in Group | Phylum | Class | Order | Family | Genus | Species |
| Blood | IgAN | Actinobacteria | Actinobacteria | Coriobacteriales | Coriobacteriaceae | Collinsella | 1 |
| Actinobacteria | Coriobacteriia | Eggerthellales | Eggerthellaceae | Unknown | 1 | ||
| Actinobacteria | Actinobacteria | Actinomycetales | Micrococcaceae | Rothia | 1a | ||
| Bacteroidetes | Bacteroidia | Bacteroidales | Tannerellaceae | Parabacteroides | 1 | ||
| Bacteroidetes | Bacteroidia | Chitinophagales | Saprospiraceae | Unknown | 1a | ||
| Proteobacteria | Gammaproteobacteria | Legionellales | Legionellaceae | Legionella | 1 | ||
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Multiaffiliation | 1a | ||
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Coprococcus | 1a | ||
| Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Ruminococcus-1 | 1a | ||
| Healthy | Proteobacteria | Gammaproteobacteria | Betaproteobacteriales | Burkholderiaceae | Delftia | 1 | |
| Stool | IgAN | Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides | 13 |
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Lachnoclostridium | 2 | ||
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Hungatella | 1 | ||
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Unknown | 1 | ||
| Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Ruminiclostridium | 1 | ||
| Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Unknown | 1 | ||
| Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Escherichia-Shigella | 1 | ||
| Healthy | Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides | 2 | |
| Bacteroidetes | Bacteroidia | Bacteroidales | Rikenellaceae | Alistipes | 2 | ||
| Bacteroidetes | Bacteroidia | Bacteroidales | Tannerellaceae | Parabacteroides | 1 | ||
| Bacteroidetes | Bacteroidia | Bacteroidales | Prevotellaceae | Prevotella 9 | 3 | ||
| Bacteroidetes | Bacteroidia | Bacteroidales | Prevotellaceae | Prevotella 2 | 1 | ||
| Bacteroidetes | Bacteroidia | Bacteroidales | Prevotellaceae | Paraprevotella | 1 | ||
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | CAG-56 | 1 | ||
| Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | NK4A214 group | 1 | ||
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | NK4A136 | 1 | ||
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Lachnoclostridium | 1 | ||
| Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | UCG-002 | 1 | ||
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Multiaffiliation/ unknown | 5 | ||
| Firmicutes | Negativicutes | Selenomonadales | Veillonellaceae | Allisonella | 1 |
Taxonomic differences generated using the linear discriminant analysis effect size (LEfSe) algorithm representing statistical and biologic differences between groups. Numbers in the species column indicate number of different species between groups. Exact species difficult to identify due to clustering and multiple affiliations of organisms. IgAN, IgA nephropathy.
Differences lost after excluding three outliers.
Table 3.
Taxonomic proportions of blood microbiota in IgA nephropathy and healthy groups
| Phylum | Class | Order | Family | Genus |
| Actinobacteria (H=20.15, I=17.17) |
Actinobacteria (H=18.33, I=15.81) |
Bifidobacteriales (H=0.005, I=0.005) |
Bifidobacteriaceae (H=0.005, I=0.005) |
Bifidobacterium (H=0.005, I=0.003) |
| Corynebacteriales (H=0.27, I=0.78) |
Corynebacteriaceae (H=0.001, I=0.005) |
|||
| Nocardiaceae (H=0.001, I=0.005) |
||||
| Micrococcales (H=7.94, H=8.05) |
Microbacteriaceae (H=0.303, I=0.004) |
|||
| Micrococcaceae (H=4.65, I=6.82) |
Arthrobacter (H=3.73, I=4.82) |
|||
| Propionibacteriales (H=2.91, I=0.34) |
Propionibacteriaceae (H=2.70, I=0.188) |
Cutibacterium (H=2.70, I=0.19) |
||
| Coriobacteriia (H=0.000, I=0.004)a |
Coriobacteriales (H=0.000, I=0.004)a |
|||
| Proteobacteria (H=64.17, I=62.74) |
Alphaproteobacteria (H=16.88, I=11.83) |
Caulobacterales (H=3.34, I=1.25) |
Caulobacteraceae (H=3.34, I=1.25) |
|
| Rhizobiales (H=2.17, I=1.28) |
Rhizobiaceae (H=0.02, I=0.31) |
Phyllobacterium (H=0.005, I=0.172) |
||
| Sphingomonadales (H=2.51, I=2.01) |
Sphingomonadaceae (H=2.51, I=2.06) |
Sphingomonas (H=1.64, I=2.06) |
||
| Gammaproteobacteria (H=42.85, I=47.14) |
Betaproteobacteriales (H=7.01, I=5.85) |
Burkholderiaceae (H=5.59, I=5.85) |
||
| Enterobacteriales (H=2.23, I=1.72) |
Enterobacteriaceae (H=2.23, I=1.72) |
Escherichia-Shigella (H=0.828, I=0.004) |
||
| Legionellales (H=0.00, I=0.74)a |
Legionellaceae (H=0.00, I=0.74)a |
Legionella (H=0.00, I=0.74)a |
||
| Pseudomonadales (H=33.96, I=37.69) |
Moraxellaceae (H=0.97, I=1.11) |
Enhydrobacter (H=0.000, I=0.003)a |
||
| Pseudomonadaceae (H=28.84, I=35.17) |
Pseudomonas (H=28.84, I=35.17) |
|||
| Bacteroidetes (H=1.61, I=5.09) |
Bacteroidia (H=1.61, I=5.09) |
Bacteroidales (H=0.02, I=0.98) |
Bacteroidaceae (H=0.008, I=0.015) |
Bacteroides (H=0.09, I=0.02) |
| Firmicutes (H=9.03, I=9.29) |
Bacilli (H=1.66, I=2.76) |
Bacillales (H=0.01, I=0.06) |
Staphylococcaceae (H=0.000, I=0.005) |
Staphylococcus (H=0.000, I=0.005) |
| Lactobacillales (H=0.003, I=0.018) |
Lactobacillaceae (H=0.001, I=0.003) |
Streptococcus (H=0.000, I=0.001) | ||
| Clostridia (H=4.94, I=7.25) |
Clostridiales (H=4.94, I=7.25) |
Lachnospiraceae (H=0.59, I=0.01) |
||
| Ruminococcaceae (H=0.01, I=0.78) |
Faecalibacterium (H=0.000, I=0.006) |
|||
| Cyanobacteria (H=0.00, I=0.03) |
Oxyphotobacteria (H=0.00, I=0.03) |
Chloroplast (H=0.00, I=0.03) |
All numbers represent median taxa proportions expressed in percentages. H, healthy; I, IgA nephropathy.
Significantly different between the IgA nephropathy and healthy groups.
