

Abstract
Decoding the identity of biomolecules from trace samples is a longstanding goal in the field of biotechnology. Advances in DNA analysis have significantly impacted clinical practice and basic research, but corresponding developments for proteins face challenges due to their relative complexity and our inability to amplify them. Despite progress in methods such as mass spectrometry and mass cytometry, single-molecule protein identification remains a highly challenging objective. Toward this end, we combine DNA nanotechnology with single-molecule force spectroscopy to create a mechanically reconfigurable DNA Nanoswitch Caliper (DNC) capable of measuring multiple coordinates on single biomolecules with atomic resolution. Using optical tweezers, we demonstrate absolute distance measurements with angstrom-level precision for both DNA and peptides, and using multiplexed magnetic tweezers, we demonstrate quantification of relative abundance in mixed samples. Measuring distances between DNA-labeled residues, we perform single-molecule fingerprinting of synthetic and natural peptides, and show discrimination, within a heterogeneous population, between different post-translational modifications. DNA Nanoswitch Calipers are a powerful and accessible tool for characterizing distances within nanoscale complexes that will enable new applications in fields such as single-molecule proteomics.
Introduction
The ability to identify and characterize proteins has led to both a deeper understanding of biological mechanisms and more rational approaches for developing therapeutics. Mass-spectrometry is a gold-standard for protein identification, but many low-abundant proteins are non-identifiable due to limitations in sensitivity, and challenges in analyzing complex mixtures1. A method for accurately and comprehensively analyzing proteins within trace samples would impact fields ranging from diagnostics to cell biology, but remains a highly challenging goal. One promising approach is through the emerging field of single-molecule proteomics, which has the potential to transform protein analysis analogously to how next-generation sequencing has transformed genomics and transcriptomics2–5, and could provide an avenue towards single-cell proteomics. Many single-molecule approaches are based on molecular fingerprints6–8, such as the distances in primary sequence between specifically labeled amino acids, which can be compared against a protein database for identification. Yet experimental realizations of this vision are currently hampered by limitations in resolution, throughput, and accuracy.
Here, we introduce DNA Nanoswitch Calipers (DNC), a high-resolution approach for rapidly measuring the spatial positions of multiple amino-acid residues within a single molecule in solution. DNC is a force spectroscopy based approach, which joins emerging fluorescence9, 10, nanopore11–14, and tunneling-current15, 16 based single-molecule proteomics methods, each with its own distinct benefits2. DNC increases measurement accuracy through three key principles: i) mechanical force is used to stretch the polypeptide chain, decreasing thermal noise and internal interactions, ii) the DNA nanostructure enables repeated interrogation of the same target molecule to increase measurement precision, and iii) the DNA nanostructure provides a precise molecular reference for converting changes in length into absolute distance measurements. By combining single-molecule force spectroscopy with mechanically reconfigurable DNA nanostructures, we have created a powerful yet accessible molecular tool for proteomic analysis.
DNA Nanoswitch Caliper Basic Concepts
The DNA Nanoswitch Caliper is a molecular device for measuring distances within single molecules that can be mechanically actuated between a looped and unlooped state. This work builds on programmable DNA nanoswitches17, self-assembled molecular tethers designed to enhance single-molecule force spectroscopy measurements18, 19. Other engineered tethers include looped constructs generated through protein engineering20, and other approaches based on DNA nanotechnology21–25. DNA nanoswitches are tethers of DNA decorated at specific sites with functionalities that can bind—either directly to each other, or through a third bridging complex. Binding causes a section of the nanoswitch to loop out, shortening the tether length; when these bonds break, the tether extends back to its original length. Here, we leverage this approach to enable not only the measurement of forces18, 19, but also the precise measurement of pairwise distances within single molecules—fingerprints that can reveal protein identity, post-translational modifications, and shape. Essentially, the change in length from a looped to an unlooped configuration reflects the size of the molecular bridge that closes the loop—the larger the bridge, the smaller the change in length. DNCs are constructed with two specific DNA regions capable of hybridizing to DNA handles on a target molecule; by grabbing and releasing a pair of handles, the caliper reports the distances between these handles by changing its end-to-end length. The caliper loop provides a reference length, enabling this change in length to be converted to an absolute distance measurement; the distance between two handles (d) is given by the effective loop length (L0) minus the measured change-in-length (ΔL), i.e. d = L0 – ΔL. Figure 1 illustrates the DNC approach: target molecules are labeled with DNA handles (Fig. 1a), distances between handles are measured with the caliper (Fig. 1b), single-molecule fingerprints are analyzed (Fig. 1c).
Fig. 1. Schematic overview of single-molecule mechanical fingerprinting with DNA Nanoswitch Calipers (DNCs).
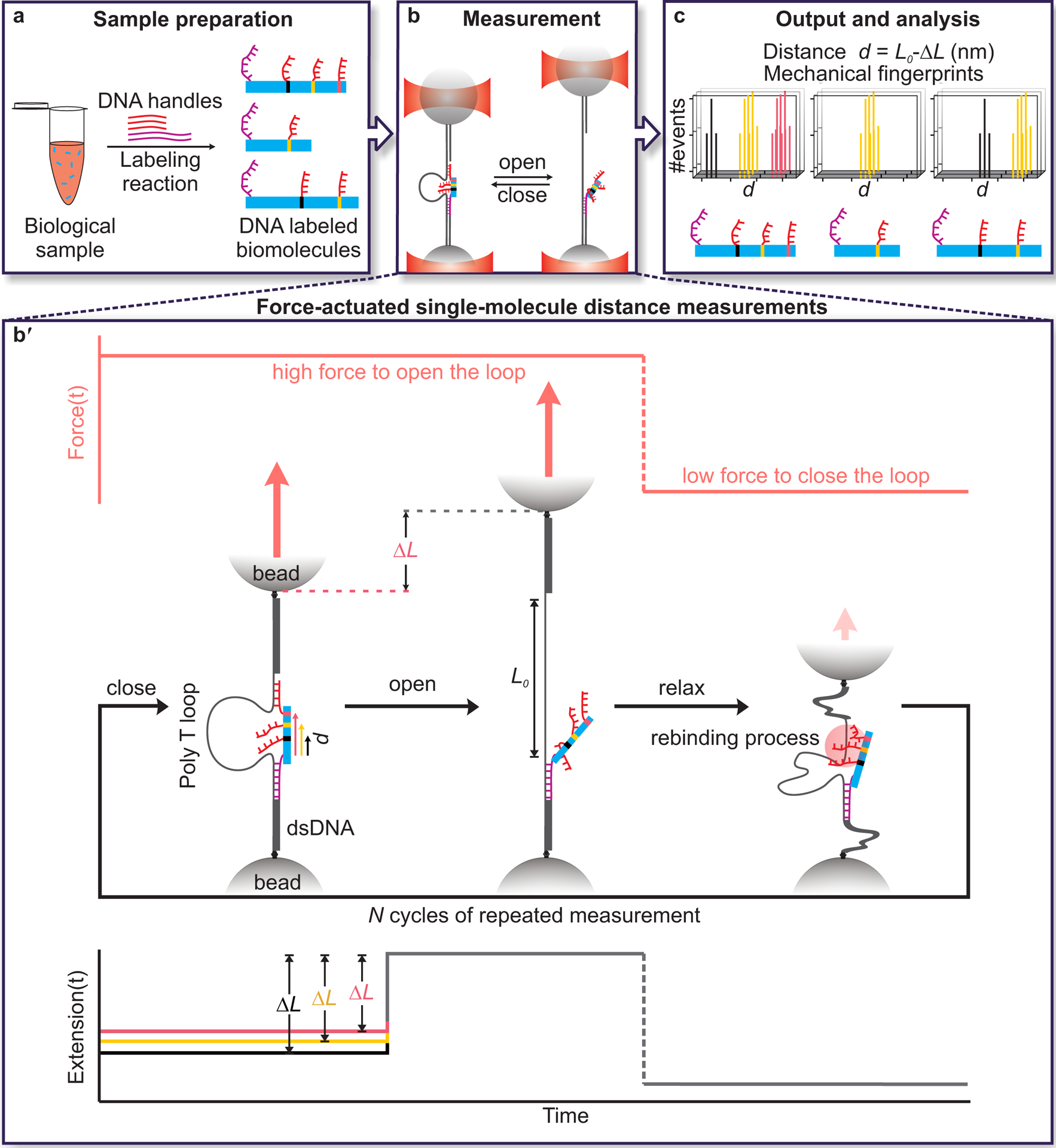
a, Samples are prepared through residue-specific labeling of biomolecules with DNA handles. Here, a long, mechanically strong handle (purple strand) and several shorter, mechanically weak handles (red strands) are attached to each target biomolecule. b, Distances between pairs of handles on target molecules are measured by grabbing and releasing them with the caliper while monitoring the change-in-length (ΔL) between looped and unlooped states. Force-actuation is carried out by tethering the caliper between two optically trapped beads. b′, Detailed view of distance measurements showing the application of force on a DNA Nanoswitch caliper in repeated cycles (top panel), the corresponding changes in conformation of the caliper (middle panel), and the corresponding changes in extension (bottom panel). The increase in length (ΔL) observed when the loop opens during the high-force period depends on the initial distance between handles on the target bridging the loop—the longer the initial distance, the shorter the ΔL. c, Changes in tether length (ΔL) are subtracted from the loop length (L0) to yield absolute distance measurements (d = L0 − ΔL). Multiple measurements are made on each single-molecule target to yield mechanical fingerprints, which can enable the identification of targets in a heterogeneous biological sample. Note that figures are not to scale.
