

Abstract
This review is focused on genetic regulators of bleeding and thrombosis with a focus on next‐generation sequencing (NGS) technologies for diagnosis and research of patients with inherited disorders. The molecular diagnosis of hemostatic phenotypes relies on the detection of genetic variants in the 99 curated disease‐causing genes implicated for bleeding, platelet, and thrombotic disorders through the use of multigene panel tests. In this review, we will provide an overview of the advantages and disadvantages of using such multigene panel tests for diagnostics. During the past decade, NGS technologies have also been used for the gene discovery of 32 novel genes involved in inherited hemostatic phenotypes. We will provide a brief overview of these genes and discuss what information (eg, linkage, consanguinity, multiple index cases with similar phenotypes, mouse models, and more) was used to support the gene discovery process. Next, we provide examples on how RNA sequencing is useful to explore disease mechanisms of novel and often unexpected genes. This review will summarize the important findings concerning NGS technologies for diagnostics and gene discovery that were presented at the ISTH 2021 conference. Finally, future perspectives in our field mainly deal with finding the needle in the haystack for some still unexplained patients and the need for exploring the noncoding gene space and rapid disease validation models.
Essentials.
Gene panel tests are used for diagnostics of inherited bleeding and thrombotic disorders.
International data sharing is essential for variant curation and gene discovery studies.
Next‐generation sequencing (NGS) contributes to gene discovery, but data interpretation remains difficult.
Data from the ISTH 2021 congress illustrates the added value of NGS methods.
1. INTRODUCTION
Pathways involved in hemostasis are very complex and involve interplays between platelets, coagulation, and fibrinolysis. 1 Their normal function is required to prevent excessive blood loss upon vessel injury (bleeding) or, on the other hand, the formation of blood clots (thrombosis). The critical balance between these pathways is regulated by diverse proteins, and genetic variants in genes that encode for these proteins are known to cause inherited forms of bleeding or thrombosis. This review focuses on insights related to this genetic regulation of bleeding and thrombosis.
Inherited bleeding disorders have a wide range of frequencies, from 1 in 1000 live births for von Willebrand disease (VWD) and 1 in 5000 males for hemophilia A, as most common inherited bleeding disorders, to just a handful of cases for many ultra‐rare platelet disorders. 2 , 3 , 4 Venous thrombosis has an overall annual incidence of about 1 in 1000 and is caused by environmental (lifestyle) effects, in association with genetic risk factors. Yet it is rare in the pediatric population, with rates of about 1 in 100 000 due to mainly genetic factors. 5 However, while significant advances have been made in understanding inherited thrombophilia (IT), many more heritable forms of thrombophilia are undiscovered, and thus, it is not possible to determine the true prevalence of IT.
To date, 99 curated disease‐causing genes (named TIER1 genes) have been identified to cause inherited bleeding, platelet, or thrombotic disorders. 6 The gene curation work was a project from the Scientific and Standardization Committee for Genetics in Thrombosis and Hemostasis (SSC‐GinTH) and the most recent gene list can be downloaded from the following web page: www.isth.org/page/GinTh_GeneLists. The TIER1 gene list is updated by the SSC‐GinTH during their session at the most recent yearly ISTH meeting. These genes are represented in Figure 1, ordered according to the pathway in which they function. A distinction is made between genes related to platelet disorders (including thrombocytopenia and platelet function disorders) and genes related to coagulation and thrombotic disorders. These TIER1 genes have a proven association with known inherited bleeding, platelet, or thrombotic disorders in humans and can be used in diagnostic laboratories for the genetic analysis of patients. 6 This is typically done nowadays with a multigene panel test as discussed in a next section. The web page, however, also contains a list of TIER2 genes that currently lack sufficient evidence to be considered as proven diagnostic‐grade genes, mostly because they were discovered in single or small pedigrees and still require confirmation studies in independent pedigrees as well as functional assays and a mouse model. Genetic studies in a research environment are critical to upgrade these TIER2 genes to the TIER1 status and have them implemented in diagnostics. The implementation of next‐generation sequencing (NGS) technologies has indeed resulted in the identification of many novel genes that regulate platelet formation and function, but for many of these genes, their exact biological role still remains unknown. However, this is also true for many of the TIER1 genes that were recently discovered but of which the exact function in megakaryocytes and platelets is still unknown (Figure 1).
FIGURE 1.
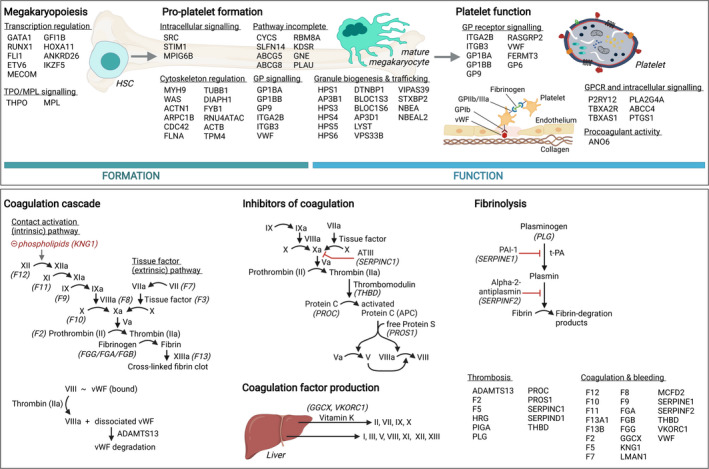
Overview of TIER1 genes and their respective roles in platelet formation and function, and the coagulation cascade. Top: identified TIER1 genes with a role in platelet formation and function, ordered by the molecular pathway in which they function. Bottom: TIER1 genes related to the coagulation cascade, coagulation inhibition, production of coagulation factors, and fibrinolysis. vWF, von Willebrand factor. Figure created with Biorender.com
In this review, we will provide an overview of how NGS techniques have been applied for diagnostics and gene discovery in hemostatic disorders. Current molecular diagnostic testing relies on high‐throughput genetic approaches using multigene panels. After a patient has been informed during a clinical consultation and consent has been given, a blood sample is drawn for a high‐throughput sequencing (HTS) diagnostic panel test. Panel test results are discussed during multidisciplinary meetings and consist of either a known diagnostic‐grade variant, which will directly lead to a molecular diagnosis, or a variant of unknown clinical significance (VUS) in either a TIER1 gene or an unknown gene, for which additional testing is required. In case the VUS is detected in a TIER1 gene, validation studies lead to variant reclassification and a subsequent molecular diagnosis. Variants detected in non‐TIER1 genes can be further studied in a research setting for gene discovery. An overview of this workflow can be found in Figure 2.
