

Abstract
The objective of this study was to develop an in‐situ gel containing lorazepam (LZM) loaded nanostructured lipid carriers (NLCs) for direct nose‐to‐brain delivery in order to increase drug therapeutic efficacy in the treatment of epilepsy. Accordingly, LZM loaded NLCs were formulated using emulsification solvent diffusion and evaporation method; then the effects of the formulation variables on different physicochemical characteristics of NLCs were investigated. Thermosensitive in‐situ gels containing LZM‐NLCs were prepared using a combination of chitosan and β‐glycerol phosphate (β‐GP). The anticonvulsant efficacy of LZM‐NLCs‐Gel was then examined using the pentylenetetrazole (PTZ) model. The optimised NLCs were spherical, showing the particle size of 71.70 ± 5.16 nm and the zeta potential of −20.06 ± 2.70 mV. The pH and gelation time for the chitosan solution with 15% (w/v) β‐GP were determined to be 7.12 ± 0.03 and 5.33 ± 0.58 min, respectively. The in‐vivo findings showed that compared with the control group and the group that received LZM‐Gel, the occurrence of PTZ‐induced seizures in the rats was significantly reduced by LZM‐NLCs‐Gel after intranasal administration. These results, therefore, suggested that the LZM‐NLCs‐Gel system could have potential applications for brain targeting through nasal route and might increase LZM therapeutic efficacy in the treatment of epilepsy.
Inspec keywords: biomedical materials, nanomedicine, cellular biophysics, electrokinetic effects, drug delivery systems, nanoparticles, brain, pH, drugs, particle size, nanofabrication, medical disorders, polymer gels
Other keywords: evaporation method, β‐glycerol phosphate, β‐GP, optimised NLCs, received LZM‐Gel, LZM therapeutic efficacy, chitosan‐based thermosensitive gel, lorazepam NLCs, nose‐to‐brain delivery, drug therapeutic efficacy, emulsification solvent diffusion, in‐vivo evaluation, in‐vitro evaluation, LZM‐NLC‐gel system, status epilepticus treatment, lorazepam loaded nanostructured lipid carriers, epilepsy treatment, physicochemical characteristics, thermosensitive in‐situ gel, anticonvulsant efficacy, pentylenetetrazole model, particle size, zeta potential, pH, gelation time, chitosan solution, PTZ‐induced seizures, intranasal administration
1 Introduction
Status epilepticus (SE) is a medical emergency defined as some continuous seizure lasting more than 5 min or a kind of repeated seizure within a 5 min period without regaining consciousness between episodes. SE needs immediate intervention because the longer the episode of seizures, the more difficult it is to control and the greater the risk of the permanent brain damage.
Benzodiazepines are regarded as the preferred choice treatment in the management of SE. The most commonly used benzodiazepines for the treatment of SE include lorazepam (LZM), diazepam and midazolam. LZM is widely used for the management of SE thanks to its longer duration of action (12 h), less respiratory depression, fewer hypotensive effects and better toleration, in comparison to diazepam [1]. It is generally administered as IV injection or an oral route. However, the IV administration requires medically trained personnel and could be associated with injection site pain and inflammation. On the other hand, in the patients with the SE seizure, severe spasm in the faciomandibular area could affect the patient's ability to swallow the LZM tablet [2, 3]. Thus, an alternative, more convenient and effective way of drug administration is highly needed.
Intranasal administration of medications is a patient‐friendly non‐invasive, cost‐effective, painless and safe route of administration that has recently received a great deal of attention by many researchers due to several advantages; from clinical perspectives, these advantages are escaping hepatic first pass metabolism, bypassing blood brain barrier to deliver therapeutics into brain through olfactory and trigeminal nerve components in nasal epithelium, ensuring the fast onset of action due to the large absorption area, and contributing to high vascularisation [4, 5, 6, 7, 8].
Nanostructured lipid carriers (NLCs) are well‐known universal nanocarriers that hold a great potential in nose to brain drug delivery because of their biocompatibility, biodegradability, low toxicity, rapid uptake by the brain, controlled drug release, possible high drug payload, possibility of loading both hydrophilic and lipophilic drugs, improved drug stability, less prepare costs in comparison to polymeric or surfactant based carriers and ease of scale‐up and sterilisation. NLCs have been previously employed for the improvement of the intranasal delivery of valproic acid [9], basic fibroblast growth factor [10], asenapine [11], rivastigmine [12] and venlafaxine [13]. The major drawback of NLCs dispersion for intranasal administration is the short residence time in the nasal cavity due to the rapid mucociliary clearance of the nasal epithelia. To overcome this limitation, different strategies including the incorporation of NLCs in a mucoadhesive in‐situ gel have been proposed.
Temperature sensitive in‐situ forming hydrogels are polymeric low viscosity aqueous solutions at room temperature which form a gel network upon in‐vivo administration as they reach body temperature, increasing the residence time of the drug in contact with the nasal mucosa. So far, several thermosensitive in‐situ gelling formulations have been developed to prolong the nasal residence time and increase the bioavailability of some drugs such as insulin [14], curcumin [15] and phenylephrine hydrochloride [16].
Chitosan, a natural polysaccharide made of glucosamine and N‐acetyl glucosamine, was selected to prepare the thermosensitive gel formulation in the present study. It shows thermoresponsive behaviour in the presence of polyol salts, with excellent properties such as biocompatibility, biodegradability, non‐toxicity, non‐immunogenicity and good nasal tolerance [14, 17, 18]. Chitosan‐based thermosensitive hydrogels were previously employed in nasal delivery of various therapeutic agents including insulin [14], doxepin [17], ibuprofen [19] and exenatide [20].
In the present study, an in‐situ gelling system based on chitosan (CS) and β‐glycerophosphate (β‐GP) containing LZM loaded NLCs was prepared to promote the delivery of the drug into brain after nasal administration. A full factorial design was, accordingly, employed to optimise the effect of different formulation variables on the properties of NLCs containing LZM, as prepared by the emulsification solvent diffusion and evaporation method. The preferred formulation was incorporated into a thermosensitive in‐situ gelling system and its performance was evaluated after intranasal administration to the rats.
