

ABSTRACT
The covalent modification of RNA molecules is a pervasive feature of all classes of RNAs and has fundamental roles in the regulation of several cellular processes. Mapping the location of RNA modifications transcriptome-wide is key to unveiling their role and dynamic behaviour, but technical limitations have often hampered these efforts. Nanopore direct RNA sequencing is a third-generation sequencing technology that allows the sequencing of native RNA molecules, thus providing a direct way to detect modifications at single-molecule resolution. Despite recent advances, the analysis of nanopore sequencing data for RNA modification detection is still a complex task that presents many challenges. Many works have addressed this task using different approaches, resulting in a large number of tools with different features and performances. Here we review the diverse approaches proposed so far and outline the principles underlying currently available algorithms.
KEYWORDS: RNA modifications, direct rna sequencing, nanopore, software, epitranscriptome
Introduction
In contrast to DNA, which contains only a dozen epigenetic modifications, RNA is decorated with almost 170 different modifications [1], collectively referred to as the ‘epitranscriptome’[2], which has emerged as an important regulatory layer of RNA metabolism and general cell homoeostasis. Our view of the epitranscriptome has dramatically changed in recent years, starting with the discovery that the fat-mass and obesity-associated (FTO) enzyme could demethylate the N6-methyladenosine (m6A) RNA modification [3]. The first assays coupling antibody immunoprecipitation to Next-Generation Sequencing (NGS) for studying RNA modifications appeared in 2012 (MeRIP-seq or m6A-seq) [4,5], providing new avenues to examine the post-transcriptional regulatory world transcriptome-wide. These pioneering works revealed that RNA modifications are far more widespread than previously thought, are subjected to dynamic regulation [6], and have a major impact on RNA processing, stability [7], translation [8,9] and localization [10]. Following studies have demonstrated that RNA modifications are key regulators of a wide range of biological processes, including cellular differentiation [11,12] sex determination [13,14] and maternal-to-zygotic transition in vertebrate embryos [15,16], among others.
Despite the major achievements of NGS technologies, one of their major limitations is that they require an initial conversion of the RNA molecule into complementary DNA (cDNA). Thus, these methods will typically erase RNA modifications. This removes the ability to directly detect the modifications in the cDNA sequences, and requires other indirect assays to infer modified positions. There are some exceptions, such as inosine [17,18], where modifications can be detected in the cDNA because they cause misincorporations during retrotranscription [17,18]. Nevertheless, to identify the majority of RNA modifications genome-wide, two alternative indirect strategies are commonly used: i) antibody-based detection, where the antibody specifically recognizes the modified ribonucleotide [4,5,19–21]; and ii) chemical-based detection, using chemical compounds that will selectively react with the modified ribonucleotide of interest, followed by reverse transcription of the RNA fragment, leading to accumulation of reads with identical ends [22–28]. However, the limited repertoire of commercial antibodies [29] or lack of chemicals that can selectively recognize RNA modifications [30] currently leaves 90% of known RNA modifications unmapped with NGS-based detection methods [29], hindering our understanding of their biological function and dynamics. Moreover, even when these reagents are available, NGS-based methods suffer from severe caveats: i) they are often not quantitative, ii) have high false-positive rates [31], iii) are inconsistent when using distinct antibodies [32], iv) do not provide isoform-specific information, v) often lack single nucleotide resolution [4,5,19] and vi) require multiple ligation steps and extensive PCR amplification, introducing strong biases in the data [23,33,34].
A promising alternative to sequencing-by-synthesis-technologies is the direct RNA nanopore sequencing (dRNA-seq) platform offered by Oxford Nanopore Technologies (ONT) (Fig. 1A). This technology allows direct sequencing of native RNA molecules; therefore, in contrast to present antibody-based or chemical-based methods, it is in principle able to detect any RNA modification of interest at single nucleotide resolution and in individual full-length native RNAs [35,36]. To date, dRNA-seq has been proven capable of detecting a wide range of RNA modification types, including N6-methyladenosine (m6A), N7-methylguanosine (m7G), 5-methylcytosine (m5C), 5-hydroxymethylcytosine (hm5C), pseudouridine (Y) and 2ʹ-O-methylations (Nm), among others [35,37–44].
Figure 1.
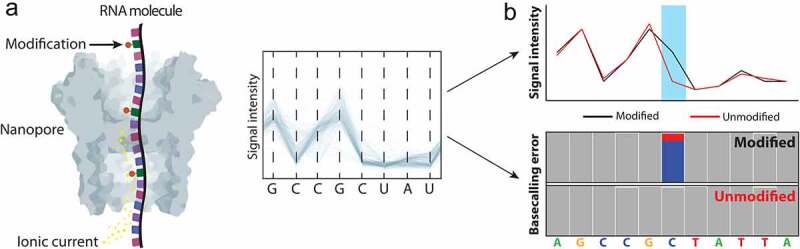
Schematic overview of direct RNA nanopore sequencing and the strategies to detect RNA modifications. (A) Direct RNA sequencing allows the sequencing of native RNA molecules. As the molecule goes through the nanopore, it causes alterations in the ionic current that is going through the nanopore. These disruptions can be converted into their corresponding nucleotide sequences using machine learning algorithms, such as hidden Markov models or recurrent neural networks. (B) Schematic representation of the two major approaches used to detect RNA modifications in nanopore sequencing data: the detection of RNA modifications in the form of alterations of raw signal intensities (upper panel), or systematic base-calling ‘errors’ (lower panel)
ONT sequencing relies on the use of membrane-embedded protein nanopores, which are coupled to highly sensitive ammeters that measure ionic current passing through the pore. As an RNA or DNA molecule translocates through the nanopore, the sensor measures disruptions in the ionic current, which can in turn be used to identify the transiting nucleotide sequences using machine learning algorithms (also known as ‘base-calling algorithms’). In particular, the level of ionic current recorded at any point in time is a function of the set of k nucleotides residing within the pore (kmer), with k typically modelled as 5 for dRNA-seq. As output, nanopore sequencing runs will produce FAST5 files, which is an HDF5 file format that contains the per-read ionic current information, as well as other metadata. Base-calling algorithms will then take as input the FAST5 files, and produce as output FASTQ files that contain the base-called sequences and quality scores for each individual read.