FIGURE 2.
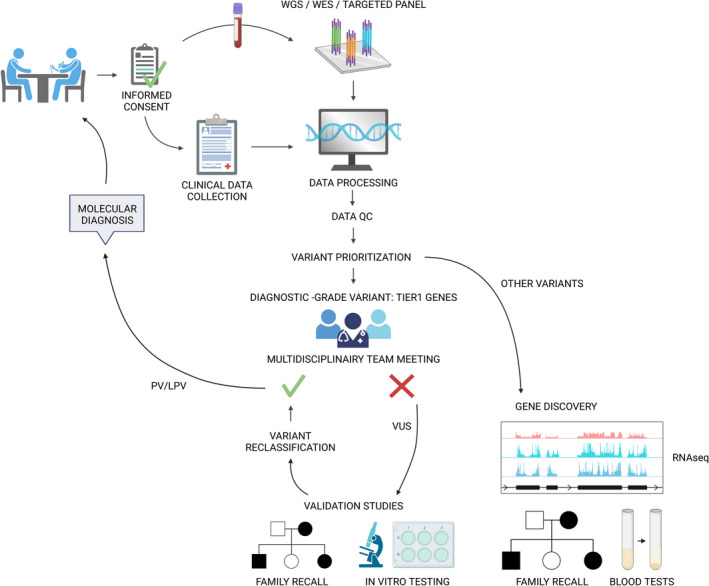
Standard workflow for NGS approaches in the diagnosis of inherited bleeding and thrombotic disorders. The standard diagnostics workflow using NGS approaches consists of several steps. After informed consent is obtained through clinical consultations, patient blood samples are submitted to multigene panel testing. After data QC, processing, and variant prioritization, candidate variants are selected in multidisciplinary meetings. PVs and LPVs in TIER1 genes are reported to the clinician, while VUSs in TIER1 genes can be further investigated using functional assays or cosegregation analysis for variant reclassification. If a virtual multigene panel was used based on WES or WGS data, these data can further be used for gene discovery using RNA sequencing, functional assays, and family recall studies. LPV, likely pathogenic variant; PV, pathogenic variant; QC, quality control; VUS, variant of unknown significance; WES, whole‐exome sequencing; WGS, whole‐genome sequencing. Figure created with Biorender.com
2. NGS FOR DIAGNOSTICS OF HEMOSTATIC DISORDERS: THE USE OF MULTIGENE PANEL TESTS
Several studies have investigated the use of multigene panels in the diagnosis of inherited bleeding and thrombotic disorders and proven its benefit compared to the previously employed method of Sanger sequencing of single genes. 7 , 8 , 9 , 10 , 11 Where the Sanger sequencing approach is a time‐consuming, work‐intensive, and difficult procedure that can screen only one gene at a time, a multigene panel test allows for the quick and easy screening of a great number of candidate genes at the same time, greatly improving diagnostic proficiency. 12 Patient inclusion for a panel test is based on both clinical history and routine laboratory testing results, to ensure that the bleeding or thrombotic disorder is indeed an inherited and not acquired defect. Given that the diagnostic rate is highly dependent on the type of disorder, carefully selected patient inclusion and exclusion criteria for panel testing are of great importance. 11 Previous studies showed that while a relatively high diagnostic rate could be obtained for thrombotic, platelet count, and coagulation disorders (48.9%, 47.8%, and 63.6%, respectively), the diagnostic rate for platelet function, and unexplained bleeding disorders remained low (26.1% and 3.1%, respectively). 10 These findings underline the need for new implicated genes to be discovered and added to genetic panel tests to improve the diagnostic rate, especially in platelet function and unexplained bleeding disorders.
The use of multigene panel testing in a clinical setting is paired with several advantages and disadvantages. An important benefit of panel testing is that the test can be focused only on clinically validated (TIER1) genes. 6 This ensures that the generated data are limited in size (as compared to whole‐exome sequencing [WES] or whole‐genome sequencing [WGS] approaches), allowing for a lower analysis burden and reduced required storage space, overall reducing testing costs. However, this focus on TIER1 genes also means that no novel gene‐disease associations or variants in TIER2 genes with limited evidence can be detected. Second, noncoding or deep intronic variants are not always detected in panel tests and are often never reported. Additionally, large structural variations, such as gene insertions, deletions, or duplications can be detected only through WGS but not through gene panel tests or WES. While gene panels also do not allow the detection of novel variants, this is of less importance in a clinical diagnostic setting and more important in a research context. However, as the current diagnostic rate using gene panel tests remains low for some patient groups with undefined clinical and laboratory phenotypes, 12 , 31 it would be highly beneficial if the panel test data for undiagnosed patients could be shared with a connected research group and used for gene discovery studies. Because of the low incidence of bleeding disorders worldwide, data sharing between clinicians and researchers is indeed of great importance to further increase our understanding of these complex and highly heterogeneous disorders. The ISTH SSC‐GinTH has developed a resource for clinical data sharing, called GoldVariants, which allows clinicians to report variants of curated genes discovered in routine diagnostics to an open access central database. 13 The GoldVariants database aims to create a well‐curated database of disease‐causing variants that is easily accessible to the public and allows for easy variant submission by community members active in diagnostics. Finally, using HTS approaches for diagnostics requires a specialized analysis approach for correct classification of detected variants, as multiple candidate variants are commonly detected for each case. 14 This specialized analysis needs to be performed by adequately trained personnel who then communicate discovered candidate variants to the treating clinician.
