

Abstract
Metabolic derangements following traumatic brain injury are poorly characterized. In this single-centre observational cohort study we combined 18F-FDG and multi-tracer oxygen-15 PET to comprehensively characterize the extent and spatial pattern of metabolic derangements.
Twenty-six patients requiring sedation and ventilation with intracranial pressure monitoring following head injury within a Neurosciences Critical Care Unit, and 47 healthy volunteers were recruited. Eighteen volunteers were excluded for age over 60 years (n = 11), movement-related artefact (n = 3) or physiological instability during imaging (n = 4). We measured cerebral blood flow, blood volume, oxygen extraction fraction, and 18F-FDG transport into the brain (K1) and its phosphorylation (k3). We calculated oxygen metabolism, 18F-FDG influx rate constant (Ki), glucose metabolism and the oxygen/glucose metabolic ratio. Lesion core, penumbra and peri-penumbra, and normal-appearing brain, ischaemic brain volume and k3 hotspot regions were compared with plasma and microdialysis glucose in patients.
Twenty-six head injury patients, median age 40 years (22 male, four female) underwent 34 combined 18F-FDG and oxygen-15 PET at early, intermediate, and late time points (within 24 h, Days 2–5, and Days 6–12 post-injury; n = 12, 8, and 14, respectively), and were compared with 20 volunteers, median age 43 years (15 male, five female) who underwent oxygen-15, and nine volunteers, median age 56 years (three male, six female) who underwent 18F-FDG PET.
Higher plasma glucose was associated with higher microdialysate glucose. Blood flow and K1 were decreased in the vicinity of lesions, and closely related when blood flow was <25 ml/100 ml/min. Within normal-appearing brain, K1 was maintained despite lower blood flow than volunteers. Glucose utilization was globally reduced in comparison with volunteers (P < 0.001). k3 was variable; highest within lesions with some patients showing increases with blood flow <25 ml/100 ml/min, but falling steeply with blood flow lower than 12 ml/100 ml/min. k3 hotspots were found distant from lesions, with k3 increases associated with lower plasma glucose (Rho −0.33, P < 0.001) and microdialysis glucose (Rho −0.73, P = 0.02). k3 hotspots showed similar K1 and glucose metabolism to volunteers despite lower blood flow and oxygen metabolism (P < 0.001, both comparisons); oxygen extraction fraction increases consistent with ischaemia were uncommon.
We show that glucose delivery was dependent on plasma glucose and cerebral blood flow. Overall glucose utilization was low, but regional increases were associated with reductions in glucose availability, blood flow and oxygen metabolism in the absence of ischaemia. Clinical management should optimize blood flow and glucose delivery and could explore the use of alternative energy substrates.
Keywords: traumatic brain injury, PET, cerebral blood flow, glucose metabolism, microdialysis
Hermanides et al. show that following TBI, glucose delivery is limited by plasma glucose and cerebral blood flow. Overall glucose utilization is low, but regional increases are associated with decreases in glucose availability, blood flow and oxygen metabolism in the absence of ischaemia.
Introduction
Acute derangements in cerebral glucose metabolism (CMRG) are common following traumatic brain injury (TBI).1-4 These have been attributed to anaerobic glycolysis in the presence of classical ischaemia, but other pathophysiological mechanisms may also be responsible.5–7 Microdialysis shows marked reductions in brain glucose8 with modest decreases in plasma glucose within the normal range resulting in metabolic crisis9 and poor outcome.10 In addition to altered relationships between plasma and brain glucose,8 substantial spatial and temporal heterogeneity in glucose utilization exists,2 with increases in non-oxidative glucose utilization seen across white matter,11 and in relation to brain lesions.2,12 The changes in brain glucose metabolism represent complex interactions between glucose availability (plasma glucose), delivery (blood flow), uptake across the blood–brain barrier (transport), entry into the glycolytic pathway (phosphorylation), and oxidative metabolism of glucose (characterized as the metabolic ratio of oxygen-to-glucose utilization). Addressing these complex inter-relationships, while accounting for spatial variations in physiology, requires appropriate tools.
PET using the glucose analogue 18F-fluorodeoxyglucose (18F-FDG) and multi-tracer oxygen-15 (O, 15O2 and C15O) allow imaging of regional CMRG and kinetic parameters,13–15 and comparison with cerebral blood flow (CBF), oxygen metabolism (CMRO2) and oxygen extraction fraction (OEF). Hattori et al.2 showed that 18F-FDG transport into brain, expressed by the rate constant K1, was reduced within the vicinity of contusions following TBI. Glucose uptake depends on supply (plasma glucose and CBF) with transport facilitated via GLUT-1.16 Reductions within the vicinity of lesions could be explained by a decrease in glucose availability, delivery, transport capacity, or a combination of these factors. Increases in glucose phosphorylation have also been seen following TBI,2,12 and tend to occur on the background of an overall reduction in phosphorylation activity (measured by the 18F-FDG rate constant k3).11
An integrated understanding of these acute metabolic derangements following clinical TBI is critical for development of treatments that improve outcome. Using a combination of 18F-FDG and 15O PET, with brain microdialysis in a subset of patients, we investigate how derangements in 18F-FDG kinetic parameters relate to visible TBI lesions, and changes in plasma and microdialysis glucose, CBF, CMRO2, and OEF increases consistent with classical ischaemia.
Materials and methods
In this single-centre observational cohort study, 26 TBI patients requiring intracranial pressure (ICP) monitoring within intensive care and 47 healthy volunteers underwent 18F-FDG and 15O PET between February 1998 and July 2014. We excluded volunteers aged >60 years, technically inadequate imaging studies and those with large variations of arterial pressure or PaCO2. Some data have been reported previously,6,17 but none for the aims of the current manuscript.
Studies were conducted in accordance with 1964 Declaration of Helsinki and later amendments following approval by Cambridgeshire Research Ethics Committee (REC 97/290) and UK Administration of Radioactive Substances Advisory Committee. Volunteers provided informed consent, and assent obtained from patient representatives with consent at follow-up if capacity was regained.
Clinical protocols
Patients
Protocolized and graded interventions used for cerebral perfusion pressure (CPP) and ICP control18 included sedation (propofol 3–5 mg/kg/h and fentanyl 1–4 µg/kg/h) and neuromuscular blockade, surgery for space-occupying lesions, drainage of CSF, vasoactive agents for CPP increment, osmotic therapy, mild hyperventilation [aimed at ∼4.5 kPa (35 mmHg)], mild-to-moderate hypothermia (33–36°C), decompressive craniectomy, and barbiturate metabolic depression. Management aimed for ICP < 20 mmHg and CPP > 65 mmHg19 with tight control of physiology during PET. Outcome was recorded using the Glasgow Outcome Score (GOS)20 at 6 months post-TBI.
Healthy volunteers
Blood pressure, pulse oximetry, and arterial blood gases were monitored to ensure physiological stability during imaging.
Imaging
PET data were acquired on a GE Advance PET scanner (GE Medical Systems). Parametric maps of CBF, cerebral blood volume (CBV), OEF, CMRO2 and 18F-FDG transport into the brain (K1), phosphorylation (k3), and influx rate constant (Ki) (Supplementary Fig. 1), together with CMRG were calculated at the voxel and regional level as previously described.21–23 A more detailed description is provided within the Supplementary material. We categorized PET studies into those performed within 24 h (early), between 2–5 days (intermediate), and 6–12 days (late) following TBI. Parametric maps were co-registered to acute anatomy using structural X-ray CT. In addition, and where available, MRI obtained at follow up were co-registered to early CT.