Base-calling algorithms typically rely on hidden Markov models (HMM) or recurrent neural networks (RNN). Early basecallers were based on the former architecture, while most recent implementations use the latter. Neural networks are composed of nodes, which can receive and carry out calculations, organized in layers. After being trained with specific datasets, they can identify patterns and generate predictions. During the basecalling process, they recognize patterns in the ionic current intensity to predict the nucleotide sequence. At present, the most widely used basecaller is Guppy, which was developed by Oxford Nanopore Technologies (ONT) and is available to all members of the ONT community. This algorithm uses an RNN to identify the underlying nucleotide sequence from the raw signal and recent works have shown that it has good performance in terms of speed and accuracy [45]. However, its training was agnostic to RNA modifications and, as a consequence, its output is limited to the four canonical bases.
To identify RNA modifications in nanopore direct RNA sequencing data, two major strategies have been proposed: i) identification of RNA modifications through the analysis of FAST5 features and ii) identification of RNA modifications through the analysis of base-called ‘error’ features (Fig. 1B). The first approach typically identifies RNA modifications in the form of altered ionic current intensities [35,37,41]. However, several works have recently pointed out that the absolute change of differential ionic current intensity between modified and unmodified reads, for some specific RNA modifications or sequence contexts, is often too small to identify RNA modifications or bin reads into two groups [44]. Therefore, additional features such as ‘trace’ (the probability reported by the base-calling software that a nucleotide matches a model for a canonical nucleotide) or ‘dwell time’ (the amount of time a kmer persists in the sensitive region of the nanopore) are needed to improve the detection of RNA modifications across a variety of sequence contexts [44,46]. The second approach to detect RNA modifications in dRNA-seq data relies on the use of systematic base-calling errors that are caused by the presence of RNA modifications. Considering the error rate of nanopore direct RNA sequencing base-calling algorithms, it is strongly encouraged to use a control condition to remove systematic errors that are unrelated to the presence of RNA modifications, as these could otherwise lead to increased false positive rates. Notably, the ‘error signature’ can also be exploited to identify which RNA modification type is present in the dRNA-seq data. For example, pseudouridine typically appears in the form of U-to-C mismatches [41], whereas m5C modifications appear in the form of insertions or deletions in the +1 position [44].
In the last few years, the scientific community has been actively engaged in the development of novel algorithms to detect and quantify RNA modifications from nanopore direct RNA sequencing data (Fig. 2). While some of the available tools have been compared to each other within the individual manuscripts, a systematic review of capabilities, advantages and limitations of each available tool is presently missing. Here, we provide a systematic review of the extant algorithms and tools that have been developed in the last few years to detect RNA modifications from dRNA-seq data. Moreover, we provide an exhaustive overview of the features, datasets and RNA modification types used to benchmark each individual algorithm, thus providing a comprehensive resource of the algorithms to date and their capabilities, strengths and limitations (Supplementary Table 1).
Figure 2.
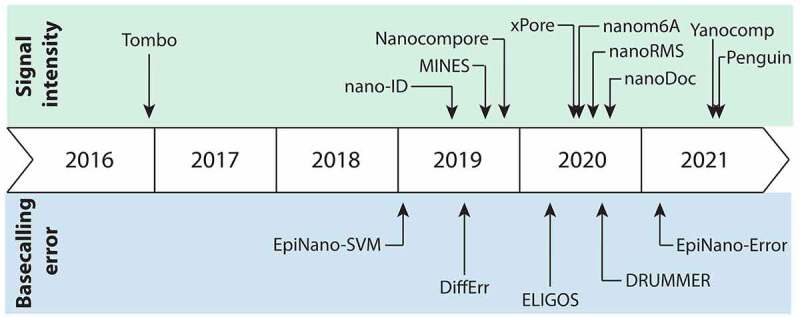
Chronological overview of the community efforts to develop tools to detect and quantify RNA modifications using direct RNA nanopore sequencing
Ionic current/signal intensity based methods
All algorithms that directly use the electrical signal to detect the presence of modifications need to access the ionic current data for each sequenced kmer. Unfortunately, the Guppy base-calling algorithm does not presently return the ionic current data matching each kmer. For this reason, a pre-processing step is necessary, where the continuous ionic current intensity data (also known as the squiggle) is segmented into discrete blocks corresponding to each kmer and then aligned to the base-called sequence. This resquiggling process permits comparisons among the ionic current for all nanopore reads associated with each particular kmer. After resquiggling, there are two basic schemes that comparative algorithms follow. The first scheme compares the observed ionic current for a kmer to the expected ionic current for the same kmer composed of only canonical bases. If there is a significant difference between observed and expected ionic currents, the kmer is labelled as containing a modified nucleotide. This approach has the advantage of being independent from a reference sample and can potentially identify any modified position in the entire transcriptome, but does not provide information about which modification was present. Alternatively, the observed ionic current could be directly compared to available signal models for modified bases when they are known (at present, the only such model available is for m5C and is only implemented in Tombo [43]). The second scheme uses additional sequencing information from a matched sample that is devoid of one or more modifications, such as a knockdown (KD) or knockout (KO) experiment for a modification writer enzyme or an in vitro transcribed RNA (IVT). This strategy has the advantage that each kmer is compared against a reference unmodified kmer in exactly the same sequence context, and that the type of modification can be identified based on how they were removed from the reference sample. The main drawbacks of this second approach are that they require sequencing at least two samples for each experiment (e.g. wild type and knockout) and that the modification must be easily removed from the reference sample. The following paragraphs will provide an overview of the tools that implement one or both these approaches.
Tombo [43] is the successor to nanoraw, the first publicly available software for detection of modified bases in nanopore DNA sequencing data. In its present implementation, Tombo automatically runs sequence alignment to the reference as well as sequence-to-signal assignment (resquiggling). The alignment is performed using minimap2 [47], whereas resquiggling is done through a multi-step procedure that performs i) signal normalization, ii) event detection and iii) sequence-to-signal assignment per se, which uses a banded dynamic time warping algorithm to find the optimal chain of matches between segmented events and the reference sequence. After these pre-processing operations, Tombo performs modified bases detection. This functionality is implemented using three alternative strategies: i) using pre-computed models of the signal for specific non-canonical bases, ii) using a de novo method that detects any non-canonical base or iii) through the comparison with an unmodified sample. For the first strategy (i.e. model-based alternate base detection), Tombo presently only provides an RNA model for m5C, which was generated from in vitro transcribed E. coli RNAs spiked-in with m5C (Marcus Stoiber, personal communication). The second approach is instead model-free and consists in calculating a statistical test for each kmer of each read comparing the measured electrically signal to the expected signal based on the canonical model. This strategy is readily applicable since it does not require a model nor a control sample; however, it does not provide information about the identity of the non-canonical bases that it detects. Furthermore, the authors warn that this method is prone to a high number of Type I errors. Lastly, the third strategy detects modifications in a sample of interest through the comparison with a reference sample. The reference sample can be a completely unmodified RNA (e.g. obtained by IVT) and in this case Tombo uses it to locally adjust the canonical model before using the de novo approach described above. As a third approach, Tombo can perform a per kmer comparison between the signal distribution in the experimental and control samples. In this case, testing is performed using a Kolmogorov-Smirnoff, Mann-Whitney or Student’s t-test. According to the authors, this is usually the most reliable Tombo mode to detect modification for direct RNA data when a reference sample is available. By default, this method requires a coverage of at least 50 reads per transcript, but a higher threshold value is recommended to properly control for the false positive rate.
