

Abstract
The phosphatidylinositol 3-kinase (PI3K)/Akt pathway plays a crucial role in various cellular processes and is aberrantly activated in cancers, contributing to the occurrence and progression of tumors. Examining the upstream and downstream nodes of this pathway could allow full elucidation of its function. Based on accumulating evidence, strategies targeting major components of the pathway might provide new insights for cancer drug discovery. Researchers have explored the use of some inhibitors targeting this pathway to block survival pathways. However, because oncogenic PI3K pathway activation occurs through various mechanisms, the clinical efficacies of these inhibitors are limited. Moreover, pathway activation is accompanied by the development of therapeutic resistance. Therefore, strategies involving pathway inhibitors and other cancer treatments in combination might solve the therapeutic dilemma. In this review, we discuss the roles of the PI3K/Akt pathway in various cancer phenotypes, review the current statuses of different PI3K/Akt inhibitors, and introduce combination therapies consisting of signaling inhibitors and conventional cancer therapies. The information presented herein suggests that cascading inhibitors of the PI3K/Akt signaling pathway, either alone or in combination with other therapies, are the most effective treatment strategy for cancer.
Subject terms: Oncogenes, Drug development
Introduction
The phosphoinositide 3-kinase (PI3K)/Akt signaling pathway is a major signaling pathway in various types of cancer.1 It controls hallmarks of cancer, including cell survival, metastasis and metabolism. The PI3K/Akt pathway also plays essential roles in the tumor environment, functioning in angiogenesis and inflammatory factor recruitment. The PI3K/Akt pathway can be aberrantly activated through various mechanisms, including different genomic alterations, such as mutations of PIK3CA, phosphatase and tensin homolog (PTEN), Akt, TSC1, and mechanistic target of rapamycin (mTOR).2 PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) to generate phosphatidylinositol-3,4,5-trisphosphate (PIP3), and PIP3 then recruits oncogenic signaling proteins, including the serine and threonine kinase Akt.3 Once active, Akt phosphorylates a number of substrates. mTOR, one of the most common downstream effectors of Akt, integrates many proteins to promote cancer progression. Members of the PI3K/Akt/mTOR pathway are often mutated and activated in cancer.4,5
The study of PI3K/Akt networks has led to the discovery of inhibitors for one or more nodes in the network, and the discovery of effective inhibitors is important for improving the survival of patients with cancer. To date, many inhibitors of the PI3K/Akt signaling pathway have been developed, some of which have been approved for the treatment of patients with cancer in the clinic. However, many issues associated with the use of pathway inhibitors, including which drugs should be used to treat specific types of cancer and whether combination therapies will improve treatment outcomes, remain to be resolved. Current research aims to learn from clinical successes and failures to improve the design of clinical therapies based on pathway inhibitors and ultimately improve clinical cancer treatment.6
Overview of the PI3K/Akt signaling pathway
PI3Ks
Three classes of PI3Ks, class I, class II, and class III, have been identified, each of which has specific substrates and distinct effectors in addition to the common substrate Akt (Table 1).3 To date, class I, class II and class III PI3Ks have been widely mentioned in former studies.7,8 Further understanding the PI3K isoforms will help to fully elucidate the biological processes in various types of cancer cells.
Table 1.
Different effectors of class I, II and III PI3Ks
| PI3K isoforms | Effectors | References | |
|---|---|---|---|
| Class I PI3K | Serine/Threonine kinases of the AGC kinase family | AKT | 183 |
| PKC | 2 | ||
| PDK-1 | 183 | ||
| SGK1 | 244 | ||
| Tyrosine kinases of the TEC family | BTK | 15 | |
| BMX | 245 | ||
| ITK | 246 | ||
| Guanine nucleotide exchange factors (GEFs) | Rac | 247 | |
| P-Rex1 | 16 | ||
| Tiam1 | 248 | ||
| Vav1 | 249 | ||
| Vav2 | 249 | ||
| Class II PI3K | Sortins | 250 | |
| Small GTPase | 251 | ||
| Myotubularins | 252 | ||
| Class III PI3K | LKB1 | 26 | |
| SGK3 | 253 | ||
Class I PI3Ks
Class I PI3Ks, heterodimers that consist of a p110 catalytic subunit and a p85 regulatory subunit, exert their functions by activating downstream tyrosine kinases, such as G protein-coupled receptors (GPCRs) and small monomeric GTPases.9 Moreover, the p85 subunit can transmit various cellular signals by laying a critical foundation for signal integration and the activation of downstream proteins.10
Class I PI3Ks are divided into four catalytic isoforms, p110α, p110β, p110γ and p110δ, and they are encoded by PIK3CA, PIK3CB, PIK3CG and PIK3CD, respectively.3 Among the four class I catalytic isoforms, PIK3CA is mentioned commonly in human cancer due to its frequent mutations.6 The time point of pathway activation varies among cancer types and patients. For example, the activation of PIK3CA mutations is an early event in breast and colon cancer.11 In contrast to PIK3CA, transforming mutations in the PIK3CB gene are rare; however, the gene is ubiquitously expressed, probably due to the distinct mode of interaction between this isoform and regulatory subunits. PIK3CD is mainly expressed in white blood cells and B cells and is indispensable for B cell follicle maturation and survival.12 Although the PIK3CG level is related to cancer growth, reduced PIK3CG expression has been reported to promote colon cancer growth and development.13 According to differences in the regulatory subunits, class I PI3Ks are further divided into class IA and class IB enzymes, and both of their catalytic subunits are formed by the PI3K catalytic core and extended N-terminally with the Ras-binding domain (RBD).9,14 In cancer, somatic genetic activation through multiple mechanisms is very common in the class IA PI3K pathway. These mechanisms include inactivation of PTEN and the p110 catalytic subunit. Activating mutations of the p110 catalytic subunits often occur in PIK3CA, while mutations in PIK3CB and PIK3CD are much less frequent.4,14
Class I PI3Ks play an important role in the development of cancers and exert their functions by regulating downstream effectors. The downstream effectors of class I PI3Ks share a pleckstrin homology (PH) domain and include serine/threonine kinases from the AGC kinase family, tyrosine kinases expressed in hepatocellular carcinoma (HCC, TEC family) and guanine nucleotide exchange factors (GEFs). Akt is also a member of the serine/threonine AGC kinase family, which will be introduced in detail below. TEC family members are key effectors of class I PI3Ks in lymphocytes. As a member of the TEC family, Bruton tyrosine kinase (BTK) has a highly selective PH domain for PIP3. Activation of the BTK signaling pathway triggers the growth of B cell malignancies. BTK is often overexpressed in chronic lymphocytic leukemia (CLL) cells, and its phosphorylation level is increased.15 GEFs are also important effectors of class I PI3Ks. PI3K activates and promotes Rac-mediated actin recombination in cancer- and growth factor-stimulated fibroblasts. In addition to modulating cell morphology and motility, this PI3K/Rac signaling axis drives an increase in glycolysis flow by releasing aldolase from actin filaments.16
Class II PI3Ks
Class II PI3Ks comprise the C2α, C2β, and C2γ catalytic isoforms and lack regulatory subunits; thus, they can be activated as monomers.17 In mammals, three class II PI3K isoforms have been identified, among which PI3KC2α and PI3KC2β are broadly expressed, while PI3KC2γ is mainly expressed in the liver.18 PI3KC2α plays a pivotal role in breast cancer progression by affecting mitotic spindle formation.19 In addition, class II PI3Ks contain additional protein-binding domains and an extended N-terminal region, which contributes to intracellular localization. Another feature of class II PI3Ks is that they do not produce PIP3 in vitro; however, they can generate PIP2 using PIP as a substrate, which is significantly different from the functions of class I and III PI3Ks.20
Because the lipid products generated by class II and class I PI3Ks are substantially different, class II PI3Ks may activate different downstream effectors.21 Sortins, small GTPases and myotubularins are three main downstream effectors of class II PI3Ks.22 Additionally, the catalytic pocket of class II PI3Ks differs from that of class I and III PI3Ks. Currently, class II PI3Ks function as major signaling enzymes and play important roles under normal and pathological conditions.22 These three isoforms of class II PI3Ks importantly contribute to various cellular activities because they synthesize unique lipid products in cancer.23
Class III PI3Ks
Class III PI3K VPS34 (also called PIK3C3) is unique, as it plays an important role in regulating autophagy and macrophage phagocytosis by binding to a protein complex composed of a regulatory subunit and a catalytic subunit. Therefore, heterodimeric class III PI3K can also regulate autophagy.7
The smallest PI3K catalytic core is the constituent element of VPS34, and it forms tetrameric complex I and tetrameric complex II.7 Complex I play a vital role in the formation and extension of autophagosomes (wrapping and separating cytoplasmic components), mainly by promoting recruitment at the phagocytosis initiation site in the endoplasmic reticulum after activation. Complex II has the advantage of increasing autophagosome-late endosome/lysosome fusion by controlling endosome maturation.24
While VPS34 does not directly regulate signal transduction, it transduces signals by regulating various protein kinases when activated by amino acids. For example, in breast cancer treatment, the use of Akt inhibitors upregulates VPS34-dependent SGK3 signaling.25 Another kinase regulated by VPS34 is intimal LKB1 liver kinase B1 (LKB1; also known as STK11), an AMPK activator and a positive regulator of cell polarity and epithelial tissue.26 VPS34 and mTOR complex 1 (mTORC1) signaling have been shown to be correlated. VPS34 exerts its function by activating acute stimuli but not by controlling the basal activity of mTORC1.27 Therefore, approaches targeting VPS34 may be a useful clinical treatment strategy. Although the widely used VPS34 inhibitor 3-methyladenine is not selective, related studies have documented the use of a highly selective inhibitor of VPS34.28
Akt
The serine and threonine kinase Akt, also known as protein kinase B (PKB), was discovered 25 years ago.29 The upstream and downstream targets of Akt have also been studied. Various diseases are induced by Akt dysfunction, including cancer.25,30 Three Akt isoforms have been identified in mammals: Akt1, Akt2 and Akt3. Akt1 and Akt2 are enriched in many tissues, such as pancreatic tissue, while Akt3 is mainly expressed in the brain, and its expression is very limited by its tissue distribution.31 Different Akt isoforms play different and vital roles in cancer; for example, Akt2 is involved in cancer cell migration and invasion, and Akt3 is associated with hormone independence.31,32 Moreover, Akt2 gene expression is amplified in pancreatic cancer, while Akt3 is overexpressed in breast and prostate cancer, and both Akt2 and Akt3 are insensitive to hormones.33
Akt is classically activated by receptor tyrosine kinases (RTKs) and GPCRs,3 and activated Akt recruits and activates one or more subtypes of class I PI3Ks on the plasma membrane. In turn, activated PI3K also phosphorylates the T ring and the C hydrophobic motif, two key residues in the core activation domain of Akt1, which regulate the corresponding residues of Akt2 and Akt3.34 In addition, IGF1 activates Akt. When IGF1 binds to IGF1R, IRS-1 and PI3K are recruited and activated. Activated PI3K converts PIP2 to PIP3, which activates phosphoinositide-dependent kinase-1 (PDK1) and then influences Akt.35 TRAF6 and TBK1 also modulate the activity of Akt.36 In conclusion, Akt activation is easily monitored, and research on class I PI3K signal transduction might thus be performed by focusing on Akt.
Major upstream activators of the PI3K/Akt signaling pathway
Upstream activation of the PI3K/Akt signaling pathway is essential for its function in cancer and other related diseases. Activation of this pathway is related to many factors, including the RTK family, Toll-like receptors (TLRs), and B-cell antigen receptors (BCRs). Major upstream components of the PI3K/Akt signaling pathway are discussed below (Fig. 1).
Fig. 1.

Upstream activation of the PI3K/Akt signaling pathway. On the one hand, ligands combined with specific RTKs (EGFR, VEGFR and FGFR) can activate class I PI3Ks via RAS; on the other hand, class I PI3Ks can be activated by BCRs through B cell adapters and by GPCRs. The FGFR substrate FRS2 is phosphorylated in combination with GRB2, SOS and GAB1 to activate class I PI3Ks. In addition to being activated by EGFR, class II PI3Ks can be activated by TCRs. While class III PI3Ks are activated by amino acids, total activated PI3K phosphorylates the third carbon of the PIP2 inositol head and transforms it into PIP3 to thereby activate AKT via PDK1 and RAC, and this transformation process can be inhibited by PTEN. In addition, IGF-1 in combination with IGF1R can recruit IRS-1 and class I PI3Ks and then participate in the conversion of PIP2 to PIP3. Moreover, mTORC2 can affect the activity of Akt by affecting the phosphorylation of Akt and then affect downstream mTORC1 via TSC1/2, and both Akt and mTORC1 can be activated by TBK1. Moreover, TRAF6 can affect the activity of Akt by affecting its ubiquitylation. Furthermore, DNA damage can affect Akt via ATM and ATR
RTKs
RTKs constitute a transmembrane protein family with intrinsic phosphotyrosine kinase activity that mainly includes epidermal growth factor receptors (EGFRs), vascular endothelial growth factor receptors (VEGFRs) and fibroblast growth factor receptors (FGFRs). RTKs remain inactive in the plasma membrane prior to their activation by ligands.37 Ligands such as homologous growth factors, cytokines, and hormones activate PI3K signaling pathways by activating RTKs.38 Furthermore, the p85 subunit of class I PI3Ks binds to phosphorylated RTK, resulting in a conformational change in the catalytic domain of PI3K (p110).
EGFRs
EGFRs are 170-kDa RTKs that phosphorylate tyrosine residues and activate class I PI3K and class II PI3K signal transduction by binding to ligands and forming homodimers or heterodimers.39,40 EGFRs belong to the RTK ErbB family, which also includes ErbB-3, ErbB-4, and HER2. HER2 forms HER2/EGFR heterodimers with EGFR, and HER2/EGFR heterodimers have greater signal transduction potential than EGFR homodimers.41 Inhibitors designed based on this feature have obvious effects on clinical efficacy and may delay drug resistance. The other family member, ErbB-3, has six tyrosine phosphorylation sites and effectively binds to PI3K.42 The ligand TGF-α also binds to EGFRs, activates the PI3K signaling pathway and promotes tumor growth and metastasis in colorectal cancer cells.43 EGFR antibodies, such as cetuximab and panitumumab, inhibit PI3K/Akt signal transduction by competitively binding to EGFRs, thus inhibiting the occurrence and development of cancer.44
VEGFRs
VEGFRs are RTKs that are categorized three main types: VEGFR-1 (Flt-1), VEGFR-2 (KDR, Flk-1) and VEGFR-3 (Flt-4). Among the three types, VEGFR-1 and VEGFR-2 mainly function in endothelial cells, while VEGFR-3 is present in the lymphatic endothelium.45 The VEGFR extracellular domain binds to VEGF, leading to the dimerization and phosphorylation of the intracellular tyrosine kinase domain and to the activation of downstream proteins. Therefore, VEGFRs vitally stabilize neovascularization and promote cell survival and migration.46 Similarly, VEGF forms VEGF/VEGFR-2 dimers, which activate the PI3K/Akt pathway to mediate tumor metastasis and angiogenesis.
