

Abstract
Pancreatic cancer is rapidly progressive and notoriously difficult to treat with cytotoxic chemotherapy and targeted agents. Recent demonstration of the efficacy of maintenance PARP inhibition in germline BRCA mutated pancreatic cancer has raised hopes that increased understanding of the DNA damage response pathway will lead to new therapies in both homologous recombination (HR) repair-deficient and proficient pancreatic cancer. Here, we review the potential mechanisms of exploiting HR deficiency, replicative stress, and DNA damage-mediated immune activation through targeted inhibition of DNA repair regulatory proteins.
Keywords: pancreatic cancer, PARP inhibitor, DNA repair, targeted therapy, Fanconi anemia/BRCA pathway, replicative stress
INTRODUCTION
Pancreatic adenocarcinoma (PDAC) has the highest mortality rate of all cancers and is soon expected to become the second leading cause of cancer-related death in the United States (1,2). In recent years, the treatment of many other advanced malignancies has significantly improved with the introduction of immunotherapy and targeted therapies. Disappointingly, with the exception of very rare patients with mismatch repair deficient disease, immunological checkpoint blockade is ineffective in PDAC (3). Efforts to therapeutically target oncogenes driving the growth of PDAC have been stymied by the almost universal presence of KRAS mutations (4–8). The molecular profiles of PDAC are typically characterized by mutations in KRAS, p53, CDKN2A, and SMAD4, without clearly actionable molecular alterations. In recent years, multiple genomic studies have identified a subgroup of patients with PDAC whose tumors bear mutations in homologous recombination (HR) pathway genes responsible for the repair of double-strand DNA breaks (4–9). Given the success of Poly (ADP-ribose) polymerase (PARP) inhibitors in breast, ovarian, and prostate cancer, the finding of a DNA repair deficiency subgroup in PDAC ultimately led to the POLO trial, which has demonstrated the effectiveness of PARP inhibitor maintenance therapy in germline BRCA1 and BRCA2 mutated PDAC (10).
Standard Systemic Treatment Options for Pancreatic Cancer
Until 2011, the standard of care front-line regimen for patients with metastatic PDAC was single agent gemcitabine (11). The objective response rate to gemcitabine monotherapy was less than 10%, and the 1-year survival rate was approximately 20% (12). A major advance in the field was the demonstration that a multidrug regimen of 5-fluorouracil (5-FU), leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX), compared with gemcitabine monotherapy, substantially improves the objective response rate (31% verse 9%) and median overall survival (11.1 months verse 6.8 months) (11). A drawback of FOLFIRINOX is the significant increase in hematological, neuropathic, and gastrointestinal toxicity; hence, FOLFIRINOX is typically offered only to patients with a good performance status. The daunting toxicity of FOLFIRINOX has led many clinicians to modify the chemotherapy doses of this regimen. A commonly used modified FOLFIRINOX regimen, which was successfully tested in the adjuvant setting (see below), decreases the dose of irinotecan and does not administer the 5-FU bolus (oxaliplatin 85 mg per square meter of body-surface area (mg/m2), irinotecan 150 mg/m2, leucovorin 400 mg/m2, fluorouracil 2,400 mg/m2 continuous infusion over 46 hours) (13).
Two years after the introduction of FOLFIRINOX, another first-line regimen consisting of gemcitabine and nanoparticle albumin-bound (nab)-paclitaxel was approved by the Food and Drug Administration. The approval of first-line gemcitabine/nab-paclitaxel was based on the randomized MPACT phase III trial, which also used single-agent gemcitabine as the comparator arm (14). Gemcitabine/nab-paclitaxel therapy resulted in a significantly higher response rate (23% versus 7%) and medial overall survival (8.5 months versus 6.7 months) compared to gemcitabine alone (14).
Because gemcitabine/nab-paclitaxel and FOLFIRINOX have not been directly compared in a prospective clinical trial, a challenge in the field is determining the optimal first-line regimen for patients with metastatic PDAC. The gemcitabine/nab-paclitaxel regimen is widely believed to be better tolerated than FOLFIRINOX (15). However, on the basis of cross-trial comparisons demonstrating a trend toward a higher response rate (31% verse 23%) and median overall survival (11.1 months versus 8.5 months) for FOLFIRINOX, some experts believe that FOLFIRINOX is the more potent regimen, although this remains controversial. An ongoing randomized multicenter phase II clinical trial of FOLFIRINOX versus gemcitabine/nab-paclitaxel in the first-line setting will address this question (NCT04469556). In current practice, oncologists typically present both the FOLFIRINOX and gemcitabine/nab-paclitaxel regimens, and patients ultimately decide which regimen to start according to side-effect profiles and preferences regarding treatment aggressiveness. Because of the toxicities associated with FOLFIRINOX, gemcitabine/nab-paclitaxel is the most common chemotherapy backbone used in clinical trials evaluating standard chemotherapy combined with an experimental agent. Although gemcitabine/nab-paclitaxel was evaluated as a first-line regimen, it is also frequently used as a second-line regimen when patients start with first-line FOLFIRINOX. Conversely, patients who are initially treated with gemcitabine/nab-paclitaxel are usually treated in the second line with a two drug 5-FU based regimen such as either 5-FU/liposomal irinotecan or 5-FU/oxaliplatin (FOLFOX) (16,17).
In recent years, FOLFIRINOX has also been incorporated into neoadjuvant and adjuvant PDAC treatment protocols. In a landmark study, Conroy et al. demonstrated that adjuvant FOLFIRINOX significantly improves the median overall survival (54 months versus 35 months) compared to the previous standard of care, single agent gemcitabine (13). In a surprising result, the randomized phase III APACT trial failed to show that adjuvant gemcitabine/nab-paclitaxel was superior to gemcitabine monotherapy (18).
The potency of FOLFIRINOX has also inspired a shift toward its use as neoadjuvant therapy before surgery (19). This shift occurred partly because of the ability of FOLFIRINOX to downstage tumors, thereby increasing the feasibility of surgical resection of the primary tumor by decreasing the cancer’s contact with essential surrounding blood vessels. In the results of the Alliance A021501 study, presented at the 2021 Gastrointestinal Cancers Symposium, patients with borderline PDAC treated with eight cycles of neoadjuvant FOLFIRINOX followed by surgery and four cycles of FOLFOX had an impressive median overall survival of 31 months (20).
The HR Pathway and Mechanisms of DNA Repair
The DNA repair system has evolved numerous repair pathways to maintain the integrity of the genome in response to exogenous and endogenous damage (21,22). Single strand DNA repair is mediated by three mechanisms: the mismatch DNA repair system, base excision repair, and nucleotide excision repair (23–25). The major mechanism of double-strand DNA repair is HR. HR is evolutionarily favored over other types of DNA repair, such as classical non-homologous end joining (cNHEJ) and alternative nonhomologous end-joining (Alt-NHEJ), because of its ability to perform high fidelity DNA repair (26).
Because of the existential threat that double-strand DNA breaks pose to the cell, a robust sensor, the MRN complex, has evolved to rapidly detect double-strand DNA breaks. The MRN complex of proteins, comprising MRE11, RAD50, and NBS1, locates and binds double-strand DNA breaks (27). It then activates the PI3K-related protein kinase (PIKK) ATM, which functions as a signal transducer that propagates the DNA damage signal to downstream effectors. ATM directly activates CHK2, p53, and γH2AX, thus leading to the activation of RAD51, BRCA1, BRCA2, and PALB2 (28).
After the MRN complex has activated the DNA damage signal transduction cascade, the DNA repair machinery is poised to repair the break. The decision about which mechanism of double-strand DNA repair is used partly depends on the cell cycle phase. If the cell is in the S or G2 phase of the cell cycle, HR is the favored approach, because the sister chromatid, generated through DNA replication, can be used as a template for high fidelity DNA repair (29). Another difference between HR and error prone cNHEJ is that a single-strand DNA 3´ overhang is an absolute requirement for HR. End resection, the process creating the single-strand DNA 3´ overhang, can be obstructed by the presence of the tumor suppressor p53-binding protein 1 (53BP1) and the shieldin complex, comprising the SHLD1, SHLD2, SHLD3, and REV7 proteins (30). In this way, the presence or absence of the shieldin complex and 53BP1 regulates the choice of cNHEJ or HR to repair double-strand DNA breaks (31).
cNHEJ has a strong likelihood of introducing DNA errors, because rather than utilizing homology-based DNA repair, it uses ligation-based DNA repair, which can lead to DNA alterations such as deletions and telomeric allelic imbalance (26,32). Unlike cNHEJ, DNA repair via HR is highly accurate because it uses the sister chromatid as a repair template. Conceptually, the first step of HR is end resection, when the MRN complex, BRCA1, and CtIP endonuclease create a single-strand DNA 3´ overhang (33). The next critical step in HR is the formation of the BRCA1, PALB2, and BRCA2 complex at the site of the DNA break (34) (Figure 1). This complex allows RAD51 to bind BRCA2 and begin to polymerize the RAD51 nucleoprotein filament with single strand DNA (29). The RAD51 nucleoprotein filament invades the sister chromatid and searches for complementary DNA to serve as a template for DNA synthesis.
Figure 1: Function of “Core HR” complex in repair of double-strand DNA breaks.
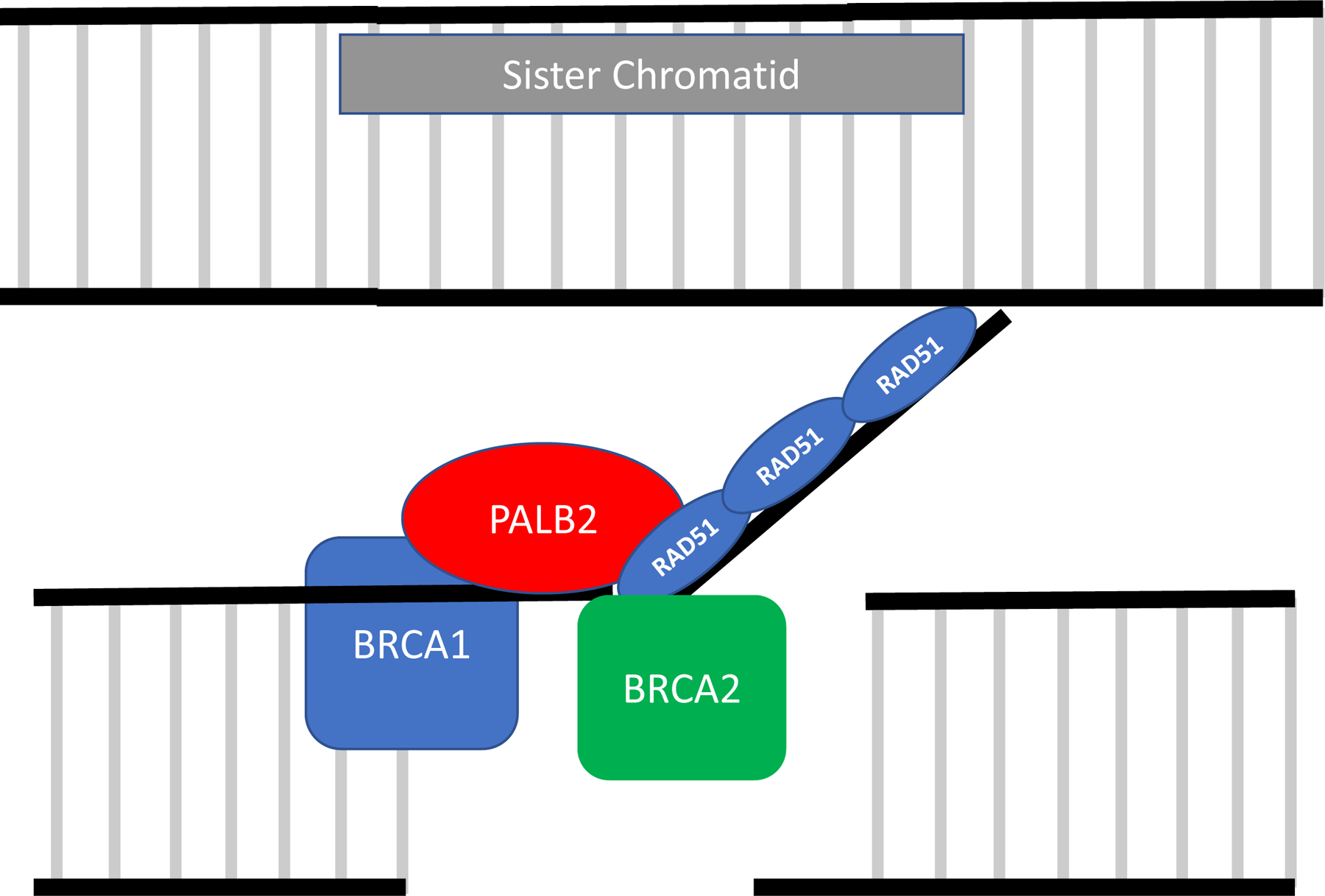
BRCA1 binds to the DNA and promotes creation of the single-strand DNA 3´ overhang. PALB2 then acts as a bridge by binding to BRCA1 and BRCA2 (forming the BRCA1-PALB2-BRCA2 complex). PALB2 and BRCA2 then bind to RAD51 allowing for the creation of the RAD51 nucleoprotein filament. The RAD51 nucleoprotein filament invades the sister chromatid and searches for complementary DNA to serve as a template for DNA synthesis.
