

Abstract
The present study aimed to develop a surface‐modified biocompatible nanostructured lipid carrier (NLCs) system using polyoxyethylene (40) stearate (POE‐40‐S) to improve the oral bioavailability of poorly water‐soluble Biopharmaceutics Classification System class‐II drug like tamoxifen (TMX). Also aimed to screen the most influential factors affecting the particle size (PS) using Taguchi (L12 (211)) orthogonal array design (TgL12 OA). Then, to optimize the TMX loaded POE‐40‐S (P) surface‐modified NLCs (TMX‐loaded‐PEG‐40‐S coated NLC (PNLCs) or PNLCs) by central composite design (CCD) using a four‐factor, five‐level model. The most influential factors affecting the PS was screened and optimized. The in‐vitro study showed that increased drug‐loading (DL) and encapsulation efficiency (EE), decreased PS and charge, sustained drug release for the prolonged period of the time with good stability and suppressed protein adsorption. The Ex‐vivo study showed that decreased mucous binding with five‐fold enhanced permeability of PNLC formulation after surface modification with POE‐40‐S. The in‐vitro cytotoxicity study showed that the blank carrier is biocompatible and cytotoxicity of the formulation was dependent on the concentration of the drug. Finally, it can be concluded that the surface‐modified PNLCs formulation was an effective, biocompatible, stable formulation in the enhancement of dissolution rate, solubility, stability with reduced mucus adhesion and increased permeability thereby which indicates its enhanced oral bioavailability.
Inspec keywords: nanoparticles, cellular biophysics, solubility, drug delivery systems, toxicology, adsorption, adhesion, dissolving, biomedical materials, encapsulation, polymers, proteins, nanomedicine, permeability, particle size, electrokinetic effects
Other keywords: water‐soluble BCS class‐II, TgL12 OA, TMX‐loaded POE‐40‐S surface‐modified NLC, surface‐modified PNLC formulation, lipid‐based NLC system, oral bioavailability, stable formulation, biocompatible formulation, blank carrier, in vitro cytotoxicity, surface modification, PNLC formulation, drug release, central composite design, orthogonal array design, encapsulation efficiency, steric stabilisation effect, particle size, dissolution rate, polyoxyethylene stearate, surface‐modified biocompatible carrier system, systemic toxicity, water‐soluble drug, tamoxifen‐loaded surface‐modified nanostructured lipid carrier
1 Introduction
Cancer nanotherapeutics was beginning to overwhelm the global research and viewed to be the revolutionary treatment regime in the medical field. One of the most promising candidates in this delivery system is the nanostructured lipid carriers (NLCs) [1, 2, 3, 4]. It can improve the biodistribution of existing anticancer drugs and prolong drug accumulation in both tumour tissue and in the blood stream [5]. However, a number of formulation and processing variables influence the overall performance of nanoparticles (NPs). Thus, it becomes challenging to study the effect of each variable and interaction among them through the conventional approach [6]. Therefore, to minimise the number of trials and to study the effect of factors in all possible combinations, Taguchi orthogonal array design was applied to formulate an optimised formulation [7]. However, it ignores interaction and concentrates on main effect estimation. Central composite design (CCD) is one of the techniques of response surface methodology (RSM) for optimisation of pharmaceutical dosage forms [8, 9] and is a handy tool in understanding the interactions among the parameters that have been optimised. Also, this method helps to optimise the effective parameters with a minimum number of experiments [10].
The present study aimed to develop a surface‐modified biocompatible carrier system using polyoxyethylene (40) stearate (POE‐40‐S) to improve the oral bioavailability of TMX, through enhancement of solubility, dissolution rate with the reduction of particle size (PS)<200 nm and stability of the formulation with steric stabilisation effect for the poorly water‐soluble biopharmaceutics classification system (BCS) class‐II drug such as tamoxifen (TMX). Since, TMX lipophilicity (high log p ‐value – 6.26) and vulnerability to enzymatic degradation restrict its oral bioavailability [11]. Besides, it was reported that the major limiting factors for lipid NP in drug absorption is the destruction of the structure by digestive enzymes and non‐specific adhesion by secreted mucins, which potentially cause the drug recrystallisation and inactive trans‐epithelial transport [12, 13, 14]. For this reason, the surface‐modified NLCs were selected to improve the oral bioavailability of TMX, which is expected to bypass the hepatic metabolism through the lymphatic absorption pathway and its surface modification with hydrophilic moiety provides long circulation of drug with steric protection and reduces macrophage uptake in vivo. Also aimed to screen the most influential factors affecting the PS along with polydispersity index (PDI), drug‐loading (DL) and encapsulation efficiency (EE) using Taguchi [L12 (211)] orthogonal array design (TgL12 OA) and to optimise the TMX‐loaded POE‐40‐S (P) surface‐modified NLCs (TMX‐loaded‐PNLCs) by CCD using a four‐factor, five‐level model.
2 Materials and methods
2.1 Materials
TMX citrate (TMX), glyceryl monostearate (GMS) and Cremophor relative humidity (RH) 40 (CRH40) were kindly provided as a gift sample from East West Pharma, Raipur, Uttarakhand. Glyceryl trimyristate, olive oil, corn oil, sunflower oil, soybean oil, Tween 80 (T80), stearic acid and polyvinyl alcohol (PVA) were procured from Sisco Research Laboratories Pvt. Ltd., Mumbai, India. Poloxamer188 (P188) and polyoxyethylene (40) stearate were purchased from Sigma (USA). Double‐distilled water was prepared in our laboratory, and all other chemicals were of analytical reagent grade.
2.2 Determination of solubility of TMX by shake flask method
An excess amount of TMX was added into various solid lipid and liquid lipid and dissolved by vortex mixing for 30 s. Then, the mixtures were shaken for 48 h at 5°C above the melting point of the lipids in a thermostatically controlled shaking water bath, followed by equilibrium for 24 h. After equilibration, the mixtures were separated by centrifugation at 3000 rpm for 10 min, and the supernatant was filtered, suitably diluted with methanol and analysed for TMX content by High‐performance liquid chromatography (HPLC) method at 278 nm [11]. The experiment was repeated in triplicates.
The HPLC system (Agilent series 1100; Data Apex, Milan, Italy) consists of a 25 cm long, 4 mm inner diameter stainless C18 ACE column and a mobile phase of methanol, water and orthophosphoric acid (60:40:0.05 v/v/v). The flow rate and ultraviolet wavelength were set at 1 ml/min and 278 nm, respectively.
2.3 Experimental design
In this study, optimisation of PS, PDI, DL and EE of TMX‐loaded‐PNLCs were performed in two stages. In the first stage, the parameters that showed significant effects on PS and loading efficiency have been identified screened by TgL12 orthogonal array design. At the second stage, the optimum values of these parameters were determined by CCD properly using a four‐factor, five‐level design [15].
2.4 Screening study
In a preliminary laboratory study by a TgL12 OA design on drug: lipid ratio (TMX: GMS – 1:10 or 1:15), surfactant type (T80 or CRH40) and co‐surfactant (P188) ratio (2:1 or 3:0), surfactant: co‐surfactant type (T80: P188 or CRH40: P188), speed of homogeniser (800 or 1300 rpm), volume of water (50 or 100 ml), lecithin: lipid ratio (30:70 or 20:80), solid lipid: liquid lipid ratio (1:1 or 1:2), lipid: POE‐40‐S ratio (0 or 1:1), PVA% (5 or 10%) and stirring time (30 or 60 min), the most important variables/factors on the PS were determined along with PDI, DL and EE (Design‐Expert, version 10.0). Table 1 shows the factors and their levels.
Table 1.
Studied variables and levels
| Studied variables with code | Levels | |
|---|---|---|
| I | II | |
| A – drug:lipid ratio | 1:10 | 1:15 |
| B – surfactant type | T80 | CRH40 |
| C – surfactant:co‐surfactant ratio | 3:0 | 2:1 |
| D – surfactant:co‐surfactant type | T80:P188 | CRH40:P188 |
| E – speed of homogeniser, rpm | 800 | 1300 |
| F – lecithin:lipid ratio | 20:80 | 30:70 |
| G – stirring time, min | 30 | 60 |
| H – solid lipid:lipid lipid | 0 | 1:1 |
| I – volume of water | 50 | 100 |
| J – lipid:POE‐40‐S ratio | 0 | 1:1 |
| K – PVA, %w/v | 5% | 10% |
2.5 Optimisation study
After selecting the most critical factors influencing PNLCs PS, the CCD was constructed using four factors [amount of drug (A), volume of liquid lipid (B), amount of PEG40 (C) and amount of stabilising agent (D)] with 30 experiments to optimise the formulation. The responses studied were PSs (Y 1), PDI (Y 2), DL (Y 3) and EE (Y 4). A statistical software ‘Design Expert’ was used to generate the combinations of these factors at different levels.
This model is described by following equation:
| (1) |
where Y is the response, β 0 is the intercept and β 1, β 2, β 3 and β 4 are the regression coefficient, X 1, X 2, X 3 and X 4 are coded levels of independent variables, X 1, X 2 and X 3 are interaction and quadratic terms, respectively [15].
2.6 Preparation of PNLCs by micro emulsion cooling technique
TMX‐loaded‐PNLCs were prepared by micro emulsion cooling technique (MECT) [16, 17, 18, 19]. The lipid phase was produced by heating a mixture of a specified amount of GMS, olive oil and soya lecithin at 75°C to form a melted oil fraction. To this mixture, 10 mg of TMX was added to make a clear homogeneous solution. The aqueous phase consisting of a specified amount of surfactant as well as PVA as stabiliser were heated to the same temperature (75°C) and was added to the lipid melt under constant magnetic stirring at a specified rpm (800 or 1300) for a specified time (30 or 60 min) so as to form a homogenous milky slurry or an o/w microemulsion in the form of a TMX‐loaded‐PNLCs dispersion. This o/w microemulsion was further cooled at room temperature to form PNLCs precipitate from it. The NLC, as well as blank NLC (BNLC) and blank PNLCs (BPNLC), were also prepared without POE‐40‐S and drug, respectively, using the same composition for further analysis. The composition of prepared PNLCs formulations was shown in Table 2 for screening and Table 3 for optimisation.
Table 2.