VEGFR-2-mediated activation of the PI3K/Akt signaling pathway is important for tumor survival. The binding of a ligand to VEGFR-2 activates PI3K and phosphorylates PIP2, resulting in the accumulation and reactivation of PIP3 to thereby activate the PI3K/Akt signaling pathway. VEGFR inhibitors used in combination with PI3K/Akt/mTOR signaling pathway inhibitors represent an effective therapeutic strategy.47 Furthermore, VEGFR-1 and VEGFR-2 were shown to interact using the siRNA method. The siRNA-mediated knockout of VEGFR-1 in endothelial cells resulted in the attenuation of VEGFR-2 promoter activity, suggesting that VEGFR-1 plays an important role in inducing and modulating multiple signaling pathways.48
FGFRs
FGFRs are very important for cancer metastasis and angiogenesis. For example, FGFR amplification in breast cancer affects the phosphorylation of downstream fibroblast growth factor receptor substrate 2 (FRS2), resulting in PI3K/Akt signaling pathway activation.49 High FGFR levels lead to PI3K/Akt pathway activation, which is involved in tumor growth. Further studies revealed that changes downstream of the FGFR pathway affect tumor cells and alter the tumor microenvironment.50 In addition, the inhibition of FGFRs may result in gene reprogramming.51
In summary, RTKs are involved in PI3K/Akt pathway activation. Mutations in RTKs potentially result in ligand-independent transduction, which can be suppressed by pathway inhibitors in non-small-cell lung cancer (NSCLC).52 The activation of class IA PI3Ks is related to RTK signaling, and p85 regulatory subunits are essential for the RTK-mediated activation of class IA PI3Ks. Therefore, RTK inhibitors might negatively regulate PI3K signaling.53 However, many types of cancer are always resistant to a single RTK inhibitor because PI3K is activated by multiple RTKs in cancer.54 Therefore, understanding RTK signaling networks will facilitate a better comprehension of the PI3K pathway and further exploration of cancer therapies.
TLRs
TLRs are expressed in immune and nonimmune cells and are important in cancer progression.55,56 TLRs can identify conserved microbial motifs in bacterial lipopolysaccharides (recognized by TLR4) and single- or double-stranded RNA (identified by TLR3).57 TLRs also distinguish and bind endogenous ligands, thereby activating the signaling pathway.58 For example, some studies found that natural TLR4 ligands activate Akt by increasing its phosphorylation in a time-dependent manner, which promotes the progression of colorectal cancer.59 Similarly, activated TLR4 protects cells from chemotherapy, leading to drug resistance in head and neck squamous cell carcinoma.60 TLR3 activation leads to cancer cell apoptosis and has antitumor effects on renal cell carcinoma and melanoma.61
BCRs
The signaling pathways activated by BCRs are crucial for the development, activation, and differentiation of B cells.62 Among them, the PI3K/Akt pathway is particularly important. In B cells, class I PI3Ks are activated by BCRs via the B-cell receptor associated protein (BCAP), which is an important step in PIP3 production and Akt activation.63 BCRs and cytoplasmic adapters profoundly affect PI3K/Akt signaling pathway activation, and Akt is not activated when B cells lack BCRs.64
The noncatalytic region of the tyrosine kinase (NCK) family comprises a set of BCR adapters that are recruited to BCR signaling complexes, which are essential factors that activate B cells to exert their function. The NCK structure is characterized by three SRC homology 3 domains at the N-terminus and an SH2 domain at the C-terminus.65 NCK affects PI3K/Akt signaling in B cells, and although Akt phosphorylation is decreased to some extent in the absence of NCK1, almost no phosphorylation occurs when both NCK1 and NCK2 are deficient.63
To date, BCR research has mainly focused on CLL. According to previous studies, BCR signaling is vital for the maintenance of cancer cell survival, and its function is downregulated by p110δ or the inhibition of BTK.66
GPCRs
GPCRs constitute the largest cell surface protein family and play an important role in cell signal transduction; additionally, they are a common target of the PI3K/Akt signaling pathway.67 GPCRs transmit signals through heterotrimeric G proteins and regulate downstream effector pathways by interacting with various small G proteins that bind directly to GPCRs and participate in the regulation of signaling networks. One characteristic of GPCRs is that they recognize and respond to chemically distinct ligands to effectively activate PI3K/Akt signaling in different cells.3,68 Moreover, GPCRs regulate cancer cell proliferation and survival, and their persistent activation affects mitotic and metabolic responses, which are the basis for tumorigenesis.
The predominant mechanisms by which GPCRs activate PI3K are tissue-specific, and many GPCR ligands, such as sphingosine 1-phosphate, activate PI3Ks.69 GPCRs activate PI3K/Akt signaling by stimulating Ras to thereby activate class I PI3Ks, which regulate cancer and many other diseases. Small GTPases also promote tumor metastasis by controlling PI3K/Akt signaling.70
PTEN
PTEN is a tumor suppressor that is key for maintaining normal physiological activity; PTEN was initially identified as a gene prone to mutations in multiple types of sporadic tumors.71 The C2 domain houses the PTEN lipid substrate and is necessary for the proper localization of PTEN on the plasma membrane; PTEN has shown affinity for the phospholipid membrane in vitro.72 Nuclear PTEN is important for the maintenance of chromosomal integrity and centromere stability.73
As a lipid phosphatase, PTEN negatively regulates the PI3K signaling pathway and transforms PIP3 into PIP2.74 When PTEN is mutated or participates in another form of inactivation, PI3K effectors, especially Akt, become activated without any exogenous oncogenic stimulus.75 In particular, PI3K phosphorylates membrane-bound PIP2 to generate PIP3. The binding of PIP3 to the PH domain anchors Akt on the plasma membrane and allows it to be phosphorylated and activated by PDK1. Usually, PTEN participates in tumor signal transduction by dephosphorylating protein targets such as focal adhesion kinase (FAK), insulin receptor substrate 1, c-SRC, and PTEN itself.76 The overactivation of Akt by PTEN is the most important carcinogenic factor in PTEN-deficient cancers. In addition, PTEN plays an important role in the control of tumor cell migration and angiogenesis.77
Major effectors downstream of the PI3K/Akt signaling pathway
Akt phosphorylates downstream effectors on serine and threonine in a sequence-dependent manner, which typically recognizes substrates containing the consensus phosphorylation motif R-X-R-X-X-S/T.78 Akt signaling promotes tumor cell survival, proliferation, growth, and metabolism by activating its downstream effectors. Here, we introduce the major targets downstream of the PI3K/Akt signaling pathway (Fig. 2).
Fig. 2.

Downstream effectors of the PI3K/Akt signaling pathway and their cellular functions. The activation of Akt signaling can promote (arrows) or inhibit (blocking arrows) the phosphorylation of downstream effectors. Downstream regulation by Akt contributes to many cellular processes, including tumor growth, tumor survival, tumor cell proliferation, cancer immunity, cancer metabolism and cancer angiogenesis
mTOR
More than 100 Akt substrates have been discovered, although not all of them have been confirmed.78 Due to its parallel regulation of different substrates, the downstream effects of Akt signaling are extensive. mTOR is reported to regulate tumor growth, survival, metabolism, and immunity. As a protein kinase, mTOR is considered an atypical member of the PI3K-related kinase family, and it is usually assembled into complexes, such as mTORC1 and mTOR complex 2 (mTORC2), to play critical roles in many biological processes.79
Many growth factors and their receptors, such as VEGF and VEGFR, act as positive regulators to transmit signals to mTOR through the PI3K/Akt pathway.80 Under normal conditions, PI3K activity is at a basal level. After stimulation with growth factors, signals are transmitted to PI3K. Then, PI3K catalyzes the production of PIP3, which binds to the PH domain of Akt. This step is limited by a negative regulator of mTOR named PTEN.81 Akt and mTOR are thought to interact, as Akt was shown to activate mTOR through the phosphorylation of tuberous sclerosis complex 2 (TSC2).82
The mTORC1 complex is composed of mTOR, mLST8, raptor, and PRAS40. mTORC1 controls cell growth in part by phosphorylating S6 kinase 1 (S6K1) and eIF-4E-binding protein 1 (4EBP1), known regulators of protein synthesis.83 Furthermore, mTORC1-mediated signaling to HIF1α and LIPIN1 increases glucose metabolism and lipid synthesis, respectively.84 mTORC2 is composed of mTOR, mLST8, SIN1 and rictor. Activated mTOR interacts with its protein subunits and forms mTORC2.85 Akt is phosphorylated by mTORC2 upon activation by growth factors. In conclusion, mTOR inhibition has enormous potential in clinical cancer therapy.
GSK3
The multifunctional serine and threonine protein kinase glycogen synthase kinase 3 (GSK3) was the first reported Akt substrate.86 Two subtypes of GSK3, GSH3α and GSK3β, have been identified. These two subtypes share 85% sequence homology, and they were originally identified to be associated with the glycogen synthesis response to insulin.87 Studies have confirmed that different GSK3 subtypes have specific functions in different tissues.88
GSK3 is considered to be expressed at the crossroads of multiple biochemical pathways in some diseases.89 The EGFR/RAS/PI3K/PTEN/Akt/GSK3/mTORC1 pathway is common in cancer, and GSK3 is one of its targets. After the Akt-induced phosphorylation of either Ser21 (α) or Ser9 (β) in N-terminal regulatory domains in response to PI3K-mediated signaling, GSK3 (both GSH3α and GSK3β) is inactivated and targeted for proteasomal degradation.86,90 The Akt-mediated phosphorylation of GSK3 produces an intramolecular pseudosubstrate that blocks the binding pocket and inhibits the substrate from approaching GSK3.91 GSK3 expression affects various biochemical processes in cancer. In addition to tumor growth, GSK3 participates in tumor metabolism by phosphorylating and inhibiting metabolic enzymes such as its substrate glycogen synthase (GS).92 Similarly, another study suggested that the GSK3 inhibitor IX increases apoptosis and alters the structures of membrane lipids.93
FOXOs
Forkhead box Os (FOXOs) are a subgroup of a forkhead box (FOX)-containing transcription factor (TF) superfamily. FOXO TFs include four direct downstream targets of Akt: FOXO1, FOXO3, FOXO4, and FOXO6. In mammals, these TFs control the expression of many target genes94 and are expressed in specific tissues. FOXO1 and FOXO4 are predominantly expressed in adipose tissue and skeletal muscle, respectively, whereas FOXO3 tends to be expressed in the brain, heart, kidney and spleen. In contrast, FOXO6 is expressed mainly in the adult brain, suggesting its important function in the nervous system.95
A major characteristic of FOXOs is the strict control of their localization in the cytoplasm and nucleus. FOXOs contain a nuclear localization signal (NLS) domain and a nuclear export signal (NES) domain, which closely regulate FOXO shuttling.96 A study found that the PI3K/Akt signaling pathway partially regulates cell survival by phosphorylating FOXOs to thereby increase their binding to the 14-3-3 protein, which masks the NLS (also blocking nuclear translocation from the cytoplasm but increasing FOXO removal from the nucleus) and leads to the final ubiquitin proteasome pathway (UPP)-dependent degradation of FOXOs.97 Therefore, the insulin/PI3K/Akt pathway is essential for adjusting FOXO levels by phosphorylation.
Similarly, the highly conserved genetic relationship between Akt and FOXOs supports the hypothesis of an important regulatory association existing between Akt and FOXO suppression. This association was first identified in C. elegans, in which the loss of FOXO family members rescued dauer-stage arrest caused by the depletion of Akt-1 and Akt-2.98 The activity and substrates of Akt are regulated by the phosphorylation of threonine 308 and serine 473,99 and the phosphorylation of serine 473 is not essential for the Akt-mediated phosphorylation of TSC2 and GSK3β; however, it is essential for the phosphorylation and inactivation of FOXOs.100 In summary, these biochemical and genetic studies have confirmed that the main phenotypes induced by Akt depletion are driven by FOXO-mediated transcription; therefore, FOXOs are downstream targets of Akt signaling in various biological reactions.
TSC2
PI3K and Akt stimulation by growth factors such as IGF1 is an evolutionarily conserved function that promotes cell growth through the Akt-mediated activation of mTORC1 and the subsequent phosphorylation and inhibition of TSC2 (also known as tuberin).82 TSC1 and TSC2 are encoded by tumor suppressor genes that are mutated in tuberous sclerosis (TSC).101 The complex comprising TSC1 and TSC2 suppresses mTORC1 activity; through a C-terminal domain and a GTPase-activating protein domain, TSC2 converts Ras-related Rheb-GTP, a potent activator of mTORC1, to Rheb-GDP,102 thereby inactivating mTORC1. However, the Akt-mediated phosphorylation of TSC2 reverses this process and is thus important for the regulation of mTORC1 and might impair the ability of TSC2 to inhibit Rheb and mTORC1.
MDM2
Decreased p53 levels have been proposed to arrest the cell cycle, while increased p53 levels induce cell apoptosis.103 The activation of PI3K/Akt signaling may be induced by the suppression of p53 through the activation of another tumor promoter, MDM2.104 MDM2 is an oncogene that induces tumorigenesis, and its mRNA level is controlled by p53 in response to oxidative stress and DNA damage.105 These findings, in addition to the finding that MDM2 forms a complex with wild-type p53, demonstrate that MDM2 exerts its oncogenic function by interacting with wild-type p53 and suppressing the transcription of target genes.106
The intracellular localization of MDM2 is posttranslationally modulated by PI3K/Akt.107 A study revealed that PI3K/Akt phosphorylates a serine in the MDM2 domain.108 Because phosphorylation is essential for the transfer of MDM2 from the cytoplasm to the nucleus, activated PI3K/Akt can induce the nuclear translocation of MDM2, bypass mitogen stimulation, and activate PI3K/Akt signaling. Therefore, after nuclear entry, MDM2 binds to the tumor suppressor p53, inhibits its transcription, and induces its degradation.108,109 Consistently, the suppression of the PI3K/Akt pathway by PTEN protects p53 from MDM2-induced degradation. Furthermore, p53 expression is positively associated with DNA damage.110
Crosstalk with other pathways
According to previous studies, no pathways exist independently. Other signaling pathways are associated with the PI3K/Akt pathway network through direct regulation or downstream targets.
Crosstalk with MAPK signaling
The RAS/RAF/MEK/ERK (MAPK) pathway is important in cellular processes such as tumor cell proliferation, survival and invasion. The mutation of components such as RAS, RAF and MEK results in dysregulation of the pathway in various types of cancer.111 Moreover, 30% of RAS GTPases are activated by mutations in these types of cancer, but inhibitors that directly target RAS proteins have not been developed. To date, research has aimed to develop inhibitors of the downstream RAF/MEK/ERK and PI3K/Akt pathways.112
According to reports, the PI3K/Akt and MEK/ERK pathways cooperate in tumor growth.113 Signaling pathways also interact with each other, and the enhancement of one signaling pathway may enhance or inhibit another pathway. For instance, the effect of PI3K/mTOR inhibitors on tumor cells is blocked by the inhibition of MEK or knockout of ERK, and the combined inhibition of MEK and PI3K/Akt/mTOR signaling thus inhibits tumor cell growth.114 In addition, MAPK pathway signals function as second messengers to attenuate PI3K/Akt signals by decreasing reactive oxygen species (ROS) levels. MEK1/2 inhibition upregulates Akt phosphorylation and MEK1/2 inhibition in response to mild hypoxia.115 These effects may provide negative feedback for the PI3K/Akt-induced activation of MAPK pathways.
