

Abstract
Target enrichment is becoming increasingly popular for phylogenomic studies. Although baits for enrichment are typically designed to target single-copy genes, paralogs are often recovered with increased sequencing depth, sometimes from a significant proportion of loci, especially in groups experiencing whole-genome duplication (WGD) events. Common approaches for processing paralogs in target enrichment data sets include random selection, manual pruning, and mainly, the removal of entire genes that show any evidence of paralogy. These approaches are prone to errors in orthology inference or removing large numbers of genes. By removing entire genes, valuable information that could be used to detect and place WGD events is discarded. Here, we used an automated approach for orthology inference in a target enrichment data set of 68 species of Alchemilla s.l. (Rosaceae), a widely distributed clade of plants primarily from temperate climate regions. Previous molecular phylogenetic studies and chromosome numbers both suggested ancient WGDs in the group. However, both the phylogenetic location and putative parental lineages of these WGD events remain unknown. By taking paralogs into consideration and inferring orthologs from target enrichment data, we identified four nodes in the backbone of Alchemilla s.l. with an elevated proportion of gene duplication. Furthermore, using a gene-tree reconciliation approach, we established the autopolyploid origin of the entire Alchemilla s.l. and the nested allopolyploid origin of four major clades within the group. Here, we showed the utility of automated tree-based orthology inference methods, previously designed for genomic or transcriptomic data sets, to study complex scenarios of polyploidy and reticulate evolution from target enrichment data sets.[Alchemilla; allopolyploidy; autopolyploidy; gene tree discordance; orthology inference; paralogs; Rosaceae; target enrichment; whole genome duplication.]
Polyploidy, or whole-genome duplication (WGD), is prevalent throughout the evolutionary history of plants (Cui et al. 2006; Jiao et al. 2011, 2012; Leebens-Mack et al. 2019). As a result, plant genomes often contain large numbers of paralogous genes from recurrent gene and genome duplication events (Lynch and Conery 2000; Panchy et al. 2016). Paralogs are defined as homologous genes that share a common ancestor as the product of gene duplication (Fitch 1970), either from small-scale duplications or WGD. One special case of WGD is allopolyploidy, where genome doubling is accompanied by hybridization between two different species. The duplicated genes in allopolyploids are not paralogs in the traditional sense and are referred to as homoeologs, which are expected to be sister to their respective ancestral lineages, rather than to each other (Smedmark et al. 2003; reviewed in Glover et al. 2016). For practical purposes, however, we refer to the product of any kind of duplications found in gene trees hereafter as paralogs, as homoeologs are indistinguishable from paralogs until diagnosed as resulting from allopolyploidy. With very few nuclear genes being truly single- or low-copy, careful evaluation of orthology is critical for phylogenetic analyses (Fitch 1970). Orthology inference has received much attention in the phylogenomic era with multiple pipelines available for this task (e.g., Li 2003; Dunn et al. 2013; Kocot et al. 2013; Yang and Smith 2014; Emms and Kelly 2019, also see Glover et al. 2019 and Fernández et al. 2020 for recent reviews). But these approaches have been mainly applied to genomic or transcriptomic data sets. So far, few studies have employed automated, phylogeny-aware orthology inference in target enrichment data sets. The most common approach for dealing with paralogy in target enrichment data sets is removing entire genes that show any evidence of potential paralogy (e.g., Nicholls et al. 2015; Jones et al. 2019; Andermann et al. 2020; but see Moore et al. 2018). Removal of entire genes might seem appropriate in target enrichment data sets in which only a small number of genes show evidence of paralogy (e.g., Larridon et al. 2020), but in some data sets, this could result in a significant reduction of the number of loci (e.g., Montes et al. 2019). More importantly, dealing with paralogs by removing entire genes assumes that target enrichment assembly pipelines (e.g., Faircloth 2016; Johnson et al. 2016; Andermann et al. 2018) have flagged all genes containing potential paralogs. It also assumes that if no sequence in a gene is flagged, all sequences are single-copy and orthologous. On the other hand, this approach also removes genes that show allelic variation instead of paralogs. Given the prevalence of WGD and reticulations, these assumptions can lead to errors in orthology inference. As paralogous genes are prevalent in plants, more appropriate orthology inference methods need to be applied to target enrichment data. The same automated approaches used for genome and transcriptome data sets can be applied for target enrichment data sets, as these are tree-based and agnostic to the data source for tree inference.
The ability to explicitly process paralogs opens the door for using target enrichment data for inferring gene duplication events and pinpointing the phylogenetic locations of putative WGDs. In the past, the phylogenetic placement of WGD events has most often been carried out using genome and transcriptome sequencing data (e.g., Li et al. 2015; Huang et al. 2016; McKain et al. 2016; Yang et al. 2018) using either the synonymous distance between paralog gene pairs (Ks; Lynch and Conery 2000) or tree-based reconciliation methods (e.g., Jiao et al. 2011; Li et al. 2015; Yang et al. 2015; Huang et al. 2016; Xiang et al. 2017; Leebens-Mack et al. 2019). Similar to orthology inference, tree-based methods used to investigate WGDs in genome and transcriptome data sets should be useful in target enrichment data sets. Target enrichment methods (e.g., Mandel et al. 2014; Weitemier et al. 2014; Buddenhagen et al. 2016) have been widely adopted to collect hundreds to over a thousand nuclear loci for plant systematics, allowing studies at different evolutionary scales (e.g., Villaverde et al. 2018), and the use of museum-preserved collections (e.g., Forrest et al. 2019). This creates new opportunities to adopt tree-based reconciliation methods to explore WGD patterns in groups for which genomic and transcriptomic resources are not available or feasible.
With at least 350 ( 1100) species worldwide, Alchemilla in the broad sense (s.l.) has been a challenging group to study due to the presence of reticulate evolution, polyploidy, and apomixis. Based on previous phylogenetic analyses, Alchemilla s.l. contains four clades: Afromilla, Aphanes, Eualchemilla, and Lachemilla (Supplementary Table S1 available on Dryad at https://doi.org/10.5061/dryad.cc2fqz660). Together they form a well-supported clade nested in the subtribe Fragariinae, which also includes the cultivated strawberry (Gehrke et al. 2008). Unlike the more commonly recognized members of the rose family (Rosaceae), Alchemilla s.l. is characterized by small flowers with no petals, and a reduced number (1–4[–5]) of stamens that have anthers with one elliptic theca on the ventral side of the connective that opens by one transverse split (Perry 1929; Soják 2008). Gehrke et al. (2008) presented the first phylogeny of Alchemilla s.l. and established the paraphyly of traditional Alchemilla (in the strict sense) as consisted of a primarily African clade, Afromilla, and a Eurasian clade, Eualchemilla. Gehrke et al. (2008) also suggested treating Afromilla and Eualchemilla, along with Aphanes and Lachemilla as a single genus based on nomenclatural stability and the lack of morphological characters to distinguish between Afromilla and Eualchemilla. The four clades within Alchemilla s.l. are mainly defined by geographic distribution, as well as the number and insertion of the stamens on the disk lining the hypanthium (Supplementary Table S1 available on Dryad). Phylogenetic analyses using at least one nuclear and one chloroplast marker (Gardner et al. 2016, Gehrke et al. 2008) found significant cytonuclear discordance regarding the relationships among the four major clades. Similar patterns, often attributed to hybridization and allopolyploidy, have been detected in other genera of Fragariinae (Lundberg 2009; Eriksson et al. 2015; Gehrke et al. 2008, Kamneva et al. 2017; Morales-Briones et al. 2018a), leaving the phylogenetic relationships of Alchemilla s.l. to the rest of Fragariinae unresolved. Unlike most members of Fragariinae that have predominantly diploid species, Alchemilla s.l. is known for high rates of polyploidy. The base chromosome number of Alchemilla s.l. is eight (
1100) species worldwide, Alchemilla in the broad sense (s.l.) has been a challenging group to study due to the presence of reticulate evolution, polyploidy, and apomixis. Based on previous phylogenetic analyses, Alchemilla s.l. contains four clades: Afromilla, Aphanes, Eualchemilla, and Lachemilla (Supplementary Table S1 available on Dryad at https://doi.org/10.5061/dryad.cc2fqz660). Together they form a well-supported clade nested in the subtribe Fragariinae, which also includes the cultivated strawberry (Gehrke et al. 2008). Unlike the more commonly recognized members of the rose family (Rosaceae), Alchemilla s.l. is characterized by small flowers with no petals, and a reduced number (1–4[–5]) of stamens that have anthers with one elliptic theca on the ventral side of the connective that opens by one transverse split (Perry 1929; Soják 2008). Gehrke et al. (2008) presented the first phylogeny of Alchemilla s.l. and established the paraphyly of traditional Alchemilla (in the strict sense) as consisted of a primarily African clade, Afromilla, and a Eurasian clade, Eualchemilla. Gehrke et al. (2008) also suggested treating Afromilla and Eualchemilla, along with Aphanes and Lachemilla as a single genus based on nomenclatural stability and the lack of morphological characters to distinguish between Afromilla and Eualchemilla. The four clades within Alchemilla s.l. are mainly defined by geographic distribution, as well as the number and insertion of the stamens on the disk lining the hypanthium (Supplementary Table S1 available on Dryad). Phylogenetic analyses using at least one nuclear and one chloroplast marker (Gardner et al. 2016, Gehrke et al. 2008) found significant cytonuclear discordance regarding the relationships among the four major clades. Similar patterns, often attributed to hybridization and allopolyploidy, have been detected in other genera of Fragariinae (Lundberg 2009; Eriksson et al. 2015; Gehrke et al. 2008, Kamneva et al. 2017; Morales-Briones et al. 2018a), leaving the phylogenetic relationships of Alchemilla s.l. to the rest of Fragariinae unresolved. Unlike most members of Fragariinae that have predominantly diploid species, Alchemilla s.l. is known for high rates of polyploidy. The base chromosome number of Alchemilla s.l. is eight (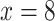 ), which differs from all other members of Fragariinae that have a base of number of seven (
), which differs from all other members of Fragariinae that have a base of number of seven (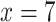 ; Dickinson et al. 2007; Lundberg et al. 2009). Ploidy levels have been well documented in Eualchemilla that shows only polyploid species (
; Dickinson et al. 2007; Lundberg et al. 2009). Ploidy levels have been well documented in Eualchemilla that shows only polyploid species (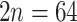 to 220–224; octoploid to 28-ploid; e.g., Turesson 1943; Izmailow 1981; Walters and Boznan 1967; Hayirhoglu-Ayaz et al. 2006). Aphanes has mainly diploid species (
to 220–224; octoploid to 28-ploid; e.g., Turesson 1943; Izmailow 1981; Walters and Boznan 1967; Hayirhoglu-Ayaz et al. 2006). Aphanes has mainly diploid species (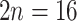 ), except for Aphanes arvensis that is hexaploid (
), except for Aphanes arvensis that is hexaploid (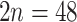 ; Montgomery 1997). Lachemilla has mostly polyploid members (
; Montgomery 1997). Lachemilla has mostly polyploid members (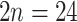 –96; triploid to 12-ploid) with a single species reported to have diploid (
–96; triploid to 12-ploid) with a single species reported to have diploid (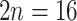 ) and triploid (
) and triploid (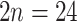 ) populations (Morales-Briones et al. 2018a). Lastly, little is known about ploidy levels in Afromilla, but so far, the two species reported were both polyploids (
) populations (Morales-Briones et al. 2018a). Lastly, little is known about ploidy levels in Afromilla, but so far, the two species reported were both polyploids (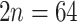 –80; octoploid and decaploid; Hjelmqvist 1956; Morton 1993). A recent phylogenomic analysis focused on Lachemilla using target enrichment data and 32 species of the group detected a high frequency of paralogs shared with Eualchemilla and Afromilla (Morales-Briones et al. 2018b). This paralog frequency suggested a possible ancient WGD event; however, the sampling was limited to one species each of Eualchemilla and Afromilla, and the location and mode of this putative WGD remained uncertain.
–80; octoploid and decaploid; Hjelmqvist 1956; Morton 1993). A recent phylogenomic analysis focused on Lachemilla using target enrichment data and 32 species of the group detected a high frequency of paralogs shared with Eualchemilla and Afromilla (Morales-Briones et al. 2018b). This paralog frequency suggested a possible ancient WGD event; however, the sampling was limited to one species each of Eualchemilla and Afromilla, and the location and mode of this putative WGD remained uncertain.
In this study, we aim to explore the utility of tree-based methods for 1) paralog processing and orthology inference and 2) detecting and placing WGDs in target enrichment data sets. To this, we used Alchemilla s.l., which has a history of prevalent polyploidy and reticulation. We sampled 68 species across the major clades of Alchemilla s.l., and included 11 additional closely related species of Fragariinae, which allowed us to 1) test for polyploid events in the origin of Alchemilla s.l. and 2) explore the reticulate evolution among major clades of Alchemilla s.l. using a target enrichment data set.
Materials and Methods
Taxon Sampling and Data Collection
We sampled 68 species representing the four major clades of Alchemilla s.l. (sensu Gehrke et al. 2008), and 11 species to represent all other genera in Fragariinae (except Chamaecallis; sensu Dobeš et al. 2015; Morales-Briones and Tank 2019). Additionally, we sampled one species each of Potentilla, Sanguisorba, and Rosa as outgroups. Voucher information is provided in Supplementary Table S2 available on Dryad. We used a Hyb-Seq approach (Weitemier et al. 2014), that combines target enrichment and genome skimming, to capture nuclear exon and off-target chloroplast (cpDNA) sequences. We used baits designed for Fragaria vesca (strawberry, also a member of Fragariinae) to target 1419 exons in 257 genes (Liston 2014; Kamneva et al. 2017). These genes were identified as single-copy orthologs among the apple (Malus domestica), peach (Prunus persica), and strawberry genomes based on reciprocal nucleotide similarity comparisons. The 257 genes resulted from first retaining only genes 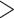 960 bp long and with
960 bp long and with 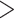 85% similarity in pairwise comparisons among the three genomes. The remaining genes were further filtered by removing exons
85% similarity in pairwise comparisons among the three genomes. The remaining genes were further filtered by removing exons 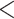 80 bp long, with GC content
80 bp long, with GC content 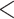 30% or
30% or 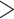 70%, and with
70%, and with 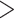 90% sequence similarity to annotated repetitive DNA in the genome, followed by removing exons with any paralogs with
90% sequence similarity to annotated repetitive DNA in the genome, followed by removing exons with any paralogs with 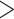 90% sequence similarity in the same genome (Liston 2014; Kamneva et al. 2017).
90% sequence similarity in the same genome (Liston 2014; Kamneva et al. 2017).