2 Methods and materials
2.1 Materials
LZM was kindly received from Alborz Darou Pharmaceutical Co (Iran). Glycerol monostearate (GMS), oleic acid and Tween 80 were obtained from Merck (Germany). Low molecular weight chitosan, pentylenetetrazole (PTZ), pluronic F127 (PF127), β‐glycerol phosphate disodium salt pentahydrate (β‐GP) and dialysis bag (molecular cutoff 12000 Da) were obtained from Sigma (USA).
2.2 Preparation of LZM loaded NLCs (LZM‐NLCs)
In the production of LZM‐NLCs three different variables (independent factors), including lipid/drug ratio (w/w), surfactant concentration (%) and oil content (%) were studied each at two levels by a full factorial design. The variables and the level of independent variables used to prepare LZM‐NLCs formulations are shown in Table 1. The experimental factors and factor levels were chosen on the basis of our preliminary studies and literature review. Eight formulations of LZM‐NLCs were designed using Design‐Expert® software (version 10, USA). The composition of different studied LZM‐NLCs is shown in Table 2. The studied dependent factors were encapsulation efficiency (EE) %, drug loading (DL) %, particle size, polydispersity index (PdI), zeta potential and the drug release efficiency during 10 h (RE10 %). Design‐Expert® software was used for the optimisation and evaluation of the statistical experimental design. Analysis of variance (ANOVA) was performed to conclude the significance of the factor and their interaction.
Table 1.
Independent variables and their level in full factorial design
| Studied variable | Level 1 | Level 2 |
|---|---|---|
| lipid/drug ratio (w/w) | 10 | 5 |
| surfactant concentration, % | 0.5 | 0.25 |
| oil content, % | 10 | 30 |
Table 2.
Composition of different studied LZM‐NLCs designed by full‐factorial experimental design
| Formulations | Lipid/drug ratio (w/w) | Surfactant concentration, % | Oil content, % |
|---|---|---|---|
| L10 S0.5 O10 | 10 | 0.5 | 10 |
| L5 S0.5 O10 | 5 | 0.5 | 10 |
| L5 S0.25 O10 | 5 | 0.25 | 10 |
| L10 S0.25 O10 | 10 | 0.25 | 10 |
| L10 S0.25 O30 | 10 | 0.25 | 30 |
| L10 S0.5 O30 | 10 | 0.5 | 30 |
| L5 S0.25 O30 | 5 | 0.25 | 30 |
| L5 S0.5 O30 | 5 | 0.5 | 30 |
L: lipid/drug ratio, S: surfactant concentration,O: oil content.
The LZM‐NLCs and blank‐NLCs were prepared by emulsification solvent diffusion and evaporation method as previously described [21, 22]. About 100 mg mixture of various amounts of oleic acid (as liquid lipid) and GMS (as solid lipid) and different quantities of LZM (10 or 20 mg) were completely dissolved into the 3 ml of acetone. The organic phase was heated at 50°C and then injected dropwise into the 30 ml of hot aqueous phase (50°C) containing different concentration of PF127 (0.25 or 0.5% w/v) as emulsifier under magnetic stirring (800 rpm). The obtained coarse emulsion was further sonicated using a probe sonicator (Bandelin, Germany) for 2 min at 50 W. Thereafter, the prepared emulsion was stirred for 2 h at room temperature to evaporate the acetone.
2.3 Particle size, PdI and zeta potential
Particle size, PdI and zeta potential were measured using Malvern nanosizer (ZEN3600, Malvern Instruments Ltd, UK) at 25°C after diluting with double distilled water. The analysis was performed as triplicate and the average values were taken.
2.4 Scanning electron microscopy (SEM)
The surface morphology of the prepared LZM‐NLCs was analysed by SEM (Quanta‐200, USA). A droplet of the formulation was placed on a carbon‐film‐coated 200 mesh copper grid and coated with thin layer of gold under argon atmosphere prior to inspection by electron microscope.
2.5 Determination of EE% and DL%
To determine EE% and DL% of LZM‐NLCs, 0.5 ml of each sample was centrifuged (Microcentrifuge Sigma 30 k, UK) at 14,000 rpm for 15 min using Amicon microcentrifugation tubes (cutoff 10,000 Da, Ireland). Then free drug which was removed from the bottom of the microcentrifugation tube was analysed by UV‐visible spectrophotometry (UV1200, Shimadzu, Tokyo, Japan) at 260 nm. The LZM EE% and DL% of the NLCs were calculated using equations 1 and 2
| (1) |
| (2) |
where total amount of lipid is blend of liquid and solid lipids.
To eliminate interference with oil and surfactant, the same procedure was done for drug free NLCs as blank for spectroscopy.
2.6 In‐vitro release study of LZM‐NLCs
The in‐vitro release study was carried out using dialysis membrane method. About 1 ml of each formulation was placed in dialysis bag, tied and immersed in a phosphate buffer solution (pH = 7.4) containing 0.1% Tween 80 at 37°C. The release medium was maintained at 37°C and stirred at 800 rpm. At predetermined time intervals, 1 ml of release medium was removed and replaced with fresh medium. Drug concentration in release medium was determined using UV‐visible spectrophotometry at 254 nm. To compare drug release profiles from NLCs, the RE10 % was determined using the following equation:
| (3) |
where yt ·dt is area under the release curve up to the time, t and y 100t is the area of the rectangle described by 100% release at the same time
2.7 Preparation of chitosan/β‐GP in‐situ hydrogel containing LZM‐NLCs (LZM‐NLCs‐Gel)
About 200 mg of chitosan powder and different amount of β‐GP (500–1500 mg) were dissolved in 8 ml aqueous hydrochloric acid (0.1 M) and 2 ml of distilled water, respectively. The β‐GP solution was slowly added to the chitosan solution placed in an ice‐water bath at 4°C under stirring. The final solution contained 2% (w/v) chitosan and 5–15% (w/v) β‐GP and maintained at 4°C for further studies. For preparation of LZM‐NLCs‐Gel (0.2% w/v), an accurate amount of freeze dried LZM‐NLCs containing 20 mg LZM was added to 10 cc of chitosan/β‐GP solution under stirring at 4°C. The gelation time of resultant LZM‐NLCs‐Gels was determined at 37°C by the test tube inverting method every 30 s. The pH values of all prepared formulations were determined using a pH meter [23, 24]. The free LZM loaded hydrogel (0.2% w/v) was prepared by the addition of an ethanolic solution of LZM to chitosan/β‐GP solution under stirring at 4°C.
