

Abstract
Background & Aims:
Throughout life, the intestinal epithelium undergoes constant self-renewal from intestinal stem cells. Together with genotoxic stressors and failing DNA repair, this self-renewal causes susceptibility towards malignant transformation. X-box binding protein 1 (XBP1) is a stress sensor involved in the unfolded protein response (UPR). We hypothesized that XBP1 acts as a signaling hub to regulate epithelial DNA damage responses.
Methods:
Data from the TCGA were analyzed for association of XBP1 with CRC survival and molecular interactions between XBP1 andp53 pathway activity. The role of XBP1 in orchestrating p53-driven DNA damage response was tested in-vitro, in mouse models of chronic intestinal epithelial DNA damage (Xbp1/H2bfl/fl, Xbp1ΔIEC, H2bΔIEC, H2b/Xbp1ΔIEC) and via orthotopic tumor organoid transplantation. Transcriptome analysis of intestinal organoids was performed to identify molecular targets of Xbp1-mediated DNA damage response.
Results:
In the TCGA dataset of CRC, low XBP1 expression was significantly associated with poor overall survival (OS) and reduced p53 pathway activity. In-vivo, H2b/Xbp1ΔIEC mice developed spontaneous intestinal carcinomas. Orthotopic tumor organoid transplantation revealed a metastatic potential of H2b/Xbp1ΔIEC-derived tumors. RNA sequencing of intestinal organoids (H2b/Xbp1fl/fl, H2bΔIEC, H2b/Xbp1ΔIEC, H2b/p53∆IEC) identified a transcriptional program downstream of p53, in which XBP1 directs DNA damage-induced Ddit4l expression. DDIT4L inhibits mTOR-mediated phosphorylation of 4E-BP1. Pharmacological mTOR inhibition suppressed epithelial hyperproliferation via 4E-BP1.
Conclusions:
Our data suggest a crucial role for XBP1 in coordinating epithelial DNA damage responses and stem cell function via a p53-DDIT4L-dependent feedback mechanism.
Keywords: DNA damage, XBP1, p53, intestinal epithelial cell, CRC
Graphical Abstract
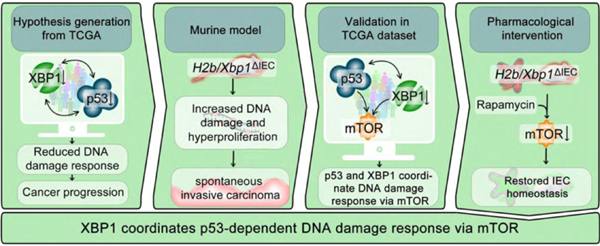
Lay summary:
Welz et al. show that XBP1 coordinates epithelial stem cell function in response to DNA damage in a mechanism involving the tumor suppressor TP53.
Introduction:
X-box binding protein 1 (XBP1) is an endoplasmic reticulum (ER)-resident protein coordinating the unfolded protein response (UPR) via translocation of a spliced sXBP1 transcript that acts as a nuclear transcription factor1. Multiple studies have underlined the role of XBP1 in various cancer types, albeit with contradictory effects on the directionality of the overall outcome. With regard to cancer (CRC), murine studies investigating the role of XBP1 in intestinal epithelial carcinogenesis point towards a tumor suppressive role of the IRE1/XBP1 axis, as epithelial specific deletion of Xbp1 results in epithelial hyperproliferation and increased tumor burden in experimental cancer models (AOM-DSS, ApcMin/+ mice)2. Yet, the exact mechanism of this effect and its importance for sporadic CRC, which represents the majority of CRC cases, remains unknown. In sporadic CRC, genomic instability and impaired DNA repair have been recognized as essential cellular properties that enable the acquisition of mutations in oncogenes or tumor-suppressor genes encoding e.g. PIK3CA and TP53, TGFβ receptors and SMADs, thereby explaining the sequential adenoma to carcinoma transformation3. In addition, proto-oncogenes such as KRAS, BRAF and frequent mutations within the Wnt-signaling pathway (APC, CTNNB1) contribute to the pathogenesis of CRC. Functionally, all these mutations converge in the activation of the Wnt, RAS-RAF-MEK-ERK or PI3K-AKT pathway4,5.
It has been shown that ribonucleotide excision repair (RER) is responsible for removing misincorporated ribonucleotides from replicating DNA, with the RNase H2 complex providing the first key step of RER by cleaving 5’ of the DNA-embedded ribonucleotide6. Intestinal ablation of Rnaseh2b in murine epithelium (H2bΔIEC) results in spontaneous DNA damage associated with proliferative exhaustion of the resident stem cell compartment7. Notably, co-deletion of the tumor suppressor p53 in H2b/p53∆IEC mice promoted spontaneous age-dependent formation of intestinal carcinomas.
A limited body of evidence indicates that XBP1 might directly be involved in regulating DNA damage response (DDR) by means of interfering with double-strand break repair or p53-driven DDR8–10. In addition, it has been demonstrated that the resolution of genotoxic stress necessitates IRE1α, the upstream protein facilitating XBP1 splicing in response to ER-stress, in a mechanism involving IRE1α-dependent decay (RIDD)11.
However, although a role of the ER stress regulator XBP1 in coordinating DNA damage repair has been proposed, a clear genetic link demonstrating XBP1 function in DDR and consecutive epithelial carcinogenesis is still missing. Here, we set out to directly assess the role of XBP1 in coordinating intestinal epithelial DDR and tumor suppression. We demonstrate that reduced XBP1 expression in CRC is associated with poor long-term patient survival and reduced p53 pathway activity. Using mice double deficient for Rnaseh2b and Xbp1 (H2b/Xbp1ΔIEC), in which both RER and ER stress control are selectively inhibited in the intestinal epithelium, we show that XBP1 maintains tumor suppression in a mechanism involving p53-dependent target gene expression.
Materials and Methods:
Mice
Floxed Xbp1 (Xbp1fl/fl) or Rnaseh2b (H2bfl/fl) mice were created as described previously2,12. Villin-cre+;Xbp1fl/fl (Xbp1ΔIEC), Villin-cre+;Rnaseh2bfl/fl (H2bΔIEC) and Villin-cre+;Rnaseh2bfl/fl/Xbp1fl/fl (H2b/Xbp1ΔIEC) mice were backcrossed for at least six generations and used at an age of 8 – 12 weeks for all experiments with appropriate genotype littermate controls. All mice were maintained in a specific pathogen–free (SPF) facility and the quarterly health report did not indicate presence of pathogenic species. Littermates of the same genotypes were cohoused. Mice were provided with food and water ad libitum and maintained in a 12-h light–dark cycle under standard conditions at Kiel University. Mice orthotopically transplanted with tumor organoids were housed in a SPF facility at the Technische Universität München. Tail or ear biopsy genomic DNA of respective mouse strains were used for genotyping. For experiments including application of Rapamycin or DSS, equal numbers (minimum n = 3 per genotype; see figure legends for details) of age-matched male and female animals were used. Procedures involving animal care were conducted conform to national and international laws and policies with appropriate permission. All experiments were performed in accordance with the guidelines for Animal Care of Kiel University and of the Institutional Animal Care and Use Committees of the Technische Universität München (Regierung von Oberbayern, Munich, Germany).