Genomics of HR-Deficient Pancreatic Cancer
In 1994, the BRCA1 and BRCA2 genes were initially discovered in studies of women with familial breast and ovarian cancer (35). Approximately a decade later, in 2007, another gene in the HR pathway, PALB2, was also found to increase susceptibility to breast cancer (36). BRCA1, BRCA2, and PALB2 are also known as Fanconi Anemia genes, corresponding to the genes FANCS, FANCD1, and FANCN respectively (37). These three genes are well-known to cooperate with several other FANC genes in a DNA interstrand crosslink repair pathway known as the Fanconi Anemia/BRCA pathway. Although BRCA1, BRCA2, and PALB2 were initially thought of primarily as breast and ovarian cancer susceptibility genes, in recent years, multiple studies have shown that these “core HR gene” mutations are also common causes of familial PDAC. Germline mutation in BRCA2 is the most common mechanism by which core HR function is compromised in familial PDAC, occurring in up to 5% of patients with PDAC (9,38). Beyond the “core HR” genes of BRCA1, BRCA2, and PALB2, germline ATM mutations have been demonstrated to increase susceptibility to PDAC and to occur in approximately 5% of patients with PDAC (4,38–40).
The realization that a subset of patients with PDAC have germline mutations in HR genes led to the recommendation by the National Comprehensive Cancer Network (NCCN) that all patients with PDAC undergo germline testing (41). Factors motivating this recommendation include the identification of high-risk family members and the therapeutic opportunities to exploit these mutations. Several centers have developed surveillance protocols for adults who carry these mutations, with the goal of identifying PDAC development early while the primary tumor remains resectable (42).
Large scale tumor sequencing efforts performed by multiple groups have demonstrated that some patients with PDAC, beyond those with germline alterations, have HR pathway somatic alterations. In an analysis of the frequency of a panel of 17 HR gene alterations, Park et al. found that in a cohort of 262 patients with PDAC, germline HR gene alterations were more frequent (15%) than somatic only HR gene alterations (4%) (38). Notably, of the 17 HR genes analyzed, most of the HR gene mutations occurred in core HR genes (BRCA1, BRCA2, or PALB2) rather than non-core HR genes (12% versus 7%).
Functional inactivation of both copies of an HR gene (i.e., biallelic inactivation) may be an important biomarker for targeting these HR gene mutations. Park et al. analyzed the frequency of biallelic inactivation of HR genes identifiable by DNA sequencing. They demonstrated that 71% of core HR gene alterations in PDAC are biallelic (4,9,38). Importantly, this finding may be an underestimate, because DNA methylation has been demonstrated to be an additional mechanism of inactivation of HR genes. In a recent study of 237 triple-negative breast cancers, Glodzik et al. have found that functional inactivation of BRCA1 by promoter hypermethylation is twice as frequent as BRCA1 pathogenic genomic alterations (43). Further study is needed to understand the importance of promoter hypermethylation of HR genes in PDAC.
Because analysis of single HR gene alterations might be insufficient to identify all patients with PDAC with HR deficiency, other biomarkers are being evaluated that may capture a greater number of HR deficient cancers. Computational analysis of large data sets of whole exome sequences has revealed that a specific nucleotide variant (SNV) substitution signature, COSMIC signature 3, is a marker of HR deficiency and is strongly correlated with functional loss of BRCA1, BRCA2, and PALB2, and RAD51C deficiency (44,45). As expected, the hallmark of COSMIC signature 3 is a genomic consequence of HR deficiency: large numbers of indels and insertions with microhomology at breakpoint junctions. The identification of COSMIC signature 3 suggested the possibility that patients with HR deficiency who do not have BRCA1, BRCA2, and PALB2 mutations might be identified in the clinic and placed on therapies that exploit their HR deficiency. Illustrating the potential value of this approach in PDAC, an analysis of 14 COSMIC signature 3- positive patients with PDAC found that the HR deficient status of only 11 of 14 of patients could be explained by the mutational status of specific DNA repair genes (4). Given the importance and challenge of identifying HR-deficient tumors, several genomic algorithms, including the Classifier of Homologous Recombination Deficiency (CHORD) and HRDetect, have been developed to identify the characteristic genomic scar caused by HR deficiency (46,47). The HRDetect algorithm was developed to incorporate the signals of multiple characteristic signatures of HR deficiency, such as COSMIC signature 3, microhomology-mediated deletions, rearrangement signatures 2 and 5, and base substitution signature 8 (47). In an analysis of whole genome sequencing using the HRDetect score, Golan et al. found evidence of HR deficiency in an additional 7–10% of patients (n=391) with PDAC who did not have a germline BRCA or PALB2 mutation (48). Similarly, in a cohort of 96 PDAC patients, HRDetect correctly identified all of the patients with known HR gene mutations and also identified HR deficiency in 5 patients with no clear HR gene mutation (47).
The challenge in using HRDetect and the COSMIC signature 3 clinically is that they are both derived from either whole genome or whole exome sequencing, whereas most CLIA-approved sequencing tests use targeted next generation sequencing (NGS) assays. A computational methodology, the SigMA algorithm, was designed to overcome this limitation. The SigMA algorithm can identify COSMIC signature 3-positive tumors that have been analyzed by targeted NGS gene panels, and the results have been found to correlate with response to niraparib and pembrolizumab in an ovarian cancer clinical trial (49,50). However, in an effort to test the feasibility of using the SigMA algorithm on clinical sequencing panels, Park et al. found that only 58% of PDAC patient samples have a mutational burden sufficient to calculate a SigMA score (38). This finding suggests that the relatively low mutational burden of PDACs would prevent the routine clinical use of the SigMA algorithm with currently available clinical grade sequencing assays.
Similar to how the COSMIC signature 3 detects HR deficiency via recognition of characteristic DNA alterations, other genomic signatures can also identify HR-deficient tumors by assaying for other characteristics of the “genomic scar” left by HR deficiency. Large-scale state transitions (LST), defined as chromosomal breaks of adjacent regions over 10 mB in length, are caused by increased use of double-strand DNA repair mechanisms, such as NHEJ, when the HR pathway is not functioning. In contrast to their findings for the SigMA algorithm, Park et al. were able to calculate an LST score from all patients in their targeted sequencing panel (38). However, the LST scores did not appear to enhance the detection of HR deficiency beyond that of mutational analysis of specific HR genes (38). In ovarian cancer, a commercial assay (Myriad’s myChoice® HRD CDx assay) that measures LST, loss of heterozygosity, and telomeric allelic imbalance has prospectively identified people with tumors harboring wild type BRCA who benefited from PARP inhibition (51,52). Golan et al. evaluated the Myriad HRD assay in their cohort of 391 PDAC cases and observed that it was predictive of platinum sensitivity. However, the authors also demonstrated that the HRDetect assay was more sensitive and specific than the Myriad HRD assay in detecting HR deficiency in PDAC (48).
Interestingly, in an analysis of PDAC chromosomal structure and rearrangement, Waddell et al. observed that PDACs can be sub-divided into four categories based on the type and complexity of chromosomal abnormalities (53). They observed that PDACs with deleterious BRCA1/2 and PALB2 mutations clustered into an “unstable subtype” that demonstrated the highest degree of chromosomal structural variation. Notably, PDACs in the unstable chromosomal subtype appeared to have increased sensitivity to platinum-based chemotherapy (53).
Subgroups of Pancreatic Cancer Defined by RNA Expression Analysis
The use of RNA expression analysis is increasingly informing key aspects of PDAC biology for stratification of PDACs into different molecular subtypes. In solid tumors, the classification of cancers into subtypes according to RNA expression was pioneered in breast cancer, with studies beginning in 2000 (54). One of these breast cancer subtypes was designated the basal subtype, because it resembles myoepithelial cells in the outer (i.e., basal) layer of the breast gland. Most triple-negative breast cancers, which have notoriously poor prognosis and are enriched in HR deficiency, fall within the basal subtype (55). A similar basal-like/squamous subtype was identified in PDAC (56). RNA signatures identifying this basal-like PDAC subtype have substantial overlap with those signatures previously defined for bladder and breast cancers, and the PDAC basal-like gene expression signature has been demonstrated to reproduce basal subtype calls in breast and bladder cancer with prognostic significance (56). The basal-like subtype of PDAC has poorer prognosis, higher grade pathology, and more frequent p53 mutations (56–58). Moreover, basal-like PDAC tumors also appear to be more resistant to standard cytotoxic chemotherapy (58,59). Whereas some studies have suggested that the basal-like subtype might be more PARP inhibitor-sensitive, other studies have not shown an association between transcriptional subtype and HR deficiency (60,61). Interestingly, a recent analysis of patient-derived pancreatic cancer cell lines has suggested that the basal-like/squamous subtype is enriched in cancers under a high degree of replicative stress and characterized by elevated expression of the cell cycle regulatory proteins CDK6, CDK7, and WEE1 (see ‘Replicative stress is a potential pancreatic cancer vulnerability’ section) (61).
Efficacy of Platinum-Based Chemotherapy in HR Deficient Pancreatic Cancer
HR deficiency creates a vulnerability to platinum chemotherapy (62). Most crosslinks caused by platinum chemotherapy are intrastrand crosslinks (approximately 90%) that are repaired by nucleotide excision repair (NER). However, interstrand DNA crosslinks occur approximately 1–2% of the time are more cytotoxic than intrastrand crosslinks because covalent binding of the two DNA strands prevents normal physiological processes such as DNA replication and transcription (62,63). Interstrand DNA crosslinks likely contribute to greater toxicity than intrastrand crosslinks, perhaps accounting for the more restricted role in cancer therapy for drugs that cause greater interstrand crosslinks, e.g. mitomycin C and nitrogen mustards (cyclophosphamide and melphalan) (62).
Mechanistic studies from patients with Fanconi anemia, a rare hereditary disorder characterized by bone marrow failure and cellular hypersensitivity to DNA interstrand crosslinking agents, have elucidated many of the steps in this repair process (37). Repair of the DNA damage caused by platinum chemotherapy is now understood to be a complex process that can involve multiple mechanisms of DNA repair including NER, HR, and translesion DNA synthesis (64,65). This complexity has made accurate predictions of platinum chemotherapy sensitivity challenging. However, the prominent role of the HR pathway in removing interstrand DNA crosslinks makes it unsurprising that HR-mutated cancers are highly sensitive to platinum chemotherapies.
Multiple studies have demonstrated that patients with PDAC with deleterious HR gene alterations who are treated with platinum-based chemotherapy have better survival outcomes than patients with HR-proficient PDACs (9,38,66–69). Furthermore, patients with HR-deficient PDAC have significantly better progression-free survival when they are treated with first-line platinum-based chemotherapy compared with non-platinum chemotherapy (12.6 months versus 4.4 months) (38). Importantly, germline BRCA-mutated PDAC patients had a higher rate of pathological complete response to neoadjuvant FOLFIRINOX as compared to BRCA wild type patients, and this translated into improved overall survival for these patients (69). The heightened sensitivity of HR-deficient PDACs to platinum-based chemotherapy was further highlighted by a recent phase 2 clinical trial of gemcitabine/cisplatin (cisplatin 25 mg/m2 and gemcitabine 600 mg/m2 on day 1 and 8 of each 21 day cycle) in patients with metastatic PDAC with germline BRCA1/2 or PALB2 mutation (70). The efficacy outcomes in this trial far surpassed those previously seen in non-selected metastatic PDAC. In addition, this trial set a benchmark for patients with metastatic PDAC with core HR gene mutations, demonstrating a 65% objective response rate, 9.7-month median progression-free survival, 16.4-month median overall survival, and 30.6% 2-year survival rate. Finally, it is important to note that less common HR gene alterations, such as RAD51C, could also predict platinum sensitivity. For example, Golan et al. describe an extraordinary PDAC responder to FOLFIRINOX who had biallelic inactivation of RAD51C (48).
