

SUMMARY
Understanding how cells are likely to evolve can guide medical interventions and bioengineering efforts that must contend with unwanted mutations. The adaptome of a cell—the neighborhood of genetic changes that are most likely to drive adaptation in a given environment—can be mapped by tracking rare beneficial variants during the early stages of clonal evolution. We used multiplex adaptome capture sequencing (mAdCap-Seq), a procedure that combines unique molecular identifiers and hybridization-based enrichment, to characterize mutations in eight Escherichia coli genes known to be under selection in a laboratory environment. We tracked 301 mutations at frequencies as low as 0.01% and inferred the fitness effects of 240 of these mutations. There were distinct molecular signatures of selection on protein structure and function for the three genes with the most beneficial mutations. Our results demonstrate how mAdCap-Seq can be used to deeply profile a targeted portion of a cell’s adaptome.
Keywords: Clonal interference, Distribution of fitness effects, Long-term evolution experiment with E. coli (LTEE), Pyruvate kinase, DNA Topisomerase I, PykF, NadR, TopA
Graphical Abstract
eTOC blurb
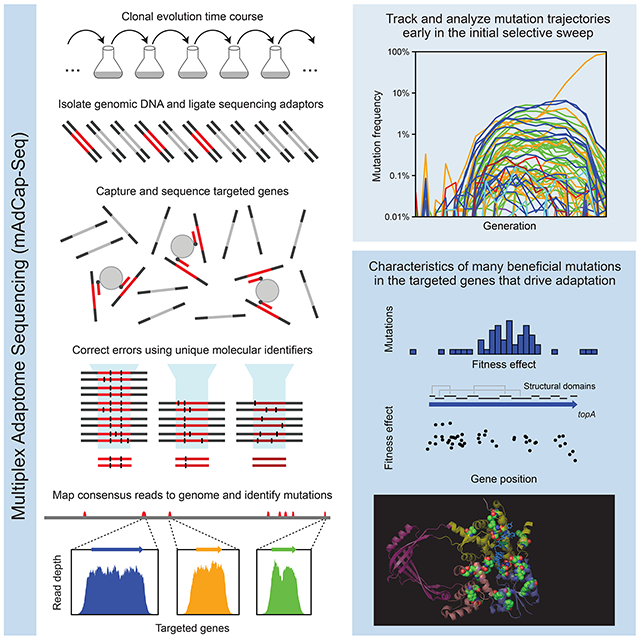
Multiplex adaptome capture sequencing (mAdCap-Seq) tracks the frequencies of beneficial mutations as they compete during clonal evolution. This study used mAdCap-Seq of six laboratory populations of E. coli to characterize 301 mutations in eight targeted genes. Analyzing this large set of beneficial mutations revealed how selection acted on the functions of the topA, nadR, and pykF genes. Using mAdCap-Seq to map how a cell is likely to evolve in a certain environment could guide medical interventions and bioengineering design.
INTRODUCTION
New mutations arise naturally in the genomes of cells during DNA replication and repair. These de novo mutations are the main drivers of adaptive evolution in clonal populations that have little or no recombination or standing genetic variation. For example, numerous lineages with different beneficial mutations arise and contend within large laboratory populations of asexual microbes before any one lineage outcompetes the ancestor and its competitors (Good et al., 2017; Lang et al., 2013; Maddamsetti et al., 2015). This ‘clonal interference’ leads to heterogeneous populations with many lineages simultaneously adapting via different sets of mutations (Desai et al., 2012; Gerrish and Lenski, 1998; Park and Krug, 2007). Often a majority of these mutations affect a small subset of genes involved in cellular processes that are under the strongest selection (Deatherage et al., 2017; Lang et al., 2013; Lind et al., 2015; Phaneuf et al., 2020). If the ‘evolvome’ is defined as the set of all spontaneous genetic changes by which a cell can potentially evolve, then the beneficial mutations that are most likely to drive adaptation in a given environment can be described as constituting its ‘adaptome’ (Ryall et al., 2012).
Human cancers and microbial infections exhibit similar genetic dynamics to those observed in these laboratory evolution experiments: single cells clonally expand as they evolve driver mutations that lead to disease progression and drug resistance. In cancer, both solid tumors and blood cancers have been shown to be genetically heterogeneous (Marusyk et al., 2012; Merlo et al., 2006; Thomas et al., 2006). De novo mutations that arise and then take over normal cell populations can lead to carcinogenesis (Genovese et al., 2014; Watson et al., 2020). Mutations in cancer cells drive neoplastic progression (Merlo et al., 2010), differences in responses to chemotherapy (Landau et al., 2013), and relapse (Ding et al., 2012). Similarly, populations of Pseudomonas aeruginosa and other bacteria that persistently infect the lungs of cystic fibrosis patients become increasingly invasive and antibiotic resistant over time (Marvig et al., 2015; Stefani et al., 2017; Winstanley et al., 2016). In both cancers and infections, the same genes are often mutated in the cells that cause disease in different individuals. Mapping the adaptomes of these cells to understand how they are likely to evolve in other patients afflicted in the same way could inform treatment decisions and improve medical outcomes.
Cells used in biomanufacturing are also prone to evolving unwanted genetic heterogeneity (Renda et al., 2014; Rugbjerg and Sommer, 2019). Typically, these cells have been highly engineered to optimize the titer of a product of interest at the expense of rapid cellular replication (Lee and Kim, 2015; Nielsen and Keasling, 2016). Therefore, there are strong selective pressures for ‘escape mutations’ that cause production to decline. Usually escape mutations directly inactivate one or more key genes in the engineered pathway. The resulting nonproducing cells can become dominant during the many cell divisions that are necessary to scale these processes up to large bioreactors (Rugbjerg et al., 2018; Sandoval et al., 2014; Zelder and Hauer, 2000). Mapping the adaptomes of engineered cells to profile the evolutionary failure modes that are most common in nonproducing mutants before attempting scale-up could guide bioengineering design decisions and thereby improve the efficiency of industrial processes.
Evolution experiments conducted in laboratory environments reproduce key aspects of microbial evolution that are observed in chronic infections and bioreactors (Barrick and Lenski, 2013; Gresham and Dunham, 2014). Certain aspects of genomic and phenotypic evolution in these controlled systems predictably occur across multiple replicate populations (Barrick, 2020; Cvijović et al., 2018; Furusawa et al., 2018; McDonald, 2019; Rainey et al., 2017), making them a useful testbed for adaptome mapping methods. In theory, tracking the frequencies of mutations during the earliest stages of clonal evolution from a single cell in these populations should allow one to map that cell’s adaptome. However, many highly beneficial mutations arise but never reach appreciable frequencies in microbial populations before they are outcompeted by lineages with other beneficial mutations (Desai et al., 2012; Gerrish and Lenski, 1998). Thus, one must be able to track extremely rare mutations to recover more of the adaptome. During the initial burst of new beneficial genetic diversity when all beneficial mutations are still rare and very few cells have accumulated multiple mutations, each mutant is effectively competing only versus the ancestor. Therefore, one can also estimate each beneficial mutation’s fitness effect directly from how rapidly its frequency increases during this critical time window.
High-throughput metagenomic DNA sequencing can be used to track rare mutations in cell populations, but most such studies of microbial evolution experiments have only been able to reliably identify mutations that are present at frequencies above ~1-10% due to limitations imposed by sequencing depth or error rates (Barrick and Lenski, 2009; Chubiz et al., 2012; Good et al., 2017; Lang et al., 2013; Traverse et al., 2013). At this point, the diversity and dynamics of the single-step beneficial mutations that constitute the clonal adaptome have typically been obscured by takeover of a few dominant mutants and further evolution of all lineages.
Several methods exist to characterize rarer mutations. Amplicon sequencing (e.g., as in FREQ-seq) is a straightforward strategy for profiling genetic variants in a targeted subset of a cell’s genome (Chubiz et al., 2012; Fischer et al., 2017; Hong et al., 2018). However, the PCR enrichment step in this method requires optimizing conditions for each targeted region and introduces biases in inferring the frequencies of mutations that alter amplicon sizes. Another approach is to add unique molecular identifiers (UMIs) to the sequenced DNA fragments before any amplification steps. This information can be used to detect PCR duplicates to more accurately estimate mutation frequencies (Hong and Gresham, 2017; Kivioja et al., 2012) and to correct sequencing errors (Schmitt et al., 2012). But, the extra depth of sequencing needed when using UMIs for error correction generally makes it infeasible to employ this approach on a genome-wide scale to track rare variants in many samples. Sequencing short barcodes inserted into the genomes of progenitor cells (BAR-seq) allows one to economically track the frequencies of extremely rare lineages derived from these cells (Levy et al., 2015; Venkataram et al., 2016); but, a limitation of this method is that the cell of interest must be genetically engineered with high efficiency to introduce a sufficient diversity of cellular barcodes, which may be difficult or impossible in certain cell types or clinical samples. Additional whole-genome sequencing is also required to identify the beneficial mutations that are linked to the winning barcodes.
Here, we describe multiplex adaptome capture sequencing (mAdCap-Seq). This method allows one to characterize many beneficial mutations in specific genes in a clonally evolving cell population. The key components of mAdCap-Seq are: (1) increasing sequencing depth in a targeted portion of a genome through hybridization-based DNA capture, (2) lowering sequencing error rates using UMIs, and (3) analyzing a time course of samples from a population during the early stages of clonal evolution. We tested mAdCap-Seq on laboratory populations that used the same ancestral strains and nearly identical culture conditions as a >70,000-generation E. coli evolution experiment (Lenski et al., 1991). We were able to directly identify diverse beneficial mutations in eight genes when they were orders of magnitude lower in frequency than could be accomplished by standard metagenomic sequencing. The molecular signatures and fitness effects of the many beneficial mutations found in three of these genes made it possible to infer the nature of selection acting on their functions in this environment. Our results demonstrate how mAdCap-Seq can be used to deeply profile a targeted portion of the adaptome of a cell.