In-vivo Treatment of Mice
For DSS treatment, mice were supplied with either 1 % of DSS (MP Biomedical) dissolved in drinking water for 6 days followed by 4 days of regular drinking water (acute models) or 1 % DSS for 2 cycles of 7 or 5 days followed by 14 days of regular drinking water (chronic models). Disease Activity Indices were obtained as described previously13. Consumption of drinking water was measured daily. For Rapamycin treatment, H2b∆IEC or H2b/Xbp1∆IEC mice were injected intraperitoneally with either 1.5 mg / kg bodyweight Rapamycin (LC Laboratories) or DMSO (Sigma Aldrich) for 7 days before being sacrificed. For all experiments, histological and histopathologic analyses were performed according to standard methods.
Histopathological Analyses of Murine Intestinal Tissue
Postmortem, the small intestine and colon were excised and cut open longitudinally. The ileum and colon were rolled up as Swiss rolls from the distal to the proximal part and fixed in 10 % formalin. Paraffin sections were cut and stained with H&E. Histological scoring displays the combined score of inflammatory cell infiltration and tissue damage as described elsewhere and was performed in a blinded fashion by two independent observers, as described previously13.
Orthotopic Organoid Transplantation
Orthotopic transplantations of organoids were performed as previously described14. Briefly, organoids were dissociated into 5 – 10 cell clusters and resuspended in a minimal medium (Advanced DMEM/F12 containing 1 x B27, 1 x N2, L-Glutamine (all from Gibco, Thermo Fisher Scientific), 10 % Matrigel (Corning), 1 % Pen/Strep and 10 μM Y-27632 (STEMCELL Technologies)). For every injection (2 – 3 per mouse), 50 dissociated organoids in a volume of 80 μl were prepared. Subsequently, the colon of the anesthetized mice was gently rinsed with PBS using a syringe and a straight oral gavage needle. Colonoscopy of mice was performed using a rigid endoscope from Karl STORZ (1.9 mm in diameter) with linear Hopkins lens optics (ColoView System). For injections of organoids into the submucosa of the colon, a flexible fine needle (Hamilton; 33 gauge, custom length of 16 inches, custom point style of 4 at 45°) was used. Injections that were correctly applied into the submucosa led to the formation of a bubble that closes the intestinal lumen.
Histological Grading of Intestinal Tumors
All identifiable adenomas and invasive adenocarcinomas were classified in analogy to the recent WHO classification of tumors of the Digestive System (fifth edition). Invasive adenocarcinomas were graded into “low-grade” and “high-grade” according to the extent of gland formation. The grade of dysplasia / intraepithelial neoplasia of small intestinal adenomas was classified into low- and high grade IEN / dysplasia based on the degree of architectural complexity, extent of nuclear stratification and severity of abnormal nuclear morphology.
Transcriptome Analysis
RNA sequencing was conducted on small intestinal organoids derived from H2b/Xbp1fl/fl, H2b∆IEC, Xbp1∆IEC, H2b/Xbp1∆IEC and H2b/p53∆IEC mice (n = 3). Samples were sequenced on Illumina HiSeq3000 using Illumina total RNA stranded TruSeq protocol (GEO Project Accession number 182353). An average of ~12 million 50-nt unpaired-end reads was sequenced for each sample. Raw reads were pre-processed using Cutadapt15 to remove adapter and low-quality sequences and then aligned to the GRCm37 reference genome with TopHat216. Gene expression values of the transcripts were computed by HTseq17, before differential gene expression (DEG) levels were analyzed and visualized by the Bioconductor package DESeq218. Generalized linear models were utilized to compare transcriptomes of H2b∆IEC, Xbp1∆IEC, H2b/Xbp1∆IEC or H2b/p53∆IEC organoids to H2b/Xbp1fl/fl organoids (p < .05). To interpret the biological significance of DEG, Gene Ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) enrichment analyses were performed using the Innate DB database19.
Multi-omics-based Network Inference Analysis
We obtained the colorectal cancer subset of The Cancer Genome Atlas (TCGA)20 collection as independent multi-omics dataset. It contains information of 101 colorectal cancer patients measured across 4 omics-levels, Transciptome, Proteome, Methylation and Mutation. This data was used as an input for KiMONo, a knowledge guided multi-omics network inference method, to infer a colorectal cancer specific multi-omics network21. The resulting network served as blueprint to detect all multi-omics features associated to either MTOR pathway genes or XBP1, TP53 and DDIT4L. In a final step, we applied KiMONo to this trimmed data, detecting effects between the multi-omics features including the MTOR pathway, XBP1, TP53 and DDIT4L.
Results
Low XBP1 expression associates with poor CRC prognosis
We correlated the expression of XBP1 with long term survival of colorectal cancer (COAD – Colon Adenocarcinoma and READ – Rectum Adenocarcinoma datasets) patients in a data set publicly available through The Cancer Genome Atlas (TCGA)20 and found that low XBP1 expression was significantly associated with poor survival (Figure 1A). To identify a potential mechanism by which XBP1 contributes to tumor suppression in CRC, we performed pathway enrichment analysis. Based on XBP1 expression (high vs. low), we estimated the activity of 14 cancer-related pathways including TNF-α, NFκB, Hypoxia, TRAIL and p53-mediated DDR using PROGENy22 and observed that low XBP1 expression was significantly associated with lower p53 pathway activity (Figure 1B). To test whether XBP1 interferes with p53-coordinated DDR in intestinal epithelial cells (IEC), we stimulated murine small intestinal iCtrl or Xbp1-silenced (iXbp1) ModeK cells with the DNA damaging agent Cytarabine A (AraC)23. We assessed canonical p53 activation in response to DNA damage by analyzing the gene expression of p53 and its target genes (Ccng1, Mdm2, Sesn2) in AraC-stimulated ModeK cells (iCtrl vs. iXbp1). iXbp1 cells presented with significantly impaired upregulation of p53-target genes in response to AraC treatment (Figure 1C), pointing to an involvement of XBP1 in p53 activation. Notably, reduced activation of p53-downstream targets in iXbp1 cells was not dependent on altered p53 protein expression in response to AraC treatment (Figure 1D). Altogether, these data point toward a role of Xbp1 in coordinating p53-driven DNA damage responses in the intestinal epithelium.