Therapeutic Targeting of HR Deficiency with PARP Inhibitors
The development of therapeutic targeting of HR deficiency has been a model for the potential of exploiting synthetic lethality in oncology (71). First demonstrated by two laboratories in 2005, cancer cells lacking functional HR are highly sensitive to PARP inhibition (72,73). Therapeutically, the advantage of this synthetic lethal relationship is that non-cancer cells, with proficient HR, are not affected by PARP inhibition, thus leading to a wide therapeutic window for these drugs. Physiologically, PARP functions by binding to single-strand DNA breaks via its DNA binding domain. After binding to single-strand DNA breaks, the PARP protein catalyzes poly ADP-ribosylation (PARylation), which results in the formation of polymers of ADP-ribose (74). The ADP-ribose polymers serve as beacons of DNA damage, thus mobilizing the DNA repair machinery (74).
The binding of PARP to single-strand DNA breaks is a critical step in the initiation of the base excision repair pathway. Hence, inhibiting PARP prevents the base excision repair pathway from acting on single-strand DNA breaks. These unrepaired single-strand DNA breaks can then progress to double-strand DNA breaks during the next replication cycle. In HR-deficient cells, the accumulation of double-strand DNA breaks causes catastrophic DNA damage leading to cell death.
In addition to the above classical mechanism of PARP inhibitor-induced cell death in HR-deficient cells, binding of a PARP inhibitor to the PARP protein can “trap” the PARP protein to the DNA and lead to cell death (75). This cytotoxicity occurs because the trapped PARP protein interferes with normal cellular processes such as DNA replication. Supporting this hypothesis is the observation that differential PARP inhibitor trapping explains the differing cytotoxicity among PARP inhibitors. Talazoparib is a very potent (IC50 = 5 nM in cytotoxicity assays) PARP inhibitor with strong PARP trapping capability, whereas veliparib has relativity modest PARP trapping activity and is a less potent PARP inhibitor (IC50 >10,000 nM in cytotoxicity assays) (76–78).
HR deficient cells also have increased dependency on Alt-EJ (also called microhomology-mediated end joining [MMEJ]), a mechanism of double-strand DNA repair that relies on small regions (2 – 20 base pairs) of homologous DNA (79,80). The PARP protein plays several critical roles in Alt-EJ, including recruitment of proteins involved in both end-resection and alignment of microhomology sequences. Hence, another potential mechanism of PARP inhibitor-mediated synthetic lethality with HR deficiency is that PARP inhibition blocks Alt-EJ (81,82). Inhibition of polymerase θ (Polθ), the protein that synthesizes DNA in Alt-EJ-mediated repair, also causes synthetic lethality in HR deficient cells and Polθ inhibitors are entering into clinical development (82,83). One of these Polθ inhibitors, novobiocin, was cytotoxic in the PDAC BRCA2 deficient CAPAN-1 cell line (84).
PARP Inhibitors in HR-Deficient Pancreatic Cancer
A key insight in the clinical developmental of PARP inhibitors has come through the analysis of patients with platinum-sensitive and resistant ovarian cancer. Investigators have noted that whereas the PARP inhibitor olaparib has impressive activity in patients with platinum-sensitive ovarian cancer, it has little to no efficacy in those with platinum-resistant ovarian cancer (85). The observation that platinum resistance is predictive of PARP inhibitor efficacy has also held true in PDAC. Initial trials of PARP inhibitors in PDAC yielded mixed results, with only rare and predominantly short-lived responses (86–88). Notably, the populations used in these initial trials were heavily pre-treated patients who had previously progressed on platinum-containing chemotherapy regimens. For example, a phase 2 trial of rucaparib in patients with metastatic PDAC with germline or somatic BRCA1/2 mutation was stopped early because of insufficient efficacy (86). A subsequent analysis of this trial demonstrated that all four responding patients had never progressed on platinum chemotherapy, whereas most non-responding patients had already progressed on platinum-based therapy (86) (Table 1). A recent report, that presented data from two parallel phase 2 studies of olaparib in previously treated PDAC patients who were HR-deficient for reasons other than a germline BRCA mutation, demonstrated that platinum-sensitive patients had a significantly increased progression-free survival compared to platinum-resistant patients (89).
Table 1:
Notable Reported PARP Inhibitor Trials in Pancreatic Adenocarcinoma
| Trial | Trial Population | OS | PFS | ORR | Notable Findings | Ref |
|---|---|---|---|---|---|---|
| Olaparib vs. PBO POLO Trial |
• Metastatic PDAC • gBRCA1/2mut • Plat. Sensitive • Maintenance after 1st line chemo |
19.0 vs. 19.2 mo p= 0.3487 | 7.4 vs. 3.8 mo p= 0.004 |
23% vs. 12% | • DOR 24.9 vs. 3.7 mo • Subgroup analysis showed no difference in outcomes for pts w/ response to plat chemo or >6 months of plat chemo |
(10,90) |
| Rucaparib Reiss Binder et al. |
• Advanced PDAC • g/sBRCA1/2mut • g/sPALB2mut • Plat. Sensitive • Maintenance after 1st line chemo |
N.A. | 9.1 mo | 36.8% | • Responses in pts w/ gPALB2 mutations (2) and sBRCA2 mutation (1) | (96) |
| Rucaparib Shroff et al. |
• Advanced PDAC • g/sBRCA1/2mut • Pts who received 1 or 2 prior lines of chemo |
N.A. | N.A. | 15.8% | • All responders were plat. sensitive • Most non-responders were plat. resistant • Trial stopped for insufficient efficacy |
(175) |
| Gem/Cis/ Veliparib vs.Gem/Cis O’Reilly et al. |
• Advanced PDAC • Untreated pts • gBRCA1/2mut or gPALB2mut |
15.5 vs.16.4 mo p = 0.6) |
10.1 vs. 9.7 mo p = 0.73 |
74% vs. 65% (p = 0.55) | • Outcomes for the gem/cis arm compare favorably to outcomes of historical, unselected pancreatic cancer pts | (70) |
| FOLFIRI/Veliparib vs. FOLFIRI SWOG S1513 |
• Metastatic PDAC • 1 prior non-irinotecan systemic therapy • Molecularly unselected pts |
5.1 vs 5.9 mo p = 0.21 |
2.1 vs 2.9 mo p = 0.05 |
N.A. | • Trial stopped early for futility • Veliparib increased toxicity |
(99) |
Abbreviations: germline (g); somatic (s); mutation (mut); median overall survival (OS); progression free survival (PFS); placebo (PBO); overall response rate (ORR); reference (ref); Platinum (Plat), chemotherapy (chemo); median duration of response (DOR); patients (pts); with (w/); Gemcitabine (Gem); Cisplatin (Cis)
Learning from the setbacks of testing PARP inhibitors in platinum-resistant patients, the phase 3 randomized, placebo-controlled Pancreas Cancer Olaparib Ongoing (POLO) trial evaluated olaparib maintenance therapy in patients with metastatic PDAC with germline BRCA1 and BRCA2 mutation who did not progress on at least 16 weeks of first-line platinum-based chemotherapy (10). Patients treated with maintenance olaparib had a 3.6-month improvement in median progression-free survival, compared with that of patients treated with the placebo control. The improvement in progression-free survival led to the Food and Drug Administration’s approval of maintenance olaparib for this indication. However, the trial, which was not powered to demonstrate an overall survival difference between the olaparib and placebo-treated arms, did not show an overall survival benefit with maintenance olaparib (10,90). The placebo control arm of the POLO study highlighted that some patients with BRCA-mutant PDAC have durable responses to platinum chemotherapy. Ten percent of placebo-treated patients showed no evidence of disease progression at 24 months (91). In addition, the objective response rate to the placebo was 10%, thus indicating the continued radiologic response to prior platinum chemotherapy after treatment cessation in some patients (91). However, among patients with a partial response while on the study drug (olaparib or placebo), those randomized to olaparib had a significantly longer median duration of response (24.9 months versus 3.7 months), demonstrating that some patients with germline BRCA-mutated PDAC benefit substantially from PARP inhibition (10).
The modest 3.6-month improvement in progression-free survival, with a lack of overall survival benefit, has led some to question the true benefit of olaparib maintenance therapy as suggested by the POLO study (10,90,91). Some experts have questioned whether the control arm of the POLO trial should have been 5-FU-based chemotherapy, rather than placebo (91). Proponents of the study argue that the favorable toxicity profile of maintenance olaparib makes it an attractive choice for patients. A central question is why maintenance PARP inhibition appears to be more effective in platinum-sensitive BRCA-mutated ovarian cancer than PDAC. Trials of maintenance PARP inhibition in platinum-sensitive BRCA-mutated ovarian cancer have shown a 12–36 month improvement in progression-free survival over placebo control, depending on the trial population (92–95). A key distinction between the ovarian and PDAC maintenance trials is the definition of platinum sensitivity. In ovarian cancer, patients were required to have had a partial radiologic response to platinum chemotherapy, whereas in the POLO study, patients with PDAC with radiologically stable disease on platinum chemotherapy were also eligible. To assess whether this distinction in the definition of platinum sensitivity was important, the POLO trial performed a planned subgroup analysis on the effects of radiologic response to platinum chemotherapy. This subgroup analysis did not show that patients with a radiologic response to platinum-based chemotherapy had improved progression-free survival with olaparib maintenance therapy compared to those with stable disease (91). Hence, although the definitions of platinum-sensitivity differ in ovarian and PDAC populations, biological differences between diseases might best explain the differing outcomes.
Beyond the POLO study, an important question is whether PARP inhibitors have activity in genomic alterations other than germline BRCA1/2 mutations. Emerging data from clinical trials and case series suggest that PARP inhibitors may be effective in both germline and somatic BRCA1/2 and PALB2 alterations (86,89,96,97). With a similar PARP inhibitor maintenance strategy to that in the POLO trial, preliminary results of a phase 2 trial demonstrated the efficacy of maintenance rucaparib in patients with advanced PDAC with tumors harboring somatic and germline BRCA1/2 and PALB2 mutations (96). In this trial, Reiss Blinder et al. observed partial responses in PDAC patients with somatic BRCA2 and germline PALB2 mutations, with the first 24 patients showing an overall response rate of 37% and a median progression-free survival of 9.1 months (96).
The activity of PARP inhibitor monotherapy in PDAC has spurred clinical investigation of combinations of PARP inhibitors and cytotoxic chemotherapy (70,98,99). Myelosuppression, a shared toxicity of cytotoxic chemotherapy and PARP inhibitors, has made these combinations challenging and has forced the use of attenuated PARP inhibitor doses in the combinations. To date, clinical trials have not shown that the addition of a PARP inhibitor enhances the activity of cytotoxic chemotherapy. In two randomized studies, one of first-line gemcitabine/cisplatin in germline BRCA and PALB2-mutated patients and the other of second-line FOLFIRI in a molecularly unselected population, the addition of the PARP inhibitor veliparib did not improve clinical outcomes (70,99).
Resistance to Platinum Chemotherapy and PARP inhibitor Monotherapy
A fundamental question regarding the use of PARP inhibitors in PDAC is understanding the mechanisms of resistance to both platinum chemotherapy and PARP inhibitors in HR-deficient patients (Figure 2). Perhaps the most widely recognized mechanism of resistance is BRCA reversion mutations, which re-establish the correct reading frame and prevent premature termination of protein translation (38,100–102). Reversion mutations in other HR genes that confer PARP inhibitor sensitivity, such as PALB2, RAD51C and RAD51D, have also been reported in ovarian and prostate cancer (103,104). In addition, reversing epigenetic modifications by promoter demethylation has been shown to cause PARP inhibitor resistance in ovarian cancer (105). In BRCA1 mutants, PARP inhibitor resistance can also develop if deleterious mutations inactivate the shieldin complex and thereby allow end resection to proceed without BRCA1 (106,107). PARP inhibitor resistance can also occur as a result of increased transport of the PARP inhibitor outside of the cell through the P-glycoprotein efflux pump (108). Another mechanism is restoring PAR formation, despite PARP inhibition, through inactivation of the enzyme PAR glycohydrolase (PARG), which ordinarily rapidly removes PAR polymers (109). Alternatively, mutations that decrease binding of the PARP protein to DNA, thereby preventing PARP inhibitor trapping, have also been shown to cause PARP inhibitor resistance in preclinical models and in a patient with ovarian cancer with de novo PARP inhibitor resistance (110). Finally, a recent study of patient derived xenografts (PDX) derived from HR deficient patients with PDAC has demonstrated that resistance to platinum chemotherapy and PARP inhibitors is associated with tumor polyploidy and a low proliferation (Ki67) index (60).
