

ABSTRACT
Epitranscriptomic modifications of stable RNAs are dynamically regulated and specific profiles of 2ʹ-O-methylation in rRNA have been associated with distinct cancer types. However, these observations pointed out the existence of at least two distinct groups: a rather large group with constitutive rRNA Nm residues exhibiting a stable level of methylation and a more restricted set of variable modifications, giving rise to the concept of ‘specialized ribosomes’. These heterogeneous ribosomes can modulate their translational properties and be key regulatory players, depending on the physiological state of the cell. However, these conclusions were drawn from a limited set of explored human cell lines or tissues, mostly related to cancer cells of the same type. Here, we report a comprehensive analysis of human rRNA Nm modification variability observed for >15 human cell lines grown in different media and conditions. Our data demonstrate that human Nm sites can be classified into four groups, depending on their observed variability. About ⅓ of rRNA 2ʹ-O-methylations are almost invariably modified at the same level in all tested samples (stable modifications), the second group of relatively invariant modifications (another ½ of the total) showing a slightly higher variance (low variable group) and two variable groups, showing an important heterogeneity. Mapping of these four classes on the human ribosome 3D structure shows that stably modified positions are preferentially located in the important ribosome functional sites, while variable and highly variable residues are mostly distributed to the ribosome periphery. Possible relationships of such stable and variable modifications to the ribosome functions are discussed.
KEYWORDS: Deep sequencing, human rRNA, epitranscriptome, regulation, variable and constitutive, 2ʹ-O-ribose methylation
Introduction
Eukaryotic ribosomal RNA (rRNA) contains a high number of post-transcriptional RNA modifications installed by both stand-alone protein enzymes (mostly for base modifications) and snoRNA-guided machinery (C/D- and H/ACA-box snoRNPs) for 2ʹ-O-methylations (Nm) and pseudouridine residues (ψ), respectively. The updated list for validated human rRNA modifications reports at least 226 potentially modified positions, including 12 base modifications, 110 Nm and 107 ψ (3 residues (28S-ψm3797, 18S-ψm1326 and hypermodified 18S-m1acp3ψ1248) are counted twice for both pseudouridine and base/ribose modification [1,2]. In contrast to yeast 5S rRNA, which contains one modified residue (ψ50), its human counterpart was reported to be unmodified, while other human rRNA species contain 4, 90 and 132 modified sites, for 5.8S, 18S and 28S rRNA, respectively. This list may still evolve, since potential modification target, which is undermodified (or not modified at all) in the analysed sample(s), may still escape identification. Several previously reported human rRNA modification sites [3,4] were not further confirmed by more recent studies [5–8], but one cannot formally exclude their existence in particular cell types or under specific growth/stress conditions.
This viewpoint matches well with several studies reporting variability of rRNA modifications in human cells and tissues. Both base and Nm/ψ residues seem to be dynamically regulated and to respond to external and internal stimuli, as well as to abnormal physiological state (or disease). Such dynamic variability gave rise to the concept of ‘specialized ribosomes’ which are able to re-program their translational activity as a part of an adaptive response to stress or various treatments [9–11]. Indeed, substantial, but non-lethal, depletion of Fibrillarin (Fib), the catalytic component of the C/D-box snoRNPs ensuring Nm formation in rRNA, led to a differential decrease of Nm levels at different rRNA sites, the 18S rRNA 2ʹ-O-methylations levels were relatively stable, while many 2ʹ-O-methylations present in 28S rRNA were strongly affected [6,7]. A parallel analysis was performed for HeLa and HCT116 cells and, with slight variations, concordant data have been obtained for both cell types. Fib depletion was found to reduce cap-dependent mRNA translation and increase IRES-driven translation initiation [6]. However, despite this correlation, the exact role of individual modified residues in rRNA remains obscure.
The existence of incompletely modified rRNA residues in different cell lines also opens a possible way for the regulation of ribosome properties in translation. It is noteworthy to mention that since no active de-modification mechanisms were described for both Nm and ψ residues in rRNA, the transition of incompletely modified rRNA to a modified state may be relatively rapid and essentially dependent on the cellular localization of the corresponding enzymatic machinery and the accessibility of the rRNA substrate. In contrast, in the absence of active de-modification, the decrease of the modification level can only be insured by a slow and progressive replacement of fully modified rRNA by a newly synthetized partially modified (or unmodified) counterpart.
Other evidences for modulation of rRNA modification come from the analysis of rRNA modification profiles in stress and disease (for review [12]). A wealth of reports suggests that both C/D- and H/ACA-box snoRNA levels vary in disease (mostly in different cancers, for review [13]) and these snoRNAs can even be considered as reliable biomarkers [14–16]. Despite these findings, such variations were only rarely related to trackable changes in rRNA modification level. However, the experimental evidence for such direct correlation of rRNA modification and the expression of the corresponding snoRNA has been obtained for 18S-Um116, which modulates proliferation of myeloid leukaemia cells and is SNORD42A-dependent [17]. Other cancer-related rRNA Nm are 28S-Cm2848/28S-Cm2863, catalysed by SNORD50 [18], the expression of which varies in diverse cancer types, including colon and prostate cancers, breast carcinoma and B-cell lymphoma (for review see [13]). Finally, reduced methylation levels at 18S-Cm1703 and 18S-Gm1328 (regulated by SNORD43 and SNORD32A, respectively) were associated with impaired clonogenic growth and depletion of SNORD43 resulted in a reduction of cell size and protein synthesis [19]. Re-programming of Nm modifications in rRNA was observed in hypoxia, and, as noticed for Fib depletion, is also associated with an increased IRES-dependent translation. Hypoxia was shown to induce a complex modulation of rRNA Nm content with a reduced methylation at 18S-Um1804, 28S-Am389, 28S-Gm1612, 28S-Cm2848, 28S-Gm4506, while an increased methylation was found at 28S-Am1858 and 28S-Cm4426 [20]. The phosphoprotein Nucleophosmin (NPM1) was identified to bind directly C/D-box snoRNAs and thus to be a key regulator of rRNA Nm content and translation modulator [21]. Five Nm residues (28S-Cm1327, 28S-Am3764, 28S-Cm3866, 28S-Um3904 and 28S-Gm4198) were significantly decreased upon NPM1 deficiency. In many instances, only relative measurements of Nm level were performed by semi-quantitative qPCR-based RTL-P approach [22,23], making comparison and assessment of real rRNA methylation level rather difficult and uncertain.