Figure 1: XBP1 expression signature associates with CRC prognosis.
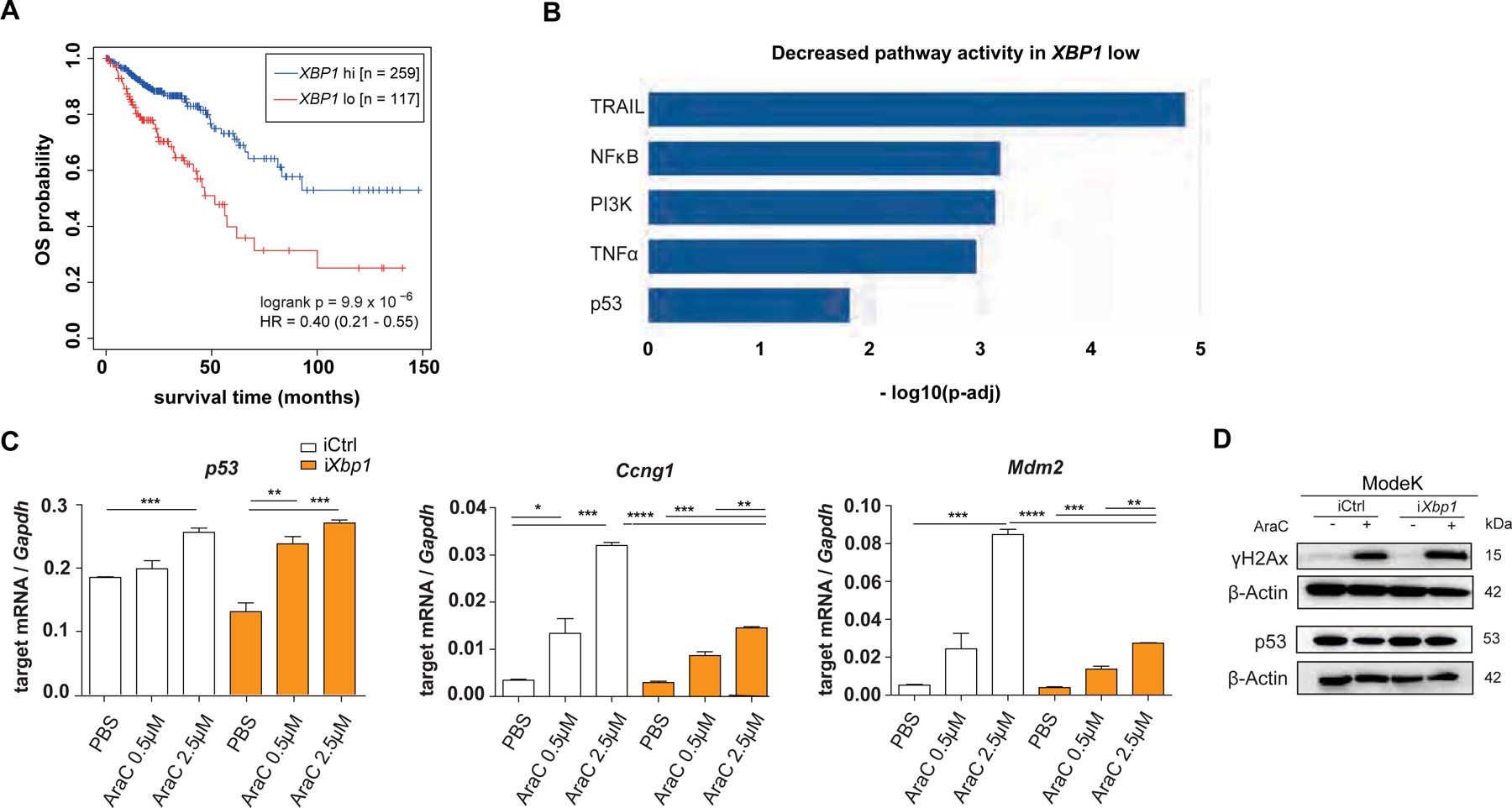
(A) RNA expression data retrieved from the TCGA database (COAD – Colon Adenocarcinoma and READ – Rectum Adenocarcinoma) were used for overall survival (OS) analysis based on high or low XBP1 expression in tumor samples. (B) PROGENy analysis based on pathway enrichment analysis within the same COAD READ dataset and shown as decreased pathway activity in XBP1 low dataset. (C) ModeK cells, treated with PBS, 0.5 μM and 2.5 μM AraC for 24 hours. qRT-PCR analysis of p53-dependent target genes (Sesn2, Ccng1, Mdm2), representative of a minimum of 3 individual experiments with n = 3 technical replicates. Relative mRNA expression is normalized to Gapdh. Data are expressed as mean ± standard error of the mean and significance was determined using an unpaired Student’s t-test. (D) ModeK cells, stimulated with 2.5 μM AraC for 24 hours. Protein lysates were probed against antibodies for p53, γH2Ax and ß-Actin as a loading control on different Western blot membranes, representative of 3 individual experiments. * p < .05, ** p < .01, *** p < .0001, **** p < .00001.