DNC Validation with ssDNA targets
To validate and characterize DNC performance, we measured the lengths of ssDNA targets using calipers actuated by optical tweezers. Calipers with loops consisting of 78 thymine bases were synthesized (Methods, Supplementary Fig. 1) and used to probe target DNA molecules, each consisting of a poly-thymine bridge flanked by a long, mechanically strong handle (40 bases) with a shearing force of ~ 60 pN at our loading rates26 and a shorter, mechanically weak handle (14 bases) (Supplementary Table 1) that could be sheared at ~26 pN (Fig. 2). These handle lengths ensured attachment integrity up to the desired shearing force, which was chosen to reduce thermal noise and secondary structure. Calipers bound with target DNA were tethered between two optically trapped beads and toggled between two force states: a lower force (~0 pN) to rebind the short handle and a higher force (~26 pN) to extend the looped DNC then shear the handle, yielding a sudden change-in-length (ΔL) (Fig. 2b). Caliper performance was characterized by measuring ΔL for target molecules with different numbers of thymine bases (4T, 5T, 6T, 7T, 10T, 15T, 20T and 30T) (Fig. 2c). As expected, the measured mean ΔLs were linearly correlated with number of thymines over a large range of designed target lengths, with systematic deviations in the shorter (<6T) and longer regimes (>20T) (Fig. 2d). When the target is very short, with a length on the order of the width of dsDNA (2 nm), steric repulsion between the two sides of the loop is expected to cause deviations in ΔL. Consistent with this, we observed this effect for the 4T and 5T targets; residual analysis from the linear fit revealed a sharp deviation at ~2.1 nm (Supplementary Fig. 2a), close to the dynamic width of dsDNA from literature27. For long targets, deviation from linearity results from the sharing of tension between the loop and the target (30T in Fig. 2d and Supplementary Note 1). We found the linear dynamic range of this particular caliper, which consists of a loop of 78 thymine bases, to be from 6 to 20 DNA bases; longer analytes can be measured, but may require non-linear effects to be taken into account. Linearity within this range is remarkable, with our data demonstrating R2 = 0.9999 and residuals on the order of tens of picometers (Figs. 2d & 2e). The intercept and slope of linear fitting respectively represent the effective loop size, L0 = 40.56±0.03 nm, and length of a single base at the given force, Lb = 0.451±0.002 nm, comparable to the literature value (0.44±0.02 nm) determined by mechanical unfolding of a DNA hairpin28. Force dependence was as expected and consistent with the worm-like chain (WLC) model29 (Supplementary Fig. 2b). Measurement repeatability is very high, with variations in mean ΔL between different single molecules indicating angstrom-level spatial precision, and single-base resolution (Supplementary Fig. 3). This precision is achieved through super-resolved force spectroscopy19, which uses repeated mechanical interrogation of a single interaction, followed by per molecule averaging or localization. Determination of L0 enables changes-in-length (Fig, 2d) to be converted to absolute distance measurements (Fig. 2e). These results demonstrate that DNA Nanoswitch Calipers can measure absolute distances within single-molecules with angstrom-level spatial precision.
Fig. 2. Characterization of DNA Nanoswitch Calipers (DNCs) with ssDNA targets.
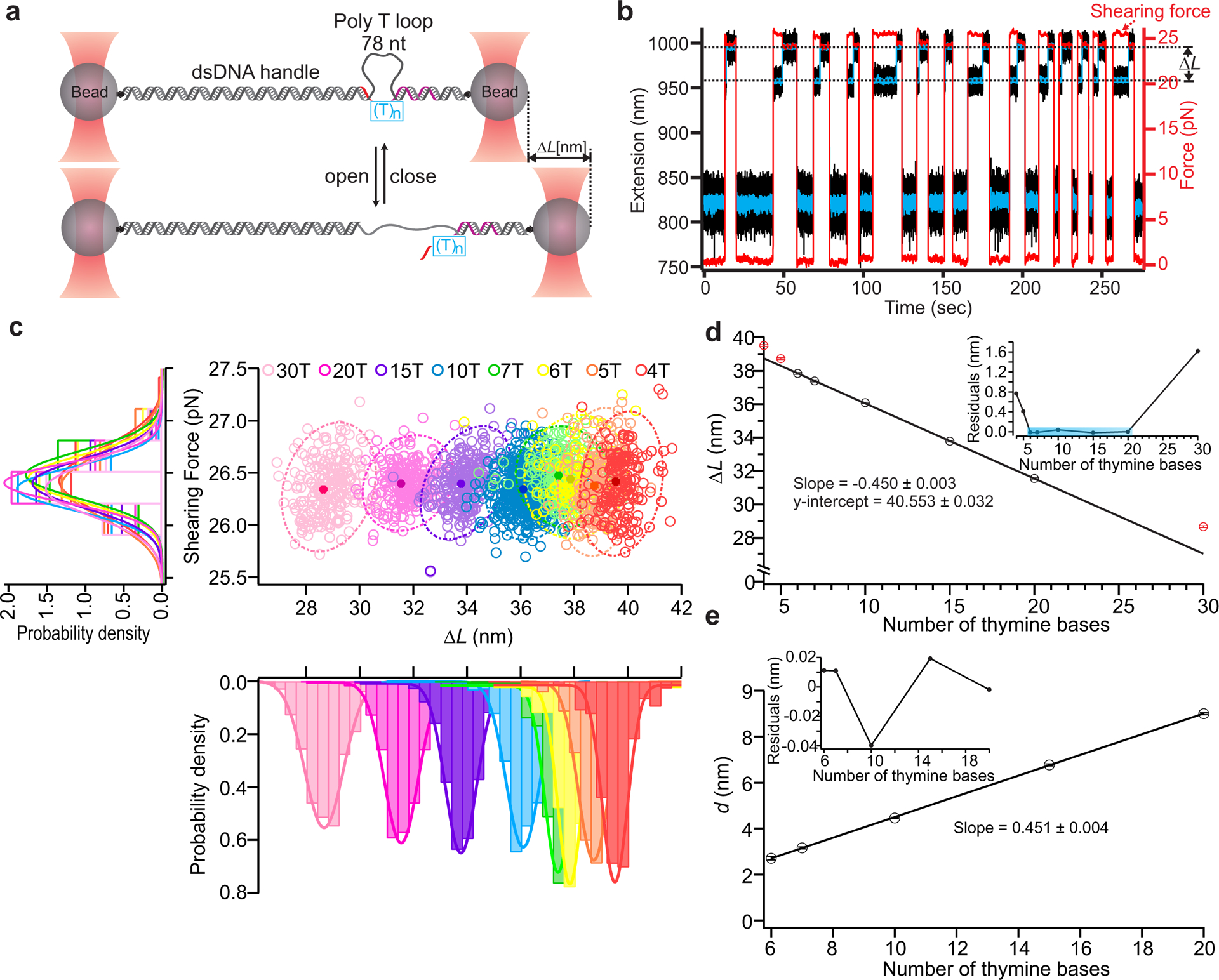
a, Calipers are stretched between two optically trapped beads and actuated by force to measure the lengths of ssDNA targets (see method). The long, mechanically strong handle (purple) and short, mechanically weak handle (red) on the opposite ends of (T)n bridges (blue box) hybridize with the caliper to form the loop (78 thymine bases). During each measurement cycle, the force switches between a low force (~0 pN) that enables rebinding of the short handle and a higher force (~26 pN) that initiates unbinding of this handle. b, Typical trajectories of force (red trace) and extension (black trace with blue trace representing data smoothed by sliding window averaging of 10) during multiple cycles of force actuation. c, Distribution analysis of the change-in-length (ΔL) during loop opening at the specific shearing force (~26 pN). Scatter plots of shearing force versus ΔL for different lengths of ssDNA targets are presented with 95% confidence ellipses superimposed (dotted lines); peak values are presented as solid circles with localization accuracy in shearing force and ΔL represented by the width in each dimension (middle panel). Histograms of the shearing force (left panel), and change-in-length (ΔL) (lower panel) for all the ssDNA targets are also presented with Gaussian fits superimposed. The number of data points in each target ranges between 126 and 622 across multiple molecules (Supplementary Table 3). d, Correlation plot of mean ΔL vs. number of thymine bases using results of Gaussian fits; a linear fit over the range of 6–20 bases is superimposed (R2 = 0.9999). The excluded points are indicated in red. Inset depicts residuals of the linear fit. e, Transformation of mean ΔL measurements within the fitting range to absolute distances measurements (d) by subtraction of the offset. Inset shows residuals in this range, which are all less than 50 picometers. Error bars represent the calculated standard error of the mean.
Calibration of DNC for peptide targets
DNC can be used to map the spatial arrangement of amino-acid residues within proteins. To demonstrate peptide fingerprinting6–8, we first measured distances within two series of synthetic peptides of varying lengths (from 10aa to 30aa for peptide series 1 and 5aa to 24 aa for peptide series 2) as well as a labeled cysteine (Supplementary Table 2). Each peptide was labeled with a long, mechanically strong handle attached to a cysteine residue at the C terminus and a short, mechanically weak handle attached to the N terminus (Fig. 3a&b top panel). Peptide series 1 was modified using procedure A30 and peptide series 2 was modified using procedure B (see Methods, Supplementary Fig. 4). Distance measurements were performed (Fig. 3a–d) as discussed above for DNA targets and as detailed in the Methods section. As before, the change-in-length ΔL was converted to an absolute distance measurement d by subtracting it from the effective loop size L0. The effective loop size for peptide targets differed from the value obtained for ssDNA targets due to differences linker length (Supplementary Fig. 5a)—notably, L0 should be calibrated for each experimental condition and setup to avoid systematic errors (see Methods). The correlation plot of d for all peptides in this collection demonstrates an excellent degree of linearity (R2 = 0.997) over a linear dynamic range of 1 to 20 amino acids (Fig. 3d). Residuals from the linear fit were slightly higher than for DNA, but still less than the length per amino acid at this force, as obtained from the slope Laa = 0.31±0.01 nm/aa (Fig. 3d) (~86% of the total expected contour length of 0.36 nm/aa31–33). The increased spread may be due to interactions between linkers and peptides with DNA strands, or within the peptide backbone and side chains. These peptide-calibration experiments provide the necessary parameters to convert between distances in primary sequence (e.g., the number of amino acids between labeled residues) and physical distances that can be measured with the calipers.
Fig. 3. Calibration of DNA Nanoswitch Calipers (DNC) for peptide targets.