Nanocompore [37] is a python package that implements a comparative strategy for modification detection where the raw electrical signal for a sample of interest is compared, kmer-wise, to the signal generated by a sample devoid of RNA modifications, e.g. a knock-out of an RNA modifying enzyme or an IVT. The pre-processing steps required to generate input data suitable for Nanocompore analysis consist of base-calling, mapping to the reference transcriptome and sequence-to-signal assignment (resquiggling) with Nanopolish [48]. Users can choose to run these steps manually or they can be executed automatically using the Nanocompore pipeline, an automated workflow implemented in Nextflow that runs the entire analysis. Nanocompore first collapses the output of Nanopolish by calculating the median signal intensity and dwell time for each kmer of each read. After grouping reads based on the reference transcript that they map to, this data is then analysed kmer-wise with a statistical test to detect differences between the experimental condition and the unmodified reference. In terms of statistics, Nanocompore implements multiple tests, giving the user the option to decide which ones to use: a two components Gaussian Mixture Modelling (GMM) followed by a logistic regression or a number of parametric/non-parametric univariate tests on signal intensity or dwell time alone. All these tests operate at the level of individual kmers. For a given kmer of a given transcript, the GMM method first aggregates median signal intensity and dwell time from all the samples provided, disregarding the experimental condition. Then a two components GMM is fit to the data using both the median signal intensity and dwell time. The rationale for this approach is that, in this bivariate space, modified sites would form two clusters, with one consisting of modified reads (from the experimental condition and, potentially, from residual modifications in the control condition) and the other of unmodified reads (from the control condition and, in addition, from a variable number of unmodified reads from the experimental condition, depending on modification stoichiometry). Alternatively, if a site is unmodified, the two clusters will have no biological interpretation and the reads from experimental and control conditions will be randomly distributed among the two. Therefore, to formally test for the presence of a modification, Nanocompore calculates whether the reads from experimental and control conditions are differentially distributed among the two clusters. To this end, first it fits a discrete logit model using the GMM cluster assignment as the response variable and condition label as the predictor. Then, it calculates the t-statistic for the predictor’s parameter. As mentioned above, Nanocompore also implements a number of parametric (t-test) or non-parametric (Kolmogorov-Smirnoff, Mann–Whitney) tests on median signal intensity or dwell time alone. Unlike the GMM method, which simultaneously captures differences on intensity, dwell time or both, these tests are univariate and as such are separately calculated for the median signal intensity and dwell time. After completing the statistical calculations Nanocompore corrects all p-values with the Benjamini-Hochberg method, saves the results in text format and generates BED tracks for the modification sites identified. Nanocompore has been applied to profile METTL3-dependent m6A sites in human cells, where the sites were validated by an orthogonal antibody-based technique (m6A individual-nucleotide-resolution cross-linking and immunoprecipitation, miCLIP [34]) and recapitulated known features of this modification. The authors also used a targeted sequencing strategy to profile m6A in the 7SK snRNA, identifying multiple novel m6A sites.
xPore [49] is a python tool that, similarly to Nanocompore, operates on signals realigned to sequence with Nanopolish and implements GMM to detect modified sites throughout the comparison of an experimental sample with an unmodified reference. Specifically, for each sample, xPore calculates the mean signal corresponding to each kmer, and then fits a multi-sample two Gaussian mixture model using the theoretical signal distribution of the unmodified kmer as a prior. Using such a prior is an important innovation of xPore as it allows assignment of the two distributions obtained from the GMM to the modified or unmodified state, thus also estimating the fraction of modified reads at each site. Finally, xPore performs a z-test on the fraction of modified reads in each sample to prioritize differentially modified sites. In their pre-print, the authors analyse a METTL3 KO human cell line to profile m6A. By comparing the xPore results with reference m6A sites obtained by m6A-Crosslinking-Exonuclease-sequencing (m6ACE-seq), the authors report a precision of 0.6 (AUC 0.86) when limiting the analysis to kmers containing a central A (NNANN). On the other hand, when the authors extended the analysis to all kmers, they reported lower precision at the same level of sensitivity. An additional feature of xPore is its intrinsic capacity to estimate modification stoichiometry, which was validated by the sequencing of mixtures of RNA from wild-type (WT) and METTL3 KO cells.
NanoRMS [44] is a python software to predict RNA modification stoichiometry in dRNA-seq datasets, and is in principle applicable to any given RNA modification type. As input, NanoRMS requires two samples in FAST5 format and a BED file with a list of sites to predict the modification stoichiometry (typically candidate RNA-modified sites). If one of the samples is a knockout condition, NanoRMS can be used to determine the absolute stoichiometry values of the second sample. Alternatively, it will report the difference in modification stoichiometry between the two samples for each candidate RNA-modified site. Briefly, NanoRMS extracts per-read features (signal intensity, dwell time and trace) from reads resquiggled with Tombo [43] and stores them in the BAM file. By default, the algorithm performs both unsupervised (k-means) and supervised (KNN) clustering of the read features to bin the reads into modified and unmodified populations. Based on this binning, NanoRMS then computes the modification stoichiometry for each RNA modified site and condition, as well as the difference in stoichiometry between the two conditions. Noteworthy, if neither of the two conditions are knockout/knockdown, NanoRMS can identify the difference in stoichiometry but not its directionality. NanoRMS was benchmarked on pseudouridine (Y) and 2-O’-methylated (Nm) sites, using both in vitro as well as in vivo constructs with different stoichiometries (3–100%). The authors found that the choice of features used to distinguish modified and unmodified reads severely affected the prediction of RNA modification stoichiometry, and that combination of ‘signal intensity’ and ‘trace’ from positions −1, 0, +1 (being 0 the modified site) led to the most accurate stoichiometry predictions in Y and Nm sites. The selected features often captured non-redundant information; for example, the authors found that certain Y sites did not lead to significant alterations in signal intensity values, but that at these positions, RNA modification information could be captured in the form of altered ‘trace’. Finally, it is important to note that stoichiometry predictions were severely affected by the choice of resquiggling algorithm. Specifically, the authors reported that Nanopolish biased up to 7X the unmodified:modified read proportion, due to its lower ability to resquiggle reads with modifications. By contrast, the authors found that Tombo was relatively robust in terms of even resquiggling of modified and unmodified reads. NanoRMS is publicly available in GitHub, and includes test data to quantify Pus1-dependent Y modifications in mRNAs, as well as a collection of R scripts for visualization of the results.