Using HTS for diagnostics also comes with several important implications for clinical management. In some cases, multiple pathogenic variants may be detected in one patient, but the clinical significance of these oligogenic variants remains largely unknown. In addition, a VUS may be detected, for which clear guidelines for variant reporting must be provided. Such variants are generally not reported to the clinician due to a lack of supporting research evidence, but may be of interest for further research. By reporting a VUS to an open‐access variant database, such as GoldVariants, additional information for variant classification can be accumulated so that the VUS can eventually be reclassified as either a (likely) pathogenic or a benign variant. A second important implication in the use of multigene panel tests is that unexpected findings may be discovered. For example, variants in some of the known Immuno Polymorphism Database–related genes also cause a higher risk for the development of leukemia, for example, RUNX1, ETV6, and ANKRD26. 14 When using multigene panel tests, patients should be informed on the possibility of unexpected findings and given the choice of whether to be informed about such variants. While other high‐throughput approaches might also result in the detection of secondary findings (ie, a clinically relevant pathogenic variant that falls outside the scope of the original test), this is avoided when using a panel test. The ISTH SSC‐GinTH has recently published updated guidelines for clinicians concerning variant reporting and ethical implications of using multigene panels in diagnostics for inherited platelet disorders. 14
3. NGS FOR GENE DISCOVERY AND INVESTIGATION OF DISEASE MECHANISMS FOR HEMOSTATIC DISORDERS: WHOLE‐EXOME, WHOLE‐GENOME, AND RNA SEQUENCING
Gene discovery using NGS technologies for hemostatic phenotypes resulted in a total of 25 TIER1 and 6 TIER2 genes for inherited platelet formation and function disorders and 1 TIER2 gene for inherited thrombosis. Only 5 TIER1 genes for inherited platelet formation and function disorders have been discovered using WGS. Noncoding discoveries are still very limited and mostly comprise large deletions. 15 The genes discovered by NGS are listed in Table 1, which also includes information in the last column about the details that were essential for the gene discovery process. It is not possible to discuss all in detail, but in this section some examples were selected that illustrate how gene discovery became possible and successful over time.
TABLE 1.
Gene discoveries using NGS technologies for bleeding and thrombotic disorders
| Gene | Year of discovery (reference) | NGS method |
TIER1 (diagnostic gene) |
TIER2 (research gene) | Hemostatic phenotype (Syndromic refers to presence of clinical phenotypes outside the blood system) | Gene discovery was supported by: |
|---|---|---|---|---|---|---|
| NBEAL2 |
2011 (PMID: 21765411, 21765412, 21765413) |
WES and RNAseq |
x | Platelet disorder | Linkage analysis and platelet RNAseq | |
| RBM8A |
2012 PMID:22366785 |
WES | x |
Platelet disorder (syndromic) |
Focus on previous CNV data (small chromosomal deletion) | |
| BLOC1S6 |
2012 PMID:22461475 |
WES | x |
Platelet disorder (syndromic) |
Single case and focus on recessive variants, similar phenotype as Bloc1s6 (or Pldn) KO mice | |
| GFI1B |
2013 PMID:23927492 |
Targeted massive parallel sequencing | x | Platelet disorder | Large pedigree and linkage analysis | |
| ACTN1 |
2013 PMID:23434115 |
WES | x | Platelet disorder | Screening multiple pedigrees with similar genotype‐phenotype, validation in additional pedigrees by Sanger sequencing | |
| THPO |
2013 PMID:2408576 for recessive 2020 PMID: 32150607 for dominant |
WES | x | Platelet disorder | Screening multiple pedigrees with similar genotype‐phenotype | |
| SLFN14 |
2014 PMID:26280575 |
WES | x | Platelet disorder | Screening multiple pedigrees with similar genotype‐phenotype | |
| STIM1 |
2014 PMID:24591628 |
WES | x |
Platelet disorder (syndromic) |
Screening affected and nonaffected cases from one pedigree, validation in additional pedigree by Sanger sequencing | |
| GNE |
2014 PMID:25257349 |
WES | x |
Platelet disorder (syndromic) |
Screening two pedigrees with similar genotype‐phenotype and focus on recessive variants | |
| RASGRP2 |
2014 PMID:24958846 |
WES | x | Platelet disorder | Analysis of homozygous variants in pedigree with consanguinity, similar phenotype as Rasgrp2 (or Caldag‐gefi) KO mutant mice | |
| PRKACG |
2014 PMID:25061177 |
WES | x | Platelet disorder | Analysis of homozygous variants in pedigree with consanguinity | |
| ETV6 |
2015 PMID:25581430 |
WES | x | Platelet disorder with predisposition to leukemia |
Screening multiple pedigrees with similar genotype‐phenotype Knowledge that somatic ETV6 variants were already shown to be implicated in leukemia |
|
| MECOM |
2015 PMID:26581901 |
WES | x |
Platelet disorder (syndromic) |
Trio sequencing and analysis of de novo variants, sequencing of additional cases for MECOM variants | |
| CDC42 |
2015 PMID:26386261 |
WES | x |
Platelet disorder (syndromic) |
Trio sequencing and analysis of de novo variants | |
| FYB1 |
2015 PMID:25876182 |