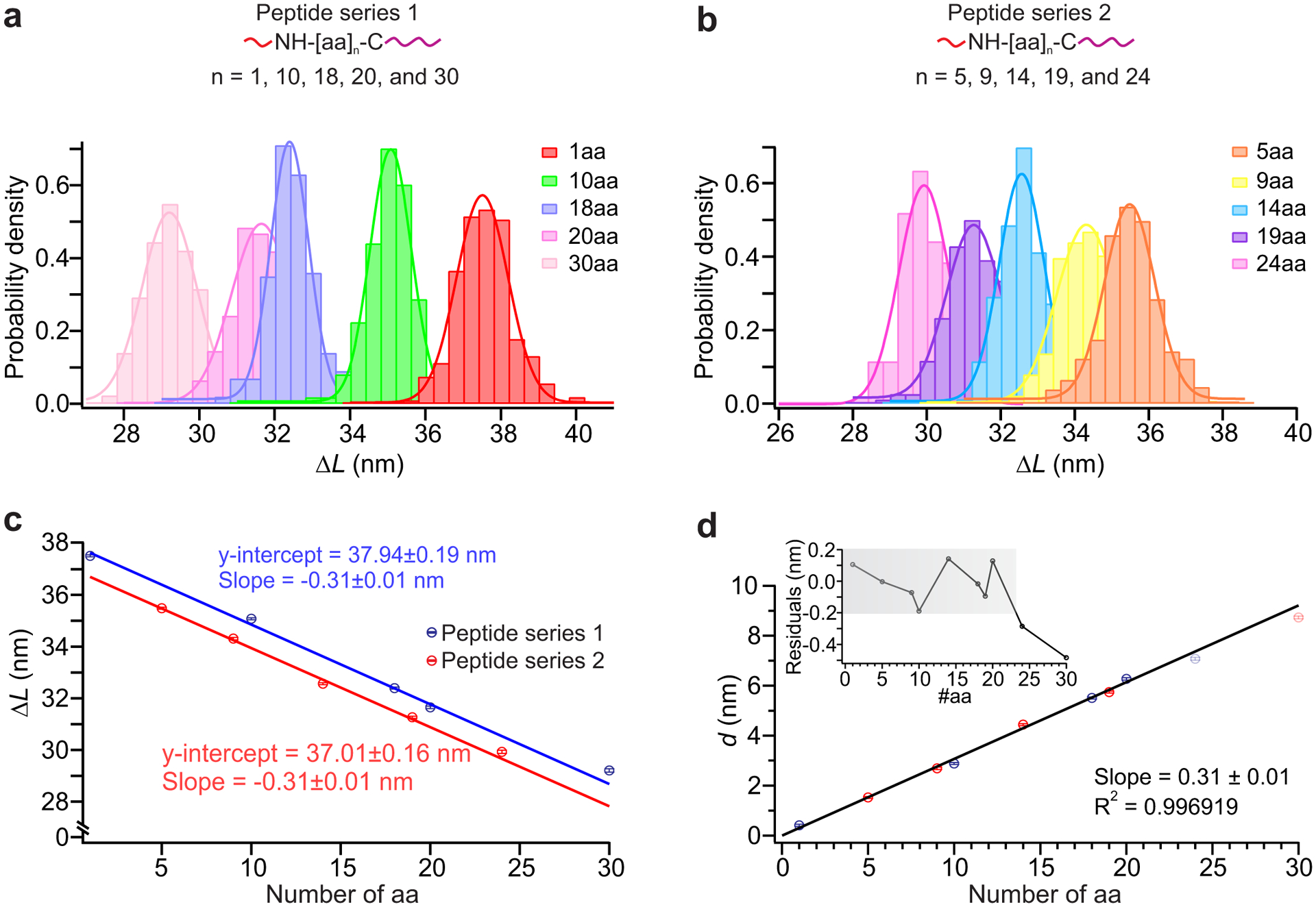
a, and b, Top panels: measurement of distances for peptides of varying lengths for peptide series 1 (a) and peptide series 2 (b) (Supplementary Table 2); each short, mechanically weak handle (red) is attached to the amino group of the N terminus and each long, mechanically strong handle (purple) is attached to the sulfhydryl group of the cysteine residue at the C terminus. The number of data points in each target ranges between 131 and 668 across multiple molecules (Supplementary Table 4). Bottom panels: histograms of the change-in-length (ΔL) measured by DNC for two different sets of peptides measured at different times presented with Gaussian fits superimposed. c, Correlation plot of mean ΔL for (a) (blue) and (b) (red) peptides vs number of amino-acid residues in each peptide, using results of Gaussian fits. Solid lines depict linear fits, with each y-intercept representing the effective loop size L0 for the given experimental conditions. d, Correlation plot of mean absolute distance d (d = L0 − ΔL) vs peptide length; a linear fit over the range 1–20aa is superimposed (R2 = 0.9969). Residuals are shown in the inset with grey shadow highlighting the linear dynamic range (the range over which the measured distance changes linearly with the peptide length) of this caliper, which is approximately 1–20aa. Error bars represent the calculated standard error of the mean.
Single-molecule peptide fingerprinting
To further demonstrate single-molecule peptide fingerprinting, we designed a synthetic peptide (Supplementary Table 2; Fig. 4a, top panel), and labeled it with DNA handles (see Methods Procedure A, Supplementary Fig. 4) to enable distance measurements between the cysteine and amine-bearing moieties (i.e. two lysines and the N terminus). Through cycles of shearing and rebinding actuated by optical tweezers, we observed stochastic rebinding of short handles at all positions (Supplementary Fig. 6). To estimate positional accuracy, we calculated the expected distances from the primary sequence (Fig. 4a top panel), using the length per amino acid Laa determined above (Fig. 3d); for lysine handles, the length of the side chain was also counted. As before, the absolute distance d between each long and short handle was determined by subtracting each ΔL from L0. The aggregated measurements (Fig. 4a) show that all distances agree closely with their expected values, deviating by less than 2 angstroms, which is smaller than the length of a single amino acid. As an additional demonstration, we measured cysteine-to-free-amine fingerprints for a peptide representing the NOXA BH3 domain of Noxa, a protein involved in p53-dependent apoptosis34, with this domain expected to form an alpha-helical secondary structure35, 36. As before, the caliper results were consistent with predictions based solely on the primary sequence with no secondary structure, with all deviations less than 2 angstroms (Fig. 4b)—consistent with denaturation and linearization by the force applied by the caliper. These results demonstrate that multiple distances in individual peptide molecules can be precisely and accurately measured using DNA Nanoswitch Calipers. We note that while the current force (~26 pN) and conditions appear sufficient to denature all peptides in this study, it is possible that other structures could require higher forces for complete denaturation—if this is the case, higher forces could be applied, and/or chemical denaturants could be introduced. Chemical denaturants could potentially also aid in the peptide labeling process, enabling residues buried in the folded structure to be labeled—this approach may be particularly useful when fingerprinting full proteins.
Fig. 4. Single-molecule peptide fingerprinting.
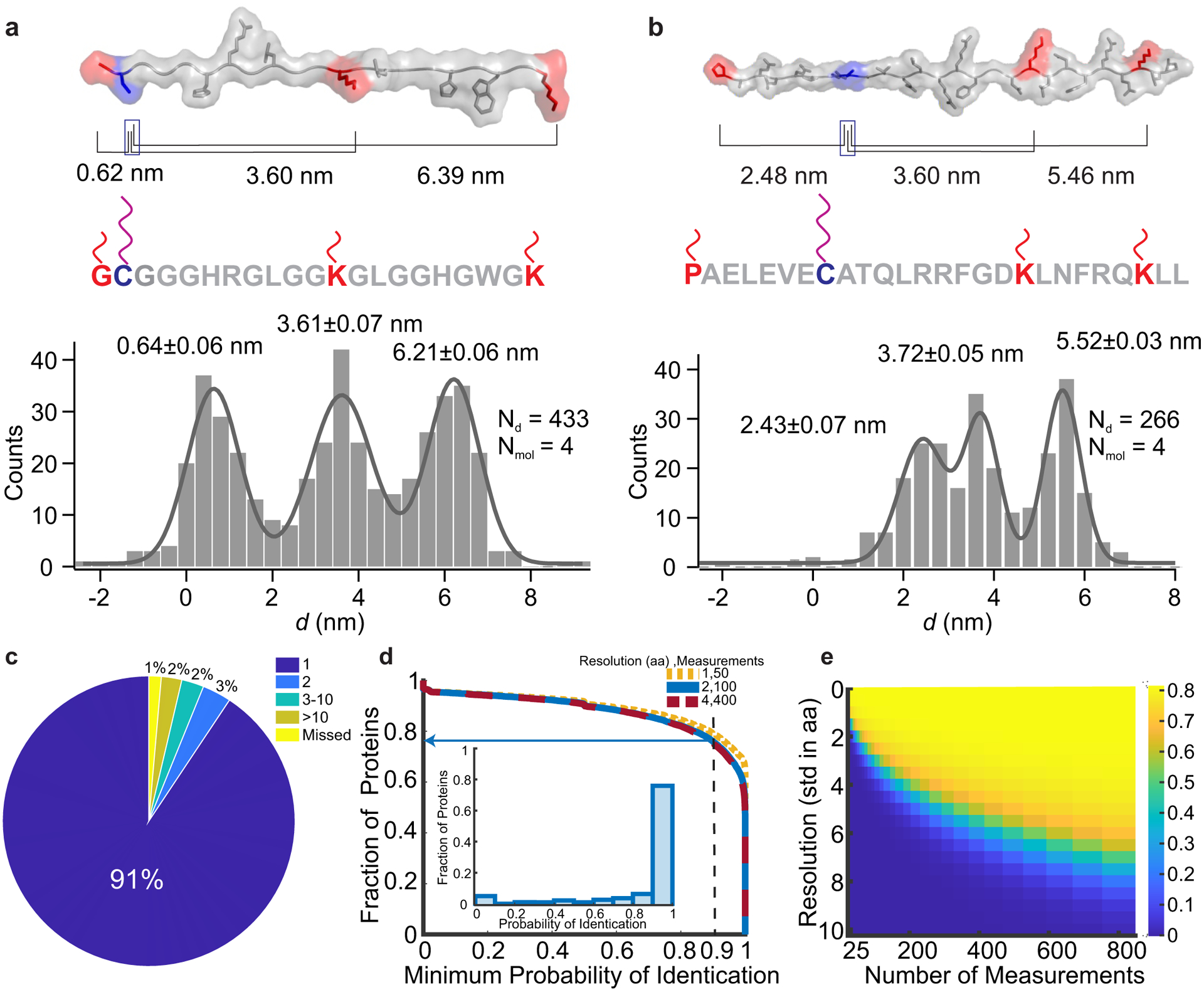
a and b, Top panels: structures of a custom-designed peptide and a NOXA BH3 peptide, respectively, that were labeled with DNA handles for caliper measurements. Amino groups (red) on the N-terminus and lysine residues were labeled with short, mechanically weak handles, while the cysteine residue (purple) was labeled with a long, mechanically strong handle. Peptide structures are rendered as linear for visualization and correspond to their putative force-denatured states during mechanical fingerprinting. Black lines indicate the expected absolute distance between the respective positions of short handles from the long handle. Bottom panels: aggregate histograms of the absolute distances (d = L0 − ΔL) with multi-peak Gaussian fits (grey curve). Representative ΔL measurements obtained on a single molecule can be seen in Supplementary Figure 6. c, Uniqueness analysis showing the number of human-protein hits in the Swiss-Prot database for each mechanical fingerprint for a caliper span of 120 aa. d, Fraction of proteins that can be identified as a function of minimum required probability of identification with arrow showing fraction of the database that can be identified with 90% certainty). Inset: Histogram of probability of identification for proteins under representative experimental conditions (caliper span: 120 aa, measurement error: 2 aa, 100 measurements per molecule). e, Heat map of fraction of proteins that can be identified with 90% certainty with a caliper span of 120 aa for a range of measurement errors and measurements per molecule.