Nanom6A [50] is a python software developed to identify m6A within RRACH 5-mers (R = G/A, H = A/U/C) according to the associated ionic current intensity signal. Specifically, dRNA-seq raw data are resquiggled with Tombo [43] and for each 5-mer of interest the mean and median ionic current intensity, its standard deviation, and the length of the signal are retrieved. A set of fully methylated and unmethylated synthetic oligos were processed according to the aforementioned protocol, and the resulting dataset was used to train and test an Extreme Gradient Boosting classifier. The authors reported an accuracy higher than 0.9 using a modification probability threshold of 0.5 to call a 5-mer as methylated within individual reads. Leveraging the same dataset, the authors investigated the ability of Nanom6A to estimate the stoichiometry of m6A sites with known stoichiometry varying from 0 to 100% and at different coverage levels. They reported a correlation of ~0.97 between expected and inferred m6A stoichiometry with just 20 reads (by default, 5-mers with a lower coverage are not processed by the algorithm). Finally, to call for the presence of m6A in a given genomic position, the authors required the corresponding kmer to be detected as methylated in at least 20 reads. By using this approach, they were able to recapitulate the 40% loss in m6A following the METTL3 KD in HEK293 cells. Moreover, modified sites detected by Nanom6A across different biological systems were compared to and partially overlapped with the sites identified by other approaches, such as miCLIP, MeRIP-seq and MAZTER-seq [51], and other methods, such as EpiNano, MINES, and DiffErr. Nanom6A is available on GitHub accompanied by a dedicated Docker image and, alternatively, a .yml file to build the required conda environment. The authors also released a toy-dataset to test the pipeline, and a manual to guide the user.
MINES [52] (m6A Identification using Nanopore Sequencing) is a python software that identifies m6A sites within DRACH motifs (D = G/A/U, R = G/A, H = A/U/C), the most common m6A sequence context [34]. MINES uses the Tombo detect modifications de novo algorithm to find sites that are likely modified, and filters the modification stoichiometry values for only those positions within a 20 nucleotide window centred on the A of each DRACH motif with at least five reads (at the time of writing this manuscript, the code has been updated to a 30 nucleotide window). This lowered the search space and reduced computational resources required to identify modified positions. These stoichiometry values are used by a random forest to classify the central A of the DRACH motif as either modified or canonical. The random forest model was trained on 70% DRACH sites identified as modified by miCLIP and an equal number of unmodified DRACH sites from HEK293T and HeLa cell lines. The model was tested with the remaining 30% of modified DRACH sites and they found their accuracy ranged from 67% to 83% and the precision ranged from 40 to 92%. Four of eighteen possible DRACH motifs accounted for the highest accuracy for identifying modifications, suggesting that the other twelve possible DRACH motifs either were false positives from miCLIP or the m6A-to-A ratio was below the limit of MINES to detect. The authors used MINES to evaluate all 28,925 DRACH sites that did not have a miCLIP signal and found that 13,034 (45.06%) were likely modified. The signal for these modified sites was compared to the signal from a METTL3 KD condition, revealing that roughly half of the sites missed by miCLIP were sensitive to the writer KD. The authors were able to leverage the long-read capabilities of nanopore sequencing to evaluate the isoform-specific m6A calls from MINES, and found that 6,168 (7.85%) sites had isoform-specific methylation. Since MINES starts with the quantifications of the modification stoichiometry from Tombo and limits its model to DRACH sites, it does not require a KD or other canonical nucleotide reference experiment (such as IVT) to identify which modifications are likely m6A.
nanoDoc [53] is a python software designed to identify RNA modifications by comparing, for each 5-mer, the raw electrical signals derived from unmodified IVT RNAs against the counterpart obtained from a sample of interest. Specifically, dRNA-seq data are resquiggled through Tombo and both ionic current intensity and dwell time are obtained for each nucleotide. After a step of data formatting and normalization, the two signals are extracted for each 5-mer and given in input to a Convolutional Neural Network (CNN) for classification with 1024 possible classes. Then, a second classifier (Deep One-Class – DOC) is defined coupling two CNNs with shared weights. One network is used to process a uniform batch of target 5-mers while the other is used to analyse a dataset composed of sequences close to the target one. After this final training step, the classification layer of the CNN is removed to obtain from the algorithm a 16-dimension vector instead of one label; i.e. from classification to dimensional reduction. For each site, the IVT data are divided into two equal batches, processed, and the resulting 16-dimension vectors are compared using the mean Euclidean distance from the five closest neighbours. The same procedure is repeated by substituting one of the two IVT batches with the corresponding data from the sample of interest. A difference in the resulting distance distributions would reveal the presence of an RNA modification; the authors convey this information through a custom score, based on their percentiles, ranging from 0 (no difference) to 1 (maximum difference). NanoDoc was applied to identify known RNA modification sites in rRNA and resulted in a global AUC of 0.96 (presence vs absence of a nucleotide analogue). They also focused on m6A analysing 81 BBABB 5-mers from a public synthetic dataset obtaining an AUC of 0.68. Finally, the tool was applied to study the epitranscriptional landscape of SARS-CoV-2. In this case, a limited overlap with orthogonal techniques was reported. Noticeably, NanoDoc is suitable to detect the presence of a generic RNA modification, however, it does not provide the identity of the base analogue. The tool is available on GitHub with limited documentation.
Penguin [54] is a python based tool which provides multiple machine learning models (e.g. Support Vector Machine, Neural Network and Random Forest) to identify 5-mers containing pseudouridine. The classifiers are based on four features extracted through Nanopolish: reference 5-mer, mean ionic current, ionic current standard deviation and dwell time. They were trained using a set of human modified locations from the literature, 2987 in total, and two dRNA-seq datasets from distinct cell lines, Hek293 and HeLa, which resulted in 13,072 and 1354 examples respectively (50% pseudouridine). Depending on the algorithm, the authors report an AUC between 0.85 and 0.93 using 80% of the available data for training and 20% for testing. A similar performance was achieved using one dRNA-seq dataset for training and the other one for testing: AUC between 0.92 and 0.95. All the analyses were performed on 5-mers with a U in third position, and using the code available on GitHub.