Xbp1 deficiency drives spontaneous intestinal carcinogenesis
To test whether absence of Xbp1 phenocopies p53 deletion and drives intestinal carcinogenesis in-vivo, we took advantage of H2b∆IEC mice, a model of chronic DNA damage in which intestinal tumor formation occurs in the context of simultaneous knockout of epithelial p53 (H2b/p53∆IEC mice). We generated mice with a conditional intestinal epithelial deletion for both Xbp1 and Rnaseh2b (hereafter H2b/Xbp1∆IEC mice) in addition to wildtype (H2b/Xbp1fl/fl) and single knockout animals (Xbp1∆IEC and H2b∆IEC). qPCR and Western blot analysis of small intestinal organoids, derived from purified IEC`s, verified epithelial-specific conditional deletion of Xbp1 and Rnaseh2b (Supplementary Figure 1). To assess the impact on spontaneous carcinogenesis, mice were aged, sacrificed at the age of 52 weeks and overall tumor burden was evaluated. Whereas there was almost a complete lack of spontaneous tumor formation in H2b/Xbp1fl/fl (n = 1/23; ‘), Xbp1∆IEC (n = 0/8) and H2b∆IEC (n = 0/9) mice, 48 % of H2b/Xbp1∆IEC mice (n = 12 / 25) developed spontaneous stenosing, predominantly small intestinal tumors (Figure 2A–C). On average, tumor bearing mice presented with n = 1.5 tumors / intestine, ranging from 0.2 to 2.5 cm in size (Figure 2D,E). Histopathologically, macroscopic tumors presented as low- / high-grade intraepithelial neoplasia or as invasive adenocarcinomas, of which the majority were high-grade adenocarcinomas (Figure 2F). Molecular phenotyping of H2b/Xbp1∆IEC tumor organoids unveiled upregulation of the Wnt target genes Ccnd1, Cd44 and Sox9, pointing to a direct interaction of XBP1 and Wnt/ß-catenin signaling 24,25. Importantly, this upregulation appeared to be dependent on autonomous release of Wnt ligands, as the pharmacological Wnt secretion inhibitor IWP-2 failed to abrogate Ccnd1, Cd44 and Sox9 expression in H2b/Xbp1∆IEC-derived tumor organoids compared to non-tumor H2b/Xbp1∆IEC-derived organoids (Supplementary Figure 2). To minimize the possibility that the observed phenotype of H2b/Xbp1∆IEC mice is primarily driven by microbial influences, we assessed both single (H2b/Xbp1∆IEC) and co-housed (H2b/Xbp1∆IEC vs. H2b/Xbp1fl/fl) 52 weeks old H2b/Xbp1∆IEC mice and found no differences in tumor incidence between groups (Supplementary Figure 3). To further investigate whether malignant properties are conserved outside of the mucosal microenvironment, we performed orthotopic colonoscopy-guided transplantation of H2b/Xbp1∆IEC-derived tumor organoids (n = 2 clones) into the colonic mucosa of immunodeficient NSG (n = 6) mice (Figure 2G). We observed that n = 6 / 6 mice presented with locally invasive carcinomas at the site of injection (Figure 2H,I). Although in this model metastases are rare events26,27, 66% (n = 4 / 6) of mice presented with liver metastasis and 100% (n = 6 / 6) of mice with lung metastasis (Figure 2J), leading to overall rapid clinical deterioration (Figure 2K).
Figure 2: Xbp1 deficiency drives spontaneous tumor development in H2b/Xbp1∆IEC mice.
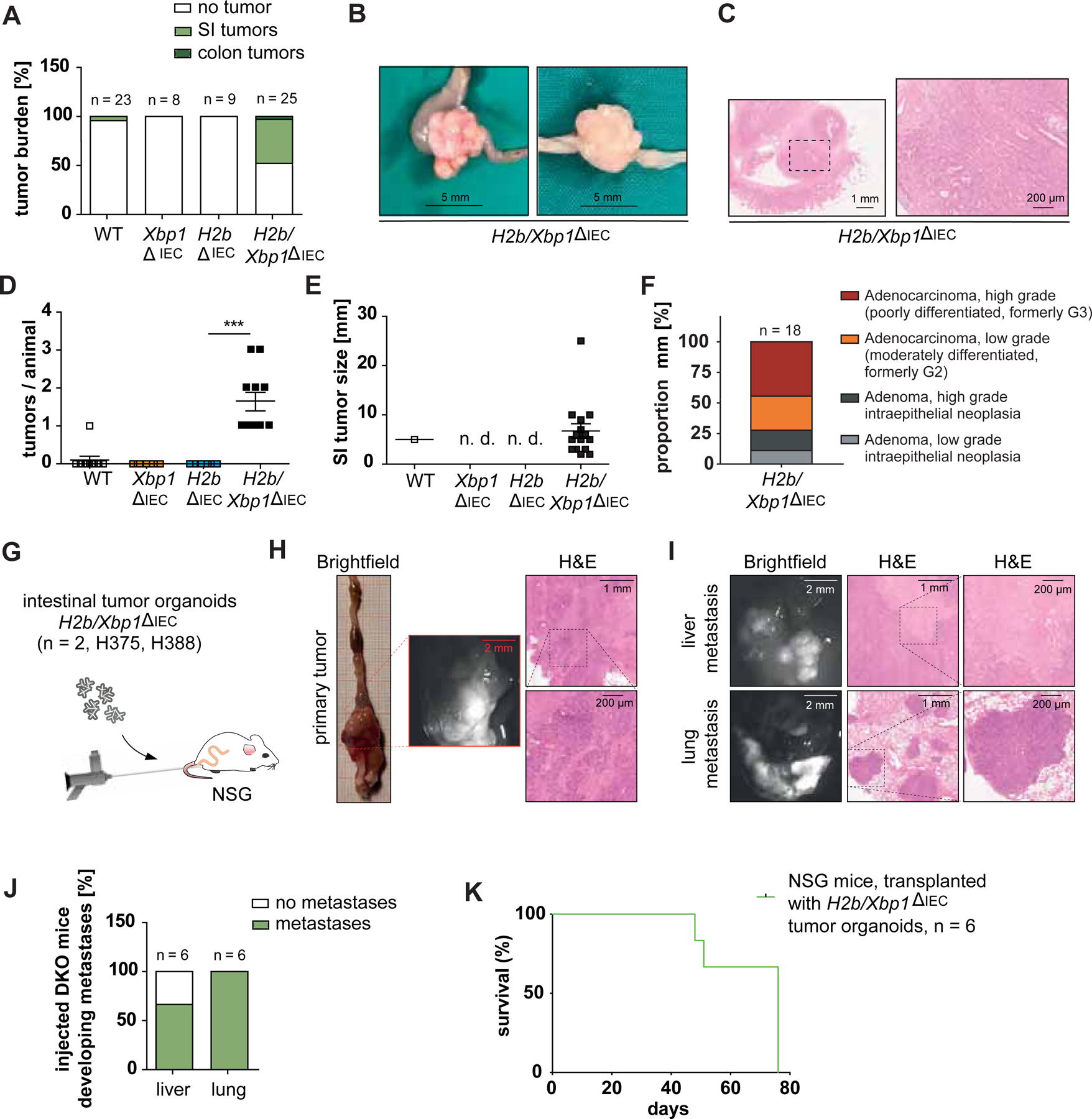
Tumor burden of 52 weeks old WT mice (n = 23, n = 6 females, 17 males), Xbp1∆IEC mice (n = 8, 1 female, 7 males), H2b∆IEC mice (n = 9, 4 females, 6 males) and H2b/Xbp1∆IEC mice (n = 25, 15 females, 10 males). (A) Percentage of mice developing tumors with (B) representative images, (C) representative H&E staining of invasive intestinal carcinomas from H2b/Xbp1∆IEC mice. (D) Mean number of SI tumors per animal, (E) size of SI tumors. (F) Histopathological grading of small intestinal mucosa from H2b∆IEC and H2b/Xbp1∆IEC mice. (G) NSG mice (n = 6, 4 females, 2 males) were orthotopically transplanted with H2b/Xbp1’IEC derived intestinal organoids (n = 2 clones). Representative picture of a primary tumor (H) and hepatic / pulmonary metastasis resulting from organoid transplantation. (I) H&E staining of tumours. (J) Percentage of hepatic and / or pulmonary metastases of implanted mice. Scale bars, Brightfield, 2 mm; H&E overview, 1 mm; H&E inset, 200 μm. (K) Kaplan-Meier survival curve of NSG mice after organoid transplantation. Data are expressed as mean ± standard error of the mean and significance was determined using an unpaired Student’s t-test. *** p < .0001. H2b/Xbp1ΔIEC organoids are abbreviated as “DKO”.