Performing multiple distance measurements on a single molecule enables the creation of a mechanical fingerprint that can be used for protein identification; e.g. measuring distances between lysines and cysteines as demonstrated above is known as CK-dist fingerprinting7. We performed a computational analysis to estimate the effectiveness of our approach (see Methods), noting that alternative fingerprinting schemes could also be implemented. First, to determine the uniqueness of each mechanical fingerprint (the distances between a specific cysteine and all lysines within range), we used the Swiss-Prot database to compare each possible fingerprint against all included entries in the human proteome5,6,8, 37. Second, to determine how identification depends on experimental parameters, we simulated our measurements and performed a Bayesian analysis to determine the probability of identification for each protein in the database. Feasibility is supported by our initial analysis of fingerprint uniqueness; in the absence of measurement error, 91% of mechanical fingerprints resolve to a single protein in the database (Fig. 4c) Under more realistic experimental limitations—such as a measurement resolution of 2 aa per cycle, consistent with our optical tweezers experiments, and 100 measurements per molecule—our analysis indicates that 76% of the proteins in the database can be identified with a probability of at least 90% (Fig. 4d)7,9. These numbers can be improved by increasing the caliper span, the maximum distance between DNA handles that can be measured by the caliper (Supplementary Fig. 7). Increasing the resolution reduces the number of measurements required to achieve similar results, whereas decreasing the resolution requires more measurements per molecule (Figs. 4d&e). These results suggest that higher-throughput can compensate for lower-resolution measurements. With our current optical tweezers assay, approximately 5 minutes are needed to collect 100 measurements per molecule. As described in the next section, a DNC assay based on multiplexed force spectroscopy could dramatically increase molecular throughput through parallelization.
To demonstrate single-molecule identification of post-translational modifications as in preceding work10, 38, we measured mechanical fingerprints of phosphorylated serines on a short-peptide mimic of the C-terminal repeating domain (CTD) of RNA polymerase II. The CTD is a repeated heptapeptide39, Tyr1-Ser2-Pro3-Thr4-Ser5-Pro6-Ser7, known to be reversibly phosphorylated on serines during each transcription cycle, which serves as a scaffold for different nuclear factors, depending on phosphorylation pattern, to guide polymerase activity40, 41. Methods that could discriminate the phosphorylation pattern in a single heptapeptide unit, a challenge for conventional approaches42, could be helpful for dissecting the role of CTD phosphorylation in RNA transcription. Toward this end, we selectively modified the phosphoserine residues of the heptapeptides using a protocol modified from previous literature10, 38, 43, conjugating them to strong DNA handles, while the N terminus was attached to a weak DNA handle as before (See Methods procedure C, Supplementary Fig. 4). We first measured the change-in-length (ΔL) for three singly phosphorylated heptapeptides (2-pS, 5-pS and 7-pS) (Supplementary Figs. 5b&c), to determine the effective loop size L0, which differed from previous peptides due to differences in linker length (Supplementary Figs. 4 & 5a). ΔL measurements were transformed into absolute distances measurements as before, then expressed as the number of amino-acid residues between handles by dividing by the slope of the calibration curve (Supplementary Fig. 5c) to give a dimensionless distance . Next, using per-molecule localization, was determined for each molecule, demonstrating clear discrimination between the three populations (Fig. 5a). We also measured distances using a mixture of two distinct phosphopeptides (5-pS and 7-pS) to demonstrate quantification of relative abundance from a heterogeneous sample, determining the distance between handles on a per-molecule basis. Analyzing 10 molecules in a mixture containing a 1:1 molar ratio of 5- and 7-phosphoheptapeptides, we recovered a ratio of 1:1, as can be seen from the non-overlapping distribution of distances at the expected lengths (Fig. 5b).
Fig. 5. Single-molecule mechanical fingerprinting of post-translational modifications in a heterogeneous mixture of peptides.
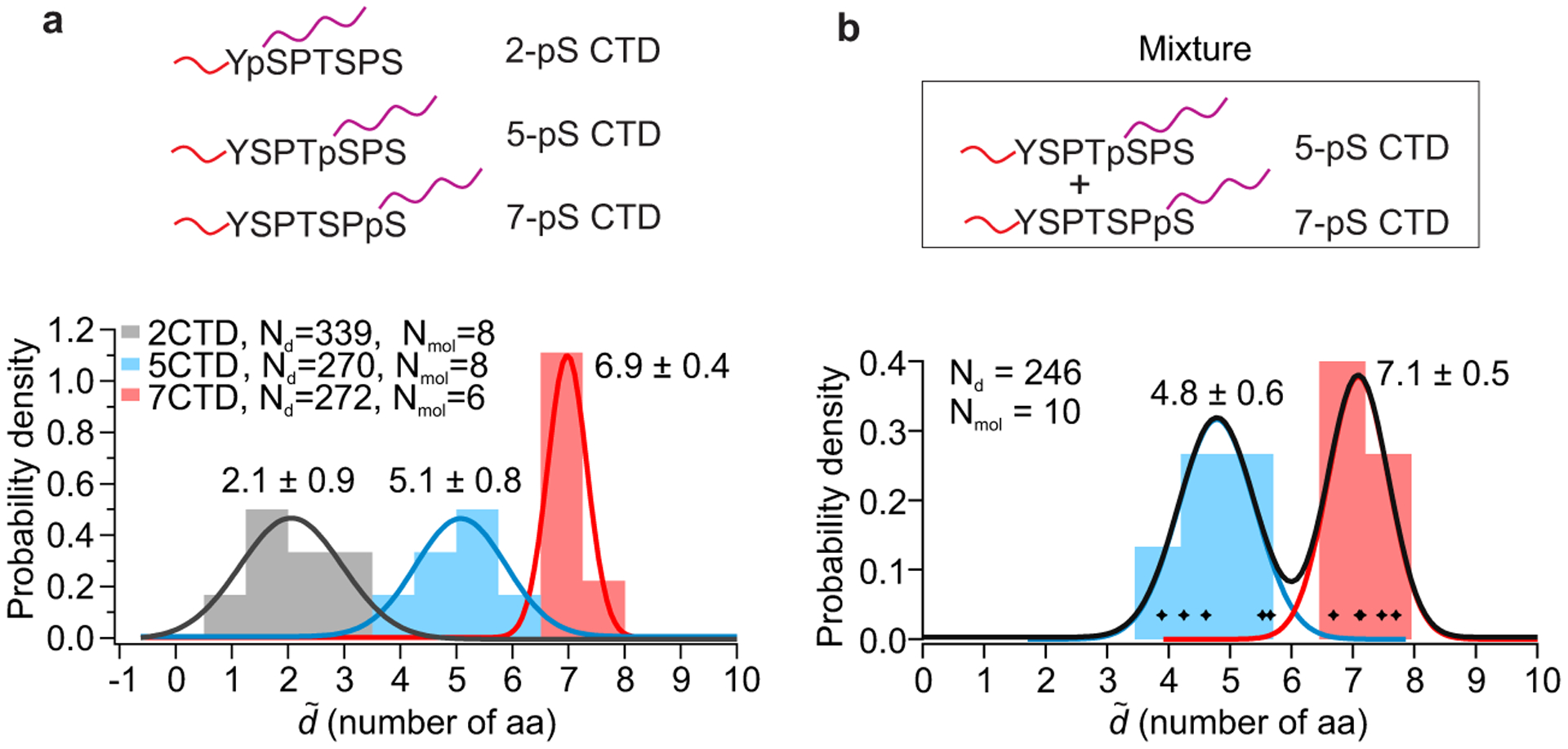
a, Characterization of post-translational modifications in C-terminal domains (CTD) of RNA Polymerase II. Long DNA handles were attached to phosphorylated serines at positions 2, 5 or 7 to measure distances from the N terminus, where a short DNA handle is attached (Top panel). Stacked histogram of the dimensionless absolute distance in units of the number of amino-acid residues between handles (, number of aa, localized per molecule) of the three different peptides is shown below, with Gaussian fits superimposed and labeled by peak value ± standard deviation. ΔL measurements presented without per molecule averaging and used for calibration can be seen in Supplementary Figures 5b&c. b, Demonstration of peptide identification in a heterogeneous sample. Histogram depicts the dimensionless absolute distance (, number of aa, localized per molecule) obtained by analyzing 10 molecules in a 1:1 mixture of 5- and 7-phosphoserine CTD peptides; molecules were classified based on the measured distance, and color-coded accordingly. Gaussian fits labeled by peak value ± standard deviation are superimposed.
Multiplexed peptide fingerprinting with parallel force spectroscopy
Methods with sufficient throughput are needed to characterize heterogeneous samples with single-molecule precision. Parallelization can be used to increase the throughput of caliper-based fingerprinting, as we demonstrate by implementing our DNC assay on a multiplexed magnetic-tweezers system23, 44–48 (see Methods). Each caliper is linked between a coverslip and a magnetic bead, which is tracked using video microscopy, enabling multiple targets to be probed in parallel (Fig. 6a). DNC constructs were anchored with a ssDNA overhang to enable strong attachment to coverslips covalently functionalized with complementary ssDNA oligos. Similarly to the optical tweezers experiments, DNC constructs were subjected to repeated cycles of force application, alternating between a low force that enabled loop closure and rebinding, and a high force that enabled loop opening, with the change in tether length at the high force used to determine target distance; this enabled multiple distance measurements to be made for each molecule (Fig. 6a, Supplementary Fig. 8).
Fig. 6. Multiplexed single-molecule mechanical fingerprinting of synthetic peptides in different heterogeneous mixtures.