Figure 2: Utilization of clinical samples to assess PARP inhibitor resistance.

Cell-free DNA and next generation sequencing (NGS) can be analyzed for HR gene reversion mutations. Whole exome sequencing can assess for HR gene reversion mutations as well as mutations in PARP, PARG, and the shieldin complex/53BP1. RNA expression analysis (RNA-Seq) can evaluate demethylation of HR genes, increased expression of p-glycoprotein pump, as well as decreased expression of shieldin complex/53BP1 and SLFN11. Immunohistochemistry (IHC) assays can assess for low SLFN11 levels and also evaluate the formation of RAD51 foci (RAD51 assay). Live cells, such as organoids, cell lines and patient-derived xenograft mouse models, can allow for fork stability testing.
Multiple preclinical studies in ovarian cancer models have demonstrated that beyond the above mechanisms, PARP inhibitor resistance occurs if the PARP inhibitor-sensitive, unstable replication fork is stabilized (111–114). Studies exploring how replication fork stability is re-established have revealed the central role that the Ataxia telangiectasia and RAD3-related (ATR) protein plays in this process by documenting the hyperactivation of ATR in PARP inhibitor- resistant cancers (28,113,115–118). Similar to ATM, ATR is a PIKK3 related protein that plays a critical role in signal transduction through the DNA damage response pathway. It is activated in response to single-strand DNA that accumulates because of DNA damage and/or stalled replication forks (28). Stalled replication forks, which cause accumulation of single-strand DNA because of the uncoupling of the DNA helicase and DNA polymerase, pose a major challenge to cells, because they can collapse if they are not stabilized (119). The exposed single-strand DNA in collapsed replication forks can lead to large deletions or chromosomal rearrangements (119).
The ability of ATR to promote PARP inhibitor resistance through replication fork stabilization has led to the hypothesis that combined ATR/PARP inhibition may be more effective than single agent PARP inhibition. In PARP inhibitor-resistant ovarian cancers, inhibition of ATR or its downstream signaling partner, CHK1, can re-sensitize BRCA1-mutated tumors to PARP inhibition (113,116,118). These preclinical data have generated enthusiasm for combining PARP and ATR inhibitors in clinical settings. However, because both ATR and PARP inhibitors cause myelosuppression, combining them is challenging. Clinical trials are currently testing multiple dosing schedules of ATR and PARP inhibitors to determine the optimal dosing regimen (Table 2).
Table 2:
Notable Ongoing Trials of DNA Repair Inhibitor Combinations in Pancreatic Adenocarcinoma
| Combination | Trial Number | Phase | Population | Trial Status | Year of Initial Posting* |
|---|---|---|---|---|---|
| ATRi/PARPi | |||||
| Olaparib/Ceralasertib | NCT03682289 | 2 | Solid tumor pts including advanced/metastatic PDAC | Recruiting | 2018 |
| ATRi/Chemotherapy | |||||
| BAY1895344/Irinotecan | NCT04514497 | 1 | Solid tumor pts including advanced/metastatic PDAC pts who have progressed on ≥1 line of therapy | Not yet recruiting | 2020 |
| BAY1895344/Gemcitabine | NCT04616534 | 1 | Solid tumor pts including advanced/metastatic PDAC pts who have progressed on ≥1 line of therapy | Not yet recruiting | 2020 |
| PARPi/Chemotherapy | |||||
| FOLFIRINOX+/−Fluzoparib | NCT04228601 | 1/2 | Advanced/Metastatic PDAC pts with gBRCA1/2 or gPALB2 mutation who have not received prior chemotherapy | Recruiting | 2020 |
| PARPi/PD1i | |||||
| Maintenance Olaparib+/−Pembrolizumab | NCT04548752 | 2 | Platinum sensitive, metastatic PDAC pts with gBRCA1/2 mutation | Recruiting | 2020 |
| Olaparib and Pembrolizumab | NCT04666740 | 2 | PDAC pts with g/sBRCA1/2 or g/sPALB2 mutations (cohort A); PDAC patients with non-BRCA/PALB2 HR gene mutations (cohort B); PDAC pts with exceptional platinum-sensitivity without HR gene alterations (cohort C) | Recruiting | 2020 |
| Multi-agent low dose chemotherapy followed by Olaparib/Pembrolizumab | NCT04753879 | 2 | Metastatic PDAC pts who have not received prior chemotherapy | Not yet recruiting | 2021 |
| Niraparib/TSR-042 | NCT04673448 | 1 | Solid tumor pts including advanced/metastatic PDAC pts, who have a g/sBRCA1/2 mutation, who have progressed on ≥1 line of therapy | Not yet recruiting | 2020 |
| Niraparib combined with nivolumab or ipilimumab | NCT03404960 | 2 | Platinum sensitive, metastatic PDAC pts | Recruiting | 2018 |
| PARPi/VEGFRi | |||||
| Niraparib/Anlotinib | NCT04764084 | 1 | Solid tumor pts including advanced/metastatic PDAC pts who have progressed on ≥1 line of therapy | Not yet recruiting | 2021 |
| WEE1i/Chemotherapy | |||||
| Gemcitabine/ Nab-paclitaxel/ adavosertib | NCT02194829 | 1/2 | Advanced/Metastatic PDAC who have not received prior chemotherapy | Active, not recruiting | 2014 |
Denotes year of initial posting on www.clinicaltrials.gov
Abbreviations: Pancreatic adenocarcinoma (PDAC); inhibitor (i); patients (pts); germline (g); homologous recombination (HR)
Replicative Stress is a Potential Pancreatic Cancer Vulnerability
Stalling or collapse of DNA replication forks, termed replicative stress, is typically compensated for by activation of cell cycle checkpoints, thus prolonging the S/G2 phase of the cell cycle (120,121). The uncontrolled cellular division in PDAC, which is stimulated by the almost universal presence of KRAS mutations together with inactivating mutations in many cell cycle checkpoint proteins such as p53 and CDKN2A, generates substantial replicative stress. Moreover, 5% and 3% of PDACs harbor MYC and CCNE1 amplifications, respectively, which are known to generate high levels of replicative stress (122). Molecular characterization of replicative stress is of great interest, because it may enable exploitation of this potential vulnerability, which can lead to cell death through genomic catastrophe(123).
The single-strand DNA generated by stalled replication forks, a defining hallmark of replicative stress, stimulates the ATR-CHK1-WEE1 pathway activation through the binding of replication protein A (RPA) to single- strand DNA (28,124). ATR interacting protein (ATRIP) binds RPA and subsequently fastens ATR to the single strand DNA. ATR then is activated through phosphorylation by the 9–1-1-TOPBP1 complex. In response to this activation, ATR propagates the DNA damage signal downstream by phosphorylating the CHK1 serine/threonine kinase (28,124). CHK1 activation slows cell cycle progression by promoting activation of the G2/M checkpoint negative regulators CDC25C and WEE1, thus allowing time for DNA replication and repair to be completed (125). One way that CHK1 promotes WEE1 activity is by preventing its ubiquitination through FAM122A sequestration (126). The combined effect of phosphorylation of CDC25C and WEE1—which activates WEE1 but blocks the activity of the CDC25C phosphatase—results in inhibition of the M phase initiating protein CDK1. Similarly, phosphorylation of CDC25C and WEE1 prolongs S phase by inactivating CDK2 (125).
Inhibitors of ATR, CHK1, and WEE1 are currently in clinical trials and may provide an opportunity to perturb the molecular mechanisms that compensate for replicative stress. The activity of single agent inhibitors of each of these proteins is limited, and the primary toxicity of all of these agents is myelosuppression (127–131). However, despite this limited single agent activity, preclinical studies suggest that inhibitors of the ATR-CHK1-WEE1 pathway may have substantial clinical activity if they are combined with agents that prime the cell by increasing replicative stress. One readily clinically available strategy to increase replicative stress is the utilization of agents that decrease the cellular concentrations of dNTPs. Scarcity of dNTPs slows replicative forks and increases replicative stress (132). Gemcitabine is an irreversible inhibitor of the enzyme ribonucleotide reductase, which produces dNTPs (133,134). Ordinarily, the ATR-CHK1-WEE1 pathway is activated in response to the replicative stress caused by gemcitabine, but inhibition of this pathway blocks the cells’ ability to compensate and drives cell death through genomic catastrophe.
In support of this hypothesis, studies of combined gemcitabine and ATR or CHK1 inhibition in PDAC models have demonstrated that the addition of an ATR or CHK1 inhibitor to gemcitabine markedly increases both replicative stress and DNA damage, as measured by increased phosphorylated RPA (pRPA) and γ-H2AX expression respectively (135,136). Both the gemcitabine/CHK1 inhibitor and gemcitabine/ATR inhibitor combinations have demonstrated synergistic cell killing in PDAC cell lines (135,136). Importantly, the gemcitabine/AZD6738 combination has shown in vivo efficacy in both the K8484 PDAC cell line allograft model and the autochthonous KRAS G12D; p53 R172H; Pdx-cre (KPC) mouse model of PDAC (135). A clinical challenge in combining gemcitabine with CHK1 or ATR inhibitors is myelosuppression. However, capitalizing on the impressive synergy of gemcitabine and CHK1 inhibitors, a trial using low dose gemcitabine (250 mg/m2) combined with the SRA737 CHK1 inhibitor has demonstrated multiple partial responses in solid tumors, despite the absence of responses in the SRA737 monotherapy trial (137,138).
The G2/M checkpoint is critical in malignancies such as PDAC, in which more than 75% of tumors have p53 pathway deficits (4,8). Lal et al. demonstrated that gemcitabine combined with the WEE1 inhibitor adavosertib (AZD1775) has enhanced activity in HR-proficient PDAC cell lines compared to HR-deficient PDAC cell lines (139). In agreement with this, Kausar et al. found that gemcitabine chemoradiation combined with adavosertib is more effective in HR-proficient PDAC cell lines than HR-deficient PDAC cell lines (140). The potential mechanism underlying the differential sensitivity of the gemcitabine/adavosertib combination may be that, compared to HR-deficient cells, HR-proficient cells do not linger in S phase and are more likely to be forced into mitosis and to undergo genomic catastrophe (139). In agreement with these findings, Drean et al. have demonstrated that in vitro and in vivo PDAC models with acquired resistance to PARP inhibitors are highly sensitive to adavosertib-mediated WEE1 inhibition (141). Further validation of the gemcitabine/WEE1 inhibitor combination has been reported in a phase 1 trial of patients with unresectable, locally advanced PDAC, in whom the combination was administered with radiation. The combination of radiation and gemcitabine/adavosertib demonstrated a favorable toxicity profile along with encouraging activity (142). Patients treated in the trial had a median progression-free survival of 9.4 months and a median overall survival of 21.7 months, results comparing favorably to those with other regimens tested in the unresectable setting. Furthermore, data from a randomized phase 2 clinical trial in platinum resistant/refractory ovarian cancer demonstrated that patients treated with the gemcitabine/adavosertib combination had superior progression-free survival compared to patients on the gemcitabine/placebo arm (143).
Biomarkers of Replicative Stress That May Aid in the Clinical Development of DNA Repair Inhibitors
Maximizing the potential of DNA repair inhibitors in pancreatic cancer will require the development of biomarkers to guide their clinical utilization (Figure 3). In recent years, preclinical efforts to identify biomarkers of replicative stress have yielded several candidates. The most widely utilized biomarker of replicative stress is pRPA (61,114,144). Inside the cell, RPA binds single-strand DNA, thereby activating the DNA response pathway. Beyond pRPA, another possible immunohistochemical marker of replicative stress is phosphorylated KAP1 (pKAP1). Like RPA, KAP1 is phosphorylated by ATR during replicative stress, thus leading to heterochromatin relaxation, which enables DNA repair and is part of the signal transduction cascade promoting cell cycle arrest and apoptosis (145).
Figure 3: Utilization of clinical samples to assess the DNA damage response.