RESULTS
Replaying the beginning of a long-term evolution experiment
We initially examined the evolution of nine replicate E. coli populations that were propagated via daily serial transfers in glucose-limited minimal medium for 500 generations. Our experiment used the same E. coli strains as the Lenski long-term evolution experiment (LTEE) and similar growth conditions (see Methods). Each population was inoculated with a 50/50 mixture of the two neutrally marked LTEE ancestor strains to visualize the initial selective sweep (Hegreness et al., 2006). An initial 30 generations of evolution occurred as these two strains were grown separately from single cells before they were combined to begin the serial transfers. Most populations maintained a roughly equal representation of descendants of both ancestral strains through the first 300 generations of the evolution experiment (Fig. 1). These dynamics are in agreement with what has previously been observed in studies of the LTEE, where few mutations reach a high frequency in the first few hundred generations of evolution (Good et al., 2017). We chose to further analyze only six of the nine populations due to constraints on how many samples we could process and sequence. Two E. coli populations (A4 and A5) were purposefully omitted because they exhibited early sweeps of one marker type, which indicates that their dynamics might have been dominated by one or a few "jackpot" mutations that occurred very early during outgrowth of these populations from single cells. The third population that was not selected for further study (A8) exhibited typical marker dynamics.
Figure 1. Replaying the first selective sweep of a long-term evolution experiment.

Nine E. coli populations were initiated from equal mixtures of two variants of the ancestral strain that differ in a neutral genetic marker for arabinose utilization (Ara). We observed the evolutionary dynamics of these populations over 500 generations of regrowth from 75 daily 1:100 serial transfers by periodically plating dilutions of each population on indicator agar. Each ancestral strain subpopulation was derived from a single colony isolate that experienced 30 generations of growth before it was combined with the opposite type to initiate the serial transfers. The ratio of Ara+ cells (pink colonies) to Ara− cells (red colonies) diverges from 1:1 when descendants of one ancestor type accumulate enough of a fitness advantage due to de novo beneficial mutations that they take over. We focused further analysis on six of the nine populations (thick lines).
Tracking the trajectories of new beneficial mutations
We next performed mAdCap-Seq on eight genes at ~25 generation increments over the entire 500 generations of the evolution experiment for four of the six populations that we examined further. These eight genes (nadR, pykF, topA, spoT, fabR, ybaL, hslU, and iclR) are known to be targets of selection in the LTEE (Good et al., 2017; Tenaillon et al., 2016). Illumina libraries containing UMIs (Schmitt et al., 2012) were prepared for sequencing and enriched for the regions of interest using solution based hybridization (Bainbridge et al., 2010). Consensus sequence reads were generated based on groups of reads with identical UMIs and aligned to the E. coli genome to predict mutations, including using split-read mapping to identify transposon insertions and large deletions (Fig. 2A). The enrichment procedure was effective: an average of 73.5% of consensus reads per sample mapped to the targeted regions that together constitute only 0.780% of the 4.63 Mbp genome. In the sample with the median number of total consensus reads, the average coverage depth across each of the eight genes of interest exceeded 5,000 (Fig. 2B). After eliminating variants that exhibited systematic biases in their frequency trajectories (see Methods), we were able to track the evolution and competition of 181 mutations, including when many were present in less than 0.1% of the cells in a population (Fig. 2C, Fig. 3).
Figure 2. Profiling many beneficial mutations in the first selective sweep by deep sequencing.

(A) Schematic of the deep sequencing approach. Genomic DNA is directly isolated from the E. coli populations and prepared for paired-end Illumina sequencing with sample barcodes and dual UMIs (colored ends attached to red/green double stranded DNA). DNA fragments matching the targeted genome regions (green centers) are captured by probes (blue) bound to magnetic beads and other sequences are washed away (red centers). Reads in pairs that have the same dual unique molecular identifiers, which implies that they were PCR amplified during library preparation from the same original genomic DNA fragment, are used to construct consensus reads to eliminate sequencing errors. Consensus reads are mapped to the reference genome to call sequence variants. (B) Enrichment of reads mapping to eight genes known to be early targets of selection in this environment from the long-term evolution experiment. The final coverage depth of consensus reads in and around these genes is shown for a typical sample (population A7 at 500 generations). (C) Frequency trajectories for mutations in the eight targeted genes as well as the sum total frequency in population A1 over the complete time course of the evolution experiment. When a mutation was not detected at a time point, its trajectory is shown as passing through a frequency of 0.0001% (outside of the plot bounds). (D) Mutation frequency trajectories for population A1 during the selective sweep window from 163 to 243 generations when mutations were first reaching detectable frequencies and outcompeting the ancestral genotype. At time points when a mutation was not detected, its frequency is shown as 0.001% (at the bottom of the plot). (E) Estimated relative fitness of population A1 in each interval between sequenced time points. The frequency trajectories of all beneficial mutations in the initial sweep shown in D were used to jointly estimate the average fitness of the entire population from the deceleration in the rate of increase of the observed mutation trajectories as genotypes with beneficial mutations became common (see Methods). This fitness trajectory fit accounts for all cells in the population, regardless of whether they have a mutation in the targeted genes or elsewhere in the genome. The red line is the maximum likelihood estimate of the population fitness trajectory. The red shading around it shows 95% confidence intervals on this value in each interval. The black line shows the increase in fitness estimated for a consensus model that was jointly fit to all mutations tracked in all six populations. The consensus population fitness trajectory was used when estimating the fitness effects of individual mutations (see Methods).
Figure 3. Frequency trajectories of mutations in the remaining populations.

The same plots described in Figure 2C-E for population A1 are shown for populations A2, A3, and A7 (top three sets of panels). For populations A6 and A9, sequencing was only performed at time points during the selective sweep window from 169 to 236 generations so only the plots corresponding to Figure 2D-E are shown (bottom two sets of panels).
Mutation trajectories in all four populations exhibited a burst of genetic diversity in the targeted genes followed by loss of this diversity. The initial dynamics are expected to be largely driven by new genotypes that each evolve a single beneficial mutation very early in the experiment. If their descendants escape stochastic loss, they will gradually increase in frequency over the first few hundred generations as they outcompete the ancestral genotype. Once the population becomes dominated by these first-step mutants, their frequency trajectories plateau because of clonal interference: they are now mainly competing against one another and are relatively evenly matched. In populations A1, A2, and A7, the total frequencies of the mutations we identified sums to 49.6-62.4% at generation 297, indicating that each population is mostly composed of genotypes with a mutation in one of the focal genes. We recovered less of the initial beneficial mutation diversity in population A3 where this sum was only 13.5%.
After around 300 generations, there is a steady decline in the frequencies of most mutations in the eight targeted genes. At this point, subpopulations of cells that have evolved multiple beneficial mutations begin to displace the genotypes that we initially tracked. Many of the most successful new genotypes are descended from cells that already had a mutation in one of the targeted genes. In these cases, the original mutations serve as markers for the further expansion of these subpopulations after a period during which their frequencies stagnate or decrease, but the new beneficial mutations responsible for this further increase in fitness are outside of the genomic regions we surveilled. The opposite situation, in which a beneficial mutation in one of the eight focal genes appears in a cell with an untracked beneficial mutation elsewhere in the genome, also occurs in a few cases. One example is a mutation in pykF that only appears after 300 generations in population A3 but then rapidly increases in frequency and becomes dominant. These dynamics indicate that its increase is accelerated by the presence of a prior, unknown beneficial mutation in the genetic background in which it evolved.
Fitness effects can be inferred from initial mutation trajectories
We next sought to calculate the fitness benefits of individual mutations by tracking how rapidly their frequencies rose early in the experiment when they were largely competing versus the ancestral genotype because all new mutations in the population were still rare. To that end, we performed mAdCap-Seq on all six populations at ~13-generation increments from 169 to 236 generations (Fig. 2D, Fig. 3). With this additional data we were able to track a total of 240 mutations as they gradually increased in frequency during the critical time window from generation 163 to 243 that captures the dynamics of the first selective sweep. In the four populations we had already sequenced (A1, A2, A3, and A7), these mutations included 120 of the 181 previously found in the complete time course data spanning 500 generations and 54 additional mutations that had not been detected when analyzing the original time course data alone. Using the new mAdCap-Seq data, we also identified 66 mutations in the two populations for which we did not have time course data at 25 generation increments across the entire 500 generations of the evolution experiment (A6 and A9). Of the 240 total mutations, 93.3% occurred in just three of the eight targeted genes: nadR, pykF, and topA (Fig. 4A).
Figure 4. Characteristics of beneficial mutations in the initial selective sweep.

(A) Total number of beneficial mutations in each of the eight targeted genes for which fitness effects could be estimated by analyzing their frequency trajectories between generations 163 and 243. (B) Distributions of fitness effects of beneficial mutations in the three genes that were the dominant targets of selection. Mutations are binned by the maximum likelihood estimates of their fitness effects. Vertical red lines show the mean of each distribution. 95% confidence limits in Figure 6A show uncertainty in the fitness effect estimated for each of these mutations.
We were able to estimate the fitness effect of each of these 240 beneficial mutations by fitting a binomial logistic model to how the counts of reads supporting the variant versus reference sequence increased over time from 163 to 243 generations. In all populations, there is initially a log-linear increase in the frequency of each mutation as the first wave of evolved cells, nearly all of which are expected to have just one of these beneficial mutations, competes against a population that is still almost entirely cells with the ancestral genotype. Then, there is a deceleration in the rate at which the frequencies of the new mutations increase around generation 196 that coincides with the onset of clonal interference. Genotypes with beneficial mutations begin to make up a sizable proportion of the population at this point, making it necessary to account for how they are increasingly competing against one another to estimate fitness effects.
We accounted for clonal interference by adding a stepwise increase in the average fitness of the entire cell population over time as an additional set of parameters to the binomial logistic model (Fig. 2E, Fig. 3). That is, we estimated how the fitness of the population, as a whole, was changing from the deceleration in the trajectories of the subset of mutations that we tracked in the targeted genes. Because overall population fitness dynamics are highly reproducible from population to population in the LTEE conditions (Lenski et al., 1991), we used one consensus model of how the fitness of the populations increased (fit from all tracked mutations in all six sequenced populations) to correct our estimates of individual mutation fitness effects for clonal interference. Most of the increase in population fitness occurs rapidly in a single step during the interval spanning 196-209 generations. This rapid change followed by stasis may seem at odds with the continuing increase in the trajectories of many beneficial mutations. However, this type of stepwise increase is a typical result of clonal dynamics in models and experiments (Gerrish and Lenski, 1998; Lenski et al., 1991). It could result from many mutations with small fitness effects, no one of which reaches an observable frequency, peaking and then being outcompeted by the more highly beneficial mutations that we are able to track, for example.