With the development of different versions of RiboMethSeq [8,24,25], see [23] for a review, a comprehensive profiling of variably modified rRNA Nm residues become possible, but was systematically performed only for some model human cell lines (e.g. HeLa/HCT116 cells) [5] as well as in two different cancer models: breast cancer tissues and B-cell lymphoma [26,27]. The latter study revealed pronounced hypomethylation of some rRNA Nm sites (namely 18S-Um354, 18S-Cm1440 and 28S-Gm4593). It is important to note that the list of the most variable positions seems to be cancer type-dependent.
Despite these findings related to disease or intentional depletion of the core components of the rRNA modification machinery, an intriguing area in the field is the ‘natural’ variability of human rRNA modification under normal physiological conditions.
The aim of our study was to broaden current knowledge of human Nm residues linked to their methylation status and their positioning in the 3D ribosome structure. Therefore, we investigated a compilation of RiboMethSeq data for >60 samples originating from various human cell lines cultivated under ‘standard’ lab growth conditions. Our data suggest that human Nm sites can be classified into four broad groups: stable, low variable, variable and highly variable. Since a high-resolution structure for human ribosome is now available, these groups were mapped to the ribosome 3D structure and proximity with functionally important ribosome sites was evaluated. This mapping shows that, except 28S-Am3739 located in immediate proximity of the decoding centre (DC), the most variable Nm sites are located relatively far away from the peptidyl-transferase (PTC) and decoding centres (DC) and are well accessible at the ribosome surface.
Results
RiboMethSeq profiling of Nm sites in rRNA
RiboMethSeq analysis pipeline dedicated to mapping and quantification of Nm residues in RNA is based on partial alkaline hydrolysis, followed by adapter ligation to RNA fragments, sequencing and bioinformatic analysis. Since ribose methylation protects 3ʹ-adjacent phosphodiester bond from nucleophilic attack by OH−, RNA fragments ending with 3ʹ-Nm and starting at the nucleotide N + 1 are underrepresented in the sequenced pool, creating a characteristic gap in 5ʹ/3ʹ-ends profile. The depth of the gap is proportional to the methylation level at the Nm site, allowing precise quantification. The initial version of RiboMethSeq used proprietary ligation protocol [24], but other versions of the approach were adapted for standard adapter ligation and further coupled to Illumina sequencing [8,25]. Due to ‘negative’ mode of modification detection (protection against cleavage at a given site compared to the environment) all RiboMethSeq protocols require rather substantial depth in sequencing coverage, and thus are mostly adapted to the analysis of highly abundant (or at least enriched) RNA species [5,6,28–30].
Selection of samples for analysis
In this work, we present a comparative analysis of rRNA Nm levels observed in different human cell lines. Over 100 samples were initially included in the screening. Since different series were prepared by different persons (or even by different research teams), and different time periods, we first performed rigorous selection of samples showing similar degradation level (as judged from Bioanalyzer capillary electrophoresis profiles and RIN number, see Supplementary Table S1 and Figure S1) and, even more importantly, concordant cleavage profiles (Supplementary Figure S2AB). Degraded RNA samples were avoided, since it was already noticed that partial RNA degradation affects the precision of RiboMethSeq quantification. In addition, inspection of correlation for cleavage profiles allowed to exclude non-concordant samples. Representative examples of good and bad correlation of the cleavage profiles are shown in Figure S2AB. When samples demonstrated non-concordant cleavage profile with other datasets for the same cell line, those were systematically excluded. While the exact origin of such inconsistent RNA cleavage profile remains unclear, it is plausible that it may be related to partial RNA unfolding due to contaminants.
Variability of human rRNA 2ʹ-O-methylations (Nm)
Analysis of 2ʹ-O-methylation profiles was performed for a large dataset of human total RNA samples obtained for >100 samples extracted from ~20 human cell lines. The coverage and correlation of raw reads for all available datasets were inspected by rRNA type (Supplementary Figure S3) and only samples showing sufficient coverage (typically >1000 reads’ extremities/rRNA position) and a good correlation coefficient were retained (see Materials and Methods for further details). The final list of retained samples included 62 RiboMethSeq datasets for multiple biological replicates of 16 cell lines: HeLa (and HeLa S3 as well), HEK293, HCT116 (± p53), Jurkat T, HMEC, HUVEC cells, as well as human fibroblasts, chondrocytes, and stem cells from bone marrow and Wharton jelly. We also included other well characterized cancer cell lines (MDA-MB-231, T47D, BT20 and MCF7) (See Supplementary Table S1 for more details). For a number of cell lines, biological replicates were prepared independently, by different labs or in different time periods, those are labelled as series (see Table S1). Calculation of RiboMethSeq MethScore (previously called ScoreC2 [25,31]) was done using the standard calculation pipeline, with ±2 nt neighbouring interval. A MethScore was calculated for all known positions of 2ʹ-O-methylation (see Supplementary Table S2 for a comprehensive list of analysed Nm sites)
A variance analysis across all tested samples demonstrated that all 2ʹ-O-methylated sites in human rRNA can be grouped in 4 distinct classes, depending on their observed variability (Figure 1 and Figure 2, see Figure S4 for a full heatmap and Figure S5 for positions ordered according to rRNA numbering). The first class (called here stable modifications, 38 Nm positions in total, 1 in 5.8S rRNA, 17 in 18S rRNA and 20 in 28S rRNA) shows an extremely low variability in their methylation levels, variance <0.001 (s.d. <0.03) (Figure 2A), this threshold of variability was selected since it is inferior to the measurement’s precision observed between technical replicates in RiboMethSeq [25]. The second group (called here low variable modifications, is composed of 54 Nm positions, 1 in 5.8S rRNA, 18 in 18S rRNA and 35 in 28S rRNA) displays a higher variability, with variance comprised between 0.001 and 0.01 (and thus 0.03 < s.d.< 0.1) (Figure 2B) and the variable group is composed of two distinct sub-classes: 14 variable positions (4 in 18S rRNA and 10 in 28S rRNA) and 4 highly variable residues (18S-Gm867, 18S-Gm1447, 28S-Cm1327 and 28S-Cm2352) with a variance exceeding 0.05 (s.d. of 0.2–0.3) (Figure 2C). As it was already reported in previous studies, the methylation level for stably modified Nm positions is relatively high (35/38 with MethScore >0.9), while low variable positions show a lower modification level (e.g. 28S-Am3846 and 28S-Am1310). For variable and highly variable Nm positions the average methylation level ranges from almost zero (18S-Cm1440) to 0.9 (28S-Cm2791) with the majority (15/18) with a methylation level > 0.5. Of note, Nm positions guided by the same C/D-box snoRNA, may display differential variability (e.g. 28S-Cm2338 and 28S-Cm2352 guided by snoRD24 belong to low and highly variable groups, respectively). This may be related to the differential activity of Fib/snoRNP complex at different rRNA sites, and thus variations of snoRNA expression may differentially affect Nm level.