2.8 Particle size and PdI of the LZM‐NLCs‐Gel
The particle size and PdI of the LZM‐NLCs after incorporation in 2% chitosan and 15% β‐GP were measured using Malvern nanosizer.
2.9 Viscosity measurement
Sol‐to‐gel behaviour of LZM‐NLCs‐Gels containing 2% chitosan and 15% β‐GP was investigated by measuring the viscosity of the sample as a function of time using a digital rotary viscometer (RVDV‐III U, Daiki Sciences Co. USA) at 37°C [23].
2.10 In‐vitro release study of LZM‐NLCs‐Gel
The in‐vitro drug release from LZM solution, gel containing free LZM (LZM‐Gel), LZM‐NLCs‐Gel was determined using dialysis bag membrane [25] as described in Section 2.6.
2.11 PTZ induced seizures in rats
All procedures were conducted according to the protocol for animal use approved by the Care and Use of Laboratory Animals at Isfahan University of Medical Sciences. Twenty‐four male Wistar rats (200 ± 20 g) were divided into four groups each consisted of six animals. Group I was intranasally administered with chitosan/β‐GP in‐situ hydrogel as a control, group II was intranasally received in‐situ gel containing LZM (1 mg/kg), group III was intranasally dosed with in‐situ gel containing LZM‐NLCs (1 mg/kg) and group IV was received intraperitoneal LZM (1 mg/kg). All animals underwent a preliminary screening test for checking the response to PTZ‐induced seizure and those showed positive response were included for experimental seizure induction at least 72 h after preliminary test. The animals were free to take standard food and water except during the test and all of the ethical regulations were taking into account both in keeping and working with animals. To induce epilepsy, 60 mg/kg PTZ was injected intraperitoneally and half an hour later the candidate drug was instilled within each of nostrils (50 µl) by polyethylene tube attached to the top of a syringe
3 Results and discussion
There are several methods for the preparation of NLCs, such as high pressure homogenisation, microemulsion, emulsification solvent diffusion and evaporation method, phase inversion, multiple emulsion, ultrasonication and membrane contractor technique [26, 27]. In the present study, the emulsification solvent diffusion and evaporation method was selected to prepare LZM‐NLCs due to several advantages including simplicity and the less requirements of the instrument [27]. To obtain uniform and small NLCs, the organic solution was added to the aqueous surfactant solution at the same temperature of 50°C. According to Hu et al. [28], particle size growth occurs at the lower temperature of dispersion medium due to the lower diffusion rate of the organic dispersion. The full factorial design was then used to optimise the level of different variables influencing the physicochemical properties of NLCs, such as lipid‐to‐drug ratio, the concentration of surfactant and the liquid oil content. The prepared LZM‐NLCs were characterised for particle size, PdI, zeta potential, EE%, DL% and RE%. The results are shown in Table 3.
Table 3.
Physical properties of different prepared LZM‐NLCs (mean ± SD, n = 3)
| Formulations | Encapsulation efficiency% | Drug loading% | Particle size, nm | PdI | Zeta potential, mV | Release efficiency% |
|---|---|---|---|---|---|---|
| L10 S0.5 O10 | 86.97 ± 1.94 | 8.00 ± 0.16 | 389.00 ± 13.73 | 0.43 ± 0.04 | −8.84 ± 1.64 | 44.22 ± 0.995 |
| L5 S0.5 O10 | 93.64 ± 0.98 | 15.78 ± 0.13 | 356.93 ± 53.05 | 0.49 ± 0.10 | −12.40 ± 0.85 | 64.86 ± 4.79 |
| L5 S0.25 O10 | 92.64 ± 0.53 | 15.63 ± 0.07 | 289.16 ± 35.85 | 0.53 ± 0.02 | −10.93 ± 1.95 | 40.68 ± 7.71 |
| L10 S0.25 O10 | 95.02 ± 1.96 | 8.67 ± 0.16 | 144.33 ± 3.02 | 0.30 ± 0.03 | −4.34 ± 0.43 | 57.18 ± 3.94 |
| L10 S0.25 O30 | 81.80 ± 5.04 | 7.56 ± 0.43 | 71.70 ± 5.16 | 0.21 ± 0.02 | −20.06 ± 2.70 | 36.50 ± 7.41 |
| L10 S0.5 O30 | 89.14 ± 4.47 | 8.18 ± 0.37 | 120.20 ± 8.76 | 0.60 ± 0.02 | −2.75 ± 1.62 | 38.72 ± 6.72 |
| L5 S0.25 O30 | 94.72 ± 2.57 | 15.92 ± 0.36 | 155.86 ± 12.10 | 0.41 ± 0.01 | −11.24 ± 2.14 | 44.88 ± 1.22 |
| L5 S0.5 O30 | 95.40 ± 0.96 | 16.02 ± 0.13 | 131.93 ± 13.07 | 0.45 ± 0.00 | −9.13 ± 1.69 | 45.31 ± 10.06 |
3.1 Particle size and PdI of LZM‐NLCs
As shown in Table 3, the particle size of LZM‐NLCs was in the range of 71.70–389.00 nm and PdI values changed from 0.21 to 0.60. The PdI is an important parameter showing the homogeneity of nanosuspension, ranging from 0 to 1. The high values of PdI represent a broad size distribution.
Analysis of the data showed that the most prominent parameter affecting the NLCs particle size was oil content (Fig. 1); by changing the oil percent from 10 to 30, the particle size was significantly decreased (P < 0.05). This observation was in agreement with the previous studies [29, 30] revealing that an increase in the oil content of the nanostructured system decreased the particle size of the NLCs effectively.
Fig. 1.