The mean fitness effect that we inferred for the 240 tracked mutations in all six populations was 9.00% with a standard deviation of 1.33%. Although the distributions of the fitness effects estimated for mutations in nadR, pykF, and topA overlap (Fig. 4B), there was a significant stratification among these genes. Mutations in nadR were 0.44% more beneficial than mutations in topA, on average, and this difference was significant (p = 0.022, one-tailed Mann–Whitney U test). In turn, mutations in topA were 0.70% more beneficial than those in pykF (p = 0.00046, one-tailed Mann–Whitney U test). The fitness effects of the 16 mutations in the other genes (spoT, fabR, ybaL, and iclR) were not significantly different from the those of the 224 mutations in nadR, pykF, and topA (p = 0.33, two-tailed Mann–Whitney U test). Thus, highly beneficial mutations are possible in these genes as well, but they occur at a much lower rate than similarly beneficial mutations in nadR, pykF, and topA.
One metric for how effectively we mapped the E. coli adaptome is the fraction of the increase in the fitness of each population that is captured by the subset of beneficial mutations tracked with mAdCap-Seq. The fitness increase of each population could be reliably estimated by generation 196, and separate estimates for each population were in close agreement with the consensus estimate that included all populations after generation 209 (Fig 1E, Fig. 2). Thus, we could make robust calculations beginning at these time points. The percentage of the fitness increase contributed by the tracked mutations ranged from a low of 10.4% in population A9 to a high of 34.9% in population A2 at generation 223. The mean across all six populations was 27.5%. This fraction was not constant across time. As evolution continued, cells with these same mutations should account for a higher and higher fraction of the population fitness if they are displacing cells with less-beneficial mutations. In line with this expectation, they accounted for only 19.8% and 18.3%, on average, of the increase in population fitness earlier in the experiment, at generations 196 and 206, respectively. Later, the frequency trajectories plateaued for the beneficial mutations we tracked. This behavior means that the fitness of the whole population had caught up and was now roughly the same as the fitness of a cell with one of these beneficial mutations. Under this assumption, the tracked mutations account for 16.3–78.8% (42.9% on average) of the ~9% fitness increase observed at generation 270 in the four populations sequenced at this time. Overall, while we could account for a considerable portion of the fitness evolution of these populations with the mutations captured by mAdCap-Seq, mutations in other genes and/or less-beneficial mutations contributed more in most cases.
Beneficial mutations reveal different signatures of selection on gene function
Of the 301 total beneficial mutations that we were able to identify using mAdCap-Seq, 272 were in the nadR, pykF, or topA genes. This large set of beneficial mutations gave us the statistical power to test for several signatures of molecular evolution to ascertain what types of changes in the function of each gene improved E. coli fitness in this environment. Each of the three genes exhibited a distinct spectrum of beneficial mutations (Fig. 5). In some cases, different types of mutations were also unevenly distributed throughout the sequences of these three commonly hit genes and had noticeably different effects on bacterial fitness (Fig. 6A).
Figure 5. Spectra of early beneficial mutations in nadR, pykF, and topA.

These three genes were the dominant targets of selection during the evolution experiment among the eight genes profiled for beneficial mutations. The total number of mutations identified in each gene is indicated above its column. The width of the bars distinguishes base substitutions (thin bars) from other types of mutations (thick bars).
Figure 6. Mutations in the three genes that were the dominant targets of selection.

(A) Nucleotide positions and properties of all mutations found in each of the three genes that were the dominant targets of selection during the evolution experiment. Colors represent the type of mutation. Symbols indicate whether each mutation was detected in the selective sweep window, the complete time course, or both. The upper panel for each gene, shows the fitness effects estimated for mutations in that gene. Error bars are 95% confidence limits. When the same genetic change was detected in multiple populations, information from all of its frequency trajectories was combined to estimate one overall fitness value and confidence limit for all of those mutations (see Methods). Thus, the symbols and error bars completely overlap for each independent occurrence of these mutations in a different population. The reading frame of the gene is shown above this panel with protein domains labeled. Vertical dashed grey lines represent the start and end of each gene. Horizontal grey lines show the extent of large deletions within the pictured region. Horizontal red lines represent the average fitness effects for all mutations in a gene. The lower panel for each gene shows mutations that were only detected in the complete time courses and therefore do not have an estimated fitness effect. Symbols in this panel are randomly jittered in the vertical direction to improve their visibility. (B) Structural context of mutations in PykF. Sites of nonsymonymous mutations are highlighted by showing space-filling models of the ancestral amino acid residues. All four subunits of the PykF homotetramer are shown. (C) Structural context of mutations in the catalytic core of TopA. Sites of nonsymonymous mutations are highlighted by showing space-filling models of the ancestral amino acid residues. Only domains D1-D4 are present in the structure. The DNA strand interacting with TopA is shown as a stick model.
The E. coli nadR gene has three distinct functions related to NAD biosynthesis: (1) the N-terminal domain is a helix-turn-helix that binds to DNA so that it can act as a negative transcriptional regulator of NAD salvage and transport pathways; (2) the internal domain is an NMN adenylyltransferase (Raffaelli et al., 1999); and (3) the C-terminal domain is predicted to have ribosylnicotinamide kinase activity (Kurnasov et al., 2003). Large deletions, frameshifts from small insertions or deletions (indels), insertions of transposable insertion sequence (IS) elements, and base substitutions creating stop codons dominate the nadR mutational spectrum (Fig. 5). These disruptive mutations, most of which are expected to result in complete loss of gene function, are significantly overrepresented versus nonsynonymous base substitutions in the first two domains of the gene compared to the remainder (13.7 odds ratio, p = 1.2 × 10−8, one-tailed Fisher's exact test) (Fig. 6A). Yet, there was not a significantly greater fitness effect for disruptive mutations compared to nonsynonymous mutations overall (p = 0.063, one-tailed Mann–Whitney U test). These results indicate that complete inactivation of nadR yields the maximum benefit possible for a mutation in this gene. Consistent with our observations from mapping its adaptome, deletion of nadR has been shown to be highly beneficial in the very similar environment of the LTEE (Barrick et al., 2009).
Pyruvate kinase 1 (pykF) catalyzes the final step of glycolysis, transferring a phosphate group from phophoenolpyruvate (PEP) to ADP to generate pyruvate and ATP. It is a key enzyme in regulating glycolytic flux (Kochanowski et al., 2013; Siddiquee et al., 2004). We observed an intermediate representation of disruptive mutations in pykF: fewer than in nadR but more than in topA (Fig. 5). Nonsynonymous base substitutions in pykF tend to have a larger fitness effect than disruptive mutations (p = 0.00031, one-tailed Mann–Whitney U test) (Fig. 6A). This finding is in agreement with a study of various pykF alleles that arose in the LTEE which found that nearly all pykF point mutations were more beneficial than deletion of the pykF gene, both in the ancestor and in evolved genetic backgrounds (Peng et al., 2018). PykF forms a homotetramer in which each polypeptide folds into three structural domains (Donovan et al., 2016; Mattevi et al., 1995). The central domain A forms the active site at the interface with domain B and the binding site for the allosteric effector fructose 1,6-bisphosphate at the interface with domain C. The nonsynonymous mutations that we observed are more concentrated than expected in domain A versus the other structural domains based on their relative lengths in the gene sequence (p = 0.0018 one-tailed binomial test) (Fig. 6B). Overall, adaptome mapping finds that complete inactivation of pykF is highly beneficial in the environment of our evolution experiment, but mutations that alter its activity—likely in ways that reduce glycolytic flux—are even more so. These results are consistent with a hypothesis that reducing pykF activity is beneficial in the similar glucose-limited conditions of the LTEE because this allows more PEP to be diverted to power import of glucose into cells via the phosphotransfer system (Woods et al., 2006).
DNA topoisomerase I (topA) relaxes negative supercoiling introduced into the chromosome by replication and transcription (Massé and Drolet, 1999). The mutations we observed in topA are almost exclusively single-base substitutions (Fig. 5). This type of adaptome signature implies that modulating the enzymatic activity of TopA provides the greatest fitness benefit. Complete loss of topA gene function is lethal to E. coli without compensatory mutations in DNA gyrase (Dinardo et al., 1982; Pruss et al., 1982). The structure of E. coli TopA consists of four N-terminal domains (D1-D4) that make up the catalytic core and five C-terminal zinc finger and ribbon domains (D5-D9) (Tan et al., 2015). The few out-of-frame indels and the large deletion that we observe truncate TopA within domains D7-D9, which interact with single-stranded DNA and with RNA polymerase but are not critical for catalysis. Considering only the catalytic core, we find that nonsynonymous mutations are more concentrated in domains D1 and D4 versus D2 and D3 than expected from their relative sizes (p = 0.00068, one-tailed binomial test) (Fig. 6C). D1 and D4 together form the ssDNA binding groove leading to the active site, and D1 also forms part of the active site at its interface with D3 (Perry and Mondragón, 2003). Several base substitutions in topA have been shown to increase positive supercoiling in evolved LTEE strains (Crozat et al., 2005, 2010). The exact reason that this change in supercoiling is beneficial is unknown, but it may be linked to increasing the expression of ribosomal RNAs (Crozat et al., 2005), altering gene regulation responses to starvation or other stresses (Crozat et al., 2010), and/or increasing the expression of genes in divergently transcribed operons (Houdaigui et al., 2019).
Recurrent beneficial mutations do not have greater fitness effects
We observed many examples of exact genetic parallelism. That is, the same mutation occurred and reached high frequency in different experimental populations. Each of these E. coli populations was founded from single cells, so we can conclude that these recurrent genetic changes are due to independent mutational events. We observed a total of 252 distinct genetic changes across all eight profiled genes and 31 of these were found in more than one population. While no single genetic change was detected in all six populations, 2, 2, 8 and 19 changes were detected in 5, 4, 3, and 2 populations, respectively. Most of these were in the three genes that were the main targets of selection (nadR, pykF, and topA), but one that occurred in three populations was in fabR. These mutations may be recurrent because they have a higher fitness benefit than other mutations, occur at a higher rate than other mutations, are more easily detected in the sequencing data, or due to some combination of these factors. We had fitness effect estimates for all of the 31 recurrent mutations and for 167 mutations that were each observed in only one population. The recurrent mutations had a 0.12% greater fitness effect, on average, compared to the singleton mutations, but this difference was not significant (p = 0.25, one-tailed Mann–Whitney U test). Thus, it is unlikely that many cases of exact genetic parallelism are due to these mutations being more beneficial than others in our dataset.