Yanocomp [55] (yet another nanopore modification comparison tool) is a python software that compares two samples against each other: one being a sample of interest and the other being a reference sample without modifications. In order to detect modified sites, Yanocomp models the mean ionic current amplitudes for a sliding window of five kmers using multivariate Gaussian Mixture Models. Yanocomp uses two Gaussian components and a third uniform component to account for outliers, which is claimed to reduce overfitting. Yanocomp has the option to model the signal distributions to make single molecule predictions of modified sites. Yanocomp was tested on a WT strain of A. Thaliana compared against a low m6A expression strain. In this context the authors reported the identification of 20,033 m6A sites, of which 84.1% were within five nucleotides of m6A sites identified by miCLIP. These m6A sites were then correlated with polyadenylation site usage, demonstrating how single-molecule modification detection can be used to understand and phase related biological processes. At present, Yanocomp has not been compared against other RNA modification detection tools, nor has it been tested on modifications other than m6A. However, the framework allows the detection of potentially any RNA modification.
Nano-ID [46] is a R/python software that, following RNA metabolic labelling, relies on the detection of RNA modifications to profile nascent RNA and quantify RNA half-lives [56]. Reads from nascent RNA are identified based on the incorporation of the exogenous nucleotide analogue 5-Ethynyluridine (5EU). A neural network is adopted for the identification of 5EU containing reads, which has to be trained on fully labelled and unlabelled datasets. The former could be obtained by dRNA-seq following 24 h 5EU labelling to create a modified dataset composed of reads containing, with high probability, at least one 5EU nucleotide (as expected according to the typical half-life of transcripts), the latter could be obtained by dRNA-seq of unlabelled RNA. These data were processed to extract information about: the associated ionic current intensity signal, the base-caller performance (i.e. confidence of the called bases), and the alignment (i.e. read length, bases occurrences, and similarity to the reference). The result is a vector of 4694 features for each read in the two samples. On an independent test set, Nano-ID achieved an AUROC of 0.94 without constraints on read length and 0.96 for reads longer than 1kb. This method represents the first attempt to merge information about ionic current intensity signal, base-calling and alignment to perform RNA modifications classification. The coupling of this method with the identification of endogenous RNA modifications, such as m6A, was proposed as a way for comprehensively quantifying the effect of those modifications on RNA fate [57]. The main drawback of Nano-ID is the lack of positional information on the modified bases. This tool is released as a collection of R scripts on GitHub, however, no documentation is available, and the set-up of the pipeline requires a certain amount of effort from the user.
Base-calling and alignment-based methods
The impact of RNA modifications on the ionic current signal indirectly affects the performance of the base-caller increasing the probability for mismatch and indels which can be detected when nanopore reads are aligned against a reference. Due to the intrinsic limited accuracy of the base-caller, a single error is not sufficient to claim the presence of a non-canonical nucleotide. However, systematic accumulation of mismatches or indels on a specific site of a transcriptional unit suggests the presence of an RNA modification. Several tools were developed to identify anomalous error frequencies compared to a background which can be profiled just for the training of the algorithm or ad hoc. As previously mentioned, the first approach is more flexible and simpler but the second one, which has a heavier experimental design, offers a better control on the results of the analyses. In general, these approaches provide the location of modified sites given multiple reads but not at single-molecule resolution.
EpiNano [39] was the first algorithm to exploit systematic base-calling ‘errors’ present in direct RNA sequencing as a strategy to detect RNA modifications in dRNA-seq data. In its initial version (1.0), EpiNano used Support Vector Machines (SVMs) to predict m6A-modified sites transcriptome-wide, which had been previously trained with m6A-modified and unmodified RRACH kmers. While this approach had the advantage of not requiring a KO condition to predict RNA modifications, the authors found that the relatively high error rate in RNA base-calling algorithms led to high false positive rates. Thus, the authors recommend using EpiNano in paired conditions (e.g. wild type and KO), to remove possible false positives. Later versions of EpiNano (version 1.2 and later) can be ran in two different modes: i) EpiNano-SVM, which uses pre-trained SVMs to predict RNA modifications in a given dataset as explained above, and ii) EpiNano-Error, which can be used to study virtually any given RNA modification, and does not require pre-trained SVM models [58], but does require paired conditions to be compared. EpiNano-Error has been recently used to identify novel pseudouridylated sites in a transcriptome-wide fashion both in yeast mitochondrial rRNAs and mRNAs, and several of them were validated using CMC-probing based orthogonal methodologies [44]. As a general rule, a minimum coverage of 30 reads is recommended to use EpiNano. However, the authors showed that the coverage required was strongly dependent on the stoichiometry of modification. EpiNano source code can be downloaded from GitHub, but can be also executed directly as part of the MasterOfPores [59] NextFlow workflow, which is aimed at facilitating the analysis of direct RNA nanopore sequencing data, and performs data pre-processing, mapping, RNA modification detection and poly(A) tail length estimation.
DiffErr [38] was developed to identify m6A sites according to differential base-calling errors between a WT and a control sample with a lower modification level (e.g. METTL3 KD). This tool applies G-tests to a 2 × 5 contingency table containing, for each sample (rows), the number of: A, C, G, U, and indels (columns). For the sites with a p-value lower than 0.05, DiffErr performs a second set of G-tests to compare replicates of the same condition, if available. If the G statistic between replicates exceeds the G statistic between conditions the site is discarded. P-values are adjusted with Benjamini-Hochberg, and ratios of mismatches from reference genome over matches are computed. Sites with a significance level smaller than 0.05, and a log2 fold change in mismatch to match ratio between WT and KD greater than 1, are finally classified as methylated. DiffErr was applied to study the m6A landscape of Arabidopsis thaliana. From this analysis, established properties of the mark emerged (e.g. known m6A motifs), and 66% of the sites identified by the algorithm resulted closer than 5 nucleotides from a miCLIP peak. DiffErr requires at least 10 reads to process a specific site, however, a more stringent threshold is recommended, and 200 reads are required to obtain a reliable classification of sites with a methylation frequency higher than 50%. This software was released on GitHub as a collection of python scripts with a set-up file to instal the required dependencies but limited documentation is available.