We observed significantly higher levels of the genus Bacteroides (P=0.01), family Bacteroidaceae, and genus Escherichia-Shigella (P=0.01), family Enterobacteriaceae, in the stool from those in the IgAN group (Table 4). Although differences were no longer significant after stratifying patients with IgAN by eGFR, they continued to remain at higher proportions in both eGFR IgAN groups when compared with the healthy controls group, despite being more prominent in the low-eGFR IgAN group. The proportion of the genus Bacteroides was much higher in patients with an eGFR of <60 ml/min, with a significantly higher ratio of the phylum Bacteroidetes to Firmicutes when compared with healthy controls. The healthy group had a significantly higher abundance of the genus Prevotella 9 (P=0.02) and the Ruminococcaceae groups NK4A214 (P=0.001) and UCG002 (P=0.04). The major taxonomic comparisons between IgAN and healthy groups up to the genus level in blood and stool are outlined in Tables 3 and 4, respectively.
Table 4.
Taxonomic proportions of stool microbiota in IgA nephropathy and healthy groups
| Phylum | Class | Order | Family | Genus |
| Actinobacteria (H=0.41, I=0.23) |
Actinobacteria (H=0.17, I=0.15) |
Bifidobacteriales (H=0.16, I=0.12) |
Bifidobacteriaceae (H=0.16, I=0.12) |
Bifidobacterium (H=0.16, I=0.12) |
| Coriobacteriia (H=0.10, I=0.06) |
Coriobacteriales (H=0.10, I=0.06) |
Coriobacteriaceae (H=0.04, I=0.01) |
Collinsella (H=0.04, I=0.01) |
|
| Eggerthellaceae (H=0.02, I=0.03) |
Adlercreutzia (H=0.000, I=0.007) |
|||
| Proteobacteria (H=3.35, I=3.34) |
Alphaproteobacteria (H=0.02, I=0.03) |
Caulobacterales (H=0.008, I=0.009) |
Caulobacteraceae (H=0.008, I=0.009) |
|
| Gammaproteobacteria (H=2.78, I=2.33) |
Betaproteobacteriales (H=1.09, I=1.75) |
Burkholderiaceae (H=1.09, I=1.75) |
Sutterella and Parasutterella | |
| Enterobacteriales (H=0.02, I=0.07) |
Enterobacteriaceae (H=2.23, I=1.72) |
Escherichia-Shigella (H=0.006, I=0.069)a |
||
| Pasteurellales (H=0.008, I=0.004) |
Pasteurellaceae (H=0.008, I=0.004) |
Haemophilus (H=0.008, I=0.004) |
||
| Pseudomonadales (H=0.01, I=0.01) |
Pseudomonadaceae (H=0.008, I=0.006) |
Pseudomonas (H=0.008, I=0.006) |
||
| Deltaproteobacteria (H=0.10, I=0.21) |
Desulfovibrionales (H=0.10, I=0.21) |
Desulfovibrionaceae (H=0.10, I=0.21) |
Bilophila (H=0.02, I=0.12) |
|
| Bacteroidetes (H=69.37, I=73.95) |
Bacteroidia (H=69.37, I=73.95) |
Bacteroidales (H=69.37, I=73.95) |
Bacteroidaceae (H=33.36, I=59.32)a |
Bacteroides (H=33.36, I=59.32)a |
| Barnesiellaceae (H=0.62, I=0.01) |
Barnesiella (H=0.62, I=0.005) |
|||
| Marinifilaceae (H=0.42, I=0.48) |
Butyricimonas and Odoribacter | |||
| Prevotellaceae (H=1.34, I=0.006) |
Prevotella 9 (H=0.173, I=0.003)a |
|||
| Rikenellaceae (H=4.58, I=2.94) |
Alistipes (H=4.00, I=2.94) |
|||
| Tannerellaceae (H=2.17, I=3.10) |
Parabacteroides (H=2.17, I=3.10) |
|||
| Firmicutes (H=26.18, I=21.05) |
Negativicutes (H=0.04, I=0.06) |
Selemonadales (H=0.04, I=0.06) |
Veillonellaceae (H=0.009, I=0.005) |
Dialister (H=0.000, I=0.005) |
| Bacilli (H=0.04, I=0.02) |
Lactobacillales (H=0.04, I=0.02) |
Staphylococcaceae (H=0.000, I=0.005) |
Staphylococcus (H=0.000, I=0.005) |
|
| Streptococcaceae (H=0.02, I=0.01) |
Streptococcus (H=0.02, I=0.01) |
|||
| Clostridia (H=25.98, I=20.68) |
Clostridiales (H=25.98, I=20.68) |
Lachnospiraceae (H=11.43, I=9.74) |
Eubacterium eligens group (H=0.24, I=0.06)a |
|
| Others: Agathobacter, Anaerostipes, Blautia, CAG-56, Coprococcus, Dorea, Fusicatenibacter, Lachnoclostridium, Lachnospira, Roseburia, other Eubacterium groups | ||||
| Christensenellaceae (H=0.16, I=0.06) |
Christensenellaceae R7 group (H=0.16, I=0.06) |
|||
| Peptostreptococcaceae (H=0.03, I=0.03) |
Romboutsia (H=0.005, I=0.002) |
|||
| Ruminococcaceae (H=12.99, I=10.84) |
NK4A214 group (H=0.05, I=0.00)a |
|||
|
UCG002 (H=1.74, I=0.58)a | ||||
| Others: Butyricicoccus, DTU089, Faecalibacterium, Flavonifractor, Oscillibacter, Oscillospira, Ruminiclostridium, other UCG groups, Ruminococcus, Subdoligranulum, UBA1819 |
All numbers represent median taxa proportions expressed in percentages. H, healthy; I, IgA nephropathy.
Significantly different between the IgA nephropathy and healthy groups.
When comparing taxonomic differences between blood and stool samples, both IgAN and healthy groups exhibited large bacterial microbiome differences at all taxonomic levels, with several blood microbiome taxa that were absent in stool (Figure 3).
Figure 3.

Taxonomic differences between blood and stool of IgA Nephropathy and Healthy Control groups. Linear discriminant analysis effect size (LEfSe) cladogram showing large microbiome differences at various taxonomic levels between stool (S) and whole blood (WB) samples in (A) healthy and (B) IgA nephropathy groups.
Correlation Analyses
Correlation of Microbiome with Clinical Parameters of IgAN
Blood and stool microbiota correlations were performed with clinical characteristics of IgAN, such as urine albumin-creatinine ratio, blood levels of eGFR, C-reactive protein, IgA level, albumin, WBC count, 16S rDNA, and histologic MEST-C (Mesangial cellularity, Endocapillary cellularity, Segmental sclerosis, Tubular atrophy, Crescent) score (26). These correlations are detailed in Table 5. Due to wide interindividual variations in eGFRs >60 ml/min in the IgAN group, we restricted our analysis to patients with CKD and an GFR of ≤60 ml/min, which included 11 patients with IgAN. eGFR levels did not correlate with 16S rDNA levels (r=−0.14, P=0.7). Notably, blood IgA levels were available only for 12 patients with IgAN, and the MEST-C score report was available only for 16 patients with IgAN, because four patients were previously biopsied at other institutions or locations from which the record could not be obtained. Given the variable timeline of biopsies in relation to study enrollment, interpretations are limited.