DISCUSSION
We used mAdCap-Seq to profile bacterial evolution during the initial stages of clonal competition when there is a burst of beneficial genetic diversity as many new subpopulations with different mutations evolve and begin to displace the ancestral genotype. We focused on eight genes known to accumulate adaptive mutations in the >70,000 generation Lenski long-term evolution experiment (LTEE) with E. coli that used nearly the same environment as our experiments. The only difference was that we added four times as much of the limiting nutrient (glucose). By combining Illumina sequencing using UMIs for error correction, hybridization-based capture of DNA encoding these genes, and dense temporal sampling, we were able to identify more beneficial mutations and track them at much lower frequencies than is possible with standard metagenomic sequencing. We detected a total of 301 mutations in the focal genes: 181 in the complete time courses of four populations and 240 during the initial selective sweep in these populations and two others, with 120 mutations overlapping between the two sets.
By densely sampling and deeply sequencing E. coli populations, we were able to characterize many beneficial mutations that never reached the normal detection limit of Illumina sequencing before they become casualties of clonal interference. Only 13 of the 181 mutations we detected in the complete time courses ever achieved a frequency of 5% or more, which can be reliably distinguished from noise without the use of UMIs or other error correction techniques, and only seven were this common for 100 or more generations, such that they were likely to be detected by a typical time-sampling scheme. Considering all of our data sets, we characterized 241 and 42 mutations that never reached 1% or 0.1% thresholds, respectively, at any sampled time point. Our success in recovering rare variants meant that we discovered more examples of beneficial mutations in the three commonly mutated genes (topA, pykF, and nadR) than have been reported in all prior studies of the evolution of the twelve LTEE populations through 60,000 generations of evolution (Barrick et al., 2009; Deatherage et al., 2015; Good et al., 2017; Ostrowski et al., 2008; Tenaillon et al., 2016; Woods et al., 2006). These large sets of mutations enabled us to identify distinct molecular signatures of adaptation in each protein.
mAdCap-Seq profiles genetic variation in specific genes. Whether and to what extent mutations in a given gene contribute to a cell’s adaptome depends on a combination of two main factors: how many mutations in that gene are sufficiently beneficial that they can compete with other top mutations (the distribution of fitness effects) and the chances that these mutations will evolve (the mutational target size). For the three genes with the most mutations in our experiment, we can rationalize the rank-order of their representation (nadR > pykF > topA) in terms of these parameters. First, mutations that we tracked in nadR are more beneficial than mutations in the other two genes, on average. Because complete loss of function of this gene is maximally beneficial, the target size for these mutations is also larger, as it includes not only base substitutions but also small indels causing frameshifts, larger deletions, and IS element insertions. The next most commonly mutated gene is pykF. The mutations we identified in pykF are actually slightly less beneficial on average than the mutations in topA. However, mutations that completely knock out topA function are not represented in the adaptome, whereas these types of mutations in pykF are highly beneficial. In this case, larger target size appears to outweigh smaller fitness effects in determining the representation of mutations in each gene.
From our experiment alone, it is unclear why mutations in the other five captured genes are rarer in the adaptome. We detected no mutations in hslU and iclR. Mutations we tracked in the other three genes (spoT, fabR, and ybaL) do not have significantly different fitness effects from those in the three genes with the most mutations (nadR, pykF, topA). However, our statistical power for detecting differences is limited by the small number of mutations detected in these genes, so we cannot definitively conclude that they are underrepresented solely due to having smaller target sizes for top-flight mutations. As described below, comparing our results to the long history of the LTEE does provide some further insights into why mutations in each of the genes that we profiled are more or less abundant in the adaptome in our evolution experiment.
We captured beneficial mutations in eight genes known to be targets of selection in the LTEE. Mutations in four of these (topA, pykF, spoT, and fabR) reach high frequencies within the first 1,000 generations of the LTEE in multiple populations (Deatherage et al., 2015; Good et al., 2017). Mutations in the other four (hslU, nadR, ybaL, and iclR) are also common in the LTEE, but they typically occur later (often within the first 2,000 to 10,000 generations) (Good et al., 2017; Tenaillon et al., 2016). Nearly all mutations in these genes in our evolution experiment were in topA, pykF, and nadR, but we also found multiple mutations that were similarly beneficial in spoT, fabR, and ybaL. Mutations in nadR were more widespread than expected in our experiment and may be more likely to completely disrupt its function than beneficial alleles that evolve in the LTEE (Ostrowski et al., 2008). Mutations in spoT and fabR were rarer than expected from the LTEE. The increased concentration of glucose in our experiment compared to the LTEE may explain these slight differences. These results are reminiscent of how changing a different aspect of the LTEE environment (temperature) re-focused the mutations of largest benefit that succeeded early onto different genes in two prior studies (Deatherage et al., 2017; Tenaillon et al., 2012). Despite the subtle difference between the LTEE and our experiment, we were still able to use mAdCap-Seq to effectively map the adaptome. We accounted for the majority of the genetic variation present after the first sweep in three of the four populations that we profiled over the entire 500 generations by capturing mutations in just eight genes.
We can ask to what extent profiling mutations while they were rare by mAdCap-Seq gave ‘early warning’ of mutations driving adaptation in these clonal cell populations. In general, we were able to begin tracking most mutations when they were above a frequency of 0.01%. This level of profiling enabled us to first detect mutations an average of 69, 150, and 290 generations before they surpassed frequencies of 0.1%, 1%, and 5%, respectively. Under the conditions of our experiment these intervals take roughly 10, 23, and 44 days, respectively. (The amount of lead time becomes disproportionately longer when requiring a mutation to reach higher frequencies due to clonal interference between beneficial mutations.) Therefore, even though we made these observations retrospectively, there would have been sufficient time to complete the DNA isolation, library preparation, sequencing, and analysis steps in mAdCap-Seq quickly enough for this approach to give early warning of the types and targets of genetic variants driving evolution of these populations. The chances and timescales of early detection would be expected to increase even more if ecological interactions or spatial structure further slowed the takeover of new variants, as has been demonstrated and discussed in other microbial evolution experiments (Baym et al., 2016; Frenkel et al., 2015; Traverse et al., 2013).
Genes in which we observe early, but unsuccessful beneficial mutations may acquire mutations again and again until they are successful in a population's evolutionary future. The extent to which this occurs is determined by the nature of epistatic interactions. In the LTEE and other microbial evolution experiments, diminishing returns epistasis dominates between beneficial mutations in different genes (Chou et al., 2011; Khan et al., 2011; Kryazhimskiy et al., 2014; Wei and Zhang, 2019; Wiser et al., 2013). That is, mutations in one gene that improve the fitness of the ancestor tend to still be beneficial to evolved genotypes containing beneficial mutations in other genes, just less so than when those other mutations are present. Subpopulations with mutations in both nadR and pykF evolve by 20,000 generations in all 12 LTEE populations, and cells that also contain a mutation in topA are found in six of the LTEE populations at this point (Tenaillon et al., 2016). By this time, mutations in ybaL and spoT are also found in nine and six LTEE populations, respectively. So, for five of the six genes in which we detected multiple mutations in the initial burst phase, it is likely that nearly all of them would have eventually accumulated beneficial mutations if we continued our experiment.
The other three genes (fabR, iclR, and hslU) likely represent other scenarios. Mutations in fabR transiently appear within the first 2,000 generations of the LTEE (Deatherage et al., 2015). They interact unfavorably with beneficial mutations in spoT and other genes, such that a fabR mutation essentially precludes further adaptation by mutating the other set of genes and vice-versa (Deatherage et al., 2015; Woods et al., 2011). We detected 9 mutations in fabR, which was more than the five we observed in ybaL. However, we predict that fabR mutations are unlikely to re-emerge and be successful in the future of these populations because of their negative interactions with other beneficial mutations. On the other hand, we detected only a single mutation in iclR and a single mutation in hslU. Of the 12 LTEE populations, 11 have sizable subpopulations with mutations in iclR and 11 have mutations in hslU by 20,000 generations, which makes them more common than mutations in spoT and ybaL in the long run. Therefore, mutations in iclR and hslU appear to either require the presence of mutations in other genes to become highly beneficial or may not be able to experience any mutations that are beneficial enough to make them competitive early on in our experiment.
The nature of epistasis and the limits that it imposes on predicting the future evolution of a cell population could be further probed using mAdCap-Seq in several ways. One could repeat the evolution experiment beginning with genotypes containing different first-step beneficial mutations and compare their adaptomes. One could also interrogate the diverse collections of cells containing different beneficial alleles that we have evolved, by taking the 150-generation populations and further evolving them under different conditions to map genotype by environment effects, for example. Such experiments might also reveal latent beneficial mutations in other genes (e.g., iclR and hslU) that were able to outcompete the ancestor in our experiment but remained below the detection limit because they were not as beneficial as mutations in topA, pykF, and nadR in this environment. There is precedent for changes in the environment reorienting selection to different subsets of the same genes. In an offshoot of the LTEE that began with a clone that had spoT, topA, and pykF mutations, selection focused further mutations on either hslU, iclR, or nadR depending on changes in temperature (Deatherage et al., 2017).
mAdCap-Seq is one among several high-throughput methods for interrogating the possibilities that a cell’s descendants can explore in its fitness landscape. Different approaches reveal complementary types of information and have different limitations. For example, deep mutational scanning uses high-throughput sequencing to simultaneously query large libraries of mutations in a single gene (Fowler and Fields, 2014). Transposon insertion sequencing (Tn-Seq) allows one to infer the fitness effects of transposon insertions in essentially every gene in a genome (van Opijnen and Camilli, 2013). While nearly all Tn-Seq mutations disrupt gene function, CRISPR-enabled trackable genome engineering can make genetic changes of other types, including those that increase expression of a target gene, and track the abundance and thereby fitness of these variants in parallel by sequencing barcodes (Garst et al., 2017). These and other related methods rely on artificially constructing variant libraries and may test mutations in a nonnatural context (e.g., in genes on a plasmid). Thus, they do not provide information about which genetic variants are accessible by spontaneous mutations.