WES | x | Platelet disorder | Analysis of homozygous variants in pedigree with consanguinity | |
| TRPM7 |
2016 PMID:27020697 |
WES | x | Platelet disorder | Search in WES data set for thrombocytopenia patients after detecting this phenotype in Trpm7 KO mice, co‐segregation in a single pedigree | |
| AP3D1 |
2016 PMID:26744459 |
WES | x |
Platelet disorder (syndromic) |
Analysis of homozygous variants in pedigree with consanguinity, similar phenotype as Ap3D1 mutant mice | |
| DIAPH1 |
2016 PMID:26912466 |
WES | x |
Platelet disorder (syndromic) |
Screening multiple pedigrees with similar genotype‐phenotype using a statistical approach | |
| SRC |
2016 PMID:26936507 |
WES and WGS | x |
Platelet disorder (syndromic) |
Large pedigree with statistical approach of genotype‐phenotype data using phenotype of Src KO mice | |
| MPIG6B |
2016 PMID:27743390 |
WES | x | Platelet disorder | Linkage analysis and detection of homozygous variants in pedigree with consanguinity within the region of interest | |
| ARPC1B |
2017 PMID:28368018 |
WES | x |
Platelet disorder (syndromic) |
Screening of 2 pedigrees with similar genotype‐phenotype and analysis of homozygous variants in pedigrees with consanguinity | |
| TPM4 |
2017 PMID:28134622 |
WES and WGS | x | Platelet disorder | Search in WES dataset for thrombocytopenia patients after detecting this phenotype in Tpm4 KO mice | |
| KDSR |
2017 PMID:28774589 |
WES | x |
Platelet disorder (syndromic) |
Screening multiple pedigrees with similar genotype‐phenotype | |
| RNU4ATAC |
2018 PMID: 29391254 |
WGS | x |
Platelet disorder (syndromic) |
Screening of two pedigrees with similar genotype‐phenotype using a statistical approach | |
| EPHB2 |
2018 PMID:30213874 |
WES | x | Platelet disorder | Detection of homozygous variants in consanguineous pedigree | |
| IKZF5 |
2019 PMID: 31217188 |
WES and WGS | x | Platelet disorder | Screening multiple pedigrees with similar genotype‐phenotype | |
| PTPRJ |
2019 PMID:30591527 |
WES | x | Platelet disorder | Single pedigree and focus on recessive variants, phenotype similar to Ptprj KO mice | |
| NFE2 |
2019 PMID:31951293 |
WES | x | Platelet disorder | Single pedigree and focus on recessive variants | |
| ABCC4 |
2020 PMID:31826245 |
WES | x | Platelet disorder | Screening multiple pedigrees with similar genotype‐phenotype combined with a red blood cell proteome analysis to detect absent protein (PEL blood group) | |
| BLOC1S5 |
2020 PMID:32565547 |
WES | x |
Platelet disorder (syndromic) |
Screening two pedigrees with similar genotype‐phenotype and focus on recessive variants, similar phenotype as Bloc1S5 mutant mice | |
| PTGS1 |
2021 PMID:32299908 |
WGS | x | Platelet disorder | Detection of homozygous variants in complex consanguineous pedigree | |
| STAB2 |
2021 PMID:33465109 |
WES | x | Thrombotic disorder | Large pedigree with cosegregation analysis |
Abbreviations: CNV, copy number variation; KO, knockout; RNAseq, RNA sequencing; WES, whole‐exome sequencing; WGS, whole‐genome sequencing.
The first gene discovered with NGS technologies in our field, simultaneously by three groups, was NBEAL2 in 2011 for causing gray platelet syndrome (GPS) using WES, linkage analysis combined Sanger sequencing and WES, and platelet RNA sequencing (RNAseq). 16 , 17 , 18 To our knowledge, this is still the only gene discovered to date with platelet RNAseq. Before its discovery, it was already known that the candidate gene for GPS would be located within a 9.4‐megabase region on chromosome 3p21 based on earlier linkage analysis studies, narrowing the RNAseq analysis to genes located in this genomic region. 19 , 20 Only a recent cell biology study could provide some insights on how NBEAL2 can regulate alpha granule formation via the endoplasmic reticulum and in the same year 2020, it was shown that NBEAL2 defects can result in a broader clinical phenotype than bleeding with evidence for autoimmune pathologies. 21 , 22 The time between gene discovery and the understanding of the function of novel genes and proteins for megakaryocyte and platelet biology and its relation with clinical phenotypes can typically take years.
In 2012, the next gene discovered using WES was RBM8A for causing thrombocytopenia with absent radius (TAR) syndrome. 23 Interestingly, it was already known since 2007 that TAR syndrome patients have a common interstitial microdeletion of 200 kb on chromosome 1q21.1 and this deletion could be de novo but also be inherited from a nonaffected parent, suggesting a recessive mode of inheritance. 24 Interestingly, coding variants on the non‐deleted allele were ruled out after Sanger sequencing the 10 genes located in this region. 24 Afterwards, the WES discovery study did detect noncoding low‐frequency variants in the 5′ untranslated region (rs139428292, minor allele frequency [MAF] in gnomAD:0.01) or first intron (rs201779890, MAF in gnomAD: 0.007) of RBM8A on the nondeleted allele that together with deletion caused TAR. 23 It was a coincidence that the noncoded regions were covered in the WES data. Today, it is still unknown how RBM8A, which codes for Y14 of the exon junction complex, important for diverse RNA processing tasks, can alter megakaryopoiesis.