Of the 82 total species, only sequences for Fragaria vesca, were from a reference genome (Shulaev et al. 2011). Twenty-two were from a previously published Hyb-Seq data set using the same bait set as this study (Morales-Briones et al. 2018b; Supplementary Table S2 available on Dryad), including 19 species of Lachemilla that did not show evidence of hybridization within Lachemilla, and one species each of Eualchemilla, Afromilla, and Aphanes. Newly generated sequence data for 55 species (Supplementary Table S2 available on Dryad) were collected as follows. Total genomic DNA was isolated from silica-dried or herbarium material with a modified CTAB method (Doyle and Doyle 1987). Probe synthesis, library preparation, hybridization enrichment, and high-throughput sequencing (HiSeq2000 instrument, 2  101 bp) were carried out at Rapid Genomics LLC (Gainesville, FL, USA). Data for the remaining four species, Drymocallis glandulosa, Potentilla indica, Rosa woodsii, and Sanguisorba menziesii were collected as described in Weitemier et al. (2014).
101 bp) were carried out at Rapid Genomics LLC (Gainesville, FL, USA). Data for the remaining four species, Drymocallis glandulosa, Potentilla indica, Rosa woodsii, and Sanguisorba menziesii were collected as described in Weitemier et al. (2014).
Read Processing and Assembly
We removed sequencing adaptors and trimmed low-quality bases (Phred scores 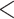 20) from raw reads with SeqyClean v.1.10.07 (Zhbannikov et al. 2017) using default settings. Plastomes were assembled using Alignreads v.2.5.2 (Straub et al. 2011) and 12 closely related plastome references (with one Inverted Repeat removed; Supplementary Table S2 available on Dryad). Plastome assemblies were annotated using Fragaria vesca as a reference in Geneious v.11.1.5 (Kearse et al. 2012). Assembly of nuclear loci was carried out with HybPiper v.1.3.1 (Johnson et al. 2016) using exons of F. vesca as references. Given the large number of paralogs detected in Lachemilla, Eualchemilla, and Afromilla, multiexon gene assemblies resulted in chimeric sequences of exons from distinct paralogs (Morales-Briones et al. 2018b). To avoid chimeric sequences that can affect orthology inference and phylogenetic analyses, assemblies were performed on each exon separately. Only exons with a reference length of
20) from raw reads with SeqyClean v.1.10.07 (Zhbannikov et al. 2017) using default settings. Plastomes were assembled using Alignreads v.2.5.2 (Straub et al. 2011) and 12 closely related plastome references (with one Inverted Repeat removed; Supplementary Table S2 available on Dryad). Plastome assemblies were annotated using Fragaria vesca as a reference in Geneious v.11.1.5 (Kearse et al. 2012). Assembly of nuclear loci was carried out with HybPiper v.1.3.1 (Johnson et al. 2016) using exons of F. vesca as references. Given the large number of paralogs detected in Lachemilla, Eualchemilla, and Afromilla, multiexon gene assemblies resulted in chimeric sequences of exons from distinct paralogs (Morales-Briones et al. 2018b). To avoid chimeric sequences that can affect orthology inference and phylogenetic analyses, assemblies were performed on each exon separately. Only exons with a reference length of 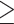 150 bp were assembled (939 exons from 257 genes). Paralog detection was carried out for all exons with the “paralog_investigator” option in HybPiper. This option flags loci with potential paralogs when multiple contigs cover at least 75% of the reference sequence length. Exon assemblies that included flagged paralogs were extracted using the “paralog_retriever” command of HybPiper and used for orthology inference.
150 bp were assembled (939 exons from 257 genes). Paralog detection was carried out for all exons with the “paralog_investigator” option in HybPiper. This option flags loci with potential paralogs when multiple contigs cover at least 75% of the reference sequence length. Exon assemblies that included flagged paralogs were extracted using the “paralog_retriever” command of HybPiper and used for orthology inference.
Orthology Inference for Nuclear Exons
To infer orthologs for phylogenetic analyses, all exons were processed as follows (Fig. 1a). Individual exons were aligned using MACSE v.2.03 (Ranwez et al. 2018) with default parameters. Codons with frameshifts (labeled with “!” by MACSE) were replaced with gaps and aligned columns with more than 90% missing data were removed using Phyx (Brown et al. 2017). Initial homolog trees were built using RAxML v.8.2.11 (Stamatakis 2014) with a GTRCAT model and clade support assessed with 100 rapid bootstrap (BS) replicates. Clades and paraphyletic grades that belonged to the same taxon were pruned by keeping only the tip with the highest number of characters in the trimmed alignment following Yang and Smith (2014). To obtain the final homolog trees, outlier tips with unusually long branches were detected and removed by maximally reducing the tree diameter with TreeShrink v.1.3.2 (Mai and Mirarab 2018). Orthology inference was carried out using two outgroup-aware strategies from Yang and Smith (2014). We set Potentilla, Sanguisorba, and Rosa as outgroups and all members of Fragariinae as ingroups. First, we used the “monophyletic outgroup” (MO) approach keeping only ortholog groups with at least 25 ingroup taxa. The MO approach filters for homolog trees with outgroup taxa being monophyletic and single-copy, and therefore filters for single- and low-copy genes. The second approach used was the “rooted ingroup” (RT), with at least 25 ingroup taxa. The RT approach iteratively searches subtrees of ingroup taxa and cuts them out as rooted trees. Both approaches root the gene tree by the outgroups, traverse the rooted tree from root to tip, and remove the side with fewer taxa (MO) or keep both sides (RT) when gene duplication is detected at any given node. In the case of MO, homolog trees with nonmonophyletic outgroups or duplicated taxa in the outgroups are discarded. If no taxon duplication is detected in a homolog tree, the MO approach outputs a one-to-one ortholog. The RT approach maximizes the number of orthologs compared to MO while not requiring monophyletic outgroups and allowing for duplicated taxa in the outgroups but removes outgroups from all orthologs. To add outgroups back to the RT orthologs for downstream analyses, we kept only RT orthologs from homologs that had a MO ortholog (i.e., using only homolog trees with monophyletic and nonduplicated outgroups for both MO and RT). Then, we used the outgroups of the MO ortholog for all the RT orthologs of the same homolog (Fig. 1b). Scripts for homolog tree estimation and orthology inference can be found at https://bitbucket.org/dfmoralesb/target_enrichment_orthology.
Figure 1.

Homolog and ortholog inference workflow used in this study. a) Flow chart of paralog processing and homolog tree inference. b) Only homologs with outgroup present and monophyletic were used for orthology inference. Monophyletic outgroups (MO) will prune single-copy genes keeping clades with at least a user-defined minimum number of ingroup taxa. Rooted ingroups (RT) will keep all subtrees with at least a user-defined minimum number of ingroups taxa. If the homolog trees can be pruned using both MO and RT, then RT orthologs are added to the same root. Homologs that lack monophyletic outgroups were excluded from further consideration.
Phylogenetic Analyses
We used concatenation and coalescent-based methods to reconstruct the phylogeny of Alchemilla s.l. Analyses were carried out in the two sets of final orthologs, MO (910 orthologs) and RT (1894 orthologs), separately. Each ortholog was aligned using MACSE with default parameters. Codons with frameshifts were replaced with gaps, aligned columns with more than 90% missing data were removed using Phyx, and alignments with at least 150 characters and 25 taxa were retained. We first estimated a maximum likelihood (ML) tree from the concatenated alignments with RAxML using a partition by gene scheme with a GTRGAMMA model for each partition. Clade support was assessed with 100 rapid bootstrap (BS) replicates. To estimate a species tree that is statistically consistent with the multispecies coalescent (MSC), we first inferred individual ML gene trees using RAxML with a GTRGAMMA model, and 100 BS replicates to assess clade support. Individual gene trees were then used to estimate a species tree using ASTRAL-III v.5.6.3 (Zhang et al. 2018) with local posterior probabilities (LPP; Sayyari and Mirarab 2016) to assess clade support.
To evaluate nuclear gene tree discordance, we calculated the internode certainty all (ICA) score to quantify the degree of conflict on each node of a species tree given individual gene trees (Salichos et al. 2014). Also, we calculated the number of conflicting and concordant bipartitions on each node of a species tree. We calculated both the ICA scores and the number of conflicting/concordant bipartitions with Phyparts (Smith et al. 2015) using individual ortholog trees with BS support of at least 50% for the corresponding node. The proportion of conflicting/concordant bipartitions was visualized using pie charts taking in consideration missing data, which is typically expected in target enrichment data sets. Pie chart visualization scripts can be found at https://bitbucket.org/dfmoralesb/target_enrichment_orthology. Additionally, to distinguish strong conflict from weakly supported branches, we evaluated tree conflict and branch support with Quartet Sampling (QS; Pease et al. 2018) using 1000 replicates. Quartet Sampling subsamples quartets from an input tree (e.g., species tree or cpDNA tree) and concatenated alignment to assess the confidence, consistency, and informativeness of each internal branch by the relative frequency of the three possible quartet topologies at each node (Pease et al. 2018).
In addition to species tree reconstruction using inferred orthologs, we used a recently developed quartet-based species tree method (ASTRAL-Pro; Zhang et al. 2020a) to estimate the phylogeny of Alchemilla s.l. ASTRAL-Pro directly uses multilabeled trees (MUL-tree) to estimate a species tree that is statistically consistent with the MSC and a birth–death gene duplication and loss model. We used all 923 final homolog trees as input for ASTRAL-Pro, ignoring trees with less than 20 taxa, and estimated LPP to assess clade support. Additionally, we calculated ICA scores and the number of conflicting/concordant bipartitions with Phyparts using homolog trees with BS support of at least 50% for the corresponding nodes.
For the plastome phylogenetic analyses, 74 partial plastome assemblies and nine reference plastome sequences were included (Supplementary Table S3 available on Dryad). Contiguous plastome sequences were aligned using MAFFT v.7.307 (Katoh and Standley 2013) with the default settings and aligned columns with more than 70% missing data were removed with Phyx. We estimated an ML tree of the plastome alignment with RAxML using a partition by coding (CDS) and noncoding regions (introns and intergenic spacers) scheme, with a GTRGAMMA model for each partition and clade support assessed with 100 rapid BS replicates. In addition, we carried out QS analysis with 1000 replicates to detect potential within-plastome conflict in the backbone of Alchemilla s.l. as recently reported in other groups (e.g., Gonçalves et al. 2019; Walker et al. 2019; Zhang et al. 2020b; Morales-Briones et al. 2021).
Mapping Whole Genome Duplications based on Homologous Gene Tree Topology
We took two alternative approaches for detecting WGD events by mapping gene duplications from gene trees onto an inferred species tree. Here, we refer to trees inferred using either concatenation or coalescent-based method as the “species tree” that we use to map individual gene trees onto.
The first approach begins by extracting orthogroups from the final homolog trees. Orthogroups are rooted ingroup lineages separated by outgroups that include the complete set of genes in a lineage from a single copy in their common ancestor. We extracted orthogroups requiring at least 50 out of 79 species in Fragariinae. Gene duplication events were then recorded on the most recent common ancestor (MRCA) on the species tree when two or more species overlapped between the two daughter clades. Each node on a species tree can be counted only once from each gene tree to avoid nested gene duplications inflating the number of recorded duplications (Yang et al. 2018; https://bitbucket.org/blackrim/clustering, “extract_clades.py” and “map_dups_mrca.py”). We mapped duplication events onto both the MO and RT trees using orthogroups from final homologs, filtering orthogroups using an average BS of at least 50%. We carried out the mapping using two sets of orthogroups, one from all homologs (923), and from the longest homologs (the single longest aligned exon per gene; 256) to avoid inflating the counts in multiexon genes.
For the second strategy of WGD mapping, we explicitly tested for polyploidy mode using GRAMPA (Thomas et al. 2017). GRAMPA uses MRCA reconciliation with MUL-trees to compare allo- and autopolyploid scenarios in singly-labeled or multilabeled trees. To reduce the computational burden of searching all possible reconciliations, we constrained searches to only among major clades of Alchemilla s.l., each of which are well supported (including the “dissected” and “lobed” clades of Eualchemilla; see results) and among genera in Fragariinae. We ran reconciliation searches using all 923 final homologs, as well as using only the 256 longest homologs against the MO and RT RAxML trees. We expected multiple WGD events within Alchemilla s.l. (see Results), but GRAMPA can only infer one WGD at a time. To disentangle nested duplication events, we also carried out similar GRAMPA reconciliations using the MO tree and sequentially excluding major groups of Alchemilla s.l. that were identified as a polyploid clade. We only used the MO tree as it differs from the RT tree only by the location of the “lobed” clade, which was the first clade identified as allopolyploid (see Results) and was removed for subsequent GRAMPA analyses. Finally, to test for a polyploid origin of Alchemilla s.l., we carried out GRAMPA constrained searches among Alchemilla s.l. and the remaining genera in Fragariinae using the MO and cpDNA trees. The backbone of Fragariinae differed between the MO (same as RT) tree and the cpDNA tree. Thus, we tested how this affected the inference of the polyploid origin of Alchemilla s.l. We also carried out similar searches but using each of the five major clades (including the “dissected” and “lobed” clades of Eualchemilla; see Results) individually.
Both approaches used here to detect WGD events use final homolog trees and as any other tree-based method they may be sensitive to tree informativeness. To explore node support across individual homologs, we ran a conflict analysis with Phyparts using individual final homologs trees. We ran the analysis using all homolog exons as well as only the longest homolog exon per gene to map against both the MO and RT trees.
Distribution of Synonymous Distance among Gene Pairs (Ks plots)
To obtain further evidence for WGD events and compare them to those inferred from gene duplication events from target enrichment data, we analyzed the distribution of synonymous distances (Ks) from RNA-seq data of four species of Alchemilla s.l. and nine species of Fragariinae (Supplementary Table S4 available on Dryad). Read processing and transcriptome assembly followed Morales-Briones et al. (2021). For each species, a Ks plot of within-species paralog pairs based on BLASTP hits was done following Yang et al. (2018; https://bitbucket.org/blackrim/clustering; “ks_plots.py”). Ks peaks were identified using a mixture model as implemented in mixtools v.1.2.0 (Benaglia et al. 2009). The optimal number of mixing components was estimated using parametric bootstrap replicates of the likelihood ratio test statistic (McLachlan and Peel 2000). We tested up to five components using 500 bootstrap replicates in mixtools. Additionally, we used between-species Ks plots to determine the relative timing of the split between two species and compare it to that of WGD events inferred with within-species Ks plots. Ks plots of between-species also followed Yang et al. (2018; “ks_between_taxa_cds.py”). Lastly, we also attempted to build Ks plots using raw homologs from the target enrichment data, but the relatively low number of genes (256) failed to produce a meaningful distribution (not shown).