Crosstalk with NF-κB signaling
The NF-κB TF family includes components such as p50 and p65; NF-κB is a heterodimer that is isolated in the cytoplasm by inhibitor of kappa B (IκB).116 Upon the phosphorylation of IκB, NF-κB is released, enabling its nuclear translocation and binding to genes involved in processes such as cell proliferation and angiogenesis in esophageal cancer.117 Some studies have suggested that Akt signaling phosphorylates IκB kinase α to activate NF-κB TFs, which are downstream of multiple signaling pathways.118
A regulatory circuit between the EGFR/PI3K/Akt/mTORC1 and IKK/NF-κB signaling pathways has been identified in cancer.119 The EGFR/PI3K/Akt/mTORC1 signaling pathway regulates the IKK/NF-κB signaling pathway, while IKK/NF-κB also regulates EGFR expression and subsequently modulates the PI3K/Akt pathway. Inhibitors targeting IKK effectively block the EGFR/PI3K/Akt and IKK/NF-κB signaling pathways, which is very important for the use of IKK inhibitors as a single drug or in combination with other inhibitors in clinical trials.119
Crosstalk with Wnt/β-catenin signaling
Although the Wnt/β-catenin and PI3K/Akt pathways have different carcinogenic mechanisms, they have been shown to be associated. The Wnt pathway is an important factor maintaining intestinal homeostasis; it regulates the self-renewal of stem cells and increases the proliferation of intestinal epithelial cells, and overactivation of the Wnt pathway may eventually lead to cancer.120,121 Some studies have shown an association between the Wnt/β-catenin and PI3K/Akt pathways in cancer. Wnt/β-catenin pathway activation is mediated by phospholipase D1PLD1 (PLD1), which downregulates ICAT via the PI3K/Akt signaling axis.122
Moreover, in breast cancer, activation of the PI3K/Akt pathway by Nectin-4 induces the activation of the Wnt pathway and then affects the proliferation of tumor stem cells, which is an important mechanism by which cancer stem cells achieve self-renewal.123 Communication between the Wnt/β-catenin and PI3K/Akt pathways has been observed in different types of human cancer, and PI3K/Akt pathway activation leads to Wnt/β-catenin pathway inhibition. In contrast, when the PI3K/Akt pathway is inhibited, the Wnt/β-catenin pathway is overactivated.124
PI3K/Akt signaling and cancer
PI3K/Akt signaling and tumorigenesis
At present, tumors are generally thought to be propagated by somatic cells, constituting the tumorigenesis process. Due to dysregulation of their self-detection function, cells are unable to identify their own mutations and “quit” dividing on time and instead replicate and reproduce with mutations, resulting in the accumulation of mutations. Moreover, autoimmune deficiencies, endocrine disorders, and other adverse stimuli also provide conditions that support the process of tumorigenesis. A study identified AMPK as a vital regulator of Akt activation by various stresses in tumorigenesis.125 Other studies have also shown that the PI3K/Akt signaling pathway regulates its downstream effectors, thereby promoting the occurrence of tumors.126
As mentioned above, FOXO is a vital target protein in the PI3K/Akt signaling pathway. After phosphorylation by PI3K/Akt signaling, the entry of FOXO into the nucleus is blocked,127 which prevents the expression of its target genes and eliminates its transcriptional effects on AR, ERG, Runx2, and other target genes, thereby promoting the occurrence of cancer.
Based on the important role of the PI3K/Akt signaling pathway in tumorigenesis, some oncogenes in the PI3K/Akt signaling pathway are positively regulated. For example, KDM5a plays a vital role in the occurrence of tumors. It promotes the formation of HCC lesions by regulating miR-433-FXYD3-PI3K-Akt signaling.128 Therefore, studies on this pathway and its related pathways may be crucial to increase the anticancer efficacies of clinical PI3K/Akt inhibitors.
PI3K/Akt signaling and tumor growth
One important feature of cancer cells is their uncontrolled proliferation, and the proliferation rate determines the type of tumor therapy. EGFRs regulate the proliferation of tumor cells through signal transduction pathways. Studies have shown that the PI3K/Akt signaling pathway affects the cell cycle by modulating its downstream targets, thereby promoting the proliferation of tumor cells.129
Akt phosphorylates cyclin-dependent kinase inhibitors and prevents p27 from translocating to the nucleus, thereby weakening its inhibitory effect on the cell cycle and directly promoting the proliferation of tumor cells.129 Akt also promotes the proliferation of tumor cells through its downstream effector p27. Since the PI3K/Akt signaling pathway can directly control the proliferation of tumor cells, some proteins are also involved in cancer cell proliferation through the PI3K/Akt pathway. Furthermore, insulin growth factor 2 (IGF2) is involved in the development of many malignancies and alters the cancer cell proliferation process by regulating the PI3K/Akt pathway.130
During cancer treatment, the PI3K/Akt signaling pathway is often targeted to inhibit cancer cell proliferation; for example, the signaling pathway is controlled by the inhibition of lncRNA TDRG1 expression in osteosarcoma cells, thus interfering with their proliferation.131
PI3K/Akt signaling and apoptosis
Apoptosis is an autonomous cell death process.132 Abnormal apoptosis and uncontrolled growth allow cancer cells to rapidly proliferate. PI3K/Akt signaling blocks the expression of proapoptotic proteins, reduces tissue apoptosis and increases the survival rate of cancer cells.133
Akt inhibits the proapoptotic factors Bad and procaspase-9 through phosphorylation and induces the expression of the proapoptotic factor Fas ligand. In addition, Akt activation is associated with resistance to increased apoptosis induced by tumor necrosis factor (TNF)-associated apoptosis-inducing ligand (TRAIL/APO-2L), a member of the TNF superfamily that has been shown to have selective antitumor activity.134 Similarly, Akt negatively regulates the function or expression of Bcl-2 homology domain 3-only proteins, which play a proapoptotic role by inactivating the original prosurvival Bcl-2 family members. In conclusion, inhibition of the PI3K/Akt signaling pathway, which has been shown to regulate cancer cell apoptosis, can serve as a new direction for future research on cancer treatment.131
PI3K/Akt signaling and drug resistance
In tumor therapy, cancer drug resistance is the main reason for treatment failure and indirectly promotes tumor progression.135 The dysregulation of PI3K/Akt signaling also plays an important role in cancer drug resistance. Furthermore, studies have demonstrated that the IGF1R/p110β/AKT/mTOR axis confers resistance to BYL-719 in PIK3CA mutant breast cancers.136 Similarly, a study showed that targeting PI3K/Akt signaling pathway components can be used to overcome drug resistance in cancer therapy.137
Chemotherapy resistance
Chemotherapies are mainly used to destroy tumor cell DNA and thereby prevent the cells from replicating, eventually affecting cell survival. During routine chemotherapy, no treatment interval exists, allowing resistant cells to be generated and leading to tumor regeneration.138 In addition, DNA destruction is prevented in drug-resistant cells due to their dormancy.139
The PI3K/Akt signaling pathway is important for the drug resistance of different types of cancer, such as lung cancer140 and esophageal cancer.141 PI3K/Akt inhibitors inhibit tumor growth and induce tumor cell apoptosis. For NSCLC cells with high Akt expression, the use of PI3K/Akt signaling pathway inhibitors increases their cell apoptosis induced by chemotherapy and reduces their resistance to chemotherapy; furthermore, inhibition of the PI3K/Akt signaling pathway effectively improves drug-induced lung cancer cell apoptosis.142 Moreover, members of the PI3K/Akt signaling pathway play an important role in antiestrogen resistance in breast cancer.143
Immunotherapeutic resistance
The discovery and application of immune checkpoint inhibitors have substantially advanced the treatment of malignant tumors. Thus far, CTLA-4, PD-1 and PD-L1 have achieved significant clinical efficacy and have been approved for the treatment of many types of cancer.144 However, the problem of immune drug resistance persists. Some patients do not respond to immunotherapy, while other relapse after immunotherapeutic treatment. The PI3K/Akt signaling pathway plays an important role in the regulation of immune checkpoints and sensitivity to immune checkpoint inhibitors. Studies have shown that the activation of the Akt signaling pathway caused by the deletion of PI3KCA mutations is strongly related to the upregulation of PD-L1 expression in the prostate gland.145 In mouse lung cancer models and human lung cancer cell lines, the Akt signaling pathway regulates the expression of PD-L1 at the protein translation level.146 In conclusion, the PI3K/Akt signaling pathway is obviously related to immune resistance, which provides a basis for combined therapeutic strategies including immune checkpoint inhibitors and PI3K pathway inhibitors in the future.
PI3K/Akt signaling and cancer metabolism
One of the main features of cancer is the occurrence of metabolic aberrations, such as changes in glycolytic pathway. Metabolic aberrations will lead to changes in signaling pathways, affecting the occurrence, development and metastasis of cancer. Under physiological conditions, the PI3K/Akt signaling pathway is activated by the actions of insulin, growth factors and cytokines to regulate metabolism in organisms.147 In cancer cells, the activation of oncogenes in the PI3K/Akt signaling pathway reprograms cellular metabolism by enhancing the activities of nutrient transporters and metabolic enzymes, thus supporting the anabolic needs of abnormally growing cells.147
PI3K/Akt signaling not only regulates metabolism-associated proteins such as SREBP and alters metabolism through phosphorylation mediated by metabolic enzymes but also indirectly alters metabolism by controlling various TFs.147 The phosphorylation of metabolic enzymes causes acute changes in the activities of metabolic pathways and the directionality of metabolic fluxes, while long-term changes in cellular metabolism are usually achieved by controlling gene expression programs.147 Akt not only directly phosphorylates a variety of metabolic enzymes and nutrient transport regulators but also regulates cellular metabolism by activating mTORC1, GSK3 and FOXO.91 The phosphorylation of TSC2 by Akt leads to Rheb-GTP aggregation, which in turn activates mTORC1 to thereby enhance glucose metabolism and lipid synthesis.84 GSK3, a key regulatory factor in cellular metabolism, participates in cellular metabolism by phosphorylating and inhibiting metabolic enzymes, such as its substrate glycogen synthase.86,87,92 FOXO TFs regulate cellular metabolism and tumor suppression.148 Akt phosphorylates FOXO and prevents it from entering the nucleus to mediate the expression of its target genes.127,149 In addition, accumulating evidence indicates that PI(3,4)P2 is not only a waste product for the removal of PI(3,4,5)P3 but also functions as a signaling molecule in cancer metabolism.150
PI3K/Akt signaling and angiogenesis
Angiogenesis in tumors mainly refers to the growth of new capillaries from the existing capillary network that supply tumor cells with nutrition. Among the three subtypes of Akt, Akt1 is the main subtype that regulates the normal physiological function of endothelial cells. After activation by VEGF, Akt promotes the proliferation, migration and survival of endothelial cells, thus affecting angiogenesis.151 This finding also provides lateral support for the conclusion that endothelial nitric oxide carbon synthase (eNOS), which controls vascular tone, is a specific substrate of Akt1 in endothelial cells.152 At the same time, the loss of Akt1 in mouse endothelial cells reduces the amount of nitric oxide on the cell membrane and affects the formation of blood vessels.153 Akt1 is also related to vascular remodeling, which is mediated by endothelial cells. Expression of the activated Akt1 allele prevents the formation of intimal lesions after arterial injury, induces pathological angiogenesis, and increases vascular permeability.154
In addition, a recent report revealed that the p110α subtype of PI3K promotes tumor angiogenesis in homogenic mouse models and that the inactivation of p110α leads to an increased vascular density, decreased vessel size and altered pericyte coverage on vessels. This decrease in vascular function is associated with increased tumor necrosis and decreased tumor growth. The role of p110α in tumor angiogenesis is mediated by several factors, such as the regulation of endothelial cell proliferation and the expression of delta-like protein 4 (DLL4). Thus, p110α regulates angiogenesis in the tumor stroma, and inactivation of p110α suppresses tumor angiogenesis and tumor growth.155
PI3K/Akt signaling and cancer metastasis
Studies have shown that cancer metastasis is the main cause of poor prognosis.156 The mechanism of cancer metastasis involves many factors, such as genetic material, surface structure, invasion, adhesion, angiogenesis and lymphangiogenesis. Two main mechanisms by which PI3K/Akt signaling promotes cancer metastasis have been identified.
First, the PI3K pathway promotes metastasis by reducing intercellular adhesion and enhancing mobility. After extracellular signaling molecules bind to specific receptors on the surface of the cell membrane, they are activated by different signal transduction pathways in the cell to modulate the activities of different TFs, resulting in differential degrees of epithelial cell phenotype transformation. The PI3K/Akt signaling pathway plays an important role in the induction of squamous cancer cell epithelial-mesenchymal transition (EMT). PI3K activation produces the second messenger PIP3 downstream of Akt activation, which activates or inhibits downstream target proteins via phosphorylation, thereby regulating cell survival, proliferation, and differentiation as well as the composition of the cytoskeleton, among other processes, inducing EMT. PI3K/Akt signaling pathway activation increases tumor cell invasion and metastasis.157 Steelman et al. reported that the continuous activation and high expression of PI3K/Akt are closely related to EMT in NSCLC.158
Second, the PI3K pathway promotes metastasis by promoting tumor neovascularization, which is required for the metastatic spread of tumors. Multiple signaling pathways have been shown to be involved in the regulation of tumor angiogenesis, among which PI3K/Akt signaling is the most important.159 PI3K forms a complex with E-cadherin, β-catenin, and VEGFR-2 and is involved in endothelial signaling mediated by VEGF through the activation of the PI3K/Akt pathway.160 The PI3K/Akt signaling pathway also promotes TNF-induced endothelial cell migration and regulates tumor angiogenesis.161 Matrix metalloproteinases (MMPs) and cyclooxygenase 2 (COX-2) also affect tumor angiogenesis. In tumor invasion and metastasis, platelet-derived growth factor (PDGF) induces MMP expression through a PI3K-mediated signaling pathway.162 Upregulation of the antiapoptotic protein Bcl-2 and activation of the PI3K/Akt signaling pathway are the main mechanisms by which COX-2 stimulates endothelial angiogenesis.163
However, not all components of the PI3K/Akt signaling pathway promote metastasis. One of the Akt isoforms, Akt1, can exert an antimetastatic effect, which is accompanied by increased ERα expression.164 Breast cancer cells undergo differentiation upon the activation of Akt1, thereby losing their metastatic potential.165 Similarly, Akt1 can inhibit metastasis in mice by regulating MMP9 and E-cadherin.166
PI3K/Akt signaling and inflammation
Virchow first proposed an association between inflammation and cancer in 1863,167 and many subsequent studies have supported this association.168 A study showed that the PI3K/Akt signaling pathway promotes the development of inflammation by affecting neutrophils, lymphocytes and other white blood cells.169 However, IL-1, IL-6, TFN-α and other inflammatory factors activate Akt and expand the range of inflammation, while Akt inhibition blocks both inflammation and tumor development.170 IL-6 activates the PI3K/Akt signaling pathway to promote the expression of B-FGF, which induces angiogenesis.171 mTOR and p70 S6K1, downstream effectors of Akt, directly act on endothelial cells to promote tumor inflammation.172
Studies have shown that the positive effect of Akt signaling on inflammatory cells is conducive to promoting the aggregation of reactive cells, and the resulting oxidative stress reaction leads to the accumulation and release of peroxides (ROS) at the tumor site.173 In addition, IL-37 is an anti-inflammatory cytokine that has been reported to induce autophagy and apoptosis by regulating PI3K/Akt/mTOR signaling.174
Inflammation also promotes cancer development by activating the PI3K/Akt signaling pathway. For example, inflammation-induced PI3K/Akt signaling regulates the permeability and migration of endothelial cells and affects cancer progression.175 In conclusion, inflammation affects the biological processes of endothelial cells in tumors through the PI3K/Akt signaling pathway, which is an important component of inflammation-induced tumor development.
PI3K/Akt signaling and immunity
Immunity is divided into innate and adaptive immunity. The immune system plays an important role in tumor detection and elimination. To date, various immunotherapies have been applied in the clinic. The PI3K/Akt signaling pathway also exerts a vital effect on the immune system. Growth factors, cytokines, and other factors activate Akt signaling in myeloid cells, thus activating downstream effectors of Akt.