Table 5.
Blood and stool microbiota correlations with clinical parameters of IgA nephropathy
| Clinical Variable | Taxonomic Level | Blood | Stool | ||
| Positive Correlation | Negative Correlation | Positive Correlation | Negative Correlation | ||
| Urine albumin-creatinine ratio | Phylum | Bacteroidetes | Bacteroidetes | ||
| Class | Clostridia | ||||
| Order | Legionellales | Bacteroidales, Enterobacteriales | Clostridiales | ||
| Family | Legionellaceae, Staphylococcaceae | Bacteroidaceae, Enterobacteriaceae | Christensenellaceae, Lachnospiraceae | ||
| Genus | Legionella | Bacteroides | CAG-56 (Lachnospiraceae) | ||
| Staphylococcus | Escherichia-Shigella | Christensenellaceae R7 group, Cutibacterium, Prevotella 9, Ruminococcaceae NK4A214 group, Ruminococcaceae UCG005, Eubacterium eligens group | |||
| GFR ≤60 ml/min (n=11) | Phylum | Cyanobacteriaa | Proteobacteriaa | ||
| Class | Coriobacteriia,a Oxyphotobacteriaa | Bacillia | |||
| Order | Bifidobacteriales,a Chloroplast,a Coriobacterialesa | Negativicutes,a Selenomonadalesa | |||
| Family | Bifidobacteriaceaea | Veillonellaceaea | |||
| Genus | Arhtrobacter a | DTU089 (family Ruminococcaceae),a Dialistera | |||
| C-reactive protein (mg/L) | Phylum | Actinobacteria | |||
| Class | Coriobacteriia | Actinobacteria | |||
| Order | Coriobacteriales | Caulobacterales | Bifidobacteriales | ||
| Family | Caulobacteriaceae | Bacteroidaceae, Bifidobacteriaceae | Marinifilaceae | ||
| Genus | Bacteroides, Bifidobacterium | CAG-56 (Lachnospiraceae) | |||
| MEST-C score (n=16)b | Class | Gammaproteobacteria | |||
| Family | Rikenellaceaea | ||||
| Genus | Alistepes,a Anaerostipesa | ||||
| Serum IgA level (mg/dl) (n=12)b | Order | Bacteroidalesa | |||
| Family | Bacteroidaceaea | Bacteroidaceaea | |||
| Genus | Bacteroides,a Butyricimonasa | ||||
| Serum albumin (g/dl) | Genus | Ruminiclostridium 6, Ruminococcus 2 | |||
| White blood cell count in blood | Family | Rhizobiaceae | |||
| Genus | CAG-56 (Lachnospiraceae), Dorea, Ruminococcaceae NK4A214 group, Eubacterium eligens group | ||||
| 16S rDNA (copies/μl blood) | Order | Micrococcales | Bifidobacteriales | ||
| Family | Bifidobacteriacaeae | ||||
MEST-C, Mesangial cellularity, Endocapillary cellularity, Segmental sclerosis, Tubular atrophy, Crescent; rDNA, ribosomal DNA.
Strong correlation with correlation coefficient ≥0.6, others without a footnote symbol (a) have a correlation coefficient <0.6.
Missing values and historical measures that may have changed with time (interpret with caution).
Correlation between Blood and Stool Microbiota
Proportions of major taxa were significantly different between blood and stool samples in both IgAN and healthy groups (Figure 4A). There was no correlation found between blood and stool samples in the major phyla Proteobacteria (r=0.21, P=0.19), Actinobacteria (r=−0.24, P=0.13), Bacteroidetes (r=−0.25, P=0.11), and Firmicutes (r=−0.07, P=0.65). Even at the genus level, the top 15 taxa observed in blood were different from those observed in stool in both IgAN and healthy groups (Figure 4B).
Figure 4.
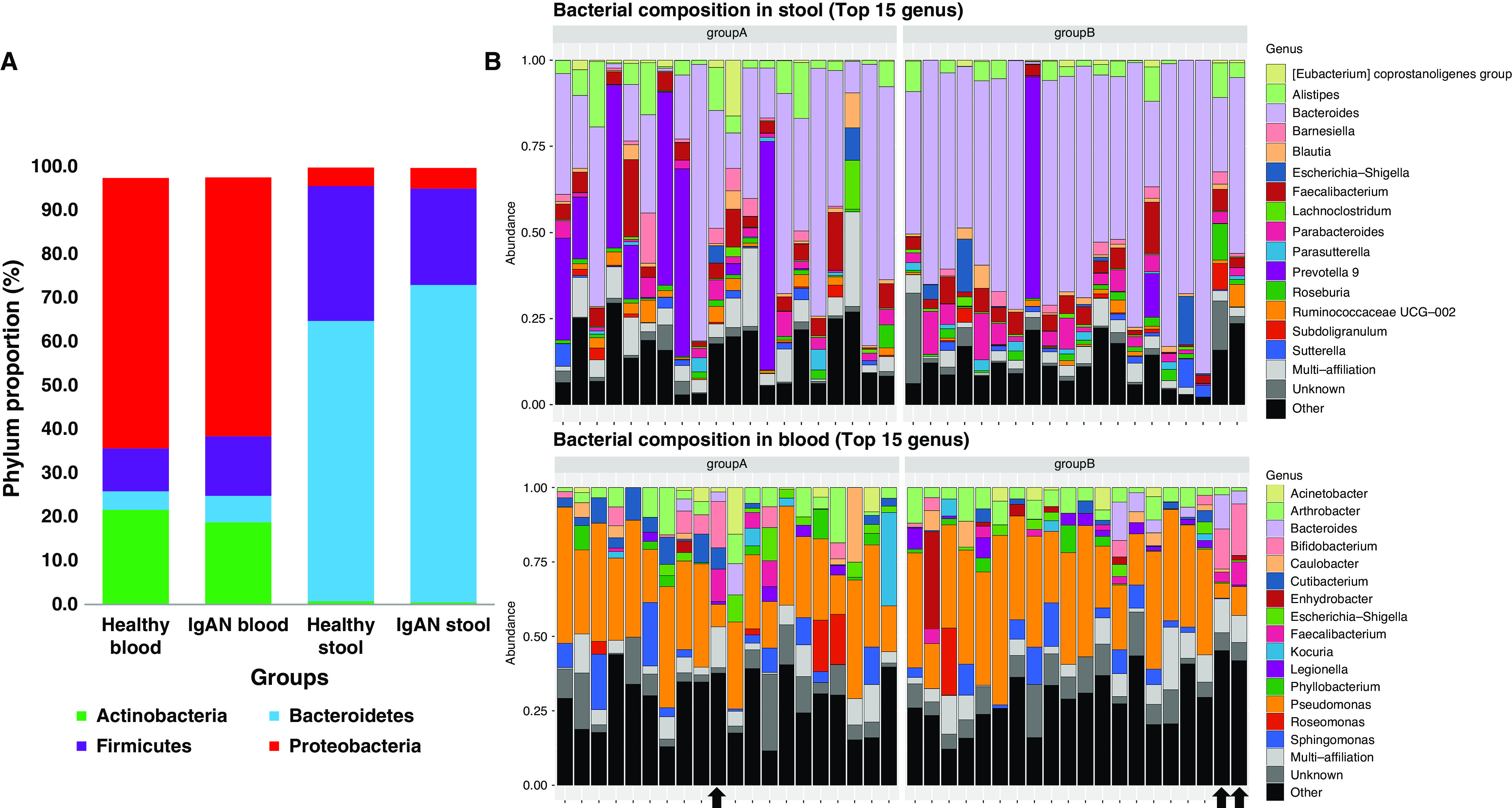
Comparison of blood and stool major phyla proportions and top 15 genera between Healthy and IgA Nephropathy groups. (A) Major phyla distribution in blood and stool samples of IgAN and healthy groups showing similar distribution within the same body site, but different distributions between blood and stool body sites. (B) Bacterial composition at the genus level, showing the top 15 genera markedly different between blood and stool of healthy (group A) and IgAN (group B) groups. Three outliers highlighted by arrows.