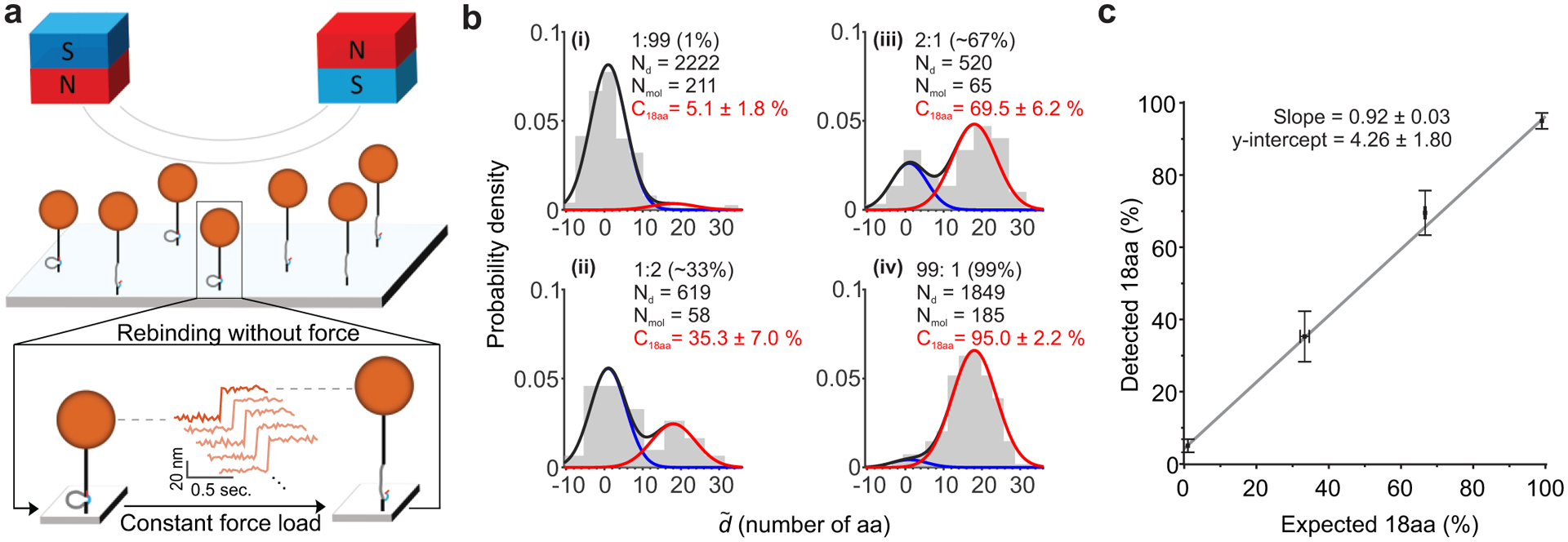
a, Schematic representation of DNA Nanoswitch Calipers distance measurements performed in parallel using magnetic tweezers. The magnetic field gradient generated by the pair of magnets above the sample cell pulls the tethered beads upward with force controlled by the magnets’ position. Cartoon below illustrates the protocol for repeated cycles of force application: each cycle consists of a constant pulling force to induce rupture of the weak handles, followed by a near zero-force reassociation period to facilitate rehybridization of the weak handles. b, Histograms of the dimensionless distance distributions , in units of the number of amino acids between handles, obtained from samples containing mixtures of peptides (18aa and 1aa) at different ratios: i) 1:99, ii) 66:33, iii) 33:66, and iv) 99:1. The solid lines plotted over the histogram show the results of maximum likelihood fitting of a mixture of two Gaussians, which was used to obtain the detected percentage of 18aa peptide in each mixture. c, Correlation between the expected and detected percentage of 18aa peptides in the heterogeneous mixtures (R2 = 0.997). Error bars along the y-axis represent estimated 68% confidence intervals, while x-error bars represent standard deviations from gel-based quantification measurements.
To characterize this assay, we performed distance measurements on CTD peptide mimics with different phosphorylated serines, as in the previous section (Supplementary Figs. 9a&b). Our magnetic tweezers results are in good agreement with optical-tweezers measurements performed at the same force levels (Supplementary Fig. 9c). As before, repeatedly interrogating each target and then averaging the results on a per-molecule basis (i.e., super-resolved force spectroscopy), improves the resolution of our calipers measurements significantly. Using this approach, we achieved a distance resolution of approximately ~1 nm with our magnetic-tweezers assay (Supplementary Fig. 9b). While lower than achieved with optical tweezers, our bioinformatics analysis shows that for protein identification a decrease in resolution can be compensated for by increasing the number of measurements per molecule (Fig. 4e), supporting this tradeoff between throughput and resolution.
As an additional demonstration of our multiplexed calipers assay, we measured the relative concentrations of two different peptides within a set of heterogeneous mixtures (Fig. 6). The DNA-labeled peptides (calibration peptides n=1 and n=18 from Fig. 3a) were mixed at four different ratios, then mechanically fingerprinted in parallel using magnetic tweezers. Distances between handles were made dimensionless by dividing by the slope of the calibration curve (Supplementary Fig. 9d). Maximum-likelihood analysis was used to determine the relative concentrations of peptides within each mixture (Fig. 6b). The resulting data shows an excellent correlation between the expected and measured ratios (Fig. 6c)9, with a linear fit (R2 = 0.997) indicating no appreciable bias within error (e.g. based on the fit, an expected ratio of 50%, would yield a detected ratio of 50 +/− 2 %).
Conclusions
We have introduced DNA Nanoswitch Calipers, a tool for measuring multiple pairwise distances within single molecules that combines force spectroscopy with nanoscale DNA devices. We have demonstrated the measurement of distances within peptides and DNA constructs with angstrom-level precision, and have applied this capability to fingerprinting peptides—showing the ability to distinguish between post-translational modifications at the single-molecule level, and characterizing the feasibility of using this strategy for protein identification. We have also demonstrated how throughput can be increased through parallelization, by implementing this assay on a multiplexed magnetic-tweezers system. This enabled us to measure the relative concentrations of different ratios of peptides within a heterogeneous mixture.
While current DNC capabilities are sufficient for a variety of applications, future improvements will expand the range of systems that can be probed. For example, to increase the maximum distance that can be measured, we have tested a caliper with a longer loop length (140 thymine bases), which corrects the previously observed deviation in ΔL for ssDNA targets longer than 30 bp (Supplementary Fig. 10). To measure long distances on full-length proteins, long loop lengths could be combined with high forces to resolve issues that could arise from internal interactions, including sequence- or distance-dependent effects. DNC could also be integrated into other force-spectroscopy approaches, to incorporate additional features or improve dynamic range, spatial precision, or throughput. For example, the throughput of our parallel magnetic-tweezers assay could be improved using targeted DNA tethering45, and the effective resolution could be improved by better accounting for force heterogeneity and off-axis movements49.
We expect that our approach will yield two significant applications in proteomics. Firstly, protein identification could be achieved by comparing DNC fingerprints against a protein database as explored above, complementing other emerging approaches for single-molecule fingerprinting and identification2, 3, 5. Furthermore, some of the concepts used for DNC could benefit these other approaches. For example, the repeated interrogation of single-molecules that we demonstrate could be applied to other methods, such as nanopore-based characterization, to increase discrimination between different targets50. Additionally, our use of force to help denature peptides or proteins, and to decrease thermal noise, could potentially be transferable to methods such as nanopore characterization50, 51. Secondly, DNA nanoswitch calipers could potentially be used to measure distances between multiple residues of a protein in its folded structure, which would enable the characterization of the dynamic 3D structures of biomolecules and biomolecular complexes, complementing existing biophysical methods for structural elucidation. DNC provides a powerful approach for characterizing distances and geometries within nanoscale complexes, with the potential to impact a wide range of fields, from proteomics and nanotechnology, to structural biology and drug discovery.
Methods
Materials
Dibenzocyclooctyne-N-hydroxysuccinimidyl ester (DBCO-NHS), N-Hydroxysuccinimide (NHS), barium hydroxide, and ethanedithiol were purchased from Sigma Aldrich. 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), (succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate) (SMCC), NHS-PEG4-Azide, and Phosphate buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 10 mM phosphate buffer, pH 7.4) were purchased from Thermo Fisher Scientific. The carboxylated silica beads (3 μm) were purchased from Microspheres-Nanospheres. Biotin-modified reverse primer, BsaI-restriction site included forward primer and other DNA oligonucleotides (see sequences in Supplementary Table 1) were purchased from Integrated DNA Technologies (IDT). The pET-26b (+) plasmid was received from EMD Millipore Sigma. All synthetic peptides were purchased from GenScript. Noxa BH3 peptide was purchased from Anaspec. (See sequences in Supplementary Table 2).
Procedure for modification of amine-functionalized oligonucleotides
A 10 μL aliquot of a 1 mM solution of the amine-functionalized oligonucleotide in water (final concentration of 100 μM) was added to 40 μL of 100 mM phosphate buffer at pH 8.0. To the resulting solution was added 50 μL of a freshly prepared 10 mM solution of the N-hydroxysuccinimidyl ester reagent in DMSO (final concentration of 5 mM) to provide a 1:1 solution of phosphate buffer:DMSO. The reaction was briefly agitated and incubated at room temperature. After 3 h, the reaction was purified by gel filtration using a Nap 5 column (GE Healthcare) according to the manufacturer’s instructions. Water was used to elute the labeled-oligonucleotide and the purified material was concentrated by lyophilization. The dried material was taken up with PBS buffer at pH 7.2 to obtain a final concentration of 100 μM of maleimide-functionalized oligonucleotide or DBCO-functionalized oligonucleotide.
Procedure A for conjugation of ssDNA handles to peptides from series 1
This procedure was used for the labeling of peptides from series 1 (10, 18, 20, and 30 aa) in order to prevent off-target labeling of the peptides with the N-hydroxysuccinimidyl ester reagent, NHS-PEG4-azide. A 2 μL aliquot of a 1 mM solution of the peptide in water (final concentration of 20 μM) was diluted with 97.8 μL of 100 mM phosphate buffer at pH 8.0 with 5 mM EDTA. To the resulting solution was added 0.2 μL of a freshly prepared 100 mM solution of NHS-PEG4-Azide in DMSO (final concentration of 200 μM). The reaction was briefly agitated and incubated at room temperature. After 2 h, the reaction was heated to 90 °C for 1 h. The reaction was cooled to room temperature30. A 45 μL aliquot of the peptide with azide modification (20 μM stock solution in phosphate buffer, final concentration of 12 μM) was diluted with 27 μL of PBS buffer at pH 7.2. To this solution was added 3 μL of the 100 μM solution of the maleimide-functionalized oligonucleotide in PBS buffer at pH 7.2 (final concentration 4 μM). After 2 h, the reaction was purified by using 0.5 mL 7 kDa MWCO Zeba spin desalting columns (Thermo Fisher Scientific) according to the manufacturer’s instructions. After two rounds of purification, 70 μL of the purified cysteine-labeled peptide conjugate with azide modification (final concentration of ~2.33 μM) was diluted with 13.8 μL PBS buffer at pH 7.2. To this solution was added 6.2 μL of the 100 μM solution of the DBCO-functionalized oligonucleotide (final concentration of ~6.9 μM) was added. The reaction was briefly agitated and incubated at room temperature. After 16 h, the reaction was purified by gel electrophoresis using a 12% urea polyacrylamide gel, which was run at 150 V for 1.5–2 h using 0.5X TBE as a running buffer. The fully DNA-labeled peptide conjugate was extracted from the gel using the “crush and soak” method by incubating the crushed gel in 0.3 M sodium acetate at pH 5.2 for 16 h on a mixer at 1000 rpm. The recovered DNA-labeled peptide conjugate was concentrated by ethanol precipitation and taken up with water to obtain a final concentration of 0.5–1 μM.