Microdialysis
Microdialysis catheters (CMA70 10-mm membrane, CMA) were inserted into the cerebral parenchyma as part of standard clinical monitoring. Vials were collected every 20 min and glucose, glutamate, lactate and pyruvate were determined using the CMA600 bedside microdialysis analyser.24 We calculated the mean microdialysis and plasma glucose during 18F-FDG PET.
Imaging analysis
Imaging data were analysed using a combination of custom-designed automated software (PETAn)25 and several software packages, including Statistical Parametric Mapping (SPM, Wellcome Department of Imaging Neuroscience, University College London), MATLAB (MathWorks, Inc), Analyze (AnalyzeDirect, Inc) and Advanced Normalization Tools (ANTs).26 Structural data (CT or T1-weighted MRI) were edited to extract a mask that identified brain tissue voxels and excluded extracranial tissue, CSF, and extra-axial haematomas. All whole brain and region of interest data were masked prior to calculation of PET parameters. In healthy volunteers a whole brain region was used incorporating mixed grey and white matter. In patients, T1-weighted MRI obtained at follow-up were non-rigidly registered with acute CT using ANTs.
Structural T1-weighted MRI acquired in healthy volunteers, and at follow-up in patients, were processed on a specific TBI pipeline27 incorporating segmentation into grey and white matter regions of interest. These regions of interest were applied to the Jacobian determinant images28 providing the amount of contraction or expansion required to normalize the subjects brain into template space (Supplementary material).
Structural regions of interest
Lesion-based regions of interest were drawn on CT using a bespoke segmentation tool (Imseg V1.8, BioMedIA, London, UK). We defined haemorrhagic contusion core and low signal intensity penumbra (Fig. 1A). In addition, we drew a 1 cm cuff of normal-appearing tissue adjacent to penumbra (peri-penumbra). When present, we drew a 10 mm circular cuff of tissue centred on the microdialysis catheter tip. All other normal-appearing tissue was defined as the ‘normal-appearing’ region of interest.
Figure 1.

Peri-lesional hyperglycolysis in the absence of cerebral ischaemia. CT, CBF, CMRO2, OEF, and 18F-FDG kinetic parameters obtained in a 29-year-old male within 24 h of severe TBI following a road traffic accident. Plasma glucose during imaging was 6.6 mmol/l. (A) CT demonstrates bilateral temporal and parietal haemorrhagic contusions. Regions of interest are defined for haemorrhagic lesion (core, red), hypodense tissue (penumbra, blue), and a 1 cm border zone of normal-appearing tissue (peri-penumbra, green). (B) Co-registered parametric maps of CBF, CMRO2, OEF, K1, Ki and k3. The regions with a critical increase in OEF above the individually calculated threshold (ischaemic brain volume) and region of increased phosphorylation activity (k3 hotspot) are both shown in red overlying the CT scan. The ischaemic brain and k3 hotspot volumes were 7 ml and 56 ml, respectively, and overlap between these two tissue classes in this patient was 0.4 ml.
Physiological regions of interest
We used the mean and standard deviation (SD) of k3 voxels within normal-appearing brain of TBI patients to identify regional hotspots. These k3 hotspot regions of interest were defined as the region where k3 voxels were found above the upper 95% confidence interval threshold calculated for each individual patient using the mean plus 1.96 SDs of normal-appearing brain voxels.
We estimated an individualized critical OEF threshold using a previously validated technique to define and calculate the ischaemic brain volume (IBV).17 A more detailed description is provided in the Supplementary material. We examined the spatial location, underlying physiology and mismatch between k3 hotspots and the IBV. MRI obtained at follow-up were co-registered to acute CT. The k3 hotspot regions of interest (minus lesion core and penumbra) were superimposed to explore possible relationships between increased k3 and tissue fate.
Statistical analyses
Data were analysed using R (R Foundation for Statistical Computing, Vienna, Austria), and expressed as median (interquartile range, IQR) unless otherwise stated. Correlations were calculated using linear regression and, in the case of small datasets, Spearman rank. For comparisons between groups, we used the Mann-Whitney U-test and Kruskal-Wallis with Dunn’s test for post hoc comparisons. The Dice similarity coefficient was used to determine the degree of spatial overlap between k3 hotspots and the IBV.29 Locally weighted scatterplot smoothing (LOWESS)30 was used to plot relationships using whole brain voxel data using a 2.5% weighted sample. Results were interpreted taking into consideration that a Bonferroni correction for use of 11 variables [K1, k2, k3, Ki, CMRG, lumped constant, CBF, CMRO2, OEF, CBV, and CMRO2/CMRG metabolic ratio (MR)] would require P < 0.005 for formal statistical significance.
Data availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Results
Additional data are presented within the Supplementary material.
Study population
We analysed data from 20 healthy volunteers following 15O PET and nine healthy volunteers following 18F-FDG PET, with no significant neurological/psychiatric illness, and 34 combined PET sessions (18F-FDG and 15O) in 26 TBI patients with stable ventilation and CPP as per protocolized clinical management. Eleven healthy volunteers were excluded for age >60 years, three for movement-related artefact and four with physiological instability during imaging (variations in PaCO2 > 0.5 kPa).
Patients had severe TBI with a Glasgow Coma Score (GCS) ≤ 8 or presented with moderate TBI (GCS 9–12), but deteriorated, requiring sedation and ventilation for management of ICP. Of the 34 PET sessions in patients, 12 were within 24 h (early), eight between 2–5 days (intermediate), and 14 between 6–12 days (late) after TBI. Eight subjects underwent imaging on two occasions, but for one of these only the CBF from the late session was analysed. Table 1 summarizes participant characteristics. The 18F-FDG healthy volunteers were older than the 15O healthy volunteers and TBI patients [56 (53–57) versus 43 (31–47) and 40 (26–52) years, respectively; both comparisons P < 0.01], while 15O healthy volunteers and TBI patients were similar in age (P = 0.95). Female participants were more common within 18F-FDG healthy volunteers compared to the 15O healthy volunteers and TBI patients [6 of 9 (67%) versus 5 of 20 (25%) and 4 of 26 (15%), respectively; both comparisons P < 0.05], while 15O healthy volunteers and TBI patients were similar in terms of gender (P = 0.47).
Table 1.
Participant characteristics
| Healthy volunteers |
TBI patients |
||
|---|---|---|---|
| 18F-FDG (n = 9) | 15O (n = 20) | Total (n = 26) | |
| Age, years | 56 (53–57) | 43 (31–47) | 40 (26–52) |
| Gender, female/male | 6/3 | 5/15 | 4/22 |
| GCS post-resuscitation | – | – | 6 (3–9) |
| Injury mechanism | |||
| Fall | – | – | 10 |
| Road traffic collision | – | – | 16 |
| Marshall classification | |||
| Diffuse injury I | – | – | 0 |
| Diffuse injury II | – | – | 5 |
| Diffuse injury III | – | – | 0 |
| Diffuse injury IV | – | – | 0 |
| Evacuated mass lesion V | – | – | 13 |
| Non-evacuated mass lesion VI | – | – | 8 |
| Primary lesion | |||
| Subdural haematoma | – | – | 5 |
| Contusion | – | – | 18 |
| DAI, IVH and tSAH | – | – | 3 |
| Craniectomy | – | – | 12 |
| Barbiturate coma | – | – | 2 |
| Hypothermia | – | – | 1 |
| Blood glucose, mmol/l | 4.6 (4.4–5.4) | – | 5.8 (5.3–6.7) |
| Outcome (GOS) | – | – | 3 (3–4)a |
Characteristics of the TBI patients who underwent combined 18F-FDG and 15O PET, and the healthy volunteers who underwent separate 18F-FDG and 15O PET sessions. Data shown are median (IQR). DAI = diffuse axonal injury; GCS = Glasgow Coma Scale; IVH = intraventricular haemorrhage; tSAH = traumatic subarachnoid haemorrhage. Blood glucose is the mean value of this variable over the course of each PET study.