It is clear from these two gene discovery studies that the chromosomal location of the candidate genes could already be specified from earlier linkage and chromosomal deletion studies before analyzing rare variants in these regions using WES. This was also true for the discovery of GFI1B for dominant thrombocytopenia in a large 4‐generation pedigree where linkage analysis identified a single genome‐wide significant region on the telomeric end of chromosome 9 and targeted massive parallel sequencing of this region detected a frameshift variant in GFI1B as the cause for the disease. 25 Other gene discoveries were aided via screening of multiple pedigrees with similar genotype‐phenotypes relationships (eg, ACTN1, THPO, SLFN14, STIM1, GNE, ETV6, and KDSR), by focusing on homozygous variants in consanguineous pedigrees (eg, RASGRP2, PRKACG, FYB1, AP3D1, MPIG6B, ARPC1B, EPHB2, and PTGS1), by Trio WES for the detection of de novo variants (eg, MECOM and CDC42), by proteomics (eg, ABCC4), by screening specific genes in patients with phenocopies of hemostatic phenotypes generated in mice models (eg, BLOC1S6, TRPM7, TPM4, PTPRJ, and BLOC1S5), cosegregation analysis in very large pedigrees (eg, STAB2) and by applying sophisticated statistical methods that rank rare variants associated with phenotypes, named the BeviMed method (eg, DIAPH1, RNU4ATAC, and IKZF5) or even more complex variant ranking methods (eg, SRC; Table 1). 26
In addition to NGS technologies for gene discovery, platelet and megakaryocyte RNAseq and single cell RNAseq were already used to gain insights in the underlying pathways and disease mechanisms associated with a gene defect. Platelets are easy to isolate while megakaryocytes can be obtained from in vitro differentiation cultures using peripheral stem cells from patients. Platelet and leukocyte RNAseq was used for patients with GPS with NBEAL2 variants. 22 It was remarkable to see that their CD4 lymphocytes contained more differentially expressed genes than their platelets (255 vs 95 genes, respectively). Though patients with GPS have an obvious platelet defect resulting in thrombocytopenia and bleeding symptoms, their immune‐related pathologies only became obvious from performing this large cohort study. Pathway analyses showed that GPS platelets contain fewer transcripts that code for “alpha granules” and “releasate” proteins. A similar strategy using platelet versus leukocyte RNAseq was applied to profile the downstream genes for the transcription factor IKZF5. 27 Patients with pathogenic IKZF5 variants present with thrombocytopenia and minimal bleeding symptoms, and RNAseq data showed 1194 differentially expressed genes in their platelets, while only 4 differentially expressed genes could be detected in their different white blood cell types. The most significantly enriched pathway among the downregulated differentially expressed genes was platelet activation, signaling, and aggregation, but further studies are required to pinpoint the function of IKZF5 in platelet formation. Finally, megakaryocyte and B‐cell RNAseq studies were performed for patients Roifman syndrome that present with spondyloepiphyseal dysplasia, growth retardation, cognitive delay, hypogammaglobulinemia, and, in some patients, thrombocytopenia due to recessive variants in the small nuclear RNA gene RNU4ATAC, which is necessary for U12‐type intron splicing. 28 Significant minor intron retention was detected for 354 megakaryocyte genes that included splicing defects in genes known to regulate platelet formation (eg, DIAPH1). The megakaryocytes for this experiment were obtained from differentiating CD34+ hematopoietic stem cells of patients and controls. Single‐cell RNAseq was used to study CD42+ megakaryocyte‐biased induced hematopoietic stem cells from induced pluripotent stem cells generated from a patient with a pathogenic variant in RUNX1 and its isogenic variant–corrected line. 29 Dominant variants in RUNX1 cause thrombocytopenia, and patients are at risk to develop leukemia. The analysis of upregulated genes in RUNX1‐deficient cells indicated enrichment for response to stress, regulation of signal transduction, and immune signaling‐related gene sets. In general, RNAseq methods can deliver a wealth of information, but data interpretation and validation is not easy, as we do not yet have access to rapid high‐throughput methods to study the effects of gene expression on platelet formation and function (see future perspectives). It might also be essential to validate the changes in gene expression observed in RNAseq data at the protein level, as platelet transcriptomics and proteomics data do not always correlate well.
4. ISTH CONGRESS REPORT
Different abstracts presented at the 2021 ISTH congress using NGS technologies contributed to our understanding of genetic disorders causing hemostatic phenotypes. We have focused only on those that deal with the use of NGS technologies for diagnostics, gene discovery, and disease mechanisms.
At the SSC‐GinTH session of the ISTH 2021 congress, the curated gene list was updated by upgrading TPM4 from TIER2 to TIER1 status thanks to the study of Marín‐Quílez et al, 30 which presented a novel third pedigree with macrothrombocytopenia due to a TPM4 loss‐of‐function variant. Their microscopy data studying platelet spreading and the localization of TPM4 supported a role for TPM4 for normal cytoskeletal remodeling.