Innate immunity
The innate immune system, which is composed of monocytes, leukocytes and macrophages, is the first line of defense against infection.176 According to some studies, inhibition of the PI3K signaling pathway reduces the secretion of proinflammatory cytokines, and the PI3K pathway is related to the movement of macrophages.177
The PI3K signaling pathway affects the secretion of proinflammatory cytokines from innate immune system cells by regulating the activity of downstream targets. It participates in the movement and adhesion of macrophages and plays an important role in cells.178 On the other hand, Akt signaling is the key signal transduction pathway activated in macrophages under various external stimuli.179 Akt signaling is activated in both M1 and M2 macrophages, but most of the current evidence suggests that Akt signaling exacerbates the M2 state. Hence, interventions targeting PI3K signaling may be an effective treatment for some immune and tumor-related diseases.180 Similarly, the PI3K/Akt pathway affects natural killer (NK) cells. A study reported reduced NK cellularity and a decreased number of CD27(high) NK cells in mice expressing p110 δ (D910A), which is a catalytically inactive form of p110 δ, and that inactivated p110 δ slightly impaired NK-mediated tumor cell cytotoxicity in vitro and in vivo.181
Adaptive immunity
The adaptive immune system mainly includes T and B cells, each of which have unique antigen receptors. PI3K signaling plays a key role in T and B cells.182 PI3Kδ has been identified as the main subtype of PI3K activated by T cell receptors (TCRs) and BCRs, and activated PI3Kδ subsequently activates Akt and other downstream signals.62
TEC family proteins, such as BTK, play a key role in PI3K signal transduction.183 BTK inhibition reduces the output of the PI3K signal in B cells.66,184 Cooperation between the PI3K signaling pathway and BTK in immune cells has also been documented. Upon the antigen activation of B cells, the cells undergo cloning, expansion and differentiation and secrete different antigen-specific antibodies. Some B cells rapidly differentiate into plasma cells, mainly producing low-affinity IgM, while others secrete high-affinity conversion antibodies. This differentiation is determined in part by the levels of PI3K/Akt signaling and FOXO TF activity.185
In CD8+ T cells, class IA PI3Ks are mainly activated by RTKs, such as TCRs and cytokine receptors. Activated PI3K/Akt signaling promotes the uptake of glucose and amino acids by CD8+ T cells for energy-demanding cellular processes, such as proliferation and cytokine synthesis and secretion.186 Moreover, research has shown that Akt inhibition effectively enhances memory T cell differentiation in cancer.187 Therefore, inhibition of the PI3K/Akt signaling pathway may affect T cell function. The PI3K/Akt signaling pathway plays an indispensable role in the immune system.
Functional role of the PI3K/Akt signaling pathway in the tumor microenvironment
The tumor microenvironment plays an important role in the occurrence, development, and metastasis of tumors and influences cancer treatment.188 The tumor microenvironment includes immune cells, endothelial cells, fibroblasts, the extracellular matrix, and signaling molecules surrounding tumor cells. PI3K/Akt and its downstream effectors are known to constitute a signaling pathway involved in tumor cell development. PI3K signals regulate cell survival, development, and proliferation by relying on extracellular signaling molecules, and signaling molecules outside tumor cells are part of the tumor microenvironment.
In the tumor microenvironment, cancer-associated fibroblasts (CAFs) are the most abundant cells in the tumor matrix and promote tumor progression by secreting several growth factors, such as FGFs.189 Hepatocyte growth factor (HGF) secreted by CAFs specifically triggers PI3K/Akt signaling to affect cancer progression, and HGF has been detected in many types of human cancer, such as ovarian cancer.190 Furthermore, CCL5 derived from CAFs has been detected in ovarian cancer cells and shown to influence drug resistance by regulating PI3K/Akt signaling.191
In addition, tumor growth depends on oxygen and nutrients delivered by new blood vessels,192 which are in turn related to endothelial cells, an important component of the tumor microenvironment. As shown in previous studies, PI3K subtypes promote the differentiation of endothelial cells and other cells,193 and different PI3K inhibitors thus produce different vascular responses. Interventions targeting PI3K in CAFs and endothelial cells in the tumor microenvironment in combination with conventional therapies have great potential in the treatment of cancer. In summary, the PI3K/Akt signaling pathway plays an essential role in multiple cancer phenotypes, as summarized in Fig. 3.
Fig. 3.
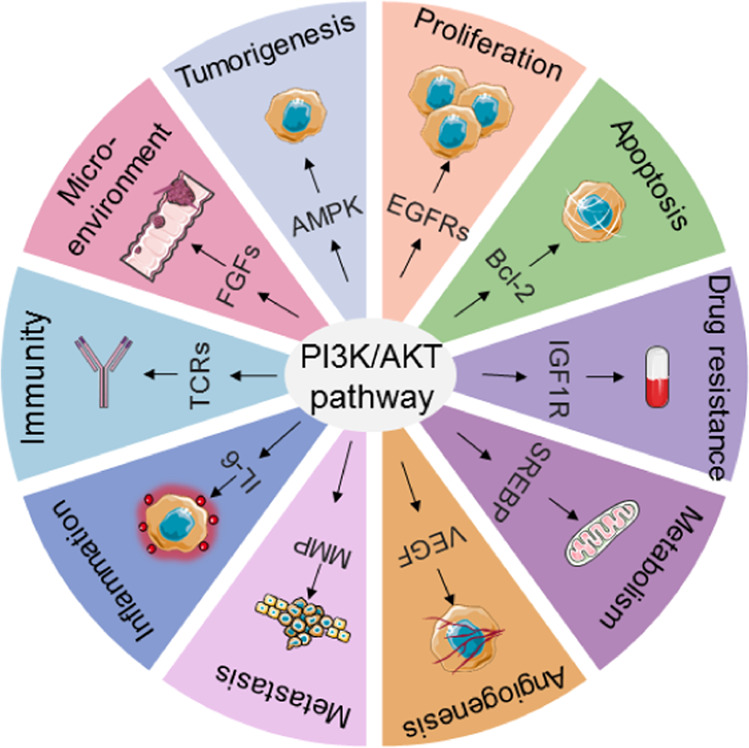
PI3K/Akt signaling and cancer. Various biological processes are regulated by the PI3K/Akt pathway via key mediators/pathways
Cancer therapies targeting the PI3K/Akt pathway
PI3K inhibitors
Because of the function of PI3K/Akt signaling in oncogenesis, various inhibitors targeting PI3K are being explored for the treatment of various types of cancer (Table 2); to date, some agents have been approved for the treatment of malignancies and for the improvement of cancer therapy.194
Table 2.
Novel agents targeting PI3K signaling in cancer
| Class | Inhibitor | Phase | Toxic effects | Reference |
|---|---|---|---|---|
| Pan‑PI3K inhibitors | GDC-0941 | I | Neutropenia、Neuropsychiatric effects (confusion, depression, anxiety)、Hepatotoxicity、Diarrhea | 254 |
| BKM-120 | II | Hyperglycemia | 255 | |
| BAY-80-6946 | Approved |
Nausea High blood sugar |
256 | |
| XL-147 | I | Rash | 257 | |
| PX-866 | II |
Diarrhea ALT/AST elevation |
258 | |
| Isoform-selective PI3K inhibitors | BYL-719 | I | Hyperglycemia | 259 |
| GDC-0032 | I | Rash、Diarrhea、Pneumonitis | 206 | |
| IPI-145 | Approved | Hyperglycemia、Rash、Diarrhea、Hypertension | 260 | |
| SAR-260301 | Preclinical | Nausea、Vomiting、Diarrhea | 261 | |
| MLN-1117 | I | Nausea | 262 | |
| Dual pan‑PI3K and mTOR inhibitors | BEZ-235 | I/II | Fatigue/asthenia、Thrombocytopenia | 263 |
| SF-1126 | I/II | Hyperglycemia | 264 | |
| GDC-0980 | I |
Maculopapular rash Symptomatic hyperglycemia |
265 | |
| PF-04691502 | II |
Fatigue Loss of appetite、Nausea、High blood sugar、Rash、Vomiting |
266 | |
| XL-765 | I | Stomatitis | 267 | |
| GSK-2126458 | I | Diarrhea | 268 | |
| PF-05212384 | II | Fatigue | 266 | |
| GSK-1059615 | Preclinical | Immunosuppression | 269 | |
| BGT-226 | I/II |
Diarrhea Nausea Loss of appetite Vomiting Fatigue |
270 |
Pan-PI3K inhibitors
Pan-PI3K inhibitors target the p110 subunits of class IA PI3Ks, which are the most widely involved subunits in cancer. These inhibitors have greater anticancer activity and fewer side effects (less toxicity) than inhibitors of other PI3K classes. Some representative small-molecule pan-PI3K inhibitors include pictilisib (GDC-0941), buparlisib (BKM120) and pilaralisib (XL147).195
GDC-0941, a thienopyrimidine derivative, was the first inhibitor to enter clinical trials. As a pan-class I PI3K inhibitor, GDC-0941 shows the same activity (IC50 = 3 nM) against p110α and p110δ enzymes, and in kinase assays, it exerts an inhibitory effect on p110β and p110γ at nanomolar concentrations.196 Experiments have shown that monotherapy or combination therapy with GDC-0941 and other agents has strong antitumor activity in mouse xenograft models of human cancer.196,197
BKM120, also known as NVP-BKM120, is another effective pan-class I PI3K inhibitor that exerts a strong inhibitory effect on p110-α and p110β enzymes, with IC50 values of 52 nM and 166 nM, respectively. It also effectively inhibits p110δ and p110γ enzymes, with IC50 values of 116 nM and 262 nM, respectively.198 Preclinical studies have shown that BKM120 hinders the growth of U-87MGGBM cells after xenotransplantation in the brain, which helps to prolong the xenotransplantation duration in host animals; most importantly, it does not induce significant adverse effects.199 According to a research report describing a phase I study of 35 patients with advanced solid tumors receiving BKM120 at 12.5–150 mg per day, the administration of BKM120 at the maximum tolerated dose of 100 mg/day was safe and exhibited preliminary antitumor activity.200
Another class I inhibitor, XL147, has high specificity for four subtypes of PI3Ks.201 Analysis of its selectivity for more than 130 protein kinases revealed that XL147 is much more selective for class I PI3Ks than other kinases. In cell experiments, XL147 inhibited the formation of PIP3 in the cell membrane and the phosphorylation of kinases such as Akt in a variety of tumor cell lines, influencing genetic alterations in the PI3K pathway.201 In mouse transplant models, the oral administration of XL147 inhibited the phosphorylation of Akt, p70S6K, and S6 for at least 24 h, and repeated administration of XL147 significantly inhibited tumor growth.201
Isoform-selective PI3K inhibitors
Compared to pan-PI3K inhibitors, isotype-specific inhibitors exhibit less off-target toxicity because they are specific to the tumor type; thus, isotype-specific inhibitors can be administered at higher doses and are more effective.202 Alpelisib (BYL-719) and taselisib (GDC-0032) are representative homospecific inhibitors.
BYL-719 is the first inhibitor to selectively target class I p110α. In a phase I trial, BYL-719 resulted in monotherapy sensitivity in patients with PI3KCA-mutant solid tumors.203 The combination of BYL-719 and fulvestrant resulted in the remission of 29% of patients with severe pretreated PI3KCA-mutant tumors and had a good safety profile in patients.204
Based on the antitumor activity of BYL-719 in phase I clinical trials, scientists conducted a phase III clinical trial of BYL-719 combined with fulvestrant in the treatment of advanced breast cancer.205 The phase III trial showed a significantly prolonged survival rate of patients with advanced breast cancer. The overall response of patients treated with BYL-719 plus fulvestrant was better than that of patients treated with the placebo plus fulvestrant. However, patients treated with BYL-719 plus fulvestrant had higher rates of hyperglycemia, rashes and diarrhea than those treated with the placebo.
GDC-0032 exhibits the same activity against p110α, p110γ and p110δ, but its inhibitory effect on p110β is 30-fold lower.206 Due to the stronger selectivity of GDC-0032, this drug has shown better efficacy in the treatment of PI3KCA-mutant tumors, with a response rate of 36% among breast cancer patients with PI3KCA-mutant tumors.207 In a phase III clinical trial, GDC-0032 was more effective than fulvestrant,208 and the results showed that the main adverse events after GDC-0032 administration were diarrhea and hyperglycemia. In some patients, the moderate improvement in progression-free survival after GDC-0032 administration occurred at the cost of significant toxicity, and GDC-0032 was thus not suitable for use in these patients. In terms of symptoms after administration, GDC-0032 produced more side effects than BYL-719, possibly because of its stronger specificity and exertion of a more obvious inhibitory effect on p110α.206
Dual pan-PI3K and mTOR inhibitors
Since both mTOR and PI3K have a p110 subunit with similar structures, treatment with dual pan-PI3K and mTOR inhibitors may exert an improved therapeutic effect due to the more efficient inhibition of the PI3K/Akt/mTOR signaling pathway. The current pan-PI3K and mTOR inhibitors mainly include SF1126, dactolisib (Bez235), voxtalisib (XL765) and GSK1059615.30 Although this type of inhibitor is less specific than isotype-specific inhibitors, it has the potential to treat a variety of tumors with a wide range of genetic abnormalities; however, it also has many unknown toxicities and side effects.
Another advantage of these inhibitors is that they completely inhibit the p110 subunit; however, they should not be used in conjunction with other PI3K and Akt inhibitors.6 Key factors affecting this class of inhibitors include whether the dose at which all p110 subunits are completely suppressed is tolerable in patients and whether the use of these inhibitors is at the expense of other inhibitor targets.6 mTORC1 inhibitors tend to activate the PI3K signaling pathway through feedback inhibition.209 Therefore, the advantages of dual pan-PI3K and mTOR inhibitors in inhibiting feedback inhibition are highlighted, and they have obvious therapeutic advantages.
Akt inhibitors
Due to the existence of three Akt subunits, most current inhibitors of Akt are pan-Akt inhibitors,74 and the research and development of specific inhibitors against the three subunits of Akt will be very challenging. Several inhibitors of Akt are currently being investigated in clinical trials, such as MK-2206, GSK-2141795, GSK-2110183, AZD5363 and GDC0068 (Table 3).
Table 3.
Current inhibitors targeting Akt signaling in cancer
| Inhibitor | Phase | Target | ClinicalTrials.gov Identifier: | Reference |
|---|---|---|---|---|
| Perifosine | II | AKT1/2/3 | 271 | |
| GSK-690693 | I | AKT1/2/3 | 272 | |
| VQD-002 | I | AKT1/2/3 | 273 | |
| AZD-5363 | III | AKT1/2/3 | NCT04493853 | |
| GDC-0068 | III | AKT1/2/3 | NCT04650581 | |
| GSK-2141795 | II | AKT1/2/3 | NCT01964924 | |
| M2698 | I | AKT1/3 | 274 | |
| GSK-2110183 | II | AKT1/2/3 | NCT01531894 | |
| MK-2206 | II | AKT1/2/3 | NCT01370070 |
MK-2206, an allosteric pan-Akt inhibitor, shows synergistic activity with cytotoxic compounds such as doxorubicin, gemcitabine, docetaxel and carboplatin in lung cancer.75 In addition, MK-2206 inhibits the phosphorylation of Akt mediated by carboplatin and gemcitabine, thereby enhancing the therapeutic effect of the drug by inhibiting tumor survival laterally.210 MK-2206 also enhances erlotinib activity in erlotinib-sensitive patients and erlotinib-resistant NSCLC cell lines.211 Recently, MK-2206 entered preclinical studies to determine its effects on acute myeloid leukemia (AML). Data obtained from mouse models in preclinical studies indicate that weekly administration is effective for the treatment of AML, which supports its use in subsequent clinical trials.212 A preclinical study on the inhibitor MK-2206 in nasopharyngeal carcinoma (NPC) is also ongoing.213 HGF was used to resensitize drug-resistant cells to the drug, revealing the therapeutic efficacy of MK-2206.211 The efficacy of MK-2206 was also reported in a phase I clinical trial in which the tumors of patients with PTEN-deficient pancreatic cancer regressed after treatment with MK-2206 alone. MK-2206 also exerts a mild therapeutic effect on patients with melanoma and endocrine tumors.214
AZD5363 is an inhibitor targeting the kinase activity of the three Akt subtypes (Akt1, 2, and 3).215 AZD5363 inhibits cancer cell proliferation and phosphorylates GSK3β and the downstream channel protein S6 in vitro. In vivo, AZD5363 inhibits tumor growth in xenograft tumor models and maintains pharmacodynamic activity for at least 24 hours.215 Preclinical sensitivity to AZD5363 is strongly associated with the presence of PIK3CA, and this trend has also been observed for other inhibitors of the PI3K/Akt/mTOR pathway.216
mTORC1 and mTORC2 inhibitors
Studies have shown that mTOR, a classical downstream effector of the PI3K oncogenic pathway, has activity ranging from 40-90% in various solid tumors.217 Therefore, the use of mTOR as a target for cancer treatment has become a research hotspot.