Two missing data-points for GOS.
Glucose availability, delivery and transport
Brain glucose is correlated with plasma glucose
Plasma glucose during PET was higher following TBI compared with healthy volunteers 5.8 (5.3–6.7) versus 4.6 (4.4–5.4) mmol/l (P < 0.001). Microdialysis data were available for 10 PET sessions from eight patients. On two occasions the microdialysis catheter was inserted within the vicinity of injured tissue. Plasma and microdialysis glucose in this subset ranged from 4.9–8.9 mmol/l and 0.4–4.1 mmol/l, respectively, and increases in plasma glucose were associated with higher microdialysis glucose (Rho = 0.64, P = 0.047; Supplementary Fig. 5). Brain tissue glucose concentration calculated using the 18F-FDG kinetic parameters was related to plasma glucose within healthy volunteers (R2 = 0.47, P = 0.04) and all TBI regions (R2 = 0.16, P < 0.001). In patients, this relationship was strongest within normal-appearing brain and peri-penumbra (R2 = 0.66, P < 0.001 and R2 = 0.69, P < 0.001, respectively), in comparison with core and penumbra (R2 = 0.06, P = 0.17 and R2 = 0.25, P < 0.01, respectively) (Supplementary Fig. 6).
Glucose delivery varies between normal-appearing, perilesional and lesion core tissue
CBF and K1 were decreased in core and penumbra as compared to peri-penumbra and normal-appearing brain in patients (P < 0.001; Table 2). When compared with healthy volunteers, normal-appearing brain in patients had lower CBF (P < 0.001), but similar K1. There were no significant changes in CBF and K1 within lesion and normal-appearing brain regions at different time points (Fig. 2 and Supplementary Table 1).
Table 2.
Regional metabolic parameters
| Patients |
Healthy volunteers |
||||||
|---|---|---|---|---|---|---|---|
| Core | Penumbra | Peri-penumbra | Normal appearing | P-valuea | P-valueb | ||
| K1, ml/ml/min | 0.039 (0.021)c,d | 0.049 (0.018)c | 0.084 (0.022)e | 0.091 (0.025) | <0.001 | 0.091 (0.007) | 0.720 |
| k2,/min | 0.154 (0.069) | 0.131 (0.048)c | 0.156 (0.052) | 0.173 (0.055) | <0.001 | 0.128 (0.029) | <0.001 |
| k3,/min | 0.044 (0.021)c,d | 0.039 (0.010)c | 0.030 (0.012) | 0.029 (0.010) | <0.001 | 0.057 (0.009) | <0.001 |
| Ki, ml/ml/min | 0.009 (0.006)c,d | 0.012 (0.004) | 0.014 (0.004) | 0.013 (0.005) | <0.001 | 0.034 (0.008) | <0.001 |
| CMRG, µmol/100 ml/min | 11.9 (5.4)c,d | 14.9 (4.3)c | 19.5 (4.1)e | 19.9 (4.9) | <0.001 | 26.3 (1.9) | <0.001 |
| LC | 0.48 (0.07)c,d | 0.47 (0.05)c | 0.43 (0.05)e | 0.41 (0.04) | <0.001 | 0.56 (0.04) | <0.001 |
| CBF, ml/100 ml/min | 12.2 (10.7)c,d | 18.1 (6.4)c | 27.4 (8.1)e | 28.6 (4.9) | <0.001 | 42.9 (11.5) | <0.001 |
| CBV, ml/100 ml | 2.7 (0.9)c,d | 2.8 (0.8)c | 3.5 (0.9)e | 4.3 (0.9) | <0.001 | 3.5 (0.5) | <0.001 |
| CMRO2, µmol/100 ml/min | 29.6 (23.8)c,d | 45.6 (20.6)c | 71.9 (27.3)e | 82.1 (30.5) | <0.001 | 139.0 (39) | <0.001 |
| OEF, % | 30.0 (20.1)c,d | 38.5 (14.4) | 42.3 (12.2) | 42.1 (10.4) | <0.001 | 43.9 (4.5) | 0.685 |
| Metabolic ratio | 2.6 (1.3)c,d | 2.9 (0.8)c | 4.0 (1.2)e | 4.0 (1.2) | <0.001 | ||
18F-FDG kinetic parameters, CMRG, lumped constant (LC), CBF, CBV, CMRO2, OEF and CMRO2/CMRG metabolic ratio for the different regions of interest in patients and healthy volunteers. Values are median (IQR).
Kruskal-Wallis test for comparison between core, penumbra, peri-penumbra and normal-appearing regions of interest within patients, with subsequent post hoc Dunn’s tests surviving correction for multiple comparisons (P < 0.005) between each lesion region of interest and normal-appearing brainc, between core and peri-penumbrad, and between peri-penumbra and penumbrae.
Mann-Whitney U-test for comparison between the normal-appearing region of interest in patients versus healthy volunteers.
Figure 2.

Spatial and temporal pattern of metabolic parameters. Box-and-whisker plots of the 18F-FDG kinetic parameters (K1, k3, Ki), CBF, CBV, OEF, CMRO2, CMRG, and CMRO2/CMRG metabolic ratio for the different regions of interest in patients within 24 h (Early; white), Days 2–5 (Intermediate; green) and Days 6–12 (Late, blue) post-injury. The horizontal line within each box denotes the median value, the lower and upper boundaries the 25th and 75th centile, the vertical lines the 10th and 90th centile, and the closed circles outlying data-points. The solid and dashed black lines represent the median and the full range of values for healthy volunteers, respectively. For the metabolic ratio the median and range in healthy volunteers is calculated from two different cohorts of subjects.
Cerebral blood flow is an important determinant of brain glucose transport
Region of interest data in patients showed a clear relationship between CBF and glucose transport (as measured by K1) across all tissue classes (R2 = 0.65, P < 0.001. Fig. 3). This relationship was driven by core and penumbra (R2 = 0.81 and 0.48, respectively, P < 0.001), where both K1 and CBF were below the lower limit of values found within healthy volunteers. There was no significant relationship within peri-penumbra and normal-appearing tissue (both R2 = 0.1, P = 0.08 and P = 0.07, respectively).
Figure 3.
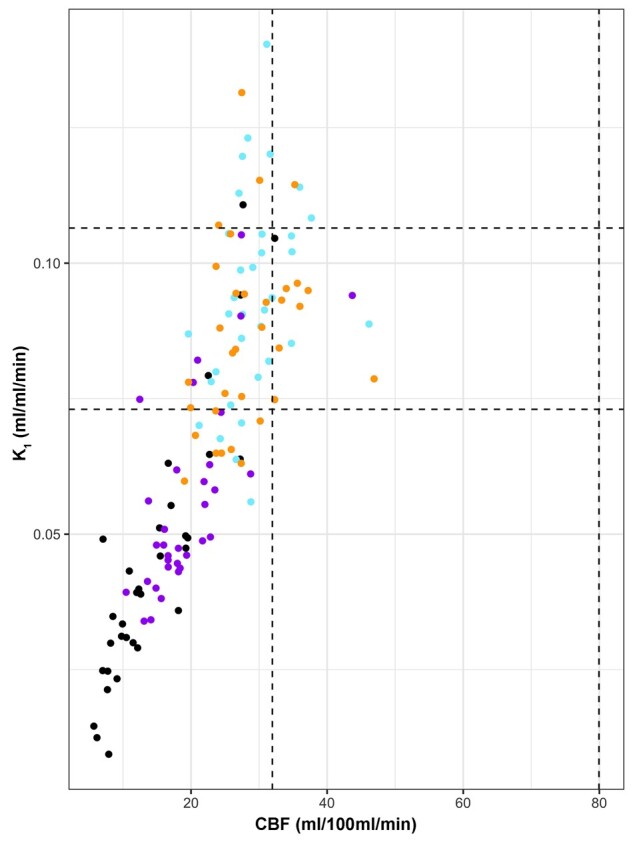
Relationship between glucose transport and CBF based on region of interest data. Scatterplot of 18F-FDG transport (K1) and CBF in patient regions of interest: lesion core (black), penumbra (purple), peri-penumbra (blue) and normal-appearing (orange). The black vertical and horizontal dotted lines indicate the full range for healthy volunteer CBF and K1 values, respectively.