Several immunohistochemical (IHC) markers can be utilized to interrogate the DNA damage response. γH2AX is a marker of DNA damage while phosphorylated RPA (pRPA) and phosphorylated KAP1 (pKAP1) are markers of replicative stress. Markers of replicative stress regulation (SLFN11, ATR, WEE1, pRAD50, pCHK1, and pCDK1) and inducers of replicative stress (CCNE1 and MYC) can also be identified by IHC assays. RNA expression analysis (RNA-Seq) can be utilized to detect signatures of replicative stress and measure levels of RNA transcripts of genes involved in replicative stress (SLFN11, ATR, WEE1, CCNE1 and MYC). Live cells (such as organoids, cell lines and patient-derived xenograft mouse models) can be harvested to perform fork stability testing.
Beyond immunohistochemical markers, when live cells (such as organoids and cell lines) are available, DNA fiber assays that assess nucleotide uptake can determine whether a replication fork is stable or unstable (114,146). Another approach is the utilization of an RNA expression signature to detect replicative stress. Dreyer et al. have recently demonstrated that PDAC patient-derived cell lines with a high replicative stress signature score show increased pRPA staining along with enhanced susceptibility to ATR and WEE1 inhibitors (61). Interestingly, these PDAC models with high replicative stress signature scores were not enriched for mutations in classical HR genes (61).
Another emerging immunohistochemical biomarker is schlafen 11 (SLFN11), a protein in the family of schlafen, meaning “sleep” in German. SLFN11 came to the attention of the oncology community in 2012, when drug sensitivity testing of two large cell line collections revealed an association between diminished SLFN11 expression and resistance to cytotoxic chemotherapies such as cisplatin, gemcitabine, irinotecan, and alkylating agents (147,148). Similarly, low levels of SLFN11 are also predictive of resistance to PARP inhibitors (149,150). SLFN11 protein levels are modulated in cancerous cells through either transcriptional regulation and/or silencing via promoter methylation (151).
Mechanistic studies have revealed that SLFN11 localizes to regions of DNA under replicative stress in an ATR-independent fashion (152). In agreement with SLFN11 functioning independently from the ATR-CHK1-WEE1 pathway in replicative stress, inhibitors of the ATR-CHK1-WEE1 pathway re-sensitize SLFN11-deficient tumors to cytotoxic chemotherapy (153). Furthermore, the degree of synergy between cytotoxic chemotherapy and inhibitors of the ATR-CHK1-WEE1 pathway appears to be greatest in SLFN11-deficient cell lines (153). Structural analysis has revealed that SLFN11 has a helicase domain (147). Recent work has demonstrated that this helicase domain exposes unstable replication forks to nucleases, such as DNA2 and MRE11, which degrade the replication fork (154). Hence, whereas ATR-CHK1-WEE1 prevent genomic catastrophe from collapsed replication forks, SLFN11 functions differently and promotes nuclease degradation of unstable replication forks. This finding has important implications, suggesting that SLNF11’s promotion of nuclease-mediated degradation of replication forks is a key step in the cancer cell death caused by cytotoxic chemotherapies and PARP inhibitors.
Therapeutic Targeting of ATM
Approximately 5–10% of patients with pancreatic cancer have either germline or somatic ATM mutations (4,38,39). The inability to therapeutically target ATM mutations has been a source of frustration for clinicians caring for patients with pancreatic cancer. Whereas ATM activates the HR pathway in response to double-strand DNA breaks, the efficacy of single-agent PARP inhibitors in this population has been disappointing (155). Unlike deleterious BRCA1/2 and PALB2 alterations, PDACs with inactivating biallelic ATM alterations do not exhibit the characteristic genomic changes, such as COSMIC signature 3, seen in HR deficient cancers (48). Hence, strategies for targeting ATM deficiency likely need to move beyond PARP inhibitor monotherapy.
ATM, ATR, and DNA-PK are a trio of homologous PIKK related proteins at the apex of the DNA response signal transduction pathway (156). Although these proteins are each activated differently, they have overlapping functions and can compensate for one another if one of the three proteins is deficient (156) (Figure 4). Consequently, inhibitors of DNA-PK and ATR have been shown to be synthetically lethal in ATM deficient tumors (157–162). Recently, a phase 1 trial of the BAY 1895344 ATR inhibitor has shown single-agent activity in patients with solid tumors with ATM deficiency(131). Importantly, the investigators defined ATM deficiency by less than 1% of the evaluated tumor cells’ nuclei staining positive for ATM (131).” Encouragingly, a recent preclinical study has also demonstrated substantial synergy in cell lines and murine PDAC models with ATM-deficiency treated with a combination of the AZD6738 ATR inhibitor and low dose gemcitabine (163). Further clinical study of ATR inhibitors in ATM-deficient PDAC is needed.
Figure 4: Synthetic lethality of ATM and ATR.

A. Double-strand DNA (dsDNA) breaks activate ATM leading to CHK2 phosphorylation. CHK2 subsequently activates p53 and promotes G1/S phase checkpoint blockade. Replicative stress, through the accumulation of single strand DNA, activates ATR. ATR phosphorylates CHK1 leading to activation of p53 and WEE1 as well as the inhibition of CDC25C, which ultimately causes S phase prolongation and G2/M checkpoint blockade. B. When there is ATM deficiency, replicative stress increases leading to compensatory activation of the ATR signaling pathway. C. Lack of signaling through both the ATR and ATM pathways prevents DNA repair and leads ultimately to cancer cell death from genomic catastrophe.
Combining DNA Repair Inhibitors with Immunotherapeutics
Development of immunotherapy in PDAC to date has been stymied by its immunosuppressive microenvironment stemming from the heightened activity of regulatory T cells (Tregs) and myeloid-derived suppressor cells, coupled with diminished activity of effector CD8+ T cells, NK cells, and dendritic cells (3). Immunological checkpoint blockade with PD1 and CTLA-4 directed therapies has been ineffective in microsatellite stable pancreatic cancers, potentially because of the scarcity of intratumoral effector CD8+ T cells and PDAC’s modest tumor mutational burden (TMB) (3). Whereas the TMB of biallelic HR-deficient PDACs is slightly higher than that of HR proficient PDACs, the median TMB is still below 10, and PD1 directed monotherapy is also ineffective in this population (38).
A major goal in PDAC immunotherapy is increasing activation and expansion of anti-tumor T cells (164). One possible way to accomplish this goal is by leveraging the ability of the innate immune system, which has evolved to defend against viral and bacterial pathogens by activating interferon-mediated immune response pathways. The cyclic GMP–AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway, which evolved to detect viral intracytoplasmic DNA, is activated when cGAS binds double-strand DNA (165). Subsequently, cGAS undergoes a conformational change that transforms it into the second messenger 2’3’-cGAMP, which activates STING, thereby resulting in the expression of interferon related genes and the eventual secretion of type I interferons (165,166). In response to type I interferons, the immune microenvironment becomes more pro-inflammatory, leading to dendritic cell activation, which in turn promotes CD8+ T cell priming and migration (165). Mechanistic studies have revealed that inhibitors of DNA damage repair, such as PARP and ATR inhibitors, activate the cGAS-STING pathway and thereby promote intratumoral CD8+ T cell infiltration and antitumor immune responses in BRCA1-deficient models of breast and ovarian cancer (167,168). A recent preclinical study has suggested that STING agonists synergistically increase cell death when combined with a PARP inhibitor in a murine model of BRCA1-deficient triple-negative breast cancer (169). In pancreatic cancer models, ATM inhibition leads to type I interferon production, although the mechanism of action is independent of cGAS/STING, suggesting a possible opportunity for combination therapy (170).
Although additional study is needed, exploration of the PARP inhibitor and STING agonist combination in pancreatic cancer is worthwhile. Multiple preclinical studies have demonstrated the ability of the STING pathway to increase the migration and activation of effector CD8+ T cells into pancreatic cancers (171–173). To date, clinical trials of STING agonists have shown that these agents are well tolerated, and consequently no toxicity barriers to PARP inhibitor/STING agonist combination are expected to exist (174). Early generation STING agonists were hampered by heterogeneity of STING alleles in the human population and pharmacokinetics limiting drug delivery to the target, but next generation STING agonists have been designed to address both of these issues (174). Given the presence of DNA repair deficiencies in a high fraction of PDAC cases, novel opportunities to synergistically target DNA repair pathways and immune checkpoints may emerge with additional study.
Conclusion
DNA repair defects are common in PDAC and represent a tractable therapeutic opportunity in this challenging disease. The efficacy of PARP inhibitors in germline BRCA-mutated PDAC provides proof-of-concept that DNA repair inhibitors can be effective in treating pancreatic cancer. However, the modest efficacy observed in the POLO trial highlights the need to better understand biomarkers that predict response to these agents. Furthermore, the limited efficacy of PARP inhibitor monotherapy highlights the need for combining DNA repair inhibitors with synergistic agents. A challenge in using combinatorial strategies will be overcoming toxicity concerns such as myelosuppression. Increasing mechanistic understanding of these pathways may lead to strategies that minimize toxicity concerns, such as low dose cytotoxic compounds and immunotherapeutic agents. Furthermore, the development of these precision medicine strategies in PDAC will require careful clinical trial design to ensure that the DNA repair inhibitors are tested in the appropriate molecularly defined populations.
Acknowledgments
Funding: This work was supported by the DFCI Hale Family Center for Pancreatic Cancer (BMW, AJA, JMC, SKD), Stand Up To Cancer (BMW, GIS, ADD, JMC), the Lustgarten Foundation (BMW, GIS, AJA, JAN, ADD, JMC), NIH P50 CA127003, and the Dana-Farber/Harvard Cancer Center Specialized Program of Research Excellence (SPORE) in Gastrointestinal Cancer (BMW, GIS, AJA, JAN, ADD, JMC, SR). BMW is also supported by NIH U01 CA210171, the Noble Effort Fund, the Wexler Family Fund, and Promises for Purple. AJA is also supported by the Doris Duke Charitable Foundation, Pancreatic Cancer Action Network, NIH-NCI K08 CA218420–02, U01 CA224146, and U01 CA250549. JAN is also supported by NIH U01 CA250549, R01 CA248857, and R01 CA205406. SR is also supported by the Hope Funds for Cancer Research, an Eleanor and Miles Shore Faculty Development Award, and Harvard Catalyst/The Harvard Clinical and Translational Science Center UL1 TR002541. JMC is also supported by the Grateful Foundation, Team Evan Schumacher, and the Haya Linde Memorial Fund.