Rapamycin
Rapamycin and its analogs (temsirolimus, everolimus, and deforolimus) are allosteric inhibitors of mTOR and constitute the first generation of mTOR inhibitors.218 Rapamycin binds to mTOR and FKBP-12 to form a ternary complex and specifically inhibits the phosphorylation of the mTORC1 protein kinase S6K1, thus inhibiting mTORC1 activity.218 Rapamycin is produced by Streptomyces sp. and possesses antifungal properties. In the 1980s, rapamycin was reported to have anticancer activity, but its clinical application was limited due to its poor water solubility and stability.218
Although rapamycin inhibits mTOR with high specificity, its efficacy in different environments depends on its dosage. Clinical doses of rapamycin may vary due to the differential sensitivities of cancer cells to rapamycin. The amount of rapamycin required to phosphorylate the substrate also differs for different mTOR substrates, which is caused by the competitive inhibition of mTOR by phosphatidic acid and rapamycin.219
ATP-competitive mTOR inhibitors
Strategies involving the targeting of mTORC1 and mTORC2 have been developed to inhibit mTOR more completely, and many ATP-competitive mTOR inhibitors have been exploited. ATP-competitive mTOR inhibitors are a class of small-molecule ATP analogs that compete with ATP to occupy mTOR kinase active sites.218 Unlike rapamycin analogs, these ATP analogs ensure the complete blockade of mTORC1 and mTORC2, thereby preventing Akt phosphorylation caused by mTORC2 and the observed resistance to rapamycin analogs. In vitro studies have shown that ATP-competitive inhibitors exert a greater inhibitory effect than rapamycin analogs.218 AZD8055, one of the most recent and potent ATP-competitive mTOR inhibitors, functions by inhibiting the phosphorylation of Akt and its downstream proteins and has been shown to induce autophagy in cancer cells and inhibit tumor growth in vivo.220 In conclusion, inhibitors targeting the PI3K/Akt pathway are promising for cancer therapy, and numerous PI3K/Akt pathway inhibitors have been developed (summarized in Table 4).
Table 4.
Therapies targeting PI3K/Akt signaling
| Agent | Targets | Inhibitors |
|---|---|---|
| Apoptosis | p53 | BKM120 |
| CAL-101 | ||
| GDC0068 | ||
| p-S6 | BYL719 | |
| GDC-0068 | ||
| Rapamycin | ||
| p38 | GSK2141795 | |
| Proliferation | p53 | Bez235 |
| Temsirolimus | ||
| p-S6 | XL765 | |
| XL147 | ||
| GSK2121183 | ||
| LY2780301 | ||
| Rapamycin | ||
| Deforolimus | ||
| CDK1 | Everolimus | |
| TGF-β1 | Bez235 | |
| Autophagy | LC3B | MK2206 |
| p-S6 | AZD5363 |
Combination therapeutic strategies
Cancer treatment has always been difficult and perplexed humans for many years, and combination therapy is an inevitable trend. A logical approach is to combine PI3K-Akt pathway inhibitors with standard targeted drugs approved for the treatment of specific malignancies. This approach will also facilitate the inclusion of PI3K/Akt inhibitors in treatment regimens for patients with early-stage disease and preclude their use for only patients with relapsed or refractory tumors. Here, we describe the combination of PI3K signaling inhibitors with growth factor inhibitors, MAPK inhibitors, and conventional therapies, such as chemotherapy, radiation therapy and immunotherapy.
Combination with GFR inhibitors
GFR alterations are some of the most common causes of cancer among various types of tumors. For example, abnormal HER2 amplification is common in breast cancer.221 GFR inhibition has several advantages for tumor therapy, as it not only blocks signal initiation and crosstalk with complementary pathways but also increases the therapeutic sensitization of tumor cells to chemotherapy and radiation.221 A trial of buparlisib in combination with paclitaxel and trastuzumab for breast cancer with abnormal HER2 amplification showed that buparlisib can be used in combination with paclitaxel and trastuzumab.222 Additionally, alpelisib in combination with cetuximab can be used for the treatment of head and neck squamous cell carcinoma.223
Combination with MAPK inhibitors
The PI3K and MAPK signaling networks have been shown to interact, which creates a potential pathway for the development of combination therapies (PI3K and MAPK pathway inhibitors) for cancer. Additionally, complete inhibition of the PI3K and MAPK signaling pathways is required for tumor control.224 The results of a clinical trial on treatment with both PI3K and MAPK signaling inhibitors support this hypothesis, as PI3K and MAPK therapies used in combination improved the treatment efficacy compared to that achieved with either inhibitor alone. However, the disadvantage of this approach is increased toxicity.225
In a phase I trial, GDC-0941 and SAR245409 were used to target PI3K in combination with GDC-0973 and AS-703026 to target MEK and showed promising results in the treatment of KRAS-mutated tumors.226,227 Further research on combinations of drugs targeting these two signaling pathways is imperative. PI3K and MAPK inhibitor combination therapies should include solid tumors that depend on MAPK pathway activation, such as BRAF-mutant and KRAS-mutant melanoma, colorectal carcinoma and ovarian cancer.226–228
Combination with chemotherapy
According to previous studies, PI3K signaling pathway inhibitors may increase the sensitivity of tumor cells to chemotherapy by changing the peripheral vascular system and tumor perfusion, thereby increasing the efficacy of systemic therapy and inducing apoptosis.229 Early clinical studies showed that PI3K pathway inhibitors are well tolerated when administered with chemotherapy; pictilisib, carboplatin, and paclitaxel have shown good antitumor activity in patients with NSCLC. Additionally, a phase II clinical trial on ipatasertib was initiated because of its positive effect on gastric cancer when used in combination with chemotherapy.230
According to recent studies, PI3K signaling inhibitors increase DNA damage and sensitize cell lines to poly (ADP-ribose) polymerase inhibitors.231 Therefore, treatments targeting the association between the PI3K pathway and DNA repair are emerging as a therapeutic strategy for BRCA1-deficient tumors. A phase I trial on buparlisib in combination with olaparib is underway in patients with triple-negative breast cancer and highly serous ovarian cancer.232 The results of these trials might further our understanding of this combination and provide new insights into strategies for overcoming resistance.
Combination with radiotherapy
Since the 21st century, radiation therapy for cancer has been continuously improved, as the damage to normal patient tissue has been reduced, allowing the therapeutic dose to be increased, and the cure rate among patients with cancer has been improved.233 Despite the improvements in radiation therapy for cancer treatment, the survival rate of patients with squamous cell carcinoma of the head and neck in the last five years was only 40%.234 With combination therapy, inhibitors of the PI3K/Akt signaling pathway not only restore the sensitivity of tumor cell growth but also increase the sensitivity of the tumor to chemotherapy, radiotherapy and hormone therapy.235 Moreover, cell cycle arrest was observed after suppression of the PI3K/Akt pathway.236 A recent study revealed that buparlisib, a class I PI3K inhibitor, could be safely combined with radiotherapy and improved the radiotherapeutic outcomes in patients with NSCLC.237 In conclusion, the combination of PI3K/Akt signaling pathway inhibitors and radiotherapy is a promising approach for cancer treatment.
Combination with immunotherapy
Although cancer is a genetic disease characterized by abnormal cell proliferation, it is also a chronic immune disease.238 Immunotherapy has recently attracted increasing attention and has indeed achieved some progress in treating cancer.238 Studies on the PI3K/Akt signaling pathway in immune cells have shown that its activation is not based on a simple on/off mechanism239; as cancer is a chronic immune disease, immunotherapies should be designed to increase the ability of immune cells to kill tumor cells such that the body exerts a direct antitumor effect. Determining whether drugs targeting the PI3K signaling pathway enhance or inhibit the efficacies of emerging immunotherapies will be critical for determining whether they can be combined. Both PI3K and mTOR inhibitors have been shown to enhance the efficacies of targeted immunotherapies in mouse tumor xenotransplantation models.240
Traditional adoptive cell therapy (ACT) is performed by isolating tumor-infiltrating lymphocytes (TILs) from biopsy materials.241 One of the main characteristics of TILs is their long-term persistence after migration. However, a major difficulty is that most TILs isolated from tumor tissues are terminally differentiated and do not form phenotypes with memory ability.187 Recently, ATC technology has been improved to enable the in vitro transduction of CD8+ T cells by the chimeric antigen receptor (CAR), with which the specific treatment of tumor antigens is not limited by major histocompatibility complex I.242 Recent reports on the efficacy of CAR-T cells for solid tumors in vivo indicate that it can be improved by PI3K inhibitors; however, these findings are preliminary, and the underlying mechanisms remain to be further elucidated.242
Conclusions and remarks
In the past few decades, numerous insights into the PI3K/Akt signaling pathway have revealed its complex networks, including its mechanism of activation, upstream and downstream targets, and types of inhibitors, thereby increasing our understanding of the occurrence and development of different types of human cancers.
The diverse functions of PI3K/Akt signaling stem from downstream factors that link cellular function to stimulated upstream factors. For instance, extracellular growth factors recruit PI3K to the cell membrane, and the PI3K p110 subunit then induces the phosphorylation of PIP2 to PIP3, which promotes the localization of Akt to the plasma membrane. After activation by PDK1 and mTORC2, Akt drives the expression of targets associated with the cell phenotype. A dynamic system exists that comprises many substrates and crosstalk among them as well as crosstalk with other major signaling networks. This system exerts significant effects on modulating the oncogenicity of cancer.
After years of basic research, the development of inhibitors has undeniably advanced the treatment of cancer, but problems still persist. The most common problems related to inhibitors is their toxicity and side effects, such as hyperglycemia, rashes and other symptoms, and these effects on patients must not be ignored. In addition, concerns about the efficacy of animal models for predicting drug toxicity and whether in vivo toxicity can be predicted by in vitro experiments have been noted. Another problem is that conventional models, including phase I, II, and III trials, may not be suitable for testing PI3K inhibitors, which is detrimental to the determination of therapeutic efficacy. Although none of the inhibitors have entered phase III clinical trials, they show promise in treating cancer.243
Fortunately, most drugs have short half-lives and are easily managed, enabling early intervention; thus, patients treated with inhibitors can undergo active and effective intervention while monitoring for early toxicity identifiers. This pathway can be effectively manipulated to treat various human diseases caused by PI3K/Akt signaling dysregulation only when researchers fully elucidate the underlying mechanisms.
Acknowledgements
The current work was supported by the National Natural Science Foundation of China (Project nos. 31961160727, 81773085, 82073196, 81973339, 81873455 and 81803551), the Guangdong Natural Science Research Grant (No. 2021A1515011158), and the Fundamental Research Funds for the Central Universities (No. 21620429).
Author contributions
Y.H., M.M.S., G.G.Z. and J.Y. wrote the manuscript. B.L., W.W.X. and K.S.C. designed and revised the manuscript. All authors have read and approved the article.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Yan He, Miao Miao Sun, Guo Geng Zhang, Jing Yang
Contributor Information
Kui Sheng Chen, Email: chenksh2002@163.com.
Wen Wen Xu, Email: xuwen6966@163.com.
Bin Li, Email: libinjnu@163.com.