Methods that characterize just those mutations that spontaneously arise in clonally evolving cell populations more specifically map adaptomes. For example, a capture-based enrichment and high-throughput sequencing method, similar to mAdCap-Seq but without UMIs, has been used to detect rare mutations in circulating tumor DNA in patient blood samples (Newman et al., 2014, 2016). FREQ-Seq is an example of a multiplexed amplicon sequencing approach that is similar to mAdCap-Seq in how it aims to track the frequencies of new beneficial mutations in specific, targeted regions of microbial genomes (Chubiz et al., 2012). When not employing UMIs for error correction, amplicon sequencing has a limited ability to differentiate extremely rare mutations from sequencing errors (Schmitt et al., 2012). Another issue is that the initial PCR steps used to create amplicons can bias detection. In our experiment with E. coli, IS element insertions are common. DNA templates with such large insertions would be highly disfavored during PCR amplification, leading one to greatly underestimate the frequencies of these mutations. By contrast, the hybridization-based capture approach utilized by mAdCap-Seq for target enrichment recovers DNA fragments with these and other types of mutations with the same efficiency. Large deletions in an evolved genome that completely remove a targeted region are an exception. These mutations create a blind spot for both amplicon and capture enrichment approaches. The lack of any reads from genomes with the deletion will lead to overestimating the frequencies of other mutations that preserve recovery of DNA from the region. While this is a general caveat that should be considered when using these approaches, large deletions are unlikely to have affected our mAdCap-Seq results. The genes we captured are rarely mutated in this way in the LTEE, and we tracked mutations early in the evolution of these populations from single cells when all new genetic variants were still rare.
BAR-seq represents a class of lineage-tracking methods that can be used to characterize spontaneous mutations through monitoring changes in the frequencies of barcode sequences inserted into the genomes of cells (Blundell and Levy, 2014). Since all high-throughput sequencing reads can be concentrated into counting barcodes that differ from one another (to the extent that sequencing errors do not affect proper assignment), these methods can track rarer subpopulations of cells than mAdCap-Seq. This means they can characterize essentially all of the beneficial mutations competing in a population. For comparison, we tracked ~60 mutations per E. coli population in the initial selective sweep, but tens of thousands of mutations have been analyzed in this way using BAR-seq on a single yeast population (Levy et al., 2015). Thus, lineage-tracking methods have much greater power for reconstructing the distribution of fitness effects of all mutations that spontaneously arise in a cell population. However, there are trade-offs relative to other approaches. Additional post-hoc whole-genome sequencing of many isolates from the evolved population is needed to link each barcode to a mutation causing the observed fitness change (Venkataram et al., 2016). But the foremost consideration is that one must genetically engineer cells to introduce barcodes into their genomes. This limitation has restricted the use of BAR-seq to populations of model organisms evolved in the laboratory so far.
The adaptome is the neighborhood of accessible genetic changes that are most likely to become dominant and contribute to ongoing adaptation of a population. We have demonstrated how mAdCap-Seq can be used to deeply profile a targeted portion of the adaptome of a bacterial cell undergoing clonal evolution in a controlled laboratory environment. Mapping clonal adaptomes, which consist solely of de novo beneficial mutations, is likely to be of particular interest and utility in systems that evolve repeatedly from a defined starting point. These range from bioreactors that are seeded with the same engineered strain in different production runs to lung infections in cystic fibrosis patients that start from similar, but not identical, strains of opportunistic pathogens. This approach could also be used to probe the fitness landscapes underlying how human infections and cancers evolve drug resistance and greater pathogenicity. Mapping out the likely evolutionary possibilities in these systems can potentially allow one to monitor for the appearance of especially costly or dangerous mutations and could even lead to developing interventions that deflect the population from these undesirable adaptive pathways.
Outside of the laboratory, some of the simplifying assumptions used in our study break down. In may be difficult or impossible to sample a new cell population found in the environment or in a patient multiple times during the critical time window before it has already evolved substantial genetic diversity. Furthermore, many microbes have mechanisms for sexual recombination that can rapidly combine multiple beneficial mutations into one genome, which may violate our assumption that double mutants are rare during the first selective sweep. While these real-world situations complicate evaluating the relative benefits of each new genetic variant from sequencing alone, mAdCap-Seq would still be a useful approach for characterizing whether mutations in specific genes are contributing to the leading edge of adaptation in these situations.
STAR★METHODS
Resource Availability
Lead Contact
Lead contact, Jeffrey E. Barrick (jbarrick@cm.utexas.edu), will provide additional information and fulfill any requests for materials.
Materials availability
Frozen E. coli populations from each sequenced time point are available.
Data and code availability
Illumina sequencing reads associated with this study have been deposited into the NCBI Sequence Read Archive (Accession: PRJNA601748). Code used for analysis and figure generation is archived on Zenodo (DOI: 10.5281/zenodo.5092871).
Experimental Model and Subject Details
Bacterial Strains and Culture Conditions
E. coli B strains REL606 and REL607 and growth conditions are derived from the Lenski long-term evolution experiment (Lenski and Travisano, 1994; Lenski et al., 1991). REL606 and REL607 differ by a mutation in an arabinose utilization gene enabling us to monitor their relative frequencies during the evolution experiment (Lenski et al., 1991). Growth was carried out in 50 mL Erlenmeyer flasks in 10 mL of Davis Minimal (DM) media supplemented with 100 mg/L glucose (DM100). DM is made by dissolving 5.3 g/L K2HPO4, 2 g/L KH2PO4, 1 g/L (NH4)2 SO4, and 500 mg/L Na3C6H5O7 (H2O)2 in water. After autoclaving, 1 mL/L of a 10% (w/v) MgSO4 stock solution (separately autoclaved) and 1 mL/L of a 0.2% (w/v) stock solution of thiamine (filter sterilized) are added. DM100 has a slightly higher concentration of glucose than the 25 mg/L glucose (DM25) used in the LTEE, but it still well below the ~1000 mg/L concentration at which nutrients other than glucose begin to limit growth in this medium. We used this higher glucose concentration to ensure we had enough cells for sequencing and archiving. Liquid cultures were grown at 37°C with orbital shaking over a one-inch diameter at 120 RPM.
Method Details
Evolution experiment
Nine colonies of E. coli B strain REL606 and nine of strain REL607 were selected at random and used to inoculate separate flasks containing 10 mL of DM25. Approximately 30 generations of growth occurred starting from the initial single cell that gave rise to each colony until saturation of these cultures. Populations A1 through A9 were founded by inoculating 10 mL of fresh DM100 with 50 μL of one REL606 culture and 50 μL of one REL607 culture. The remaining culture volume for all 18 founding colonies was archived in 15 mL conical tubes at −80°C with 2 mL dimethyl sulfoxide (DMSO) added as cryoprotectant. Every 24 hours, 100 μL of overnight culture was transferred to 10 mL of fresh DM100, and the remaining culture volume was archived in the same way. This procedure was continued through 75 daily transfers. Periodically 1 μL of culture was diluted 10,000-fold in sterile saline and plated on tetrazolium arabinose (TA) agar to allow growth of ~200 colonies. TA agar is made by adding 10 g/L tryptone, 1 g/L yeast extract, 5 g/L NaCl, and 16 g/L agar to water and autoclaving. Then, a separately autoclaved solution of 10 g/L arabinose in water is added. The combined volume of these two solutions is such that it yields the indicated final concentrations of each component. Roughly 4/5 and 1/5 of the total volume are used for the two solutions, respectively. As the solution cools, 1 mL/L of 5% (w/v) triphenyl tetrazolium chloride (filter sterilized) is added. On TA plates, the mutation in the arabinose utilization gene makes REL606 (Ara−) colonies red and REL607 (Ara+) colonies pink (Lenski et al., 1991). The ratio of red to pink colonies was used to monitor these populations for selective sweeps (Hegreness and Kishony, 2007; Woods et al., 2011).
DNA isolation and library preparation
Genomic DNA (gDNA) was isolated from frozen population samples by first thawing each 15 mL conical tube on ice. Of the ~12 mL total volume of culture plus cryoprotectant, 1.2 mL was transferred to a 2 mL cryovial and refrozen. The remaining ~10.8 mL was centrifuged at 6,500 × g at 4°C for 15 minutes. The resulting cell pellets were transferred with a volume of remaining solution to 1.7 mL Eppendorf tubes. Then, gDNA was isolated using the PureLink Genomic DNA Mini kit (Life Technologies). For each sample, 1 μg of gDNA was randomly fragmented on a Covaris S2 Focused Ultrasonicator.
Illumina libraries were constructed using the Kappa Biosystems LTP Library Preparation Kit with the following modifications. Following the end-repair step, fragmented DNA was T-tailed (rather than A-tailed) in a 50 μl reaction including 10 mM dTTP and 5 units of Klenow fragment, exo− (New England Biolabs). In the adapter ligation step, modified Illumina adapters containing 12-base unique molecular identifiers were ligated to the T-tailed fragments as previously described (Schmitt et al., 2012). Adapters used here differ slightly from those used in (Schmitt et al., 2012) as full-length adapter sequences containing unique external sample barcodes were directly ligated to the T-tailed dsDNA inserts to reduce the risk of cross-contamination between samples. The full list of DNA sequence adaptors is provided in Table S1.
Probe design and target capture
Oligonucleotide probes consisting of 60-base xGen Lockdown probes (Integrated DNA Technologies) were designed to tile across each of the eight genes of interest including upstream promoter elements. Probes for each gene were compared to the entire E. coli B strain REL606 reference genome (GenBank: NC_012967.1) (Jeong et al., 2009) using BLASTN (Camacho et al., 2009). The starting positions of all probes in a set were shifted by one base at a time until every probe had only a single significant predicted binding location (match with E-value < 2×10−5). The sequences of the final set of 242 probes are provided in Table S2.
Capture was performed using a SeqCap EZ Hybridization and Wash Kit (NimbleGen). The procedure in the SeqCap EZ Library SR User Guide v3.0 (NimbleGen) was followed with several modifications. First, 18 to 20 population samples with different sample barcodes were pooled together in a single capture reaction that contained a total of 1 μg of library DNA from all samples, 1 mmol of a universal blocking oligo, and 1 mmol of a degenerate sample barcode blocking oligo. The sequences of these blocking oligos are provided in Table S3. Second, after hybridization for 72 h, DNA fragments hybridized to the biotinylated probes were recovered using MyOne Streptavidin C1 Dynabeads (Life Technologies). Third, a final 8-cycle PCR step was performed with HiFi Hotstart DNA Polymerase (Kappa Biosystems).