Outlying Patients
Three subjects (one healthy control and two patients with IgAN) did not pass the technical quality control of sequencing and had abnormal taxonomic profiles with a high proportion of Lachnospiraceae and Ruminococcaceae families from the Clostridia class and Firmicutes phylum, when compared with remaining 37 subjects (Figure 5). We conducted our statistical analysis both including and excluding these three outliers in the blood samples; the α- and α-diversities, and correlation analyses, remained largely unchanged. Only five out of ten OTUs in blood remained significantly different between the two groups (Table 2). Significance was lost after stratification by eGFR.
Figure 5.
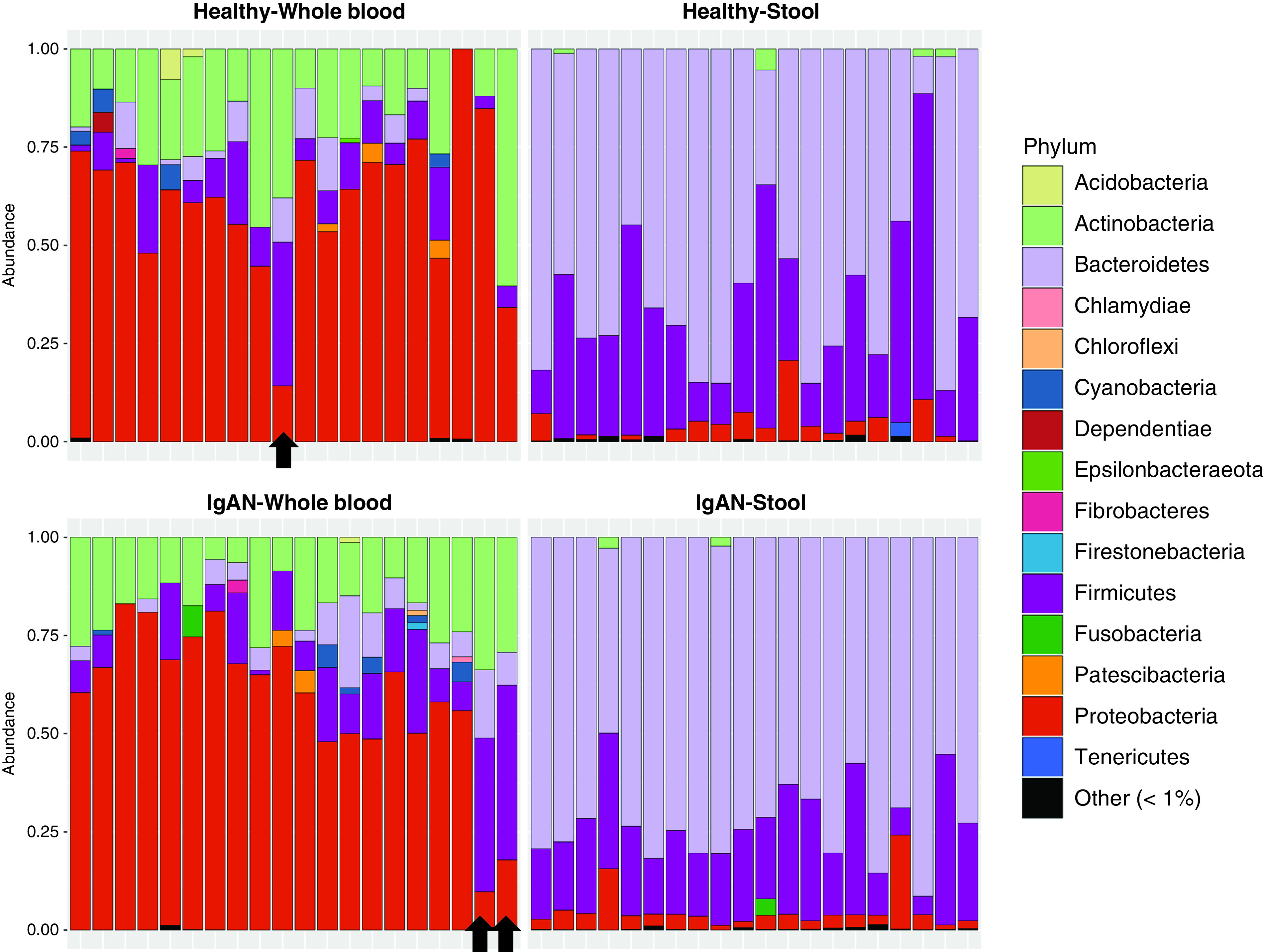
Blood and stool phyla composition of IgA Nephropathy and Healthy control subjects. Phylum distribution in blood and stool of IgAN and healthy groups, with three outliers highlighted by arrows.
Outlying Proportions of Blood Microbiota
Several patients with IgAN had certain blood microbiota in disproportionately high abundance that were not statistically significant between groups due to being absent in most subjects. We excluded the three outliers for this observation. Most subjects had Streptococcus and Staphylococcus genera proportion undetectable to <1%, but four patients with IgAN had Streptococcus proportions of 2%–6%, and one patient with IgAN had a Staphylococcus proportion of 10%. Several other minor outlying genera in different patients with IgAN included Parabacteroides, Phyllobacterium, Parasutterella, Ruminococcus UCG005 and UCG013, Romboutsia, and Sphingomonas.