Results
Assembly and Orthology Inference
The number of assembled exons per species (with 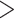 75% of the target length) ranged from 632 (Alchemilla fissa) to 934 (Dasiphora fruticosa) out of 939 single-copy exon references from F. vesca, with an average of 873 exons (Supplementary Table S5 available on Dryad). The number of exons with paralog warnings ranged from 10 in Drymocallis glandulosa to 746 in Alchemilla mollis (Supplementary Table S5 available on Dryad). The number of exon alignments with
75% of the target length) ranged from 632 (Alchemilla fissa) to 934 (Dasiphora fruticosa) out of 939 single-copy exon references from F. vesca, with an average of 873 exons (Supplementary Table S5 available on Dryad). The number of exons with paralog warnings ranged from 10 in Drymocallis glandulosa to 746 in Alchemilla mollis (Supplementary Table S5 available on Dryad). The number of exon alignments with 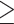 25 species was 923 from 256 genes. The orthology inference resulted in 914 MO orthologs (Supplementary Table S6 available on Dryad), and 1906 RT orthologs (Supplementary Table S6 available on Dryad). The trimmed alignments of the MO orthologs ranged from 136 to 5740 characters with a mean of 425 characters (median
25 species was 923 from 256 genes. The orthology inference resulted in 914 MO orthologs (Supplementary Table S6 available on Dryad), and 1906 RT orthologs (Supplementary Table S6 available on Dryad). The trimmed alignments of the MO orthologs ranged from 136 to 5740 characters with a mean of 425 characters (median 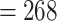 ). The concatenated alignment of the MO orthologs, with at least 150 aligned characters and 25 species for each exon, included 910 exons and 387,042 characters with a matrix occupancy of 66%. The trimmed alignments of the RT orthologs ranged from 136 to 5740 characters with a mean of 394 characters (median
). The concatenated alignment of the MO orthologs, with at least 150 aligned characters and 25 species for each exon, included 910 exons and 387,042 characters with a matrix occupancy of 66%. The trimmed alignments of the RT orthologs ranged from 136 to 5740 characters with a mean of 394 characters (median 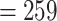 ). The concatenated alignment of the RT orthologs, with at least 150 aligned characters and 25 species, included 1894 exons and 746,562 characters with a matrix occupancy of 54%. The cpDNA alignment included 124,079 characters with a matrix occupancy of 77%.
). The concatenated alignment of the RT orthologs, with at least 150 aligned characters and 25 species, included 1894 exons and 746,562 characters with a matrix occupancy of 54%. The cpDNA alignment included 124,079 characters with a matrix occupancy of 77%.
Nuclear Phylogenetic Analyses
All nuclear analyses recovered the monophyly of Alchemilla s.l. with maximum support (i.e., bootstrap percentage [BS] 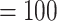 , local posterior probabilities [LPP]
, local posterior probabilities [LPP] 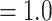 ; Fig. 2; Supplementary Fig. S1 available on Dryad), most informative gene trees being concordant with this node (858 out of 863 for MO; 977/984 for RT; 912/932 for ASTRAL-Pro; ICA
; Fig. 2; Supplementary Fig. S1 available on Dryad), most informative gene trees being concordant with this node (858 out of 863 for MO; 977/984 for RT; 912/932 for ASTRAL-Pro; ICA 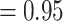 ), and full QS support (1.0/–/1.0; i.e., all sampled quartets supported that node). Five major clades were identified within Alchemilla s.l.: Afromilla, Aphanes, Eualchemilla-“dissected”, Eualchemilla-“lobed,” and Lachemilla. The relationships among these clades showed high levels of discordance and varied among the MO and RT results.
), and full QS support (1.0/–/1.0; i.e., all sampled quartets supported that node). Five major clades were identified within Alchemilla s.l.: Afromilla, Aphanes, Eualchemilla-“dissected”, Eualchemilla-“lobed,” and Lachemilla. The relationships among these clades showed high levels of discordance and varied among the MO and RT results.
Figure 2.

a) Maximum likelihood phylogeny of Alchemilla s.l. inferred from RAxML analysis of the concatenated 910-nuclear exon supermatrix from the “monophyletic outgroup” (MO) orthologs. Bootstrap support (BS) and Local posterior probability (LLP) are shown above branches. Nodes with full support (BS 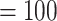 /LLP
/LLP 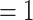 ) are noted with an asterisk (*). Em dashes (—) denoted alternative topology compared to the ASTRAL tree (not shown). Quartet Sampling (QS) scores for major clades are shown below branches. QS scores in blue indicate strong support and red scores indicate weak support. QS scores: Quartet concordance/Quartet differential/Quartet informativeness. QS score
) are noted with an asterisk (*). Em dashes (—) denoted alternative topology compared to the ASTRAL tree (not shown). Quartet Sampling (QS) scores for major clades are shown below branches. QS scores in blue indicate strong support and red scores indicate weak support. QS scores: Quartet concordance/Quartet differential/Quartet informativeness. QS score  1/—/1 denotes maximum support. Pie charts for major clades represent the proportion of exon ortholog trees that support that clade (blue), the proportion that support the main alternative bifurcation (green), the proportion that support the remaining alternatives (red), and the proportion (conflict or support) that have
1/—/1 denotes maximum support. Pie charts for major clades represent the proportion of exon ortholog trees that support that clade (blue), the proportion that support the main alternative bifurcation (green), the proportion that support the remaining alternatives (red), and the proportion (conflict or support) that have 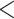 50% bootstrap support (gray). Gene trees with missing data that were uninformative for the node were ignored. Branch lengths are in number of substitutions per site (scale bar on the bottom). Inset: b) Summary maximum likelihood phylogeny inferred from RAxML analysis of the concatenated 1,894-nuclear exon supermatrix from the “rooted ingroup” orthologs (RT). BS and LLP are shown above branches and QS scores below the branches. Branch lengths are in number of substitutions per site. See Supplementary Fig. S1 available on Dryad for expanded tree.; c) Summary ASTRAL-Pro tree inferred from 923 multilabeled exon homolog trees. LLP are shown next to nodes. Branch lengths are in coalescent units. See Supplementary Fig. S2 available on Dryad for expanded tree; d) Summary maximum likelihood phylogeny inferred from RAxML analysis of concatenated partial plastomes. BS and LLP are shown above branches and QS scores below the branches. Branch lengths are in number of substitutions per site. See Supplementary Fig. S4 available on Dryad for expanded tree.
50% bootstrap support (gray). Gene trees with missing data that were uninformative for the node were ignored. Branch lengths are in number of substitutions per site (scale bar on the bottom). Inset: b) Summary maximum likelihood phylogeny inferred from RAxML analysis of the concatenated 1,894-nuclear exon supermatrix from the “rooted ingroup” orthologs (RT). BS and LLP are shown above branches and QS scores below the branches. Branch lengths are in number of substitutions per site. See Supplementary Fig. S1 available on Dryad for expanded tree.; c) Summary ASTRAL-Pro tree inferred from 923 multilabeled exon homolog trees. LLP are shown next to nodes. Branch lengths are in coalescent units. See Supplementary Fig. S2 available on Dryad for expanded tree; d) Summary maximum likelihood phylogeny inferred from RAxML analysis of concatenated partial plastomes. BS and LLP are shown above branches and QS scores below the branches. Branch lengths are in number of substitutions per site. See Supplementary Fig. S4 available on Dryad for expanded tree.
Analyses of the MO orthologs using concatenation and coalescent-based approaches resulted in similar topologies for the backbone of Alchemilla s.l. (Fig. 2a). The monophyly of the five major clades each received maximum support (BS 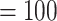 ; LPP
; LPP 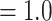 ) and had most trees being concordant (except for the two clades of Eualchemilla). Eualchemilla was paraphyletic and split into the “dissected” and “lobed” clades. Monophyly of the “dissected” clade was supported by 118 out of 429 informative trees (ICA
) and had most trees being concordant (except for the two clades of Eualchemilla). Eualchemilla was paraphyletic and split into the “dissected” and “lobed” clades. Monophyly of the “dissected” clade was supported by 118 out of 429 informative trees (ICA 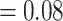 ) and strong QS score (0.87/0.34/1), while the “lobed” clade was supported by 73 out of 420 informative trees (ICA
) and strong QS score (0.87/0.34/1), while the “lobed” clade was supported by 73 out of 420 informative trees (ICA  0.06) and strong QS score (0.61/0.98/0.99). In both cases, the “dissected” and “lobed” clades had a relatively small percentage of supporting trees, but the conflict analysis and QS score did not reveal any well-supported alternative topology. Aphanes was recovered as sister to the Eualchemilla-“lobed” clade with relatively low support (BS
0.06) and strong QS score (0.61/0.98/0.99). In both cases, the “dissected” and “lobed” clades had a relatively small percentage of supporting trees, but the conflict analysis and QS score did not reveal any well-supported alternative topology. Aphanes was recovered as sister to the Eualchemilla-“lobed” clade with relatively low support (BS 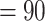 , LPP
, LPP 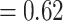 ), 60 concordant trees (out of 430 informative gene trees; ICA
), 60 concordant trees (out of 430 informative gene trees; ICA 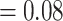 ), and a weak QS score (0.016/0.95/0.98) with similar frequencies for the two discordant alternative topologies. The Eualchemilla-“dissected” clade was recovered as sister to Eualchemilla-“lobed”
), and a weak QS score (0.016/0.95/0.98) with similar frequencies for the two discordant alternative topologies. The Eualchemilla-“dissected” clade was recovered as sister to Eualchemilla-“lobed” 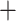 Aphanes with maximum support, 279 concordant trees (out of 482 informative gene trees; ICA
Aphanes with maximum support, 279 concordant trees (out of 482 informative gene trees; ICA 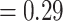 ), and full QS score. Afromilla was recovered as sister to the clade consisted of Eualchemilla (“dissected” and “lobed”) and Aphanes with high to low support (BS
), and full QS score. Afromilla was recovered as sister to the clade consisted of Eualchemilla (“dissected” and “lobed”) and Aphanes with high to low support (BS 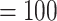 , LPP
, LPP 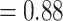 ), only 146 concordant trees (out of 413 informative gene trees; ICA
), only 146 concordant trees (out of 413 informative gene trees; ICA 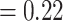 ), and weak QS support (0.2/0.44/0.99) with a skew in discordance suggesting a possible alternative topology (Lachemilla sister to Eualchemilla
), and weak QS support (0.2/0.44/0.99) with a skew in discordance suggesting a possible alternative topology (Lachemilla sister to Eualchemilla 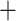 Aphanes). Lastly, Lachemilla was recovered as the sister to the rest of Alchemilla s.l.
Aphanes). Lastly, Lachemilla was recovered as the sister to the rest of Alchemilla s.l.
Analysis of the RT orthologs using concatenation and coalescent-based approaches both recovered the same major clades, but they differed in the relationship among these five clades (Fig. 2b; Supplementary Fig. S1 available on Dryad). In both analyses, Lachemilla, Afromilla, and Aphanes had maximum support (BS 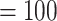 ; LPP
; LPP 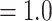 ) and had most trees being concordant. Eualchemilla was recovered as monophyletic and composed of the “dissected” and “lobed” clades. The monophyly of Eualchemilla had high to low support (BS
) and had most trees being concordant. Eualchemilla was recovered as monophyletic and composed of the “dissected” and “lobed” clades. The monophyly of Eualchemilla had high to low support (BS 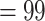 , LPP
, LPP 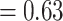 ), only 231 concordant trees (out of 819 informative gene trees; ICA
), only 231 concordant trees (out of 819 informative gene trees; ICA 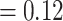 ), and weak QS support (0.023/0.87/0.98) with similar frequencies for the two discordant alternative topologies. Similar to the MO analyses, the “dissected” and “lobed” clades each had low number of concordant trees (218 out of 557 [ICA
), and weak QS support (0.023/0.87/0.98) with similar frequencies for the two discordant alternative topologies. Similar to the MO analyses, the “dissected” and “lobed” clades each had low number of concordant trees (218 out of 557 [ICA 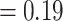 ] and 136 out of 707 [ICA
] and 136 out of 707 [ICA 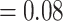 ], respectively), and strong QS support (0.98/0/1 and 0.62/0.17/0.99, respectively). Eualchemilla was recovered as sister of Aphanes with maximum support 348 concordant trees (out of 728 informative trees; ICA
], respectively), and strong QS support (0.98/0/1 and 0.62/0.17/0.99, respectively). Eualchemilla was recovered as sister of Aphanes with maximum support 348 concordant trees (out of 728 informative trees; ICA 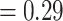 ), and full QS support. The RAxML concatenated tree (Fig. 2b; Supplementary Fig. S1a available on Dryad) placed Afromilla as sister to Eualchemilla
), and full QS support. The RAxML concatenated tree (Fig. 2b; Supplementary Fig. S1a available on Dryad) placed Afromilla as sister to Eualchemilla 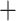 Aphanes with maximum support (BS
Aphanes with maximum support (BS 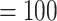 ), 212 concordant gene trees (out of 771 informative trees; ICA
), 212 concordant gene trees (out of 771 informative trees; ICA 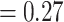 ), and weak QS support (0.18/0.66/0.99) with no significant skew between the two discordant alternatives. Lachemilla was placed as sister to the rest of Alchemilla s.l. The ASTRAL tree in turn (Supplementary Fig. S1b available on Dryad), retrieved Lachemilla as sister to Eualchemilla
), and weak QS support (0.18/0.66/0.99) with no significant skew between the two discordant alternatives. Lachemilla was placed as sister to the rest of Alchemilla s.l. The ASTRAL tree in turn (Supplementary Fig. S1b available on Dryad), retrieved Lachemilla as sister to Eualchemilla 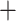 Aphanes with no support (LPP
Aphanes with no support (LPP 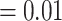 ), 247 concordant trees (out of 953 informative trees; ICA
), 247 concordant trees (out of 953 informative trees; ICA 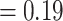 ), and QS counter-support (
), and QS counter-support ( 0.21/0.29/0.99), showing that the majority of the quartets supported one alternative topology (Afromilla sister to Eualchemilla
0.21/0.29/0.99), showing that the majority of the quartets supported one alternative topology (Afromilla sister to Eualchemilla 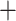 Aphanes). In this case, Afromilla was placed as sister to the rest of Alchemilla s.l.
Aphanes). In this case, Afromilla was placed as sister to the rest of Alchemilla s.l.