Whole-brain voxel-wise analysis showed that where CBF was maintained within the normal range, K1 was higher than within healthy volunteers, but reductions in CBF below 25 ml/100 ml/min were associated with steep reductions in K1 (Fig. 4 and Supplementary Fig. 7).
Figure 4.

Relationships between 18F-FDG kinetic parameters and CBF based on voxel data. For each PET imaging session in patients the relationship between CBF and 18F-FDG transport (K1; left) and phosphorylation activity (k3; right) is plotted for voxels across the whole brain. The fitted light blue lines represent modelling of the relationship between each parameter using locally weighted scatterplot smoothing (LOWESS), with the 95% confidence interval shown in grey. The blue vertical and horizontal dotted lines indicate the full range for healthy volunteer CBF and 18F-FDG kinetic parameters, respectively.
Glucose utilization
Regional glucose utilization and phosphorylation rates are discordant and variable
Data are displayed in Table 2. There was a general decrease in phosphorylation activity (k3) in TBI patients with normal-appearing brain showing lower activity than healthy volunteers 0.029 (0.024–0.034) versus 0.057 (0.055–0.063)/min, P < 0.001. In patients, k3 was highest within core 0.044 (0.036–0.057)/min with values significantly higher than peri-penumbra 0.030 (0.025–0.037)/min and normal-appearing brain (P < 0.001 for both comparisons). Penumbra k3 values 0.039 (0.033–0.043)/min were higher than normal-appearing brain (P = 0.002). Increases in k3 were associated with a fall in metabolic ratio, particularly within core and penumbra where metabolic ratio was <4 (Table 2 and Supplementary Fig. 8).
Glucose metabolism and Ki were lowest in core and highest in normal-appearing brain, but significantly lower when compared to healthy volunteers (P < 0.001 for both comparisons). When dephosphorylation (k4) was included within regional kinetic modelling the results were similar (Supplementary Table 2). A similar pattern was found for CBF, CBV, CMRO2 and metabolic ratio, with lowest values found within the core, and greater values found with increasing distance from the lesion core. The temporal pattern of regional data is displayed in Fig. 2 and Supplementary Table 1. There was a non-significant trend for higher k3 values at late time points post-injury within core (P = 0.09), and higher metabolic ratio values within all regions of interest at early time points (which did not survive correction for multiple comparisons). In comparison, regional CMRO2 and OEF showed higher values at early times points post-injury, with CMRO2 values for normal-appearing brain (P = 0.003) and OEF values in penumbra (P = 0.004) remaining significant after correction for multiple comparisons.
Phosphorylation activity is dependent on the adequacy of cerebral blood flow
The voxel-wise relationship between k3 and CBF showed variability both within and between subjects (Supplementary Fig. 7), but demonstrated clear patterns across the dataset. k3 remained constant across the range of CBF values found in healthy volunteers, but showed a small rise at CBF ∼20 ml/100 ml/min, followed by a precipitous fall with CBF <12 ml/100 ml/min (Fig. 4). A similar relationship was seen between k3 and CMRO2 (Supplementary Fig. 9).
Relationships between plasma and microdialysis glucose and phosphorylation activity
Across all structural regions in patients, a lower plasma glucose was associated with an increase in k3 and Ki (Rho = −0.33 and −0.50, respectively, P < 0.001). Using the modelled 18F-FDG kinetic parameters, we calculated the proportion of brain tissue glucose phosphorylated across all patient regions and related this to plasma and calculated brain glucose concentration. The fraction of tissue glucose undergoing phosphorylation increases when plasma glucose falls to <6–8 mmol/l and/or brain glucose <1–2 mmol/l, particularly within the vicinity of brain lesions (Supplementary Fig. 10).
For 10 PET sessions within eight patients we found microdialysis glucose of 1.6 (0.9–2.8) mmol/l, lactate 4.4 (1.9–6.5) mmol/l, pyruvate 138.4 (95.8–181.9) µmol/l, glutamate 10.4 (4.2–22.4) µmol/l and the lactate/pyruvate ratio 28.3 (17.4–49.3). Lower microdialysis glucose was associated with an increase in k3 (Rho = −0.73, P = 0.02), (Supplementary Fig. 11) as well as Ki (Rho = −0.71, P = 0.03). There were no significant relationships between microdialysis glucose and glutamate, pyruvate, lactate or lactate/pyruvate ratio. No microdialysis catheters were inserted in areas where k3 or OEF values were above hotspot or IBV thresholds.
Hyperglycolysis is found in perilesional and normal brain, and is largely independent of ischaemia
Cerebral physiology differed between k3 hotspots and the ischaemic brain volume
The volume of brain meeting the criteria for a k3 hotspot was larger than IBV: 64 (55–96) versus 7 (2–19) ml (P < 0.001, Mann-Whitney U-test), and the volume of k3 hotspot and IBV did not differ between early, intermediate and late groups. Similar proportions of k3 hotspots [34 (13–44)%] and IBV [29 (15–54)%] were within 1 cm of a lesion (P = 0.75), but regions of physiological derangement consistent with both tissue classes were seen within normal-appearing brain. Evidence of overlap between k3 hotspots and IBV was uncommon [0 (0–1) ml], with a Dice similarity co-efficient of 0.01 (0–0.02), and even where such overlap was substantial, it represented the minority of each physiological tissue class (Fig. 5). More commonly, k3 increases were found in the absence of ischaemia, both within the vicinity of injured (Fig. 1) and normal-appearing brain (Supplementary Fig. 12).
Figure 5.

Evidence of cerebral ischaemia and hyperglycolysis. CT, CBF, CMRO2, OEF, and 18F-FDG kinetic parameters obtained in a 40-year-old female within 24 h of severe TBI resulting from a fall. Imaging was obtained following evacuation of a subdural haematoma. Plasma glucose during PET was 3.7 mmol/l. (A) CT demonstrates residual subdural blood with minimal midline shift and underlying haemorrhagic contusions. Regions of interest are defined for haemorrhagic lesion (core, red), hypodense tissue (penumbra, blue), and a 1 cm border zone of normal-appearing tissue (peri-penumbra, green). (B) Co-registered parametric maps of CBF, CMRO2, OEF, K1, Ki and k3. The regions with a critical increase in OEF above the individually calculated threshold (ischaemic brain volume) and increased phosphorylation activity (k3 ‘hotspot’) are both shown in red overlying the CT scan. The ischaemic brain volume and k3 hotspot volume were 96 ml and 174 ml, respectively, and the volume of overlap between these two tissue classes in this patient was 38 ml.
The underlying physiology in k3 hotspots and the IBV is summarized in Supplementary Table 3. Despite lower CBF in k3 hotspots [25.0 (21.9–29.5) ml/100 ml/min] compared to healthy volunteers [42.9 (37.0–48.5)] and normal-appearing brain in patients [28.6 (26.4–31.4)], there was a signature of maintained K1 and higher CMRG. Metabolic ratio in k3 hotspots [2.7 (2.1–3.3)] was lower than that found within IBV [5.2 (4.0–5.8)] and normal-appearing brain in patients [4.0 (3.5–4.6)].