References
- 1.Lawrence MS, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hennessy BT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Disco. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 3.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010;11:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 4.Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer. 2015;15:7–24. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanker AB, Kaklamani V, Arteaga CL. Challenges for the clinical development of PI3K inhibitors: Strategies to improve their impact in solid tumors. Cancer Disco. 2019;9:482–491. doi: 10.1158/2159-8290.CD-18-1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat. Rev. Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 7.Backer JM. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem J. 2016;473:2251–2271. doi: 10.1042/BCJ20160170. [DOI] [PubMed] [Google Scholar]
- 8.Kriplani N, Hermida MA, Brown ER, Leslie NR. Class I PI 3-kinases: function and evolution. Adv. Biol. Regul. 2015;59:53–64. doi: 10.1016/j.jbior.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Burke JE. Structural basis for regulation of phosphoinositide kinases and their involvement in human disease. Mol. Cell. 2018;71:653–673. doi: 10.1016/j.molcel.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Carnero A, et al. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets. 2008;8:187–198. doi: 10.2174/156800908784293659. [DOI] [PubMed] [Google Scholar]
- 11.Gerstung M, et al. The evolutionary history of 2,658 cancers. Nature. 2020;578:122–128. doi: 10.1038/s41586-019-1907-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herman SE, et al. Phosphatidylinositol 3-kinase-δ inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116:2078–2088. doi: 10.1182/blood-2010-02-271171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasaki T, et al. Colorectal carcinomas in mice lacking the catalytic subunit of PI(3)Kgamma. Nature. 2000;406:897–902. doi: 10.1038/35022585. [DOI] [PubMed] [Google Scholar]
- 14.Tsolakos N, et al. Quantitation of class IA PI3Ks in mice reveals p110-free-p85s and isoform-selective subunit associations and recruitment to receptors. Proc. Natl Acad. Sci. USA. 2018;115:12176–12181. doi: 10.1073/pnas.1803446115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gunderson AJ, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Disco. 2016;6:270–285. doi: 10.1158/2159-8290.CD-15-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Damoulakis G, et al. P-Rex1 directly activates RhoG to regulate GPCR-driven Rac signalling and actin polarity in neutrophils. J. Cell Sci. 2014;127:2589–2600. doi: 10.1242/jcs.153049. [DOI] [PubMed] [Google Scholar]
- 17.Virbasius JV, Guilherme A, Czech MP. Mouse p170 is a novel phosphatidylinositol 3-kinase containing a C2 domain. J. Biol. Chem. 1996;271:13304–13307. doi: 10.1074/jbc.271.23.13304. [DOI] [PubMed] [Google Scholar]
- 18.Braccini L, et al. PI3K-C2γ is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 2015;6:7400. doi: 10.1038/ncomms8400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gulluni F, et al. Mitotic spindle assembly and genomic stability in breast cancer require PI3K-C2α scaffolding function. Cancer Cell. 2017;32:444–459.e447. doi: 10.1016/j.ccell.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 20.Franco I, et al. PI3K class II α controls spatially restricted endosomal PtdIns3P and Rab11 activation to promote primary cilium function. Dev. Cell. 2014;28:647–658. doi: 10.1016/j.devcel.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pirola L, et al. Activation loop sequences confer substrate specificity to phosphoinositide 3-kinase alpha (PI3Kalpha). Functions of lipid kinase-deficient PI3Kalpha in signaling. J. Biol. Chem. 2001;276:21544–21554. doi: 10.1074/jbc.M011330200. [DOI] [PubMed] [Google Scholar]
- 22.Gulluni F, et al. Class II PI3K functions in cell biology and disease. Trends Cell Biol. 2019;29:339–359. doi: 10.1016/j.tcb.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 23.Marat AL, Haucke V. Phosphatidylinositol 3-phosphates-at the interface between cell signalling and membrane traffic. Embo J. 2016;35:561–579. doi: 10.15252/embj.201593564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stjepanovic G, Baskaran S, Lin MG, Hurley JH. Vps34 kinase domain dynamics regulate the autophagic PI3-kinase complex. Mol. Cell. 2017;67:528–534.e523. doi: 10.1016/j.molcel.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vasudevan KM, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Farrell F, et al. Class III phosphatidylinositol-3-OH kinase controls epithelial integrity through endosomal LKB1 regulation. Nat. Cell Biol. 2017;19:1412–1423. doi: 10.1038/ncb3631. [DOI] [PubMed] [Google Scholar]
- 27.Juhász G, et al. The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J. Cell Biol. 2008;181:655–666. doi: 10.1083/jcb.200712051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gulati P, et al. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008;7:456–465. doi: 10.1016/j.cmet.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl Acad. Sci. USA. 1987;84:5034–5037. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment. Annu Rev. Med. 2016;67:11–28. doi: 10.1146/annurev-med-062913-051343. [DOI] [PubMed] [Google Scholar]
- 31.Murthy SS, Tosolini A, Taguchi T, Testa JR. Mapping of AKT3, encoding a member of the Akt/protein kinase B family, to human and rodent chromosomes by fluorescence in situ hybridization. Cytogenet Cell Genet. 2000;88:38–40. doi: 10.1159/000015481. [DOI] [PubMed] [Google Scholar]
- 32.Arboleda MJ, et al. Overexpression of AKT2/protein kinase Bbeta leads to up-regulation of beta1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 2003;63:196–206. [PubMed] [Google Scholar]
- 33.Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc. Natl Acad. Sci. USA. 2001;98:10983–10985. doi: 10.1073/pnas.211430998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stokoe D, et al. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- 35.Yoshida T, Delafontaine P. Mechanisms of IGF-1-mediated regulation of skeletal muscle hypertrophy and atrophy. Cells. 1970;9:2020. doi: 10.3390/cells9091970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Z, et al. Tetrathiomolybdate treatment leads to the suppression of inflammatory responses through the TRAF6/NFkappaB pathway in LPS-stimulated BV-2 microglia. Front Aging Neurosci. 2018;10:9. doi: 10.3389/fnagi.2018.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spangle JM, Roberts TM. Epigenetic regulation of RTK signaling. J. Mol. Med. 2017;95:791–798. doi: 10.1007/s00109-017-1546-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sangwan V, Park M. Receptor tyrosine kinases: role in cancer progression. Curr. Oncol. 2006;13:191–193. [PMC free article] [PubMed] [Google Scholar]
- 39.Fukuoka M, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) J. Clin. Oncol. 2003;21:2237–2246. doi: 10.1200/JCO.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 40.Arcaro A, et al. Class II phosphoinositide 3-kinases are downstream targets of activated polypeptide growth factor receptors. Mol. Cell Biol. 2000;20:3817–3830. doi: 10.1128/mcb.20.11.3817-3830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jhawer M, et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008;68:1953–1961. doi: 10.1158/0008-5472.CAN-07-5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soltoff SP, et al. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol. Cell Biol. 1994;14:3550–3558. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spano JP, et al. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann. Oncol. 2005;16:102–108. doi: 10.1093/annonc/mdi006. [DOI] [PubMed] [Google Scholar]
- 44.Liu B, et al. Fibroblast growth factor and insulin-like growth factor differentially modulate the apoptosis and G1 arrest induced by anti-epidermal growth factor receptor monoclonal antibody. Oncogene. 2001;20:1913–1922. doi: 10.1038/sj.onc.1204277. [DOI] [PubMed] [Google Scholar]
- 45.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat. Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 46.Byrne AM, Bouchier-Hayes DJ, Harmey JH. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF) J. Cell Mol. Med. 2005;9:777–794. doi: 10.1111/j.1582-4934.2005.tb00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mantha AJ, et al. Targeting the mevalonate pathway inhibits the function of the epidermal growth factor receptor. Clin. Cancer Res. 2005;11:2398–2407. doi: 10.1158/1078-0432.CCR-04-1951. [DOI] [PubMed] [Google Scholar]
- 48.Kou R, Greif D, Michel T. Dephosphorylation of endothelial nitric-oxide synthase by vascular endothelial growth factor. Implications for the vascular responses to cyclosporin A. J. Biol. Chem. 2002;277:29669–29673. doi: 10.1074/jbc.M204519200. [DOI] [PubMed] [Google Scholar]
- 49.Reis-Filho JS, et al. FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin. Cancer Res. 2006;12:6652–6662. doi: 10.1158/1078-0432.CCR-06-1164. [DOI] [PubMed] [Google Scholar]
- 50.Reuter JA, et al. Modeling inducible human tissue neoplasia identifies an extracellular matrix interaction network involved in cancer progression. Cancer Cell. 2009;15:477–488. doi: 10.1016/j.ccr.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dey JH, et al. Targeting fibroblast growth factor receptors blocks PI3K/AKT signaling, induces apoptosis, and impairs mammary tumor outgrowth and metastasis. Cancer Res. 2010;70:4151–4162. doi: 10.1158/0008-5472.CAN-09-4479. [DOI] [PubMed] [Google Scholar]
- 52.Engelman JA, et al. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc. Natl Acad. Sci. USA. 2005;102:3788–3793. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bianco R, et al. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene. 2003;22:2812–2822. doi: 10.1038/sj.onc.1206388. [DOI] [PubMed] [Google Scholar]
- 54.Berns K, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 55.Kaczorowski DJ, et al. Mechanisms of Toll-like receptor 4 (TLR4)-mediated inflammation after cold ischemia/reperfusion in the heart. Transplantation. 2009;87:1455–1463. doi: 10.1097/TP.0b013e3181a36e5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat. Med. 2007;13:552–559. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 57.Fujio Y, et al. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000;101:660–667. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 59.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 60.Szczepanski MJ, et al. Triggering of Toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res. 2009;69:3105–3113. doi: 10.1158/0008-5472.CAN-08-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salaun B, et al. TLR3 can directly trigger apoptosis in human cancer cells. J. Immunol. 2006;176:4894–4901. doi: 10.4049/jimmunol.176.8.4894. [DOI] [PubMed] [Google Scholar]
- 62.Okkenhaug K, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 2002;297:1031–1034. doi: 10.1126/science.1073560. [DOI] [PubMed] [Google Scholar]
- 63.Aiba Y, et al. Regulation of B-cell development by BCAP and CD19 through their binding to phosphoinositide 3-kinase. Blood. 2008;111:1497–1503. doi: 10.1182/blood-2007-08-109769. [DOI] [PubMed] [Google Scholar]
- 64.Castello A, et al. Nck-mediated recruitment of BCAP to the BCR regulates the PI(3)K-Akt pathway in B cells. Nat. Immunol. 2013;14:966–975. doi: 10.1038/ni.2685. [DOI] [PubMed] [Google Scholar]
- 65.Jones N, et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature. 2006;440:818–823. doi: 10.1038/nature04662. [DOI] [PubMed] [Google Scholar]
- 66.Bojarczuk K, et al. BCR signaling inhibitors differ in their ability to overcome Mcl-1-mediated resistance of CLL B cells to ABT-199. Blood. 2016;127:3192–3201. doi: 10.1182/blood-2015-10-675009. [DOI] [PubMed] [Google Scholar]
- 67.Marinissen MJ, Gutkind JS. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharm. Sci. 2001;22:368–376. doi: 10.1016/s0165-6147(00)01678-3. [DOI] [PubMed] [Google Scholar]
- 68.Rozengurt E, Sinnett-Smith J, Kisfalvi K. Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clin. Cancer Res. 2010;16:2505–2511. doi: 10.1158/1078-0432.CCR-09-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu CY, et al. Andrographolide inhibits TNFα-induced ICAM-1 expression via suppression of NADPH oxidase activation and induction of HO-1 and GCLM expression through the PI3K/Akt/Nrf2 and PI3K/Akt/AP-1 pathways in human endothelial cells. Biochem Pharm. 2014;91:40–50. doi: 10.1016/j.bcp.2014.06.024. [DOI] [PubMed] [Google Scholar]
- 70.Ballou LM, et al. Galphaq binds to p110alpha/p85alpha phosphoinositide 3-kinase and displaces Ras. Biochem J. 2006;394:557–562. doi: 10.1042/BJ20051493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Soria JC, et al. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin. Cancer Res. 2002;8:1178–1184. [PubMed] [Google Scholar]
- 72.Stambolic V, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 73.Shi W, et al. Dysregulated PTEN-PKB and negative receptor status in human breast cancer. Int J. Cancer. 2003;104:195–203. doi: 10.1002/ijc.10909. [DOI] [PubMed] [Google Scholar]
- 74.Nunnery SE, Mayer IA. Management of toxicity to isoform α-specific PI3K inhibitors. Ann. Oncol. 2019;30:x21–x26. doi: 10.1093/annonc/mdz440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Papadimitrakopoulou V. Development of PI3K/AKT/mTOR pathway inhibitors and their application in personalized therapy for non-small-cell lung cancer. J. Thorac. Oncol. 2012;7:1315–1326. doi: 10.1097/JTO.0b013e31825493eb. [DOI] [PubMed] [Google Scholar]
- 76.Agoulnik IU, Hodgson MC, Bowden WA, Ittmann MM. INPP4B: the new kid on the PI3K block. Oncotarget. 2011;2:321–328. doi: 10.18632/oncotarget.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 78.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. 2014;4:64. doi: 10.3389/fonc.2014.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–183. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 82.Inoki K, et al. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 83.Fingar DC, et al. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–1487. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Murugan AK. mTOR: Role in cancer, metastasis and drug resistance. Semin Cancer Biol. 2019;59:92–111. doi: 10.1016/j.semcancer.2019.07.003. [DOI] [PubMed] [Google Scholar]
- 85.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 86.Cross DA, et al. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 87.Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 1980;107:519–527. [PubMed] [Google Scholar]
- 88.Kaidanovich-Beilin O, Woodgett JR. GSK-3: Functional insights from cell biology and animal models. Front Mol. Neurosci. 2011;4:40. doi: 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McCubrey JA, et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget. 2014;5:2881–2911. doi: 10.18632/oncotarget.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J. 1993;296(Pt 1):15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405. doi: 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Parker PJ, Caudwell FB, Cohen P. Glycogen synthase from rabbit skeletal muscle; effect of insulin on the state of phosphorylation of the seven phosphoserine residues in vivo. Eur. J. Biochem. 1983;130:227–234. doi: 10.1111/j.1432-1033.1983.tb07140.x. [DOI] [PubMed] [Google Scholar]
- 93.Acikgoz E, et al. Glycogen synthase kinase-3 inhibition in glioblastoma multiforme cells induces apoptosis, cell cycle arrest and changing biomolecular structure. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019;209:150–164. doi: 10.1016/j.saa.2018.10.036. [DOI] [PubMed] [Google Scholar]
- 94.van der Vos KE, Coffer PJ. The extending network of FOXO transcriptional target genes. Antioxid. Redox Signal. 2011;14:579–592. doi: 10.1089/ars.2010.3419. [DOI] [PubMed] [Google Scholar]
- 95.Fu Z, Tindall DJ. FOXOs, cancer and regulation of apoptosis. Oncogene. 2008;27:2312–2319. doi: 10.1038/onc.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ma J, Matkar S, He X, Hua X. FOXO family in regulating cancer and metabolism. Semin Cancer Biol. 2018;50:32–41. doi: 10.1016/j.semcancer.2018.01.018. [DOI] [PubMed] [Google Scholar]
- 97.Matsuzaki H, et al. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc. Natl Acad. Sci. USA. 2003;100:11285–11290. doi: 10.1073/pnas.1934283100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Paradis S, Ruvkun G. Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev. 1998;12:2488–2498. doi: 10.1101/gad.12.16.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alessi DR, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. Embo J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 100.Guertin DA, et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 101.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N. Engl. J. Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 102.Manning BD, Cantley LC. Rheb fills a GAP between TSC and TOR. Trends Biochem Sci. 2003;28:573–576. doi: 10.1016/j.tibs.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 103.Schwartz D, Rotter V. p53-dependent cell cycle control: response to genotoxic stress. Semin Cancer Biol. 1998;8:325–336. doi: 10.1006/scbi.1998.0095. [DOI] [PubMed] [Google Scholar]