Footnotes
Disclosures
JMC received research funding to his institution from Abbvie, Merus, Roche, and Bristol Myers Squibb. He received research funding from Merck, Astrazeneca, Esperas Pharma, and Tesaro; received consulting fees from Bristol Myers Squibb; and received travel funding from Bristol Myers Squibb. BMW receives grant support from Celgene and Eli Lilly, and has consulted for BioLineRx, Celgene, and GRAIL. GIS has received research funding from Eli Lilly, Merck KGaA/EMD-Serono, Merck, and Sierra Oncology. He has served on advisory boards for Pfizer, Eli Lilly, G1 Therapeutics, Roche, Merck KGaA/EMD-Serono, Sierra Oncology, Bicycle Therapeutics, Fusion Pharmaceuticals, Cybrexa Therapeutics, Astex, Almac, Ipsen, Bayer, Angiex, Daiichi Sankyo, Seattle Genetics, Boehringer Ingelheim, ImmunoMet, Asana, Artios, Atrin, Concarlo Holdings, Syros, Zentalis, and CytomX. In addition, he holds a patent entitled “Dosage regimen for sapacitabine and seliciclib,” also issued to Cyclacel Pharmaceuticals, and a pending patent entitled “Compositions and Methods for Predicting Response and Resistance to CDK4/6 Inhibition,” together with Liam Cornell. OA has received research support from Merck and speaker fees for activities supported by educational grants from BMS and Merck. AJA has consulted for Oncorus, Inc., Arrakis Therapeutics, and Merck & Co., Inc, and has research funding from Mirati Therapeutics, Deerfield, Inc., and Novo Ventures that is unrelated to this work. ADD receives research funding from Eli Lilly and Merck KGaA-EMD Serono; has served on advisory boards for Eli Lilly, Merck KGaA-EMD Serono, Sierra Oncology, and Formation Biologics; and holds equity in Ideaya Inc, Cyteir Therapeutics, and Cedilla Therapeutics, Inc. SKD receives research funding from Novartis, Bristol Myers Squibb, and Eli Lilly and is a co-founder of Kojin Therapeutics.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70(1):7–30 doi 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74(11):2913–21 doi 10.1158/0008-5472.Can-14-0155. [DOI] [PubMed] [Google Scholar]
- 3.Balachandran VP, Beatty GL, Dougan SK. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019;156(7):2056–72 doi 10.1053/j.gastro.2018.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, et al. Real-time genomic characterization of advanced pancreatic cancer to enable precision medicine. Cancer Discovery 2018. doi 10.1158/2159-8290.Cd-18-0275. [DOI] [PMC free article] [PubMed]
- 5.Jones S, Zhang X, Parsons DW, Lin JC-H, Leary RJ, Angenendt P, et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008;321(5897):1801–6 doi 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nature communications 2015;6:6744 doi 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531(7592):47–52 doi 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 8.Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer cell 2017;32(2):185–203.e13 doi 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yurgelun MB, Chittenden AB, Morales-Oyarvide V, Rubinson DA, Dunne RF, Kozak MM, et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genetics in medicine : official journal of the American College of Medical Genetics 2019;21(1):213–23 doi 10.1038/s41436-018-0009-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. New England Journal of Medicine 2019. doi 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed]
- 11.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. New England Journal of Medicine 2011;364(19):1817–25 doi 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 12.Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 1997;15(6):2403–13 doi 10.1200/jco.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 13.Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul J-L, et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. New England Journal of Medicine 2018;379(25):2395–406 doi 10.1056/NEJMoa1809775. [DOI] [PubMed] [Google Scholar]
- 14.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369(18):1691–703 doi 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muranaka T, Kuwatani M, Komatsu Y, Sawada K, Nakatsumi H, Kawamoto Y, et al. Comparison of efficacy and toxicity of FOLFIRINOX and gemcitabine with nab-paclitaxel in unresectable pancreatic cancer. Journal of gastrointestinal oncology 2017;8(3):566–71 doi 10.21037/jgo.2017.02.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang-Gillam A, Li CP, Bodoky G, Dean A, Shan YS, Jameson G, et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016;387(10018):545–57 doi 10.1016/s0140-6736(15)00986-1. [DOI] [PubMed] [Google Scholar]
- 17.Pelzer U, Schwaner I, Stieler J, Adler M, Seraphin J, Dörken B, et al. Best supportive care (BSC) versus oxaliplatin, folinic acid and 5-fluorouracil (OFF) plus BSC in patients for second-line advanced pancreatic cancer: a phase III-study from the German CONKO-study group. Eur J Cancer 2011;47(11):1676–81 doi 10.1016/j.ejca.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 18.Tempero MA, Reni M, Riess H, Pelzer U, O’Reilly EM, Winter JM, et al. APACT: phase III, multicenter, international, open-label, randomized trial of adjuvant nab-paclitaxel plus gemcitabine (nab-P/G) vs gemcitabine (G) for surgically resected pancreatic adenocarcinoma. Journal of Clinical Oncology 2019;37(15_suppl):4000- doi 10.1200/JCO.2019.37.15_suppl.4000. [DOI] [Google Scholar]
- 19.Janssen QP, O’Reilly EM, van Eijck CHJ, Groot Koerkamp B. Neoadjuvant Treatment in Patients With Resectable and Borderline Resectable Pancreatic Cancer. Front Oncol 2020;10:41 doi 10.3389/fonc.2020.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katz MHG. Alliance A021501: Preoperative mFOLFIRINOX or mFOLFIRINOX plus hypofractionated radiation therapy (RT) for borderline resectable (BR) adenocarcinoma of the pancreas. In: Matthew H. G. Katz QSJPMJMHMCBMWSARdWMLHSSBW, x, Reilly, The University of Texas Md Anderson Cancer Center HTX, Mayo Clinic RMN, Miami Cancer Institute MFL, et al. , editors 2021; Gastrointestinal Cancers Symposium. American Society of Clinical Oncology. [Google Scholar]
- 21.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature 2012;481(7381):287–94 doi 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 22.Cleary JM, Aguirre AJ, Shapiro GI, D’Andrea AD. Biomarker-Guided Development of DNA Repair Inhibitors. Molecular cell 2020;78(6):1070–85 doi 10.1016/j.molcel.2020.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fishel R Mismatch repair. J Biol Chem 2015;290(44):26395–403 doi 10.1074/jbc.R115.660142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 2017;58(5):235–63 doi 10.1002/em.22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schärer OD. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol 2013;5(10):a012609 doi 10.1101/cshperspect.a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao B, Rothenberg E, Ramsden DA, Lieber MR. The molecular basis and disease relevance of non-homologous DNA end joining. Nat Rev Mol Cell Biol 2020;21(12):765–81 doi 10.1038/s41580-020-00297-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bian L, Meng Y, Zhang M, Li D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: implications for cancer treatment. Molecular Cancer 2019;18(1):169 doi 10.1186/s12943-019-1100-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams RM, Zhang X. Roles of ATM and ATR in DNA double strand breaks and replication stress. Prog Biophys Mol Biol 2020. doi 10.1016/j.pbiomolbio.2020.11.005. [DOI] [PubMed]
- 29.Wright WD, Shah SS, Heyer WD. Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem 2018;293(27):10524–35 doi 10.1074/jbc.TM118.000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018;560(7716):117–21 doi 10.1038/s41586-018-0340-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Molecular cell 2013;49(5):872–83 doi 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nature Reviews Molecular Cell Biology 2017;18(8):495–506 doi 10.1038/nrm.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao F, Kim W, Kloeber JA, Lou Z. DNA end resection and its role in DNA replication and DSB repair choice in mammalian cells. Experimental & Molecular Medicine 2020;52(10):1705–14 doi 10.1038/s12276-020-00519-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sy SMH, Huen MSY, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proceedings of the National Academy of Sciences 2009;106(17):7155–60 doi 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miki Y, Swensen J, Shattuck-Eidens D, Futreal P, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994;266(5182):66–71 doi 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 36.Erkko H, Xia B, Nikkilä J, Schleutker J, Syrjäkoski K, Mannermaa A, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature 2007;446(7133):316–9 doi 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- 37.Ceccaldi R, Sarangi P, D’Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nature Reviews Molecular Cell Biology 2016;17(6):337–49 doi 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- 38.Park W, Chen J, Chou JF, Varghese AM, Yu KH, Wong W, et al. Genomic Methods Identify Homologous Recombination Deficiency in Pancreas Adenocarcinoma and Optimize Treatment Selection. Clinical Cancer Research 2020. doi 10.1158/1078-0432.Ccr-20-0418. [DOI] [PMC free article] [PubMed]
- 39.Russell R, Perkhofer L, Liebau S, Lin Q, Lechel A, Feld FM, et al. Loss of ATM accelerates pancreatic cancer formation and epithelial-mesenchymal transition. Nature communications 2015;6:7677- doi 10.1038/ncomms8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roberts NJ, Norris AL, Petersen GM, Bondy ML, Brand R, Gallinger S, et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov 2016;6(2):166–75 doi 10.1158/2159-8290.Cd-15-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tempero MA. NCCN Guidelines Updates: Pancreatic Cancer. J Natl Compr Canc Netw 2019;17(5.5):603–5 doi 10.6004/jnccn.2019.5007. [DOI] [PubMed] [Google Scholar]
- 42.Aslanian HR, Lee JH, Canto MI. AGA Clinical Practice Update on Pancreas Cancer Screening in High-Risk Individuals: Expert Review. Gastroenterology 2020;159(1):358–62 doi 10.1053/j.gastro.2020.03.088. [DOI] [PubMed] [Google Scholar]
- 43.Glodzik D, Bosch A, Hartman J, Aine M, Vallon-Christersson J, Reuterswärd C, et al. Comprehensive molecular comparison of BRCA1 hypermethylated and BRCA1 mutated triple negative breast cancers. Nature communications 2020;11(1):3747- doi 10.1038/s41467-020-17537-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet 2017;49(10):1476–86 doi 10.1038/ng.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500(7463):415–21 doi 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen L WM Martens J, Van Hoeck A, Cuppen E. Pan-cancer landscape of homologous recombination deficiency. Nature communications 2020;11(1):5584 doi 10.1038/s41467-020-19406-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nature medicine 2017;23(4):517–25 doi 10.1038/nm.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Golan T, O’Kane GM, Denroche RE, Raitses-Gurevich M, Grant RC, Holter S, et al. Genomic Features and Classification of Homologous Recombination Deficient Pancreatic Ductal Adenocarcinoma. Gastroenterology 2021. doi 10.1053/j.gastro.2021.01.220. [DOI] [PubMed]
- 49.Gulhan DC, Lee JJ, Melloni GEM, Cortes-Ciriano I, Park PJ. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet 2019;51(5):912–9 doi 10.1038/s41588-019-0390-2. [DOI] [PubMed] [Google Scholar]
- 50.Farkkila A, Gulhan DC, Casado J, Jacobson CA, Nguyen H, Kochupurakkal B, et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nature communications 2020;11(1):1459 doi 10.1038/s41467-020-15315-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore KN, Secord AA, Geller MA, Miller DS, Cloven N, Fleming GF, et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): a multicentre, open-label, single-arm, phase 2 trial. The Lancet Oncology 2019;20(5):636–48 doi 10.1016/s1470-2045(19)30029-4. [DOI] [PubMed] [Google Scholar]
- 52.Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. New England Journal of Medicine 2019;381(25):2416–28 doi 10.1056/NEJMoa1911361. [DOI] [PubMed] [Google Scholar]
- 53.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518(7540):495–501 doi 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature 2000;406(6797):747–52 doi 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 55.Botti G, Cantile M, Collina F, Cerrone M, Sarno S, Anniciello A, et al. Morphological and pathological features of basal-like breast cancer. Translational Cancer Research 2019:S503–S9. [DOI] [PMC free article] [PubMed]
- 56.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 2015;47(10):1168–78 doi 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531(7592):47–52 doi 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 58.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nature medicine 2011;17(4):500–3 doi 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aung KL, Fischer SE, Denroche RE, Jang GH, Dodd A, Creighton S, et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clinical cancer research : an official journal of the American Association for Cancer Research 2018;24(6):1344–54 doi 10.1158/1078-0432.Ccr-17-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Y, Park JYP, Pacis A, Denroche RE, Jang GH, Zhang A, et al. A Preclinical Trial and Molecularly Annotated Patient Cohort Identify Predictive Biomarkers in Homologous Recombination-deficient Pancreatic Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2020;26(20):5462–76 doi 10.1158/1078-0432.Ccr-20-1439. [DOI] [PubMed] [Google Scholar]