The temporal pattern of changes in physiology within k3 hotspots and the IBV are shown with Supplementary Fig. 13 and Supplementary Table 4. Within k3 hotspots, CMRO2, OEF and metabolic ratio tended to reduce with time post-TBI, but only comparisons for OEF survived correction for multiple comparisons (P = 0.002).
Association between the ischaemic brain volume and k3 hotspots and outcome
Within 24 h, IBV showed a non-significant trend with worse outcome quantified using the GOS (Rho = −0.51, P = 0.11), and there was no association across the whole cohort (Rho = −0.28; P = 0.13). The volume of k3 hotspot was not associated with the GOS (Rho = −0.18, P = 0.31).
Eight patients underwent follow-up MRI at >3 months post-injury (median 9, range 4–35 months), of which seven had T1-weighted volumetric images for comparison with similar data acquired within the nine 18F-FDG PET healthy volunteers. Mean normalized cortical grey matter volume was similar (0.42 versus 0.39, P = 0.05) in patients and healthy volunteers, while white matter volume was lower in patients (0.42 versus 0.47, P < 0.01). The mean Jacobian determinant values for cortical grey and white matter regions in patients compared with healthy volunteers were 0.04 versus −0.01 (P = 0.01) and 0.02 versus −0.01 (P = 0.06), respectively, with higher values indicative of smaller volumes requiring expansion to match the brain template. Early increases in k3 outside of lesion core and penumbra were often found in close proximity to areas showing evidence of late structural injury on MRI obtained at follow-up (Fig. 6). There was no consistent voxel-wise relationship between k3 and volume contraction using Jacobian determinant values across the normal-appearing brain in TBI patients (Supplementary Fig. 14).
Figure 6.

Association between early metabolic derangements and tissue fate. Imaging obtained in a 17-year-old male with moderate TBI following a road traffic accident with initial GCS of 12, but who required sedation and ventilation for control of raised ICP. Co-registered CBF, CMRO2, OEF, 18F-FDG kinetic parameters and CT imaging obtained on Day 8 following TBI. Plasma glucose during imaging was 5.4 mmol/l. The CT demonstrates left frontal and temporal haemorrhagic contusions and is shown with the T1-weighted MRI obtained at 9 months post-injury; the MRI has been non-rigidly registered to the CT. Both structural images are displayed with the region of k3 hotspot outlined in red, but both lesion core and penumbra have been excluded from the hotspot to ensure that only increases in k3 outside of the lesion identified on CT imaging are shown. The volume of brain within the k3 hotspot in this subject was 149 ml and, in comparison with brain that appeared structurally normal, had CBF 19.6 versus 19.6 ml/100 ml/min, CMRO2 55.2 versus 60.2 µmol/100 ml/min, OEF 37.8 versus 42.4%, K1 0.097 versus 0.087 ml/ml/min, Ki 0.018 versus 0.012 ml/ml/min and k3 0.058 versus 0.024/min, respectively. The T1-weighted MRI demonstrates established lesions within the left frontal and temporal brain regions in close proximity with k3 increases outside of lesion core and penumbra identified on the CT image.
Discussion
We used 18F-FDG and multi-tracer 15O PET together with brain microdialysis in a cohort of acute TBI patients to image the impact of glucose availability, delivery and utilization on cerebral metabolism. Brain glucose delivery (K1) is dependent on both plasma glucose and the adequacy of CBF, particularly within regions where CBF <25 ml/100 ml/min. An overall global reduction in glucose utilization following TBI appears to be driven by low glucose delivery and low phosphorylation activity, but regional hotspots were found within, and distant from, visible brain lesions. Such increases were explained by preserved glucose delivery (K1) despite reductions in CBF, and in plasma and brain glucose. Increases in glucose utilization were typically accompanied by CMRO2 reductions, but without OEF increases consistent with ischaemia. These metabolic derangements represent a potential therapeutic target.
Glucose availability, delivery and transport
We confirm previous findings that brain glucose is associated with serum glucose, but variable across the injured brain,8,31 and that K1 is similar to healthy volunteers in normal-appearing brain and reduced around lesions following TBI.2 However, we also show for the first time, that K1 was related to CBF, particularly at CBF <25 ml/100 ml/min (Figs 3 and 4). Microdialysis shows a linear relationship between plasma and brain glucose in non-injured areas after TBI,8,31 but glucose transport is impaired within the injured brain.8 CBF may be the rate-limiting factor for glucose transport into the brain, but we found that normal-appearing brain in TBI patients had similar K1 to that found within healthy volunteers despite lower CBF, suggesting that increases in GLUT-1 expression may also contribute.32
Glucose utilization
In comparison with healthy volunteers, TBI patients showed a global decrease in glucose utilization, but derangements in kinetic parameters were heterogeneous across the injured brain, in keeping with past reports,33 with reductions attributed to a combination of sedation,34,35 decreased consciousness and/or TBI itself.36 All patients who underwent PET imaging received variable rate infusions of sedatives and analgesics to facilitate metabolic suppression as part of intensive care management and ICP-directed therapy, but despite this, there were regional increases in glucose utilization. The metabolic ratio was reduced within visible brain lesions, but also within normal-appearing brain, as reported previously.15,37 Lower values within lesion core and penumbra imply increased anaerobic glycolysis, but the lack of OEF increase is against classical ischaemia and is suggestive of microvascular ischaemia and mitochondrial dysfunction.7
Whilst visible lesions had the lowest Ki and CMRG, this was driven by low glucose delivery (K1 and CBF) rather than a reduction in k3, which was higher than that found within normal-appearing brain. A sharp fall in k3 was associated with CBF <12 ml/100 ml/min and cerebral oxygen metabolism consistent with irreversible tissue injury (<40 µmol/100 ml/min).38 Prior to reaching this threshold, increases in k3 within individual subjects (Supplementary Fig. 7) were seen below the lower limit of CBF found in healthy volunteers (<25 ml/100 ml/min). Such findings support measures to preserve or optimize CBF and glucose delivery within the vicinity of brain lesions to maintain metabolism and prevent neuronal loss. In addition, studies highlight that alternative substrates may be utilized as an energy source, via the tricarboxylic acid cycle, following TBI and are a potential therapeutic approach.39–42
Increases in k3 and Ki in TBI patients were associated with both lower plasma glucose and microdialysis glucose, raising the possibility that low brain glucose is driven by increased consumption as well as reduced delivery. Our data show that the proportion of brain glucose utilized increases as plasma and brain glucose falls below 6–8 and 1–2 mmol/l, respectively, particularly within the vicinity of lesions (Supplementary Fig. 10). Other studies have shown CMRG increases with tight glycaemic control, associated with an increase in lactate/pyruvate ratio indicative of metabolic stress.43 We found no association between microdialysis lactate or lactate/pyruvate ratio and k3 in our current study, and have previously shown an increase in glucose metabolism to both lactate and pyruvate with no change in the lactate/pyruvate ratio following TBI.44 Randomized controlled trials of intensive insulin therapy in TBI show no benefit,45–47 and though a recent meta-analysis is inconclusive,48 our data suggest that plasma glucose levels should be carefully controlled and not be allowed to fall below 6–8 mmol/l, ideally in combination with monitoring of brain glucose to ensure values of 1–2 mmol/l are maintained across the injured brain.