Xbp1 deficiency fuels epithelial DNA damage and compromises DNA damage-dependent stem cell suppression in-vivo
To further investigate spontaneous carcinogenesis in H2b/Xbp1∆IEC mice, we assessed morphological changes in the small intestine of H2b/Xbp1∆IEC and H2b∆IEC non-tumor bearing mice of the same experimental group (52 weeks). Macroscopically, H2b/Xbp1∆IEC animals presented with decreased body weight and increased small intestinal length (Supplementary Figure 4A).
Epithelial DNA damage, cell death and proliferation were assessed using immunohistochemical and fluorescence stainings for Ki67, BrdU, γH2Ax and TUNEL of the small intestine of non-tumor bearing aged mice. Compared to H2b∆IEC mice, H2b/Xbp1∆IEC animals presented with heightened levels of DNA damage (Figure 3A,B), augmented epithelial cell death (Figure 3C,D) and elevated epithelial proliferation (Figure 3E,F). We also found that expansion of basal crypt proliferation in H2b/Xbp1∆IEC mice coincided with a nearly complete reconstitution of Paneth cells, pointing towards a role of Paneth cells in providing the niche for intestinal stem cells in the context of epithelial DNA damage (Figure 3G–J). Notably, young (8 – 12 weeks old) mice of the indicated genotypes presented with a nearly complete phenocopy of aged mice, albeit to a lesser degree (Supplementary Figure 4B, 5). We further validated that Xbp1 knockdown in-vitro enhanced DNA damage-induced epithelial cell death (Supplementary Figure 6A–G) and that young H2b/Xbp1∆IEC intestinal organoids, compared to H2b∆IEC mice intestinal organoids, presented with increased colony forming numbers, thereby reflecting the observed in vivo phenotype (Supplementary Figure 6H–I).
Figure 3: Xbp1 deficiency limits intestinal epithelial regeneration in the context of DNA damage in-vivo.
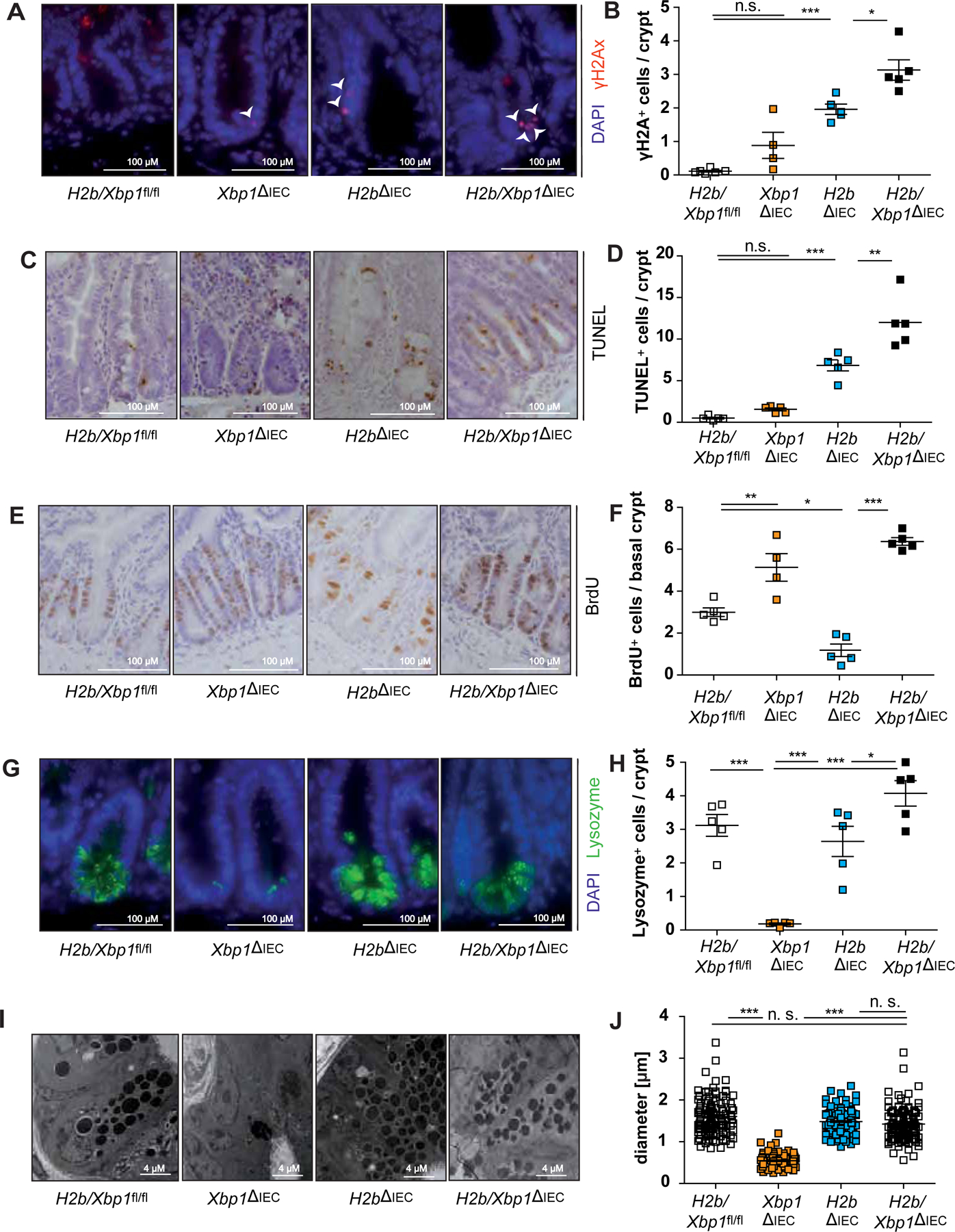
Representative images of the SI of 52 weeks old H2b/Xbp1fl/fl (n = 5, 1 female, 4 male), Xbp1∆IEC (n = 5, 2 females, 3 males), H2b∆IEC (n = 5, 2 females, 3 males) and H2b/Xbp1∆IEC (n = 5, 2 females, 3 males) mice, stained for (A) γH2Ax, with white arrowheads indicating γH2Ax+-nuclei, (C) TUNEL, (E) BrdU and (G) Lysozyme and the respective statistical analysis in which (B) γH2Ax+, (D),TUNEL+, (F) BrdU+ and (H) Lysozyme+ cells were assessed in a total of 50 crypts. (I) Representative images and (J) diameter measurements of Paneth cell granule. Data are expressed as mean ± standard error of the mean and significance was determined using one-way ANOVA. * p < .05, ** p < .01, *** p < .0001.