Figure 1.
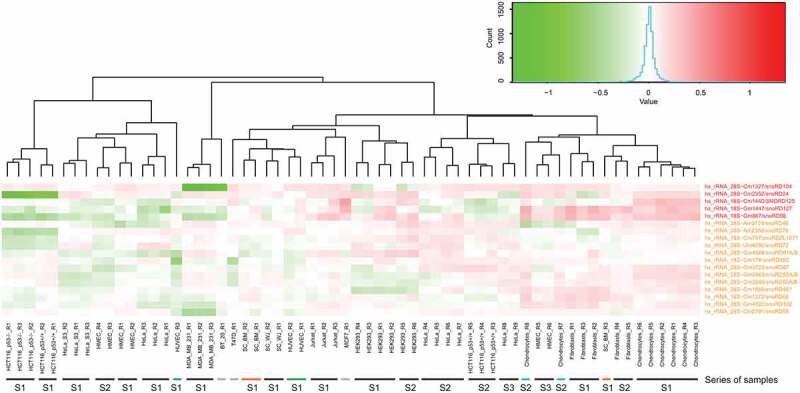
Heatmap for variable and highly variable Nm positions in human rRNA. Differential heatmap shows MethScore values row-normalized to the average value for each line, positive (red) and negative (green) variations of Nm level by position are shown). The scale of MethScore variation is shown by the colour gradient (colour key). Identity of the human cell line is shown at the bottom (S indicates the series, R stands for biological replicate), type and location of the modified residues, as well as associated C/D-box snoRNA (if known) are indicated at the right. Only Nm residues from variable (orange) and highly variable (red) groups are shown. The low variable position 28S−Am2388 which clusters together with the variable group (see Figure S4), is omitted
Figure 2.
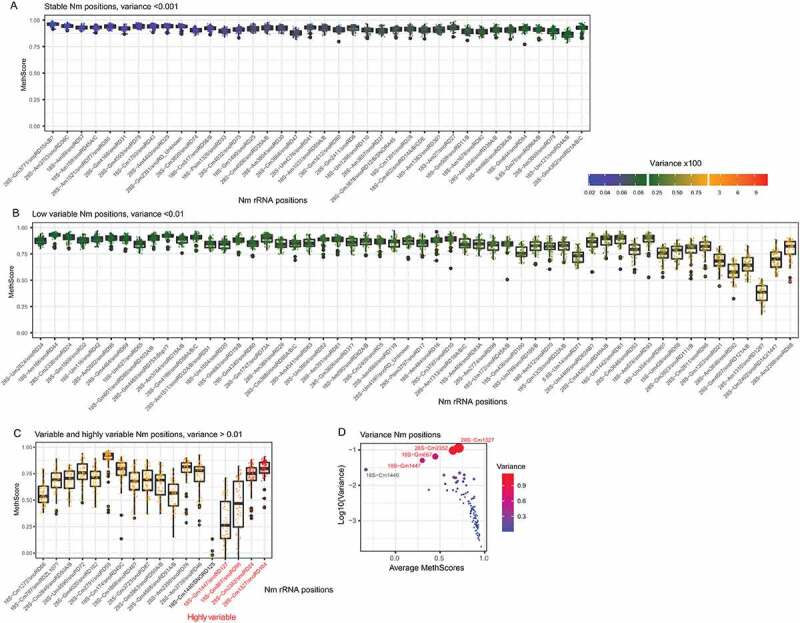
Global level and variability of all known Nm sites in human rRNA across the analysed human cell lines. Positions are sorted according to the calculated variance, from lowest to highest, boxplot shows the average level of the MethScore (central bar), quartiles (box borders and whiskers) and eventual outliers (black dots). Colour scale for individual data points represents the level of the variance, the scale is shown. Panels A-C: stable, low variable and variable groups, the last five positions in the variable group show high variability (variance >0.1, highly variable). Panel D shows a scatterplot for Average MethScore (X-axis) and log10 of the variance (Y-axis), shown also by colour code and by diameter of the dots
Using this large human rRNA RiboMethSeq dataset, we calculated a correlation value of MethScore across all available samples. As anticipated, multiple Nm residues (52/110) show a mutual positive correlation (Figure 3 and Figure S6, shown in blue), and thus vary in the same direction, this is likely related to a coordinated regulation of C/D-box snoRNA expression or natural variations of Fib expression, which would affect a subset(s) of sites in a coordinated manner. Noteworthy, positively correlated Nm positions form 4–5 distinct groups (see Figure 3 and Figure S6). In contrast, few modified positions showed negative correlation with the others, their methylation level varies in an opposite direction to the others (Figure 3, shown in red, group A1). This is the case for 14 positions (4 in 18S and 10 in 28S rRNA) (18S−Um172, 18S−Cm174, 18S−Gm436, 18S−Am576 and 28S−Am1310, 28S−Cm1327, 28S−Cm1868, 28S−Am2774, 28S−Cm2791, 28S−Cm2848, 28S−Gm2863, 28S−Am3846, 28S−Um4468, 28S−Gm4607), these opposite variations may represent an adaptive mechanism(s) intended to functionally compensate the Nm loss at some other important/functional rRNA sites. Remarkably, many of these anti-correlated rRNA Nm positions mostly belong to variable and highly variable groups and are located in a relative proximity one to another in the sequence of 18S and 28S rRNAs (see below for 3D mapping, Figure S7).