TMX‐loaded‐PNLCs TgL12 OA composition and response
| Drug:lipid ratio | Surfactant type | Surfactant:co‐surfactant ratio | Speed of homogeniser | Lecithin:GMS ratio | Stirring time, min | Solid lipid: liquid lipid | The volume of an aqueous phase | Lipid:PEG40‐S ratio | Poly vinyl alcohol | PS | PDI | DL | EE |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rpm | S | Ml | % | Nm | % | % | |||||||
| {1} | {−1} | {1} | {1} | {−1} | {1} | {1} | {−1} | {1} | {−1} | 3880 | 1 | 5.84 | 74.6 |
| {1} | {−1} | {−1} | {1} | {1} | {−1} | {1} | {1} | {−1} | {−1} | 2088 | 0.95 | 6.3 | 70.8 |
| {−1} | {−1} | {−1} | {−1} | {−1} | {−1} | {−1} | {−1} | {−1} | {−1} | 2518 | 1 | 8.48 | 74.2 |
| {1} | {1} | {−1} | {−1} | {−1} | {1} | {−1} | {1} | {1} | {−1} | 2169 | 1 | 5.93 | 75.7 |
| {−1} | {1} | {1} | {1} | {1} | {1} | {−1} | {1} | {−1} | {−1} | 2017 | 0.83 | 5.59 | 74.3 |
| {1} | {1} | {−1} | {1} | {1} | {−1} | {−1} | {−1} | {1} | {1} | 455.3 | 0.56 | 6.37 | 71.5 |
| {−1} | {−1} | {−1} | {−1} | {1} | {1} | {1} | {1} | {1} | {1} | 661.5 | 0.58 | 9.68 | 75.2 |
| {1} | {1} | {1} | {−1} | {−1} | {−1} | {1} | {1} | {−1} | {1} | 1553 | 0.59 | 5.87 | 75 |
| {1} | {−1} | {1} | {−1} | {1} | {1} | {−1} | {−1} | {−1} | {1} | 3243 | 1 | 6.68 | 75.2 |
| {−1} | {1} | {1} | {−1} | {1} | {−1} | {1} | {−1} | {1} | {−1} | 1752 | 0.67 | 9.64 | 74.8 |
| {−1} | {−1} | {1} | {1} | {−1} | {−1} | {−1} | {1} | {1} | {1} | 1233 | 0.8 | 8.47 | 74.1 |
| {−1} | {1} | {−1} | {1} | {−1} | {1} | {1} | {−1} | {−1} | {1} | 172 | 0.33 | 8.38 | 73.3 |
Table 3.
TMX‐loaded‐PNLCs CCD composition and response
| Factor 1 | Factor 2 | Factor 3 | Factor 4 | Response 1 | Response 2 | Response 3 | Response 4 | |
|---|---|---|---|---|---|---|---|---|
| Run | A:amount of drug, mg | B:volume of liquid lipid, ml | C:amount of PEG40‐S, mg | D:amount of stabilising agent, mg | DL | EE | PS | PDI |
| 1 | 0.000 | 0.000 | 0.000 | 0.000 | 18.91 | 81.5 | 181 | 0.417 |
| 2 | 0.000 | 0.000 | 0.000 | 2.000 | 17.15 | 82.85 | 179 | 0.5 |
| 3 | −1.000 | 1.000 | −1.000 | −1.000 | 07.14 | 61.6 | 165 | 0.296 |
| 4 | 0.000 | −2.000 | 0.000 | 0.000 | 16.63 | 79.8 | 170 | 0.605 |
| 5 | 0.000 | 0.000 | 0.000 | −2.000 | 16.33 | 78.15 | 188.1 | 0.671 |
| 6 | 0.000 | 0.000 | −2.000 | 0.000 | 17.25 | 83.45 | 168.34 | 0.473 |
| 7 | 1.000 | 1.000 | 1.000 | 1.000 | 13.21 | 88.8 | 173.9 | 0.393 |
| 8 | 0.000 | 0.000 | 0.000 | 0.000 | 18.33 | 83.95 | 181 | 0.417 |
| 9 | 1.000 | −1.000 | 1.000 | −1.000 | 27.04 | 89.13 | 273 | 0.735 |
| 10 | 0.000 | 0.000 | 0.000 | 0.000 | 17.89 | 82.05 | 181 | 0.417 |
| 11 | −2.000 | 0.000 | 0.000 | 0.000 | 00.00 | 0.00 | 176.34 | 0.958 |
| 12 | 1.000 | 1.000 | −1.000 | 1.000 | 31.89 | 85.1 | 259.04 | 0.388 |
| 13 | 0.000 | 0.000 | 0.000 | 0.000 | 19.4 | 81.5 | 181 | 0.417 |
| 14 | −1.000 | −1.000 | 1.000 | 1.000 | 6.91 | 55.3 | 228.8 | 0.901 |
| 15 | −1.000 | −1.000 | 1.000 | −1.000 | 6.91 | 55.3 | 174 | 0.723 |
| 16 | 1.000 | 1.000 | 1.000 | −1.000 | 33.08 | 88.3 | 202.8 | 0.672 |
| 17 | −1.000 | −1.000 | −1.000 | 1.000 | 6.91 | 55.3 | 170.9 | 0.815 |
| 18 | 0.000 | 0.000 | 2.000 | 0.000 | 19.4 | 77.65 | 312.3 | 0.699 |
| 19 | 1.000 | −1.000 | −1.000 | −1.000 | 30.9 | 85.1 | 600 | 0.856 |
| 20 | 0.000 | 0.000 | 0.000 | 0.000 | 19.4 | 77.65 | 181 | 0.417 |
| 21 | 2.000 | 0.000 | 0.000 | 0.000 | 46.77 | 91.6 | 421 | 0.805 |
| 22 | −1.000 | 1.000 | −1.000 | 1.000 | 05.8 | 55.3 | 185 | 0.426 |
| 23 | 1.000 | 1.000 | −1.000 | −1.000 | 31.89 | 85.1 | 323.2 | 0.396 |
| 24 | −1.000 | 1.000 | 1.000 | −1.000 | 07.4 | 55.3 | 172.2 | 0.886 |
| 25 | 0.000 | 0.000 | 0.000 | 0.000 | 19.4 | 77.65 | 181 | 0.417 |
| 26 | 0.000 | 2.000 | 0.000 | 0.000 | 20.17 | 80.8 | 179.8 | 0.461 |
| 27 | −1.000 | −1.000 | −1.000 | −1.000 | 6.91 | 55.3 | 216 | 0.427 |
| 28 | 1.000 | −1.000 | −1.000 | 1.000 | 32.44 | 86.53 | 524 | 0.678 |
| 29 | −1.000 | 1.000 | 1.000 | 1.000 | 07.85 | 62.9 | 378 | 0.404 |
| 30 | 1.000 | −1.000 | 1.000 | 1.000 | 31.9 | 85.1 | 253 | 0.441 |
2.7 Characterisation of PNLCs
The mean PS (Z‐ave), PDI and zeta potential (ζ) of the prepared TMX‐loaded‐PNLCs were obtained by the photon correlation spectroscopy technique (PCS) using Zetasizer Nano Series equipment (Malvern Instruments, Worcestershire, UK) [20]. Fourier transform infrared spectroscopy (FTIR) (4000–400 cm−1) was performed using a Perkin‐Elmer system 2000 spectrophotometer to understand if there is any interaction between the drug and excipients. Differential scanning calorimetry (DSC) was carried out to study the physical state of the formulation using Perkin‐Elmer Instruments, Norwalk, USA. The operating conditions were sample weight, 5 mg; heating rate of 10 °C/min [21]. Powder X‐ray diffractometer (PXRD) (Multiflex, M/s. Rigaku, Tokyo, Japan) studies were also performed to confirm the crystal form on the samples by exposing them to nickel‐filtered copper (Cu) Ka radiation (40 kV, 30 mA) and scanned from 2° to 70°, 2θ at a step size of 0.045° and step time of 0.5 s [10]. Besides, scanning electron microscopy (SEM) (JSM 6390, JEOL DATUM Ltd., Tokyo, Japan) [22] and an energy‐filtering transmission electron microscope (TEM) (Libra120, Carl Zeiss, Göttingen, Germany) [23] were used to observe the morphology of the formulation.
2.8 Evaluation of EE and DL
To determine the EE and DL in the lipid NP, 10 mg freeze‐dried TMX‐loaded‐PNLCs was dissolved in methanol, and this nanodispersion was centrifuged at 15,000 rpm for 45 min [21]. The supernatant and the filtrate were diluted suitably and analysed for TMX content by HPLC method. Then, the EE% and DL% were calculated by using the following equation:
.
2.9 Storage stability studies
Freeze‐dried TMX‐loaded‐PNLCs were evaluated for stability as per the international council for harmonisation of technical requirements for pharmaceuticals for human use (ICH) guidelines Q1A (R2) (2003). TMX‐loaded‐PNLCs (equivalent to 10 mg) were transferred in amber coloured glass vials, sealed and was stored upright in stability chamber. Physical and chemical stability of TMX‐loaded‐PNLCs was evaluated for 6 months by storing them at 30 ± 2°C/65 ± 5% RH and 40 ± 2°C/75 ± 5% RH. Samples were withdrawn at specified time intervals (0, 1, 3 and 6 months of storage) and assessed for changes in PS, PDI and DC in addition to physical appearance and ease of reconstitution [21].
2.10 In vitro release study
In vitro release of pure TMX drug and TMX from TMX‐loaded‐PNLCs were performed in 0.1 N HCl pH 1.2 and phosphate buffer saline (PBS) pH 7.4 by the dialysis‐bag method (12,000 Da) [24]. The bags were soaked in double‐distilled water for 24 h before use. The TMX‐loaded‐PNLCs and NLCs were placed in a dialysis bag, with the two ends fixed by clamps and transferred into 60 ml of buffer medium containing conical flasks. The experiment was done in the thermostatic shaker at 37°C at a rate of 100 times/min. At different time intervals, 1 ml of the medium was removed by filtration and replaced with fresh medium to maintain sink conditions. The filtrate was analysed by HPLC method at 278 nm. Obtained TMX‐loaded‐PNLCs data were fitted into zero‐order, first‐order, Higuchi and Korsmeyer–Peppas [25] mathematical models for evaluation of drug release kinetics [26].
2.11 In vitro cytotoxicity study
The cytotoxicity of TMX solution, TMX‐loaded‐PNLCs, NLCs, BNLCs and BPNLCs against Michigan Cancer Foundation‐7 (Breast cancer cells) (MCF‐7) cells were measured using the [3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide] (MTT) assay. Briefly, the cells were seeded into 96‐well plates at a cell density of 5 × 104 cells/ml (20 µl/well) and incubated for adherence at 37°C in 5% carbon dioxide, 20 µl of drug solution or a formulation was added into the wells. The concentrations of TMX solution and BPNLCs, TMX‐loaded‐PNLCs (equivalent to the same amount of TMX) were 1, 10, 50 and 100 ng/ml in different multiwell plates. After 24 h of incubation, 20 µl MTT solution (5 mg/ml) was added to each well, and the plates were incubated for a further 2 h. The solution in each well‐containing media, unbound MTT and dead cells were removed by suction, and 100 µl of dimethyl sulfoxide (DMSO) was added to each well. The plates were then shaken, and the optical density was read using a microplate reader at a test wavelength of 570 nm. Cells incubated in culture medium alone served as a control for cell viability (untreated wells).