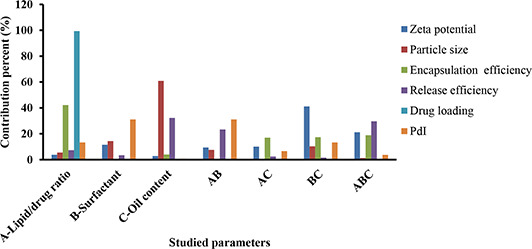
Contribution percent of different studied parameters and their interactions on particle size, PdI, zeta potential, RE, DL and EE of LZM‐NLC
An increment of the oil content led to a decrease in the viscosity inside NLCs, consequently reducing the surface tension; this, in turn, led to a reduction of the NLCs particle size. Furthermore, the ANOVA test showed that surfactant concentration and the interactions of oil content with the surfactant concentration had a considerable effect on the particle size of NLCs. It could be concluded from the ANOVA results that surfactant concentration and the interaction between surfactant concentration and lipid‐to‐drug ratio or oil content influenced the PdI values. An increase in the surfactant concentration in the external phase corresponded to an increase in PdI and particle size. Since at the high concentration of the surfactant, the possibility of the compression of the surfactant molecules at the particles surface with the formation of loops and tails was increased, bridging occurred between the primary nanoparticles, leading to the formation of larger particles with less uniformity [31]. This finding may also be related to the influence of the surfactant concentration on the mixing rate of two phases upon NLCs preparation [32]. By increasing the surfactant, a reduction of the mixing rate could give rise to the formation of larger and less uniform NLCs.
The higher drug content in the matrix of nanoparticles (although not significant) increased particle size and PdI of nanoparticles, possibly due to the higher viscosity of the molten oil phase since the melting point of the drug was higher than the mixing temperature [33]. Similarly, Fathi et al. [34] and Liu et al. [35] showed that the particle size of hesperetin‐NLC and lutein‐NLC was enlarged by increasing the drug content of NLCs.
3.2 Zeta potential of LZM‐NLCs
Zeta potential, a useful tool to predict the physical stability of nanoparticles, indicates the degree of the repulsive forces existing between the particles in the dispersion [36]. The zeta potential values of the prepared NLC formulations were in the range of −2.75 to −20.06 mV. As a guideline, a zeta potential of at least ±30 mV for electrostatically stabilised system is desired to obtain a physically stable suspension. However, in electrostatic and sterically stabilised system, even lower zeta potentials are sufficient to achieve stable suspension [37, 38]. Even though, the obtained zeta potential was not high enough for a sufficient electrostatic stabilisation. However, PF127 can provide additional steric stabilisation to retain stability of NLCs. Similar findings was reported by other researchers when non‐ionic surfactant was used as stabiliser [37, 38, 39]. In the study by Sis and Birinci [40], dispersion of carbon black powders in aqueous medium was investigated by using anionic surfactant and a series of non‐ionic surfactants. They demonstrated that non‐ionic surfactants could better stabilise nanoparticles in aqueous medium than anionic surfactants possibly due to the molecular structure and bulk properties of the surfactants.
Fig. 1 shows that the surfactant concentration in the interaction with the oil content had the most impact on the zeta potential of LZM‐NLCs. According to the results obtained from the experiments for zeta potential, there was no significant change in zeta potential as a function of the factors studied.
3.3 EE% and DL% of LZM‐NLCs
The EE of LZM‐NLCs varied from 81.80 to 95.02%, while DL% was in the range of 7.56–16.02%. Analysis of data revealed that the lipid‐to‐drug ratio was the most effective factor on EE and DL% of the prepared NLCs (Fig. 1); as such, increasing drug content in the formulation led to a significant increase in EE and DL% (P < 0.05). ANOVA test also showed the significant effect of the oil content in interaction with lipid to drug ratio and the oil content in interaction with surfactant concentration on the EE of LZM‐NLCs.
3.4 In‐vitro release study of LZM‐NLCs
The in‐vitro release profiles of LZM from NLCs are shown in Fig. 2. For all formulations, a drug release pattern was biphasic, which could be characterised by an initial stage burst release due to drug‐enriched shell around the particles [2, 41]; this was followed by a sustained release period. The biphasic drug release could be supportive to deliver a loading dose for the faster onset of action at the initial stage, while subsequently sustaining the release of drug could be guaranteed for a long period of time [42].
Fig. 2.
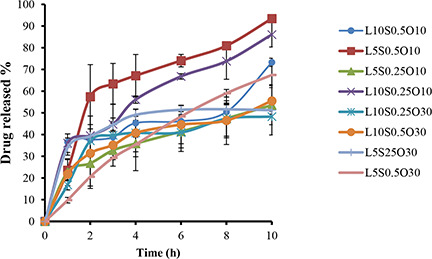
In‐vitro release profiles of LZM from NLCs (mean ± SD, n = 3)
To compare the drug release profiles from NLCs, RE10 % was calculated. RE10 % was found to be in a range from 36.50 to 64.86%. As shown in Fig. 1, RE10 % was mostly affected by the oil content. Lipid‐to‐drug ratio in interaction with the surfactant concentration also had a significant effect on RE10 %. Increasing the oil content from 10 to 30% led to a reduction in RE10 %. However, while increasing the oil content had no significant effect on EE, a more homogeneous entrapment of LZM throughout the systems could result in the decline of RE [29]. Similar results were obtained by Sharif Makhmal Zadeh et al. [43], who proved that increasing the oil content resulted in the decrease of the drug release rate. Conversely, the increment of the surfactant concentration (though not significant) in the formulation led to the increase of RE, possibly due to the increased drug solubility in the water phase [44]. This finding was in accordance with the results reported by Rahman et al. [41]. Lipid‐to‐drug weight ratio also had a negligible effect on RE10 % (p > 0.05).
3.5 Optimisation
Five dependent variables were optimised using Design‐Expert® software. Optimisation was carried out to obtain the levels of each variable, which maximised the absolute value of the zeta potential, while minimising the particle size, PdI, and targeting EE%, DL% and RE10 % in the range of the obtained data. Based on the obtained results, LZM‐NLC coded L10 S0.25 O30 fulfilled the requirements of optimisation by the desirability of 0.997. L10 S0.25 O30 was prepared using 10 mg of LZM, 70 mg of GMS, 30 mg oleic acid when PF127 concentration in aqueous phase was 0.25%. The optimised formulation had a particle size of 71.70 ± 5.16 nm, the PdI of 0.21 ± 0.02, the EE of 81.80 ± 5.04%, the DL% of 7.56 ± 0.43, the zeta potential of −20.06 ± 2.70 mV and the release efficiency of 36.50 ± 7.41%. As can be seen by SEM images (Fig. 3), the optimised LZM‐NLCs were monodispersed spherical particles with a narrow size distribution. To prevent the aggregation and instability of nanoparticles during storage, the optimised nanosuspensions of LZM‐NLCs were freeze‐dried. The freeze‐dried LZM‐NLCs formed a solid residue easily redispersed in water by gentle hand agitation; they had an approximately similar particle size (72.93 ± 8.92 nm) and zeta potential as LZM‐NLCs did (−16.35 ± 4.45 mV).