- 104.Abraham AG, O’Neill E. PI3K/Akt-mediated regulation of p53 in cancer. Biochem Soc. Trans. 2014;42:798–803. doi: 10.1042/BST20140070. [DOI] [PubMed] [Google Scholar]
- 105.Proctor CJ, Gray DA. Explaining oscillations and variability in the p53-Mdm2 system. BMC Syst. Biol. 2008;2:75. doi: 10.1186/1752-0509-2-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Momand J, et al. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 107.Chang H, et al. Luteolin prevents H2O2-induced apoptosis in H9C2 cells through modulating Akt-P53/Mdm2 signaling pathway. Biomed. Res. Int. 2016;2016:5125836. doi: 10.1155/2016/5125836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl Acad. Sci. USA. 2001;98:11598–11603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhou BP, et al. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell Biol. 2001;3:973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- 110.Vecino R, et al. The MDM2-p53 pathway is involved in preconditioning-induced neuronal tolerance to ischemia. Sci. Rep. 2018;8:1610. doi: 10.1038/s41598-018-19921-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lauth M. RAS and Hedgehog–partners in crime. Front Biosci. 2011;16:2259–2270. doi: 10.2741/3852. [DOI] [PubMed] [Google Scholar]
- 112.Frémin C, Meloche S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J. Hematol. Oncol. 2010;3:8. doi: 10.1186/1756-8722-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Niederst MJ, Engelman JA. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci. Signal. 2013;6:re6. doi: 10.1126/scisignal.2004652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Toulany M, et al. ERK2-dependent reactivation of Akt mediates the limited response of tumor cells with constitutive K-RAS activity to PI3K inhibition. Cancer Biol. Ther. 2014;15:317–328. doi: 10.4161/cbt.27311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Reardon DB, et al. Dominant negative EGFR-CD533 and inhibition of MAPK modify JNK1 activation and enhance radiation toxicity of human mammary carcinoma cells. Oncogene. 1999;18:4756–4766. doi: 10.1038/sj.onc.1202849. [DOI] [PubMed] [Google Scholar]
- 116.Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-kappaB by the Akt/PKB kinase. Curr. Biol. 1999;9:601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- 117.Li B, Li YY, Tsao SW, Cheung AL. Targeting NF-kappaB signaling pathway suppresses tumor growth, angiogenesis, and metastasis of human esophageal cancer. Mol. Cancer Ther. 2009;8:2635–2644. doi: 10.1158/1535-7163.MCT-09-0162. [DOI] [PubMed] [Google Scholar]
- 118.Ozes ON, et al. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 119.Li Z, et al. A positive feedback loop involving EGFR/Akt/mTORC1 and IKK/NF-kB regulates head and neck squamous cell carcinoma proliferation. Oncotarget. 2016;7:31892–31906. doi: 10.18632/oncotarget.7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cheng X, et al. Therapeutic potential of targeting the Wnt/β-catenin signaling pathway in colorectal cancer. Biomed. Pharmacother. 2019;110:473–481. doi: 10.1016/j.biopha.2018.11.082. [DOI] [PubMed] [Google Scholar]
- 121.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 122.Gottardi CJ, Gumbiner BM. Role for ICAT in beta-catenin-dependent nuclear signaling and cadherin functions. Am. J. Physiol. Cell Physiol. 2004;286:C747–C756. doi: 10.1152/ajpcell.00433.2003. [DOI] [PubMed] [Google Scholar]
- 123.Siddharth S, et al. Nectin-4 is a breast cancer stem cell marker that induces WNT/β-catenin signaling via Pi3k/Akt axis. Int J. Biochem Cell Biol. NLM. 2017;89:85–94. doi: 10.1016/j.biocel.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 124.Kang DW, et al. Phospholipase D1 inhibition linked to upregulation of ICAT blocks colorectal cancer growth hyperactivated by Wnt/β-Catenin and PI3K/Akt signaling. Clin. Cancer Res. 2017;23:7340–7350. doi: 10.1158/1078-0432.CCR-17-0749. [DOI] [PubMed] [Google Scholar]
- 125.Han F, et al. The critical role of AMPK in driving Akt activation under stress, tumorigenesis and drug resistance. Nat. Commun. 2018;9:4728. doi: 10.1038/s41467-018-07188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yan Y, Huang H. Interplay among PI3K/AKT, PTEN/FOXO and AR signaling in prostate cancer. Adv. Exp. Med Biol. 2019;1210:319–331. doi: 10.1007/978-3-030-32656-2_14. [DOI] [PubMed] [Google Scholar]
- 127.Greer EL, Brunet A. FOXO transcription factors in ageing and cancer. Acta Physiol. 2008;192:19–28. doi: 10.1111/j.1748-1716.2007.01780.x. [DOI] [PubMed] [Google Scholar]
- 128.Ma YS, et al. KDM5A silencing transcriptionally suppresses the FXYD3-PI3K/AKT axis to inhibit angiogenesis in hepatocellular cancer via miR-433 up-regulation. J Cell Mol. Med. 2021;25:4040–4052. doi: 10.1111/jcmm.16371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shin I, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat. Med. 2002;8:1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- 130.Xu WW, et al. IGF2 induces CD133 expression in esophageal cancer cells to promote cancer stemness. Cancer Lett. 2018;425:88–100. doi: 10.1016/j.canlet.2018.03.039. [DOI] [PubMed] [Google Scholar]
- 131.Huang Y, et al. LncRNA TDRG1 promotes proliferation, invasion and epithelial-mesenchymal transformation of osteosarcoma through PI3K/AKT signal pathway. Cancer Manag. Res. 2020;12:4531–4540. doi: 10.2147/CMAR.S248964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.D’Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019;43:582–592. doi: 10.1002/cbin.11137. [DOI] [PubMed] [Google Scholar]
- 133.Datta SR, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 134.Chen X, et al. Constitutively active Akt is an important regulator of TRAIL sensitivity in prostate cancer. Oncogene. 2001;20:6073–6083. doi: 10.1038/sj.onc.1204736. [DOI] [PubMed] [Google Scholar]
- 135.Rozengurt E, Soares HP, Sinnet-Smith J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol. Cancer Ther. 2014;13:2477–2488. doi: 10.1158/1535-7163.MCT-14-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Leroy C, et al. Activation of IGF1R/p110β/AKT/mTOR confers resistance to α-specific PI3K inhibition. Breast Cancer Res. 2016;18:41. doi: 10.1186/s13058-016-0697-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001;61:3986–3997. [PubMed] [Google Scholar]
- 138.St Croix B, Man S, Kerbel RS. Reversal of intrinsic and acquired forms of drug resistance by hyaluronidase treatment of solid tumors. Cancer Lett. 1998;131:35–44. doi: 10.1016/s0304-3835(98)00199-2. [DOI] [PubMed] [Google Scholar]
- 139.Vives M, et al. Metronomic chemotherapy following the maximum tolerated dose is an effective anti-tumour therapy affecting angiogenesis, tumour dissemination and cancer stem cells. Int J. Cancer. 2013;133:2464–2472. doi: 10.1002/ijc.28259. [DOI] [PubMed] [Google Scholar]
- 140.Liu LZ, et al. AKT1 amplification regulates cisplatin resistance in human lung cancer cells through the mammalian target of rapamycin/p70S6K1 pathway. Cancer Res. 2007;67:6325–6332. doi: 10.1158/0008-5472.CAN-06-4261. [DOI] [PubMed] [Google Scholar]
- 141.Li B, et al. Competitive binding between Id1 and E2F1 to Cdc20 regulates E2F1 degradation and thymidylate synthase expression to promote esophageal cancer chemoresistance. Clin. Cancer Res. 2016;22:1243–1255. doi: 10.1158/1078-0432.CCR-15-1196. [DOI] [PubMed] [Google Scholar]
- 142.Poh TW, Pervaiz S. LY294002 and LY303511 sensitize tumor cells to drug-induced apoptosis via intracellular hydrogen peroxide production independent of the phosphoinositide 3-kinase-Akt pathway. Cancer Res. 2005;65:6264–6274. doi: 10.1158/0008-5472.CAN-05-0152. [DOI] [PubMed] [Google Scholar]
- 143.Stoica GE, et al. Effect of estradiol on estrogen receptor-alpha gene expression and activity can be modulated by the ErbB2/PI 3-K/Akt pathway. Oncogene. 2003;22:7998–8011. doi: 10.1038/sj.onc.1206769. [DOI] [PubMed] [Google Scholar]
- 144.Syn NL, Teng MWL, Mok TSK, Soo RA. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017;18:e731–e741. doi: 10.1016/S1470-2045(17)30607-1. [DOI] [PubMed] [Google Scholar]
- 145.Parsa AT, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007;13:84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 146.Lastwika KJ, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016;76:227–238. doi: 10.1158/0008-5472.CAN-14-3362. [DOI] [PubMed] [Google Scholar]
- 147.Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer. 2020;20:74–88. doi: 10.1038/s41568-019-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hornsveld M, Dansen TB, Derksen PW, Burgering BMT. Re-evaluating the role of FOXOs in cancer. Semin Cancer Biol. 2018;50:90–100. doi: 10.1016/j.semcancer.2017.11.017. [DOI] [PubMed] [Google Scholar]
- 149.Brunet A, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 150.Gozzelino L, et al. PI(3,4)P2 signaling in cancer and metabolism. Front. Oncol. 2020;10:360. doi: 10.3389/fonc.2020.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chen J, et al. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat. Med. 2005;11:1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lee MY, et al. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc. Natl Acad. Sci. USA. 2014;111:12865–12870. doi: 10.1073/pnas.1408472111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ackah E, et al. Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. J. Clin. Invest. 2005;115:2119–2127. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Phung TL, et al. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell. 2006;10:159–170. doi: 10.1016/j.ccr.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Soler A, et al. Inhibition of the p110α isoform of PI3-kinase stimulates nonfunctional tumor angiogenesis. J. Exp. Med. 2013;210:1937–1945. doi: 10.1084/jem.20121571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Xu WW, et al. Direct targeting of CREB1 with imperatorin inhibits TGFbeta2-ERK signaling to suppress esophageal cancer metastasis. Adv. Sci. 2020;7:2000925. doi: 10.1002/advs.202000925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Grille SJ, et al. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. 2003;63:2172–2178. [PubMed] [Google Scholar]
- 158.Steelman LS, et al. Akt as a therapeutic target in cancer. Expert Opin. Ther. Targets. 2008;12:1139–1165. doi: 10.1517/14728222.12.9.1139. [DOI] [PubMed] [Google Scholar]
- 159.Kerbel RS. Tumor angiogenesis. N. Engl. J. Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Katso R, et al. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev. Cell Dev. Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- 161.Zhang R, et al. Etk/Bmx transactivates vascular endothelial growth factor 2 and recruits phosphatidylinositol 3-kinase to mediate the tumor necrosis factor-induced angiogenic pathway. J. Biol. Chem. 2003;278:51267–51276. doi: 10.1074/jbc.M310678200. [DOI] [PubMed] [Google Scholar]
- 162.Kanaki T, et al. Functional analysis of aortic endothelial cells expressing mutant PDGF receptors with respect to expression of matrix metalloproteinase-3. Biochem. Biophys. Res. Commun. 2002;294:231–237. doi: 10.1016/S0006-291X(02)00468-0. [DOI] [PubMed] [Google Scholar]
- 163.Thiel A, et al. Expression of cyclooxygenase-2 is regulated by glycogen synthase kinase-3beta in gastric cancer cells. J. Biol. Chem. 2006;281:4564–4569. doi: 10.1074/jbc.M512722200. [DOI] [PubMed] [Google Scholar]
- 164.Dillon RL, et al. Akt1 and akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression. Cancer Res. 2009;69:5057–5064. doi: 10.1158/0008-5472.CAN-08-4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hutchinson JN, et al. Activation of Akt-1 (PKB-alpha) can accelerate ErbB-2-mediated mammary tumorigenesis but suppresses tumor invasion. Cancer Res. 2004;64:3171–3178. doi: 10.1158/0008-5472.can-03-3465. [DOI] [PubMed] [Google Scholar]
- 166.Riggio M, et al. AKT1 and AKT2 isoforms play distinct roles during breast cancer progression through the regulation of specific downstream proteins. Sci. Rep. 2017;7:44244. doi: 10.1038/srep44244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 168.Allin KH, Bojesen SE, Nordestgaard BG. Inflammatory biomarkers and risk of cancer in 84,000 individuals from the general population. Int J. Cancer. 2016;139:1493–1500. doi: 10.1002/ijc.30194. [DOI] [PubMed] [Google Scholar]
- 169.Xue G, et al. Integrated Akt/PKB signaling in immunomodulation and its potential role in cancer immunotherapy. J. Natl. Cancer Inst. 2015;107:djv171. doi: 10.1093/jnci/djv171. [DOI] [PubMed] [Google Scholar]
- 170.Eräsalo H, et al. Natural stilbenoids have anti-inflammatory properties in vivo and down-regulate the production of inflammatory mediators NO, IL6, and MCP1 possibly in a PI3K/Akt-dependent manner. J. Nat. Prod. 2018;81:1131–1142. doi: 10.1021/acs.jnatprod.7b00384. [DOI] [PubMed] [Google Scholar]
- 171.Ribatti D, Crivellato E. Immune cells and angiogenesis. J. Cell Mol. Med. 2009;13:2822–2833. doi: 10.1111/j.1582-4934.2009.00810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Shi Y, et al. Signal pathways involved in activation of p70S6K and phosphorylation of 4E-BP1 following exposure of multiple myeloma tumor cells to interleukin-6. J. Biol. Chem. 2002;277:15712–15720. doi: 10.1074/jbc.M200043200. [DOI] [PubMed] [Google Scholar]
- 173.Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic. Biol. Med. 2010;49:1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Huo R, et al. Cabazitaxel-induced autophagy via the PI3K/Akt/mTOR pathway contributes to A549 cell death. Mol. Med. Rep. 2016;14:3013–3020. doi: 10.3892/mmr.2016.5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Morbidelli L, Donnini S, Ziche M. Role of nitric oxide in the modulation of angiogenesis. Curr. Pharm. Des. 2003;9:521–530. doi: 10.2174/1381612033391405. [DOI] [PubMed] [Google Scholar]
- 176.Solana R, et al. Innate immunosenescence: effect of aging on cells and receptors of the innate immune system in humans. Semin Immunol. 2012;24:331–341. doi: 10.1016/j.smim.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 177.Soond DR, et al. PI3K p110delta regulates T-cell cytokine production during primary and secondary immune responses in mice and humans. Blood. 2010;115:2203–2213. doi: 10.1182/blood-2009-07-232330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Marshall NA, et al. Immunotherapy with PI3K inhibitor and Toll-like receptor agonist induces IFN-γ+IL-17+ polyfunctional T cells that mediate rejection of murine tumors. Cancer Res. 2012;72:581–591. doi: 10.1158/0008-5472.CAN-11-0307. [DOI] [PubMed] [Google Scholar]
- 179.Covarrubias AJ, Aksoylar HI, Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol. 2015;27:286–296. doi: 10.1016/j.smim.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Weichhart T, Hengstschläger M, Linke M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015;15:599–614. doi: 10.1038/nri3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Guo H, Samarakoon A, Vanhaesebroeck B, Malarkannan S. The p110 delta of PI3K plays a critical role in NK cell terminal maturation and cytokine/chemokine generation. J. Exp. Med. 2008;205:2419–2435. doi: 10.1084/jem.20072327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.So L, Fruman DA. PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem J. 2012;442:465–481. doi: 10.1042/BJ20112092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fruman DA, et al. The PI3K pathway in human disease. Cell. 2017;170:605–635. doi: 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Saito K, et al. BTK regulates PtdIns-4,5-P2 synthesis: importance for calcium signaling and PI3K activity. Immunity. 2003;19:669–678. doi: 10.1016/s1074-7613(03)00297-8. [DOI] [PubMed] [Google Scholar]
- 185.Limon JJ, Fruman DA. Akt and mTOR in B cell activation and differentiation. Front Immunol. 2012;3:228. doi: 10.3389/fimmu.2012.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Chang CH, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.van der Waart AB, et al. Inhibition of Akt signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood. 2014;124:3490–3500. doi: 10.1182/blood-2014-05-578583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Xu WW, et al. Cancer cell-secreted IGF2 instigates fibroblasts and bone marrow-derived vascular progenitor cells to promote cancer progression. Nat. Commun. 2017;8:14399. doi: 10.1038/ncomms14399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yu M, Tannock IF. Targeting tumor architecture to favor drug penetration: a new weapon to combat chemoresistance in pancreatic cancer? Cancer Cell. 2012;21:327–329. doi: 10.1016/j.ccr.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 190.Meng Q, et al. Role of PI3K and AKT specific isoforms in ovarian cancer cell migration, invasion and proliferation through the p70S6K1 pathway. Cell Signal. 2006;18:2262–2271. doi: 10.1016/j.cellsig.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 191.Zhou B, et al. Cisplatin-induced CCL5 secretion from CAFs promotes cisplatin-resistance in ovarian cancer via regulation of the STAT3 and PI3K/Akt signaling pathways. Int J. Oncol. 2016;48:2087–2097. doi: 10.3892/ijo.2016.3442. [DOI] [PubMed] [Google Scholar]