2.12 Protein adsorption
Bovine serum albumin (BSA) was used to evaluate the protein adsorption of the TMX‐loaded‐PNLCs, NLCs, BNLCs and BPNLCs formulations. The lipid formulations were suspended in PBS (pH 7.4) at a concentration of 10 mg/ml and incubated with BSA solution (0.8 mg/ml) at 37ο C under constant agitation. After 2 h, the mixture was centrifuged at 13,000 rpm for 30 min. The amount of BSA in the supernatant was measured by the Lowry method. Results were expressed as the amount of adsorbed protein (mg)/mg of the NPs.
2.13 Ex vivo evaluation of PNLC
To detect the interaction between lipid NP and mucins as well as intestinal permeation of optimised PNLC formulation, ex vivo intestinal fluids and sac were prepared using Sprague–Dawley female rats. All studies were in accordance with the Guidelines of Institutional Animal Ethics Committee (IAEC) of SRM College of Pharmacy, SRMIST, India with registration no. 66/02/C/CPCSEA. Prior to experimentation, six rats (250 ± 10 g) were fasted for 24 h but allowed free access to water. The rats were orally given 2 ml water by gavage before collection of intestinal fluids so as to obtain more physiological liquids. After 30 min, the rats were sacrificed by an excess dose of anaesthesia [thiopental sodium (50 mg/kg) administrated intraperitoneally]. The intestine from the duodenum to ileum of each rat was carefully isolated from the mesentery following ligation of both ends. The external wall and two ends of the isolated intestines were rinsed with physiological saline.
2.14 Ex vivo mucin binding study
In this method, intestinal fluids were squeezed out from one end by hand and centrifuged at 10,000 rpm for 15 min to collect the mucin containing clear liquids. The PS of ex vivo intestinal fluids was measured with a Zetasizer Nano ZS after appropriate dilution. To investigate the capture of lipid NPs by mucins, TMX‐loaded‐NLC (TNLCs) (100 μl) were added into 100 μl intestinal fluids and incubated at 37°C. At different time intervals (0.25, 0.5, 1.0 and 2.0 h), the PS of the co‐incubation system was determined in real time [12].
2.15 Ex vivo intestinal permeation study
The intestinal permeation of optimised PNLC and NLC formulation was compared with the raw TMX dispersion using a non‐everted sac technique [27]. After isolation of intestine, the jejunum was quickly cut into segments, each about 5–7 cm in length. The segments were washed immediately with oxygenated saline solution (0.9%, weight/volume, sodium chloride) using a syringe equipped with a blunt end. Sac of each segment was tied on one side with a silk suture and filled with raw TMX or formulations (equivalent to 10 mg of the drug). Then, the sacs were tied on the other side and placed in a glass conical flask containing 10 ml of Ringer's solution. The entire system was maintained at 37°C in a shaking water bath operated at 100 rpm and aerated using a laboratory aerator. Samples were withdrawn from outside the sac, and the medium was totally replaced by fresh medium every 15 min for 2 h. Samples were analysed by HPLC.
3 Results and discussion
3.1 Selection of matrix lipids
A selection of suitable lipids and other excipients used as matrix lipid is significant to develop a biocompatible surface‐modified NLCs. Since they will directly influence the performance of the carrier system such as toxicity and biocompatibility, DL and EE, drug expulsion during storage etc. [28, 29, 30]. The solubility of TMX in various solid lipids (GMS, stearic acid and glyceryl trimyristate) and liquid lipid (olive oil, corn oil, sunflower oil and soybean oil) was studied and shown in Fig. 1. From this study, TMX showed excellent solubility in GMS as solid lipid and olive oil as liquid lipid was selected for further formulation development studies. The increased solubility of TMX in GMS could be due to its inherent self‐emulsifying property. Moreover, the hydroxyl group of glycerides in GMS and CH2 –O–phenyl group of TMX could form a molecular complex, which tends to further enhance TMX solubility [21]. After selection of NLCs matrix materials, it is important to identify the product and process variables [31]. Hence, based on the literature review, the fishbone diagram was made, and 11 factors were selected for Taguchi design.
Fig. 1.
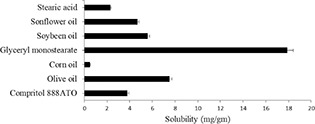
Solubility of TMX in various solid lipid and liquid lipid
3.2 Screening of influential factors and process variables
On the basis of factors (studied variables) and corresponding levels, a TgL12 OA design was generated. The generated designs were prepared, and its results were tabulated (Table 2).
Results from Table 2 are then used to construct the % contribution plot to identify the ranking of significant factors affecting the objective response (Figs. 2 a –d). In the case of small in PS and PDI, the ranking of significant factors was K > C>B > A>E > H>J > G>F > I and K > B>H > A>C > E>I > G>F > J, respectively, whereas for high DL and EE, it was A > E>J > H>B > K>I > C>G > F and E > G>C > F>A > J>I > H>B > K, respectively. Analysis of variance (ANOVA) of the experimental data for TgL12 OA design confirms that the model was significant (Model Prob > F<0.05) (Table 4 ). The goodness of fit of the model was expressed by the coefficient of determination R 2, which indicating that 94.62, 96.12, 81.30 and 98.32% of the variability in the response could be explained by the model. Besides, the value of R indicating a good agreement existed between the experimental and predicted values of PS, PDI, DL and EE, respectively.
Fig. 2.
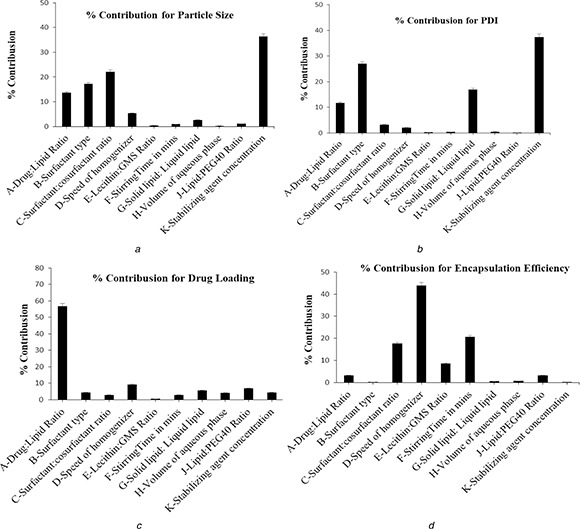
% Contribution plot for
(a) PS, (b) PDI, (c) DL, (d) EE
Table 4.
TgL12 OA design ANOVA results
| Source model | Sum of squares | df | Mean square | F value | p ‐Value Prob>F | R 2 value | R value | |
|---|---|---|---|---|---|---|---|---|
| PS | 2165.29 | 5 | 433.06 | 21.10 | 0.0010 | 0.9462 | 0.9013 | significant |
| PDI | 0.54 | 5 | 0.11 | 29.70 | 0.0004 | 0.9612 | 0.9288 | significant |
| DL | 21.37 | 5 | 4.27 | 5.22 | 0.0342 | 0.8130 | 0.6573 | significant |
| EE | 24.43 | 6 | 4.07 | 48.77 | 0.0003 | 0.9832 | 0.9630 | significant |
3.3 TgL12 OA design optimisation result
The screened factors meant that decreasing drug:lipid ratio decreases the PS due to decreased dispersion viscosity [32]. Also, small in PS was obtained using CRH40 at 1300 rpm. On the other hand, the addition of P188 as a co‐surfactant along with either T80 or CRH40 and incorporation of lecithin within the lipid matrices showed no significant reduction in PS. However, the factor stirring time has a significant influence only on the EE at level 2 than other response. This is because long‐time stirring enough to dissolve the viscosity of the formulation and its increased EE. Besides, the addition of POE‐40‐S at two different levels within the lipid matrices led to no significant influence on the PS and PDI reduction. However, shown increased DL and EE, which may be due to its hydrophilic nature or its coating renders the poor permeability to the drug from core to external phase or prevent the drug leakage from core to external phase. Moreover, the addition of liquid lipid with the solid lipid in the formulation increases the DL, which may be due to the creation of imperfection in the crystal lattice. From the screening analysis result such as % contribution plot and ANOVA, the most influencing factor for the formulation of PNLCs with small in PS and increased DL, EE was identified, when 1:10 ratio of drug:lipid, 3% of CRH40 surfactant at 1300 rpm for 60 min, 20:80 ratio of lecithin:lipid, 1:1 ratio of solid lipid:lipid lipid and 100 ml of 10% PVA. However, each response was influenced by different factors and its interaction effect was not known. As well its risk assessment clearly showed the amount of drug, amount of POE‐40‐S, the concentration of stabilising agent and volume of liquid lipid to be the highly influential variables with small in PS and increased DL, EE. Hence, subsequently, these factors were screened for further experimental optimisation design using four‐factor, five‐level and 30 runs CCD.
3.4 Optimisation of influential factors and process variables
In this study, the statistical parameters computed by Design‐Expert software for CCD indicated that the quadratic polynomial model was the best fit for the experimental data for all responses, and the comparative values of R 2, standard deviation (SD), Adj R‐squared, Pred R‐squared and % coefficient of variation (CV) along with the regression equation generated for the selected responses are given in Tables 5 and 6. ANOVA of the experimental data confirms that the model was significant (Model Prob> F<0.05) [33]. ANOVA identifies the significant factors that affect the responses. For DL, the amount of drug and for EE, the volume of liquid lipid was identified as significant model terms, whereas for PS and PDI, the amount of drug and volume of liquid lipid was identified as significant model terms. Moreover, the predicted R‐squared value was found to be in reasonable agreement with adjusted R‐squared value, which indicates that the model has predicted the responses well. This model can be used to navigate the design space. Only statistically significant (p <0.05) coefficients are included in the equations.
Table 5.
CCD ANOVA results
| DL | EE | PS | PDI | |
|---|---|---|---|---|
| Standard deviation | 0.85 | 3.65 | 2.47 | 0.045 |
| CV, % | 4.37 | 4.80 | 0.95 | 7.58 |
| R‐squared | 0.9969 | 0.9579 | 0.9998 | 0.9760 |
| Adj R‐squared | 0.9936 | 0.9158 | 0.9996 | 0.9454 |
| Pred R‐squared | 0.9816 | 0.7365 | 0.9940 | 0.7482 |
| Adeq precision | 68.155 | 13.584 | 215.781 | 18.157 |
Table 6.