Fig. 3.

SEM image of optimised LZM‐NLCs
3.6 Characteristics of LZM‐NLCs‐Gel
Chitosan is soluble in the pH value below its pK a (pH < 6.2) due to the charge repulsion caused by the protonation of its free amine groups. By simply adding an alkali to raise the pH of the chitosan solution (above 6.2), a gel‐like structure could be obtained. When the chitosan solution is neutralised by β‐GP, the system remains in solution, even at neutral pH values within a physiologically acceptable range (pH 6.8 to 7.2). This could be due to the protective hydration of the chitosan chain upon the addition of β‐GP at room temperature. However, the system could be solidified into a gel as temperature is increased to body temperature due to more chitosan–chitosan hydrophobic and hydrogen interactions [45, 46]. Characteristics of thermosensitive chitosan/β‐GP solution containing LZM‐NLCs are presented in Table 4. As illustrated, the time of gelation at body temperature (37°C) was obviously shortened when the β‐GP concentration was increased to an optimal amount of 15% w/v. This finding has been described by Ghasemi Tahrir et al. [24] and Deng et al. [47]; it could be due to the fact that β‐GP decreases the apparent charge density of chitosan and establishes more hydrophobic interactions between the polymer chains. The gelation time of the prepared hydrogel containing 15% β‐GP occurred over 5 min, which was well below the time for the mucociliary clearance (Table 4). Fig. 4 reveals that the CS/β‐GP hydrogel could undergo the sol–gel transition into non‐flowing opaque hydrogel.
Table 4.
Characteristics of thermosensitive chitosan/β‐GP solution containing LZM‐NLCs (mean ± SD, n = 3)
| Samples | Chitosan (%W/V) | β‐GP (%W/V) | Gelation time at 37°C, min | pH |
|---|---|---|---|---|
| 1 | 2 | 5 | 46.66 ± 2.89 | 6.34 ± 0.06 |
| 2 | 2 | 10 | 33.33 ± 2.89 | 6.8 ± 0.06 |
| 3 | 2 | 15 | 5.33 ± 0.58 | 7.12 ± 0.03 |
Fig. 4.

Overview of
(a) LZM‐NLCs‐gel, (b) LZM‐NLCs‐solution
The viscosity of LZM‐NLCs‐Gel (2% chitosan/15% β‐GP) at 37°C as a function of time is shown in Fig. 5. The increase in the viscosity of the solution over the time was indicative of gel formation.
Fig. 5.
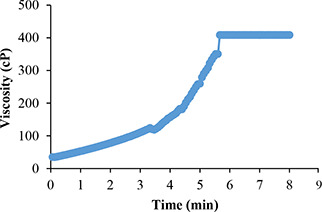
Viscosity as a function of time at 37°C for LZM‐NLCs‐Gel
The particle size and PdI of LZM‐NLCs after dispersing in hydrogel (2% chitosan/15% β‐GP) were about 165.46 ± 9.25 nm and 0.3 ± 0.03, respectively. This increase in particle size could be related to adsorption of sticky chitosan on NLCs surface, which was in accordance with Hao et al. study [48], who showed an increase in particle size after incorporation of Resina Draconis‐loaded SLNs in poloxamer‐based hydrogels.
3.7 In‐vitro release of LZM‐NLCs‐Gel
The release profiles of the LZM solution (free LZM), LZM‐Gel and LZM‐NLCs‐Gel are shown in Fig. 6. The drug release rate from LZM‐Gel was reduced, as compared to the free LZM, mainly due to the obstruction effect of chitosan‐β‐GP hydrogel. Incorporation of LZM‐NLCs in the gel resulted in the further reduction of the release rate, as compared with LZM‐Gel, thereby indicating that NLCs had a remarkable effect on extending the LZM release. In the gel containing NLCs, the drug molecules must be released out of NLCs at first; then they are diffused through and out of the gel matrix. A similar pattern of drug release has been observed by other researchers [18, 49].
Fig. 6.
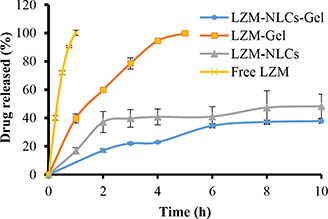
Release profiles of free LZM, LZM‐NLCs, LZM‐Gel and LZM‐NLCs‐Gel (mean ± SD, n = 3)
3.8 In‐vivo efficacy study
As shown in Table 5, in the control (gel) group, all six rats showed epileptic seizure after 2 min of PTZ injection. The symptoms were elapsed for 5 min in one rat, 10 min in two rats and 20 min in three ones in this group. Stretching movements and permanent contractions in trunk, which were spread to the hands and feet of the rats, were also evident. Salivation occurred in two rats. In the group receiving LZM‐NLCs‐Gel, muscle contractions in the hands and feet were less frequent and weaker in comparison to the control group and lasted 1 min in one‐third (33%) of the rats. In the group treated with LZM‐Gel, movements occurred in the hands of two rats, while in two other rats, movements were in trunk and feet, while two rats had no symptoms of epileptic seizure. In the group receiving LZM intraperitoneally, only one rat represented movements in trunk that lasted for 30 s.
Table 5.