Discussion
This study simultaneously measured the blood and gut microbiomes in patients with IgAN, and compared them to healthy controls to identify potential bacterial microbiota that may be implicated in the pathogenesis of IgAN. We found a higher quantity of blood 16S rDNA in the IgAN group. No significant difference in the α-diversity was observed between the IgAN and healthy groups in either blood or stool. The β-diversity was largely similar between the IgAN and healthy groups, except for ten OTU differences in blood and 42 OTU differences in the gut. Interestingly, we detected a striking difference between the blood and gut microbiota across all subjects, without any direct correlation in corresponding phyla between the two samples, confirming that the blood microbiome does not directly reflect the gut microbiome, as observed previously (27,28).
A higher quantity of 16S rDNA was detected in blood of the IgAN group when compared with the healthy controls group, even after adjustment for eGFR. Because this blood microbiome does not seem to reflect the gut microbiome, other body sites—including the oropharynx and respiratory tract—may also be possible sources, as is suggested by the observation of certain taxa in blood that were not observed in stool. A strong correlation between 16S rDNA quantity and WBC count could suggest a leukocytic response to the invading microbiome. Alternatively, the increased 16S rDNA quantity could be an outcome of a higher mean WBC count in the IgAN group, because the majority of the blood microbiome has been observed from within the buffy coat (18). Given the cross-sectional nature of this study, it is difficult to establish a temporal cause-effect relationship.
α-Diversity was similar between the IgAN and healthy groups in both blood and stool (Figure 2A). Previous studies have shown a direct correlation of α-diversity with stronger immunity, whereas lower diversity has been associated with diseases, including advanced CKD (17,29). α-Diversity studies in early CKD stages are limited and may be minimally affected, as in our study findings. In both IgAN and healthy groups, a significantly higher α-diversity is observed in stool compared with blood, suggesting a larger variety or complexity of bacteria in the gut compared with blood. Overall, α-diversity does not seem to suggest a pathogenic role in IgAN.
β-Diversity measures the overall microbial community within a sample. A large overlap between the IgAN and healthy groups in both blood and stool suggests that the majority of bacterial taxa (except a few differences) were similar between the two groups (Figure 2B). Lack of an exact overlap indicates there were subtle bacterial differences between the two groups, which may correlate with a diseased state. Again, striking differences in β-diversity between blood and stool in both groups indicate that the bacterial communities residing in the gut and blood are remarkably different, as observed previously (27,28). Consistent with the findings of the Human Microbiome Project (22), β-diversity (bacterial community) tends to be more similar between individuals within the same body site than between different body sites within an individual, suggesting the microbial communities tend to adapt to specific body sites.
Among several other taxonomic differences, a higher prevalence of the class Coriobacteriia and Bacilli in blood; genera Legionella, Enhydrobacter, Staphylococcus, and Streptococcus in blood; and genera Bacteroides and Escherichia-Shigella in stool were observed in the IgAN group compared with healthy controls. These genera have been implicated previously as bacterial antigens in IgAN (30–32). The genera Prevotella, Ruminococcus NK4214 group, Barnesiella, Bifidobacterium, and Coprococcus in stool have been observed to have a higher prevalence among healthy individuals when compared with those with IgAN in a Chinese population (13,14). In accordance with these studies, we observe a similar trend, although not all of them achieved statistical significance.
Other minor genera with high proportions in the blood of certain patients with IgAN, but not in healthy subjects, included Parabacteroides, Parasutterella, Phyllobacterium, Romboutsia, Sphingomonas, and a few genera from the Ruminococcaceae family. However, they were neither statistically significant nor uniform across all patients with IgAN. Interindividual variations were observed among patients with IgAN: each patient had two to five distinct genera, with collective proportions as high as 5%–15%, which differed from other subjects (represented as “others” in Figure 4B). Given the heterogeneity of IgAN with a multihit pathogenesis and different microbes previously implicated, these microbiota may not be statistically significant across all patients with IgAN, but may still hold clinical significance within an individual. He et al. (33) observed that certain human genetic variants may be associated with certain microbiota. This can explain some of the observed heterogeneity. The combination of the above genera in blood and stool, which have consistently shown differences between patients with IgAN and healthy subjects, can be used as biomarkers in a predictive model for diagnosis and prognosis of IgAN. Correlation of microbiota with clinical parameters of IgAN can further strengthen this model and serve as potential personalized intervention targets.
Striking differences between stool and blood microbiota with lack of correlation between corresponding taxa could suggest other potential sources of origin of certain blood microbiota. Piccolo et al. (34) compared the salivary microbiota between patients with IgAN and healthy controls, and observed Firmicutes as the dominant phyla, with proportions of 30%–40% in both groups, and a lower Firmicutes/Proteobacteria ratio in patients with IgAN. Microbiome measurement from tonsillar crypts of patients with IgAN who underwent tonsillectomy showed no significant differences from those without IgAN (35). Further, the seven predominant genera observed were markedly different from the blood microbiome genera observed in our study (Figure 4B). Subsequently, Park et al. (36) conducted another study comparing the microbiome from tonsillar swabs from healthy controls with patients with IgAN, those with diabetic nephropathy, and those with membranous nephropathy, and observed several microbiome differences between each of the groups; in particular, higher abundances of Rahnella, Ruminococcus_g2, and Clostridium_g21 genera were found when compared with healthy controls. Overall, the microbiome composition from the saliva and tonsils of patients with IgAN is different than what we observe in blood, suggesting the blood microbiome may not reflect the tonsillar microbiome.
This study was not designed to examine the mechanisms accounting for the differences between the blood and stool microbiome, but learning why such striking differences exist will improve our understanding of the physiologic mechanisms involved in the regulation of the human blood microbiome, and explain the differences between the blood and stool microbiota observed in this study. Such differences were observed across all subjects, and were not specific to those with IgAN. Whittle et al. (37) compared their patients’ blood microbiome data with stool, oral cavity, and skin microbiome data from the Human Microbiome Project, and showed that the blood microbiome more closely resembled that of the skin and oral cavity, rather than the gut. Despite this similarity in microbiota, the proportions of the phyla Firmicutes (30%–40% in skin and oropharynx compared with % in blood) and Proteobacteria (30% in skin and oropharynx compared with 70%–80% in blood) were remarkably different (38,39). These studies suggest that skin and oral microbiota entering into the blood may undergo further regulation to maintain a characteristic composition of the blood microbiome dominated by the phylum Proteobacteria followed by the phyla Actinobacteria and Firmicutes, as was also observed in other blood microbiome studies (18,37,40,41). Although several prior studies have observed gut translocation of bacteria into blood during a dysbiotic state, the proportions of gut and blood microbiota have not been compared (8,42,43). Studies comparing the blood and stool microbiome have found striking differences in the proportions of major phyla, similarly to our study findings (26,27). These studies suggest that microbiota from different body sites likely undergo further regulation after entering the blood, which could explain the differences observed between the blood and stool microbiome without eliminating the possibility of gut bacterial translocation into blood.