Regression coefficients and ANOVA of the model parameters
| Source | DL | EE | PS | PDI | ||||
|---|---|---|---|---|---|---|---|---|
| p ‐Value | Coefficient | p ‐Value | Coefficient | p ‐Value | Coefficient | p ‐Value | Coefficient | |
| Model | <0.0001 | <0.0001 | <0.0001 | <0.0001 | ||||
| intercept | 18.89 | 80.72 | 181.00 | 0.42 | ||||
| A – amount of drug | <0.0001 | 12.29 | <0.0001 | 15.34 | <0.0001 | 45.30 | 0.0047 | −0.041 |
| B – volume of liquid lipid | 0.0022 | 0.69 | 0.3489 | 0.72 | <0.0001 | −30.33 | 0.0004 | −0.058 |
| C – amount of PEG40 | 0.2046 | 0.24 | 0.9650 | −0.033 | <0.0001 | −30.76 | 0.0003 | 0.063 |
| D – amount of PVA | 0.0765 | 0.35 | 0.6381 | 0.36 | 0.0319 | −2.04 | 0.0151 | −0.033 |
| AB | 0.0391 | 0.52 | 0.4080 | −0.78 | <0.0001 | −55.01 | 0.1675 | −0.023 |
| AC | 0.4069 | −0.19 | 0.5834 | 0.51 | <0.0001 | −68.64 | <0.0001 | −0.11 |
| AD | 0.0357 | 0.53 | 0.8193 | −0.21 | <0.0001 | −20.61 | <0.0001 | −0.12 |
| BC | 0.0140 | 0.64 | 0.7072 | 0.35 | <0.0001 | 49.22 | 0.0026 | 0.063 |
| BD | 0.1183 | −0.38 | 0.7677 | 0.28 | 0.0958 | 1.52 | 0.0240 | −0.040 |
| CD | 0.1050 | 0.40 | 0.5503 | 0.56 | <0.0001 | 11.39 | 0.0481 | −0.032 |
| A2 | 0.0006 | 0.94 | <0.0001 | −6.01 | <0.0001 | 37.28 | <0.0001 | 0.11 |
| B2 | 0.7097 | −0.063 | 0.3831 | −0.64 | <0.0001 | 14.80 | 0.0132 | 0.026 |
| C2 | 0.6292 | −0.081 | 0.4304 | −0.57 | <0.0001 | 48.14 | 0.0010 | 0.039 |
| D2 | 0.0124 | −0.48 | 0.4206 | −0.59 | 0.3641 | 0.69 | 0.0010 | 0.039 |
The quadratic polynomial equations in terms of coded levels for the four responses are as follows:
in which A, B, C and D were the descriptors for four independent variables.
A positive value in the regression equation represented an effect that favoured the optimisation due to synergism, while a negative value indicated an inverse relationship or antagonistic effect between the variables and the responses [34]. As can be seen in Table 6, DL was positively influenced by A, B, C and D. However, the EE was positively influenced by A, B, D and negatively influenced by C, whereas PS was positively influenced by A and negatively influenced by B, C and D. As well, PDI was positively influenced by C and negatively influenced by A, B and D. Also, there were significant interactive parameters (p <0.05) in all the responses, which were the interaction between either factor A, B, C or D.
3.5 Effects on DL and EE
Table 3 showed the DL and EE values ranged from 5.8 to 46.77% and 55.3 to 91.6%, respectively. As shown in Table 6, only the factor amount of drug was statistically significant (p < 0.0001) in the improvement of DL and EE. This influence provided a positive effect, suggesting a directly proportional relationship with the DL and EE. Therefore, as shown in Figs. 3 a and b, DL and EE improved with increases in the amount for drug. This can be explained by the fact that high drug concentration may be sufficient to achieve saturation of the lipid matrix, as well as higher lipid content prevents the escape of drug to outer milieu by effectively enclosing it [35]. Besides, the high loading ability of the PNLCs may be due to the hydrophobicity of TMX (log p ‐value of 4.0 and above). Also, it was observed that the addition of an increased volume of liquid lipid content influences the DL (p < 0.05) (Fig. 3 a). This was in accordance with previously reported results that the NLC consists of solid lipid (GMS) and liquid lipid (oil), have less perfect crystals with many imperfections offering enough space to accommodate TMX molecules, which ultimately improves DL capacity. However, the addition of POE‐40‐S showed no significant effect (p > 0.05) on the enhancement of DL and EE (data not shown). It may be due to its hydrophilicity, which disturbs the solubility of the drug in the lipid matrix and tends to pull the drug out of the lipid phase.
Fig. 3.
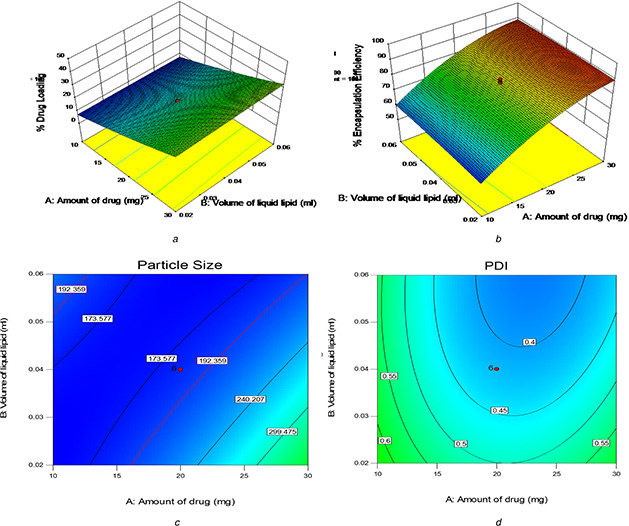
Three‐dimensional response surface plots (%DL and %EE) and contour plots (PS and PDI )
As well, it is evident from Table 6 that EE significantly increased with increasing the amount of drug with increasing the volume of liquid lipid and stabilizing agent (SA) but with decreasing POE‐40‐S. Besides, it was observed from the contour plot that the interaction effect influences the enhancement of EE at a high level of volume of liquid lipid, along with low level to medium level of POE‐40‐S (79.5–80.5%) and with a medium level of SA (79.5–81%) (data not shown), whereas the interaction effect of medium level of POE‐40‐S and medium to a high level of SA showed enhancement of EE about 80–80.5% (data not shown). Besides, it was reported that the higher amount of liquid lipid in NLC enhances the solubilisation capacity of total lipid phase, thus entrapping more drug during formulation [9]. Moreover, long‐chain fatty acids attached to the glyceride resulted in increased accommodation of lipophilic drugs [36].
3.6 Effects on PS and PDI
Table 3 showed the PS and PDI values ranged from 165 to 600 nm and 0.296 to 0.958, respectively. As shown in Table 7, the factor amount of drug, volume of liquid lipid and amount of POE‐40‐S were statistically significant (p < 0.0001) in the reduction of PS. This influence provided a positive effect in the case of amount of drug, suggesting the incorporation of the drug in the NLC matrix increases the PS and also detailed its poor homogeneity and negative effect in the case of volume of liquid lipid and amount of POE‐40‐S suggesting the increase of liquid lipid and PEG‐40‐S reduced the viscosity and surface tension inside the NLCs leading to the formation of homogeneously distributed, smaller nanosized particles [37, 38]. As POE‐40‐S was the surfactant bearing long hydrophilic polyoxyethylene chains of 2000 Da molecular weight, high concentration and long hydrophilic chains helped to reduce surface tension at o/w emulsion interfaces and resulted in reduced PS [39]. Besides, it was reported that the excess liquid lipid inhibits crystallisation of the solid lipid leading to smaller particles [9, 40]. Also, it was observed that the addition of the amount of PVA influences the PS (p < 0.05). This influence provided a negative effect in the case of the amount of PVA suggesting the increase of the amount of PVA reduced the aggregation of NLCs leading to the formation of homogeneously distributed, smaller nanosized particles due to its stabilising effect. Also, it was observed from the contour plot that the interaction effect influences the reduction of PS<200 nm at medium level of amount of drug, along with low level to high level of volume of liquid lipid (173–192 nm) (Fig. 3 c) and with high level of POE‐40‐S (173–192 nm) (data not shown), whereas the interaction effect of the high level of volume of liquid lipid with the high level of POE‐40‐S showed a reduction of PS<197 nm (data not shown). Besides, the interaction effect of the high level of POE‐40‐S with medium to a high level of SA also showed a reduction of PS<197 nm (data not shown), whereas in the case of PDI, the factor amount of drug, the volume of liquid lipid, amount of PEG40 and amount of PVA showed statistically significant effect (p < 0.05) in the small PDI value. However, the factor amount of drug, volume of liquid lipid and amount of PVA provided a negative effect suggesting favour of forming NPs with more homogenous size distributions (monodisperse NPs) in nanodispersion, while the factor amount of PEG‐40‐S provided a positive effect suggesting high PDI value and indicates the heterogeneity of the nanoPS (particles of different sizes) in nanodispersion. Moreover, it was observed from the contour plot that the interaction effect influences the reduction of PDI at the medium level of amount of drug, along with the medium level of volume of liquid lipid (Fig. 3 d), amount of PEG40 and amount of PVA (0.4–0.45) (data not shown). However, the interaction effect of the high level of volume of liquid lipid with a low level of POE‐40‐S and with the high level of SA showed the reduction of PDI<0.4 (data not shown). Besides, the interaction effect of the medium level of POE‐40‐S with a medium level of SA showed the reduction of PDI<0.4 (data not shown).
Table 7.
Predicted and observed values of the optimised formulation
| Response variables | Predicted value | Observed value | % Bias | |
|---|---|---|---|---|
| % DL | — | 33.95 | 33.54 | 1.21 |
| % EE | — | 89.83 | 90.1 | 0.3 |
| PS | — | 167.58 | 161.2 | 3.81 |
| PDI | — | 0.287 | 0.30 | 4.52 |
| Bias was calculated as (predicted value − observed value)/predicted value × 100%. | ||||
| Formulation | DL, % | EE, % | PS, nm | PDI |
| PNLC | 33.54 | 90.1 | 161.2 | 0.30 |
| TNLC | 25.01 | 89.46 | 308 | 0.269 |
3.7 Optimisation and validation
After analysing the polynomial equations depicting the independent variables and responses, the formulation was optimised using numerical optimisation technique by desirability approach, targeting the prescriptive criteria of maximum DL, maximum EE, low PS and low PDI. On the basis of the desirability function (0.898), the composition of optimum formulation was predicted as the amount of drug 30 mg, volume of lipid 0.060 ml, amount of PEO‐40‐S 264.34 mg and amount of PVA 149.92 mg. The predicted values of PS, PDI, EE and DL were 167.58 nm, 0.287, 89.83 and 33.95%, respectively. Then, the predicted model as a new batch of PNLC according to the optimal formulation factors levels was prepared. The obtained results were in good agreement with the predicted values (Table 7) and shown a low percentage bias, suggesting that the optimised formulation was reliable and reasonable. It can be concluded that a high desirability value (0.898) could be obtained with the composition of optimum formulation, whereas the non‐optimised NLC formulation showed the PS of 308 nm, DL of 25.01%, EE of 89.46% and PDI of 0.269. This significant size difference in the NLC and PNLC clearly showed its optimisation effect as well as the effect of POE‐40‐S on the PS reduction. The poly(ethylene)glycol (PEG) chains of these NPs could act as a tighter mesh on the outside of these nano‐vesicles, thus limiting the increase of PS [41]. In addition, the PEG chains on NP surfaces are hydrophilic, thereby preventing their interactions with blood proteins (opsonins) and reduces subsequent opsonisation and phagocytosis by the RES [42].