The ASTRAL-Pro analysis using multilabeled homolog trees recovered the same backbone topology of Alchemilla s.l. as the concatenated ML analysis from the RT orthologs (Fig. 2c; Supplementary Figs. S2 and S3 available on Dryad). All five major clades had the maximum support (LPP 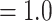 ). Eualchemilla, composed of the “dissected” and “lobed” clades, had moderate support (LPP
). Eualchemilla, composed of the “dissected” and “lobed” clades, had moderate support (LPP 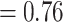 ) and only 415 concordant trees (out of 1106 informative trees; ICA
) and only 415 concordant trees (out of 1106 informative trees; ICA 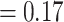 ). The “dissected” and “lobed” clades had low numbers of concordant trees (379 out of 941 [ICA
). The “dissected” and “lobed” clades had low numbers of concordant trees (379 out of 941 [ICA 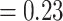 ] and 65 out of 824 [ICA
] and 65 out of 824 [ICA 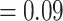 ], respectively) but did not show signal of any alternative topology. Aphanes was placed as the sister of Eualchemilla with maximum support (LPP
], respectively) but did not show signal of any alternative topology. Aphanes was placed as the sister of Eualchemilla with maximum support (LPP 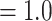 ), 426 concordant trees (out of 952 trees; ICA
), 426 concordant trees (out of 952 trees; ICA 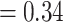 ), and no support for any major alternative topology. Afromilla was recovered as sister to Eualchemilla
), and no support for any major alternative topology. Afromilla was recovered as sister to Eualchemilla 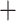 Aphanes with low support (LPP
Aphanes with low support (LPP 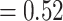 ), 492 concordant trees (out of 953 trees; ICA
), 492 concordant trees (out of 953 trees; ICA 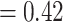 ), and no support for any alternative topology.
), and no support for any alternative topology.
Chloroplast Phylogenetic Analyses
The chloroplast ML tree (Fig. 2d; Supplementary Fig. S4 available on Dryad) recovered a well-supported backbone Alchemilla s.l. where the monophyly of Aphanes, Afromilla, and Lachemilla, had maximum or near-maximum support (i.e., bootstrap percentage [BS] 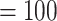 , QS support
, QS support  [1.0/–/1.0]). Eualchemilla, composed of the “dissected” and “lobed” clades, also had the maximum support. The “dissected” and “lobed” clades had strong support (BS
[1.0/–/1.0]). Eualchemilla, composed of the “dissected” and “lobed” clades, also had the maximum support. The “dissected” and “lobed” clades had strong support (BS 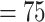 , QS
, QS 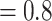 /0.43/0.88 and BS
/0.43/0.88 and BS 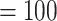 , QS
, QS 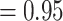 /0.25/0.92, respectively). Aphanes and Eualchemilla formed, with maximum support, a clade as in the nuclear analyses. In turn, Afromilla and Lachemilla were recovered as sister clades with maximum support, which differed from the nuclear analyses.
/0.25/0.92, respectively). Aphanes and Eualchemilla formed, with maximum support, a clade as in the nuclear analyses. In turn, Afromilla and Lachemilla were recovered as sister clades with maximum support, which differed from the nuclear analyses.
Mapping Whole Genome Duplications
By mapping the most recent common ancestor (MRCA) of gene duplication events from orthogroup trees onto the MO and RT trees, we found four nodes in Alchemilla s.l. that each had an elevated proportion of gene duplications (Fig. 3a,b; Supplementary Fig. S5 available on Dryad). This trend was consistent regardless of using all 923 homolog exons (868 after orthogroup inference and BS filtering) or using only the 256 longest homolog exons per gene (250 after orthogroup inference and BS filtering; Supplementary Fig. S5 available on Dryad). Therefore, here we describe the results only for the latter. These four clades include (Fig. 3a; Supplementary Fig. S5 available on Dryad): 1) the MRCA of Alchemilla s.l. (86.0% of the 250 genes show evidence of duplication), 2) the MRCA of Eualchemilla, Aphanes, and Afromilla (34.4%), 3) the MRCA of Eualchemilla 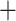 Aphanes (MO: 18.4%; RT:15.6% ), and 4) the MRCA of Lachemilla (18.4%). These four nodes have an elevated proportion of gene duplications compared to all other nodes in Fragariinae (Fig. 3b). These proportions of gene duplications are also consistent with the number of paralogs counted from the final homolog trees (after pruning of clades or paraphyletic grades of the same species; Fig. 3c). Interestingly, although deeply nested in Alchemilla s.l., Aphanes had a lower number of paralogs than the rest of Alchemilla s.l., resembling the other members of Fragariinae (Fig. 3c).
Aphanes (MO: 18.4%; RT:15.6% ), and 4) the MRCA of Lachemilla (18.4%). These four nodes have an elevated proportion of gene duplications compared to all other nodes in Fragariinae (Fig. 3b). These proportions of gene duplications are also consistent with the number of paralogs counted from the final homolog trees (after pruning of clades or paraphyletic grades of the same species; Fig. 3c). Interestingly, although deeply nested in Alchemilla s.l., Aphanes had a lower number of paralogs than the rest of Alchemilla s.l., resembling the other members of Fragariinae (Fig. 3c).
Figure 3.

Orthogroup gene duplication mapping results. a) Summarized cladogram of Alchemilla s.l. from the ASTRAL analysis of “monophyletic outgroup” (MO) ortholog trees. Percentages next to nodes denote the proportion of duplicated genes when using orthogroups from the longest homologs (250 after orthogroup inference and filtering). Nodes with elevated proportions of gene duplications are numbered 1–4 as referenced in the main text. See Supplementary Fig. S5 available on Dryad for the full tree. b) Histogram of percentages of gene duplication per branch. c) Number of paralogs per taxa in the final homolog trees. In final homologs, clades and paraphyletic grades of the same species were pruned, leaving only one tip per species. Each locus is represented by the longest homolog (the single longest aligned exon per gene; 256 total).
Bootstrap support for exon homologs was informative (BS 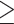 50%) at most nodes, especially regarding the relationship among the major clades of Alchemilla s.l. (Supplementary Fig. S6 available on Dryad). Therefore, uninformative homolog trees were unlikely to affect the results from WGD detection analysis overall. The proportion of uninformative nodes (BS
50%) at most nodes, especially regarding the relationship among the major clades of Alchemilla s.l. (Supplementary Fig. S6 available on Dryad). Therefore, uninformative homolog trees were unlikely to affect the results from WGD detection analysis overall. The proportion of uninformative nodes (BS 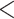 50%) was at most 30% in the worst case (Eualchemilla
50%) was at most 30% in the worst case (Eualchemilla 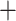 Aphanes
Aphanes 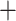 Afromilla) when using all homolog exons. This proportion reduces significantly when using only the longest homolog exons (Supplementary Fig. S6 available on Dryad).
Afromilla) when using all homolog exons. This proportion reduces significantly when using only the longest homolog exons (Supplementary Fig. S6 available on Dryad).
Similar to the results of MRCA mapping, the GRAMPA analyses recovered the same results when using all 923 homologs or only the longest homologs (256). GRAMPA reconciliations using all major clades of Alchemilla s.l. recovered optimal multilabeled trees with the best score (i.e., lowest reconciliation score; Supplementary Fig. S7 available on Dryad) where the “lobed” clade of Eualchemilla was of an allopolyploid origin, but the putative parental lineages varied between the MO and RT trees. The reconciliations using the MO tree (reconciliation score [RS]  70,250; Fig. 4a; Supplementary Fig. S8 available on Dryad) recovered a MUL-tree where the “lobed” clade was of allopolyploid origin between an unsampled or extinct lineage sister to Aphanes and an unsampled or extinct lineage (“lineage” for short hereafter) sister to “dissected”
70,250; Fig. 4a; Supplementary Fig. S8 available on Dryad) recovered a MUL-tree where the “lobed” clade was of allopolyploid origin between an unsampled or extinct lineage sister to Aphanes and an unsampled or extinct lineage (“lineage” for short hereafter) sister to “dissected” 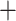 Aphanes. In turn, the reconciliations using the RT tree (RS
Aphanes. In turn, the reconciliations using the RT tree (RS  70,721; Supplementary Fig. S8 available on Dryad) recovered a MUL-tree where the “lobed” clade was of allopolyploid origin between a “lineage” sister to the “dissected” clade, and also a “lineage” sister to “dissected”
70,721; Supplementary Fig. S8 available on Dryad) recovered a MUL-tree where the “lobed” clade was of allopolyploid origin between a “lineage” sister to the “dissected” clade, and also a “lineage” sister to “dissected” 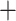 Aphanes. Alternative MUL-trees had higher (worse) RSs (70,482 for MO and 70,739 for RT; Supplementary Fig. S7 available on Dryad). The GRAMPA reconciliations performed on the MO tree with removal of major clades of Alchemilla s.l. inferred as allopolyploid resulted in MUL-trees that suggested additional polyploidy events (Fig. 4b–d). First, we removed the “lobed” clade, and this resulted in the recovery of Afromilla as an allopolyploid clade (RS
Aphanes. Alternative MUL-trees had higher (worse) RSs (70,482 for MO and 70,739 for RT; Supplementary Fig. S7 available on Dryad). The GRAMPA reconciliations performed on the MO tree with removal of major clades of Alchemilla s.l. inferred as allopolyploid resulted in MUL-trees that suggested additional polyploidy events (Fig. 4b–d). First, we removed the “lobed” clade, and this resulted in the recovery of Afromilla as an allopolyploid clade (RS  127,836). Afromilla’s parental lineages were a “lineage” sister to Aphanes
127,836). Afromilla’s parental lineages were a “lineage” sister to Aphanes 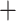 the “dissected” clade, and a “lineage” sister to all remaining Alchemilla s.l. (Fig. 4b). Alternative MUL-tree reconciliations had scores of 127,869 or higher (Supplementary Fig. S7 available on Dryad). The further removal of Afromilla resulted in recovery of the “dissected” clade as allopolyploid (RS
the “dissected” clade, and a “lineage” sister to all remaining Alchemilla s.l. (Fig. 4b). Alternative MUL-tree reconciliations had scores of 127,869 or higher (Supplementary Fig. S7 available on Dryad). The further removal of Afromilla resulted in recovery of the “dissected” clade as allopolyploid (RS  167,545). The “dissected” clade had as parental lineages the “lineage” sister to Aphanes and the “lineage” sister to all remaining Alchemilla s.l. except for Lachemilla (Fig. 4c). Other reconciliation alternatives had scores of 167,612 or higher (Supplementary Fig. S7 available on Dryad). Finally, the removal of the “dissected” clade resulted in the Lachemilla being recovered also as an allopolyploid clade (RS
167,545). The “dissected” clade had as parental lineages the “lineage” sister to Aphanes and the “lineage” sister to all remaining Alchemilla s.l. except for Lachemilla (Fig. 4c). Other reconciliation alternatives had scores of 167,612 or higher (Supplementary Fig. S7 available on Dryad). Finally, the removal of the “dissected” clade resulted in the Lachemilla being recovered also as an allopolyploid clade (RS  181,302). The parental lineages of Lachemilla were a “lineage” sister to Aphanes and a “lineage” sister to all remaining Alchemilla s.l. (Fig. 4d). Alternative MUL-tree reconciliations had scores of 181,564 or higher (Supplementary Fig. S7 available on Dryad).
181,302). The parental lineages of Lachemilla were a “lineage” sister to Aphanes and a “lineage” sister to all remaining Alchemilla s.l. (Fig. 4d). Alternative MUL-tree reconciliations had scores of 181,564 or higher (Supplementary Fig. S7 available on Dryad).
Figure 4.

Summary of optimal multilabeled tree (MUL-tree) inferred from GRAMPA analyses. a) MUL-tree from reconciliations of homologs against the ASTRAL tree inferred from “monophyletic outgroup” (MO) orthologs including all taxa. Red branches denote the allopolyploid origin of the “lobed” clade of Eualchemilla. b) MUL-tree after removing the “lobed” clade of Eualchemilla as in a). Green branches denote the allopolyploid origin of Afromilla. c) MUL-tree after removing Afromilla as in b). Blue branches denote the allopolyploid origin of the “dissected” clade of Eualchemilla. d). MUL-tree after further removing the “dissected” clade as in c). Yellow lines denote the allopolyploid origin of Lachemilla. e) MUL-tree from reconciliations of constrained homologs on the MRCA of Alchemilla s.l. against the cpDNA tree. Orange branches denote the autopolyploid origin Alchemilla s.l. f) Putative summary network of all reticulation events in Alchemilla s.l. Colored curved branches denote different polyploid events as in (a–e). Dashed curved lines represent the chloroplast donor (cpDNA) in allopolyploid events.
The GRAMPA results from the analyses with constrained searches among Alchemilla s.l. and other Fragariinae recovered different modes of polyploidy when using the MO tree or the cpDNA tree. The MO tree had Farinopsis, Sibbaldianthe 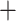 Sibbaldia, Comarum, and Fragaria forming a grade sister to Alchemilla s.l., while Drymocallis, Chamaerhodos, Potaninia, and Dasiphora form a clade that is sister to all other Fragariinae (Fig. 2). The reconciliations using the MO tree suggested an allopolyploid event for the clade composed of Alchemilla s.l., Farinopsis, Sibbaldianthe, and Sibbaldia (RS
Sibbaldia, Comarum, and Fragaria forming a grade sister to Alchemilla s.l., while Drymocallis, Chamaerhodos, Potaninia, and Dasiphora form a clade that is sister to all other Fragariinae (Fig. 2). The reconciliations using the MO tree suggested an allopolyploid event for the clade composed of Alchemilla s.l., Farinopsis, Sibbaldianthe, and Sibbaldia (RS  339,755; Supplementary Fig. S9 available on Dryad). The parental lineages of this clade were a “lineage” sister to Comarum, and a “lineage” sister to the grade formed of Comarum and Fragaria (Supplementary Fig. S9 available on Dryad). Alternative MUL-trees had scores of 340,053 or higher (Supplementary Fig. S7 available on Dryad). The reconciliations using individual major clades of Alchemilla s.l. resulted in identical patterns as in the full constrained analysis (Supplementary Fig. S10 available on Dryad). The cpDNA tree had Alchemilla s.l. as part of a grade formed along with Farinopsis, Comarum, and Sibbaldianthe
339,755; Supplementary Fig. S9 available on Dryad). The parental lineages of this clade were a “lineage” sister to Comarum, and a “lineage” sister to the grade formed of Comarum and Fragaria (Supplementary Fig. S9 available on Dryad). Alternative MUL-trees had scores of 340,053 or higher (Supplementary Fig. S7 available on Dryad). The reconciliations using individual major clades of Alchemilla s.l. resulted in identical patterns as in the full constrained analysis (Supplementary Fig. S10 available on Dryad). The cpDNA tree had Alchemilla s.l. as part of a grade formed along with Farinopsis, Comarum, and Sibbaldianthe 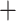 Sibbaldia, while Fragaria was recovered as sister to the clade composed of Drymocallis, Chamaerhodos, Potaninia, and Dasiphora, which is sister to all other Fragariinae (Supplementary Fig. S4 available on Dryad). The reconciliations on the cpDNA tree suggested an autopolyploid origin of Alchemilla s.l. (RS
Sibbaldia, while Fragaria was recovered as sister to the clade composed of Drymocallis, Chamaerhodos, Potaninia, and Dasiphora, which is sister to all other Fragariinae (Supplementary Fig. S4 available on Dryad). The reconciliations on the cpDNA tree suggested an autopolyploid origin of Alchemilla s.l. (RS  363,987; Fig. 4e; Supplementary Fig. S9 available on Dryad). Alternative MUL-trees had scores starting at 364,594 (Supplementary Fig. S7 available on Dryad). The analyses using individual major clades of Alchemilla s.l. recovered identical patterns as in the full constrained analysis, except for Aphanes that resulted in a singly-labeled tree (Supplementary Fig. S10 available on Dryad).