The blood microbiome could also be representing previously reported dormant and cell wall–deficient, nonculturable, L-form bacteria that have been reported to possess the ability of undergoing pleomorphic adaptation to its milieu (44–46). A characteristic composition of the blood microbiome phyla differing from those reported at other body sites suggests that microbiota in blood may be undergoing pleomorphic adaptation, similarly to L-form bacteria. The intestinal wall, immune system, and liver have been hypothesized to play key roles in the filtering of microbes and regulating the composition of the blood microbiome (47). Further studies are needed to explore these regulating mechanisms.
Our study has several limitations. Being a cross-sectional study with a small sample size, causality and generalizability of microbiome differences is limited. Our subjects with IgAN are heterogenous, with varying renal function and varying biopsy specimen findings and time points. Three outlying blood microbiome samples were excluded, as discussed earlier. We did not measure galactosylated IgA levels. Factors influencing the microbiome (including diet) have not been measured. Finally, our results are based on 16S metagenomics sequencing of the bacterial DNA and this limits the interpretation of viability and potential functional or causal roles of these bacteria in IgAN. Nevertheless, we observe important microbiome differences between groups, and between gut and blood samples, and confirm that the blood microbiome does not directly reflect the gut.
In conclusion, our study demonstrates higher quantities of bacterial DNA in the blood of patients with IgAN, with several blood and gut microbiota differences between IgAN and healthy groups and important correlations with clinical parameters of IgAN that could have potential diagnostic, prognostic, and therapeutic implications in the future. The striking differences between the gut and blood microbiota suggest that gut microbiota relevant in IgAN may not be mediating their effects via translocation into the blood. The characteristic phylum composition of the blood microbiome compared with previously reported microbiome compositions at other body sites suggests internal regulation of the invading microbiome into blood, which could explain the differences observed between the blood and stool microbiome, without eliminating the possibility of gut bacterial translocation. Further large-scale, longitudinal research studies are needed to understand factors influencing these microbiome changes and determine their functional or causal roles in IgAN.
Acknowledgment
Personal thanks for statistical advice to Dr. Lisa Yanek, member of the Biostatistics, Epidemiology and Data Management (BEAD) core consulting service at Johns Hopkins University.
Disclosures
A. S. Allegretti reports having consultancy agreements with Cymabay Therapeutics and Mallinckrodt Pharmaceuticals, and receiving research funding from Mallinckrodt Pharmaceuticals. A. Fasano reports receiving personal fees from AbbVie, Innovate Biopharmaceuticals, Mead Johnson Nutrition, Takeda, and uBiome; and receiving other funding from Alba Therapeutics; outside the submitted work. S. U. Nigwekar reports having consultancy agreements with Allena Pharma, Becker Professional Education, Epizon Pharma, and Laboratoris Sanifit; receiving research funding from Allena Pharma and Hope Pharma; receiving honoraria from Guidepoint and Sanofi-Aventis; and serving as a scientific advisor for, or member of, Vifor Pharma. N. Tolkoff-Rubin reports having consultancy agreements with, and receiving honoraria from, Best Doctors. All remaining authors have nothing to disclose.
Funding
The study was internally supported by Massachusetts General Hospital Nephrology Division funds.
Author Contributions
A. S. Allegretti was responsible for validation; A. S. Allegretti, A. Fasano, and N. Tolkoff-Rubin provided supervision; A. S. Allegretti, S. Kalim, B. Lelouvier, S. U. Nigwekar, N. Tolkoff-Rubin, and N. B. Shah reviewed and edited the manuscript; A. S. Allegretti, S. Kalim, S. U. Nigwekar, N. Tolkoff-Rubin, and N. B. Shah conceptualized the study; M. Dalal, B. Lelouvier, and F. Servant were responsible for software; M. Dalal and F. Servant were responsible for formal analysis; S. Kalim, S. Krinsky, and S. U. Nigwekar were responsible for resources; S. Krinsky and N. B. Shah were responsible for project administration; B. Lelouvier was responsible for data curation; B. Lelouvier, S. Krinsky, F. Servant, and N. B. Shah were responsible for methodology; N. B. Shah wrote the original draft; N. Tolkoff-Rubin was responsible for funding acquisition; A. S. Allegretti approved the final version of the manuscript; and all authors approved the final version of the manuscript, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Supplemental Material
This article contains the following supplemental material online at http://kidney360.asnjournals.org/lookup/suppl/doi:10.34067/KID.0000132021/-/DCSupplemental.