3.8 Fourier transform infrared spectroscopy
The FTIR spectra of pure TMX showed characteristic peaks at 1218 cm−1 (N–H‐stretching vibration), 1630–1582 cm−1 [aromatic C = C (stretch)], 699 cm−1 (monosubstituted aromatic benzene) and 1729.8 cm−1 (carbonyl bond of citrate) [21, 43]. It was observed that the O–H stretching of the terminal hydroxy group of POE‐40‐S characteristic absorption band at 3384 cm−1 and peaks at 2924 and 2856 cm−1 (C–H stretch of CH2), 1106 cm−1 (C–O–C PEG‐S) and 1737 cm−1 (C = O stretching vibration) [44]. Also, FTIR spectra of PNLC exhibited retention of all the major characteristic peaks of TMX (Fig. 4).
Fig. 4.
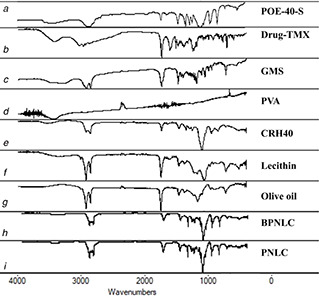
FTIR spectra of
(a) POE‐40‐S, (b) Drug‐TMX, (c) GMS, (d) PVA, (e) CRH40, (f) Lecithin, (g) Olive oil, (h) BPNLC, (i) PNLC
These results suggested that there was no drug degradation or drug excipient molecular interaction. However, the formulation exhibited similar peaks but with a negligible shift, which may be due to the addition of surfactant.
3.9 Differential scanning calorimetry
The DSC curves of pure TMX, BPNLC and PNLCs formulation were shown in Figs. 5 a –c. It was evident from the DSC profile that pure TMX exhibited a sharp endothermic peak at 148.61°C, which indicates its crystalline nature. In the case of BPNLC and PNLCs formulation, no endothermic peak was observed in the melting range of drug indicating the absence of crystallinity and the drug being molecularly dispersed in a lipid matrix.
Fig. 5.
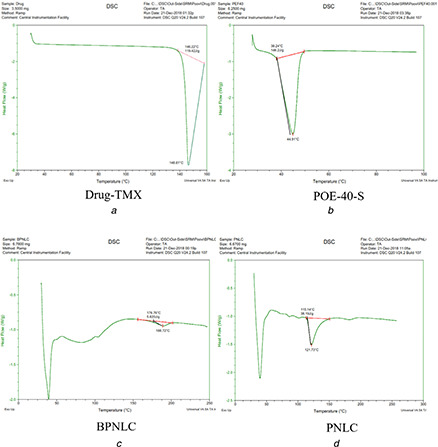
DSC thermogram of
(a) TMX, (b) PEG‐40‐S, (c) BPNLC, (d) PNLC
3.10 Powder X‐ray diffractometer
The diffraction pattern of pure TMX showed several characteristics peaks between 5.33° and 47.68° (2θ), indicating its crystalline nature. These characteristic peaks of the drug disappeared in the lyophilised BPNLC and PNLCs formulation indicating that the drug was not in crystalline nature and showed amorphous dispersion of TMX in the lipid matrix due to the less‐ordered structure of these systems because of the amount of oil content (Figs. 6 a –d). The results were a good agreement with the results obtained by the DSC measurements. Also, it is evident that the surface coating with showing characteristic diffraction pattern of POE‐40‐S peaks on the PNLC and BPNLC formulation, whereas it did not appear in the NLC formulation.
Fig. 6.
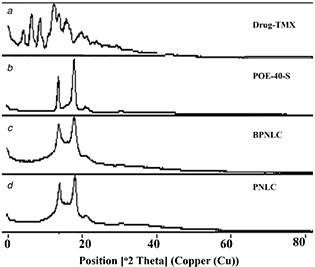
XRD patterns of
(a) Drug‐TMX, (b) POE ‐40‐S, (c) BPNLC, (d) PNLC
3.11 Scanning electron microscopy
The SEM image showed that the pure TMX was crystalline in nature (Fig. 7 a) and the BPNLC and PNLCs formulation obtained in the formulation was fairly smooth, spherical in shape and a dense lipid matrix (Figs. 7 b and c). Besides, there is no visible difference between the surface morphology of BPNLC and PNLCs formulation. Also, the crystalline nature of TMX was absent in the SEM image of BPNLC and PNLCs formulation, which suggests the absence of unencapsulated undissolved drug crystals in the dispersions.
Fig. 7.

SEM images of TMX, BPNLC and PNLCs
(a) TMX, (b) BPNLC, (c) PNLC, (d) PNLC
3.12 Transmission electron microscope
TEM study showed the discrete non‐aggregated particles of lyophilised PNLCs formulation with spherical shape and smooth surface after dilution with water, as shown in Figs. 8 a –c, suggest a core–shell model with a drug‐enriched core. It confirms nano‐size (<200 nm) of prepared PNLCs formulation. It was reported that PS below 200 nm promotes lymphatic uptake of lipid NPs [45]. Besides, it was also reported that the PEG chains at moderate and high density were obliged to stretch away from the surface, which formed brush configuration in the formulation of lipid NPs [46].
Fig. 8.
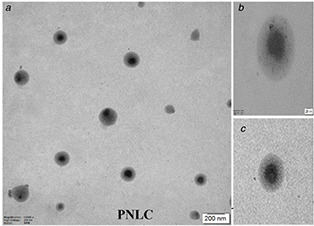
TEM images of PNLCs
3.13 In vitro release study
In vitro results revealed that TMX and PNLCs formulations passing through the strongly acidic environment of the stomach (pH 1.2) tend to release a high amount of drug (13.85% of TMX and 61.29% of PNLCs), thereby indicating significant TMX solubility in the gastric environment (Fig. 9 a). Since, TMX has pKa of 8.87, which specifies that it is a weak base and soluble only in acidic pH, whereas at pH 7.4, TMX and PNLCs formulation shown that initial burst drug release for 8 h followed by slow and sustained/prolonged release up to 72 h (7.69% of TMX and 77.87% of PNLCs). The increased release of the TMX from the PNLC formulation suggests more lipophilic as well as its hydrophilic nature of the matrix. Besides, irrespective of pH effect of the medium and pKa effect of TMX drug in the PNLC formulation due to its increased surface area (small in PS) along with its surfactant effect, long‐chain fatty acid effect and POE‐40‐S combined effect the drug diffusion and release pattern was significantly enhanced. Also, it was reported that the long hydrophilic polyoxyethylene chains of 2000 Da molecular weight polymer possess the surfactant activity [39]. However, this property is highly favourable for sustained/prolonged drug delivery of PNLC formulation and can be utilised to prolong the effect of TMX with a consequent reduction in the frequency of drug administration and dose‐related toxic effects.
Fig. 9.
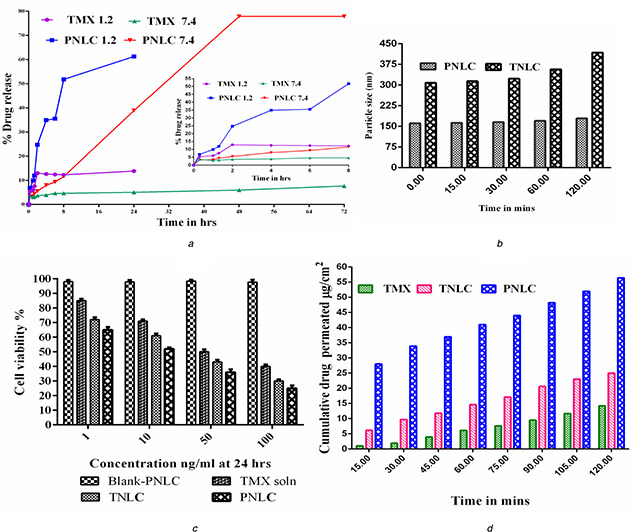
(a) In vitro release profile, (b) Ex vivo mucin binding interaction effect between TNLC, PNLC and mucins, (c) % Cell viability data, (d) Cumulative percentage of TMX permeated from TNLC through non‐everted rat intestine at 37°C
3.14 In vitro release kinetics
From the results (Table 8), based on high ‘r 2’ values, it was concluded that the release of PNLCs formulation at pH 1.2 and 7.4 was predominantly controlled by zero‐order Higuchi model. Then, based on ‘n’ values, at pH 1.2, the mechanism was diffusion and dissolution‐controlled drug release (n = 12.656, super case II type of release or non‐Fickian model) and at pH 7.4, the mechanism was diffusion and swelling‐controlled drug release (n = 0.677, non‐Fickian model or anomalous transport). Besides, at pH 1.2, there was a significant difference in the r 2 values of the zero‐order and the Hixson–Crowell equation. Hence, the erosion mechanism is not involved in the release pattern of PNLCs formulation at pH 1.2, whereas at pH 7.4, it showed erosion mechanism.
Table 8.