Previous 18F-FDG PET studies show hyperglycolysis following TBI,12 driven by regional phosphorylation increases.2 We delineated the burden and spatial pattern of k3 hotspots and compared this with evidence of ischaemia (IBV). The k3 hotspot regions were found both in the vicinity of, and distant from visible lesions. The volume of k3 hotspots was larger than the IBV, the underlying physiology different, and there was little spatial overlap between these two tissues classes. While ischaemia and hyperglycolysis sometimes overlapped within 24 h of injury (Fig. 5), this was uncommon. The typical metabolic signature of a k3 hotspot was higher CMRG with preserved K1 and OEF, despite lower CBF, CMRO2 and metabolic ratio. A late (Day 6–12 post-TBI) trend for lower CMRO2, OEF and metabolic ratio within k3 hotspots was suggestive of worsening or persistent metabolic derangements and risk of neuronal loss with CMRO2 values close to the threshold for tissue viability (<40 µmol/100 ml/min).38 While such changes could reflect differences in intensive care management at different phases of recovery following TBI, all received standardized care. This pattern of metabolic derangements is also consistent with microvascular ischaemia7 and/or mitochondrial dysfunction,49 and could be associated with such pathophysiological processes as spreading depolarization,50 cytotoxic oedema,51 and neuroinflammation.52 Such processes are common following TBI,7 and are consistent with our finding of a low metabolic ratio secondary to a decrease in oxygen metabolism within regions showing increases in glucose utilization. In patients, follow-up imaging of brain metabolism, with structural and functional tissue outcome evaluated using MRI, and functional outcome assessments would be required to definitively address the significance of early metabolic derangements.
We compared 18F-FDG PET obtained within a group of TBI patients to identical data obtained in healthy volunteers using standard kinetic modelling.53–55 Previously published values for 18F-FDG kinetic parameters in healthy volunteers and TBI patients2,56 are similar to the data we report, and the available test-retest reproducibility data suggest a co-efficient of variation of up to 10–15% for glucose metabolism.57 While test-retest variability is important in assessing the significance of interventional or serial studies within the same subject, in this observational study we demonstrate regional changes in 18F-FDG kinetic parameters and glucose metabolism between TBI subjects and healthy volunteers of up to 50% or more across the injured brain. At the voxel level, to avoid high variability in the 18F-FDG kinetic parameters, we used a basis function version of a two-tissue compartment model, which assumes irreversible phosphorylation of 18F-FDG.22,54,58 Modelling the data on a regional basis with the inclusion of dephosphorylation (k4) showed similar results (Supplementary Table 2). In contrast to Hattori et al.,2 we found no differences in k4 between patient regions on interest, or in comparison with healthy volunteers. The lumped constant within normal-appearing brain in TBI patients (0.41) was lower than that found within healthy volunteers (0.56), and is comparable to previous reports.36 While this value is used to calculate CMRG, we also report the influx rate constant (Ki).
Outcome impact
We have previously shown that early ischaemia (<24 h post-TBI) was associated with poor outcome.17 We were unable to confirm this finding, perhaps because IBV was not increased within 24 h of injury in the current cohort. k3 hotspots were found throughout the first 12 days post-TBI, but there was no association with poor outcome, as defined by GOS. Those patients who underwent follow-up MRI demonstrated lower normalized global white matter volume compared with healthy volunteers and a trend for lower cortical grey and white matter volumes. While k3 hotspots in structurally normal brain (on CT) may be associated with MRI visible lesions at follow-up, the relationship between k3 and volume contraction was variable. Metabolic derangements resulting in both increases and decreases in k3 may be associated with late structural injury and further study is required to confirm the significance of these derangements.
Limitations
We examined the impact of TBI on glucose delivery and metabolism using PET obtained within a typical patient cohort, rather than examine the temporal pattern and evolution within individual subjects. As such, we did not acquire follow-up MRI in all subjects and the analyses of the possible relationship between early metabolic derangements and structural outcome presented are illustrative of how future studies could address this issue. Future sequential imaging studies incorporating MRI and spectroscopy,59 diffusion tensor imaging and multi-tracer PET (15O, 18F-FDG, and SV2A)60 could delineate evidence of pan-necrosis, cortical atrophy and selective neuronal loss related to early metabolic derangements, and correlate these with functional deficits resulting in poor outcome post TBI.
Acute MRI was not available for TBI patients and evidence of structural injury was defined on the basis of early CT. While we may have missed structural changes not visible on CT, we were able to demonstrate significant physiological differences between structural regions of interest and used PET to define metabolic regions of interest for analyses.
Conclusions
The data presented in this study confirm previous work61 and show that, up to 12 days post-injury, there is an overall reduction in oxygen and glucose metabolism across the brain. Our results show that K1 is preserved within normal-appearing brain in the face of CBF reduction and this may help maintain glucose delivery, and mitigate the impact of modest plasma glucose reductions. However, this compensation fails when CBF and/or glucose delivery are critically low. Despite this, such regions can show high k3 values, providing evidence of metabolically active tissue, particularly within the vicinity of brain lesions at risk of expansion. Regions of increased glucose utilization were associated with non-ischaemic reductions in CBF and CMRO2 consistent with microvascular ischaemia and/or mitochondrial dysfunction. The primary physiological target in TBI has been the maintenance of CBF to ensure oxygen delivery, but we show that it may be just as important to ensure adequate glucose delivery. Where CBF elevation is either impossible or ineffective, our data provide a physiological framework for maintaining glucose at high normal levels (8–10 mmol/l) to optimize substrate delivery. Finally, these data make a case for the development and evaluation of alternative substrates (such as ketones) whose delivery is independent of transporters used to deliver glucose.42
Supplementary Material
Acknowledgements
Aspects of the data collection and sharing for this project were provided by the Cambridge Centre for Ageing and Neuroscience (CamCAN). We thank all our patients and their families, and healthy volunteers for participating in this study.
Funding
J.H. reports grants from Horizon 2020, Novonordisk Ltd, European Society of Anaesthesiology, The Netherlands Organisation for Health Research and Development, and the University of Amsterdam research fund. K.L.H.C. reports grants from the NIHR (Biomedical Research Centre Cambridge, and Invention for Innovation) and the UK Medical Research Council. P.J.H. reports grants from the Royal College of Surgeons of England, British Brain and Spine Foundation, Academy of Medical Sciences/Health Foundation, and NIHR (Research Professorship, Cambridge BRC, and Global Health Research Group on Neurotrauma). V.F.J.N. is supported by an Academy of Medical Sciences/Health Foundation Clinician Scientist Fellowship. T.D.F., F.I.A., J.P.C. and D.K.M. report grants from the UK Medical Research Council. J.P.C. reports grants from the Royal College of Anaesthetists/British Journal of Anaesthesia, National Institute of Academic Anaesthesia, Addenbrooke’s Charities, the Wellcome Trust, Beverley and Raymond Sackler, and the Academy of Medical Sciences/Health Foundation. CamCAN funding was provided by the UK Biotechnology and Biological Sciences Research Council (grant number BB/H008217/1), together with support from the UK Medical Research Council and University of Cambridge, UK. This work was supported by researchers at the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre. Cambridge University Hospitals NHS Foundation Trust and the University of Cambridge acted as the sponsor for this study, with responsibility for study conduct and management. Neither the funders nor the sponsor had input into study design; data collection, data analysis, data interpretation; writing of the report; or the decision to submit the paper for publication.
Competing interests
A.K.G. reports a paid consultancy from Pressura Neuro Ltd. V.F.J.N. reports a grant with Rioche Pharmaceuticals. D.K.M. reports paid consultancy, research grants, or membership of Data Monitoring Committee for GlaxoSmithKline Ltd, Ornim Medical, Neurovive Ltd, Calico Ltd, NeuroTrauma Sciences LLC, and Pfizer Ltd. All other authors declare no competing interests.
Supplementary material
Supplementary material is available at Brain online.