- 61.Dreyer SB, Upstill-Goddard R, Paulus-Hock V, Paris C, Lampraki EM, Dray E, et al. Targeting DNA Damage Response and Replication Stress in Pancreatic Cancer. Gastroenterology 2021;160(1):362–77.e13 doi 10.1053/j.gastro.2020.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rycenga HB, Long DT. The evolving role of DNA inter-strand crosslinks in chemotherapy. Curr Opin Pharmacol 2018;41:20–6 doi 10.1016/j.coph.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nature Reviews Cancer 2011;11(7):467–80 doi 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hashimoto S, Anai H, Hanada K. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes and Environment 2016;38(1):9 doi 10.1186/s41021-016-0037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Olivieri M, Cho T, Álvarez-Quilón A, Li K, Schellenberg MJ, Zimmermann M, et al. A Genetic Map of the Response to DNA Damage in Human Cells. Cell 2020;182(2):481–96.e21 doi 10.1016/j.cell.2020.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goldstein JB, Zhao L, Wang X, Ghelman Y, Overman MJ, Javle MM, et al. Germline DNA Sequencing Reveals Novel Mutations Predictive of Overall Survival in a Cohort of Patients with Pancreatic Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2020;26(6):1385–94 doi 10.1158/1078-0432.Ccr-19-0224. [DOI] [PubMed] [Google Scholar]
- 67.Pishvaian MJ, Blais EM, Brody JR, Lyons E, DeArbeloa P, Hendifar A, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. The Lancet Oncology 2020;21(4):508–18 doi 10.1016/s1470-2045(20)30074-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Golan T, Kanji ZS, Epelbaum R, Devaud N, Dagan E, Holter S, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer 2014;111(6):1132–8 doi 10.1038/bjc.2014.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Golan T, Barenboim A, Lahat G, Nachmany I, Goykhman Y, Shacham-Shmueli E, et al. Increased Rate of Complete Pathologic Response After Neoadjuvant FOLFIRINOX for BRCA Mutation Carriers with Borderline Resectable Pancreatic Cancer. Ann Surg Oncol 2020;27(10):3963–70 doi 10.1245/s10434-020-08469-8. [DOI] [PubMed] [Google Scholar]
- 70.O’Reilly EM, Lee JW, Zalupski M, Capanu M, Park J, Golan T, et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin With or Without Veliparib in Patients With Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2020;38(13):1378–88 doi 10.1200/jco.19.02931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang A, Garraway LA, Ashworth A, Weber B. Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discov 2020;19(1):23–38 doi 10.1038/s41573-019-0046-z. [DOI] [PubMed] [Google Scholar]
- 72.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434(7035):913–7 doi http://www.nature.com/nature/journal/v434/n7035/suppinfo/nature03443_S1.html. [DOI] [PubMed] [Google Scholar]
- 73.Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434(7035):917–21 doi http://www.nature.com/nature/journal/v434/n7035/suppinfo/nature03445_S1.html. [DOI] [PubMed] [Google Scholar]
- 74.Wei H, Yu X. Functions of PARylation in DNA Damage Repair Pathways. Genomics, Proteomics & Bioinformatics 2016;14(3):131–9 doi 10.1016/j.gpb.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Science Translational Medicine 2016;8(362):362ps17–ps17 doi 10.1126/scitranslmed.aaf9246. [DOI] [PubMed] [Google Scholar]
- 76.Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, et al. BMN 673, a Novel and Highly Potent PARP1/2 Inhibitor for the Treatment of Human Cancers with DNA Repair Deficiency. Clinical Cancer Research 2013;19(18):5003–15 doi 10.1158/1078-0432.ccr-13-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Murai J, Huang S-YN, Renaud A, Zhang Y, Ji J, Takeda S, et al. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Molecular cancer therapeutics 2014;13(2):433–43 doi 10.1158/1535-7163.mct-13-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 2012;72(21):5588–99 doi 10.1158/0008-5472.Can-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ranjha L, Howard SM, Cejka P. Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma 2018;127(2):187–214 doi 10.1007/s00412-017-0658-1. [DOI] [PubMed] [Google Scholar]
- 80.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS genetics 2008;4(6):e1000110 doi 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dutta A, Eckelmann B, Adhikari S, Ahmed KM, Sengupta S, Pandey A, et al. Microhomology-mediated end joining is activated in irradiated human cells due to phosphorylation-dependent formation of the XRCC1 repair complex. Nucleic acids research 2017;45(5):2585–99 doi 10.1093/nar/gkw1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Patterson-Fortin J, D’Andrea AD. Exploiting the Microhomology-Mediated End-Joining Pathway in Cancer Therapy. Cancer Res 2020;80(21):4593–600 doi 10.1158/0008-5472.Can-20-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015;518(7538):258–62 doi 10.1038/nature14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou J, Gelot C, Pantelidou C, Li A, Yücel H, Davis RE, et al. Polymerase Theta Inhibition Kills Homologous Recombination Deficient Tumors. bioRxiv 10.1101/2020.05.23.111658. [DOI] [PMC free article] [PubMed]
- 85.Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2010;28(15):2512–9 doi 10.1200/jco.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 86.Shroff RT, Hendifar A, McWilliams RR, Geva R, Epelbaum R, Rolfe L, et al. Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. 2018(2):1–15 doi 10.1200/po.17.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015;33(3):244–50 doi 10.1200/jco.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de Bono J, Ramanathan RK, Mina L, Chugh R, Glaspy J, Rafii S, et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. 2017;7(6):620–9 doi 10.1158/2159-8290.CD-16-1250 %J Cancer Discovery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Javle M, Shacham-Shmueli E, Xiao L, Varadhachary G, Halpern N, Fogelman D, et al. Olaparib Monotherapy for Previously Treated Pancreatic Cancer With DNA Damage Repair Genetic Alterations Other Than Germline BRCA Variants: Findings From 2 Phase 2 Nonrandomized Clinical Trials. JAMA Oncology 2021. doi 10.1001/jamaoncol.2021.0006. [DOI] [PMC free article] [PubMed]
- 90.Golan T Overall survival from the phase 3 POLO trial: Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. In: Talia Golan PHMREVCTMMJHJOPDHDAD-YOACR-SGT, xFc, l EMO, x, Reilly DMKCKSGYLHLK, The Oncology Institute SMCaT-HTAUTAI, et al. , editors 2021; Gastrointestinal Cancers Symposium. American Society of Clinical Oncology. [Google Scholar]
- 91.Nishikawa G, Booth C, Prasad V. Olaparib for BRCA mutant pancreas cancer: Should the POLO trial change clinical practice? Cancer 2020;126(18):4087–8 doi 10.1002/cncr.32979. [DOI] [PubMed] [Google Scholar]
- 92.Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. The Lancet Oncology 2017;18(9):1274–84 doi 10.1016/s1470-2045(17)30469-2. [DOI] [PubMed] [Google Scholar]
- 93.Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;390(10106):1949–61 doi 10.1016/S0140-6736(17)32440-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med 2016;375(22):2154–64 doi 10.1056/NEJMoa1611310. [DOI] [PubMed] [Google Scholar]
- 95.Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med 2018;379(26):2495–505 doi 10.1056/NEJMoa1810858. [DOI] [PubMed] [Google Scholar]
- 96.Binder KAR, Mick R, O’Hara M, Teitelbaum U, Karasic T, Schneider C, et al. Abstract CT234: A Phase II, single arm study of maintenance rucaparib in patients with platinum-sensitive advanced pancreatic cancer and a pathogenic germline or somatic mutation in BRCA1, BRCA2 or PALB2. 2019;79(13 Supplement):CT234-CT doi 10.1158/1538–7445.AM2019-CT234 % J Cancer Research. [DOI] [PubMed] [Google Scholar]
- 97.Borazanci E, Korn R, Liang WS, Guarnieri C, Haag S, Snyder C, et al. An Analysis of Patients with DNA Repair Pathway Mutations Treated with a PARP Inhibitor. Oncologist 2020;25(1):e60–e7 doi 10.1634/theoncologist.2018-0905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pishvaian MJ, Wang H, He AR, Hwang JJ, Smaglo BG, Kim SS, et al. A Phase I/II Study of Veliparib (ABT-888) in Combination with 5-Fluorouracil and Oxaliplatin in Patients with Metastatic Pancreatic Cancer. Clinical Cancer Research 2020;26(19):5092–101 doi 10.1158/1078-0432.Ccr-20-1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chiorean EG, Guthrie KA, Philip PA, Swisher EM, Jalikis F, Pishvaian MJ, et al. Randomized phase II study of second-line modified FOLFIRI with PARP inhibitor ABT-888 (Veliparib) (NSC-737664) versus FOLFIRI in metastatic pancreatic cancer (mPC): SWOG S1513. Journal of Clinical Oncology 2019;37(15_suppl):4014- doi 10.1200/JCO.2019.37.15_suppl.4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weigelt B, Comino-Mendez I, de Bruijn I, Tian L, Meisel JL, Garcia-Murillas I, et al. Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-Free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23(21):6708–20 doi 10.1158/1078-0432.Ccr-17-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pishvaian MJ, Biankin AV, Bailey P, Chang DK, Laheru D, Wolfgang CL, et al. BRCA2 secondary mutation-mediated resistance to platinum and PARP inhibitor-based therapy in pancreatic cancer. British Journal Of Cancer 2017;116:1021 doi 10.1038/bjc.2017.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pettitt SJ, Frankum JR, Punta M, Lise S, Alexander J, Chen Y, et al. Clinical BRCA1/2 Reversion Analysis Identifies Hotspot Mutations and Predicted Neoantigens Associated with Therapy Resistance. Cancer Discov 2020;10(10):1475–88 doi 10.1158/2159-8290.Cd-19-1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Goodall J, Mateo J, Yuan W, Mossop H, Porta N, Miranda S, et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discovery 2017;7(9):1006–17 doi 10.1158/2159-8290.Cd-17-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI, et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discovery 2017;7(9):984–98 doi 10.1158/2159-8290.Cd-17-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kondrashova O, Topp M, Nesic K, Lieschke E, Ho G-Y, Harrell MI, et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nature communications 2018;9(1):3970- doi 10.1038/s41467-018-05564-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nature cell biology 2018;20(8):954–65 doi 10.1038/s41556-018-0140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov 2013;3(1):68–81 doi 10.1158/2159-8290.Cd-12-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proceedings of the National Academy of Sciences of the United States of America 2008;105(44):17079–84 doi 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer cell 2018;33(6):1078–93.e12 doi 10.1016/j.ccell.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 110.Pettitt SJ, Krastev DB, Brandsma I, Dréan A, Song F, Aleksandrov R, et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nature communications 2018;9(1):1849 doi 10.1038/s41467-018-03917-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rondinelli B, Gogola E, Yücel H, Duarte AA, van de Ven M, van der Sluijs R, et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nature cell biology 2017;19:1371 doi 10.1038/ncb362610.1038/ncb3626https://www.nature.com/articles/ncb3626#supplementary-informationhttps://www.nature.com/articles/ncb3626#supplementary-information. [DOI] [PubMed] [Google Scholar]
- 112.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016;535(7612):382–7 doi 10.1038/nature18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes & development 2017;31(3):318–32 doi 10.1101/gad.290957.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hill SJ, Decker B, Roberts EA, Horowitz NS, Muto MG, Worley MJ Jr., et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov 2018;8(11):1404–21 doi 10.1158/2159-8290.Cd-18-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim H, George E, Ragland R, Rafial S, Zhang R, Krepler C, et al. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23(12):3097–108 doi 10.1158/1078-0432.Ccr-16-2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Parmar K, Kochupurakkal BS, Lazaro J-B, Wang ZC, Palakurthi S, Kirschmeier PT, et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clinical Cancer Research 2019;25(20):6127 doi 10.1158/1078-0432.CCR-19-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kim H, George E, Ragland RL, Rafail S, Zhang R, Krepler C, et al. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in <em>BRCA</em>-Mutant Ovarian Cancer Models. Clinical Cancer Research 2017;23(12):3097 doi 10.1158/1078-0432.CCR-16-2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nature communications 2020;11(1):3726- doi 10.1038/s41467-020-17127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Alexander JL, Orr-Weaver TL. Replication fork instability and the consequences of fork collisions from rereplication. Genes & development 2016;30(20):2241–52 doi 10.1101/gad.288142.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gelot C, Magdalou I, Lopez BS. Replication stress in Mammalian cells and its consequences for mitosis. Genes 2015;6(2):267–98 doi 10.3390/genes6020267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dobbelstein M, Sorensen CS. Exploiting replicative stress to treat cancer. Nat Rev Drug Discov 2015;14(6):405–23 doi 10.1038/nrd4553. [DOI] [PubMed] [Google Scholar]