Xbp1 deficiency impairs DNA damage-driven susceptibility to experimental enteritis
We have previously shown that loss of epithelial Xbp1 drives small intestinal ileitis, which can be further exacerbated in a model of DSS exposure13,28. As H2b/Xbp1∆IEC mice presented with increased small intestinal cell death and DNA damage, we tested whether Xbp1 and Rnaseh2 deletions synergistically fuel intestinal inflammation using an acute and chronic DSS exposure model. As indicated by weight loss curve (Figure 4A,C) and colon length (Figure 4B,D), H2b/Xbp1∆IEC animals suffered from increased susceptibility towards acute and chronic DSS exposure. To specifically investigate whether impaired ER stress resolution and chronic DNA damage predominantly drive small intestinal inflammation, we assessed disease severity in DSS-treated H2b/Xbp1∆IEC animals by histopathological scoring of the inflamed small intestine from acute (Supplementary Figure 7A,B) and chronic DSS exposure (Supplementary Figure 7C,D). In both models, we observed increased histopathological disease severity in H2b/Xbp1∆IEC animals, compared to control animals. Of note, we did not observe tumor formation in the chronic enteritis model. We further delineated the molecular entity of small intestinal inflammation from the chronic DSS exposure model and noticed enhanced epithelial DNA damage (Figure 4E,F), increased cell death (Figure 4G,H) and epithelial proliferation in the small intestine of H2b/Xbp1∆IEC mice, as assessed by crypt BrdU staining (Figure 4I,J). Altogether, our data implicate that Xbp1 deficiency further aggravates chronic DNA damage-driven enteritis.
Figure 4: Deletion of Xbp1 heightens susceptibility towards DSS-induced colitis.
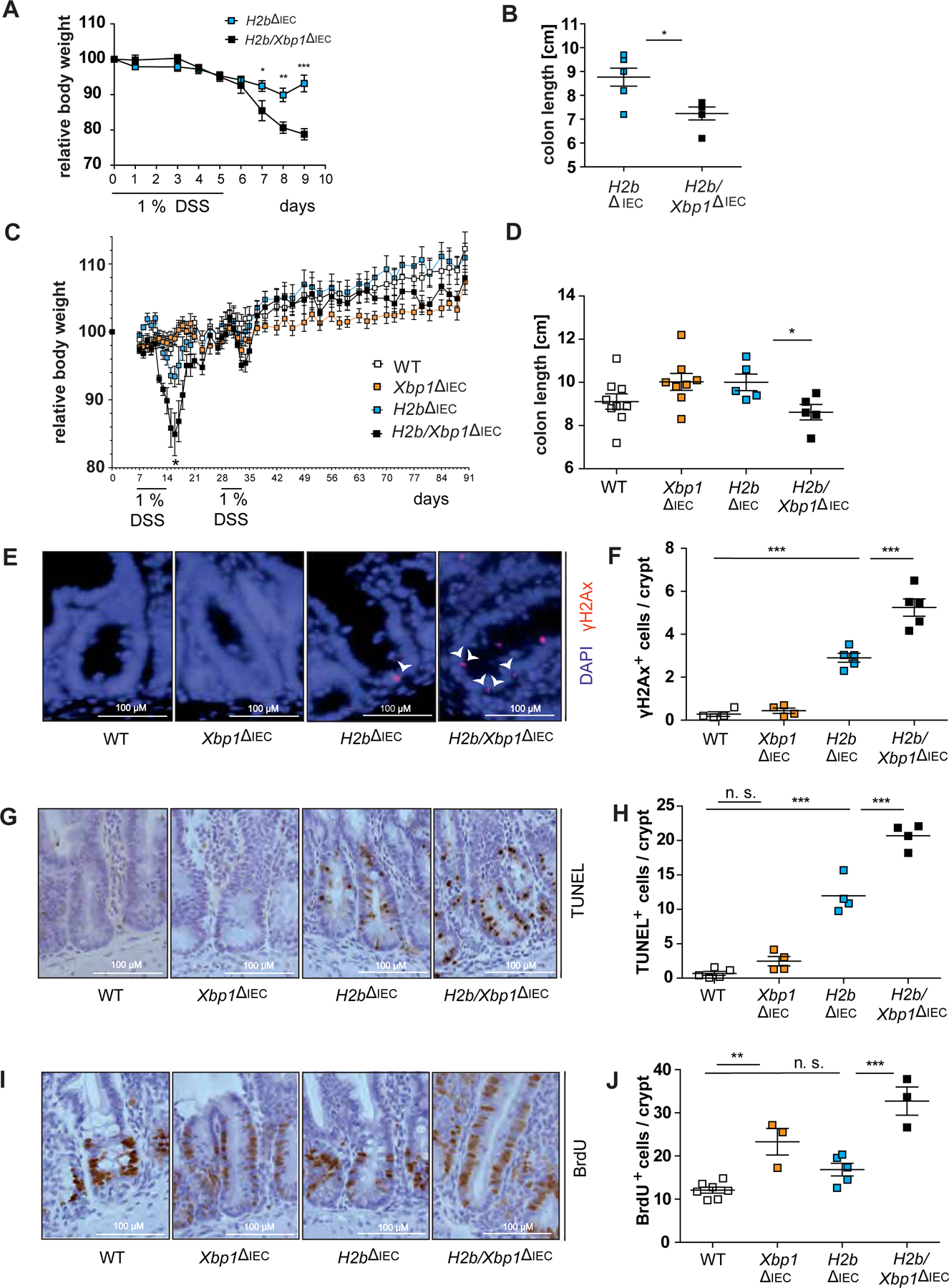
(A) Weight curve and (B) colon length from acute DSS-induced colitis with age-matched H2b∆IEC (n = 6, 1 female, 5 males) and H2b/Xbp1∆IEC (n = 6, 3 females, 3 males) animals. (C) Weight curve and (D) colon length from chronic DSS-induced colitis with age-matched WT (n = 9, 5 females, 4 males), Xbp1∆IEC (n = 8, 4 females, 4 males), H2b∆IEC (n = 5, 2 females, 3 males) and H2b/Xbp1∆IEC (n = 5, 4 females, 1 male) mice. Representative images of the SI for (E) γH2Ax, with white arrowheads indicating γH2Ax+-nuclei, (G) TUNEL and (I) BrdU and the accompanying statistical analyses of (F), γH2Ax+, (H) TUNEL+ and (J) BrdU+ cells in the respective genotypes. Data are expressed as mean ± standard error of the mean and significance was determined using unpaired Student’s t-test (A-D) or one-way ANOVA (E-J). * p < .05, ** p < .01, *** p < .0001.