(A) Comparisons of beta diversities by ordination analysis of the 16S rRNA gene sequencing data using Bray-Curtis, (B) UniFrac with alpha parameters = 0.6; and (C) Jensen-Shannon divergence dissimilarity distances in the blood samples and negative controls (NC-PCR and NC-Ext). Download Supplementary Figure 1, PDF file, 267 KB (266.7KB, pdf)
Box and whisker plot demonstrating significant differences in median 16S rDNA quantity between IgA nephropathy and healthy groups. Download Supplementary Figure 2, PDF file, 267 KB (266.7KB, pdf)
References
- 1.Wyatt RJ, Julian BA: IgA nephropathy. N Engl J Med 368: 2402–2414, 2013. 10.1056/NEJMra1206793 [DOI] [PubMed] [Google Scholar]
- 2.Suzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, Wyatt RJ, Scolari F, Mestecky J, Gharavi AG, Julian BA: The pathophysiology of IgA nephropathy. J Am Soc Nephrol 22: 1795–1803, 2011. 10.1681/ASN.2011050464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, Fasel D, Lata S, Prakash S, Shapiro S, Fischman C, Snyder HJ, Appel G, Izzi C, Viola BF, Dallera N, Del Vecchio L, Barlassina C, Salvi E, Bertinetto FE, Amoroso A, Savoldi S, Rocchietti M, Amore A, Peruzzi L, Coppo R, Salvadori M, Ravani P, Magistroni R, Ghiggeri GM, Caridi G, Bodria M, Lugani F, Allegri L, Delsante M, Maiorana M, Magnano A, Frasca G, Boer E, Boscutti G, Ponticelli C, Mignani R, Marcantoni C, Di Landro D, Santoro D, Pani A, Polci R, Feriozzi S, Chicca S, Galliani M, Gigante M, Gesualdo L, Zamboli P, Battaglia GG, Garozzo M, Maixnerová D, Tesar V, Eitner F, Rauen T, Floege J, Kovacs T, Nagy J, Mucha K, Pączek L, Zaniew M, Mizerska-Wasiak M, Roszkowska-Blaim M, Pawlaczyk K, Gale D, Barratt J, Thibaudin L, Berthoux F, Canaud G, Boland A, Metzger M, Panzer U, Suzuki H, Goto S, Narita I, Caliskan Y, Xie J, Hou P, Chen N, Zhang H, Wyatt RJ, Novak J, Julian BA, Feehally J, Stengel B, Cusi D, Lifton RP, Gharavi AG: Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet 46: 1187–1196, 2014. 10.1038/ng.3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E: Dysbiosis and the immune system. Nat Rev Immunol 17: 219–232, 2017. 10.1038/nri.2017.7 [DOI] [PubMed] [Google Scholar]
- 5.Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, David E, Savidor A, Korem T, Herzig Y, Pevsner-Fischer M, Shapiro H, Christ A, Harmelin A, Halpern Z, Latz E, Flavell RA, Amit I, Segal E, Elinav E: Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163: 1428–1443, 2015. 10.1016/j.cell.2015.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang B, Yao M, Lv L, Ling Z, Li L: The human microbiota in health and disease. Engineering 3: 71–82, 2017. 10.1016/J.ENG.2017.01.008 [DOI] [Google Scholar]
- 7.Vaziri ND, Wong J, Pahl M, Piceno YM, Yuan J, DeSantis TZ, Ni Z, Nguyen TH, Andersen GL: Chronic kidney disease alters intestinal microbial flora. Kidney Int 83: 308–315, 2013. 10.1038/ki.2012.345 [DOI] [PubMed] [Google Scholar]
- 8.Vaziri ND, Zhao YY, Pahl MV: Altered intestinal microbial flora and impaired epithelial barrier structure and function in CKD: The nature, mechanisms, consequences and potential treatment. Nephrol Dial Transplant 31: 737–746, 2016. 10.1093/ndt/gfv095 [DOI] [PubMed] [Google Scholar]
- 9.Anders HJ, Andersen K, Stecher B: The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int 83: 1010–1016, 2013. 10.1038/ki.2012.440 [DOI] [PubMed] [Google Scholar]
- 10.Coppo R: The gut-renal connection in IgA nephropathy. Semin Nephrol 38: 504–512, 2018. 10.1016/j.semnephrol.2018.05.020 [DOI] [PubMed] [Google Scholar]
- 11.Salerno-Goncalves R, Safavie F, Fasano A, Sztein MB: Free and complexed-secretory immunoglobulin A triggers distinct intestinal epithelial cell responses. Clin Exp Immunol 185: 338–347, 2016. 10.1111/cei.12801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Angelis M, Montemurno E, Piccolo M, Vannini L, Lauriero G, Maranzano V, Gozzi G, Serrazanetti D, Dalfino G, Gobbetti M, Gesualdo L: Microbiota and metabolome associated with immunoglobulin A nephropathy (IgAN). PLoS One 9: e99006, 2014. 10.1371/journal.pone.0099006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong Z, Tan J, Tan L, Tang Y, Qiu Z, Pei G, Qin W: Modifications of gut microbiota are associated with the severity of IgA nephropathy in the Chinese population. Int Immunopharmacol 89[Pt B]: 107085, 2020. 10.1016/j.intimp.2020.107085 [DOI] [PubMed] [Google Scholar]
- 14.Hu X, Du J, Xie Y, Huang Q, Xiao Y, Chen J, Yan S, Gong Z, Ouyang S: Fecal microbiota characteristics of Chinese patients with primary IgA nephropathy: A cross-sectional study. BMC Nephrol 21: 97, 2020. 10.1186/s12882-020-01741-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J; CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration): A new equation to estimate glomerular filtration rate. Ann Intern Med 150: 604–612, 2009. 10.7326/0003-4819-150-9-200905050-00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang X, Bushman FD, FitzGerald GA: Rhythmicity of the intestinal microbiota is regulated by gender and the host circadian clock. Proc Natl Acad Sci U S A 112: 10479–10484, 2015. 10.1073/pnas.1501305112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shah NB, Allegretti AS, Nigwekar SU, Kalim S, Zhao S, Lelouvier B, Servant F, Serena G, Thadhani RI, Raj DS, Fasano A: Blood microbiome profile in CKD: A pilot study. Clin J Am Soc Nephrol 14: 692–701, 2019. 10.2215/CJN.12161018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Païssé S, Valle C, Servant F, Courtney M, Burcelin R, Amar J, Lelouvier B: Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 56: 1138–1147, 2016. 10.1111/trf.13477 [DOI] [PubMed] [Google Scholar]
- 19.Schierwagen R, Alvarez-Silva C, Servant F, Trebicka J, Lelouvier B, Arumugam M: Trust is good, control is better: Technical considerations in blood microbiome analysis. Gut 69: 1362–1363, 2020. 10.1136/gutjnl-2019-319123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lluch J, Servant F, Païssé S, Valle C, Valière S, Kuchly C, Vilchez G, Donnadieu C, Courtney M, Burcelin R, Amar J, Bouchez O, Lelouvier B: The characterization of novel tissue microbiota using an optimized 16S metagenomic sequencing pipeline. PLoS One 10: e0142334, 2015. 10.1371/journal.pone.0142334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Escudié F, Auer L, Bernard M, Mariadassou M, Cauquil L, Vidal K, Maman S, Hernandez-Raquet G, Combes S, Pascal G: FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics 34: 1287–1294, 2018. 10.1093/bioinformatics/btx791 [DOI] [PubMed] [Google Scholar]