Procedure B for conjugation of ssDNA handles to peptides from series 2, custom-designed peptide, and Noxa BH3 peptide
A 4 uL aliquot of a 1 mM solution of the peptide in water (final concentration of 50 μM) was diluted with 68 μL of 100 mM PBS buffer at pH 7.2. To this solution was added 8 μl of the 100 μM solution of the maleimide-functionalized oligonucleotide in PBS buffer at pH 7.2 (final concentration of 10 μM). The reaction was briefly agitated and incubated at room temperature. After 2 h, the reaction was purified by using 0.5 mL 7 kDa MWCO Zeba spin desalting columns (Thermo Fisher Scientific) according to the manufacturer’s instructions and the buffer was exchanged to 100 mM phosphate buffer at pH 8.0. A 32 μL aliquot of the purified cysteine-labeled peptide conjugate (final concentration of ~4 μM) was diluted with 47.2 μL of 100 mM phosphate buffer at pH 8.0. To this solution was added 0.8 μL of a 100 mM solution of NHS-PEG4-Azide in DMSO (final concentration of 1 mM). The reaction was briefly agitated and incubated at room temperature. After 1–2 h, the reaction was purified by using 0.5 mL 7 kDa MWCO Zeba spin desalting columns (Thermo Fisher Scientific) according to the manufacturer’s instructions and the buffer was exchanged to PBS buffer at pH 7.2. After two rounds of purification, 70 μL of the purified cysteine-labeled peptide conjugate with azide modification (final concentration of ~2.33 μM) was diluted with 13.8 μL PBS buffer at pH 7.2. To this solution was added 6.2 μL of the 100 μM solution of the DBCO-functionalized oligonucleotide (final concentration of ~6.9 μM) was added. The reaction was briefly agitated and incubated at room temperature. After 16 h, the reaction was purified by gel electrophoresis using a 12% urea polyacrylamide gel, which was run at 150 V for 1.5–2 h using 0.5X TBE as a running buffer. The fully DNA-labeled peptide conjugate was extracted from the gel using the “crush and soak” method by incubating the crushed gel in 0.3 M sodium acetate at pH 5.2 for 16 h on a mixer at 1000 rpm. The recovered DNA-labeled peptide conjugate was concentrated by ethanol precipitation and taken up with water to achieve a final concentration of 0.5–1 μM.
Procedure C for the conjugation of ssDNA handles to phosphoserine peptides
The phosphoserine CTD peptide (1 mM stock solution in water, final concentration of 100 μM) was diluted in a solution of 4:3:1 water:acetonitrile:isopropanol. To the resulting solution was added a freshly prepared solution of Ba(OH)2 (200 mM stock solution in water, final concentration 40 mM)10, 38, 43. The reaction was briefly agitated to ensure proper mixing and incubated at room temperature or 37 °C on a mixer at 1000 rpm. After 2 h, 0.2 μL of ethanedithiol was added to 200 μL of the reaction solution. The reaction was incubated at 37 °C for 1 h. The reaction was quenched by the addition of 200 μL 1 M acetic acid. Then the reaction was concentrated using a rotary evaporator to remove excess ethanedithiol. The resulting material was taken up in 200 μL of water and concentrated. This step was performed two times. The dried material was taken up with 200 μL of water to achieve a concentration of ~100 μM of the thiol-modified CTD peptide. The single-stranded DNA handles were attached to the thiol-modified CTD peptide using procedure B for conjugation of ssDNA handles to peptides described above.
Synthesis of DNA Nanoswitch Caliper (DNC) construct
The DNA nanoswitch calipers were created by the ligation of two modified DNA constructs. Briefly, one DNA construct was prepared by the PCR amplification of a 2820 bp region of the pET-26b (+) plasmid. The forward primer was designed with a BsaI restriction site (green font), 5′- AAA AAA GGTCTC GGC ATG ATA GCG CCC GGA AGA GAG, and the reverse primer was labeled with two biotins at the 5′ end (5′-Biotin -GGG TTC GTG CAC ACA GCC CAG CTT). After PCR and purification (Blue pippin, Sage Science), the construct was digested with BsaI (New England Biolabs) to produce a sticky end (Supplementary Fig. 1a). The other DNA construct was prepared by annealing the loop oligonucleotide, 5′-digoxigenin modified oligonucleotide (DNC1), and other strands (DNC2 and DNC3) (Supplementary Fig. 1a) at equimolar concentration with the splint oligonucleotide at a 10-fold molar excess. For DNC constructs used in magnetic-tweezers experiments, oligonucleotides DNC4 and DNC5 were added instead of DNC1 and DNC2, respectively. The mixture was heated at 95 °C for 5 min and slowly annealed to the room temperature. Then, the annealed mixture was ligated to the dsDNA construct prepared by PCR amplification using T4 DNA ligase (New England Biolabs) at 16 °C for 16 h and the ligase was heat deactivated at 65 °C for 20 min. The desired product was confirmed by running agarose gel (0.8%) electrophoresis and purified by using Blue pippin (Sage Science) (Supplementary Fig. 1b).
Single-molecule distance measurement using optical tweezers
The single-molecule mechanical experiments were performed in a home-built dual trap optical tweezers derived from the previous micropipette optical tweezers18, 52. Briefly, a second optical trap was generated by splitting the 1064 nm laser beam into two using a polarizing beam splitter in the style adopted by Moffit et al.53. Where, one beam remained stationary as in the previous setup, and the second beam was driven by a finely controlled steerable mirror (Mad City Labs, Madison WI). The experiments were performed in phosphate buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 10 mM phosphate buffer, pH 7.4) with 0.1 mg/mL Roche Blocking solution (Roche) at room temperature (23 °C). The sample was prepared as described below to use in optical tweezers (Supplementary Fig. 1c). Briefly, 2 μL of the DNC construct (70 pM) was incubated with 0.5 μL of 1% (w/v) solution of streptavidin coated silica beads (3.0 μm diameter) for 30 min to immobilize the DNC construct on the surface of the beads by biotin-streptavidin interaction. Then, the incubated sample was diluted to 5 μL with the experimental buffer and washed twice by spinning down the beads using a benchtop centrifuge. The bead suspension was concentrated to 1 μL and added to the target DNA oligonucleotides or DNA-labeled peptides at a few-fold molar excess in order to bind them on the DNC construct by a strand-displacement reaction of the splint oligonucleotide. The mixture was incubated on a slow rotator at room temperature for at least 30 min and the beads were washed to remove excess analyte, splint oligo and any non-specific bindings on the bead surface. Then the DNC-modified beads were diluted to 5 μL to make the concentration of beads ~0.1% (w/v).
The sample cell was constructed by sandwiching a double-sided Kapton tape (1 mm, DuPont) between a glass slide and a cover glass. Both glass slides and cover glass were washed with 70% ethanol and dried with Argon flow. One side of the Kapton tape was attached to the surface of the glass slide and the tape was cut by a razor blade to obtain the desired numbers of channels with appropriate width. Then the channel was covered by a cover glass. Each channel was passivated with Roche Blocking solution, (10 mg/mL) for ~30 min and then flushed with the experimental buffer before injecting the samples. Then, a 1 μL aliquot of the DNC-modified beads was injected through one channel and 1 μL of the 0.1% (w/v) anti-digoxigenin coated silica beads (3 μm) was injected through the next channel. The openings were sealed with grease. The two different beads were separately trapped in the laser foci and one bead was moved closer to the other by using a steerable mirror to tether the DNC construct between the two optically trapped beads as shown in Fig. 1. The tethered DNC was confirmed to be a single molecule by estimating the contour length of the whole caliper (~1 μm) and by a single breakage of the tether. The DNC tether was extended up to 30 pN and relaxed back to 0 pN at the loading rate of 1000 nm/s. Calipers were confirmed to consist of a target molecule by observing a sudden change-in-extension due to shearing of the weak handle.
After confirmation of a target molecule, the experiment was switched to force-jump mode between ~0 pN and ~26 pN, where the lower force allowed the rebinding of a weak handle and the higher force caused shearing of the handle. By repetitive cycling of the forces, large statistics of transitions were recorded for every individual caliper. The data were recorded at 1400 Hz using a LabVIEW program 2013 (National Instruments Corporation, Austin, TX).
Data analysis
In optical tweezers, the data were acquired from LabVIEW and analyzed by custom-written MATLAB scripts. The statistical analyses and graphing of the measured data points were done using IGOR Pro. The unlooping of the nanoswitch caliper was determined by the sudden change-in-length (ΔL) observed during the waiting time at the shearing force (Fig. 2b). The tension experienced by the tethered molecule dropped suddenly as soon as the loop opened (see red curve in Fig. 2b). The compliance in ΔL due to the drop-in force was corrected by taking the slope tangent to the shearing force in the force vs. extension curve. In order to convert these changes in length (ΔL) to absolute distance measurements (d) we must subtract them from the effective loop length (L0) at the corresponding experimental conditions, i.e., d = L0 − ΔL. As depicted in Figure 2e of the main text, L0 can be determined by measuring ΔL for analytes of different lengths and performing a linear regression to find the change-in-length that would result for a zero-length analyte, i.e., L0 is the offset of the linear fit. We note that since the L0 can depend on many parameters, including linker length and the actuation/shearing force, we determined its value empirically for each experimental condition. To get the most accurate results, we also recommend re-measuring L0 if modifications have been made to the instrumentation, or if extended periods of time have elapsed between measurements (e.g., many months), to reduce the chance that drift or systematic deviations could affect the accuracy of the results.