XBP1 coordinates DNA damage response via DDIT4L / 4E-BP1
H2b/Xbp1∆IEC display a phenotype of ongoing epithelial proliferation despite overt DNA damage, reminiscent of the phenotype of H2b/p53∆IEC mice7. As our in-vitro data indicate a role for XBP1 in coordinating DNA damage-induced p53 responses, we tested whether XBP1 and p53 are part of a common signaling pathway to coordinate DDR in IEC. We performed RNA sequencing of intestinal organoids generated from H2b/Xbp1fl/fl, H2b∆IEC, Xbp1∆IEC, H2b/Xbp1∆IEC and H2b/p53∆IEC animals. Acquired data were used to identify unique and overlapping gene networks between H2b/Xbp1∆IEC and H2b/p53∆IEC intestinal organoids in comparison to the H2b∆IEC transcriptome (Figure 5A). 353 (down: 139; up: 214) differentially expressed genes were detected to be common between H2b/Xbp1∆IEC and H2b/p53∆IEC (Figure 5B). In contrast, 3,435 genes were uniquely up- or downregulated in H2b/p53∆IEC, and 1024 genes were uniquely up- or downregulated in H2b/Xbp1∆IEC intestinal organoids (Figure 5B), when compared to H2b∆IEC cultures. Based on ranked lists of the top 50 differentially downregulated transcripts from the individual comparisons, we identified Ano3, Ddit4l, Fam212 and Mcam as saliently downregulated genes shared between the respective genotypes (H2b/Xbp1∆IEC and H2b/p53∆IEC vs. H2b∆IEC) (Supplementary Figure 8).
Figure 5: XBP1 regulates regenerative DDR in a DDIT4L - mTOR-dependent manner.
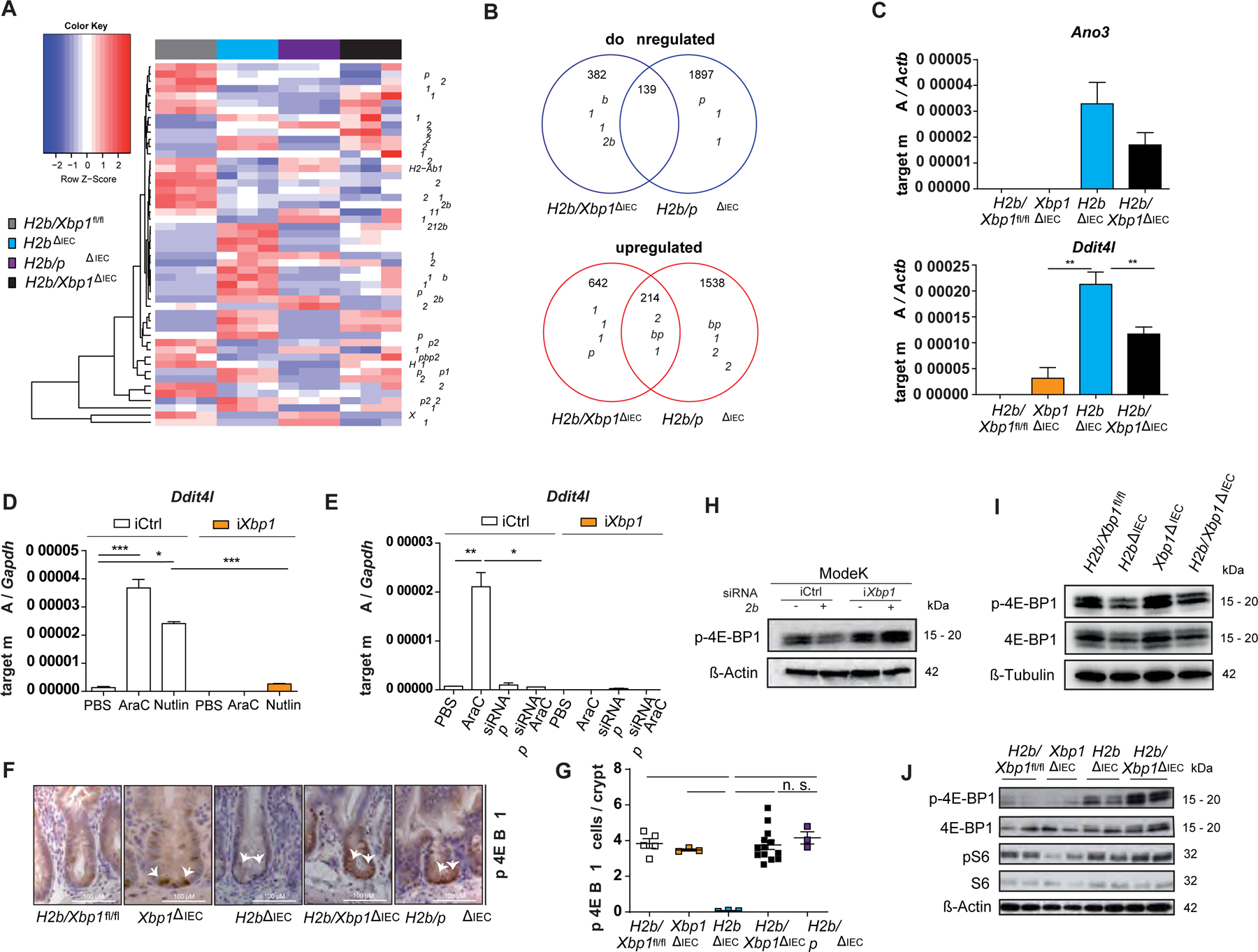
(A) RNA sequencing of n = 3 organoid samples from n=3 individual animals. (B) Venn diagram of uniquely and commonly regulated genes in intestinal organoids of H2b/Xbp1ΔIEC and H2b/p53ΔIEC vs. H2bΔIEC mice. (C) qRT-PCR analysis of small intestinal crypt cells of 52 weeks old H2b/Xbp1fl/fl (n = 3, 3 males), Xbp1∆IEC (n = 6, 1 female, 5 males), H2b∆IEC (n = 6, 2 females, 4 males) and H2b/Xbp1∆IEC (n = 5, 2 females, 3 males) mice. qRT-PCR analysis of (D) ModeK cells, treated with PBS, 2.5 μM AraC or 50 μM Nutlin for 24 hours and (E) of ModeK cells transfected with siRNA Ctrl and p53 and treated with PBS or 2.5 μM AraC for 24 hours, both experiments representative of a minimum of 3 individual experiments with n = 3 technical replicates. Relative mRNA expression is normalized to Actb (β-actin) or Gapdh. Representative images (F) and (G) statistical analysis of the p-4E-BP1-stained SI of 52 weeks old H2b/Xbp1fl/fl (n = 3, 3 males), Xbp1∆IEC (n = 3, 1 female, 2 males), H2b∆IEC (n = 3, 1 female, 2 males), H2b/Xbp1∆IEC (n = 13, 8 females, 5 males) and H2b/p53∆IEC (n = 3, 3 males) mice. Western blots, representative of each n = 3 individual experiments, of (H) ModeK cells transfected with siRNA against Rnaseh2b for 24 hours, (I) untreated intestinal organoids of the indicated genotypes and (J) small intestinal crypt cells from age-matched H2b/Xbp1fl/fl (2 females), Xbp1∆IEC (2 females), H2b∆IEC (1 female, 1 male) and H2b/Xbp1∆IEC (2 females) mice after exposure to chronic DSS colitis. ß-Actin and ß-Tubulin served as loading controls. Data are expressed as mean ± standard error of the mean and significance was determined using an unpaired Student’s t-test (D,E) or one-way ANOVA(G) * p < .05, ** p < .01, *** p < .0001, **** p < .00001.