Diverse abstracts discussed the use of multigene panel tests to diagnose patients with an inherited hemostatic phenotype, and we have selected some to illustrate the large differences in diagnostic rates. 31 , 32 , 33 , 34 The Spanish multicenter study screened 254 index patients with an inherited platelet defect using a targeted multigene panel and an overall diagnostic rate of 63% was obtained (including patients receiving a report containing a VUS). 31 The diagnostic rate was higher for patients with thrombocytopenia compared to those with a platelet function defect, with a remarkably higher rate for patients with a clear phenotype compared to patients with an indistinct phenotype. A total of 161 different variants could be detected in 45 genes for 160 index cases, and 19% were classified as a VUS using American College of Medical Genetics and Genomics guidelines. The German GPOH/GTH Study Group “ThromKid‐Plus” on Inherited Platelet Disorders used a multigene panel for the screening of 35 patients and obtained a diagnostic rate of 26% (excluding patients with only a VUS). 32 Another multigene panel test for bleeding, platelet, and thrombotic disorders was described by Vilalta et al 33 to screen 79 patients. Pathogenic and likely pathogenic variants were detected in 48% of the patients, while 22% carried a VUS, for which additional studies are required. A similar multigene panel test for bleeding, platelet, and thrombotic disorders was used by Martinho et al 34 for the analysis of 43 genes in 496 patients. Variants were detected in 73% of patients with a coagulation disorder with bleeding, in 56% with an inherited platelet defect, and in 34% of the patients with thrombosis. Double and compound heterozygous variants could explain different intra/interfamilial patient phenotypes. As discussed previously, a difference in diagnostic rate using a multigene panel test typically depends on differences in variant interpretation rules (including VUS in a diagnostic report or not) and the stringency in patient inclusion criteria. 12 Of interest was the presentation from Zaninetti et al 35 that suggested using immunofluorescence on a blood smear as an effective diagnostic tool for 20 different inherited platelet disorders and being a useful tool to help in VUS interpretation. For 94 patients of 70 pedigrees, the microscopic analysis identified alterations suggestive of a specific platelet disorder for 53% of the patients, while genetic testing diagnosed a specific platelet disorder for 60% of the pedigrees. In 7 of the 19 pedigrees with a VUS, the microscopic analysis detected phenotypic changes typical for pathogenic variants. Variant classification and data sharing are very important within the process of variant curation but also for our understanding in gene‐phenotype correlations. 13 The Clinical Genome Resource (ClinGen) convened the Coagulation Factor Deficiency Variant Curation Expert Panel to develop rule specifications for curating variants in the F8/F9 genes. 36 The rule specifications were presented for a pilot study of 80 F9 and F8 variants with examples of variant classifications. Similar variant curation work from ClinGen for the Glanzmann thrombasthenia genes was recently published. 37 The numerous VUSs will be reclassified by ClinGen, and sharing data is very important for their curation work. 13
The use of NGS technologies also improved our understanding of rare disease phenotypes and disease mechanisms. Examples of these could be found in diverse meeting abstracts with a focus on (i) inherited hemostatic phenotypes in humans and (ii) the use of NGS approaches. A detailed clinical and platelet phenotype description was provided for a novel large pedigree with the gain‐of‐function E527K variant in SRC detected in WES data. 38 Similar to patients from three other unrelated pedigrees, thrombocytopenia, alpha granule deficiency, and platelet dysfunction toward collagen activation was detected. 38 , 39 , 40 , 41 A detailed clinical investigation now also revealed that most p.E527K carriers have a syndromic phenotype that includes recurrent infections, immuno‐allergic problems, bone disease, and neurological symptoms. The SRC E527K disease mechanism was further studied in defective megakaryocytes using RNAseq and proteomics. 42 Interestingly, the top pathway that was downregulated in E527K megakaryocytes using both omics data sets represented interferon type 1 signaling, and this could recently be validated in patient‐derived megakaryocytes. 43 Endothelial colony‐forming cells derived from patients with type 1 von Willebrand disease and healthy controls under basal and phorbol 12‐myristate 13‐acetate activated conditions were compared using mRNA and miRNA sequencing. 44 No biological processes were differentially regulated in the unstimulated state while 12 biological processes (including coagulation and platelet activation) were differentially regulated in the stimulated state. In addition, miRNA profiling for coagulation and hemostasis identified miR‐23b and miR‐26b as potential modulators of genes related to fibrinolysis.
5. FUTURE PERSPECTIVES
NGS technologies have delivered numerous VUSs in diagnostic studies and a number of TIER2 genes via research studies that today cannot be used for clinical diagnostics. Improved computational and experimental tools that can predict variant pathogenicity and the development of rapid cell‐based and functional assays that can study gene functions will become very important for future research in this field. This is still an underexplored domain. An interesting method named CRIMSON (CRISPR‐edited megakaryocytes for rapid screening of platelet gene functions) was recently published as a potential tool for the rapid and systematic approach to screen genes for platelet functions in CD34+ cell‐derived megakaryocytes. 45 To our knowledge, we still do not have methods available that would allow high‐throughput screening of platelet formation or function after genetic modifications. The focus of initial in vitro and in vivo validation studies will lie on the molecular characterization of the VUSs and TIER2 genes discovered through NGS technologies so far, in order to reclassify these variants and accumulate sufficient evidence to promote TIER2 genes to TIER1 status.
Future studies will also need to focus on the noncoding domains in WGS data sets. It is currently unknown how important variants in these domains are for patients with inherited bleeding, thrombotic, and platelet disorders. An initial attempt was recently made in WGS data for patients with an inherited platelet disorder but with a focus on large deletions and noncoding variants near TIER1 genes. 15 It will become much more difficult to explore other regions, and a strategy could be to supplement WGS data with platelet RNAseq data from these patients and study genes with an altered expression.
We conclude that NGS approaches have greatly contributed to both diagnostics and gene discovery studies in inherited bleeding and thrombotic disorders and will continue to do so in future research, especially for the validation and reclassification of VUSs and for exploring the role of noncoding variants in the development of hemostatic disorders. However, additional advances in high‐throughput screening assays for variant validation are still needed. Finally, it is of great importance that diagnostic centers have access to clear and uniform guidelines on variant interpretation and communication as well as good resources for data sharing of VUSs with the international society.