Figure 3.
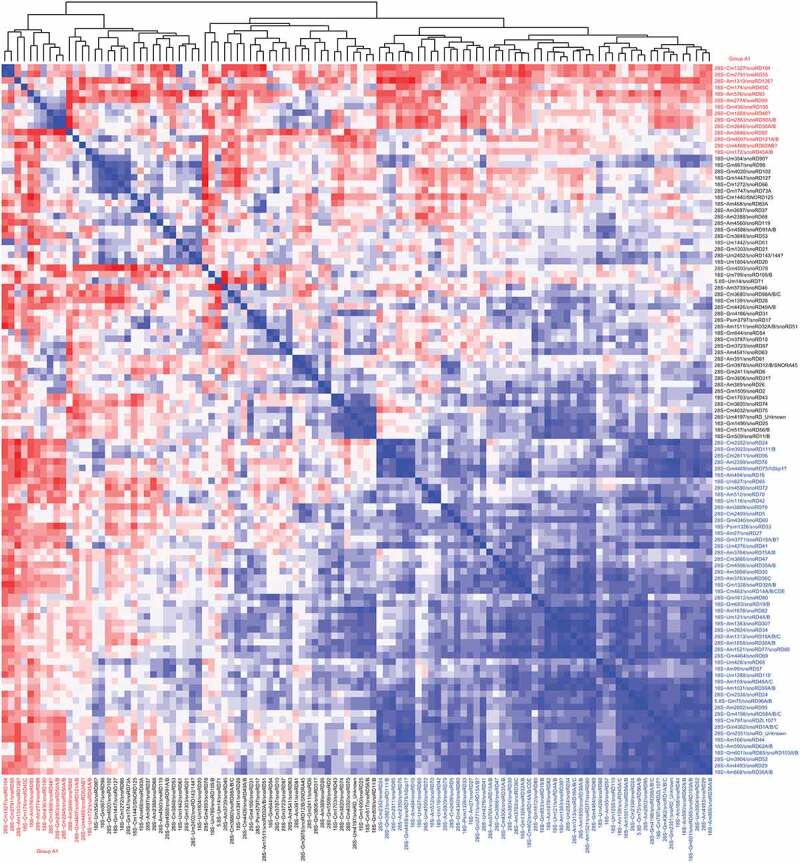
Correlation heatmap for MethScores of all Nm residues across all 62 analysed samples. Positive correlation (variations in the same direction) is shown by blue colours, negative correlation (variations in the opposite directions) is in red. The intensity of the colour represents the level of correlation coefficient. Residues of the negatively correlated group (group A1) are highlighted in red
Locations of variable Nm in the 3D ribosome structure
In order to appreciate spatial locations of rather constant (stable/low variable) and variable (or highly variable) rRNA modifications and their proximity to important ribosome functional sites, those residues have been mapped to the 3D ribosome structure obtained by X-ray crystallography and CryoEM (PDB 6EK0 was used for visualization). The 3D maps of Nm rRNA modification variability are presented on Figure 4. Position of each individual sphere representing a modified residue corresponds to the geometric centre of the modified nucleotide, the diameter of the sphere is proportional to the degree of variability (4 classes are shown).
Figure 4.