363,987; Fig. 4e; Supplementary Fig. S9 available on Dryad). Alternative MUL-trees had scores starting at 364,594 (Supplementary Fig. S7 available on Dryad). The analyses using individual major clades of Alchemilla s.l. recovered identical patterns as in the full constrained analysis, except for Aphanes that resulted in a singly-labeled tree (Supplementary Fig. S10 available on Dryad).
To further explore WGD events using alternative data sources, we analyzed Ks plots from genomes (Fragaria vesca) and transcriptomes across Fragariinae. The distribution of synonymous distances in the transcriptomes of four species of Eualchemilla (one “dissected” and three “lobed”) shared three optimal mixing components with a Ks mean at approximately 0.1, 0.34, and 1.67, respectively (Supplementary Fig. S11a,b available on Dryad). The first two components partially overlapped and corresponded to at least two WGD events in all four sampled species of Eualchemilla that happened before the splits between the lobed vs. the dissected clades (Ks 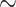 0.02; Supplementary Fig. S12a available on Dryad). The third shared component corresponds to a whole-genome triplication event early in the core eudicots (Jiao et al. 2012; Supplementary Fig. S11a,c available on Dryad). All nine species from the remaining genera in Fragariinae had two optimal mixing components. One component is a Ks peak at 1.61–1.78 corresponding to the whole genome triplication event early in eudicot (Supplementary Fig. S11a,c available on Dryad). In the case of the diploid species, the second component represents a small and very young (
0.02; Supplementary Fig. S12a available on Dryad). The third shared component corresponds to a whole-genome triplication event early in the core eudicots (Jiao et al. 2012; Supplementary Fig. S11a,c available on Dryad). All nine species from the remaining genera in Fragariinae had two optimal mixing components. One component is a Ks peak at 1.61–1.78 corresponding to the whole genome triplication event early in eudicot (Supplementary Fig. S11a,c available on Dryad). In the case of the diploid species, the second component represents a small and very young (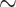 0.05) peak, most likely the product of small-scale recent gene duplications (Supplementary Fig. S11a,b available on Dryad). The only two polyploid species from the other genera in Fragariinae, Comarum palustre (
0.05) peak, most likely the product of small-scale recent gene duplications (Supplementary Fig. S11a,b available on Dryad). The only two polyploid species from the other genera in Fragariinae, Comarum palustre (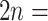 28–64) and Sibbaldianthe bifurca (
28–64) and Sibbaldianthe bifurca (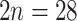 ), had a single additional significant component at 0.11 and 0.08, respectively (Supplementary Fig. S11a,b available on Dryad). The Ks plots between species of Eualchemilla and Fragariinae species outside of Alchemilla s.l., and between species of Fragariinae showed that the WGD events detected in Eualchemilla were not shared with genera outside of Alchemilla s.l. (Supplementary Fig. S12b available on Dryad). Likewise, the WGD events in Comarum palustre and Sibbaldianthe bifurca occurred after the split of the two species (Supplementary Fig. S12c available on Dryad).
), had a single additional significant component at 0.11 and 0.08, respectively (Supplementary Fig. S11a,b available on Dryad). The Ks plots between species of Eualchemilla and Fragariinae species outside of Alchemilla s.l., and between species of Fragariinae showed that the WGD events detected in Eualchemilla were not shared with genera outside of Alchemilla s.l. (Supplementary Fig. S12b available on Dryad). Likewise, the WGD events in Comarum palustre and Sibbaldianthe bifurca occurred after the split of the two species (Supplementary Fig. S12c available on Dryad).
Discussion
Processing Paralogs in Target Enrichment Data Sets
The increased use of target enrichment methods in combination with reduced sequencing costs and higher read coverage have facilitated the recovery of paralogs in such data sets. Paralogy is sometimes viewed as a nuisance for phylogenetic reconstruction and is commonly aimed to be reduced in the early stages of experimental design, by targeting only single- or low-copy genes during the selection of loci (e.g., Chamala et al. 2015; Nicholls et al. 2015; Gardner et al. 2016; Kamneva et al. 2017). Still, the recovery of paralogs is inevitable when working with groups where WGDs are prevalent, especially in plants, leading to various strategies to remove them prior to phylogenetic analyses. Commonly used target enrichment assembly pipelines (e.g., Faircloth 2016; Johnson et al. 2016; Andermann et al. 2018) use different criteria to flag assembled loci with putative paralogs that are later filtered or processed prior to phylogenetic analysis. The most used common approach for dealing with paralogous loci in target enrichment data sets is removing the entire gene that shows any signal of potential paralogy (e.g., Montes et al. 2019; Bagley et al. 2020; Crowl et al. 2020). The removal of paralogous genes can significantly reduce the size of target enrichment data sets and most often disregard the reason why a gene was flagged for putative paralogy (i.e., allelic variation, gene duplication, or fragments of the same gene). Orthology inference should be carried out for all loci in target enrichment data, as relying on settings in assembly pipelines does not guarantee that nonremoved or nonflagged genes are orthologous. Furthermore, removing paralogs before phylogenetic inference eliminates valuable information that could have been used to detect and place WGD events using target enrichment data. Other approaches either retain or remove contigs based on the distinction being putative allelic variation (flagged sequences from monophyletic conspecific groups) or putative paralogs (paralogs from the same species are nonmonophyletic) in combination with study-specific threshold or random selection (e.g., Villaverde et al. 2018; Liu et al. 2019; Stubbs et al. 2020), or manual processing (e.g., García et al. 2017; Karimi et al. 2020). As data set size increases, manual processing becomes prohibitive.
The presence of WGDs also poses some challenges for locus assembly. Target enrichment design commonly includes multicontig targets that assembly pipelines attempt to assemble into single contigs (e.g., Faircloth 2016) or “supercontigs” composed of multiple exons and partially assembled introns (e.g., Johnson et al. 2016). In groups like Alchemilla s.l., where multiple, nested WGD events led to a prevalence of paralogs, “supercontigs” can produce chimeric assemblies (Morales-Briones et al. 2018b). Instead, we assembled the exons individually to minimize chimeric genes, at the cost of working with some short exons that contribute little phylogenetic information, which can affect orthology inference and downstream analyses. Therefore, it is important to take this into consideration during target enrichment experimental design, and to preferentially target long exons when possible, especially in groups where WGD is expected. An alternative strategy to avoid chimeric supercontigs when gene duplications are frequent is to perform a preliminary orthology inference in single exon-based trees and then use the inferred orthologs as a reference to reassemble the loci into “supercontigs” (e.g., Karimi et al. 2020; Gardner et al. 2021). Another aspect to take into consideration during or right after assembly is allele phasing. While phasing heterozygous loci, from population or individual variation, have been shown to have minimal impact in phylogenetic reconstruction in target enrichment data (e.g., Kates et al. 2018), the effect on unphased or merged loci in cases of WGD can be larger and be a source of phylogenetic error (Oxelman et al. 2017; Rothfels 2021). Here, we were interested in ancient WGD in Alchemilla s.l. and relied on enough sequencing coverage and sequence dissimilarity to assemble separate paralogs (homoeologs in the case of allopolyploidy) that can be flagged as such by HybPiper. While we obtained a large number of deep paralogs across Alchemilla s.l. (Fig. 3c; Supplementary Table S5 available on Dryad), there is still the possibility that some exon assemblies included merged sequences from paralogs with high sequence similarity. Paralog merger should be more problematic in cases of recent allopolyploidy or neoallopolyploidy taxa. To this end, recently developed tools are promising in phasing gene copies into polyploid subgenomes using phylogenetic and similarity approaches (e.g., Freyman et al. 2020, Nauheimer et al. 2021).
The utility of paralogs for phylogenetic reconstruction in target enrichment data sets is gaining increased attention (e.g., Johnson et al. 2016; Gardner et al. 2021). A few studies have considered tree-based orthology inference to process affected loci (e.g., García et al. 2017; Moore et al. 2018; Morales-Briones et al. 2018b), but in some cases, the orthology approaches used on those studies cannot be easily transferred or applied to other data sets. Here, we demonstrated the utility of automated, tree-based orthology inference methods (Yang and Smith 2014), originally designed for genomic or transcriptomic data sets, to infer orthology from paralog-flagged and unflagged loci in a target enrichment data set. Our approach facilitates the automated inference of orthologs while maximizing the number of loci retained for downstream analyses. These methods are agnostic of the data source and should work for any type of target enrichment data set (e.g., anchored phylogenomics, exon capture; Hyb-Seq, ultraconserved elements).
Orthology inference methods used here (Yang and Smith 2014) are a powerful tool for target enrichment data sets. In the case of allopolyploidy, however, these methods can introduce bias in the distribution of ortholog trees inferred. In the case of MO, each time a gene duplication event is detected, the side with a smaller number of taxa is removed. When allopolyploidy occurs, MO may bias towards one subgenome due to 1) bias in gene loss between subgenomes and 2) bias in bait design. In the case of Alchemilla s.l., this is less likely as baits were designed in outgroups. If baits are designed according to ingroup taxa, depending on which taxa were used they can have a higher affinity to one subgenome instead of another, and 3) unequal sampling of parental lineages. If one parental lineage is more densely sampled than the other or one parental lineage is unsampled, the two subgenomes will be in species-rich versus species-poor clades respectively in gene trees. One could alternatively preserve a random side each time a gene duplication event is identified. However, in practice, the side with a smaller number of taxa often contains misassembled or misplaced sequences. The RT method of separating duplicated gene copies, on the other hand, keeps any subtree with sufficient number of taxa, but removes outgroups, and worked best when hierarchical outgroups were included in the taxon sampling. Therefore, both MO and RT lose information, especially in cases with complex, nested polyploidy.
Recently developed methods based on quartet similarity (Zhang et al. 2020a), Robinson–Foulds distances (Molloy and Warnow 2020), or maximum likelihood (Morel et al. 2021) can directly estimate a species tree that is consistent with the MSC from multicopy genes without inferring orthologs (for a recent review, see Smith and Hahn 2021). However, their behavior on complex data sets using archival materials is yet to be explored. For example, these methods do not define ingroup–outgroup relationships a priori, and correctly inferring the root of homolog trees can be challenging with missing data, or when WGD occurs near the root. In addition, none of these above species tree reconstruction methods (Molloy and Warnow 2020; Zhang et al. 2020a; Morel et al. 2021), or species tree methods in general, were designed to handle reticulate relationships, in which the underlying phylogenetic relationship is, in fact, a network. Depending on the topological distance of subgenomes, the inferred “species tree” may not represent any subgenome history and instead represent an “average” between subgenomes. Finally, most current methods for evaluating node support still require orthologous gene trees as input. In such cases, tools like Phyparts can still be used to visualize gene tree discordance and calculate ICA scores using MUL-trees.
Phylogenetic Implications in Alchemilla s.l.
Previous phylogenetic studies established the monophyly of Alchemilla s.l. and four major clades of the group (Gehrke et al. 2008, 2016), but the relationship among them and the placement of Alchemilla s.l. within Fragariinae remain unresolved. Our nuclear and plastid analyses both confirmed the monophyly of Alchemilla s.l. and its sister relationship to Farinopsis, as previously shown by Morales-Briones and Tank (2019) based on plastome sequences only. Gehrke et al. (2008) identified two well-supported clades within Eualchemilla that were distinguished by leaf shape, namely the “dissected” and “lobed” clades. Most species of Eualchemilla have a leaf shape consistent with their clade placement, but some had different leaf shapes that were attributed to their hybrid origin between the two clades (Gehrke et al. 2008). More recently, Gehrke et al. (2016) and Morales-Briones and Tank (2019) found that Eualchemilla is not monophyletic in analyses that included the external transcribed spacer (ETS) of the nuclear ribosomal DNA (nrDNA) cistron. Both studies found Aphanes nested between the “dissected” and “lobed” clades of Eualchemilla. Our analyses of the nuclear loci supported the monophyly of “dissected” and “lobed” clades, but the monophyly of Eualchemilla had low support (Fig. 2; Supplementary Fig. S1 available on Dryad). The analysis using only the MO orthologs even weakly supported the “lobed” clade as sister of Aphanes (Fig. 2a). In contrast, our plastome analysis recovered a well-supported, monophyletic Eualchemilla, as well as well-supported “dissected” and “lobed” clades. Both nuclear and plastid analyses strongly supported the clade composed of Aphanes and both clades of Eualchemilla (Fig. 2; Supplementary Fig. S4 available on Dryad), a relationship that is consistent with previous nuclear and plastid analyses (Gehrke et al. 2008, 2016; Morales-Briones and Tank 2019). Given the revealed hybridization in the evolution and early divergence within Alchemilla s.l., the nonmonophyly of Eualchemilla could be explained by ancient gene flow or allopolyploid origins of the “dissected” and “lobed” clades (Fig. 4a,c; see below). Besides the well-supported relationship of Eualchemilla  Aphanes, our nuclear analysis showed high levels of conflict among other major clades in Alchemilla s.l. (Fig. 2; Supplementary Fig. S1 available on Dryad) which could also be explained by additional ancient allopolyploid events (Fig. 4; see below).
Aphanes, our nuclear analysis showed high levels of conflict among other major clades in Alchemilla s.l. (Fig. 2; Supplementary Fig. S1 available on Dryad) which could also be explained by additional ancient allopolyploid events (Fig. 4; see below).
Ancient Polyploidy and Reticulation in Alchemilla s.l.