Sequencing
Paired-end 101- or 125-base sequencing of the final libraries was performed on an Illumina HiSeq 2000 at the University of Texas at Austin the Genome Sequencing and Analysis Facility (GSAF). Read sequences have been deposited into the NCBI Sequence Read Archive (PRJNA601748).
Quantification and Statistical Analysis
Read processing
Raw reads were used to generate Consensus Sequence Reads (CSR) using custom Python scripts that carried out the following steps. First, the beginning of each read was evaluated for the presence of the expected 5′-end tag, consisting of a random 12-base unique molecular identifier (UMI) followed by four fixed bases (5′-N12CAGT). Read pairs lacking the correct 5′-end tag on either read were discarded. Across all samples, 80.2% of read pairs had both UMIs. For remaining read pairs, the UMIs found on each end were concatenated to create a 24-base dual-UMI that uniquely identifies the original DNA fragment that was amplified and sequenced. To group all reads corresponding to the same initial DNA molecule, a FASTA file of all dual-UMIs was used as input into the ustacks program from the Stacks software pipeline (Version 1.48) (Catchen et al., 2013) with the following options: a single read was sufficient to seed a stack, a single mismatch within the dual-UMI was allowed in assigning a read to a stack, secondary reads and haplotypes were disabled, and stacks with high coverage were preserved. Then, CSRs were generated for all dual-UMI groups sequenced at least twice by taking the straight consensus of all reads that were merged into that stack. If no base exceeded 50% frequency at a given position in this set of reads, then that base was set as unknown (N). Of the read pairs with valid dual-UMIs, 41.6% were incorporated into consensus reads across all samples. The average number of dual-UMI read pairs utilized to create each consensus read was 2.46, which gives an overall yield of consensus reads per read in a pair with a valid dual-UMI 16.9%.
Variant calling
We used the breseq pipeline (Barrick et al., 2014; Deatherage and Barrick, 2014; Deatherage et al., 2015) (version 0.26.0) to call single-nucleotide variants (SNVs) and structural variants (SVs) from the CSRs. This pipeline used Bowtie2 (version 2.2.5) for read mapping (Langmead and Salzberg, 2012). We divided the genome sequence of the ancestral E. coli REL606 strain into two types of reference regions for mapping in this analysis. The eight regions of the genome tiled with probes—extended with hundreds of bases of flanking sequence on both sides—were input as "targeted" sequences, and the remainder of the genome with the identical eight regions masked to degenerate N bases was supplied as a "junction-only" reference (to which reads are mapped without variant calling). All 116 samples were analyzed using breseq in polymorphism prediction mode with all bias, minimum allele frequency, and read-count filters disabled. Evidence items in the Genome Diff (GD) files for all samples were combined using the gdtools utility program to generate a single merged GD file with each piece of evidence listed a single time, regardless of how many times it was detected in different samples. We then re-ran breseq using the same parameters except that this GD file was supplied as an input user-evidence file to force output of variant and reference information for these putative variants in every sample. Then, we extracted the number of variant reads supporting each putative variant allele and the total number of reads at that reference location from the GD file output by breseq. Subsequent statistical tests and fitting steps were performed in R (version 4.0.0) (R Core Team, 2016) using the ggplot2 package for data visualization (Wickham, 2016).
Since this original analysis was conducted at the level of breseq evidence (i.e., single columns of read pileups on the reference genome or instances of new sequence junctions), we next merged sets of observations that were consistent with a single mutational event when they also had frequency trajectories that tracked together. For example, a three-base deletion has separate evidence items for the first, second, and third missing bases at this stage in the analysis. To identify candidates for merging evidence into a single mutational event, we analyzed data from each complete time course (generation 30 to 530) and selective sweep window (generation 163 to 243) separately. We only considered mutations that exceeded a threshold frequency of 0.03% at some time during each time course as candidates for merging.
Read alignment (RA) evidence items were merged when they were located within 6 base pairs of one another and the normalized Canberra distance between the vectors of their frequency observations across all time points was ≤ 0.1. All RA evidence pairs of this kind were found to co-occur in the same sequencing reads. For these cases, the read counts for the first linked mutation were used to represent the entire event. For example, if a deletion of three base pairs was predicted from evidence of missing bases at positions x, y, and z; then the frequency of missing the first base (x) was assigned to the entire three-base deletion mutation.
For new junction (JC) evidence we performed the same merging procedure but allowed linked mutations to be within a larger window of 20 base pairs and within a normalized Canberra distance of 0.5. JC pairs passing these criteria were only merged if they were also consistent with an IS-element insertion in terms of their relative orientation and spacing. In this case the variant and total read counts were added together for the two different junctions, as the junctions on each side of the inserted IS element provide independent information for estimating the frequency of this type of mutation. We allowed unpaired JC evidence passing the filters to also predict IS element mutations. This situation may indicate that there was an IS-mediated deletion between an element that inserted within the gene and another element from the same family located outside of the targeted region or more complex chromosomal rearrangements involving a newly inserted IS element (Raeside et al., 2014).
Time course filtering and fitness effect estimation
After merging evidence of genetic variants into lists of putative mutations, we further eliminated some of these from consideration using several filtering steps. For the complete time courses, we first required that non-zero frequencies be observed for a mutation in samples from two different time points. We next applied a filter to eliminate spurious variants that can be recognized as arising from systematic sequencing or read alignment errors because they do not exhibit the correlated changes in frequency over time expected for the frequency trajectories of real mutations (Lang et al., 2013). Specifically, we required that the time-series of estimated frequencies for a mutation over all analyzed time points have an autocorrelation value ≥ 0.55.
For the analysis of mutation trajectories during the selective sweep window, we eliminated putative mutations for which there was great uncertainty in the estimated fitness effect or evidence that its trajectory reflected multiple beneficial mutations occurring in the same genome. Specifically, we required that a mutation was first observed at generation 196 or earlier and that its estimated frequency was ≥ 10−4 in every sample that was sequenced from generation 223 to 243. Then, we fit a binomial logistic model with slope and y-intercept terms to the time courses of counts of variant and reference (total minus variant) observations for each mutation. We used a negative offset in the model of the number of generations up to each time point so that the slope represents one plus the selection coefficient that is characteristic of the subpopulation with that mutation. We filtered out any mutations for which this fit had an AIC < 200, a p-value for the slope differing from zero of > 0.005, or a y-intercept < −20. The fitness effects that we report for mutations are the selection coefficients fit from the model divided by the natural logarithm of two so that they are expressed per generations of binary cell division. One plus the fitness effect is the relative fitness of a cell with that mutation. These values can be directly compared to experimental measurements of relative fitness and mutation fitness effects made using co-culture competition assays (Lenski et al., 1991; Tenaillon et al., 2016).
This procedure for determining fitness effects assumes that the trajectories reflect competition purely against the ancestral strain. However, we detected a consistent deviation from linearity for all mutation trajectories after generation 196. The rates at which the frequencies of all mutations were increasing decelerated, indicating that the overall population fitness had improved to a degree that it reduced their effective advantage versus their competitors. To account for this change we fit additional parameters defining a stepwise increase in the average relative fitness of the population within each interval between sequenced samples from generation 196 onward. The increase in population fitness reduces the effective time basis used in the model to determine the slope to the number of generations in each interval divided by the average relative fitness during that interval. We determined the population fitness values that minimized the AIC of this modified binomial logistic model. The figures show the best stepwise increases in population fitness between the sequenced time points from generation 196 onward fitting to the trajectories of all mutations in a given population at the same time. We performed 1000 bootstrap resamplings of the mutations in each population to estimate 95% confidence intervals on the estimated population fitness values in each interval for that population.
We combined information across multiple populations in two ways to further improve the estimates of mutation fitness effects. First, there was considerable uncertainty in the estimates of the stepwise population fitness increases for each population considered alone. Because the actual population fitness trajectories of all populations are expected to be highly similar to one another, we fit a consensus stepwise increase in population fitness over time that best improved the fits for all mutations from all populations. Second, we observed 27 cases in which the exact same change in a gene's sequence was observed and passed our filtering criteria in multiple experimental populations. Because each population was started from single cells, we can be sure that these are independent observations of the same mutation. Therefore, we fit one consensus fitness effect (slope) for each of these recurrent mutations across all populations. We still allowed the y-intercept for each of these mutations to vary from population to population because this parameter is related to how early the mutation evolved, which is expected to be different in each replicate population.
Protein structure analysis
Structural domains in NadR, PykF, and TopA were defined according to UniProt and papers reporting x-ray crystal structures. Mutations in PykF were mapped onto Protein Data Bank structure 4YNG (Donovan et al., 2016). Mutations in TopA were mapped onto Protein Data Bank structure 1MW8 (Perry and Mondragón, 2003). Protein structures were visualized using Pymol v2.3.5 (Schrödinger LLC).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Klenow fragment exo− | New England Biolabs | M0212S |
| MyOne Streptavidin C1 Dynabeads | Life Technologies | 65001 |
| Critical commercial assays | ||
| PureLink Genomic DNA Mini kit | Life Technologies | K210011 |
| LTP Library Preparation Kit | Kappa Biosystems | Kk8232 |
| SeqCap EZ Hybridization and Wash Kit | NimbleGen | 05 634 261 001 |
| KAPA Library Amplification Kit | Kappa Biosystems | KK2611 |
| Deposited data | ||
| Raw Illumina Sequencing Data | This paper | NCBI SRA: PRJNA601748 |
| Experimental models: Organisms/strains | ||
| E. coli: Strain background: REL606 | Lenski et al., 1991 | N/A |
| E. coli: Strain background: REL607 | Lenski et al., 1991 | N/A |
| Oligonucleotides | ||
| 21 Illumina Sequencing Adapters containing 12-base MI: See Table S1 | This paper | N/A |
| 242 60-base xGen Lockdown probes: see Table S2 | This paper | N/A |
| 2 Blocking Oligos: see Table S3 | This paper | N/A |
| Software and algorithms | ||
| BLASTN | Camacho et al., 2009 | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| CSR generation python script | This paper | DOI: 10.5281/zenodo.5092871 |
| Stacks v1.48 | Catchen et al., 2013 | https://catchenlab.life.illinois.edu/stacks/ |
| breseq v0.26.0 | Deatherage and Barrick, 2014 | https://github.com/barricklab/breseq |
| Bowtie2 v2.2.5 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| R v4.0.0 | R Core Team, 2016 | https://www.r-project.org/ |
| Custom breseq post-processing Python script | This paper | DOI: 10.5281/zenodo.5092871 |
| Statistical testing and modeling of trajectories R script | This paper | DOI: 10.5281/zenodo.5092871 |
| Pymol v2.3.5 | Schrodinger LLC | Pymol.org |
| Pymol figure visualization script | This paper | DOI: 10.5281/zenodo.5092871 |
| Other | ||
| REL606.6.GENES.1400-flanking.gff3 | This paper | DOI: 10.5281/zenodo.5092871 |
| REL606.6.GENES.MASKED.gff3 | This paper | DOI: 10.5281/zenodo.5092871 |
Highlights.