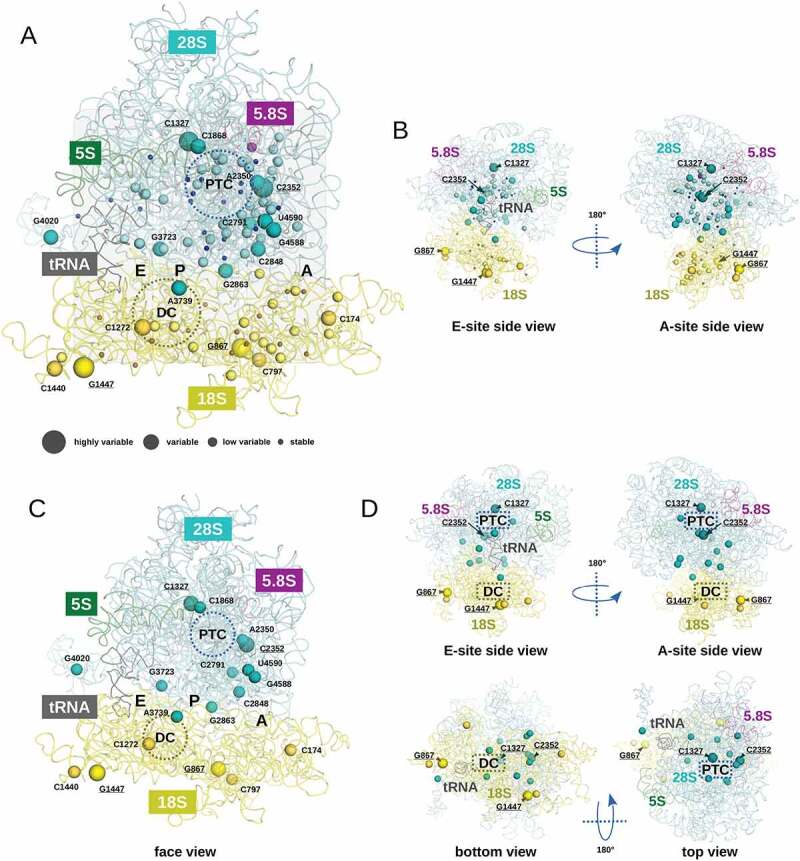
Visualization of the variation rates for Nm residues in the human ribosome 3D structure. RNA traces found in PDB entry 6EK0 are displayed using a ribbon representation. 5S, 5.8S, 18S, 28S and transfer RNAs are coloured in green, magenta, yellow, cyan and black, respectively. Four classes of modified nucleotides were defined. Stable, low variable, variable and highly variable nucleotides are represented using tiny, small, medium and large spheres, respectively. Colouring of the sphere follows the RNA ribbon colour. In panel A, all modified positions are represented with spheres on a face view of the human ribosome. The A, P and E regions of the ribosome are highlighted in grey boxes. Regions corresponding to the peptidyl transferase centre (PTC) and to the decoding centre (DC) are highlighted by circles. For display convenience, only variable and highly variable Nm residues are labelled using the conventional numbering. Highly variable modified nucleotides are underlined. In panel B, two side views of the structure shown in panel A are proposed. Only highly variable positions are labelled. In panel C, only variable (medium spheres) and highly variable (large spheres) positions are reported on the same face view than the one displayed in panel A. In panel D, four orientations of the structure displayed in panel C are proposed (two side views, one bottom and one top view). To provide orientation landmarks, highly variable Nm positions are labelled
Figure 4A shows the global distribution of Nm residues in human rRNA. The ribosomal A site contains the highest number of Nm nucleotides and many of them display substantial variation. In contrast, the E site (visible by position of tRNA in the structure) is only sparsely modified. With very few exceptions, stable and low variable residues are located rather close (or in) to PTC and DC, while variable and highly variable residues are almost excluded (Figure 4B). Only two variable residues 18S-Cm1272 and 28S-Am3739 are in a close proximity to the DC. The ribosome surface at the access cleft to the A site is relatively rich in Nm modifications, and 6 of them show substantial variability (positions 28S-Cm2352, 28S-Cm2848, 28S-Gm2863, 28S-Gm4588 and 28S-Um4590, see Figure 4A). Inspection of the 3D structure of the large ribosomal subunit (LSU) points out that the PTC itself is relatively poor in Nm residues, but a number of stable and low variable modifications are located in its immediate surrounding, including also 4 variable positions (Figure 4B). The most variable Nm in 18S rRNA are located at the surface of the SSU, this is also the case for variable and highly variable locations in 28S rRNA/LSU.
Mapping of positively and negatively correlating Nm nucleotides to 3D ribosome structure shows that the majority of Nm positions showing anticorrelation with the others (group A1, in red, Figure S5) are located in the proximity of the A-site and PTC, as mentioned above, some of these anti-correlating Nm positions are also most variable. Four anticorrelating Nm residues in 18S rRNA are at the extremities of the SSU and well accessible to interactions.
Cumulative variability map of human rRNA modifications
Finally, in order to get better insights into the global variability of rRNA modification positions, maps for Nm residues and our recently published observations on variable ψ levels in human rRNA [32] have been merged together. Since samples included into datasets were different, this comparison does not allow a thorough analysis of a direct correlation between Nm and ψ level variations, but rather to have an overview of the proximity for intrinsically stable and variable Nm and ψ residues, presented at the same scale. All, except two (28S-Am3739 and 18S-ψ1004) sites known to be important for critical ribosome functions and located in a close proximity to PTC, DC and intersubunit bridge, show only very limited variability, pointing out that variations of such vitally important rRNA modifications are not tolerated. Visualization of these Nm and ψ residues on the 3D ribosome structure (Supplementary Figure S8) confirms this observation. Ribosomal A-site is very rich in both Nm and ψ residues and these types of rRNA modification are well clustered together. Both variable and highly variable Nm and ψ residues congregate in the A-site and DC, as well as in a close proximity of the PTC (Figure S8, right).
Discussion
Origins of the variability in rRNA modification profiles
The precise origin of variability for rRNA modification is still poorly understood, mostly due to the extremely complex regulation of snoRNA-guided rRNA modification machinery in eukaryotic cells (reviewed in [12,33–35]). Indeed, the expression of individual snoRNA guides (both C/D-box for Nm and H/ACA-box for ψ) was shown to vary in different human cell lines and also in response to treatments or stresses (reviewed in [13,36–41]). In addition, the expression of protein catalysts involved in rRNA modification (fibrillarin, Fib for Nm and dyskerin, DKC for ψ) is likely to be regulated independently from snoRNA expression. Moreover, the efficiency or steady-state level of rRNA modification may be dependent on the stability of snoRNA/catalyst interaction, on the presence of other structural non-catalytic proteins in snoRNP, or on the efficiency of snoRNP complex assembly [42,43]. All these factors are certainly important for the observed outcome of rRNA modification in the living cell.
The majority of Nm sites (92/110 = 83.6%) falls into stable and low variable groups (Table 1), but proportion in each group depends on rRNA. Modifications of human 5.8S rRNA are rather stable, while the proportion of variable and highly variable sites is a bit lower in 18S rRNA (6/41 Nm) than in 28S rRNAs (12/67 Nm). In this study, we selected human cell lines only grown under ‘standard/normal’ laboratory conditions and not subjected to various types of stress or treatment. It is likely that the variations we observed here represent only a subset of more global and complex modulation of human rRNA modifications which can be only detected under particular conditions. However, it is highly conceivable that alterations in media composition (such as pH or ionic strength), partial oxygen pressure, culture growth temperature or other common laboratory variations may affect the rRNA modification pattern. For instance, while hypoxia was shown to modulate rRNA Nm content at various locations [20], only one Nm residue (28S-Cm2848, catalysed by snoRD50A/B), shown to be affected in hypoxia also shows variation in our data, the others belong to stable or low variable groups.