DRUMMER [42], similarly to DiffErr, identifies m6A sites by comparing a WT and a low modified sample in terms of matches/mismatches frequency by means of a G-test on the 2 × 5 contingency table previously described. A site is classified as methylated if the G-test adjusted p-value is lower than 10−2 (Bonferroni’s correction method), and the matches over mismatches ratio decreases at least one-fold in the WT sample compared to the control one. m6A calls obtained with DRUMMER on a viral RNA dataset were compared with Nanocompore calls: DRUMMER identified 204 m6A sites, and 162 of these were also detected by Nanocompore, which reported 41 additional calls. More than 90% of DRUMMER predicted sites were closer than 5 nucleotides to the minimum m6A motif AC. Finally, replicates are considered independent samples, and the authors recommended a coverage higher or equal than 100 reads in both WT and control conditions. This python-based tool was released on GitHub with a yml file to build the correct conda environment to run the analysis, the required documentation, and a test dataset.
ELIGOS [40] identifies modified sites comparing matches and mismatches (i.e. substitutions, insertions and deletions) from a sample of interest against either a control with an expected differential modification level (e.g. IVT RNAs, cDNA, a generic second sample), or a background model obtained from the pre-processing of an unmodified IVT dataset. Specifically, for each sample involved in the analysis, the mismatches frequency is computed for all the 5-mers in the genome covered by at least 50 reads using a reference sequence as groundtruth. Since each base is shared by five 5-mers, the corresponding alignment statistics are merged. Then, to identify differential error profiles, Fisher’s exact test is applied to the 2 × 2 contingency table obtained comparing the sample of interest against the desired control. This procedure is performed both at the single base resolution and merging the alignment statistics over the two adjacent bases for a total of three tests per nucleotide. All the resulting p-values are adjusted using the Benjamini–Hochberg method, and the maximum odd-ratio for each base determines the final result of the analysis. The authors tested the ability of ELIGOS to identify a set of 9 different RNA modifications exploiting synthetic modified IVT RNAs. Using the background model provided by the tool, the software reached an AUROC above 0.7 for all the RNA modifications except m5C (~ 0.54), with a performance up-to ~0.92 for Inosine. Although the builtin background model of ELIGOS can not detect the modification type, the authors also applied it to study m6A in yeast and human comparing WT samples against low-methylated counterparts obtained through the KD of m6A writers. Known m6A motifs and the expected spatial distribution of this mark emerged from ELIGOS results. ELIGOS source code, a dedicated Docker image, and a detailed manual are available through GitHub.
Discussion and Perspectives
The epitranscriptome has emerged as a fundamental regulatory layer of RNA metabolism, and an increasing number of biological processes have been shown to depend on the dynamic modification of RNAs [11–16,60]. The recent surge in studies exploring the epitranscriptome has been largely enabled by the development of high throughput methods for modification profiling based on NGS. However, it is becoming apparent that many of these indirect methods suffer from intrinsic limitations which hamper their performance and resolution. The direct RNA sequencing paradigm offered by the ONT platform provides a way to directly profile RNA modifications, overcoming several shortcomings of Sequencing-By-Synthesis techniques. For this reason, a significant amount of literature related to the development of computational methods for RNA modification profiling with dRNA-seq has been quickly produced since the first public release of this technology in 2017.
A common rationale shared by most available tools is to detect RNA modifications by comparing the reads obtained from a sample of interest against a reference. Such reference can either be released together with the algorithm as a model that captures the modification features, or it can be created ad hoc for each experiment from a control sample. The first approach is cheaper, but modification detection is limited to the RNA modifications included in the training dataset. Furthermore, since these models are generally trained on a single modification, there is often no guarantee that they will not misclassify other modifications that are chemically similar to the one used for training. Conversely, the tools based on an experimental reference sample are more flexible since they can identify different RNA modifications according to the control used. This flexibility comes at the cost of a more expensive experimental design, but these methods tend to be more robust to eventual condition-specific confounding factors, such as difficult to base-call kmers or damaged RNA.
This type of comparative analyses can be based on different features derived from the ionic current intensity or on the alignment of base-called reads. The methods exploiting the first class of features can reach single molecule resolution, but they require the error-prone signal-to-sequence alignment (resquiggling), which in many cases requires transcriptome alignments and is thus limited to known transcripts (see Supplementary Table 1). Unlike methods that rely on the ionic current, methods based on base-calling errors do not require this computationally expensive step, but can be more sensitive to confounding factors and lack single-molecule resolution since classification is based on an ensemble of reads. Notably, an attempt to merge information derived from both the aforementioned classes of features has also been proposed [46].
A further limitation of nanopore-based methods for modification detection is the present challenge to study more than one, or at most a few, modifications at the same time. It is becoming increasingly clear that the repertoire of RNA modifications includes multiple modification types that can potentially co-occur at the same time in the same RNA molecule. Although mass spectrometry data indicates that certain classes of RNA only contain a limited number of modification types (e.g. mRNAs), other classes (e.g. tRNAs) are affected by over ten different types of modifications. This scenario is even more complex when considering that distinct modifications might functionally interact with each other, for example leading to situations where the presence of one modified nucleotide is required for or prevents the deposition of another modification on the same RNA [61]. Studying these complex mechanisms will require techniques that allow to profile multiple modifications – ideally all of them – at the same time. Since nanopore direct RNA sequencing data has the potential to contain information about all modifications, an ideal strategy would be to develop a single classifier that directly detects all possible modifications. However, the large number of modifications combined with high electrical noise and a narrow feature space, make this an impossible strategy to pursue at present. However, future technical improvements on multiple fronts could make this problem more tractable. Specifically, we envisage that the combination of a) improved machine learning models for basecalling modifications, b) reduction in the noise of raw signal, c) development of more sensitive nanopores and d) an expanded feature space that uses additional signal properties to detect modifications, will allow the future development of a unified platform that concurrently detects a large number of modifications with single-molecule resolution.
Nanopore sequencing is still a relatively new technology, and ONT has been rapidly updating the performance of their platforms through changes to the enzymes, chemistry and software. This rapid pace of performance improvements makes nanopore sequencing more attractive, as per-read and consensus accuracy, throughput, and read lengths periodically improve, but it also causes downstream analysis tools, including those designed for RNA modification detection, to quickly become obsolete. The error profiles of the base-called reads change with each new version of the base-calling algorithm, thus requiring that the tools that rely on errors update their models. Similarly, the models based on ionic current features must either change or also become obsolete each time ONT changes the nanopore, enzyme motor or buffer conditions. Therefore, it is important to use caution when analysing datasets generated with different versions of the ONT platform than the tools were designed for. This may lead to artefacts or inaccurate conclusions that are a result of the dataset-to-tool version mismatch, and not to the biology of the dataset.