Magnetic-tweezers instrumentation
The multiplexed single-molecule experiments were performed using a custom magnetic-tweezers system adapted from previous work, including Lipfert et al., 200954, Dulin et al., 201555, and De Vlaminck et al., 201256. Briefly, a pair of aligned 5 mm nickel-plated cube neodymium magnets separated by a 1 mm gap housed in a steel holder was positioned above the flow cell with its movement and distance from the sample chamber controlled by a motorized translation stage (MTS50-Z8, Thorlabs) (Supplementary Fig. 8). We used a collimated 660 nm fiber-coupled LED (M660F1, Thorlabs) placed above the magnets to illuminate the sample, passing the light through the 1 mm gap between the magnets. The sample image was magnified using a 100X objective (Nikon Plan fluor 100X/1.30 oil DIC H) and imaged onto a 5.5-megapixel CMOS camera (Andor Zyla 5.5 CL10, Oxford Instrument) using a 200 mm lens (AC254–200-A, Thorlabs).
The vertical Z position of the sample chamber was controlled by a multi-axis piezo flexure nanopositioning stage (P-517.CD control with E-712 controller, Physik Instrumente). The piezo nanopositioning stage was mounted on a linear X-Y positioning stage (M-686.164 each axis control with a C-866 PILine controller, Physik Instrumente) to move the sample chamber, enabling us to scan different fields-of-view.
We developed custom software in LabVIEW (National Instrument) to control the magnet movement, sample position, and camera acquisition for data collection. Per field-of-view of 2500 × 2100 pixels, we recorded at 50 Hz and stored the images to physical memory, with bead tracking performed after acquisition (Supplementary Fig. 8). We constructed a calibrated diffraction pattern look-up table for each bead to determine the position in the direction of the pulling force. To construct the look-up table, we first applied over 20 pN of force for 1 minute to rupture all the weak handles, followed by scanning the sample in Z moving at 10 nm steps across 4 μm while radial diffraction patterns were recorded. To track the bead position in Z using the look-up table, we determined the minimum of a second-degree polynomial fit of the chi-square values of the measured radial diffraction pattern to the stored look-up table patterns. We used the ratio of the refractive-index of water and oil of 0.88 (nwater/noil = 1.33/1.51 = 0.88)57 as the correction factor for the focal shift in the extension calibration.
Magnetic-tweezers sample cell preparation
We functionalize the surface of coverslips with DNA oligonucleotides complementary to the DNA nanoswitch caliper at one of the tethering ends. First, the coverslips (12-518-105C, Gold Seal #1, 24 mm × 30 mm, Thermo Fisher) were cleaned by 20 minutes of sonication in isopropanol then by another 20 minutes of sonication in Milli-Q water. After the coverslips were dried, the coverslips were plasma cleaned using a low-pressure plasma system (ATTO Plasma Cleaner, Diener electronic GmbH). The coverslips were then submerged in anhydrous heptane containing 0.25% (v/v) of APTES ((3-Aminopropyl) triethoxysilane, 440140, Millipore Sigma). After adding amino groups to the surface, the coverslips were cleaned in acetone, followed by Milli-Q water before drying. The sample cell was constructed using double-sided Kapton tape sandwiched between the coverslip and microslide (2947–75X25, 25 mm × 72 mm, Corning). Two 1 mm diameter ports were drilled into the microslide that served as the solution inlet and outlet before the channel assembly.
With the sample cell assembled, the surface of the coverslip was functionalized with PEG and DNA oligonucleotide. Briefly, we loaded a functionalization solution consisting of 0.1 M potassium tetraborate solution (pH 8.4) containing 10% w/w monoPEG-SVA (mPEG-Succinimidyl Valerate, MW 5000, Laysan Bio Inc.), 2 μM DBCO-PEG4-NHS (Dibenzocyclooctyne-PEG4-N-hydroxysuccinimidyl ester, 764019, Millipore Sigma), and 50 nM 5’-Azide DNA oligonucleotide (manufactured by IDT). After one hour of incubation, we loaded another round of freshly prepared functionalization solution. One hour after completing the second round of functionalization, the channel was washed with DNase/RNase-Free Distilled Water (10977015, Thermo Fisher Scientific) before drying in a vacuum chamber where we stored the functionalized sample cells.
Magnetic tweezers DNA Nanoswitch Caliper tethering
The final stage in sample preparation for the magnetic tweezers assay was carried out by first incubating the channel with 1X TBS Tween-20 (28360, Thermo Fisher Scientific) for 20 minutes to rehydrate PEG and DNA oligonucleotides on the surface. The channel was then further incubated with a surface passivation solution consisted of 10 mg/mL Blocking Reagent (11096176001, Roche) dissolved in PBS for 40 minutes. After the passivation step, the channel was washed with a TBS buffer containing 10 mM MgCl2 (pH 8.0), then 100 pM of DNA nanoswitch caliper construct was added and incubated for 30 minutes, during which caliper constructs became tethered to the surface through DNA hybridization. After caliper tethering, the flow channel was washed with 20 μL of the TBS buffer containing 10 mM MgCl2 before loading the channel with 1 nM of the target analyte. After 1 hour of target analyte incubation, where the tethered caliper captured the target analyte, the channel was washed with the experiment buffer of 1X PBS containing 0.1 mg/mL Blocking Reagent (11096176001, Roche) and 0.05% (v/v) Tween-20. For each experiment, M-270 Streptavidin Dynabeads (2.8 μm in diameter, Thermo Fisher Scientific) were first washed with passivation solution, followed by extensive washing with the experimental buffer before loading them into the sample cell. The loading of the beads into the sample channel and removal of the excess non-tethered beads was assisted with a syringe pump (PHD ULTRA Syringe Pumps, Harvard Apparatus), where the flow rate was set to 20 μl/min. The inlet and outlet of the channel were sealed with silicone base vacuum grease (1597418, Dow Corning) before mounting the channel onto the magnetic tweezers system.
Magnetic tweezers force protocol and data analysis
To actuate the tethered DNA Nanoswitch Calipers in parallel using magnetic force, we switch the position of the magnet from 1.3 mm to 10 mm from the coverslip each cycle to apply a high and near-zero force to the tethers, respectively (Supplementary Fig. 8). The average high force for the experiment was ~21.3 pN with up to 9% variation among the beads estimated from the 3% CV size variation as tethered beads were not calibrated individually. Due to variations in the position of the motorized magnet stage from cycle to cycle, we estimate an additional 1% force variation between cycles, based on the precision of the stage.
The duration of high force was set to be 9 seconds in each cycle, allowing us to measure the change in length (ΔL) at a constant force that resulted when the weak handles stochastically ruptured (Supplementary Fig. 8). The change in length was calculated by taking the difference in the average position before and after the transition. To avoid incorporating transitions that did not result from weak-handle rupture of a single tethered per bead, we examined the force-extension curve and filtered out tethers with contour lengths that were at least 20% lower than the expected length of ~1 μm, which could be due to multiple or nonspecific tethering. We also evaluated the persistent length to filter out beads with potentially multiple tethers (Supplementary Fig. 8). In the final data set, we only included tethered beads that passed these filters and that had at least three identified transitions each collected within one force cycle to further filter out beads that rarely produced robust transitions or had instances of multiple transitions per cycle.
Proteomics analysis
The reviewed human UniProtKB proteome containing 20,353 canonical human proteins was used37, consistent with previous fingerprint analysis5,6,7. For each sequence, the cysteines were used as anchor points where the mechanically strong handle from the DNC would bind. For each cysteine, a fingerprint was made based on the absolute distance to any lysines within the caliper span, with all other amino acid information discarded. For example, the sequence MKLCSKAVKV would become C020010, with each digit representing the number of lysines that distance away from the cysteine anchor in amino acid space.
The caliper span is a key parameter governing the mechanical fingerprints obtained with DNA Nanoswitch Calipers. It is the maximum distance between DNA handles that can be measured by the caliper, which is set by the loop size. The caliper span can be estimated and expressed in terms of the number of amino acids by dividing the measured effective loop size L0 by the empirical length per amino acid (e.g. by dividing the intercept by the slope in the calibration curve of Figures 3c&d), which yields a caliper span for this particular caliper of ~120 aa. In addition to our standard caliper which has a caliper span of ~120 aa, we also created long-looped calipers with almost twice this caliper span (Supplementary Fig. 10). Correspondingly, we performed our analysis using two different caliper spans: 120 aa and 215 aa.
For an initial feasibility analysis, we looked to see how diverse the cysteine fragments were by grouping all the sequences by fingerprint. Cysteine fragments with a group of one are unique to a single protein in the database and fragments in the “missed” category did not have any lysines to pull with the caliper.
To estimate identification with measurement error and experimental limitations, a Bayesian framework was applied. A probability distribution was assigned at each distance with a lysine representing the probability of making a distance measurement given that cysteine lysine pair in the caliper. A gaussian probability was assigned to each lysine at the true distance with a standard deviation set to the specified resolution representing experimental measurement error. We note that in this analysis we are using units of amino acids for the measurement resolution, making this quantity dimensionless—these values represent the spatial resolution per cycle of a single distance measurement divided by the length per amino acid under these conditions. Probabilities less than 0.0001 were rounded to zero to simplify the calculation. We assumed every lysine on a given fragment had equal probability of binding to the DNC and being measured. For each cysteine fragment, data was simulated by randomly sampling from its assigned probability mass function the specified number of times, with each sample corresponding to a measurement and shearing release of the lysine handle by the DNC. We then calculate the posterior probability of identifying a fragment given the simulated data measurement by
The likelihood of a given data set given a fragment is the product of the probabilities of each sampled point from the fragment’s assigned probability mass function.
The prior distribution p(fragment) is assumed to be uniform for all fragments within the database. Since we are interested in protein identification, the posterior probability of each fragment in a protein given a single simulated data set was summed together. This summed posterior probability represents the probability of identifying a specific protein in the database given data measured from a fragment from that protein. This probability was averaged for each fragment in the protein to estimate the probability of identification for each protein. The histogram shown in the inset of Figure 4d is the probability of identification for all proteins in the database for a particular experimental condition of interest (caliper span: 120 aa, measurement error: 2 aa, 100 measurements per molecule). This same data can also be represented as a cumulative distribution, which we plot in Figure 4d in a slightly different way by plotting 1 minus the cumulative probability of identification vs. the probability of identification. This curve represents the fraction of the database that can be identified with a desired minimum probability. To explore how identification capabilities depend on experimental conditions, a wide range of resolutions (between 0–10 aa standard deviation) and measurements per molecule (25–800, log distributed) were simulated. The fraction of proteins that can be identified with at least 90% probability was plotted in the heat map for each experimental condition. To explore how changes in caliper span affect the ability to identify proteins, we performed additional analysis for a longer caliper span of 215 aa (Supplementary Fig. 7).