Whole-genome duplications are frequent across Rosaceae (Dickinson et al. 2007; Xiang et al. 2017), and allopolyploidy has been suggested as the primary source for the cytonuclear discordance in Fragariinae (Lundberg et al. 2009; Gehrke et al. 2016; Morales-Briones and Tank 2019). We recovered four nodes in Alchemilla s.l. with a high percentage of gene duplications (Fig. 3a; Supplementary Fig. S5 available on Dryad). One of the nodes showing a high percentage of gene duplication (18.4%) was the MRCA of Aphanes and both clades of Eualchemilla (node 3 in Fig. 3a; Supplementary Fig. S5 available on Dryad). This duplication event agreed with the MRCA of the ancestral lineages inferred with GRAMPA for the allopolyploid origin of the “lobed” clade of Eualchemilla (Fig. 4a). Moreover, the GRAMPA reconciliations after the removal of the “lobed” clade and Afromilla inferred a scenario where the “dissected” clade is of allopolyploid origin with one of the parental lineages as sister to Aphanes (Fig. 4c). Although there is some uncertainty about the placement of the parental lineages of the “dissected” clade, due to the removal of major clades for the GRAMPA analyses, the cpDNA tree suggest that it is likely sister to the parental lineage of “lobed” clade that is also sister to Aphanes. Ks plots of all species of the “dissected” and “lobed” clades had two peaks that are not shared with members of Fragariinae (Supplementary Figs. S11 and S12 available on Dryad), suggesting that at least two WGD events have happened between the stem of Alchemilla s.l. and the split of the “dissected” and the “lobed” clades. The between-species Ks plots of “dissected” vs. “lobed” species (Supplementary Fig. S12 available on Dryad) showed that the split between these two groups is more recent than the WGD events, suggesting very close time of origin (or single origin) of both clades. Still, the sister relationship of the “dissected” and “lobed” clades is not supported by nuclear genes, suggesting that the two clades of Eualchemilla might have had independent allopolyploid origins, while sharing the same or a closely related chloroplast donor (cpDNA; Fig. 4f).
The GRAMPA reconciliation, after the removal of the “lobed” clade, recovered an allopolyploid origin of Afromilla (Fig. 4b) with the MRCA of the ancestral lineages at the MRCA of the remaining Alchemilla s.l. Similarly, the further removal of both Afromilla and the “dissected” clade recovered Lachemilla as allopolyploid, with the MRCA of parental lineages mapped to the MRCA of the remaining Alchemilla s.l. (Fig. 4d). In the case of Lachemilla, because of the removal of major clades for the GRAMPA analyses, there lacks information in the placement of its parental lineages. Still, Afromilla and Lachemilla are sisters in the cpDNA tree (Fig 1c), suggesting these two share the same or a closely related chloroplast donor (Fig. 4f). The elevated percentage of orthogroups that show evidence of gene duplication (34.4%) at the MRCA of Afromilla, Eualchemilla, and Aphanes (node 2 in Fig. 3a) further support the allopolyploid origin of Afromilla.
Finally, the node with the highest percentage of duplicated genes (86%) was placed at the MRCA of Alchemilla s.l. (node 1 in Fig. 3a). The GRAMPA analysis using the MO tree showed an allopolyploid event for the clade that included Alchemilla s.l., Farinopsis salesoviana, Sibbaldia, and Sibbaldianthe (Supplementary Fig. S9 available on Dryad). However, an allopolyploid origin of Farinopsis salesoviana, Sibbaldia, and Sibbaldianthe is not supported by chromosome numbers, orthogroup gene duplication counts, or Ks plots. All members of Fragariinae, except Alchemilla s.l., mainly consists of diploid species and base chromosome number of seven ( ), including Sibbaldia and Sibbaldianthe. On the other hand, Alchemilla s.l. has a base number of eight (
), including Sibbaldia and Sibbaldianthe. On the other hand, Alchemilla s.l. has a base number of eight ( ) and contains mostly species with high ploidy levels (octoploid to 24-ploid), with the exception of most species of Aphanes (
) and contains mostly species with high ploidy levels (octoploid to 24-ploid), with the exception of most species of Aphanes (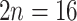 ) and one species of Lachemilla (L. mandoniana,
) and one species of Lachemilla (L. mandoniana, 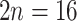 ). Also, our gene duplication counts show low percentages (1.6%) of gene duplication for the MRCA of the GRAMPA-inferred allopolyploid clade or the MRCA (3.6%) of the inferred parental lineages (Fig. 3). Previous phylotranscriptomic analyses of Rosaceae (Xiang et al. 2017) that included one species each of the “dissected” and “lobed” clades of Eualchemilla, found 33.2% of genes showed evidence of gene duplication at the MRCA of these two clades but did not recover any other node with elevated gene duplications within Fragariinae. The Ks plots of the four species of Alchemilla s.l. all showed peaks with similar Ks means, but these peaks were not shared with species of Sibbaldia or Sibbadianthe (Supplementary Fig. S11 available on Dryad). Furthermore, the between-species Ks plots showed that the WGD events detected in Alchemilla s.l. were more recent than the split with members of Fragariinae (Supplementary Fig. S12 available on Dryad). Although the chromosome number and Ks data for Farinopsis salesoviana are not available, all the above evidence suggest an unlikely allopolyploid origin of the clade consisting of Farinopsis, Sibbaldia, Sibadianthe, and Alchemilla sl. On the other hand, the GRAMPA reconciliations against the cpDNA tree resulted in an optimal MUL-tree where Alchemilla s.l. had an autopolyploid origin (Fig. 4e). This scenario is compatible with the high percentage of gene duplication at the MRCA of Alchemilla s.l. and the low percentage of gene duplication in the backbone of the rest of Fragariinae. Another compatible scenario is an allopolyploid origin of Alchemilla s.l. where both parental lineages are missing or extinct, but this scenario is indistinguishable from autopolyploidy. The atypical high proportion of gene duplication at the MRCA of Alchemilla s.l. can be explained by the autopolyploid event at this branch. In addition, given the short branch lengths among major clades within Alchemilla s.l., gene tree estimation error (e.g., uninformative genes), incomplete lineage sorting (ILS), allopolyploid events among major clades of Alchemilla s.l., and/or homoeologous exchanges among subgenomes (Edger et al. 2018; McKain et al. 2018) can all contribute to additional gene duplication events mapped to the MRCA of Alchemilla s.l.
). Also, our gene duplication counts show low percentages (1.6%) of gene duplication for the MRCA of the GRAMPA-inferred allopolyploid clade or the MRCA (3.6%) of the inferred parental lineages (Fig. 3). Previous phylotranscriptomic analyses of Rosaceae (Xiang et al. 2017) that included one species each of the “dissected” and “lobed” clades of Eualchemilla, found 33.2% of genes showed evidence of gene duplication at the MRCA of these two clades but did not recover any other node with elevated gene duplications within Fragariinae. The Ks plots of the four species of Alchemilla s.l. all showed peaks with similar Ks means, but these peaks were not shared with species of Sibbaldia or Sibbadianthe (Supplementary Fig. S11 available on Dryad). Furthermore, the between-species Ks plots showed that the WGD events detected in Alchemilla s.l. were more recent than the split with members of Fragariinae (Supplementary Fig. S12 available on Dryad). Although the chromosome number and Ks data for Farinopsis salesoviana are not available, all the above evidence suggest an unlikely allopolyploid origin of the clade consisting of Farinopsis, Sibbaldia, Sibadianthe, and Alchemilla sl. On the other hand, the GRAMPA reconciliations against the cpDNA tree resulted in an optimal MUL-tree where Alchemilla s.l. had an autopolyploid origin (Fig. 4e). This scenario is compatible with the high percentage of gene duplication at the MRCA of Alchemilla s.l. and the low percentage of gene duplication in the backbone of the rest of Fragariinae. Another compatible scenario is an allopolyploid origin of Alchemilla s.l. where both parental lineages are missing or extinct, but this scenario is indistinguishable from autopolyploidy. The atypical high proportion of gene duplication at the MRCA of Alchemilla s.l. can be explained by the autopolyploid event at this branch. In addition, given the short branch lengths among major clades within Alchemilla s.l., gene tree estimation error (e.g., uninformative genes), incomplete lineage sorting (ILS), allopolyploid events among major clades of Alchemilla s.l., and/or homoeologous exchanges among subgenomes (Edger et al. 2018; McKain et al. 2018) can all contribute to additional gene duplication events mapped to the MRCA of Alchemilla s.l.
Although deeply nested in Alchemilla s.l., remarkably, Aphanes showed a significantly lower number of paralogs than the rest of Alchemilla s.l. (Fig. 3). The relatively low number of paralogs, its chromosome numbers being mainly diploid, and the best GRAMPA reconciliation resulting in a singly-labeled tree (Supplementary Fig. S10 available on Dryad), together suggest that Aphanes is a functional diploid clade. One plausible scenario is that postpolyploid diploidization (reviewed in Mandáková and Lysak 2018) occurred after the autopolyploidy event at the MRCA of Alchemilla s.l. After diploidization, Afromilla, Eualchemilla (“lobed” and “dissected” clades), and Lachemilla each originated from allopolyploid events, while Aphanes descended from a diploidized ancestor that did not duplicate further (Fig. 4f). The orthogroup gene duplication mapping recovered Aphanes being nested in a clade with elevated proportions of gene duplication (Fig. 3a,b, nodes 2 and 3). This can be explained by the lineage leading to Aphanes contributed to subgenomes in allopolyploidy events, while Aphanes remained in its diploid condition.
GRAMPA has been shown to be useful for identifying multiple, nested polyploidy events one at a time (e.g., Thomas et al. 2017; Guo et al. 2021; Koenen et al. 2021). However, like any tree-based approach, GRAMPA is sensitive to gene tree estimation error or ILS (Thomas et al. 2017). Its reconciliation assumes that the user-defined “species tree” topology represents the evolutionary history of one of the subgenomes. In complex allopolyploid scenarios like Alchemilla s.l., the inferred “species tree” can instead represent an “average” between subgenomes and misled the reconciliations. Furthermore, potential recent reticulations events could bias GRAMPA to infer false allopolyploidy reconciliations when restricting the searches to deeper nodes. Given that GRAMPA is a parsimony-based reconciliation approach, and although we showed that the optimal reconciliation had visibly lower (and therefore better) reconciliation scores than the second best in most cases (Supplementary Fig. S7 available on Dryad), it is difficult to evaluate the uncertainty of the reconciled MUL-trees. Lastly, it is possible that the early diversification of Alchemilla s.l. was a hybrid swarm instead of multiple, distinguishable allopolyploid events. In addition to GRAMPA, methods to infer species networks in the presence of ILS (e.g., Solís-Lemus and Ané 2016; Wen et al. 2018) could also be used to explore the prevalence of ancient hybridization in Alchemilla s.l. Although these methods are under continuous improvement, currently they are only tractable in simple scenarios with few reticulation events (Hejase and Liu 2016; Kamneva and Rosenberg 2017). Similarly, the signal of the  -Statistic (Green et al. 2010; Durand et al. 2011), commonly used to detect introgression, can be lost or distorted in the presence of multiple reticulations (Elworth et al. 2019). Complex reticulate scenarios like Alchemilla s.l. are likely to face these problems and have phylogenetic network and
-Statistic (Green et al. 2010; Durand et al. 2011), commonly used to detect introgression, can be lost or distorted in the presence of multiple reticulations (Elworth et al. 2019). Complex reticulate scenarios like Alchemilla s.l. are likely to face these problems and have phylogenetic network and  -statistic identifiability issues as seen in other groups (e.g., Morales-Briones et al. 2021).
-statistic identifiability issues as seen in other groups (e.g., Morales-Briones et al. 2021).
Conclusions
In this study, we have shown the utility of target enrichment data sets in combination with tree-based methods for orthology inference and WGD investigation. Here, we used Alchemilla s.l. to highlight the importance of processing paralogs, rather than discarding them before phylogenetic analysis, to shed light on the complex polyploidy and reticulate histories. We showed evidence that the entire Alchemilla s.l. is the product of an ancient autopolyploidy event, and that Afromilla, Eualchemilla (“lobed” and “dissected” clades), and Lachemilla originated from subsequent and nested ancient allopolyploid events. Our results from analyzing target enrichment data corroborated with previously published chromosome numbers and distribution of Ks values from transcriptomes. Our analyses have several important implications for future target enrichment projects, including 1) design baits to obtain a relatively large number of loci as this is required for accurate species tree and networks estimation in complex scenarios (e.g., higher levels of ILS; Solís-Lemus and Ané 2016; Nute et al. 2018), 2) target long individual loci to improve the information content of individual gene trees for proper tree-based orthology inference and identifying gene duplication events, and 3) design baits to minimize lineage-specific and paralog-specific capture efficiency and missing data. Furthermore, in target enrichment, unlike genome or transcriptome data, only several hundreds of genes are typically recovered with levels of missing data that varies by lineage and are nonrandom. This limits the utility of target enrichment for generating Ks plots and creates the need to carefully scrutinize the variation in the percentage of gene duplications among nodes. In the end, even with these limitations, target enrichment is overall a valuable and cost-effective approach of genomic subsampling to explore patterns of reticulation and WGD, especially in groups for which whole genome or transcriptome data are not possible to generate, including from museum/herbarium specimens. As research continues to deepen in other clades across the Tree of Life using similar target enrichment methods, we expect that other complex patterns of duplication and reticulation, as those shown here in Alchemilla s.l. will continue to emerge.
Acknowledgments
The authors thank M. Fishbein, J. Mandel, B. Carstens, and three anonymous reviewers for providing helpful comments. This work was supported by the University of Minnesota, Graduate Student Research Grants from the Botanical Society of America, American Society of Plant Taxonomists, International Association of Plant Taxonomists, the University of Idaho Stillinger Herbarium Expedition Funds to D.F.M.-B., and a National Science Foundation Doctoral Dissertation Improvement Grant to D.C.T. for D.F.M.-B. (DEB-1502049).
Supplementary Material
Data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.cc2fqz660.