mAdCap-Seq tracks many competing beneficial mutations in a clonal population
Identified 301 mutations by mAdCap-Seq of eight genes in six E. coli populations
Measured the fitness benefits of 240 mutations directly from their trajectories
The functions of the topA, nadR, and pykF genes evolved in distinctive ways
ACKNOWLEDGEMENTS
The authors acknowledge several anonymous reviewers for helpful feedback and the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing high-performance computing resources. Funding: this work was supported by the Cancer Prevention & Research Institute of Texas (CPRIT) (RP130124), the National Institutes of Health (R00GM087550 and R01GM088344), the National Science Foundation (CBET-1554179 and DEB-1813069), and the NSF BEACON Center for the Study of Evolution in Action (DBI-0939454). D.E.D. acknowledges support from a University of Texas at Austin CPRIT research traineeship (RP101501). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
J.E.B. is the owner of Evolvomics LLC. D.E.D. has been a paid consultant for Evolvomics LLC.
REFERENCES
- Bainbridge MN, Wang M, Burgess DL, Kovar C, Rodesch MJ, D’Ascenzo M, Kitzman J, Wu Y-Q, Newsham I, Richmond TA, et al. (2010). Whole exome capture in solution with 3 Gbp of data. Genome Biol. 11, R62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE (2020). Limits to predicting evolution: insights from a long-term experiment with Escherichia coli. In Evolution in Action: Past, Present and Future, Banzhaf W, Cheng BHC, Deb K, Holekamp KE, Lenski RE, Ofria C, Pennock RT, Punch WF, and Whittaker DJ, eds. (Cham: Springer; ), pp. 63–76. [Google Scholar]
- Barrick JE, and Lenski RE (2009). Genome-wide mutational diversity in an evolving population of Escherichia coli. Cold Spring Harb. Symp. Quant. Biol 74, 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, and Lenski RE (2013). Genome dynamics during experimental evolution. Nat. Rev. Genet 14, 827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Yu DS, Yoon SH, Jeong H, Oh TK, Schneider D, Lenski RE, and Kim JF (2009). Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461, 1243–1247. [DOI] [PubMed] [Google Scholar]
- Barrick JE, Colburn G, Deatherage DE, Traverse CC, Strand MD, Borges JJ, Knoester DB, Reba A, and Meyer AG (2014). Identifying structural variation in haploid microbial genomes from short-read resequencing data using breseq. BMC Genomics 15, 1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baym M, Lieberman TD, Kelsic ED, Chait R, Gross R, Yelin I, and Kishony R (2016). Spatiotemporal microbial evolution on antibiotic landscapes. Science 353, 1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell JR, and Levy SF (2014). Beyond genome sequencing: Lineage tracking with barcodes to study the dynamics of evolution, infection, and cancer. Genomics 104, 417–430. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, and Madden TL (2009). BLAST+: architecture and applications. BMC Bioinformatics 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen J, Hohenlohe PA, Bassham S, Amores A, and Cresko WA (2013). Stacks: an analysis tool set for population genomics. Mol. Ecol 22, 3124–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou H-H, Chiu H-C, Delaney NF, Segrè D, and Marx CJ (2011). Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science 332, 1190–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubiz LM, Lee M-C, Delaney NF, and Marx CJ (2012). FREQ-Seq: a rapid, cost-effective, sequencing-based method to determine allele frequencies directly from mixed populations. PLoS ONE 7, e47959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozat E, Philippe N, Lenski RE, Geiselmann J, and Schneider D (2005). Long-term experimental evolution in Escherichia coli. XII. DNA topology as a key target of selection. Genetics 169, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozat E, Winkworth C, Gaffe J, Hallin PF, Riley MA, Lenski RE, and Schneider D (2010). Parallel genetic and phenotypic evolution of DNA superhelicity in experimental populations of Escherichia coli. Mol. Biol. Evol 27, 2113–2128. [DOI] [PubMed] [Google Scholar]
- Cvijović I, Nguyen Ba AN, and Desai MM (2018). Experimental studies of evolutionary dynamics in microbes. Trends Genet. 34, 693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deatherage DE, and Barrick JE (2014). Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol. Biol 1151, 165–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deatherage DE, Traverse CC, Wolf LN, and Barrick JE (2015). Detecting rare structural variation in evolving microbial populations from new sequence junctions using breseq. Front. Genet 5, 468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deatherage DE, Kepner JL, Bennett AF, Lenski RE, and Barrick JE (2017). Specificity of genome evolution in experimental populations of Escherichia coli evolved at different temperatures. Proc. Natl. Acad. Sci. U. S. A 114, E1904–E1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai MM, Walczak AM, and Fisher DS (2012). Genetic diversity and the structure of genealogies in rapidly adapting populations. Genetics 193, 565–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinardo S, Voelkel KA, Sternglanz R, Reynolds AE, and Wright A (1982). Escherichia coli DNA topoisomerase I mutants have compensatory mutations in DNA gyrase genes. Cell 31, 43–51. [DOI] [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. (2012). Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481, 506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan KA, Atkinson SC, Kessans SA, Peng F, Cooper TF, Griffin MDW, Jameson GB, and Dobson RCJ (2016). Grappling with anisotropic data, pseudo-merohedral twinning and pseudo-translational noncrystallographic symmetry: A case study involving pyruvate kinase. Acta Crystallogr. Sect. D Struct. Biol 72, 512–519. [DOI] [PubMed] [Google Scholar]
- Fischer S, Greipel L, Klockgether J, Dorda M, Wiehlmann L, Cramer N, and Tümmler B (2017). Multilocus amplicon sequencing of Pseudomonas aeruginosa cystic fibrosis airways isolates collected prior to and after early antipseudomonal chemotherapy. J. Cyst. Fibros 16, 346–352. [DOI] [PubMed] [Google Scholar]
- Fowler DM, and Fields S (2014). Deep mutational scanning: a new style of protein science. Nat. Methods 11, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel EM, McDonald MJ, Van Dyken JD, Kosheleva K, Lang GI, and Desai MM (2015). Crowded growth leads to the spontaneous evolution of semistable coexistence in laboratory yeast populations. Proc. Natl. Acad. Sci 112, 11306–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa C, Horinouchi T, and Maeda T (2018). Toward prediction and control of antibiotic-resistance evolution. Curr. Opin. Biotechnol 54, 45–49. [DOI] [PubMed] [Google Scholar]
- Garst AD, Bassalo MC, Pines G, Lynch SA, Halweg-Edwards AL, Liu R, Liang L, Wang Z, Zeitoun R, Alexander WG, et al. (2017). Genome-wide mapping of mutations at single-nucleotide resolution for protein, metabolic and genome engineering. Nat. Biotechnol 35, 48–55. [DOI] [PubMed] [Google Scholar]
- Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrish PJ, and Lenski RE (1998). The fate of competing beneficial mutations in an asexual population. Genetica 102–103, 127–144. [PubMed] [Google Scholar]
- Good BH, McDonald MJ, Barrick JE, Lenski RE, and Desai MM (2017). The dynamics of molecular evolution over 60,000 generations. Nature 551, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham D, and Dunham MJ (2014). The enduring utility of continuous culturing in experimental evolution. Genomics 104, 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegreness M, and Kishony R (2007). Analysis of genetic systems using experimental evolution and whole-genome sequencing. Genome Biol. 8, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegreness M, Shoresh N, Hard D, and Kishony R (2006). An equivalence principle for the incorporation of favorable mutations in asexual populations. Science 311, 1615–1617. [DOI] [PubMed] [Google Scholar]
- Hong J, and Gresham D (2017). Incorporation of unique molecular identifiers in TruSeq adapters improves the accuracy of quantitative sequencing. Biotechniques 63, 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Brandt N, Abdul-Rahman F, Yang A, Hughes T, and Gresham D (2018). An incoherent feedforward loop facilitates adaptive tuning of gene expression. eLife 7, e32323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houdaigui B. El, Forquet R, Hindré T, Schneider D, Nasser W, Reverchon S, and Meyer S (2019). Bacterial genome architecture shapes global transcriptional regulation by DNA supercoiling. Nucleic Acids Res. 47, 5648–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Barbe V, Lee CH, Vallenet D, Yu DS, Choi S-H, Couloux A, Lee S-W, Yoon SH, Cattolico L, et al. (2009). Genome sequences of Escherichia coli B strains REL606 and BL21(DE3). J. Mol. Biol 394, 644–652. [DOI] [PubMed] [Google Scholar]
- Khan AI, Dinh DM, Schneider D, Lenski RE, and Cooper TF (2011). Negative epistasis between beneficial mutations in an evolving bacterial population. Science 332, 1193–1196. [DOI] [PubMed] [Google Scholar]
- Kivioja T, Vähärautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, and Taipale J (2012). Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods 9, 72–74. [DOI] [PubMed] [Google Scholar]
- Kochanowski K, Volkmer B, Gerosa L, Van Rijsewijk BRH, Schmidt A, and Heinemann M (2013). Functioning of a metabolic flux sensor in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A 110, 1130–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryazhimskiy S, Rice DP, Jerison ER, and Desai MM (2014). Global epistasis makes adaptation predictable despite sequence-level stochasticity. Science 344, 1519–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurnasov OV, Polanuyer BM, Ananta S, Sloutsky R, Tam A, Gerdes SY, and Osterman AL (2003). Ribosylnicotinamide Kinase Domain of NadR Protein: Identification and Implications in NAD Biosynthesis. J. Bacteriol 185, 698–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, et al. (2013). Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152, 714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GI, Rice DP, Hickman MJ, Sodergren E, Weinstock GM, Botstein D, and Desai MM (2013). Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature 500, 571–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, and Kim HU (2015). Systems strategies for developing industrial microbial strains. Nat. Biotechnol 33, 1061–1072. [DOI] [PubMed] [Google Scholar]