Comparative anti‐convulsant activity following intranasal administration of Gel, LZM‐Gel and LZM‐NLCs‐Gel and intraperitoneal administration of LZM in rats (mean ± SD, n = 6)
| Groups | Route of administration | Prevalence of epileptic seizure, % | Lag time of incidence, min | Severity of symptoms in trunk (0‐3) | Severity of symptoms in hands and feet (0‐3) | Duration of symptoms, min |
|---|---|---|---|---|---|---|
| Gel (control) | intranasal | 100 | (1.2–2.2) 1.8 | 3 | 3 | (5–20) 15.2 |
| LZM‐Gel | intranasal | 66a | (10–12.6) 11.3 | 1 | 2 | (0.9–1.3) 1.1 |
| LZM‐NLCs‐Gel | intranasal | 33a | (12.9–17.3) 15.1 | 0 | 1 | (0.5–1) 0.75 |
| IP‐LZM | intraperitoneal | 16.5a , b | 14.8 | 1 | 0 | 0.5(0.5) |
One‐way ANOVA has been used to detect epileptic seizure prevalence. Number of animals per group is 6.
a There was a significant difference (P < 0.001) compared to the control group.
b There was a significant difference (P < 0.01) compared to LZM‐Gel and LZM‐NLC‐Gel groups.
The prevalence of epileptic seizure in the groups treated with LZM‐Gel, LZM‐NLCs‐Gel and intraperitoneal LZM was significantly lower than that of the control group. As can be seen in Table 5, administration of LZM‐NLCs‐Gel increased the lag time of seizure incidence more than other groups. The prevalence of epileptic seizure and severity of symptoms in trunks, hands and feet, as well as the duration of symptoms, were considerably reduced by using LZM‐NLCs‐Gel, as compared with Gel‐LZM. The obtained results, therefore, suggested that the incorporation of NLCs into in‐situ gels could efficiently increase the LZM therapeutic efficacy in the treatment of epilepsy. This could be due to the tiny size of NLCs, which permitted LZM to be transported transcellularly through olfactory neurones to the brain via various endocytic pathways of neuronal cells found in the olfactory membrane [50]. Consistent with our findings, Eskandari et al. [9] have reported that NLCs containing valproic acid were more effective than free drugs after treatment via the intranasal route due to the higher brain/plasma concentration ratio. Intranasal administration of lamotrigine NLCs also showed a significant improvement in latency and duration in the tonic hind limb extension, indicating the increased nose‐to‐brain drug delivery via nanoformulations, as compared to the drug solution of an equivalent dose. Several studies revealed that NLCs [13, 50, 51] and chitosan in‐situ gel [14] were safe for nasal administration. Despite this, further pharmacokinetic and histopathological studies need to be carried out to determine its potential applications and safety in the treatment of epilepsy.
4 Conclusion
In the current study, we evaluated the potential application of a thermosensitive gel loaded with LZM‐NLCs as a nasal drug delivery system for the treatment of epilepsy. LZM‐NLCs were successfully prepared via the emulsification solvent diffusion and evaporation method and then optimised using the full‐factorial experimental design. The oil content was found to be the most effective parameter on the size of the particles and in‐vitro drug release efficiency, while EE and zeta potential were more affected by the lipid/drug ratio and the surfactant concentration in the interaction with the oil content, respectively. The optimum condition suggested for the production of NLCs included the lipid‐to‐drug ratio of 10, the surfactant concentration of 0.25% and the liquid oil content of 30%. The resultant NLCs were dispersed into a thermosensitive hydrogel composed of chitosan and β‐GP. The optimum gel formulation consisted of 2% chitosan and 15% β‐GP turned into the gel in 5.33 ± 0.58 min at the physiological temperature. The in‐vivo study in PTZ induced convulsion model illustrated the potential of LZM‐NLCs‐Gel for the treatment of epilepsy as this formulation inhibited either the occurrence of seizure or diminished the seizure intensity.
5 Acknowledgments
The authors thank the Research Vice Chancellery of Isfahan University of Medical Sciences for supporting this work.
6 References
- 1. Manno E.M.: ‘Status epilepticus: current treatment strategies’, Neurohospitalist., 2011, 1, (1), pp. 23 –31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abdelbary G. Fahmy R.H.: ‘Diazepam‐loaded solid lipid nanoparticles: design and characterization’, AAPS Pharm. Sci. Tech., 2009, 10, (1), pp. 211 –219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nour S.A. Abdelmalak N.S. Naguib M.J. et al.: ‘Intranasal brain‐targeted clonazepam polymeric micelles for immediate control of status epilepticus: in vitro optimization, ex vivo determination of cytotoxicity, in vivo biodistribution and pharmacodynamics studies’, Drug. Deliv., 2016, 23, (9), pp. 3681 –3695 [DOI] [PubMed] [Google Scholar]
- 4. Pardeshi C.V. Belgamwar V.S.: ‘N, N, N‐trimethyl chitosan modified flaxseed oil based mucoadhesive neuronanoemulsions for direct nose to brain drug delivery’, Int. J. Biol., 2018, 120, pp. 2560 –2571 [DOI] [PubMed] [Google Scholar]
- 5. Hao J. Zhao J. Zhang S. et al.: ‘Fabrication of an ionic‐sensitive in situ gel loaded with resveratrol nanosuspensions intended for direct nose‐to‐brain delivery’, Colloids Surf. B. Biointerfaces, 2016, 147, pp. 376 –386 [DOI] [PubMed] [Google Scholar]
- 6. Mittal D. Md S. Hasan Q. et al.: ‘Brain targeted nanoparticulate drug delivery system of rasagiline via intranasal route’, Drug Deliv., 2016, 23, (1), pp. 130 –139 [DOI] [PubMed] [Google Scholar]