References
- Andermann T., Cano Á., Zizka A., Bacon C., Antonelli A. 2018. SECAPR—a bioinformatics pipeline for the rapid and user-friendly processing of targeted enriched Illumina sequences, from raw reads to alignments. PeerJ. 6:e5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andermann T., Torres Jiménez M.F., Matos-Maraví P., Batista R., Blanco-Pastor J.L., Gustafsson A.L.S., Kistler L., Liberal I.M., Oxelman B., Bacon C.D., Antonelli A. 2020. A guide to carrying out a phylogenomic target sequence capture project. Front. Genet. 10:1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagley J.C., Uribe-Convers S., Carlsen M.M., Muchhala N. 2020. Utility of targeted sequence capture for phylogenomics in rapid, recent angiosperm radiations: Neotropical Burmeistera bellflowers as a case study. Mol. Phylogenet. Evol. 152:106769. [DOI] [PubMed] [Google Scholar]
- Benaglia T., Chauveau D., Hunter D.R., Young D. 2009. mixtools?: an R package for analyzing finite mixture models. J. Stat. Softw. 32:1–29. [Google Scholar]
- Brown J.W., Walker J.F., Smith S.A. 2017. Phyx - phylogenetic tools for unix. Bioinformatics 33:1886–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buddenhagen C., Lemmon A.R., Lemmon E.M., Bruhl J., Cappa J., Clement W.L., Donoghue M.J., Edwards E.J., Hipp A.L., Kortyna M., Mitchell N., Moore A., Prychid C.J., Segovia-Salcedo M.C., Simmons M.P., Soltis P.S., Wanke S., Mast A. 2016. Anchored phylogenomics of angiosperms I: assessing the robustness of phylogenetic estimates. bioRxiv. 10.1101/086298. [DOI]
- Chamala S., García N., Godden G.T., Krishnakumar V., Jordon-Thaden I.E., De Smet R., Barbazuk W.B., Soltis D.E., Soltis P.S. 2015. MarkerMiner 1.0: A new application for phylogenetic marker development using angiosperm transcriptomes. Appl. Plant Sci. 3:1400115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowl A.A., Manos P.S., McVay J.D., Lemmon A.R., Lemmon E.M., Hipp A.L. 2020. Uncovering the genomic signature of ancient introgression between white oak lineages (Quercus). New Phytol. 226:1158–1170. [DOI] [PubMed] [Google Scholar]
- Cui L., Wall P.K., Leebens-Mack J.H., Lindsay B.G., Soltis D.E., Doyle J.J., Soltis P.S., Carlson J.E., Arumuganathan K., Barakat A., Albert V.A., Ma H., dePamphilis C.W. 2006. Widespread genome duplications throughout the history of flowering plants. Genome Res. 16:738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson T.A., Lo E., Talent N. 2007. Polyploidy, reproductive biology, and Rosaceae: understanding evolution and making classifications. Plant Syst. Evol. 266:59–78. [Google Scholar]
- Dobeš C., Lückl A., Kausche L., Scheffknecht S., Prohaska D., Sykora C., Paule J. 2015. Parallel origins of apomixis in two diverged evolutionary lineages in tribe Potentilleae (Rosaceae): origin of apomixis in Potentilleae. Bot. J. Linn. Soc. 177:214–229. [Google Scholar]
- Doyle J.J., Doyle J.L. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19:11–15. [Google Scholar]
- Dunn C.W., Howison M., Zapata F. 2013. Agalma: an automated phylogenomics workflow. BMC Bioinformatics 14:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand E.Y., Patterson N., Reich D., Slatkin M. 2011. Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 28:2239–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edger P.P., McKain M.R., Bird K.A., VanBuren R. 2018. Subgenome assignment in allopolyploids: challenges and future directions. Curr. Opin. Plant Biol. 42:76–80. [DOI] [PubMed] [Google Scholar]
- Elworth R.A.L., Ogilvie H.A., Zhu J., Nakhleh L. 2019. Advances in computational methods for phylogenetic networks in the presence of hybridization. In: Warnow T., editor. Bioinformatics and Phylogenetics. Computational Biology, vol 29. Cham, Switzerland: Springer. p. 317–360. [Google Scholar]
- Emms D.M., Kelly S. 2019. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson T., Lundberg M., Töpel M., Östensson P., Smedmark J.E.E. 2015. Sibbaldia: a molecular phylogenetic study of a remarkably polyphyletic genus in Rosaceae. Plant Syst. Evol. 301:171–184. [Google Scholar]
- Faircloth B.C. 2016. PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinforma. Oxf. Engl. 32:786–788. [DOI] [PubMed] [Google Scholar]
- Fernández R., Gabaldon T., Dessimoz C. 2020. Orthology: Definitions, Prediction, and Impact on Species Phylogeny Inference. In: Scornavacca C., Delsuc F., Galtier N., editors. Phylogenetics in the genomic era. No commercial publisher | Authors open access book. p. 2.4:1-2.4:14. [Google Scholar]
- Fitch W.M. 1970. Distinguishing homologous from analogous proteins. Syst. Biol. 19:99–113. [PubMed] [Google Scholar]
- Forrest L.L., Hart M.L., Hughes M., Wilson H.P., Chung K.-F., Tseng Y.-H., Kidner C.A. 2019. The limits of Hyb-Seq for herbarium specimens: impact of preservation techniques. Front. Ecol. Evol. 7:439. [Google Scholar]
- Freyman W.A., Johnson M.G., Rothfels C.J. 2020. Homologizer: phylogenetic phasing of gene copies into polyploid subgenomes. bioRxiv. 10.1101/2020.10.22.351486. [DOI] [PubMed]
- García N., Folk R.A., Meerow A.W., Chamala S., Gitzendanner M.A., de Oliveira R.S., Soltis D.E., Soltis P.S. 2017. Deep reticulation and incomplete lineage sorting obscure the diploid phylogeny of rain-lilies and allies (Amaryllidaceae tribe Hippeastreae). Mol. Phylogenet. Evol. 111:231–247. [DOI] [PubMed] [Google Scholar]
- Gardner E.M., Johnson M.G., Pereira J.T., Ahmad Puad A.S., Arifiani D., Sahromi, Wickett N.J., Zerega N.J.C. 2021. Paralogs and off-target sequences improve phylogenetic resolution in a densely-sampled study of the breadfruit genus (Artocarpus, Moraceae). Syst. Biol. 70:558–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner E.M., Johnson M.G., Ragone D., Wickett N.J., Zerega N.J.C. 2016. Low-coverage, whole-genome sequencing of Artocarpus camansi (Moraceae) for phylogenetic marker development and gene discovery. Appl. Plant Sci. 4:1600017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrke B., Bräuchler C., Romoleroux K., Lundberg M., Heubl G., Eriksson T. 2008. Molecular phylogenetics of Alchemilla, Aphanes and Lachemilla (Rosaceae) inferred from plastid and nuclear intron and spacer DNA sequences, with comments on generic classification. Mol. Phylogenet. Evol. 47:1030–1044. [DOI] [PubMed] [Google Scholar]
- Gehrke B., Kandziora M., Pirie M.D. 2016. The evolution of dwarf shrubs in alpine environments: a case study of Alchemilla in Africa. Ann. Bot. 117:121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover N.M., Redestig H., Dessimoz C. 2016. Homoeologs: what are they and how do we infer them? Trends Plant Sci. 21: 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover N., Dessimoz C., Ebersberger I., Forslund S.K., Gabaldón T., Huerta-Cepas J., Martin M.-J., Muffato M., Patricio M., Pereira C., da Silva A.S., Wang Y., Sonnhammer E., Thomas P.D. 2019. Advances and applications in the quest for orthologs. Mol. Biol. Evol. 36:2157–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves D.J.P., Simpson B.B., Ortiz E.M., Shimizu G.H., Jansen R.K. 2019. Incongruence between gene trees and species trees and phylogenetic signal variation in plastid genes. Mol. Phylogenet. Evol. 138:219–232. [DOI] [PubMed] [Google Scholar]
- Green R.E., Krause J., Briggs A.W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M.H.Y., Hansen N.F., Durand E.Y., Malaspinas A.S., Jensen J.D., Marques-Bonet T., Alkan C., Prufer K., Meyer M., Burbano H.A., Good J.M., Schultz R., Aximu-Petri A., Butthof A., Hober B., Hoffner B., Siegemund M., Weihmann A., Nusbaum C., Lander E.S., Russ C., Novod N., Affourtit J., Egholm M., Verna C., Rudan P., Brajkovic D., Kucan Z., Gusic I., Doronichev V.B., Golovanova L.V., Lalueza-Fox C., de la Rasilla M., Fortea J., Rosas A., Schmitz R.W., Johnson P.L.F., Eichler E.E., Falush D., Birney E., Mullikin J.C., Slatkin M., Nielsen R., Kelso J., Lachmann M., Reich D., Paabo S. 2010. A draft sequence of the neandertal genome. Science 328:710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X., Mandáková T., Trachtová K., Özüdoðru B., Liu J., Lysak M.A. 2021. Linked by ancestral bonds: multiple whole-genome duplications and reticulate evolution in a Brassicaceae tribe. Mol. Biol. Evol. 38:1695–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayirhoglu-Ayaz S., Ýnceer H., Frost-Olsen P. 2006. Chromosome counts in the genus Alchemilla (Rosaceae) from SW Europe. Folia Geobot. 41:335–344. [Google Scholar]
- Hejase H.A., Liu K.J. 2016. A scalability study of phylogenetic network inference methods using empirical datasets and simulations involving a single reticulation. BMC Bioinformatics 17:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmqvist H. 1956. The embryology of some African Alchemilla species. Bot. Not. 109:21–32. [Google Scholar]
- Huang C.-H., Zhang C., Liu M., Hu Y., Gao T., Qi J., Ma H. 2016. Multiple polyploidization events across Asteraceae with two nested events in the early history revealed by nuclear phylogenomics. Mol. Biol. Evol. 33:2820–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izmailow R. 1981. Karyological studies in species of Alchemilla L. from the series Calycinae Bus. (section Brevicaulon Rothm.). Acta Biol. Cracoviensia Ser. Bot. 23:117–130. [Google Scholar]
- Jiao Y., Wickett N.J., Ayyampalayam S., Chanderbali A.S., Landherr L., Ralph P.E., Tomsho L.P., Hu Y., Liang H., Soltis P.S., Soltis D.E., Clifton S.W., Schlarbaum S.E., Schuster S.C., Ma H., Leebens-Mack J., dePamphilis C.W. 2011. Ancestral polyploidy in seed plants and angiosperms. Nature 473:97–100. [DOI] [PubMed] [Google Scholar]
- Jiao Y., Leebens-Mack J., Ayyampalayam S., Bowers J.E., McKain M.R., McNeal J., Rolf M., Ruzicka D.R., Wafula E., Wickett N.J., Wu X., Zhang Y., Wang J., Zhang Y., Carpenter E.J., Deyholos M.K., Kutchan T.M., Chanderbali A.S., Soltis P.S., Stevenson D.W., McCombie R., Pires J., Wong G., Soltis D.E., dePamphilis C.W. 2012. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 13:R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M.G., Gardner E.M., Liu Y., Medina R., Goffinet B., Shaw A.J., Zerega N.J.C., Wickett N.J. 2016. HybPiper: extracting coding sequence and introns for phylogenetics from high-throughput sequencing reads using target enrichment. Appl. Plant Sci. 4:1600016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K.E., Fér T., Schmickl R.E., Dikow R.B., Funk V.A., Herrando-Moraira S., Johnston P.R., Kilian N., Siniscalchi C.M., Susanna A., Slovák M., Thapa R., Watson L.E., Mandel J.R. 2019. An empirical assessment of a single family-wide hybrid capture locus set at multiple evolutionary timescales in Asteraceae. Appl. Plant Sci. 7:e11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamneva O.K., Rosenberg N.A. 2017. Simulation-based evaluation of hybridization network reconstruction methods in the presence of incomplete lineage sorting. Evol. Bioinforma. 13:117693431769193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamneva O.K., Syring J., Liston A., Rosenberg N.A. 2017. Evaluating allopolyploid origins in strawberries (Fragaria) using haplotypes generated from target capture sequencing. BMC Evol. Biol. 17:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi N., Grover C.E., Gallagher J.P., Wendel J.F., Ané C., Baum D.A. 2020. Reticulate evolution helps explain apparent homoplasy in floral biology and pollination in baobabs (Adansonia; Bombacoideae; Malvaceae). Syst. Biol. 69:462–478. [DOI] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kates H.R., Johnson M.G., Gardner E.M., Zerega N.J.C., Wickett N.J. 2018. Allele phasing has minimal impact on phylogenetic reconstruction from targeted nuclear gene sequences in a case study of Artocarpus. Am. J. Bot. 105:404–416. [DOI] [PubMed] [Google Scholar]
- Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C., Thierer T., Ashton B., Meintjes P., Drummond A. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocot K.M., Citarella M.R., Moroz L.L., Halanych K.M. 2013. PhyloTreePruner: a phylogenetic tree-based approach for selection of orthologous sequences for phylogenomics. Evol. Bioinforma. Online 9:429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenen E.J.M., Ojeda D.I., Bakker F.T., Wieringa J.J., Kidner C., Hardy O.J., Pennington R.T., Herendeen P.S., Bruneau A., Hughes C.E. 2021. The origin of the legumes is a complex paleopolyploid phylogenomic tangle closely associated with the Cretaceous-Paleogene (K-Pg) mass extinction event. Syst. Biol. 70:508–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larridon I., Villaverde T., Zuntini A.R., Pokorny L., Brewer G.E., Epitawalage N., Fairlie I., Hahn M., Kim J., Maguilla E., Maurin O., Xanthos M., Hipp A.L., Forest F., Baker W.J. 2020. Tackling rapid radiations with targeted sequencing. Front. Plant Sci. 10:1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leebens-Mack J.H., Barker M.S., Carpenter E.J., Deyholos M.K., Gitzendanner M.A., Graham S.W., Grosse I., Li Z., Melkonian M., Mirarab S., Porsch M., Quint M., Rensing S.A., Soltis D.E., Soltis P.S., Stevenson D.W., Ullrich K.K., Wickett N.J., DeGironimo L., Edger P.P., Jordon-Thaden I.E., Joya S., Liu T., Melkonian B., Miles N.W., Pokorny L., Quigley C., Thomas P., Villarreal J.C., Augustin M.M., Barrett M.D., Baucom R.S., Beerling D.J., Benstein R.M., Biffin E., Brockington S.F., Burge D.O., Burris J.N., Burris K.P., Burtet-Sarramegna V., Caicedo A.L., Cannon S.B., Çebi Z., Chang Y., Chater C., Cheeseman J.M., Chen T., Clarke N.D., Clayton H., Covshoff S., Crandall-Stotler B.J., Cross H., dePamphilis C.W., Der J.P., Determann R., Dickson R.C., Di Stilio V.S., Ellis S., Fast E., Feja N., Field K.J., Filatov D.A., Finnegan P.M., Floyd S.K., Fogliani B., García N., Gâteblé G., Godden G.T., Goh F. (Qi Y., Greiner S., Harkess A., Heaney J.M., Helliwell K.E., Heyduk K., Hibberd J.M., Hodel R.G.J., Hollingsworth P.M., Johnson M.T.J., Jost R., Joyce B., Kapralov M.V., Kazamia E., Kellogg E.A., Koch M.A., Von Konrat M., Könyves K., Kutchan T.M., Lam V., Larsson A., Leitch A.R., Lentz R., Li F.-W., Lowe A.J., Ludwig M., Manos P.S., Mavrodiev E., McCormick M.K., McKain M., McLellan T., McNeal J.R., Miller R.E., Nelson M.N., Peng Y., Ralph P., Real D., Riggins C.W., Ruhsam M., Sage R.F., Sakai A.K., Scascitella M., Schilling E.E., Schlösser E.-M., Sederoff H., Servick S., Sessa E.B., Shaw A.J., Shaw S.W., Sigel E.M., Skema C., Smith A.G., Smithson A., Stewart C.N., Stinchcombe J.R., Szövényi P., Tate J.A., Tiebel H., Trapnell D., Villegente M., Wang C.-N., Weller S.G., Wenzel M., Weststrand S., Westwood J.H., Whigham D.F., Wu S., Wulff A.S., Yang Y., Zhu D., Zhuang C., Zuidof J., Chase M.W., Pires J.C., Rothfels C.J., Yu J., Chen C., Chen L., Cheng S., Li J., Li R., Li X., Lu H., Ou Y., Sun X., Tan X., Tang J., Tian Z., Wang F., Wang J., Wei X., Xu X., Yan Z., Yang F., Zhong X., Zhou F., Zhu Y., Zhang Y., Ayyampalayam S., Barkman T.J., Nguyen N., Matasci N., Nelson D.R., Sayyari E., Wafula E.K., Walls R.L., Warnow T., An H., Arrigo N., Baniaga A.E., Galuska S., Jorgensen S.A., Kidder T.I., Kong H., Lu-Irving P., Marx H.E., Qi X., Reardon C.R., Sutherland B.L., Tiley G.P., Welles S.R., Yu R., Zhan S., Gramzow L., Theißen G., Wong G.K.-S., One Thousand Plant Transcriptomes Initiative. 2019. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574:679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13:2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Baniaga A.E., Sessa E.B., Scascitelli M., Graham S.W., Rieseberg L.H., Barker M.S. 2015. Early genome duplications in conifers and other seed plants. Sci. Adv. 1:e1501084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston A. 2014. 257 nuclear genes for Rosaceae phylogenomics. figshare. Dataset. 10.6084/m9.figshare.1060394.v1 [DOI]