Supplementary Material
Acknowledgements
The authors would like to thank Prof. Stephen Buratowski, Dr. Marie Bao and all members of the Wong and Shih Labs for helpful discussions and comments on the manuscript.
Funding:
This work was funded by support from ONR Award N000141510073, Smith Family Foundation Odyssey Award, NIH NIGMS R35 GM119537 (W.P.W.), and the Wyss Institute at Harvard. E.K. acknowledges support from the Human Frontier Science Program (LT001077/2015-C).
Footnotes
Code availability
Code used to analyze data in this paper are available from the corresponding authors upon reasonable request.
Supplementary information is available in the online version of the paper. Reprints and permission information is available online at www.nature.com/reprints. Correspondence and requests for materials should be addressed to W.S. and W.W.
Competing interests
W.M.S. and W.P.W. have filed patent applications for various aspects of this work.
Data availability
Data supporting the findings of this paper are available from the supplementary files and the corresponding authors upon reasonable request. Source data are provided with this paper.
References
- 1.Aebersold R & Mann M Mass spectrometry-based proteomics. Nature 422, 198–207 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Restrepo-Pérez L, Joo C & Dekker C Paving the way to single-molecule protein sequencing. Nat. Nanotechnol 13, 786–796 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Callahan N, Tullman J, Kelman Z & Marino J Strategies for Development of a Next-Generation Protein Sequencing Platform. Trends Biochem. Sci 45, 76–89 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Timp W & Timp G Beyond mass spectrometry, the next step in proteomics. Sci. Adv 6, eaax8978 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alfaro JA et al. The emerging landscape of single-molecule protein sequencing technologies. Nat. Methods 18, 604–617 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swaminathan J, Boulgakov AA & Marcotte EM A Theoretical Justification for Single Molecule Peptide Sequencing. PLoS Comput. Biol 11, e1004080 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yao Y, Docter M, van Ginkel J, de Ridder D & Joo C Single-molecule protein sequencing through fingerprinting: computational assessment. Phys Biol. 12, 055003 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Ohayon S, Girsault A, Nasser M, Shen-Orr S & Meller A Simulation of single-protein nanopore sensing shows feasibility for whole-proteome identification. PLoS Comput. Biol 15, e1007067 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Ginkel J et al. Single-molecule peptide fingerprinting. Proc. Natl Acad. Sci. USA 115, 3338 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swaminathan J et al. Highly parallel single-molecule identification of proteins in zeptomole-scale mixtures. Nat Biotechnol, 10.1038/nbt.4278 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosen CB, Rodriguez-Larrea D & Bayley H Single-molecule site-specific detection of protein phosphorylation with a nanopore. Nat Biotechnol 32, 179–181 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kennedy E, Dong Z, Tennant C & Timp G Reading the primary structure of a protein with 0.07 nm3 resolution using a subnanometre-diameter pore. Nat. Nanotechnol 11, 968–976 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Restrepo-Pérez L, Wong CH, Maglia G, Dekker C & Joo C Label-Free Detection of Post-translational Modifications with a Nanopore. Nano Letters 19, 7957–7964 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ouldali H et al. Electrical recognition of the twenty proteinogenic amino acids using an aerolysin nanopore. Nat Biotechnol 38, 176–181 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Y et al. Single-molecule spectroscopy of amino acids and peptides by recognition tunnelling. Nat. Nanotechnol 9, 466–473 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohshiro T et al. Detection of post-translational modifications in single peptides using electron tunnelling currents. Nat. Nanotechnol 9, 835–840 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Koussa MA, Halvorsen K, Ward A & Wong WP DNA nanoswitches: a quantitative platform for gel-based biomolecular interaction analysis. Nat. Methods 12, 123 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halvorsen K, Schaak D & Wong WP Nanoengineering a single-molecule mechanical switch using DNA self-assembly. Nanotechnology 22, 494005 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Yang D, Ward A, Halvorsen K & Wong WP Multiplexed single-molecule force spectroscopy using a centrifuge. Nat. Commun 7, 11026 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J, Zhang C-Z, Zhang X & Springer TA A mechanically stabilized receptor–ligand flex-bond important in the vasculature. Nature 466, 992–995 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfitzner E et al. Rigid DNA Beams for High-Resolution Single-Molecule Mechanics. Angew. Chem. Int. Ed 52, 7766–7771 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kilchherr F et al. Single-molecule dissection of stacking forces in DNA. Science 353 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Kostrz D et al. A modular DNA scaffold to study protein–protein interactions at single-molecule resolution. Nat. Nanotechnol 14, 988–993 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Gosse C, Strick TR & Kostrz D Molecular scaffolds: when DNA becomes the hardware for single-molecule investigations. Curr. Opin. Chem. Biol 53, 192–203 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Ma X et al. Interactions between PHD3-Bromo of MLL1 and H3K4me3 Revealed by Single-Molecule Magnetic Tweezers in a Parallel DNA Circuit. Bioconjug Chem. 30, 2998–3006 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Hatch K, Danilowicz C, Coljee V & Prentiss M Demonstration that the shear force required to separate short double-stranded DNA does not increase significantly with sequence length for sequences longer than 25 base pairs. Phys. Rev. E 78, 011920 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Watson JD & Crick FHC Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature 171, 737–738 (1953). [DOI] [PubMed] [Google Scholar]
- 28.Woodside MT et al. Nanomechanical measurements of the sequence-dependent folding landscapes of single nucleic acid hairpins. Proc. Natl Acad. Sci. USA 103, 6190–6195 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bustamante C, Marko JF, Siggia ED & Smith S Entropic Elasticity of λ-Phage DNA. Science 265, 1599–1600 (1994). [DOI] [PubMed] [Google Scholar]
- 30.Abello N, Kerstjens HAM, Postma DS & Bischoff R Selective Acylation of Primary Amines in Peptides and Proteins. J. Proteome Res 6, 4770–4776 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Halvorsen K, Zhang C-Z, Wong WP & Springer TA Mechanoenzymatic Cleavage of the Ultralarge Vascular Protein Von Willebrand Factor. Science 324, 1330–1334 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oesterhelt F et al. Unfolding pathways of individual bacteriorhodopsins. Science 288, 143–146 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Carrion-Vazquez M et al. The mechanical stability of ubiquitin is linkage dependent. Nat. Struct. Mol. Biol 10, 738–743 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Oda E et al. Noxa, a BH3-Only Member of the Bcl-2 Family and Candidate Mediator of p53-Induced Apoptosis. Science 288, 1053 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Czabotar PE et al. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc. Natl Acad. Sci 104, 6217 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sattler M et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275, 983–986 (1997). [DOI] [PubMed] [Google Scholar]
- 37.Apweiler R et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 32, D115–D119 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adamczyk M, Gebler JC & Wu J Selective analysis of phosphopeptides within a protein mixture by chemical modification, reversible biotinylation and mass spectrometry. Rapid Commun. Mass Spectrom 15, 1481–1488 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Meinhart A & Cramer P Recognition of RNA polymerase II carboxy-terminal domain by 3′-RNA-processing factors. Nature 430, 223–226 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Phatnani HP & Greenleaf AL Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 20, 2922–2936 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Kim M, Suh H, Cho EJ & Buratowski S Phosphorylation of the yeast Rpb1 C-terminal domain at serines 2, 5, and 7. J. Biol. Chem 284, 26421–26426 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eick D & Geyer M The RNA Polymerase II Carboxy-Terminal Domain (CTD) Code. Chem. Rev 113, 8456–8490 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Knight ZA et al. Phosphospecific proteolysis for mapping sites of protein phosphorylation. Nat Biotechnol 21, 1047–1054 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Ribeck N & Saleh OA Multiplexed single-molecule measurements with magnetic tweezers. Rev. Sci. Instrum 79, 094301 (2008). [DOI] [PubMed] [Google Scholar]
- 45.De Vlaminck I et al. Highly Parallel Magnetic Tweezers by Targeted DNA Tethering. Nano Letters 11, 5489–5493 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Cnossen JP, Dulin D & Dekker NH An optimized software framework for real-time, high-throughput tracking of spherical beads. Rev. Sci. Instrum 85, 103712 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Smith SB, Finzi L & Bustamante C Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science 258, 1122 (1992). [DOI] [PubMed] [Google Scholar]
- 48.Danilowicz C, Greenfield D & Prentiss M Dissociation of Ligand−Receptor Complexes Using Magnetic Tweezers. Anal. Chem 77, 3023–3028 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Shon MJ, Rah S-H & Yoon T-Y Submicrometer elasticity of double-stranded DNA revealed by precision force-extension measurements with magnetic tweezers. Sci. Adv 5, eaav1697 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sen Y-H, Jain T, Aguilar CA & Karnik R Enhanced discrimination of DNA molecules in nanofluidic channels through multiple measurements. Lab Chip 12, 1094–1101 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keyser UF et al. Direct force measurements on DNA in a solid-state nanopore. Nat. Phys 2, 473–477 (2006). [Google Scholar]
- 52.Mulhall EM et al. Single-molecule force spectroscopy reveals the dynamic strength of the hair-cell tip-link connection. Nat. Commun 12, 849 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bustamante C, Chemla YR & Moffitt JR High-Resolution Dual-Trap Optical Tweezers with Differential Detection: Instrument Design. Cold Spring Harb. 2009, pdb.ip73 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Lipfert J, Hao X & Dekker NH Quantitative modeling and optimization of magnetic tweezers. Biophys. J 96, 5040–5049 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dulin D et al. High Spatiotemporal-Resolution Magnetic Tweezers: Calibration and Applications for DNA Dynamics. Biophys. J 109, 2113–2125 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Vlaminck I, Henighan T, van Loenhout MTJ, Burnham DR & Dekker C Magnetic Forces and DNA Mechanics in Multiplexed Magnetic Tweezers. PLOS ONE 7, e41432 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu Z et al. A force calibration standard for magnetic tweezers. Rev. Sci. Instrum 85, 123114 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the findings of this paper are available from the supplementary files and the corresponding authors upon reasonable request. Source data are provided with this paper.