- 7. Jafarieh O. Md S. Ali M. et al.: ‘Design, characterization, and evaluation of intranasal delivery of ropinirole‐loaded mucoadhesive nanoparticles for brain targeting’, Drug Dev. Ind. Pharm., 2015, 41, (10), pp. 1674 –1681 [DOI] [PubMed] [Google Scholar]
- 8. Wermeling D.P.: ‘Intranasal delivery of antiepileptic medications for treatment of seizures’, Neurotherapeutics, 2009, 6, (2), pp. 352 –358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eskandari S. Varshosaz J. Minaiyan M. et al.: ‘Brain delivery of valproic acid via intranasal administration of nanostructured lipid carriers: in vivo pharmacodynamic studies using rat electroshock model’, Int. J. Nanomed., 2011, 6, p. 363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao Y.‐Z. Li X. Lu C.‐T. et al.: ‘Gelatin nanostructured lipid carriers‐mediated intranasal delivery of basic fibroblast growth factor enhances functional recovery in hemiparkinsonian rats’, Nanomedicine, 2014, 10, (4), pp. 755 –764 [DOI] [PubMed] [Google Scholar]
- 11. Singh S.K. Dadhania P. Vuddanda P.R. et al.: ‘Intranasal delivery of asenapine loaded nanostructured lipid carriers: formulation, characterization, pharmacokinetic and behavioural assessment’, RSC Adv., 2016, 6, (3), pp. 2032 –2045 [Google Scholar]
- 12. Wavikar P.R. Vavia P.R.: ‘Rivastigmine‐loaded in situ gelling nanostructured lipid carriers for nose to brain delivery’, J. Liposome Res., 2015, 25, (2), pp. 141 –149 [DOI] [PubMed] [Google Scholar]
- 13. Shah B. Khunt D. Bhatt H. et al.: ‘Intranasal delivery of venlafaxine loaded nanostructured lipid carrier: risk assessment and QbD based optimization’, J. Drug Deliv. Sci. Technol., 2016, 33, pp. 37 –50 [Google Scholar]
- 14. Wu J. Wei W. Wang L.‐Y. et al.: ‘A thermosensitive hydrogel based on quaternized chitosan and poly (ethylene glycol) for nasal drug delivery system’, Biomaterials, 2007, 28, (13), pp. 2220 –2232 [DOI] [PubMed] [Google Scholar]
- 15. Chen X. Zhi F. Jia X. et al.: ‘Enhanced brain targeting of curcumin by intranasal administration of a thermosensitive poloxamer hydrogel’, J. Pharm. Pharmacol., 2013, 65, (6), pp. 807 –816 [DOI] [PubMed] [Google Scholar]
- 16. Xu X. Shen Y. Wang W. et al.: ‘Preparation and in vitro characterization of thermosensitive and mucoadhesive hydrogels for nasal delivery of phenylephrine hydrochloride’, Eur. J. Pharm. Biopharm., 2014, 88, (3), pp. 998 –1004 [DOI] [PubMed] [Google Scholar]
- 17. Naik A. Nair H.: ‘Formulation and evaluation of thermosensitive biogels for nose to brain delivery of doxepin’, BioMed. Res. Int., 2014, 2014 pp. 1 –10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peng Y. Li J. Li J. et al.: ‘Optimization of thermosensitive chitosan hydrogels for the sustained delivery of venlafaxine hydrochloride’, Int. J. Pharm., 2013, 441, (1‐2), pp. 482 –490 [DOI] [PubMed] [Google Scholar]
- 19. Gholizadeh H. Cheng S. Pozzoli M. et al.: ‘Smart thermosensitive chitosan hydrogel for nasal delivery of ibuprofen to treat neurological disorders’, Expert Opin. Drug Deliv., 2019, 16, (4), pp. 453 –466 [DOI] [PubMed] [Google Scholar]
- 20. Li Y. He J. Lyu X. et al.: ‘Chitosan‐based thermosensitive hydrogel for nasal delivery of exenatide: effect of magnesium chloride’, Int. J. Pharm., 2018, 553, (1‐2), pp. 375 –385 [DOI] [PubMed] [Google Scholar]
- 21. Taymouri S. Alem M. Varshosaz J. et al.: ‘Biotin decorated sunitinib loaded nanostructured lipid carriers for tumor targeted chemotherapy of lung cancer’, J. Drug. Deliv. Sci. Technol., 2019, 50, pp. 237 –247 [Google Scholar]
- 22. Varshosaz J. Eskandari S. Tabakhian M.: ‘Production and optimization of valproic acid nanostructured lipid carriers by the Taguchi design’, Pharm. Dev. Technol., 2010, 15, (1), pp. 89 –96 [DOI] [PubMed] [Google Scholar]
- 23. Hosny K.M.: ‘Preparation and evaluation of thermosensitive liposomal hydrogel for enhanced transcorneal permeation of ofloxacin’, AAPS PharmSciTech., 2009, 10, (4), pp. 1336 –1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ghasemi Tahrir F. Ganji F. Mani A.R. et al.: ‘In vitro and in vivo evaluation of thermosensitive chitosan hydrogel for sustained release of insulin’, Drug Deliv., 2016, 23, (3), pp. 1038 –1046 [DOI] [PubMed] [Google Scholar]
- 25. Kong X. Xu W. Zhang C. et al.: ‘Chitosan temperature‐sensitive gel loaded with drug microspheres has excellent effectiveness, biocompatibility and safety as an ophthalmic drug delivery system’, Exp. Ther. Med., 2018, 15, (2), pp. 1442 –1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khosa A. Reddi S. Saha R.N.: ‘Nanostructured lipid carriers for site‐specific drug delivery’, Biomed. Pharmacother., 2018, 103, pp. 598 –613 [DOI] [PubMed] [Google Scholar]
- 27. Li Q. Cai T. Huang Y. et al.: ‘A review of the structure, preparation, and application of NLCs, PNPs, and PLNs’, Nanomaterials (Basel), 2017, 7, (6), p. 122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu F.‐Q. Jiang S.‐P. Du Y.‐Z. et al.: ‘Preparation and characterization of stearic acid nanostructured lipid carriers by solvent diffusion method in an aqueous system’, Biointerfaces, 2005, 45, (3–4), pp. 167 –173 [DOI] [PubMed] [Google Scholar]