Table 1.
Number and classification of variability classes of Nm rRNA sites
| rRNA/variability class | 5.8S rRNA | 18S rRNA | 28S rRNA |
|---|---|---|---|
| Stable (var <0.001) | 1 | 17 | 20 |
| Low variable (var <0.01) | 1 | 18 | 35 |
| Variable (var <0.1) | 0 | 4 | 10 |
| Highly variable | 0 | 2 | 2 |
| Total | 2 | 41 | 67 |
Natural and pathological variability of rRNA modification
Modulation of rRNA modification profile was already observed under various conditions and in relation to pathology, mostly in different cancer types. The Nm content at different rRNA sites varies in disease and can be artificially manipulated by modulation of Fib expression. The rRNA Nm profile observed upon Fib overexpression was not analysed by RiboMethSeq, but in two independent studies, partial Fib depletion was shown to preferentially affect the Nm content of 28S rRNA, while 18S rRNA modification profile remained relatively constant [6,7]. These results should be interpreted with a great care, since Fib depletion is only transient and partial, a more extensive or prolonged reduction of Fib level was shown to be lethal for human cells. Moreover, a preferential decrease of 28S rRNA modifications may result from a differential stability or turnover of 18S and 28S rRNAs, or, as it was also suggested, from inequal accessibility of the modification sites during ribosome subunit assembly. Globally, the list of Fib-sensitive rRNA modification sites correlates rather well with the variability of Nm modifications observed in different human cell lines. Only 28S-Am1858 (snoRD38A/B), 28S-Am3703 (snoRD36C) and 28S-Um4276 (snoRD41) which are stable in our dataset, were found to be decreased upon Fib-depletion [6,7]. Variable and highly variable rRNA Nm modifications were also decreased upon transient Fib depletion. On the other hand, NPM1 deficiency was reported to negatively affect the levels of 5 Nm residues (28S-Cm1327, 28S-Am3764, 28S-Cm3866, 28S-Um3904 and 28S-Gm4198) [21]. Only 28S-Cm1327 (snoRD104) in this list was found to be highly variable in our dataset, other positions are either stable or low variable.
Other relevant observations of rRNA modification alterations come from analysis of cancer cells and tissues. In many studies, deregulation of snoRNA expression was also observed, and, in some instances, these variations were directly correlated with modulated levels of Nm or ψ residues. The most prominent disease-related correlations are 28S-Cm2848/Gm2863 (catalysed by snoRD50A/B) which are both increased in colon cancer [13,18], as well as 18S-Cm1703 (snoRD43) and 18S-Gm1328 (snoRD32A) shown to be affected in leukaemia [19]. In this study, both 28S-Cm2848 and 28S-Gm2863 were found to be rather variable in different human cell lines, in contrast, both leukaemia-related 18S-Cm1703 and 18S-Gm1328 belong to the stable and low variable groups, showing a limited correlation between ‘natural’ and ‘disease-related’ variations of rRNA modifications. Strikingly, many of Nm sites in this disease-related list show a negative correlation with the majority of human rRNA Nm positions.
Recently, a RiboMethSeq analysis was applied to large clinical cohorts of breast cancer tissues and diffuse large B-cell lymphoma samples [26,27]. Since identical analytical technique (albeit in different flavours) was applied in both studies, the results can be compared almost directly. With some noticeable exceptions, the degree of Nm level variations in these two cancer types correlates well with variations, observed here in human cell lines. This may be also related to the fact that many cell lines used here are also either immortalized or originate from different aggressive cancer types. However, 28S-Gm4593 (snoRD78) found to be variable in cancer is almost stable in our dataset, the same observation is made for 28S-Cm4032 (snoRD75), 18S-Um1288 (snoRD110), 18S-Am27 (snoRD27), 18S-Am1678 (snoRD82) and 18S-Am668 (snoRD36A/B). In contrast, we found that the 2ʹ-O-methylation level of 18S-ψm1326 (snoRD unknown) and 28S-Cm2791 (snoRD55) are variable in our selection, while only little variations at these positions were found in a large collection of breast cancer samples [26].
The variability of H/ACA-box snoRNA guides, and the levels of the corresponding ψ residues was also noticed, and in many instances were associated with cancer development. For instance, snoRA24 is downregulated in hepatocellular carcinoma [44] and the absence of snoRA24-driven ψ residues (18S-ψ609/863/1045) perturbs the aminoacylated-tRNA selection and pre-translocation complex dynamics.
While down- and up-regulation of snoRNA guides is very well documented, the molecular mechanisms linking rRNA modifications with the ribosome functions in translation still remain obscure. A global reduction of Nm by Fib depletion modulated the balance between cap-dependent and IRES-driven initiation of mRNA translation [6], but the precise function(s) of a given modified residue was not systematically addressed. The residues 18S-Cm462 (snoRD14A/B/CDE), 28S-Cm4506 (snoRD35A/B), 28S-Um2824 (snoRD34) were shown to affect protein synthesis [19], and belongs to the stable and low variable groups in this study.
Inspection of the variability for modified residues known to be located at the ribosome functional centres show that only a single nucleotide in the variable group, 28S-Am3739 (snoRD46), which is located at the helix H69 hairpin loop and may influence tRNA binding in the P-site [26], all the others show an extremely stable modification level in all samples. This may indirectly reflect the functional importance of rRNA modifications for ribosome functions, and show that deviations from a constitutive rRNA modification profile are not well tolerated.