RELATIONSHIP DISCLOSURE
The authors report no conflicts of interest.
AUTHOR CONTRIBUTIONS
FVD, VL, and KF contributed equally to the writing of the article.
Ver Donck F, Labarque V, Freson K. Hemostatic phenotypes and genetic disorders. Res Pract Thromb Haemost. 2021;5:e12637. doi: 10.1002/rth2.12637
Handling Editor: Dr Johnny Mahlangu
Funding information
This work was supported by KULeuven BOF grant C14/19/096, FWO grants G072921N and 1115222N, and research grants from Novo Nordisk, CSL Behring, Bayer, and Swedish Orphan Biovitrum AB (Sobi).
Handling Editor: Dr Johnny Mahlangu
REFERENCES
- 1. Li X, Sim MMS, Wood JP. Recent insights into the regulation of coagulation and thrombosis. Arterioscler Thromb Vasc Biol. 2020;40:E119‐E125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Palla R, Peyvandi F, Shapiro AD. Rare bleeding disorders: diagnosis and treatment. Blood. 2015;26(125):2052‐2061. [DOI] [PubMed] [Google Scholar]
- 3. Sivapalaratnam S, Collins J, Gomez K. Diagnosis of inherited bleeding disorders in the genomic era. Br J Haematol. 2017;179:363‐376. [DOI] [PubMed] [Google Scholar]
- 4. Heremans J, Freson K. High‐throughput sequencing for diagnosing platelet disorders: lessons learned from exploring the causes of bleeding disorders. Int J Lab Hematol. 2018;40(suppl 1):89‐96. [DOI] [PubMed] [Google Scholar]
- 5. Khan S, Dickerman JD. Hereditary thrombophilia. Thromb J. 2006;12(4):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Megy K, Downes K, Simeoni I, et al. Curated disease‐causing genes for bleeding, thrombotic, and platelet disorders: communication from the SSC of the ISTH. J Thromb Haemost. 2019;17:1253‐1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bastida JM, del Rey M, Lozano ML, et al. Design and application of a 23‐gene panel by next‐generation sequencing for inherited coagulation bleeding disorders. Haemophilia. 2016;1(22):590‐597. [DOI] [PubMed] [Google Scholar]
- 8. Lee EJ, Dykas DJ, Leavitt AD, et al. Whole‐exome sequencing in evaluation of patients with venous thromboembolism. Blood Adv. 2017;29(1):1224‐1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Haan HG, Bezemer ID, Doggen CJM, et al. Multiple SNP testing improves risk prediction of first venous thrombosis. Blood. 2012;19(120):656‐663. [DOI] [PubMed] [Google Scholar]
- 10. Downes K, Megy K, Duarte D, et al. Diagnostic high‐throughput sequencing of 2,396 patients with bleeding, thrombotic and platelet disorders. Blood. 2019;5(134):2082‐2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Van Laer C, Jacquemin M, Peerlinck K, Freson K. ThromboGenomics implementation in Belgium. Belg J Hematol. 2020;12:99‐105. [Google Scholar]
- 12. Ver Donck F, Downes K, Freson K. Strengths and limitations of high‐throughput sequencing for the diagnosis of inherited bleeding and platelet disorders. J Thromb Haemost. 2020;4(18):1839‐1845. [DOI] [PubMed] [Google Scholar]
- 13. Megy K, Downes K, Morel‐Kopp M‐C, et al. GoldVariants, a resource for sharing rare genetic variants detected in bleeding, thrombotic, and platelet disorders: Communication from the ISTH SSC Subcommittee on Genomics in Thrombosis and Hemostasis. J Thromb Haemost. 2021;1(19):2612‐2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Downes K, Borry P, Ericson K, et al. Clinical management, ethics and informed consent related to multi‐gene panel‐based high throughput sequencing testing for platelet disorders: communication from the SSC of the ISTH. J Thromb Haemost. 2020;1(18):2751‐2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Turro E, Astle WJ, Megy K, et al. Whole‐genome sequencing of patients with rare diseases in a national health system. Nature. 2020;2(583):96‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Albers CA, Cvejic A, Favier R, et al. Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat Genet. 2011;43:735‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gunay‐Aygun M, Falik‐Zaccai TC, Vilboux T, et al. NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet γ‐granules. Nat Genet. 2011;43:732‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kahr WHA, Hinckley J, Li L, et al. Mutations in NBEAL2, encoding a BEACH protein, cause gray platelet syndrome. Nat Genet. 2011;17(43):738‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fabbro S, Kahr WHA, Hinckley J, et al. Homozygosity mapping with SNP arrays confirms 3p21 as a recessive locus for gray platelet syndrome and narrows the interval significantly. Blood. 2011;24(117):3430‐3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gunay‐Aygun M, Zivony‐Elboum Y, Gumruk F, et al. Gray platelet syndrome: natural history of a large patient cohort and locus assignment to chromosome 3p. Blood. 2010;2(116):4990‐5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lo RW, Li L, Pluthero FG, Leung R, Eto K, Kahr WHA. The endoplasmic reticulum protein SEC22B interacts with NBEAL2 and is required for megakaryocyte a‐granule biogenesis. Blood. 2020;6(136):715‐725. [DOI] [PubMed] [Google Scholar]