- 122.Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov 2018;8(9):1096–111 doi 10.1158/2159-8290.Cd-18-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dobbelstein M, Sørensen CS. Exploiting replicative stress to treat cancer. Nature Reviews Drug Discovery 2015;14:405 doi 10.1038/nrd4553. [DOI] [PubMed] [Google Scholar]
- 124.Rundle S, Bradbury A, Drew Y, Curtin NJ. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers 2017;9(5):41 doi 10.3390/cancers9050041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ghelli Luserna di Rorà A, Cerchione C, Martinelli G, Simonetti G. A WEE1 family business: regulation of mitosis, cancer progression, and therapeutic target. J Hematol Oncol 2020;13(1):126- doi 10.1186/s13045-020-00959-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li F, Kozono D, Deraska P, Branigan T, Dunn C, Zheng XF, et al. CHK1 Inhibitor Blocks Phosphorylation of FAM122A and Promotes Replication Stress. Molecular cell 2020;80(3):410–22.e6 doi 10.1016/j.molcel.2020.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Plummer ER, Kristeleit RS, Cojocaru E, Haris NM, Carter L, Jones RH, et al. A first-in-human phase I/II trial of SRA737 (a Chk1 Inhibitor) in subjects with advanced cancer. 2019;37(15_suppl):3094- doi 10.1200/JCO.2019.37.15_suppl.3094. [DOI] [Google Scholar]
- 128.Hong DS, Moore KN, Bendell JC, Karp DD, Wang JS-Z, Ulahannan SV, et al. A phase Ib study of prexasertib, a checkpoint kinase (CHK1) inhibitor, and LY3023414, a dual inhibitor of class I phosphatidylinositol 3-kinase (PI3K) and the mammalian target of rapamycin (mTOR) in patients with advanced solid tumors. 2019;37(15_suppl):3091- doi 10.1200/JCO.2019.37.15_suppl.3091. [DOI] [Google Scholar]
- 129.Lee JM, Nair J, Zimmer A, Lipkowitz S, Annunziata CM, Merino MJ, et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. The Lancet Oncology 2018;19(2):207–15 doi 10.1016/s1470-2045(18)30009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Leijen S, van Geel RM, Pavlick AC, Tibes R, Rosen L, Razak AR, et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2016;34(36):4371–80 doi 10.1200/jco.2016.67.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yap TA, Tan DSP, Terbuch A, Caldwell R, Guo C, Goh BC, et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov 2020. doi 10.1158/2159-8290.Cd-20-0868. [DOI] [PMC free article] [PubMed]
- 132.Ubhi T, Brown GW. Exploiting DNA Replication Stress for Cancer Treatment. Cancer Res 2019;79(8):1730–9 doi 10.1158/0008-5472.Can-18-3631. [DOI] [PubMed] [Google Scholar]
- 133.Brunton L, Chabner B, Knollman B. Goodman and Gilman’s Phamracological Basis of Therapeutics. 12th ed: McGraw-Hill Companies, Inc.; 2011. [Google Scholar]
- 134.Moore EC, Cohen SS. Effects of arabinonucleotides on ribonucleotide reduction by an enzyme system from rat tumor. The Journal of biological chemistry 1967;242(9):2116–8. [PubMed] [Google Scholar]
- 135.Wallez Y, Dunlop CR, Johnson TI, Koh SB, Fornari C, Yates JWT, et al. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Molecular cancer therapeutics 2018;17(8):1670–82 doi 10.1158/1535-7163.Mct-18-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Koh S-B, Courtin A, Boyce RJ, Boyle RG, Richards FM, Jodrell DI. CHK1 Inhibition Synergizes with Gemcitabine Initially by Destabilizing the DNA Replication Apparatus. Cancer Research 2015;75(17):3583–95 doi 10.1158/0008-5472.Can-14-3347. [DOI] [PubMed] [Google Scholar]
- 137.Banerji U, Plummer ER, Moreno V, Ang JE, Quinton A, Drew Y, et al. A phase I/II first-in-human trial of oral SRA737 (a Chk1 inhibitor) given in combination with low-dose gemcitabine in subjects with advanced cancer. Journal of Clinical Oncology 2019;37(15_suppl):3095- doi 10.1200/JCO.2019.37.15_suppl.3095. [DOI] [Google Scholar]
- 138.Plummer ER, Kristeleit RS, Cojocaru E, Haris NM, Carter L, Jones RH, et al. A first-in-human phase I/II trial of SRA737 (a Chk1 Inhibitor) in subjects with advanced cancer. Journal of Clinical Oncology 2019;37(15_suppl):3094- doi 10.1200/JCO.2019.37.15_suppl.3094. [DOI] [Google Scholar]
- 139.Lal S, Zarei M, Chand SN, Dylgjeri E, Mambelli-Lisboa NC, Pishvaian MJ, et al. WEE1 inhibition in pancreatic cancer cells is dependent on DNA repair status in a context dependent manner. Scientific reports 2016;6:33323 doi 10.1038/srep33323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kausar T, Schreiber JS, Karnak D, Parsels LA, Parsels JD, Davis MA, et al. Sensitization of Pancreatic Cancers to Gemcitabine Chemoradiation by WEE1 Kinase Inhibition Depends on Homologous Recombination Repair. Neoplasia 2015;17(10):757–66 doi 10.1016/j.neo.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Drean A, Williamson CT, Brough R, Brandsma I, Menon M, Konde A, et al. Modeling Therapy Resistance in BRCA1/2-Mutant Cancers. Molecular cancer therapeutics 2017;16(9):2022–34 doi 10.1158/1535-7163.Mct-17-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cuneo KC, Morgan MA, Sahai V, Schipper MJ, Parsels LA, Parsels JD, et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2019:Jco1900730 doi 10.1200/jco.19.00730. [DOI] [PMC free article] [PubMed]
- 143.Lheureux S, Cristea MC, Bruce JP, Garg S, Cabanero M, Mantia-Smaldone G, et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021;397(10271):281–92 doi 10.1016/s0140-6736(20)32554-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, et al. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 2015;208(5):563–79 doi 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cheng C-T, Kuo C-Y, Ann DK. KAPtain in charge of multiple missions: Emerging roles of KAP1. World J Biol Chem 2014;5(3):308–20 doi 10.4331/wjbc.v5.i3.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Quinet A, Carvajal-Maldonado D, Lemacon D, Vindigni A. Chapter Three - DNA Fiber Analysis: Mind the Gap! In: Eichman BF, editor. Methods in Enzymology. Volume 591: Academic Press; 2017. p 55–82. [DOI] [PubMed] [Google Scholar]
- 147.Zoppoli G, Regairaz M, Leo E, Reinhold WC, Varma S, Ballestrero A, et al. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proceedings of the National Academy of Sciences of the United States of America 2012;109(37):15030–5 doi 10.1073/pnas.1205943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603 doi 10.1038/nature1100310.1038/nature11003https://www.nature.com/articles/nature11003#supplementary-informationhttps://www.nature.com/articles/nature11003#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Murai J, Feng Y, Yu GK, Ru Y, Tang S-W, Shen Y, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016;7(47):76534–50 doi 10.18632/oncotarget.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clinical Cancer Research 2017;23(2):523–35 doi 10.1158/1078-0432.Ccr-16-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nogales V, Reinhold WC, Varma S, Martinez-Cardus A, Moutinho C, Moran S, et al. Epigenetic inactivation of the putative DNA/RNA helicase SLFN11 in human cancer confers resistance to platinum drugs. Oncotarget 2016;7(3):3084–97 doi 10.18632/oncotarget.6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Murai J, Tang SW, Leo E, Baechler SA, Redon CE, Zhang H, et al. SLFN11 Blocks Stressed Replication Forks Independently of ATR. Molecular cell 2018;69(3):371–84.e6 doi 10.1016/j.molcel.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Winkler C, Armenia J, Jones GN, Tobalina L, Sale MJ, Petreus T, et al. SLFN11 informs on standard of care and novel treatments in a wide range of cancer models. Br J Cancer 2020. doi 10.1038/s41416-020-01199-4. [DOI] [PMC free article] [PubMed]
- 154.Deans AJ. Fanconi anemia, put to sleep. Blood 2021;137(3):286–8 doi 10.1182/blood.2020008462. [DOI] [PubMed] [Google Scholar]
- 155.Ganesan S, Garber J. Poly (ADP-Ribose) Polymerase Inhibitor Activity in Prostate Cancers Harboring Mutations in DNA Repair Genes: Who Benefits? JCO precision oncology 2020(4):1034–7 doi 10.1200/po.20.00269. [DOI] [PubMed] [Google Scholar]
- 156.Bradbury A, Hall S, Curtin N, Drew Y. Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacology & therapeutics 2020;207:107450 doi 10.1016/j.pharmthera.2019.107450. [DOI] [PubMed] [Google Scholar]
- 157.Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 2011;7(7):428–30 doi 10.1038/nchembio.573. [DOI] [PubMed] [Google Scholar]
- 158.Perkhofer L, Schmitt A, Romero Carrasco MC, Ihle M, Hampp S, Ruess DA, et al. ATM Deficiency Generating Genomic Instability Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Therapy-Induced DNA Damage. Cancer Research 2017;77(20):5576–90 doi 10.1158/0008-5472.Can-17-0634. [DOI] [PubMed] [Google Scholar]
- 159.Riabinska A, Daheim M, Herter-Sprie GS, Winkler J, Fritz C, Hallek M, et al. Therapeutic Targeting of a Robust Non-Oncogene Addiction to PRKDC in ATM-Defective Tumors. Science Translational Medicine 2013;5(189):189ra78–ra78 doi 10.1126/scitranslmed.3005814. [DOI] [PubMed] [Google Scholar]
- 160.Menezes DL, Holt J, Tang Y, Feng J, Barsanti P, Pan Y, et al. A Synthetic Lethal Screen Reveals Enhanced Sensitivity to ATR Inhibitor Treatment in Mantle Cell Lymphoma with ATM Loss-of-Function. Molecular Cancer Research 2015;13(1):120–9 doi 10.1158/1541-7786.Mcr-14-0240. [DOI] [PubMed] [Google Scholar]
- 161.Min A, Im S-A, Jang H, Kim S, Lee M, Kim DK, et al. AZD6738, A Novel Oral Inhibitor of ATR, Induces Synthetic Lethality with ATM Deficiency in Gastric Cancer Cells. Molecular cancer therapeutics 2017;16(4):566–77 doi 10.1158/1535-7163.Mct-16-0378. [DOI] [PubMed] [Google Scholar]
- 162.Wengner AM, Siemeister G, Lücking U, Lefranc J, Wortmann L, Lienau P, et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage–Inducing or Repair–Compromising Therapies in Preclinical Cancer Models. Molecular cancer therapeutics 2020;19(1):26–38 doi 10.1158/1535-7163.Mct-19-0019. [DOI] [PubMed] [Google Scholar]
- 163.Dunlop CR, Wallez Y, Johnson TI, Bernaldo de Quirós Fernández S, Durant ST, Cadogan EB, et al. Complete loss of ATM function augments replication catastrophe induced by ATR inhibition and gemcitabine in pancreatic cancer models. British journal of cancer 2020;123(9):1424–36 doi 10.1038/s41416-020-1016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bear AS, Vonderheide RH, O’Hara MH. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer cell 2020;38(6):788–802 doi 10.1016/j.ccell.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yum S, Li M, Frankel AE, Chen ZJ. Roles of the cGAS-STING Pathway in Cancer Immunosurveillance and Immunotherapy. Annual review of cancer biology 2019;3(1):323–44 doi 10.1146/annurev-cancerbio-030518-055636. [DOI] [Google Scholar]
- 166.Kwon J, Bakhoum SF. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer discovery 2019. doi 10.1158/2159-8290.Cd-19-0761. [DOI] [PMC free article] [PubMed]
- 167.Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, et al. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer discovery 2019;9(6):722–37 doi 10.1158/2159-8290.Cd-18-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ding L, Kim H-J, Wang Q, Kearns M, Jiang T, Ohlson CE, et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Reports 2018;25(11):2972–80.e5 doi 10.1016/j.celrep.2018.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pantelidou C, Jadhav H, Kothari A, Liu R, Guerriero JL, Shapiro GI. STING agonism enhances anti-tumor immune responses and therapeutic efficacy of PARP inhibition in BRCA-associated breast cancer. bioRxiv 10.1101/2021.01.26.428337. [DOI] [PMC free article] [PubMed]
- 170.Zhang Q, Green MD, Lang X, Lazarus J, Parsels JD, Wei S, et al. Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Res 2019;79(15):3940–51 doi 10.1158/0008-5472.Can-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jing W, McAllister D, Vonderhaar EP, Palen K, Riese MJ, Gershan J, et al. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J Immunother Cancer 2019;7(1):115 doi 10.1186/s40425-019-0573-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Vonderhaar EP, Barnekow NS, McAllister D, McOlash L, Eid MA, Riese MJ, et al. STING activated tumor-intrinsic type I IFN signaling promotes CXCR3 dependent antitumor immunity in pancreatic cancer. Cell Mol Gastroenterol Hepatol 2021. doi 10.1016/j.jcmgh.2021.01.018. [DOI] [PMC free article] [PubMed]
- 173.Romero JM, Grünwald B, Jang GH, Bavi PP, Jhaveri A, Masoomian M, et al. A Four-Chemokine Signature Is Associated with a T-cell-Inflamed Phenotype in Primary and Metastatic Pancreatic Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2020;26(8):1997–2010 doi 10.1158/1078-0432.Ccr-19-2803. [DOI] [PubMed] [Google Scholar]
- 174.Motedayen Aval L, Pease JE, Sharma R, Pinato DJ. Challenges and Opportunities in the Clinical Development of STING Agonists for Cancer Immunotherapy. J Clin Med 2020;9(10) doi 10.3390/jcm9103323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Shroff RT, Hendifar A, McWilliams RR, Geva R, Epelbaum R, Rolfe L, et al. Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO precision oncology 2018;2018 doi 10.1200/po.17.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]