Glossary
- CBF
cerebral blood flow
- CBV
cerebral blood volume
- CMRG
cerebral glucose metabolism
- CMRO2
oxygen metabolism
- GOS
Glasgow Outcome Score
- IBV
ischaemic brain volume
- ICP
intracranial pressure
- OEF
oxygen extraction fraction
- TBI
traumatic brain injury
References
- 1. Coles JP, Cunningham AS, Salvador R, et al. Early metabolic characteristics of lesion and nonlesion tissue after head injury. J Cereb Blood Flow Metab. 2009;29(5):965–975. [DOI] [PubMed] [Google Scholar]
- 2. Hattori N, Huang SC, Wu HM, et al. Acute changes in regional cerebral (18)F-FDG kinetics in patients with traumatic brain injury. J Nucl Med. 2004;45(5):775–783. [PubMed] [Google Scholar]
- 3. Wu HM, Huang SC, Hattori N, et al. Subcortical white matter metabolic changes remote from focal hemorrhagic lesions suggest diffuse injury after human traumatic brain injury. Neurosurgery. 2004;55(6):1306–1315; discussion 1316–1317. [DOI] [PubMed] [Google Scholar]
- 4. Wu HM, Huang SC, Vespa P, Hovda DA, Bergsneider M.. Redefining the pericontusional penumbra following traumatic brain injury: Evidence of deteriorating metabolic derangements based on positron emission tomography. J Neurotrauma. 2013;30(5):352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vespa P, Bergsneider M, Hattori N, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: A combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab. 2005;25(6):763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abate MG, Trivedi M, Fryer T, et al. Early derangements in oxygen and glucose metabolism following head injury: The ischemic penumbra and pathophysiological heterogeneity. Neurocrit Care. 2008;9(3):319–325. [DOI] [PubMed] [Google Scholar]
- 7. Veenith TV, Carter EL, Geeraerts T, et al. Pathophysiologic mechanisms of cerebral ischemia and diffusion hypoxia in traumatic brain injury. JAMA Neurology. 2016;73(5):542–550. [DOI] [PubMed] [Google Scholar]
- 8. Magnoni S, Tedesco C, Carbonara M, Pluderi M, Colombo A, Stocchetti N.. Relationship between systemic glucose and cerebral glucose is preserved in patients with severe traumatic brain injury, but glucose delivery to the brain may become limited when oxidative metabolism is impaired: Implications for glycemic control. Crit Care Med. 2012;40(6):1785–1791. [DOI] [PubMed] [Google Scholar]
- 9. Vespa P, Bergsneider M, Hattori N, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: A combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab. 2005;25(6):763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stein NR, McArthur DL, Etchepare M, Vespa PM.. Early cerebral metabolic crisis after TBI influences outcome despite adequate hemodynamic resuscitation. Neurocrit Care. 2012;17(1):49–57. [DOI] [PubMed] [Google Scholar]
- 11. Wu HM, Huang SC, Hattori N, et al. Selective metabolic reduction in gray matter acutely following human traumatic brain injury. J Neurotrauma. 2004;21(2):149–161. [DOI] [PubMed] [Google Scholar]
- 12. Bergsneider M, Hovda DA, Shalmon E, et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: A positron emission tomography study. J Neurosurg. 1997;86(2):241–251. [DOI] [PubMed] [Google Scholar]
- 13. Coles JP. Imaging of cerebral blood flow and metabolism. Curr Opin Anaesthesiol. 2006;19(5):473–480. [DOI] [PubMed] [Google Scholar]
- 14. Huang SC, Phelps ME, Hoffman EJ, Sideris K, Selin CJ, Kuhl DE.. Noninvasive determination of local cerebral metabolic rate of glucose in man. The Am J Physiol. 1980;238(1):E69–E82. [DOI] [PubMed] [Google Scholar]
- 15. Jalloh I, Carpenter KL, Helmy A, Carpenter TA, Menon DK, Hutchinson PJ.. Glucose metabolism following human traumatic brain injury: Methods of assessment and pathophysiological findings. Metab Brain Dis. 2015;30(3):615–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Simpson IA, Carruthers A, Vannucci SJ.. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J Cereb Blood Flow Metab. 2007;27(11):1766–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Launey Y, Fryer TD, Hong YT, et al. Spatial and temporal pattern of ischemia and abnormal vascular function following traumatic brain injury. JAMA Neurology. 2019;77(3):339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Menon DK. Cerebral protection in severe brain injury: Physiological determinants of outcome and their optimisation. Br Med Bull. 1999;55(1):226–258. [DOI] [PubMed] [Google Scholar]
- 19.Brain Trauma Foundation; American Association of Neurological Surgeons; Congress of Neurological Surgeons. Guidelines for the management of severe traumatic brain injury. J Neurotrauma. 2007;24(Suppl 1):S1–S106. [DOI] [PubMed] [Google Scholar]
- 20. Jennett B, Bond M.. Assessment of outcome after severe brain damage. A practical scale. Lancet. 1975;305(7905):480–484. [DOI] [PubMed] [Google Scholar]
- 21. Coles JP, Fryer TD, Smielewski P, et al. Incidence and mechanisms of cerebral ischemia in early clinical head injury. J Cereb Blood Flow Metab. 2004;24(2):202–211. [DOI] [PubMed] [Google Scholar]
- 22. Hong YT, Fryer TD.. Kinetic modelling using basis functions derived from two-tissue compartmental models with a plasma input function: General principle and application to [18F]fluorodeoxyglucose positron emission tomography. Neuroimage. 2010;51(1):164–172. [DOI] [PubMed] [Google Scholar]
- 23. Kuwabara H, Evans AC, Gjedde A.. Michaelis-Menten constraints improved cerebral glucose metabolism and regional lumped constant measurements with [18F]fluorodeoxyglucose. J Cereb Blood Flow Metab. 1990;10(2):180–189. [DOI] [PubMed] [Google Scholar]
- 24. Hutchinson PJ, Gupta AK, Fryer TF, et al. Correlation between cerebral blood flow, substrate delivery, and metabolism in head injury: A combined microdialysis and triple oxygen positron emission tomography study. J Cereb Blood Flow Metab. 2002;22(6):735–745. [DOI] [PubMed] [Google Scholar]
- 25. Smielewski P, Coles JP, Fryer TD, Minhas PS, Menon DK, Pickard JD.. Integrated image analysis solutions for pet datasets in damaged brain. J Clin Monit Comput. 2002;17(7-8):427–440. [DOI] [PubMed] [Google Scholar]
- 26. Avants BB, Epstein CL, Grossman M, Gee JC.. Symmetric diffeomorphic image registration with cross-correlation: Evaluating automated labeling of elderly and neurodegenerative brain. Med Image Anal. 2008;12(1):26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ledig C, Heckemann RA, Hammers A, et al. Robust whole-brain segmentation: Application to traumatic brain injury. Med Image Anal. 2015;21(1):40–58. [DOI] [PubMed] [Google Scholar]
- 28. Cole JH, Jolly A, de Simoni S, et al. Spatial patterns of progressive brain volume loss after moderate-severe traumatic brain injury. Brain. 2018;141(3):822–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peeters SG, Zegers CM, Lieuwes NG, et al. A comparative study of the hypoxia PET tracers [(1)(8)F]HX4, [(1)(8)F]FAZA, and [(1)(8)F]FMISO in a preclinical tumor model. Int J Radiat Oncol Biol Phys. 2015;91(2):351–359. [DOI] [PubMed] [Google Scholar]
- 30. Cleveland W. Robust locally weighted regression and smoothing scatterplots. J Am Stat Assoc. 1979;74(368):829–836. [Google Scholar]