Researchers wishing to use nanopore dRNA-seq to profile RNA modifications have to face the task of choosing which approach to follow and which software to use. This review attempts to provide a systematic comparison of all the methods available, but the choice is still hard due to the lack of clear performance comparisons. We would recommend users wishing to use any of these methods to thoroughly assess performance in their experimental setting and – when possible – to use multiple algorithms and compare the results. A systematic and independent benchmark of all methods available is urgently needed and we envisage that it could be achieved by a community effort inspired by well known machine learning competition platforms, where participants are provided with training data and the platform independently manages the test of the algorithms. The performance should be measured according to high-quality datasets including as many confounding factors affecting real biological samples as possible, e.g. presence of multiple modifications, and collected across different biological conditions. The benchmark should ideally also include datasets produced with different set-ups of the sequencing platform.
To conclude, in the last few years several valuable algorithms have been developed to detect RNA modifications from dRNA-seq data. Most of them showed good performance at detecting m6A as well as other modifications, however a systematic comparison of their sensitivity and specificity is still lacking. All these efforts and the resulting knowledge will eventually converge in a base-caller able to directly convert the output of the sequencer into a sequence using an extended dictionary that includes not only canonical bases but a vast number of non-canonical RNA bases. This will be a tremendous resource to shed light on the complex domain of RNA modifications.
Supplementary Material
Funding Statement
This paper was based upon work from COST Action CA16120 EPITRAN, supported by COST (European Cooperation in Science and Technology). AD-T is supported by an FPI Severo-Ochoa fellowship by the Spanish Ministry of Economy, Industry and Competitiveness (MEIC). This work was partly supported by funds from the Spanish Ministry of Economy, Industry and Competitiveness (MEIC) (PGC2018-098152-A-100 to EMN), and by funds from the Italian Association for Cancer Research (AIRC, project IG 2020, ID. 24784 to MP). We acknowledge the support of the MEIC to the EMBL partnership, Centro de Excelencia Severo Ochoa and CERCA Programme/Generalitat de Catalunya.
Supplemental data
Supplemental data for this article can be accessed here.
Disclosure statement
EMN, LM and TL have received reimbursement of travel or accommodation expenses to speak at Oxford Nanopore Technologies conferences. TL is a consultant for STORM Therapeutics Limited.
References
- [1].Boccaletto P, Bagiński B, MODOMICS: an Operational Guide to the Use of the RNA Modification Pathways Database [Internet], Picardi E. editor. RNA Bioinformatics. 2021;New York NY:Springer US. cited 2021 Jul 1. 481–505. Available from. https://link.springer.com/ 10.1007/978-1-0716-1307-8_26 [DOI] [PubMed] [Google Scholar]
- [2].Saletore Y, Meyer K, Korlach J, et al. The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biol. 2012;13:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jia G, Fu Y, Zhao X, et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Meyer KD, Saletore Y, Zumbo P, et al. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell. 2012;149:1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. [DOI] [PubMed] [Google Scholar]
- [6].Roundtree IA, Evans ME, Pan T, et al. RNA Modifications in Gene Expression Regulation. Cell. 2017;169:1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lee Y, Choe J, Park OH, et al. Molecular Mechanisms Driving mRNA Degradation by m6A Modification. Trends Genet. 2020;36:177–188. [DOI] [PubMed] [Google Scholar]
- [8].Mao Y, Dong L, Liu X-M, et al. m6A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat Commun. 2019;10:5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yu J, Chen M, Huang H, et al. Dynamic m6A modification regulates local translation of mRNA in axons. Nucleic Acids Res. 2018;46:1412–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Madugalle SU, Meyer K, Wang DO, et al. RNA N6-Methyladenosine and the Regulation of RNA Localization and Function in the Brain. Trends Neurosci. 2020;43:1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Geula S, Moshitch-Moshkovitz S, Dominissini D, et al. m 6 A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015;347:1002–1006. [DOI] [PubMed] [Google Scholar]
- [12].Batista PJ, Molinie B, Wang J, et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15:707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lence T, Akhtar J, Bayer M, et al. m6A modulates neuronal functions and sex determination in Drosophila. Nature. 2016;540:242–247. [DOI] [PubMed] [Google Scholar]
- [14].Haussmann IU, Bodi Z, Sanchez-Moran E, et al. m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. 2016;540:301–304. [DOI] [PubMed] [Google Scholar]
- [15].Zhao BS, Wang X, Beadell AV, et al. m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature. 2017;542:475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yang Y, Wang L, Han X, et al. RNA 5-Methylcytosine Facilitates the Maternal-to-Zygotic Transition by Preventing Maternal mRNA Decay. Mol Cell. 2019;75(1188–1202):e11. [DOI] [PubMed] [Google Scholar]
- [17].Bazak L, Haviv A, Barak M, et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014;24:365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Levanon EY, Eisenberg E, Yelin R, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol. 2004;22:1001–1005. [DOI] [PubMed] [Google Scholar]
- [19].Delatte B, Wang F, Ngoc LV, et al. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science. 2016;351:282–285. [DOI] [PubMed] [Google Scholar]
- [20].Li X, Xiong X, Zhang M, et al. Base-Resolution Mapping Reveals Distinct m1A Methylome in Nuclear- and Mitochondrial-Encoded Transcripts. Mol Cell. 2017;68:993–1005 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Safra M, Sas-Chen A, Nir R, et al. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature. 2017;551:251–255. [DOI] [PubMed] [Google Scholar]
- [22].Pandolfini L, Barbieri I, Bannister AJ, et al. METTL1 Promotes let-7 MicroRNA Processing via m7G Methylation. Mol Cell. 2019;74:1278–1290.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Marchand V, Ayadi L, Ernst FGM, et al. Profiling of m 7 G and m 3 C RNA Modifications at Single Nucleotide Resolution. Angew Chem Int Ed. 2018;57:16785–16790. [DOI] [PubMed] [Google Scholar]