TNLCs formulation release kinetics at pH 1.2 and 7.4
| pH | Zero order r 2 | First order r 2 | Higuchi r 2 | Korsmeyer–Peppas n | Hixson–Crowell r 2 |
|---|---|---|---|---|---|
| pH 1.2 | 0.869 | 0.8534 | 0.6332 | 12.656 | 0.9785 |
| pH 7.4 | 0.9827 | 0.9764 | 0.9219 | 0.677 | 0.9839 |
3.15 Storage stability study
At definite time intervals, in the freeze‐dried PNLCs formulation, no change in physical appearance (fluffy cake appearance after freeze drying) and its dispersing tendency with water on reconstitution was observed (Table 9). Also, no sign in aggregation growth and drug precipitation was observed. Besides, the zeta potential value for the optimised formulation was found to be −39.2 ± 5.3 mv, and it is sufficiently high to form stable colloidal nanosuspension. Furthermore, no drastic change in all responses was observed at both temperatures. Therefore, it can be concluded that the room temperature is a more favourable condition than the accelerated condition for freeze‐dried PNLCs formulation for a longer period. Blank NLC and BPNLC also were prepared and it showed its zeta potential values as −47 and −38 mv, respectively. The decreased zeta potential value in PNLC and BPNL is due to surface modification with steric stabiliser POE‐40‐S, which neutralises the surface charge or a shift in the shear plane of the particle [47]. It was reported that negatively charged carriers show higher lymphatic uptake than neutral or positively charged surfaces, which could be due to the fact that the interstitial matrix contains a net negative charge. Therefore, in the interstitium, anionic carrier particles encounter electrostatic repulsion and move more quickly [48].
Table 9.
Storage stability effect on freeze‐dried PNLCs formulation (6 months)
| Period and condition | DL | EE | PS | PDI | zeta potential (ZP) | Physical appearance and dispersing tendency |
|---|---|---|---|---|---|---|
| initial | 25.24 ± 0.23 | 90.1 ± 0.82 | 161 ± 1.97 | 0.34 ± 0.22 | −39.2 ± 5.3 | fluffy cake with dispersing tendency on reconstitution with water |
| 30°±2°C/65±5% RH | ||||||
| 1 month | 25.21 ± 0.25 | 87.52 ± 0.63 | 161.29 ± 3.10 | 0.34 ± 0.45 | −39.2 ± 4.3 | fluffy cake with dispersing tendency on reconstitution with water |
| 3 months | 25.20 ± 0.27 | 87.12 ± 0.49 | 162.98 ± 2.54 | 0.35 ± 0.12 | −38.7 ± 3.9 | |
| 6 months | 24.97 ± 0.29 | 86.99 ± 0.71 | 165.01 ± 1.46 | 0.39 ± 0.86 | −31.7 ± 3.1 | |
| 40°±2°C/75±5% RH | ||||||
| 1 month | 24.81 ± 0.41 | 85.71 ± 0.67 | 163.74 ± 3.08 | 0.36 ± 0.53 | −38.4 ± 273 | |
| 3 months | 23.71 ± 0.66 | 84.01 ± 0.72 | 165.33 ± 2.14 | 0.37 ± 0.21 | −37.2 ± 4.7 | |
| 6 months | 21.87 ± 0.72 | 83.99 ± 0.53 | 167.08 ± 1.97 | 0.37 ± 0.98 | −33.5 ± 2.2 | |
3.16 In vitro cytotoxicity study
Cytotoxicity of TNLCs, PNLCs and BNLCs was evaluated in order to study the concentration‐dependent effect on cell viability against MCF‐7 breast cancer cell line after 24 h incubation using MTT assay in comparison with TMX solution. It was found that the concentration‐dependent cell viability with TMX (Fig. 9 c). In this cell viability assay, TNLCs and PNLCs showed high cytotoxicity against MCF‐7 cell line compared with the simple TMX solution. The enhanced toxicity of TNLCs and PNLC may be due to the preferential uptake of nanosized particles than that of the coarse particles (simple TMX solution). The possible mechanism underlying the enhanced efficacies of TNLCs and PNLC against MCF‐7 could be explained that NLC may carry the drug into the cancer cells either by drug diffusion through the cell membrane or internalisation of drug‐carrier systems by endocytosis releasing the active inside the cells [49, 50]. However, the drug, in both cases, could be prone to go out of the cells through P‐glycoprotein (P‐gp) efflux pump mechanism [49]. However, the presence of P‐gp inhibitor CRH40 and POE‐40‐S as an excipient in the formulation showed its extent of cell viabilities and antitumour activity. Since, the P‐gp inhibitor (POE‐40‐S) inhibits the efflux of the drug outside the cells and increases the drug accumulation in tumour cells, therefore, which enhances the drug cytotoxicity (Safwat et al.). In this study, the PNLC showed the highest antitumour activity than TNLC. It could be due to the increased inhibitory effect of POE‐40‐S on P‐gp‐mediated TMX efflux mechanism, which increases the drug accumulation in the tumour cells and increases the drug cytotoxicity than CRH40 present in the TNLC formulation [49, 51, 52, 53]. Also, it was observed that the antitumour activity of TMX is not affected when it is incorporated into the lipid‐based NLCs system. This result is in good agreement with previous reports [1, 4, 54], whereas the BPNLCs exhibited no cytotoxicity on MCF‐7 cells as compared with TMX solution, TNLCs and PNLCs formulations suggest that the cause of cell death is primarily from the effect of TMX present in NLCs [47, 54]. Also, which indicates the lipid and surfactant used of NLCs were biocompatible and the possibility of its use as colloidal drug carriers in vivo [55, 56].
3.17 Protein adsorption
The protein adsorption of BNLCs, BPNLCs, TNLCs and PNLCs formulation was evaluated using BSA, the most abundant protein in serum, as a model plasma protein [57]. Since, protein adsorption occurred when the NPs came in contact with the plasma, which induced the recognition of macrophages and reduction of the tumour accumulation of the NPs [46]. The amount of protein adsorption of TNLC and PNLC after 2 h incubation was found to be 750, 300, 540 and 460 µg. It clearly indicated that the protein adsorptions of BPNLC and PNLC are significantly lower than that of BNLC and TNLC, which may be due to its improved surface hydrophilicity. It was in accordance with the previous result [57, 58]. Besides, the brush configuration of PEG chains in PNLC might effectively block the movement of protein to the vicinity of the NPs, which was mainly controlled by random Brownian motion, leading to the reduced BSA adsorption [59].
3.18 Ex vivo mucin binding
The in vivo fates of NPs most likely also play a key role in oral drug absorption. A better understanding of interactions between NPs and free mucin is highly helpful in predicting the in vivo fate and performance of nanocarriers [12]. Fig. 9 b showed the PS changes of TNLC and PNLC with time in the ex vivo intestinal fluids. The TNLC and PNLC PS increased as the incubation time increased. However, it remained in the nanometric range and did not show any appreciable change in the PS of TNLC (from 308 to 418 nm) and PNLC (from 161 to 179.2 nm), probably due to a more effective charge and steric stabilisation effects of CRH40 and POE‐40‐S [60]. Although, the extent of steric stabilisation effect is more significant in the PNLC formulation due to its surface modification with POE‐40‐S and its brush configuration, which helps to avoid the adhesion and steric inhibition by the mucin fibre mesh.
Besides, it assures that the NLC carrier shields the drug from gut enzymes and avoid its degradation, thereby which helps to improve the intestinal absorption and the oral bioavailability of the formulation. Zhang et al. [12] reported that surface modification of lipid NP with PEG reduces the enzymes accessibility and mucins binding due to its shielding effect of highly hydrated PEG corona [13, 14]. Plapied et al. [61] also reported the similar finding that the PEGylated NP helps to maintain the intact nano‐architecture of lipid NPs over a relatively long time and reduce the adhesion of free mucins through transporting across the gastrointestinal tract (GIT). Besides, similar finding was reported by many searchers using pig intestinal mucus [62, 63], pig gastric mucus [64], human respiratory mucus (Schuster et al.) [65], human cervicovaginal mucus [66, 67], mouse vaginal mucus [68] and bovine vitreous ex vivo [69].
3.19 Ex vivo intestinal permeation
The non‐everted rat intestinal sacs technique was used in this method to determine the ex vivo intestinal permeation of the TNLCs formulation compared with the pure TMX. Fig. 9 d revealed the cumulative amount of TMX permeated over a period of 2 h. The amount of drug permeated from the TNLCs and PNLC was nearly two‐fold and five‐fold higher than that permeated from the pure TMX, respectively. Such an enhancement in the permeation of the TNLCs and PNLC formulation could be attributed to the reduction of PS with increased surface areas and bioenhancing activities of the NLCs excipients, which lead to the rapid dissolution of TMX in the intestinal sac and subsequent rapid diffusion. Besides, the presence of CRH40 and POE‐40‐S in the formulation helps to reduce the efflux pump P‐gp and enhances the permeability of TNLCs and PNLC formulation than pure TMX. The high permeability of PNLC than TNLC formulation is due to POE‐40‐S steric effect with the mucus layer and P‐gp inhibitor effect in the intestine. Cu and Saltzman [70] also reported that the surface modification of polymer particles using PEG mimics ‘stealth’ migration of viruses that infect mucosal tissues and avoid the entrapment in the gel by minimising strong interactions with mucus constituents. As well, Liu et al. [42] and Zhu et al. [71] reported that the PEG‐40 stearate could competitively inhibit the activity of P‐gp efflux pumps by saturating the P‐gp receptors as a substrate and decrease its efflux activity in the P‐gp ATPase activity study. Moreover, it was reported that the lipid‐based nanocarriers formulated by using various digestive lipids such as phospholipids, glycosides, cholesterol esters and fatty acids enhance the intestinal permeability of the drug by increasing its transport from intestine, and the presence of surfactants decrease the intestinal efflux by blocking the P‐gp efflux pump located in the enterocytes of the GIT [72, 73].
4 Conclusion
The TMX‐loaded surface‐modified long‐circulating biocompatible carrier using POE‐40‐S was successfully prepared by MECT using solvent‐free preparation technique and biocompatible excipients generally recognized as safe (GRAS). Screening by TgL12 OA and optimisation by CCD helps to identify the most influential factors affecting the variables and interaction effect with minimum experimentation thus saving considerable time, efforts and resources for further in‐depth study. Besides, the surface modification of NLC with POE‐40‐S not only helps to reduce the PS growth through its tighter mesh formation on the NP but also reduces the interaction with blood proteins, mucous membrane, efflux pump P‐gp and enhances the intestinal permeability. As well, the in vitro cytotoxicity study of TMX solution, TNLCs and PNLCs formulations showed that the concentration‐dependent cytotoxicity and its antitumour activity was not affected when it is incorporated into the lipid‐based NLCs system. Moreover, no cytotoxicity was found in the case of BNLC and PNLC, which suggests that the carrier is biocompatible. Furthermore, it was found that the surface‐modified PNLCs formulation can overcome the major limiting factors for lipid NPs in drug absorption such as digestive enzymatic degradation, non‐specific adhesion by secreted mucin, poor trans‐epithelial transport. Finally, it can be concluded that the surface‐modified PNLCs formulation was an effective, biocompatible, stable formulation in the enhancement of dissolution rate, solubility, stability with reduced mucus adhesion and increased permeability, thereby which indicates its enhanced oral bioavailability in vivo.