- 22.Human Microbiome Project Consortium : Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214, 2012. 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJ: Counting the uncountable: Statistical approaches to estimating microbial diversity [published correction appears in Appl Environ Microbiol 68: 448, 2002]. Appl Environ Microbiol 67: 4399–4406, 2001. 10.1128/AEM.67.10.4399-4406.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lozupone CA, Hamady M, Kelley ST, Knight R: Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73: 1576–1585, 2007. 10.1128/AEM.01996-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C: Metagenomic biomarker discovery and explanation. Genome Biol 12: R60, 2011. 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markowitz G: Glomerular disease: Updated Oxford classification of IgA nephropathy: A new MEST-C score. Nat Rev Nephrol 13: 385–386, 2017. 10.1038/nrneph.2017.67 [DOI] [PubMed] [Google Scholar]
- 27.Lelouvier B, Servant F, Païssé S, Brunet AC, Benyahya S, Serino M, Valle C, Ortiz MR, Puig J, Courtney M, Federici M, Fernández-Real JM, Burcelin R, Amar J: Changes in blood microbiota profiles associated with liver fibrosis in obese patients: A pilot analysis. Hepatology 64: 2015–2027, 2016. 10.1002/hep.28829 [DOI] [PubMed] [Google Scholar]
- 28.Serena G, Davies C, Cetinbas M, Sadreyev RI, Fasano A: Analysis of blood and fecal microbiome profile in patients with celiac disease. Hum Microb J 11: 100049, 2019. 10.1016/j.humic.2018.12.001 [DOI] [Google Scholar]
- 29.Mosca A, Leclerc M, Hugot JP: Gut microbiota diversity and human diseases: Should we reintroduce key predators in our ecosystem? Front Microbiol 7: 455, 2016. 10.3389/fmicb.2016.00455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirabayashi A, Yorioka N, Oda H, Sekiguchi Y, Kuramoto A, Okushin S, Yamakido M: Involvement of bacterial antigens in immunoglobulin A nephropathy. Hiroshima J Med Sci 45: 113–117, 1996. http://www.ncbi.nlm.nih.gov/pubmed/9119709. Accessed March 5, 2020 [PubMed] [Google Scholar]
- 31.Rollino C, Vischini G, Coppo R: IgA nephropathy and infections. J Nephrol 29: 463–468, 2016. 10.1007/s40620-016-0265-x [DOI] [PubMed] [Google Scholar]
- 32.Fergusson RJ, Mine LT: Legionnaires’ disease and IgA nephropathy. Scott Med J 31: 114–116, 1986. 10.1177/003693308603100214 [DOI] [PubMed] [Google Scholar]
- 33.He J-W, Zhou X-J, Li Y-F, Wang YN, Liu LJ, Shi SF, Xin XH, Li RS, Falchi M, Lv JC, Zhang H: Associations of genetic variants contributing to gut microbiota composition in immunoglobin A nephropathy. mSystems 6: e00819-20, 2021. 10.1128/mSystems.00819-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piccolo M, De Angelis M, Lauriero G, Montemurno E, Di Cagno R, Gesualdo L, Gobbetti M: Salivary microbiota associated with immunoglobulin A nephropathy. Microb Ecol 70: 557–565, 2015. 10.1007/s00248-015-0592-9 [DOI] [PubMed] [Google Scholar]
- 35.Watanabe H, Goto S, Mori H, Higashi K, Hosomichi K, Aizawa N, Takahashi N, Tsuchida M, Suzuki Y, Yamada T, Horii A, Inoue I, Kurokawa K, Narita I: Comprehensive microbiome analysis of tonsillar crypts in IgA nephropathy. Nephrol Dial Transplant 32: 2072–2079, 2017. 10.1093/ndt/gfw343 [DOI] [PubMed] [Google Scholar]
- 36.Park JI, Kim TY, Oh B, Cho H, Kim JE, Yoo SH, Lee JP, Kim YS, Chun J, Kim BS, Lee H: Comparative analysis of the tonsillar microbiota in IgA nephropathy and other glomerular diseases. Sci Rep 10: 16206, 2020. 10.1038/s41598-020-73035-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whittle E, Leonard MO, Harrison R, Gant TW, Tonge DP: Multi-method characterization of the human circulating microbiome. Front Microbiol 9: 3266, 2019. 10.3389/fmicb.2018.03266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grice EA, Segre JA: The skin microbiome [published correction appears in Nat Rev Microbiol 9: 626, 2011]. Nat Rev Microbiol 9: 244–253, 2011. 10.1038/nrmicro2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, Lakshmanan A, Wade WG: The human oral microbiome. J Bacteriol 192: 5002–5017, 2010. 10.1128/JB.00542-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schierwagen R, Alvarez-Silva C, Madsen MSA, Kolbe CC, Meyer C, Thomas D, Uschner FE, Magdaleno F, Jansen C, Pohlmann A, Praktiknjo M, Hischebeth GT, Molitor E, Latz E, Lelouvier B, Trebicka J, Arumugam M: Circulating microbiome in blood of different circulatory compartments. Gut 68: 578–580, 2018 [DOI] [PubMed] [Google Scholar]
- 41.Olde Loohuis LM, Mangul S, Ori APS, Jospin G, Koslicki D, Yang HT, Wu T, Boks MP, Lomen-Hoerth C, Wiedau-Pazos M, Cantor RM, de Vos WM, Kahn RS, Eskin E, Ophoff RA: Transcriptome analysis in whole blood reveals increased microbial diversity in schizophrenia. Transl Psychiatry 8: 96, 2018. 10.1038/s41398-018-0107-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sato J, Kanazawa A, Ikeda F, Yoshihara T, Goto H, Abe H, Komiya K, Kawaguchi M, Shimizu T, Ogihara T, Tamura Y, Sakurai Y, Yamamoto R, Mita T, Fujitani Y, Fukuda H, Nomoto K, Takahashi T, Asahara T, Hirose T, Nagata S, Yamashiro Y, Watada H: Gut dysbiosis and detection of “live gut bacteria” in blood of Japanese patients with type 2 diabetes. Diabetes Care 37: 2343–2350, 2014. 10.2337/dc13-2817 [DOI] [PubMed] [Google Scholar]
- 43.Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, Di Sabatino A, Caprioli F, Bottiglieri L, Oldani A, Viale G, Penna G, Dejana E, Rescigno M: A gut-vascular barrier controls the systemic dissemination of bacteria. Science 350: 830–834, 2015. 10.1126/science.aad0135 [DOI] [PubMed] [Google Scholar]
- 44.Potgieter M, Bester J, Kell DB, Pretorius E: The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol Rev 39: 567–591, 2015. 10.1093/femsre/fuv013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Markova ND: L-form bacteria cohabitants in human blood: Significance for health and diseases. Discov Med 23: 305–313, 2017 [PubMed] [Google Scholar]
- 46.Errington J, Mickiewicz K, Kawai Y, Wu LJ: L-form bacteria, chronic diseases and the origins of life. Philos Trans R Soc Lond B Biol Sci 371: 20150494, 2016. 10.1098/rstb.2015.0494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castillo DJ, Rifkin RF, Cowan DA, Potgieter M: The healthy human blood microbiome: Fact or fiction? Front Cell Infect Microbiol 9: 148, 2019. 10.3389/fcimb.2019.00148 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Comparisons of beta diversities by ordination analysis of the 16S rRNA gene sequencing data using Bray-Curtis, (B) UniFrac with alpha parameters = 0.6; and (C) Jensen-Shannon divergence dissimilarity distances in the blood samples and negative controls (NC-PCR and NC-Ext). Download Supplementary Figure 1, PDF file, 267 KB (266.7KB, pdf)
Box and whisker plot demonstrating significant differences in median 16S rDNA quantity between IgA nephropathy and healthy groups. Download Supplementary Figure 2, PDF file, 267 KB (266.7KB, pdf)