- [24].Carlile TM, Rojas-Duran MF, Zinshteyn B, et al. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014;515:143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Schwartz S, Bernstein DA, Mumbach MR, et al. Transcriptome-wide Mapping Reveals Widespread Dynamic-Regulated Pseudouridylation of ncRNA and mRNA. Cell. 2014;159:148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lovejoy AF, Riordan DP, Brown PO. Transcriptome-Wide Mapping of Pseudouridines: pseudouridine Synthases Modify Specific mRNAs in S cerevisiae. PLoS ONE. 2014;9:e110799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dai Q, Moshitch-Moshkovitz S, Han D, et al. Nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat Methods. 2017;14:695–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Galvanin A, Ayadi L, Helm M, et al., Mapping and Quantification of tRNA 2′-O-Methylation by RiboMethSeq [Internet], Wajapeyee N, Gupta R. editors. Epitranscriptomics. 2019;New York:Springer New York. cited 2021 Jul 1. 273–295. Available from. http://link.springer.com/ 10.1007/978-1-4939-8808-2_21 [DOI] [PubMed] [Google Scholar]
- [29].Novoa EM, Mason CE, Mattick JS. Charting the unknown epitranscriptome. Nat Rev Mol Cell Biol. 2017;18:339–340. [DOI] [PubMed] [Google Scholar]
- [30].Motorin Y, Helm M. Methods for RNA Modification Mapping Using Deep Sequencing: established and New Emerging Technologies. Genes (Basel). 2019;10:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Anreiter I, Mir Q, Simpson JT, et al. New Twists in Detecting mRNA Modification Dynamics. Trends Biotechnol. 2021;39:72–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Grozhik AV, Olarerin-George AO, Sindelar M, et al. Antibody cross-reactivity accounts for widespread appearance of m1A in 5ʹUTRs. Nat Commun. 2019;10:5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lahens NF, Kavakli I, Zhang R, et al. IVT-seq reveals extreme bias in RNA sequencing. Genome Biol. 2014;15:R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Linder B, Grozhik AV, Olarerin-George AO, et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Garalde DR, Snell EA, Jachimowicz D, et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat Methods. 2018;15:201–206. [DOI] [PubMed] [Google Scholar]
- [36].Workman RE, Tang AD, Tang PS, et al. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat Methods. 2019;16:1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Leger A, Amaral PP, Pandolfini L, et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. bioRxiv 2019:843136. doi: 10.1101/843136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Parker MT, Knop K, Sherwood AV, et al. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m6A modification. eLife. 2020;9:e49658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Liu H, Begik O, Lucas MC, et al. Accurate detection of m6A RNA modifications in native RNA sequences. Nat Commun. 2019;10:4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jenjaroenpun P, Wongsurawat T, Wadley TD, et al. Decoding the epitranscriptional landscape from native RNA sequences. Nucleic Acids Res. 2021;49:e7–e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Smith AM, Jain M, Mulroney L, et al. Reading canonical and modified nucleobases in 16S ribosomal RNA using nanopore native RNA sequencing. PLOS ONE. 2019;14:e0216709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Price AM, Hayer KE, Abr M, et al. Direct RNA sequencing reveals m6A modifications on adenovirus RNA are necessary for efficient splicing. Nat Commun. 2020;11:6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Stoiber M, Quick J, Egan R, et al., De novo Identification of DNA Modifications Enabled by Genome-Guided Nanopore Signal Processing. bioRxiv 2017; 094672; doi: 10.1101/094672 [DOI] [Google Scholar]
- [44].Begik O, Lucas MC, Pryszcz LP, et al., Quantitative profiling of pseudouridylation dynamics in native RNAs with nanopore sequencing, Nat Biotechnol. 2021. 10.1038/s41587-021-00915-6 [DOI] [PubMed] [Google Scholar]
- [45].Wick RR, Judd LM, Holt KE. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019;20:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Maier KC, Gressel S, Cramer P, et al. Native molecule sequencing by nano-ID reveals synthesis and stability of RNA isoforms. Genome Res. 2020;30:1332–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Minimap LH. 2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Loman NJ, Quick J, Simpson JT. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods. 2015;12:733–735. [DOI] [PubMed] [Google Scholar]
- [49].Pratanwanich PN, Yao F, Chen Y, et al., Identification of differential RNA modifications from nanopore direct RNA sequencing with xPore, Nat Biotechnol. 2021; 10.1038/s41587-021-00949-w [DOI] [PubMed] [Google Scholar]
- [50].Gao Y, Liu X, Wu B, et al. Quantitative profiling of N6-methyladenosine at single-base resolution in stem-differentiating xylem of Populus trichocarpa using Nanopore direct RNA sequencing. Genome Biol. 2021;22:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Garcia-Campos MA, Edelheit S, Toth U, et al. Deciphering the “m6A Code” via Antibody-Independent Quantitative Profiling. Cell. 2019;178(731–747):e16. [DOI] [PubMed] [Google Scholar]
- [52].Lorenz DA, Sathe S, Einstein JM, et al., Direct RNA sequencing enables m6A detection in endogenous transcript isoforms at base specific resolution, RNA, 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ueda H, nanoDoc: RNA modification detection using Nanopore raw reads with Deep One-Class Classification. bioRxiv 2020. doi: 10.1101/2020.09.13.295089 [DOI] [Google Scholar]
- [54].Hassan D, Acevedo D, Daulatabad SV, et al., Penguin: a Tool for Predicting Pseudouridine Sites in Direct RNA Nanopore Sequencing Data. bioRxiv 2021. doi: 10.1101/2021.03.31.437901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Parker MT, Barton GJ, Simpson GG; Yanocomp: robust prediction of m6A modifications in individual nanopore direct RNA reads. bioRxiv 2021. doi: 10.1101/2021.03.31.437901 [DOI] [Google Scholar]
- [56].Furlan M, de Pretis S, Pelizzola M. Dynamics of transcriptional and post-transcriptional regulation. Brief Bioinform. 2020; 22(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Furlan M, Tanaka I, Leonardi T, et al. RNA Sequencing for the Study of Synthesis, Processing, and Degradation of Modified Transcripts. Front Genet. 2020;11:394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Liu H, Begik O, Novoa EM, EpiNano: detection of m6A RNA Modifications Using Oxford Nanopore Direct RNA Sequencing [Internet], McMahon M. editor. RNA Modifications, New York NY:Springer US; 2021; cited 2021 Jul 1]. 31–52.Available from https://link.springer.com/ 10.1007/978-1-0716-1374-0_3 [DOI] [PubMed] [Google Scholar]
- [59].Cozzuto L, Liu H, Pryszcz LP, et al. MasterOfPores: a Workflow for the Analysis of Oxford Nanopore Direct RNA Sequencing Datasets. Front Genet. 2020;11:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. 2020;20:303–322. [DOI] [PubMed] [Google Scholar]
- [61].Xiang J-F, Yang Q, Liu C-X, et al. N6-Methyladenosines Modulate A-to-I RNA Editing. Mol Cell. 2018;69:126–135.e6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.