Materials and methods
Selection of samples for RiboMethSeq analysis
Total RNA samples were extracted using TRIzol or column-based extraction kits from ‘untreated’ cultures of human cell lines cultivated under optimal/standard (recommended for a given cell line) conditions. To avoid biases, variably treated cell samples were systematically excluded, even when rRNA modification profile was not visibly altered. RNA integrity was assessed by Bioanalyzer 2200 RNA Pico Chip (See Supplementary Figure S1 for representative profiles). RIN numbers for samples are given in the Supplementary Table S1. Substantial RNA degradation may unpredictably affect RiboMethSeq analysis, so only few samples with RIN number <8.0 were retained for further analysis. However, RNA degradation seems not to be the primary source of variability, since differently degraded RNA samples (see HCT116 +/+ p53 R3/R4/R5) still give rather concordant results. Correlation of cumulated 5ʹ/3ʹ-counts between samples of the same cell type was inspected and samples with aberrant cleavage profiles (related to uncontrolled RNA degradation prior to analysis and possible presence of contaminants affecting RNA cleavage) were excluded (min correlation coefficient of 0.85, see Supplementary Figure S2AB). A complete list of retained human cell lines is presented in Supplementary Table S1.
RiboMethSeq and bioinformatic analysis
RiboMethSeq analysis was performed essentially as described previously [25,29] and bioinformatic analysis was done using ±2 nt interval, as described in [31]. ScoreC2 (called here MethScore) was used as a measure of Nm level at a given nucleotide. The MethScore can display negative values for unmethylated nucleotides and reaches 1.0 for fully methylated positions. Correlation coefficients were calculated according to Pearson, the analysis was performed using R packages Hmisc and corrplot, the clusters ordered using the hclust option.
Visualization of the human ribosome and mapping of modified positions
The PDB entry 6EK0 describing the 3D structure of the human ribosome in complex with the initiator tRNAMet in the E site was used to visualize modified positions in ribosomal RNAs [45]. All representations were drawn with PyMOL 2.3 (https://pymol.org/2/). The modified positions were represented using spheres, the geometrical centre of the nucleotide corresponding to the centre of the sphere. For the sake of clarity, only ribosomal RNA traces were displayed and ribosomal proteins were omitted from the representations.
Supplementary Material
Acknowledgements
We are grateful to all our collaborators for generously providing biological material and/or total RNA samples included in this comparative study.
Funding Statement
This work was supported by EpiARN and ViroMOD FRCR projects from Grand Est Region (France), as well as PRC ‘MetRibo’ and PRCI ‘D-erase’ projects funded by ANR. This paper was based upon work from COST Action CA16120 EPITRAN, supported by COST (European Cooperation in Science and Technology).
Availability statement
Raw sequencing data were generated at the Epitranscriptomics and RNA Sequencing (EpiRNA-Seq) Core Facility, Nancy, France and uploaded to European Nucleotide Archive under accession numbers PRJEB43738, PRJEB35565 and PRJEB46620. The derived data supporting the findings of this study as well as computer code are available on request from the corresponding author [YM].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Author contributions
Study design - YM and VM, wet lab experiments – VM, data analysis - WR and YM, mapping and visualization of the rRNA modifications -MQ.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Taoka M, Nobe Y, Yamaki Y, et al. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res. 2018;46:9289–9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ayadi L, Galvanin A, Pichot F, et al. RNA ribose methylation (2ʹ-O-methylation): occurrence, biosynthesis and biological functions. Biochim Biophys Acta Gene Regul Mech. 2019;1862:253–269. [DOI] [PubMed] [Google Scholar]
- [3].Maden BE. Locations of methyl groups in 28 S rRNA of xenopus laevis and man. Clustering in the conserved core of molecule. J Mol Biol. 1988;201:289–314. [DOI] [PubMed] [Google Scholar]
- [4].Maden EH, Wakeman JA. Pseudouridine distribution in mammalian 18 S ribosomal RNA. A major cluster in the central region of the molecule. Biochem J. 1988;249:459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Krogh N, Jansson MD, Häfner SJ, et al. Profiling of 2ʹ-O-Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res. 2016;44:7884–7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Erales J, Marchand V, Panthu B, et al. Evidence for rRNA 2ʹ-O-methylation plasticity: control of intrinsic translational capabilities of human ribosomes. Proc Natl Acad Sci U S A. 2017;114:12934–12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sharma S, Marchand V, Motorin Y, et al. Identification of sites of 2ʹ-O-methylation vulnerability in human ribosomal RNAs by systematic mapping. Sci Rep. 2017;7:11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gumienny R, Jedlinski DJ, Schmidt A, et al. High-throughput identification of C/D box snoRNA targets with CLIP and RiboMeth-seq. Nucleic Acids Res. 2017;45:2341–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shi Z, Fujii K, Kovary KM, et al. Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs genome-wide. Mol Cell. 2017;67(71–83.e7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Norris K, Hopes T, Aspden JL. Ribosome heterogeneity and specialization in development. Wiley Interdiscip Rev RNA. 2021;12:e1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ferretti MB, Karbstein K. Does functional specialization of ribosomes really exist? RNA. 2019;25:521–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Monaco PL, Marcel V, Diaz -J-J, et al. 2ʹ-O-methylation of ribosomal RNA: towards an epitranscriptomic control of translation? Biomolecules. 2018;8(4):106. doi: 10.3390/biom8040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Janin M, Coll-SanMartin L, Esteller M. Disruption of the RNA modifications that target the ribosome translation machinery in human cancer. Mol Cancer. 2020;19:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tosar JP, García-Silva MR, Cayota A. Circulating SNORD57 rather than piR-54265 is a promising biomarker for colorectal cancer: common pitfalls in the study of somatic piRNAs in cancer. RNA. 2021;27:403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Su J, Liao J, Gao L, et al. Analysis of small nucleolar RNAs in sputum for lung cancer diagnosis. Oncotarget. 2016;7:5131–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schulten H-J, Bangash M, Karim S, et al. Comprehensive molecular biomarker identification in breast cancer brain metastases. J Transl Med. 2017;15:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pauli C, Liu Y, Rohde C, et al. Site-specific methylation of 18S ribosomal RNA by SNORD42A is required for acute myeloid leukemia cell proliferation. Blood. 2020;135:2059–2070. [DOI] [PubMed] [Google Scholar]