- 31. Rostami E, Bellander BM.. Monitoring of glucose in brain, adipose tissue, and peripheral blood in patients with traumatic brain injury: A microdialysis study. J Diabetes Sci Technol. 2011;5(3):596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koepsell H. Glucose transporters in brain in health and disease. Pflugers Arch. 2020;472(9):1299–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bergsneider M, Hovda DA, McArthur DL, et al. Metabolic recovery following human traumatic brain injury based on FDG-PET: Time course and relationship to neurological disability. J Head Trauma Rehabil. 2001;16(2):135–148. [DOI] [PubMed] [Google Scholar]
- 34. Kaisti KK, Metsahonkala L, Teras M, et al. Effects of surgical levels of propofol and sevoflurane anesthesia on cerebral blood flow in healthy subjects studied with positron emission tomography. Anesthesiology. 2002;96(6):1358–1370. [DOI] [PubMed] [Google Scholar]
- 35. Schlunzen L, Juul N, Hansen KV, Cold GE.. Regional cerebral blood flow and glucose metabolism during propofol anaesthesia in healthy subjects studied with positron emission tomography. Acta Anaesthesiol Scand. 2012;56(2):248–255. [DOI] [PubMed] [Google Scholar]
- 36. Hattori N, Huang SC, Wu HM, et al. Correlation of regional metabolic rates of glucose with Glasgow coma scale after traumatic brain injury. J Nucl Med. 2003;44(11):1709–1716. [PubMed] [Google Scholar]
- 37. Glenn TC, Kelly DF, Boscardin WJ, et al. Energy dysfunction as a predictor of outcome after moderate or severe head injury: Indices of oxygen, glucose, and lactate metabolism. J Cereb Blood Flow Metab. 2003;23(10):1239–1250. [DOI] [PubMed] [Google Scholar]
- 38. Cunningham AS, Salvador R, Coles JP, et al. Physiological thresholds for irreversible tissue damage in contusional regions following traumatic brain injury. Brain. 2005;128(8):1931–1942. [DOI] [PubMed] [Google Scholar]
- 39. Gallagher CN, Carpenter KL, Grice P, et al. The human brain utilizes lactate via the tricarboxylic acid cycle: A 13C-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain. 2009;132(Pt 10):2839–2849. [DOI] [PubMed] [Google Scholar]
- 40. Jalloh I, Helmy A, Howe DJ, et al. Focally perfused succinate potentiates brain metabolism in head injury patients. J Cereb Blood Flow Metab. 2017;37(7):2626–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jalloh I, Helmy A, Howe DJ, et al. A comparison of oxidative lactate metabolism in traumatically injured brain and control brain. J Neurotrauma. 2018;35(17):2025–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Prins ML. Cerebral metabolic adaptation and ketone metabolism after brain injury. J Cereb Blood Flow Metab. 2008;28(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vespa P, McArthur DL, Stein N, et al. Tight glycemic control increases metabolic distress in traumatic brain injury: A randomized controlled within-subjects trial. Crit Care Med. 2012;40(6):1923–1929. [DOI] [PubMed] [Google Scholar]
- 44. Hutchinson PJ, O’Connell MT, Seal A, et al. A combined microdialysis and FDG-PET study of glucose metabolism in head injury. Acta Neurochir (Wien). 2009;151(1):51–61; discussion 61. [DOI] [PubMed] [Google Scholar]
- 45. Bilotta F, Caramia R, Cernak I, et al. Intensive insulin therapy after severe traumatic brain injury: A randomized clinical trial. Neurocrit Care. 2008;9(2):159–166. [DOI] [PubMed] [Google Scholar]
- 46. Coester A, Neumann CR, Schmidt MI.. Intensive insulin therapy in severe traumatic brain injury: A randomized trial. J Trauma. 2010;68(4):904–911. [DOI] [PubMed] [Google Scholar]
- 47. Green DM, O’Phelan KH, Bassin SL, Chang CWJ, Stern TS, Asai SM.. Intensive versus conventional insulin therapy in critically ill neurologic patients. Neurocrit Care. 2010;13(3):299–306. [DOI] [PubMed] [Google Scholar]
- 48. Hermanides J, Plummer MP, Finnis M, Deane AM, Coles JP, Menon DK.. Glycaemic control targets after traumatic brain injury: A systematic review and meta-analysis. Crit Care. 2018;22(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Osteen CL, Giza CC, Hovda DA.. Injury-induced alterations in N-methyl-D-aspartate receptor subunit composition contribute to prolonged 45calcium accumulation following lateral fluid percussion. Neuroscience. 2004;128(2):305–322. [DOI] [PubMed] [Google Scholar]
- 50. Hartings JA, Andaluz N, Bullock metabolic ratio, et al. Prognostic value of spreading depolarizations in patients with severe traumatic brain injury. JAMA Neurology. 2020;77(4):489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Unterberg AW, Stover J, Kress B, Kiening KL.. Edema and brain trauma. Neuroscience. 2004;129(4):1021–1029. [DOI] [PubMed] [Google Scholar]
- 52. Needham EJ, Helmy A, Zanier ER, Jones JL, Coles AJ, Menon DK.. The immunological response to traumatic brain injury. J Neuroimmunol. 2019;332:112–125. [DOI] [PubMed] [Google Scholar]
- 53. Phelps ME, Huang SC, Hoffman EJ, Selin C, Sokoloff L, Kuhl DE.. Tomographic measurement of local cerebral glucose metabolic rate in humans with (F-18)2-fluoro-2-deoxy-D-glucose: Validation of method. Ann Neurol. 1979;6(5):371–388. [DOI] [PubMed] [Google Scholar]
- 54. Sokoloff L, Reivich M, Kennedy C, et al. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: Theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem. 1977;28(5):897–916. [DOI] [PubMed] [Google Scholar]
- 55. Reivich M, Kuhl D, Wolf A, et al. The [18F]fluorodeoxyglucose method for the measurement of local cerebral glucose utilization in man. Circ Res. 1979;44(1):127–137. [DOI] [PubMed] [Google Scholar]
- 56. Heiss WD, Pawlik G, Herholz K, Wagner R, Göldner H, Wienhard K.. Regional kinetic constants and cerebral metabolic rate for glucose in normal human volunteers determined by dynamic positron emission tomography of [18F]-2-fluoro-2-deoxy-D-glucose. J Cereb Blood Flow Metab. 1984;4(2):212–223. [DOI] [PubMed] [Google Scholar]
- 57. Reivich M, Alavi A, Wolf A, et al. Use of 2-deoxy-D[1-11C] glucose for the determination of local cerebral glucose metabolism in humans: Variation within and between subjects. J Cereb Blood Flow Metab. 1982;2(3):307–319. [DOI] [PubMed] [Google Scholar]
- 58. Kuwabara H, Gjedde A.. Measurements of glucose phosphorylation with FDG and PET are not reduced by dephosphorylation of FDG-6-phosphate. J Nucl Med. 1991;32(4):692–698. [PubMed] [Google Scholar]
- 59. Stovell MG, Mada MO, Carpenter TA, et al. Phosphorus spectroscopy in acute TBI demonstrates metabolic changes that relate to outcome in the presence of normal structural MRI. J Cereb Blood Flow Metab. 2020;40(1):67–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Finnema SJ, Nabulsi NB, Eid T, et al. Imaging synaptic density in the living human brain. Sci Transl Med. 2016;8(348):348ra96. [DOI] [PubMed] [Google Scholar]
- 61. Xu Y, McArthur DL, Alger JR, et al. Early nonischemic oxidative metabolic dysfunction leads to chronic brain atrophy in traumatic brain injury. J Cereb Blood Flow Metab. 2010;30(4):883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.