- 22. Sims MC, Mayer L, Collins J, et al. Novel manifestations of immune dysregulation and granule defects in gray platelet syndrome. Blood. 2020;22(136):1956‐1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Albers CA, Paul DS, Schulze H, et al. Compound inheritance of a low‐frequency regulatory SNP and a rare null mutation in exon‐junction complex subunit RBM8A causes TAR syndrome. Nat Genet. 2012;44:435‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Klopocki E, Schulze H, Strauß G, et al. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia‐absent radius syndrome. Am J Hum Genet. 2007;80:232‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stevenson WS, Morel‐Kopp MC, Chen Q, et al. GFI1B mutation causes a bleeding disorder with abnormal platelet function. J Thromb Haemost. 2013;11:2039‐2047. [DOI] [PubMed] [Google Scholar]
- 26. Greene D, Richardson S, Turro E. A fast association test for identifying pathogenic variants involved in rare diseases. Am J Hum Genet. 2017;6(101):104‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lentaigne C, Greene D, Sivapalaratnam S, et al. Germline mutations in the transcription factor IKZF5 cause thrombocytopenia. Blood. 2019;5(134):2070‐2081. [DOI] [PubMed] [Google Scholar]
- 28. Heremans J, Garcia‐Perez JE, Turro E, et al. Abnormal differentiation of B cells and megakaryocytes in patients with Roifman syndrome. J Allergy Clin Immunol. 2018;1(142):630‐646. [DOI] [PubMed] [Google Scholar]
- 29. Estevez B, Borst S, Jarocha D, et al. RUNX‐1 haploinsufficiency causes a marked deficiency of megakaryocyte‐biased hematopoietic progenitor cells. Blood. 2021;13(137):2662‐2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marín‐Quílez A, Fernández‐Infante C, Manrique Gonzalo MÁ, et al. Characterization of a new family with lifelong macrothrombocytopenia caused by a novel nonsense variant in TPM4 [abstract]. Res Pract Thromb Haemost. 2021;5(suppl 2):147‐148. [Google Scholar]
- 31. Bastida JM, Lozano ML, Benito R, et al. GEAPC Study Group . High throughput sequencing for the molecular diagnosis of inherited platelet disorders in Spain. Experience of the GEAPC group [abstract]. Res Pract Thromb Haemost. 2021;5(suppl 2):52–53. [Google Scholar]
- 32. Schulze H, Manukjan G, Klopocki E, Andres O, GPOH/GTH Study Group ‘ThromKid‐Plus’ on Inherited Platelet Disorders . Targeted high‐throughput sequencing for genetic classification of patients with inherited platelet disorders [abstract]. Res Pract Thromb Haemost. 2020;4(suppl 1):766–767. [Google Scholar]
- 33. Vilalta N, Tirado I, Romero L, et al. Next generation sequencing in patients with bleeding, thrombotic and platelet disorders: a single centre experience [abstract]. Res Pract Thromb Haemost. 2021;5(suppl 2):281–282. [Google Scholar]
- 34. Martinho P, Silva Pinto C, Oliveira C, et al. Inherited coagulopathies study by massive sequencing (NGS) experience of a diagnostic Portuguese center [abstract]. Res Pract Thromb Haemost. 2020;4(suppl 1):338. [Google Scholar]
- 35. Zaninetti C, Rivera J, Leinøe E, et al. Diagnosing inherited platelet disorders: immunofluorescence on the blood smear in comparison with genetic testing [abstract]. Res Pract Thromb Haemost. 2021;5(suppl 2):144–145. [Google Scholar]
- 36. Lee K, Chen X, Ding Q, et al. Optimizing variant curation guidelines to improve clinical genetic testing for hemophilia A and B [abstract]. Res Pract Thromb Haemost. 2021;5(suppl 2):88–89. [Google Scholar]
- 37. Ross JE, Zhang BM, Lee K, et al. Specifications of the variant curation guidelines for ITGA2B/ITGB3: ClinGen platelet disorder variant curation panel. Blood Adv. 2021;26(5):414‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Revilla N, Palma‐Barqueros V, Galera A, et al. Clinical and biological assessment of the largest family with SRC‐RT due to p. E527K gain‐of‐function variant [abstract]. Res Pract Thromb Haemost. 2021;5(suppl 2):145–146. [Google Scholar]
- 39. Turro E, Greene D, Wijgaerts A, et al. A dominant gain‐of‐function mutation in universal tyrosine kinase SRC causes thrombocytopenia, myelofibrosis, bleeding, and bone pathologies. Sci Transl Med. 2016;2(8):328‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. De Kock L, Thys C, Downes K, et al. De novo variant in tyrosine kinase SRC causes thrombocytopenia: case report of a second family. Platelets. 2019;3(30):931‐934. [DOI] [PubMed] [Google Scholar]
- 41. Barozzi S, Di Buduo CA, Marconi C, et al. Pathogenetic and clinical study of a patient with thrombocytopenia due to the p. E527K gain‐of‐function variant of SRC. Haematologica. 2021;1(106):918‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. De Kock L, Ver Donck F, Thys C, Van Geet C, Freson K. Transcriptomics and proteomics to study the effect of hyperactive SRC kinase during megakaryopoiesis [abstract]. Res Pract Thromb Haemost. 2021;5(suppl 2):159–160.33537540 [Google Scholar]
- 43. De Kock L, Ver Donck F, Thys C, et al. Combined transcriptome and proteome profiling of SRC kinase activity in healthy and E527K defective megakaryocytes. Haematologica. 2021. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kloosterman R, Zago‐Schmitt M, Grabell J, et al. Transcriptional analysis of the response to stimulation in ECFCs derived from type 1 VWD patients [abstract]. Res Pract Thromb Haemost. 2021;5(suppl 2):156–157. [Google Scholar]
- 45. Montenont E, Bhatlekar S, Jacob S, et al. CRISPR‐edited megakaryocytes for rapid screening of platelet gene functions. Blood Adv. 2021;3(5):2362‐2374. [DOI] [PMC free article] [PubMed] [Google Scholar]