- Lenski RE, and Travisano M (1994). Dynamics of adaptation and diversification: a 10,000-generation experiment with bacterial populations. Proc. Natl. Acad. Sci. U. S. A 91, 6808–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenski RE, Rose MR, Simpson SC, and Tadler SC (1991). Long-term experimental evolution in Escherichia coli. I. Adaptation and divergence during 2,000 generations. Am. Nat 138, 1315–1341. [Google Scholar]
- Levy SF, Blundell JR, Venkataram S, Petrov DA, Fisher DS, and Sherlock G (2015). Quantitative evolutionary dynamics using high-resolution lineage tracking. Nature 519, 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind PA, Farr AD, and Rainey PB (2015). Experimental evolution reveals hidden diversity in evolutionary pathways. eLife 4, e07074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddamsetti R, Lenski RE, and Barrick JE (2015). Adaptation, clonal interference, and frequency-dependent interactions in a long-term evolution experiment with Escherichia coli. Genetics 200, 619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusyk A, Almendro V, and Polyak K (2012). Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 12, 323–334. [DOI] [PubMed] [Google Scholar]
- Marvig RL, Sommer LM, Molin S, and Johansen HK (2015). Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet 47, 57–64. [DOI] [PubMed] [Google Scholar]
- Massé E, and Drolet M (1999). Relaxation of transcription-induced negative supercoiling is an essential function of Escherichia coli DNA topoisom erase I. J. Biol. Chem 274, 16654–16658. [DOI] [PubMed] [Google Scholar]
- Mattevi A, Valentini G, Rizzi M, Speranza ML, Bolognesi M, and Coda A (1995). Crystal structure of Escherichia coli pyruvate kinase type I: molecular basis of the allosteric transition. Structure 3, 729–741. [DOI] [PubMed] [Google Scholar]
- McDonald MJ (2019). Microbial experimental evolution – a proving ground for evolutionary theory and a tool for discovery. EMBO Rep. 20, e46992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo LMF, Pepper JW, Reid BJ, and Maley CC (2006). Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 6, 924–935. [DOI] [PubMed] [Google Scholar]
- Merlo LMF, Shah NA, Li X, Blount PL, Vaughan TL, Reid BJ, and Maley CC (2010). A comprehensive survey of clonal diversity measures in Barrett’s esophagus as biomarkers of progression to esophageal adenocarcinoma. Cancer Prev. Res. (Phila) 3, 1388–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Bratman SV, To J, Wynne JF, Eclov NCW, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, et al. (2014). An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med 20, 548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, Stehr H, Liu CL, Bratman SV, Say C, et al. (2016). Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol 34, 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J,and Keasling JD (2016). Engineering cellular metabolism. Cell 164, 1185–1197. [DOI] [PubMed] [Google Scholar]
- van Opijnen T, and Camilli A (2013). Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nat. Rev. Microbiol 11, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski EA, Woods RJ, and Lenski RE (2008). The genetic basis of parallel and divergent phenotypic responses in evolving populations of Escherichia coli. Proc. R. Soc. B 275, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-C, and Krug J (2007). Clonal interference in large populations. Proc. Natl. Acad. Sci. U. S. A 104, 18135–18140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng F, Widmann S, Wünsche A, Duan K, Donovan KA, Dobson RCJ, Lenski RE, and Cooper TF (2018). Effects of beneficial mutations in pykF gene vary over time and across replicate populations in a long-term experiment with bacteria. Mol. Biol. Evol 35, 202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry K, and Mondragón A (2003). Structure of a complex between E. coli DNA topoisomerase I and single-stranded DNA. Structure 11, 1349–1358. [DOI] [PubMed] [Google Scholar]
- Phaneuf PV, Yurkovich JT, Heckmann D, Wu M, Sandberg TE, King ZA, Tan J, Palsson BO, and Feist AM (2020). Causal mutations from adaptive laboratory evolution are outlined by multiple scales of genome annotations and condition-specificity. BMC Genomics 21, 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruss GJ, Manes SH, and Drlica K (1982). Escherichia coli DNA topoisomerase I mutants: Increased supercoiling is corrected by mutations near gyrase genes. Cell 31, 35–42. [DOI] [PubMed] [Google Scholar]
- R Core Team (2016). R: A Language and Environment for Statistical Computing (Vienna, Austria: R Foundation for Statistical Computing; ). http://www.R-project.org/ [Google Scholar]
- Raeside C, Gaffé J, Deatherage DE, Tenaillon O, Briska AM, Ptashkin RN, Cruveiller S, Médigue C, Lenski RE, Barrick JE, et al. (2014). Large chromosomal rearrangements during a long-term evolution experiment with Escherichia coli. MBio 5, e01377–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaelli N, Lorenzi T, Mariani PL, Emanuelli M, Amici A, Ruggieri S, and Magni G (1999). The Escherichia coli NadR regulator is endowed with nicotinamide mononucleotide adenylyltransferase activity. J. Bacteriol 181, 5509–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey PB, Remigi P, Farr AD, and Lind PA (2017). Darwin was right: where now for experimental evolution? Curr. Opin. Genet. Dev 47, 102–109. [DOI] [PubMed] [Google Scholar]
- Renda BA, Hammerling MJ, and Barrick JE (2014). Engineering reduced evolutionary potential for synthetic biology. Mol. Biosyst 10, 1668–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugbjerg P, and Sommer MOA (2019). Overcoming genetic heterogeneity in industrial fermentations. Nat. Biotechnol 37, 869–876. [DOI] [PubMed] [Google Scholar]
- Rugbjerg P, Myling-Petersen N, Porse A, Sarup-Lytzen K, and Sommer MOA (2018). Diverse genetic error modes constrain large-scale bio-based production. Nat. Commun 9, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryall B, Eydallin G, and Ferenci T (2012). Culture history and population heterogeneity as determinants of bacterial adaptation: the adaptomics of a single environmental transition. Microbiol. Mol. Biol. Rev 76, 597–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval CM, Ayson M, Moss N, Lieu B, Jackson P, Gaucher SP, Homing T, Dahl RH, Denery JR, Abbott DA, et al. (2014). Use of pantothenate as a metabolic switch increases the genetic stability of farnesene producing Saccharomyces cerevisiae. Metab. Eng 25, 215–226. [DOI] [PubMed] [Google Scholar]
- Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, and Loeb LA (2012). Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. U. S. A 109, 14508–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiquee KAZ, Arauzo-Bravo MJ, and Shimizu K (2004). Effect of a pyruvate kinase (pykF-gene) knockout mutation on the control of gene expression and metabolic fluxes in Escherichia coli. FEMS Microbiol. Lett 235, 25–33. [DOI] [PubMed] [Google Scholar]
- Stefani S, Campana S, Cariani L, Carnovale V, Colombo C, Lleo MM, Iula VD, Minicucci L, Morelli P, Pizzamiglio G, et al. (2017). Relevance of multidrug-resistant Pseudomonas aeruginosa infections in cystic fibrosis. Int. J. Med. Microbiol 307, 353–362. [DOI] [PubMed] [Google Scholar]
- Tan K, Zhou Q, Cheng B, Zhang Z, Joachimiak A, and Tse-Dinh YC (2015). Structural basis for suppression of hypemegative DNA supercoiling by E. coli topoisomerase I. Nucleic Acids Res. 43, 11031–11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon O, Rodríguez-Verdugo A, Gaut RL, McDonald P, Bennett AF, Long AD, and Gaut BS (2012). The molecular diversity of adaptive convergence. Science 335, 457–461. [DOI] [PubMed] [Google Scholar]
- Tenaillon O, Barrick JE, Ribeck N, Deatherage DE, Blanchard JL, Dasgupta A, Wu GC, Wielgoss S, Cruveiller S, Médigue C, et al. (2016). Tempo and mode of genome evolution in a 50,000-generation experiment. Nature 536, 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RK, Nickerson E, Simons JF, Jänne PA, Tengs T, Yuza Y, Garraway LA, LaFramboise T, Lee JC, Shah K, et al. (2006). Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat. Med 12, 852–855. [DOI] [PubMed] [Google Scholar]
- Traverse CC, Mayo-Smith LM, Poltak SR, and Cooper VS (2013). Tangled bank of experimentally evolved Burkholderia biofilms reflects selection during chronic infections. Proc. Natl. Acad. Sci. U. S. A 110, E250–E259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataram S, Dunn B, Li Y, Agarwala A, Chang J, Ebel ER, Geiler-Samerotte K, Hérissant L, Blundell JR, Levy SF, et al. (2016). Development of a comprehensive genotype-to-fitness map of adaptation-driving mutations in yeast. Cell 166, 1585–1596.E22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CJ, Papula AL, Poon GYP, Wong WH, Young AL, Druley TE, Fisher DS, and Blundell JR (2020). The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science 367, 1449–1454. [DOI] [PubMed] [Google Scholar]
- Wei X, and Zhang J (2019). Patterns and mechanisms of diminishing returns from beneficial mutations. Mol. Biol. Evol 36, 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis (New York: Springer-Verlag; ). [Google Scholar]
- Winstanley C, O’Brien S, and Brockhurst MA (2016). Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol. 24, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiser MJ, Ribeck N, and Lenski RE (2013). Long-term dynamics of adaptation in asexual populations. Science 342, 1364–1367. [DOI] [PubMed] [Google Scholar]
- Woods R, Schneider D, Winkworth CL, Riley MA, and Lenski RE (2006). Tests of parallel molecular evolution in a long-term experiment with Escherichia coli. Proc. Natl. Acad. Sci. U. S. A 103, 9107–9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods RJ, Barrick JE, Cooper TF, Shrestha U, Kauth MR, and Lenski RE (2011). Second-order selection for evolvability in a large Escherichia coli population. Science 331, 1433–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelder O, and Hauer B (2000). Environmentally directed mutations and their impact on industrial biotransformation and fermentation processes. Curr. Opin. Microbiol 3, 248–251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Illumina sequencing reads associated with this study have been deposited into the NCBI Sequence Read Archive (Accession: PRJNA601748). Code used for analysis and figure generation is archived on Zenodo (DOI: 10.5281/zenodo.5092871).