- [18].Pacilli A, Ceccarelli C, Treré D, et al. SnoRNA U50 levels are regulated by cell proliferation and rRNA transcription. Int J Mol Sci. 2013;14:14923–14935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhou F, Liu Y, Rohde C, et al. AML1-ETO requires enhanced C/D box snoRNA/RNP formation to induce self-renewal and leukaemia. Nat Cell Biol. 2017;19:844–855. [DOI] [PubMed] [Google Scholar]
- [20].Metge BJ, Kammerud SC, Pruitt HC, et al. Hypoxia re-programs 2ʹ-O-Me modifications on ribosomal RNA. iScience. 2021;24:102010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nachmani D, Bothmer AH, Grisendi S, et al. Germline NPM1 mutations lead to altered rRNA 2ʹ-O-methylation and cause dyskeratosis congenita. Nat Genet. 2019;51:1518–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dong Z-W, Shao P, Diao L-T, et al. RTL-P: a sensitive approach for detecting sites of 2ʹ-O-methylation in RNA molecules. Nucleic Acids Res. 2012;40:e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Motorin Y, Marchand V. Detection and analysis of RNA Ribose 2ʹ-O-methylations: challenges and solutions. Genes (Basel). 2018;9(12):642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Birkedal U, Christensen-Dalsgaard M, Krogh N, et al. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew Chem Int Ed Engl. 2015;54:451–455. [DOI] [PubMed] [Google Scholar]
- [25].Marchand V, Blanloeil-Oillo F, Helm M, et al. Illumina-based RiboMethSeq approach for mapping of 2ʹ-O-Me residues in RNA. Nucleic Acids Res. 2016;44:e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Marcel V, Kielbassa J, Marchand V, et al. Ribosomal RNA 2′ O-methylation as a novel layer of inter-tumour heterogeneity in breast cancer. NAR Cancer Internet] 2021. [cited 2021 May 31]; 3. DOI: 10.1093/narcan/zcab006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Krogh N, Asmar F, Côme C, et al. Profiling of ribose methylations in ribosomal RNA from diffuse large B-cell lymphoma patients for evaluation of ribosomes as drug targets. NAR Cancer Internet] 2020; 2. [cited 2021 May 31]. DOI: 10.1093/narcan/zcaa035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Marchand V, Pichot F, Thüring K, et al. Next-generation sequencing-based ribomethseq protocol for analysis of tRNA 2ʹ-O-methylation. Biomolecules. 2017;7(1):13. [Google Scholar]
- [29].Ayadi L, Motorin Y, Marchand V. Quantification of 2ʹ-O-Me residues in RNA using next-generation sequencing (Illumina RiboMethSeq protocol). Methods Mol Biol. 2018;1649:29–48. [DOI] [PubMed] [Google Scholar]
- [30].Krogh N, Kongsbak-Wismann M, Geisler C, et al. Substoichiometric ribose methylations in spliceosomal snRNAs. Org Biomol Chem. 2017;15:8872–8876. [DOI] [PubMed] [Google Scholar]
- [31].Pichot F, Marchand V, Ayadi L, et al. Holistic optimization of bioinformatic analysis pipeline for detection and quantification of 2ʹ-O-methylations in RNA by RiboMethSeq. Front Genet. 2020;11:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Marchand V, Pichot F, Neybecker P, et al. HydraPsiSeq: a method for systematic and quantitative mapping of pseudouridines in RNA. Nucleic Acids Res. 2020;48(19):e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lui L, Lowe T. Small nucleolar RNAs and RNA-guided post-transcriptional modification. Essays Biochem. 2013;54:53–77. [DOI] [PubMed] [Google Scholar]
- [34].Watkins NJ, Bohnsack MT. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA. 2012;3:397–414. [DOI] [PubMed] [Google Scholar]
- [35].Henras AK, Plisson-Chastang C, Humbert O, et al. Synthesis, function, and heterogeneity of snoRNA-guided posttranscriptional nucleoside modifications in eukaryotic ribosomal RNAs. Enzymes. 2017;41:169–213. [DOI] [PubMed] [Google Scholar]
- [36].Boisvert F-M, van Koningsbruggen S, Navascués J, et al. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007;8:574–585. [DOI] [PubMed] [Google Scholar]
- [37].Sloan KE, Warda AS, Sharma S, et al. Tuning the ribosome: the influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017;14:1138–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dimitrova DG, Teysset L, Carré C. RNA 2ʹ-O-methylation (Nm) modification in human diseases. Genes (Basel). 2019;10(2):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. 2020;20:303–322. [DOI] [PubMed] [Google Scholar]
- [40].Yanas A, Liu KF. RNA modifications and the link to human disease. Methods Enzymol. 2019;626:133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mourksi N-E-H, Morin C, Fenouil T, et al. snoRNAs offer novel insight and promising perspectives for lung cancer understanding and management. Cells. 2020;9(3):541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Omer AD, Ziesche S, Ebhardt H, et al. In vitro reconstitution and activity of a C/D box methylation guide ribonucleoprotein complex. Proc Natl Acad Sci USA. 2002;99:5289–5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Reichow SL, Hamma T, Ferré-D’Amaré AR, et al. The structure and function of small nucleolar ribonucleoproteins. Nucleic Acids Res. 2007;35:1452–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].McMahon M, Contreras A, Holm M, et al. A single H/ACA small nucleolar RNA mediates tumor suppression downstream of oncogenic RAS. Elife. 2019;8:e48847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Natchiar SK, Myasnikov AG, Hazemann I, et al. Visualizing the role of 2ʹ-OH rRNA methylations in the human ribosome structure. Biomolecules. 2018;8(4):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.