5 Acknowledgments
The author wishes to acknowledge the sponsorship given by the Department of Pharmaceutics, College of Pharmacy, Mother Theresa Post Graduate and Research Institute of Health Sciences, (A Government of Puducherry Institution), Puducherry 605 006, India and thankful for giving study period to receive her Ph.D. Also, we are thankful to East West Pharma (Raipur, Uttarakhand, India) for providing gift sample of drug.
6 References
- 1. How C.W. Rasedee A. Manickam S. et al.: ‘Tamoxifen‐loaded nanostructured lipid carrier as a drug delivery system: characterization, stability assessment and cytotoxicity’, Colloids Surf B, Biointerfaces, 2013, 112, pp. 393 –399 [DOI] [PubMed] [Google Scholar]
- 2. Agrawal Y. Petkar K.C. Sawant K.K.: ‘Development, evaluation and clinical studies of acitretin loaded nanostructured lipid carriers for topical treatment of psoriasis’, Int. J. Pharm., 2010, 401, (1), pp. 93 –102 [DOI] [PubMed] [Google Scholar]
- 3. Chinsriwongkul A. Chareanputtakhun P. Ngawhirunpat T. et al.: ‘Nanostructured lipid carriers (nlc) for parenteral delivery of an anticancer drug’, AAPS Pharmscitech, 2012, 13, (1), pp. 150 –158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Poovi G. Thangavel Mahalingam V. Narayanasamy D.: ‘Solid lipid nanoparticles and nanostructured lipid carriers: a review of the effect of physicochemical formulation factors in the optimization process, different preparation technique, characterization, and toxicity’, Curr. Nanosci., 2018, 14, pp. 1 –18 [Google Scholar]
- 5. Gao D. Tang S. Tong Q.: ‘Oleanolic acid liposomes with polyethylene glycol modification: promising antitumor drug delivery’, Int. J. Nanomed., 2012, 7, p. 3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kharia A.A. Singhai A.K.: ‘Effective parameters for formulation of gastro adhesive nanoparticles: screening by design‐of‐experiments approach’, J. Microencapsul., 2014, 31, (4), pp. 399 –405 [DOI] [PubMed] [Google Scholar]
- 7. Sonam C.H. Kumar V.: ‘Taguchi design for optimization and development of antibacterial drug‐loaded plga nanoparticles’, Int. J. Biol. Macromol., 2014, 64, pp. 99 –105 [DOI] [PubMed] [Google Scholar]
- 8. Zhang X. Liu J. Qiao H. et al.: ‘Formulation optimization of dihydroartemisinin nanostructured lipid carrier using response surface methodology’, Powder Technol., 2010, 197, (1–2), pp. 120 –128 [Google Scholar]
- 9. Jain K. Sood S. Gowthamarajan K.: ‘Optimization of artemether‐loaded nlc for intranasal delivery using central composite design’, Drug Deliv., 2015, 22, (7), pp. 940 –954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dudhipala N. Veerabrahma K.: ‘Pharmacokinetic and pharmacodynamic studies of nisoldipine‐loaded solid lipid nanoparticles developed by central composite design’, Drug Dev. Ind. Pharm., 2015, 41, (12), pp. 1968 –1977 [DOI] [PubMed] [Google Scholar]
- 11. Elnaggar Y.S. El‐Massik M.A. Abdallah O.Y.: ‘Self‐nanoemulsifying drug delivery systems of tamoxifen citrate: design and optimization’, Int. J. Pharm., 2009, 380, (1–2), pp. 133 –141 [DOI] [PubMed] [Google Scholar]
- 12. Zhang X. Chen G. Zhang T. et al.: ‘Effects of pegylated lipid nanoparticles on the oral absorption of one bcs Ii drug: a mechanistic investigation’, Int. J. Nanomed., 2014, 9, p. 5503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paliwal R. Rai S. Vaidya B. et al.: ‘Effect of lipid core material on characteristics of solid lipid nanoparticles designed for oral lymphatic delivery’, Nanomed. Nanotechnol. Biol. Med., 2009, 5, (2), pp. 184 –191 [DOI] [PubMed] [Google Scholar]
- 14. Yuan H. Chen C.‐Y. Chai G.‐H. et al.: ‘Improved transport and absorption through gastrointestinal tract by pegylated solid lipid nanoparticles’, Mol. Pharm., 2013, 10, (5), pp. 1865 –1873 [DOI] [PubMed] [Google Scholar]
- 15. Varshosaz J. Ghaffari S. Khoshayand M.R. et al.: ‘Development and optimization of solid lipid nanoparticles of amikacin by central composite design’, J. Liposome Res., 2010, 20, (2), pp. 97 –104 [DOI] [PubMed] [Google Scholar]
- 16. Koziara J.M. Lockman P.R. Allen D.D. et al.: ‘Paclitaxel nanoparticles for the potential treatment of brain tumors’, J. Control. Release, 2004, 99, (2), pp. 259 –269 [DOI] [PubMed] [Google Scholar]
- 17. Koziara J. Oh J. Akers W. et al.: ‘Blood compatibility of cetyl alcohol/polysorbate‐based nanoparticles’, Pharm. Res., 2005, 22, (11), pp. 1821 –1828 [DOI] [PubMed] [Google Scholar]
- 18. Mumper R.J. Jay M.: ‘Microemulsions as precursors to solid nanoparticles’, Google Patents, 2006.
- 19. Ganesan P. Narayanasamy D.: ‘Lipid nanoparticles: different preparation techniques, characterization, hurdles, and strategies for the production of solid lipid nanoparticles and nanostructured lipid carriers for oral drug delivery’, Sustain. Chem. Pharm., 2017, 6, pp. 37 –56 [Google Scholar]
- 20. de Carvalho S.M. Noronha C.M. Floriani C.L. et al.: ‘Optimization of Α‐tocopherol loaded solid lipid nanoparticles by central composite design’, Ind. Crops Prod., 2013, 49, pp. 278 –285 [Google Scholar]
- 21. Shete H. Patravale V.: ‘Long‐chain lipid‐based tamoxifen nlc. Part I: preformulation studies, formulation development and physicochemical characterization’, Int. J. Pharm., 2013, 454, (1), pp. 573 –583 [DOI] [PubMed] [Google Scholar]
- 22. Pardeshi C.V. Rajput P.V. Belgamwar V.S. et al.: ‘Novel surface modified solid lipid nanoparticles as intranasal carriers for ropinirole hydrochloride: application of factorial design approach’, Drug Deliv., 2013, 20, (1), pp. 47 –56 [DOI] [PubMed] [Google Scholar]
- 23. Cho H.‐J. Park J.W. Yoon I.‐S. et al.: ‘Surface‐modified solid lipid nanoparticles for oral delivery of docetaxel: enhanced intestinal absorption and lymphatic uptake’, Int. J. Nanomed., 2014, 9, p. 495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jain A.K. Swarnakar N.K. Godugu C. et al.: ‘The effect of the oral administration of polymeric nanoparticles on the efficacy and toxicity of tamoxifen’, Biomaterials, 2011, 32, (2), pp. 503 –515 [DOI] [PubMed] [Google Scholar]
- 25. Barzegar‐Jalali M. Adibkia K. Valizadeh H. et al.: ‘Kinetic analysis of drug release from nanoparticles’, J. Pharm. Pharm. Sci., 2008, 11, (1), pp. 167 –177 [DOI] [PubMed] [Google Scholar]
- 26. Ravi P.R. Aditya N. Kathuria H. et al.: ‘Lipid nanoparticles for oral delivery of raloxifene: optimization, stability, in vivo evaluation and uptake mechanism’, Eur. J. Pharm. Biopharm., 2014, 87, (1), pp. 114 –124 [DOI] [PubMed] [Google Scholar]
- 27. Ibrahim W.M. Alomrani A.H. Yassin A.E.B.: ‘Novel sulpiride‐loaded solid lipid nanoparticles with enhanced intestinal permeability’, Int. J. Nanomed., 2014, 9, p. 129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mehnert W. Mäder K.: ‘Solid lipid nanoparticles: production, characterization and applications’, Adv. Drug Deliv. Rev., 2001, 47, (2), pp. 165 –196 [DOI] [PubMed] [Google Scholar]
- 29. Müller R.H. Radtke M. Wissing S.A.: ‘Solid lipid nanoparticles (sln) and nanostructured lipid carriers (nlc) in cosmetic and dermatological preparations’, Adv. Drug Deliv. Rev., 2002, 54, pp. S131 –S155 [DOI] [PubMed] [Google Scholar]
- 30. Pardeike J. Weber S. Haber T. et al.: ‘Development of an itraconazole‐loaded nanostructured lipid carrier (nlc) formulation for pulmonary application’, Int. J. Pharm., 2011, 419, (1–2), pp. 329 –338 [DOI] [PubMed] [Google Scholar]
- 31. Manjunath K. Reddy J.S. Venkateswarlu V.: ‘Solid lipid nanoparticles as drug delivery systems’, Methods Find. Exp. Clin. Pharmacol., 2005, 27, (2), pp. 127 –144 [DOI] [PubMed] [Google Scholar]
- 32. Zhang C. Gu C. Peng F. et al.: ‘Preparation and optimization of triptolide‐loaded solid lipid nanoparticles for oral delivery with reduced gastric irritation’, Molecules, 2013, 18, (11), pp. 13340 –13356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saha P. Chowdhury S. Gupta S. et al.: ‘Assessment on the removal of malachite green using tamarind fruit shell as biosorbent’, CLEAN – Soil Air Water, 2010, 38, (5–6), pp. 437 –445 [Google Scholar]
- 34. Negi L.M. Jaggi M. Talegaonkar S.: ‘A logical approach to optimize the nanostructured lipid carrier system of irinotecan: efficient hybrid design methodology’, Nanotechnology, 2012, 24, (1), p. 015104 [DOI] [PubMed] [Google Scholar]
- 35. Gaba B. Fazil M. Khan S. et al.: ‘Nanostructured lipid carrier system for topical delivery of terbinafine hydrochloride’, Bull. Faculty Pharm. Cairo Univ., 2015, 53, (2), pp. 147 –159 [Google Scholar]
- 36. Dudhipala N. Veerabrahma K.: ‘Candesartan cilexetil loaded solid lipid nanoparticles for oral delivery: characterization, pharmacokinetic and pharmacodynamic evaluation’, Drug Deliv., 2016, 23, (2), pp. 395 –404 [DOI] [PubMed] [Google Scholar]
- 37. Behbahani E.S. Ghaedi M. Abbaspour M. et al.: ‘Optimization and characterization of ultrasound assisted preparation of curcumin‐loaded solid lipid nanoparticles: application of central composite design, thermal analysis and X‐ray diffraction techniques’, Ultrason. Sonochem., 2017, 38, pp. 271 –280 [DOI] [PubMed] [Google Scholar]