- 192.Han Y, Wang X, Wang B, Jiang G. The progress of angiogenic factors in the development of leukemias. Intractable Rare Dis. Res. 2016;5:6–16. doi: 10.5582/irdr.2015.01048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Okkenhaug K, Graupera M, Vanhaesebroeck B. Targeting PI3K in cancer: impact on tumor cells, their protective stroma, angiogenesis, and immunotherapy. Cancer Disco. 2016;6:1090–1105. doi: 10.1158/2159-8290.CD-16-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.De Santis MC, et al. Targeting PI3K signaling in cancer: challenges and advances. Biochim Biophys. Acta Rev. Cancer. 2019;1871:361–366. doi: 10.1016/j.bbcan.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 195.Akinleye A, et al. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J. Hematol. Oncol. 2013;6:88. doi: 10.1186/1756-8722-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Folkes AJ, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med Chem. 2008;51:5522–5532. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 197.Junttila TT, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15:429–440. doi: 10.1016/j.ccr.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 198.Burger MT, et al. Identification of NVP-BKM120 as a potent, selective, orally bioavailable class I PI3 kinase inhibitor for treating cancer. ACS Med. Chem. Lett. 2011;2:774–779. doi: 10.1021/ml200156t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Netland IA, et al. Treatment with the PI3K inhibitor buparlisib (NVP-BKM120) suppresses the growth of established patient-derived GBM xenografts and prolongs survival in nude rats. J. Neurooncol. 2016;129:57–66. doi: 10.1007/s11060-016-2158-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bendell JC, et al. Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2012;30:282–290. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 201.Foster P, et al. The selective PI3K inhibitor XL147 (SAR245408) inhibits tumor growth and survival and potentiates the activity of chemotherapeutic agents in preclinical tumor models. Mol. Cancer Ther. 2015;14:931–940. doi: 10.1158/1535-7163.MCT-14-0833. [DOI] [PubMed] [Google Scholar]
- 202.Li M, et al. Phosphoinositide 3-kinase gamma inhibition protects from anthracycline cardiotoxicity and reduces tumor growth. Circulation. 2018;138:696–711. doi: 10.1161/CIRCULATIONAHA.117.030352. [DOI] [PubMed] [Google Scholar]
- 203.Juric D, et al. Phosphatidylinositol 3-kinase α-selective inhibition with Alpelisib (BYL719) in PIK3CA-altered solid tumors: results from the first-in-human study. J. Clin. Oncol. 2018;36:1291–1299. doi: 10.1200/JCO.2017.72.7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Juric D, et al. Alpelisib plus Fulvestrant in PIK3CA-altered and PIK3CA-wild-type estrogen receptor-positive advanced breast cancer: a phase 1b clinical trial. JAMA Oncol. 2019;5:e184475. doi: 10.1001/jamaoncol.2018.4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.André F, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 2019;380:1929–1940. doi: 10.1056/NEJMoa1813904. [DOI] [PubMed] [Google Scholar]
- 206.Ndubaku, C. O. et al. Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): a β-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J. Med. Chem. 56, 4597–4610 (2013). [DOI] [PubMed]
- 207.Juric D, et al. Phase I dose-escalation study of Taselisib, an oral PI3K inhibitor, in patients with advanced solid tumors. Cancer Disco. 2017;7:704–715. doi: 10.1158/2159-8290.CD-16-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Verret B, et al. Efficacy of PI3K inhibitors in advanced breast cancer. Ann. Oncol. 2019;30:x12–x20. doi: 10.1093/annonc/mdz381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.O’Reilly KE, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hirai H, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010;9:1956–1967. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 211.Lara PN, Jr., et al. Phase II study of the AKT inhibitor MK-2206 plus Erlotinib in patients with advanced non-small cell lung cancer who previously progressed on Erlotinib. Clin. Cancer Res. 2015;21:4321–4326. doi: 10.1158/1078-0432.CCR-14-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Konopleva MY, et al. Preclinical and early clinical evaluation of the oral AKT inhibitor, MK-2206, for the treatment of acute myelogenous leukemia. Clin. Cancer Res. 2014;20:2226–2235. doi: 10.1158/1078-0432.CCR-13-1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ma BB, et al. Preclinical evaluation of the AKT inhibitor MK-2206 in nasopharyngeal carcinoma cell lines. Invest N. Drugs. 2013;31:567–575. doi: 10.1007/s10637-012-9896-5. [DOI] [PubMed] [Google Scholar]
- 214.Yap TA, et al. First-in-class phase I trial of a selective Akt inhibitor, MK2206 (MK), evaluating alternate day (QOD) and once weekly (QW) doses in advanced cancer patients (pts) with evidence of target modulation and antitumor activity. J. Clin. Oncol. 2010;28:3009–3009. [Google Scholar]
- 215.Davies BR, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol. Cancer Ther. 2012;11:873–887. doi: 10.1158/1535-7163.MCT-11-0824-T. [DOI] [PubMed] [Google Scholar]
- 216.Li J, et al. The AKT inhibitor AZD5363 is selectively active in PI3KCA mutant gastric cancer, and sensitizes a patient-derived gastric cancer xenograft model with PTEN loss to Taxotere. J. Transl. Med. 2013;11:241. doi: 10.1186/1479-5876-11-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Menon S, Manning BD. Common corruption of the mTOR signaling network in human tumors. Oncogene. 2008;27(Suppl 2):S43–S51. doi: 10.1038/onc.2009.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Popova NV, Jucker M, et al. The role of mTOR signaling as a therapeutic target in cancer. Int. J. Mol. Sci. 2021;22:1743. doi: 10.3390/ijms22041743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Mukhopadhyay S, et al. The enigma of Rapamycin dosage. Mol. Cancer Ther. 2016;15:347–353. doi: 10.1158/1535-7163.MCT-15-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Chresta CM, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70:288–298. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 221.Zhang H, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J. Clin. Invest. 2007;117:2051–2058. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Zambrano CC, et al. Phase lb study of buparlisib (BKM120) plus either paclitaxel (PTX) in advanced solid tumors (aST) or PTX plus trastuzumab (TZ) in HER2+ breast cancer (BC) J. Clin. Oncol. 2014;32:627–627. [Google Scholar]
- 223.Razak ARA, et al. Phase lb/ll study of the PI3Kα inhibitor BYL719 in combination with cetuximab in recurrent/metastatic squamous cell cancer of the head and neck (SCCHN) J. Clin. Oncol. 2014;32:6044–6044. [Google Scholar]
- 224.Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother. Pharm. 2013;71:1395–1409. doi: 10.1007/s00280-013-2121-1. [DOI] [PubMed] [Google Scholar]
- 225.Shimizu T, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin. Cancer Res. 2012;18:2316–2325. doi: 10.1158/1078-0432.CCR-11-2381. [DOI] [PubMed] [Google Scholar]
- 226.LoRusso P, et al. A first-in-human phase Ib study to evaluate the MEK inhibitor GDC-0973, combined with the pan-PI3K inhibitor GDC-0941, in patients with advanced solid tumors. J. Clin. Oncol. 2012;30:2566–2566. [Google Scholar]
- 227.Heist RS, et al. Combination of a MEK inhibitor, pimasertib (MSC1936369B), and a PI3K/mTOR inhibitor, SAR245409, in patients with advanced solid tumors: Results of a phase Ib dose-escalation trial. J. Clin. Oncol. 2013;31:2530–2530. [Google Scholar]
- 228.Bedard PL, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin. Cancer Res. 2015;21:730–738. doi: 10.1158/1078-0432.CCR-14-1814. [DOI] [PubMed] [Google Scholar]
- 229.Bender A, et al. PI3K inhibitors prime neuroblastoma cells for chemotherapy by shifting the balance towards pro-apoptotic Bcl-2 proteins and enhanced mitochondrial apoptosis. Oncogene. 2010;30:494–503. doi: 10.1038/onc.2010.429. [DOI] [PubMed] [Google Scholar]
- 230.Bang Y-J, et al. JAGUAR: A randomized phase II study of the AKT inhibitor ipatasertib (GDC-0068) versus placebo in combination with mFOLFOX6 chemotherapy in patients (pts) with locally advanced or metastatic HER2-negative gastric (G) or gastroesophageal junction (GEJ) adenocarcinoma. J. Clin. Oncol. 2014;32:TPS4147–TPS4147. [Google Scholar]
- 231.Ibrahim YH, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Disco. 2012;2:1036–1047. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Matulonis U, et al. Phase I study of oral BKM120 and oral olaparib for high-grade serous ovarian cancer (HGSC) or triple-negative breast cancer (TNBC) J. Clin. Oncol. 2014;32:2510–2510. [Google Scholar]
- 233.Verellen D, et al. Innovations in image-guided radiotherapy. Nat. Rev. Cancer. 2007;7:949–960. doi: 10.1038/nrc2288. [DOI] [PubMed] [Google Scholar]
- 234.Keam B, et al. In vitro anticancer activity of PI3K alpha selective inhibitor BYL719 in head and neck cancer. Anticancer Res. 2015;35:175–182. [PubMed] [Google Scholar]
- 235.Burris HA., 3rd Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother. Pharm. 2013;71:829–842. doi: 10.1007/s00280-012-2043-3. [DOI] [PubMed] [Google Scholar]
- 236.Chang L, et al. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014;5:e1437. doi: 10.1038/cddis.2014.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.McGowan DR, et al. Buparlisib with thoracic radiotherapy and its effect on tumour hypoxia: a phase I study in patients with advanced non-small cell lung carcinoma. Eur. J. Cancer. 2019;113:87–95. doi: 10.1016/j.ejca.2019.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 239.Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 2009;9:324–337. doi: 10.1038/nri2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Jiang Q, et al. mTOR kinase inhibitor AZD8055 enhances the immunotherapeutic activity of an agonist CD40 antibody in cancer treatment. Cancer Res. 2011;71:4074–4084. doi: 10.1158/0008-5472.CAN-10-3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Perkins MR, et al. Manufacturing an enhanced CAR T cell product by inhibition of the PI3K/Akt pathway during T Cell expansion results in improved in vivo efficacy of anti-BCMA CAR T cells. Blood. 2015;126:1893–1893. [Google Scholar]
- 243.Morii Y, et al. Perifosine enhances the potential antitumor effect of 5-fluorourasil and oxaliplatin in colon cancer cells harboring the PIK3CA mutation. Eur. J. Pharm. 2021;898:173957. doi: 10.1016/j.ejphar.2021.173957. [DOI] [PubMed] [Google Scholar]
- 244.Di Cristofano A. SGK1: the dark side of PI3K signaling. Curr. Top. Dev. Biol. 2017;123:49–71. doi: 10.1016/bs.ctdb.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Chen S, et al. BMX-mediated regulation of multiple tyrosine kinases contributes to castration resistance in prostate cancer. Cancer Res. 2018;78:5203–5215. doi: 10.1158/0008-5472.CAN-17-3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Reynolds LF, et al. Vav1 transduces T cell receptor signals to the activation of phospholipase C-gamma1 via phosphoinositide 3-kinase-dependent and -independent pathways. J. Exp. Med. 2002;195:1103–1114. doi: 10.1084/jem.20011663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Hu H, et al. Phosphoinositide 3-kinase regulates glycolysis through mobilization of aldolase from the actin cytoskeleton. Cell. 2016;164:433–446. doi: 10.1016/j.cell.2015.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Cain RJ, Vanhaesebroeck B, Ridley AJ. The PI3K p110alpha isoform regulates endothelial adherens junctions via Pyk2 and Rac1. J. Cell Biol. 2010;188:863–876. doi: 10.1083/jcb.200907135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Arana E, et al. Activation of the small GTPase Rac2 via the B cell receptor regulates B cell adhesion and immunological-synapse formation. Immunity. 2008;28:88–99. doi: 10.1016/j.immuni.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 250.Cheng S, et al. PtdIns(4,5)P2 and PtdIns3P coordinate to regulate phagosomal sealing for apoptotic cell clearance. J. Cell Biol. 2015;210:485–502. doi: 10.1083/jcb.201501038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Cheng S, et al. Autophagy genes coordinate with the class II PI/PtdIns 3-kinase PIKI-1 to regulate apoptotic cell clearance in C. elegans. Autophagy. 2013;9:2022–2032. doi: 10.4161/auto.26323. [DOI] [PubMed] [Google Scholar]
- 252.Zou W, et al. Caenorhabditis elegans myotubularin MTM-1 negatively regulates the engulfment of apoptotic cells. PLoS Genet. 2009;5:e1000679. doi: 10.1371/journal.pgen.1000679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Gasser JA, et al. SGK3 mediates INPP4B-dependent PI3K signaling in breast cancer. Mol. Cell. 2014;56:595–607. doi: 10.1016/j.molcel.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Sarker D, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015;21:77–86. doi: 10.1158/1078-0432.CCR-14-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Armstrong AJ, et al. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM-120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur. J. Cancer. 2017;81:228–236. doi: 10.1016/j.ejca.2017.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Patnaik A, et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann. Oncol. 2016;27:1928–1940. doi: 10.1093/annonc/mdw282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Edelman G, et al. A phase I dose-escalation study of XL147 (SAR245408), a PI3K inhibitor administered orally to patients (pts) with advanced malignancies. J. Clin. Oncol. 2010;28:3004–3004. [Google Scholar]
- 258.Hotte SJ, et al. A phase II study of PX-866 in patients with recurrent or metastatic castration-resistant prostate cancer: Canadian cancer trials group study IND205. Clin. Genitourin. Cancer. 2019;17:201–208.e201. doi: 10.1016/j.clgc.2019.03.005. [DOI] [PubMed] [Google Scholar]
- 259.Fritsch C, et al. Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol. Cancer Ther. 2014;13:1117–1129. doi: 10.1158/1535-7163.MCT-13-0865. [DOI] [PubMed] [Google Scholar]
- 260.Dunbar J, et al. Pharmacokinetics and pharmacodynamics of IPI-145, a potent inhibitor of phosphoinositide-3-kinase-δ,γ, following single- and multiple-dose administration in healthy subjects and patients with advanced hematologic malignancies. Blood. 2012;120:4853–4853. [Google Scholar]
- 261.Bédard PL, et al. First-in-human trial of the PI3Kβ-selective inhibitor SAR260301 in patients with advanced solid tumors. Cancer. 2018;124:315–324. doi: 10.1002/cncr.31044. [DOI] [PubMed] [Google Scholar]
- 262.Juric D, et al. A first-in-human, phase I, dose-escalation study of TAK-117, a selective PI3Kα isoform inhibitor, in patients with advanced solid malignancies. Clin. Cancer Res. 2017;23:5015–5023. doi: 10.1158/1078-0432.CCR-16-2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Peyton JD, et al. A dose-escalation study with the novel formulation of the oral pan-class I PI3K inhibitor BEZ235, solid dispersion system (SDS) sachet, in patients with advanced solid tumors. J. Clin. Oncol. 2011;29:3066–3066. [Google Scholar]
- 264.Mahadevan D, et al. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur. J. Cancer. 2012;48:3319–3327. doi: 10.1016/j.ejca.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Wagner AJ, et al. A first-in-human phase I study to evaluate GDC-0980, an oral PI3K/mTOR inhibitor, administered QD in patients with advanced solid tumors. J. Clin. Oncol. 2011;29:3020–3020. [Google Scholar]
- 266.Del Campo JM, et al. A randomized phase II non-comparative study of PF-04691502 and gedatolisib (PF-05212384) in patients with recurrent endometrial cancer. Gynecol. Oncol. 2016;142:62–69. doi: 10.1016/j.ygyno.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 267.Brana I, et al. A phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics of XL765 (SAR245409), a PI3K/TORC1/TORC2 inhibitor administered orally to patients (pts) with advanced malignancies. J. Clin. Oncol. 2010;28:3030–3030. [Google Scholar]
- 268.Munster P, et al. First-in-human phase I study of GSK2126458, an oral pan-class I phosphatidylinositol-3-kinase inhibitor, in patients with advanced solid tumor malignancies. Clin. Cancer Res. 2016;22:1932–1939. doi: 10.1158/1078-0432.CCR-15-1665. [DOI] [PubMed] [Google Scholar]
- 269.Garlich JR, et al. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res. 2008;68:206–215. doi: 10.1158/0008-5472.CAN-07-0669. [DOI] [PubMed] [Google Scholar]
- 270.Minami H, et al. Phase I study of BGT226, a pan-PI3K and mTOR inhibitor, in Japanese patients with advanced solid cancers. Cancer Chemother. Pharm. 2019;84:337–343. doi: 10.1007/s00280-019-03883-6. [DOI] [PubMed] [Google Scholar]
- 271.Argiris A, et al. A phase II trial of perifosine, an oral alkylphospholipid, in recurrent or metastatic head and neck cancer. Cancer Biol. Ther. 2006;5:766–770. doi: 10.4161/cbt.5.7.2874. [DOI] [PubMed] [Google Scholar]
- 272.Rhodes N, et al. Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res. 2008;68:2366–2374. doi: 10.1158/0008-5472.CAN-07-5783. [DOI] [PubMed] [Google Scholar]
- 273.Sampath D, et al. Phase I clinical, pharmacokinetic, and pharmacodynamic study of the Akt-inhibitor triciribine phosphate monohydrate in patients with advanced hematologic malignancies. Leuk. Res. 2013;37:1461–1467. doi: 10.1016/j.leukres.2013.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Machl A, et al. M2698 is a potent dual-inhibitor of p70S6K and Akt that affects tumor growth in mouse models of cancer and crosses the blood-brain barrier. Am. J. Cancer Res. 2016;6:806–818. [PMC free article] [PubMed] [Google Scholar]