- Liu Y., Johnson M.G., Cox C.J., Medina R., Devos N., Vanderpoorten A., Hedenäs L., Bell N.E., Shevock J.R., Aguero B., Quandt D., Wickett N.J., Shaw A.J., Goffinet B. 2019. Resolution of the ordinal phylogeny of mosses using targeted exons from organellar and nuclear genomes. Nat. Commun. 10:1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg M., Töpel M., Eriksen B., Nylander J.A.A., Eriksson T. 2009. Allopolyploidy in Fragariinae (Rosaceae): comparing four DNA sequence regions, with comments on classification. Mol. Phylogenet. Evol. 51:269–280. [DOI] [PubMed] [Google Scholar]
- Lynch M., Conery J.S. 2000. The evolutionary fate and consequences of duplicate genes. Science 290:1151–1155. [DOI] [PubMed] [Google Scholar]
- Mai U., Mirarab S. 2018. TreeShrink: fast and accurate detection of outlier long branches in collections of phylogenetic trees. BMC Genomics 19:272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandáková T., Lysak M.A. 2018. Post-polyploid diploidization and diversification through dysploid changes. Curr. Opin. Plant Biol. 42:55–65. [DOI] [PubMed] [Google Scholar]
- Mandel J.R., Dikow R.B., Funk V.A., Masalia R.R., Staton S.E., Kozik A., Michelmore R.W., Rieseberg L.H., Burke J.M. 2014. A target enrichment method for gathering phylogenetic information from hundreds of loci: an example from the Compositae. Appl. Plant Sci. 2:1300085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauheimer L., Weigner N., Joyce E., Crayn D., Clarke C., Nargar K. 2021. HybPhaser: a workflow for the detection and phasing of hybrids in target capture datasets. bioRxiv. 10.1101/2020.10.27.354589. [DOI] [PMC free article] [PubMed]
- McKain M.R., Estep M.C., Pasquet R., Layton D.J., Vela Díaz D.M., Zhong J., Hodge J.G., Malcomber S.T., Chipabika G., Pallangyo B., Kellogg E.A. 2018. Ancestry of the two subgenomes of maize. bioRxiv. 10.1101/352351. [DOI]
- McKain M.R., Tang H., McNeal J.R., Ayyampalayam S., Davis J.I., dePamphilis C.W., Givnish T.J., Pires J.C., Stevenson D.W., Leebens-Mack J.H. 2016. A phylogenomic assessment of ancient polyploidy and genome evolution across the Poales. Genome Biol. Evol. 8:1150–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan G., Peel D. 2000. Finite mixture models. New York: Wiley. [Google Scholar]
- Molloy E.K., Warnow T. 2020. FastMulRFS: fast and accurate species tree estimation under generic gene duplication and loss models. Bioinformatics 36:i57–i65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes J.R., Peláez P., Willyard A., Moreno-Letelier A., Piñero D., Gernandt D.S. 2019. Phylogenetics of Pinus subsection Cembroides Engelm. (Pinaceae) inferred from low-copy nuclear gene sequences. Syst. Bot. 44:501–518. [Google Scholar]
- Montgomery L. 1997. Contributions to a cytological catalogue of the British and Irish flora, 5. Watsonia. 21:365–368. [Google Scholar]
- Moore A.J., Vos J.M.D., Hancock L.P., Goolsby E., Edwards E.J. 2018. Targeted enrichment of large gene families for phylogenetic inference: phylogeny and molecular evolution of photosynthesis genes in the Portullugo Clade (Caryophyllales). Syst. Biol. 67:367–383. [DOI] [PubMed] [Google Scholar]
- Morales-Briones D.F., Romoleroux K., Koláø F., Tank D.C. 2018a. Phylogeny and evolution of the neotropical radiation of Lachemilla (Rosaceae): uncovering a history of reticulate evolution and implications for infrageneric classification. Syst. Bot. 43:17–34. [Google Scholar]
- Morales-Briones D.F., Liston A., Tank D.C. 2018b. Phylogenomic analyses reveal a deep history of hybridization and polyploidy in the Neotropical genus Lachemilla (Rosaceae). New Phytol. 218:1668–1684. [DOI] [PubMed] [Google Scholar]
- Morales-Briones D.F., Tank D.C. 2019. Extensive allopolyploidy in the neotropical genus Lachemilla (Rosaceae) revealed by. Am. J. Bot. 106:415–437. [DOI] [PubMed] [Google Scholar]
- Morales-Briones D.F., Kadereit G., Tefarikis D.T., Moore M.J., Smith S.A., Brockington S.F., Timoneda A., Yim W.C., Cushman J.C., Yang Y. 2021. Disentangling sources of gene tree discordance in phylogenomic datasets: testing ancient hybridizations in Amaranthaceae s.l. Syst. Biol. 70:219–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel B., Schade P., Lutteropp S., Williams T.A., Szöllõsi G.J., Stamatakis A. 2021. SpeciesRax: a tool for maximum likelihood species tree inference from gene family trees under duplication, transfer, and loss. bioRxiv. 10.1101/2021.03.29.437460. [DOI] [PMC free article] [PubMed]
- Morton J. 1993. Chromosome numbers and polyploidy in the flora of Cameroons Mountain. Opera Bot. 121:159–172. [Google Scholar]
- Nicholls J.A., Pennington R.T., Koenen E.J.M., Hughes C.E., Hearn J., Bunnefeld L., Dexter K.G., Stone G.N., Kidner C.A. 2015. Using targeted enrichment of nuclear genes to increase phylogenetic resolution in the neotropical rain forest genus Inga (Leguminosae: Mimosoideae). Front. Plant Sci. 6:710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nute M., Chou J., Molloy E.K., Warnow T. 2018. The performance of coalescent-based species tree estimation methods under models of missing data. BMC Genomics 19:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchy N., Lehti-Shiu M., Shiu S.-H. 2016. Evolution of gene duplication in plants. Plant Physiol. 171:2294–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease J.B., Brown J.W., Walker J.F., Hinchliff C.E., Smith S.A. 2018. Quartet sampling distinguishes lack of support from conflicting support in the green plant tree of life. Am. J. Bot. 105:385–403. [DOI] [PubMed] [Google Scholar]
- Perry L.M. 1929. A tentative revision of Alchemilla § Lachemilla. Contrib. Gray Herb. Harv. Univ. 84:1–57. [Google Scholar]
- Oxelman B., Brysting A.K., Jones G.R., Marcussen T., Oberprieler C., Pfeil B.E. 2017. Phylogenetics of allopolyploids. Annu. Rev. Ecol. Evol. Syst. 48:543–557. [Google Scholar]
- Ranwez V., Douzery E.J.P., Cambon C., Chantret N., Delsuc F. 2018. MACSE v2: toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol. 35:2582–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothfels C.J. 2021. Polyploid phylogenetics. New Phytol. 230: 66–72. [DOI] [PubMed] [Google Scholar]
- Salichos L., Stamatakis A., Rokas A. 2014. Novel information theory-based measures for quantifying incongruence among phylogenetic trees. Mol. Biol. Evol. 31:1261–1271. [DOI] [PubMed] [Google Scholar]
- Sayyari E., Mirarab S. 2016. Fast coalescent-based computation of local branch support from quartet frequencies. Mol. Biol. Evol. 33:1654–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulaev V., Sargent D.J., Crowhurst R.N., Mockler T.C., Folkerts O., Delcher A.L., Jaiswal P., Mockaitis K., Liston A., Mane S.P., Burns P., Davis T.M., Slovin J.P., Bassil N., Hellens R.P., Evans C., Harkins T., Kodira C., Desany B., Crasta O.R., Jensen R.V., Allan A.C., Michael T.P., Setubal J.C., Celton J.-M., Rees D.J.G., Williams K.P., Holt S.H., Rojas J.J.R., Chatterjee M., Liu B., Silva H., Meisel L., Adato A., Filichkin S.A., Troggio M., Viola R., Ashman T.-L., Wang H., Dharmawardhana P., Elser J., Raja R., Priest H.D., Bryant D.W., Fox S.E., Givan S.A., Wilhelm L.J., Naithani S., Christoffels A., Salama D.Y., Carter J., Girona E.L., Zdepski A., Wang W., Kerstetter R.A., Schwab W., Korban S.S., Davik J., Monfort A., Denoyes-Rothan B., Arus P., Mittler R., Flinn B., Aharoni A., Bennetzen J.L., Salzberg S.L., Dickerman A.W., Velasco R., Borodovsky M., Veilleux R.E., Folta K.M. 2011. The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43:109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedmark J.E.E., Eriksson T., Evans R.C., Campbell C.S. 2003. Ancient allopolyploid speciation in Geinae (Rosaceae): evidence from nuclear granule-bound starch synthase (GBSSI) gene sequences. Syst. Biol. 52:374–385. [DOI] [PubMed] [Google Scholar]
- Smith M.L., Hahn M.W. 2021. New approaches for inferring phylogenies in the presence of paralogs. Trends Genet. 37:174–187. [DOI] [PubMed] [Google Scholar]
- Smith S.A., Moore M.J., Brown J.W., Yang Y. 2015. Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evol. Biol. 15:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soják J. 2008. Notes on Potentilla XXI. A new division of the tribe Potentilleae (Rosaceae) and notes on generic delimitations. Bot. Jahrb. Für Syst. Pflanzengesch. Pflanzengeogr. 127:349–358. [Google Scholar]
- Solís-Lemus C., Ané C. 2016. Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting. PLoS Genet. 12:e1005896–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8 - a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub S.C., Fishbein M., Livshultz T., Foster Z., Parks M., Weitemier K., Cronn R.C., Liston A. 2011. Building a model: developing genomic resources for common milkweed (Asclepias syriaca) with low coverage genome sequencing. BMC Genomics 12:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs R.L., Folk R.A., Xiang C.-L., Chen S., Soltis D.E., Cellinese N. 2020. A phylogenomic perspective on evolution and discordance in the alpine-arctic plant clade Micranthes (Saxifragaceae). Front. Plant Sci. 10:1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G.W.C., Ather S.H., Hahn M.W. 2017. Gene-tree reconciliation with MUL-trees to resolve polyploidy events. Syst. Biol. 66: 1007–1018. [DOI] [PubMed] [Google Scholar]
- Turesson G. 1943. Variation in the apomictic microspecies of Alchemilla vulgaris L. Bot. Not. 4:413–427. [Google Scholar]
- Villaverde T., Pokorny L., Olsson S., Rincón-Barrado M., Johnson M.G., Gardner E.M., Wickett N.J., Molero J., Riina R., Sanmartín I. 2018. Bridging the micro- and macroevolutionary levels in phylogenomics: Hyb-Seq solves relationships from populations to species and above. New Phytol. 220:636–650. [DOI] [PubMed] [Google Scholar]
- Walker J.F., Walker-Hale N., Vargas O.M., Larson D.A., Stull G.W. 2019. Characterizing gene tree conflict in plastome-inferred phylogenies. PeerJ. 7:e7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters S., Boznan V. 1967. Alchemilla faeroensis (Lange) Buser and A. alpina L. Proc. Bot. Soc. Br. Isles. 7:83. [Google Scholar]
- Weitemier K., Straub S.C.K., Cronn R.C., Fishbein M., Schmickl R., McDonnell A., Liston A. 2014. Hyb-Seq: combining target enrichment and genome skimming for plant phylogenomics. Appl. Plant Sci. 2:1400042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen D., Yu Y., Zhu J., Nakhleh L. 2018. Inferring phylogenetic networks using PhyloNet. Syst. Biol. 67:735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Huang C.-H., Hu Y., Wen J., Li S., Yi T., Chen H., Xiang J., Ma H. 2017. Evolution of Rosaceae fruit types based on nuclear phylogeny in the context of geological times and genome duplication. Mol. Biol. Evol. 34:262–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Moore M.J., Brockington S.F., Mikenas J., Olivieri J., Walker J.F., Smith S.A. 2018. Improved transcriptome sampling pinpoints 26 ancient and more recent polyploidy events in Caryophyllales, including two allopolyploidy events. New Phytol. 217: 855–870. [DOI] [PubMed] [Google Scholar]
- Yang Y., Moore M.J., Brockington S.F., Soltis D.E., Wong G.K.-S., Carpenter E.J., Zhang Y., Chen L., Yan Z., Xie Y., Sage R.F., Covshoff S., Hibberd J.M., Nelson M.N., Smith S.A. 2015. Dissecting molecular evolution in the highly diverse plant clade caryophyllales using transcriptome sequencing. Mol. Biol. Evol. 32:2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Smith S.A. 2014. Orthology inference in nonmodel organisms using transcriptomes and low-coverage genomes: improving accuracy and matrix occupancy for phylogenomics. Mol. Biol. Evol. 31:3081–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Rabiee M., Sayyari E., Mirarab S. 2018. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Scornavacca C., Molloy E.K., Mirarab S. 2020a. ASTRAL-Pro: quartet-based species-tree inference despite paralogy. Mol. Biol. Evol. 37:3292–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R., Wang Y.-H., Jin J.-J., Stull G.W., Bruneau A., Cardoso D., De Queiroz L.P., Moore M.J., Zhang S.-D., Chen S.-Y., Wang J., Li D.-Z., Yi T.-S. 2020b. Exploration of plastid phylogenomic conflict yields new insights into the deep relationships of leguminosae. Syst. Biol. 69:613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhbannikov I.Y., Hunter S.S., Foster J.A., Settles M.L. 2017. SeqyClean: a pipeline for high-throughput sequence data preprocessing. In: Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, ACM-BCB 17. New York (NY): Association for Computing Machinery. p. 407–416. [Google Scholar]