- 38. Sanad R.A. AbdelMalak N.S. Badawi A.A.: ‘Formulation of a novel oxybenzone‐loaded nanostructured lipid carriers (nlcs)’, AAPS Pharmscitech, 2010, 11, (4), pp. 1684 –1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Y. Wu W.: ‘ In situ evading of phagocytic uptake of stealth solid lipid nanoparticles by mouse peritoneal macrophages’, Drug Deliv., 2006, 13, (3), pp. 189 –192 [DOI] [PubMed] [Google Scholar]
- 40. Lin X. Li X. Zheng L. et al.: ‘Preparation and characterization of monocaprate nanostructured lipid carriers’, Colloids Surf. A, Physicochem. Eng. Aspects, 2007, 311, (1–3), pp. 106 –111 [Google Scholar]
- 41. Juillerat‐Jeanneret L.: ‘The targeted delivery of cancer drugs across the blood–brain barrier: chemical modifications of drugs or drug‐nanoparticles?’, Drug Discov. Today, 2008, 13, (23–24), pp. 1099 –1106 [DOI] [PubMed] [Google Scholar]
- 42. Liu Z. Okeke C.I. Zhang L. et al.: ‘Mixed polyethylene glycol‐modified breviscapine‐loaded solid lipid nanoparticles for improved brain bioavailability: preparation, characterization, and in vivo cerebral microdialysis evaluation in adult Sprague–Dawley rats’, AAPS Pharmscitech, 2014, 15, (2), pp. 483 –496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gamberini M.C. Baraldi C. Tinti A. et al.: ‘Vibrational study of tamoxifen citrate polymorphism’, J. Mol. Struct., 2007, 840, (1–3), pp. 29 –37 [Google Scholar]
- 44. Li X. Lin X. Zheng L. et al.: ‘Effect of poly (ethylene glycol) stearate on the phase behavior of monocaprate/Tween80/water system and characterization of poly (ethylene glycol) stearate‐modified solid lipid nanoparticles’, Colloids Surf. A, Physicochem. Eng. Aspects, 2008, 317, (1–3), pp. 352 –359 [Google Scholar]
- 45. Shah M.K. Madan P. Lin S.: ‘Preparation, in vitro evaluation and statistical optimization of carvedilol‐loaded solid lipid nanoparticles for lymphatic absorption via oral administration’, Pharm. Dev. Technol., 2014, 19, (4), pp. 475 –485 [DOI] [PubMed] [Google Scholar]
- 46. Zhang S. Tang C. Yin C.: ‘Effects of poly (ethylene glycol) grafting density on the tumor targeting efficacy of nanoparticles with ligand modification’, Drug Deliv., 2015, 22, (2), pp. 182 –190 [DOI] [PubMed] [Google Scholar]
- 47. Müller R.H. Mäder K. Gohla S.: ‘Solid lipid nanoparticles (sln) for controlled drug delivery – a review of the state of the art’, Eur. J. Pharm. Biopharm., 2000, 50, (1), pp. 161 –177 [DOI] [PubMed] [Google Scholar]
- 48. Khan A.A. Mudassir J. Mohtar N. et al.: ‘Advanced drug delivery to the lymphatic system: lipid‐based nanoformulations’, Int. J. Nanomed., 2013, 8, p. 2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Safwat S. Ishak R.A. Hathout R.M. et al.: ‘Nanostructured lipid carriers loaded with simvastatin: effect of peg/glycerides on characterization, stability, cellular uptake efficiency and in vitro cytotoxicity’, Drug Dev. Ind. Pharm., 2017, 43, (7), pp. 1112 –1125 [DOI] [PubMed] [Google Scholar]
- 50. Wang L. Luo Q. Lin T. et al.: ‘Pegylated nanostructured lipid carriers (peg–nlc) as a novel drug delivery system for biochanin A’, Drug Dev. Ind. Pharm., 2015, 41, (7), pp. 1204 –1212 [DOI] [PubMed] [Google Scholar]
- 51. Bansal T. Akhtar N. Jaggi M. et al.: ‘Novel formulation approaches for optimising delivery of anticancer drugs based on P‐glycoprotein modulation’, Drug Discov. Today, 2009, 14, (21–22), pp. 1067 –1074 [DOI] [PubMed] [Google Scholar]
- 52. Srivalli K.M.R. Lakshmi P.: ‘Overview of P‐glycoprotein inhibitors: a rational outlook’, Braz. J. Pharm. Sci., 2012, 48, (3), pp. 353 –367 [Google Scholar]
- 53. Lin Y. Shen Q. Katsumi H. et al.: ‘Effects of labrasol and other pharmaceutical excipients on the intestinal transport and absorption of Rhodamine123, a P‐glycoprotein substrate, in rats’, Biol. Pharm. Bull., 2007, 30, (7), pp. 1301 –1307 [DOI] [PubMed] [Google Scholar]
- 54. Shete H. Chatterjee S. De A. et al.: ‘Long‐chain lipid‐based tamoxifen nlc. Part Ii: pharmacokinetic, biodistribution and in vitro anticancer efficacy studies’, Int. J. Pharm., 2013, 454, (1), pp. 584 –592 [DOI] [PubMed] [Google Scholar]
- 55. Wang D. Zhao P. Cui F. et al.: ‘Preparation and characterization of solid lipid nanoparticles loaded with total flavones of Hippophae rhamnoides (tfh)’, PDA J. Pharm. Sci. Technol., 2007, 61, (2), pp. 110 –120 [PubMed] [Google Scholar]
- 56. Baek J.‐S. Cho C.‐W.: ‘Surface modification of solid lipid nanoparticles for oral delivery of curcumin: improvement of bioavailability through enhanced cellular uptake, and lymphatic uptake’, Eur. J. Pharm. Biopharm., 2017, 117, pp. 132 –140 [DOI] [PubMed] [Google Scholar]
- 57. Kim C. Sung S. Lee E. et al.: ‘Sterically stabilized ripl peptide‐conjugated nanostructured lipid carriers: characterization, cellular uptake, cytotoxicity, and biodistribution’, Pharmaceutics, 2018, 10, (4), p. 199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tziampazis E. Kohn J. Moghe P.V.: ‘Peg‐variant biomaterials as selectively adhesive protein templates: model surfaces for controlled cell adhesion and migration’, Biomaterials, 2000, 21, (5), pp. 511 –520 [DOI] [PubMed] [Google Scholar]
- 59. Chen H. Yuan L. Song W. et al.: ‘Biocompatible polymer materials: role of protein–surface interactions’, Prog. Polym. Sci., 2008, 33, (11), pp. 1059 –1087 [Google Scholar]
- 60. Cirri M. Maestrini L. Maestrelli F. et al.: ‘Design, characterization and in vivo evaluation of nanostructured lipid carriers (nlc) as a new drug delivery system for hydrochlorothiazide oral administration in pediatric therapy’, Drug Deliv., 2018, 25, (1), pp. 1910 –1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Plapied L. Duhem N. des Rieux A. et al.: ‘Fate of polymeric nanocarriers for oral drug delivery’, Curr. Opin. Colloid Interface Sci., 2011, 16, (3), pp. 228 –237 [Google Scholar]
- 62. Abdulkarim M. Agulló N. Cattoz B. et al.: ‘Nanoparticle diffusion within intestinal mucus: three‐dimensional response analysis dissecting the impact of particle surface charge, size and heterogeneity across polyelectrolyte, pegylated and viral particles’, Eur. J. Pharm. Biopharm., 2015, 97, pp. 230 –238 [DOI] [PubMed] [Google Scholar]
- 63. Groo A.‐C. Mircheva K. Bejaud J. et al.: ‘Development of 2d and 3d mucus models and their interactions with mucus‐penetrating paclitaxel‐loaded lipid nanocapsules’, Pharm. Res., 2014, 31, (7), pp. 1753 –1765 [DOI] [PubMed] [Google Scholar]
- 64. Griffiths P.C. Cattoz B. Ibrahim M.S. et al.: ‘Probing the interaction of nanoparticles with mucin for drug delivery applications using dynamic light scattering’, Eur. J. Pharm. Biopharm., 2015, 97, pp. 218 –222 [DOI] [PubMed] [Google Scholar]
- 65. Schuster B.S. Suk J.S. Woodworth G.F. et al.: ‘Nanoparticle diffusion in respiratory mucus from humans without lung disease’, Biomaterials, 2013, 34, (13), pp. 3439 –3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xu Q. Boylan N.J. Cai S. et al.: ‘Scalable method to produce biodegradable nanoparticles that rapidly penetrate human mucus’, J. Control. Release, 2013, 170, (2), pp. 279 –286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xu Q. Ensign L.M. Boylan N.J. et al.: ‘Impact of surface polyethylene glycol (peg) density on biodegradable nanoparticle transport in mucus ex vivo and distribution in vivo ’, ACS Nano, 2015, 9, (9), pp. 9217 –9227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ensign L.M. Tang B.C. Wang Y.‐Y. et al.: ‘Mucus‐penetrating nanoparticles for vaginal drug delivery protect against herpes simplex virus’, Sci. Transl. Med., 2012, 4, (138), p. 138ra179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xu Q. Boylan N.J. Suk J.S. et al.: ‘Nanoparticle diffusion in, and microrheology of, the bovine vitreous ex vivo ’, J. Control. Release, 2013, 167, (1), pp. 76 –84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cu Y. Saltzman W.M.: ‘Controlled surface modification with poly (ethylene) glycol enhances diffusion of plga nanoparticles in human cervical mucus’, Mol. Pharm., 2008, 6, (1), pp. 173 –181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhu S. Huang R. Hong M. et al.: ‘Effects of polyoxyethylene (40) stearate on the activity of P‐glycoprotein and cytochrome P450’, Eur. J. Pharm. Sci., 2009, 37, (5), pp. 573 –580 [DOI] [PubMed] [Google Scholar]
- 72. Singh A. Neupane Y.R. Mangla B. et al.: ‘Nanostructured lipid carriers for oral bioavailability enhancement of exemestane: formulation design, in vitro, ex vivo and in vivo studies’, J. Pharm. Sci., 2019, 108, pp. 3382 –3395 [DOI] [PubMed] [Google Scholar]
- 73. Singh A. Neupane Y.R. Panda B.P. et al.: ‘Lipid‐based nanoformulation of lycopene improves oral delivery: formulation optimization, ex vivo assessment and its efficacy against breast cancer’, J. Microencapsul., 2017, 34, (4), pp. 416 –429 [DOI] [PubMed] [Google Scholar]