- 29. Sanad R.A. Abdelmalak N.S. Elbayoomy T.S. et al.: ‘Formulation of a novel oxybenzone‐loaded nanostructured lipid carriers (NLCs)’, AAPS PharmSciTech., 2010, 11, (4), pp. 1684 –1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gaba B. Fazil M. Khan S. et al.: ‘Nanostructured lipid carrier system for topical delivery of terbinafine hydrochloride’, Bull. Fac. Pharm. Cairo. Univ., 2015, 53, (2), pp. 147 –159 [Google Scholar]
- 31. Tiyaboonchai W. Tungpradit W. Plianbangchang P.: ‘Formulation and characterization of curcuminoids loaded solid lipid nanoparticles’, Int. J. Pharm., 2007, 337, (1–2), pp. 299 –306 [DOI] [PubMed] [Google Scholar]
- 32. Varshosaz J. Taymouri S. Jafari E. et al.: ‘Formulation and characterization of cellulose acetate butyrate nanoparticles loaded with nevirapine for HIV treatment’, J. Drug Deliv. Sci. Technol., 2018, 48, pp. 9 –20 [Google Scholar]
- 33. Yuan H. Wang L.‐L. Du Y.‐Z. et al.: ‘Preparation and characteristics of nanostructured lipid carriers for control‐releasing progesterone by melt‐emulsification’, Colloids Surf. B Biointerfaces., 2007, 60, (2), pp. 174 –179 [DOI] [PubMed] [Google Scholar]
- 34. Fathi M. Varshosaz J. Mohebbi M. et al.: ‘Hesperetin‐loaded solid lipid nanoparticles and nanostructure lipid carriers for food fortification: preparation, characterization, and modeling’, Food Bioproc. Tech., 2013, 6, (6), pp. 1464 –1475 [Google Scholar]
- 35. Liu C.‐H. Wu C.‐T.: ‘Optimization of nanostructured lipid carriers for lutein delivery’, Colloids Surf. A Physicochem. Eng. Asp., 2010, 353, (2‐3), pp. 149 –156 [Google Scholar]
- 36. Taymouri S. Varshosaz J. Hassanzadeh F. et al.: ‘Optimisation of processing variables effective on self‐assembly of folate targeted synpronic‐based micelles for docetaxel delivery in melanoma cells’, IET Nanobiotechnol., 2015, 9, (5), pp. 306 –313 [DOI] [PubMed] [Google Scholar]
- 37. Lim S.‐J. Kim C.‐K.: ‘Formulation parameters determining the physicochemical characteristics of solid lipid nanoparticles loaded with all‐trans retinoic acid’, Int. J. Pharm., 2002, 243, (1‐2), pp. 135 –146 [DOI] [PubMed] [Google Scholar]
- 38. Liu J. Hu W. Chen H. et al.: ‘Isotretinoin‐loaded solid lipid nanoparticles with skin targeting for topical delivery’, Int. J. Pharm., 2007, 328, (2), pp. 191 –195 [DOI] [PubMed] [Google Scholar]
- 39. Varshosaz J. Tabbakhian M. Mohammadi M.Y.: ‘Formulation and optimization of solid lipid nanoparticles of buspirone HCl for enhancement of its oral bioavailability’, J. Liposome Res., 2010, 20, (4), pp. 286 –296 [DOI] [PubMed] [Google Scholar]
- 40. Sis H. Birinci M.: ‘Effect of nonionic and ionic surfactants on zeta potential and dispersion properties of carbon black powders’, Colloids Surf. A, Physicochem. Eng. Aspects, 2009, 341, (1‐3), pp. 60 –67 [Google Scholar]
- 41. Rahman H.S. Rasedee A. How C.W. et al.: ‘Zerumbone‐loaded nanostructured lipid carriers: preparation, characterization, and antileukemic effect’, Int. J. Nanomedicine, 2013, 8, pp. 2769 –2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang W. Li X. Ye T. et al.: ‘Design, characterization, and in vitro cellular inhibition and uptake of optimized genistein‐loaded NLC for the prevention of posterior capsular opacification using response surface methodology’, Int. J. Pharm., 2013, 454, (1), pp. 354 –366 [DOI] [PubMed] [Google Scholar]
- 43. Sharif Makhmal Zadeh B. Niro H. Rahim F. et al.: ‘Ocular delivery system for propranolol hydrochloride based on nanostructured lipid carrier’, Sci. Pharm., 2018, 86, (2), p. 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Abdel‐Mottaleb M.M. Neumann D. Lamprecht A.: ‘In vitro drug release mechanism from lipid nanocapsules (LNC)’, Int. J. Pharm., 2010, 390, (2), pp. 208 –213 [DOI] [PubMed] [Google Scholar]
- 45. Ruel‐Gariepy E. Chenite A. Chaput C. et al.: ‘Characterization of thermosensitive chitosan gels for the sustained delivery of drugs’, Int. J. Pharm., 2000, 203, (1‐2), pp. 89 –98 [DOI] [PubMed] [Google Scholar]
- 46. Zhou H.Y. Jiang L.J. Cao P.P. et al.: ‘Glycerophosphate‐based chitosan thermosensitive hydrogels and their biomedical applications’, Carbohydr. Polym., 2015, 117, pp. 524 –536 [DOI] [PubMed] [Google Scholar]
- 47. Deng A. Kang X. Zhang J. et al.: ‘Enhanced gelation of chitosan/beta‐sodium glycerophosphate thermosensitive hydrogel with sodium bicarbonate and biocompatibility evaluated’, Mater. Sci. Eng. C. Mater. Biol. Appl., 2017, 78, pp. 1147 –1154 [DOI] [PubMed] [Google Scholar]
- 48. Hao J. Wang X. Bi Y. et al.: ‘Fabrication of a composite system combining solid lipid nanoparticles and thermosensitive hydrogel for challenging ophthalmic drug delivery’, Colloids Surf. B, Biointerfaces, 2014, 114, pp. 111 –120 [DOI] [PubMed] [Google Scholar]
- 49. Varshosaz J. Taymouri S. Minaiyan M. et al.: ‘Development and in vitro/in vivo evaluation of HPMC/chitosan gel containing simvastatin loaded self‐assembled nanomicelles as a potent wound healing agent’, Drug. Dev. Ind. Pharm., 2018, 44, (2), pp. 276 –288 [DOI] [PubMed] [Google Scholar]
- 50. Devkar T.B. Tekade A.R. Khandelwal K.R.: ‘Surface engineered nanostructured lipid carriers for efficient nose to brain delivery of ondansetron HCl using Delonix regia gum as a natural mucoadhesive polymer’, Colloids. Surf. B. Biointerfaces, 2014, 122, pp. 143 –150 [DOI] [PubMed] [Google Scholar]
- 51. Madane R.G. Mahajan H.S.: ‘Curcumin‐loaded nanostructured lipid carriers (NLCs) for nasal administration: design, characterization, and in vivo study’, Drug Deliv., 2016, 23, (4), pp. 1326 –1334 [DOI] [PubMed] [Google Scholar]