DNA damage inducible transcript 4-like (DDIT4L), also known as REDD2, acts as an upstream inhibitor of mTOR signaling in various cell types, including cardiomyocytes, muscle cells and neurons29,30. As mTOR is known to direct cell proliferation via the eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1), we hypothesized that regulation of DDIT4L might be involved in the formation of the hyperproliferative H2b/Xbp1∆IEC phenotype. Indeed, downregulation of Ddit4l transcript levels was preserved in small intestinal crypts of aged H2b/Xbp1∆IEC mice (Figure 5C). To delineate the crosstalk between XBP1 and p53 in this context, we treated ModeK cells (iCtrl vs. iXbp1, transfected with siRNA against p53) with AraC or the endogenous MDM2 inhibitor Nutlin. Nutlin induces nuclear translocation of p53 and leads to subsequent activation of p53-dependent gene expression, thereby serving as a positive control of p53 activation in the absence of DNA damage. We confirmed that i) Xbp1-deficient cell showed impaired Ddit4l upregulation in response to Nutlin or Ara-C treatment (Figure 5D) and ii) Ddit4l expression essentially requires p53 (Figure 5E), further underscoring the crosstalk between p53 and Xbp1 in coordinating Ddit4l expression. Hence, we reasoned that high Ddit4l expression in H2b∆IEC mice might inhibit mTOR activity, hereby putting a halt on stem cell proliferation, whereas downregulated Ddit4l levels in H2b/Xbp1∆IEC mice might evoke uncontrolled proliferation. As mTOR inhibition acts downstream via dephosphorylation of 4E-BP1, we assessed the phosphorylation status of 4E-BP1 in all four genotypes. Indeed, p-4E-BP1 levels were significantly decreased in the crypt region of H2b∆IEC animals, but restored in H2b/Xbp1∆IEC and H2b/p53∆IEC mice (Figure 5F,G). This finding was further confirmed by Western blots using protein lysates of ModeK cells transfected with siRNA against Rnaseh2b (Figure 5H), of small intestinal organoids (Figure 5I) or of isolated small intestinal crypts (Figure 5J). Again, Rnaseh2b-silenced ModeK cells or H2b∆IEC animals displayed reduced p-4E-BP1 levels and phosphorylation was increased upon additional Xbp1 deficiency. We additionally proved that DDIT4L and p53 coordinate 4E-BP1 phosphorylation, as knockdown of p53 or Ddit4l in ModeK cells heightened p-4E-BP1 levels, whilst AraC-induced DNA damage decreased p-4E-BP1 levels in a DDIT4L-dependent manner (Supplementary Figure 9).
Multi-omics network analysis of colorectal cancer infers interaction between XBP1 and TP53 via MTOR signaling pathway
Based on these data, we concluded that XBP1 cooperates with p53 to control cellular proliferation via DDIT4L-dependent mTOR signaling. To gain deeper insights whether XBP1 and TP53 are molecularly connected via the MTOR pathway in the clinical setting of colorectal cancer, we used the TCGA dataset, in which 101 CRC patients were assessed across 4 different omics-levels: Transcriptome (16,115 transcripts), Proteome (130 proteins), Mutations (39,674 positions) and Methylation (2,043 sites). Utilizing the KiMONo network inference algorithm21, we were able to trim the multidimensional features with possible impact on XBP1, DDIT4L, TP53 and the MTOR pathway to 120 genes, 52 proteins, 70 mutation sites and 346 methylation sites. Out of these 588 features, KiMONo identified 47 features statistically affecting XBP1 and 80 features associated to TP53 and DDIT4L (Figure 6). Interestingly, we identified the gene expression of MDM2, a canonical p53 target gene, to be strongly affected by both, XBP1 and p53, which is in line with the observed association of XBP1 gene expression and p53 pathway activity shown in Figure 1. In addition, of 65 transcripts, which were correlated with XBP1, DDIT4L and p53 levels, 29 were related to the MTOR pathway, including EIF4E2, which codes for the transcription factor regulated by 4E-BP1. Altogether, this network inference algorithm in a CRC dataset strongly infers MTOR signaling as a crucial molecular executor of XBP1 and p53-mediated tumor suppression.
Figure 6: Inferred multi-omic network based on TCGA colorectal cancer subset using KiMONo algorithm.
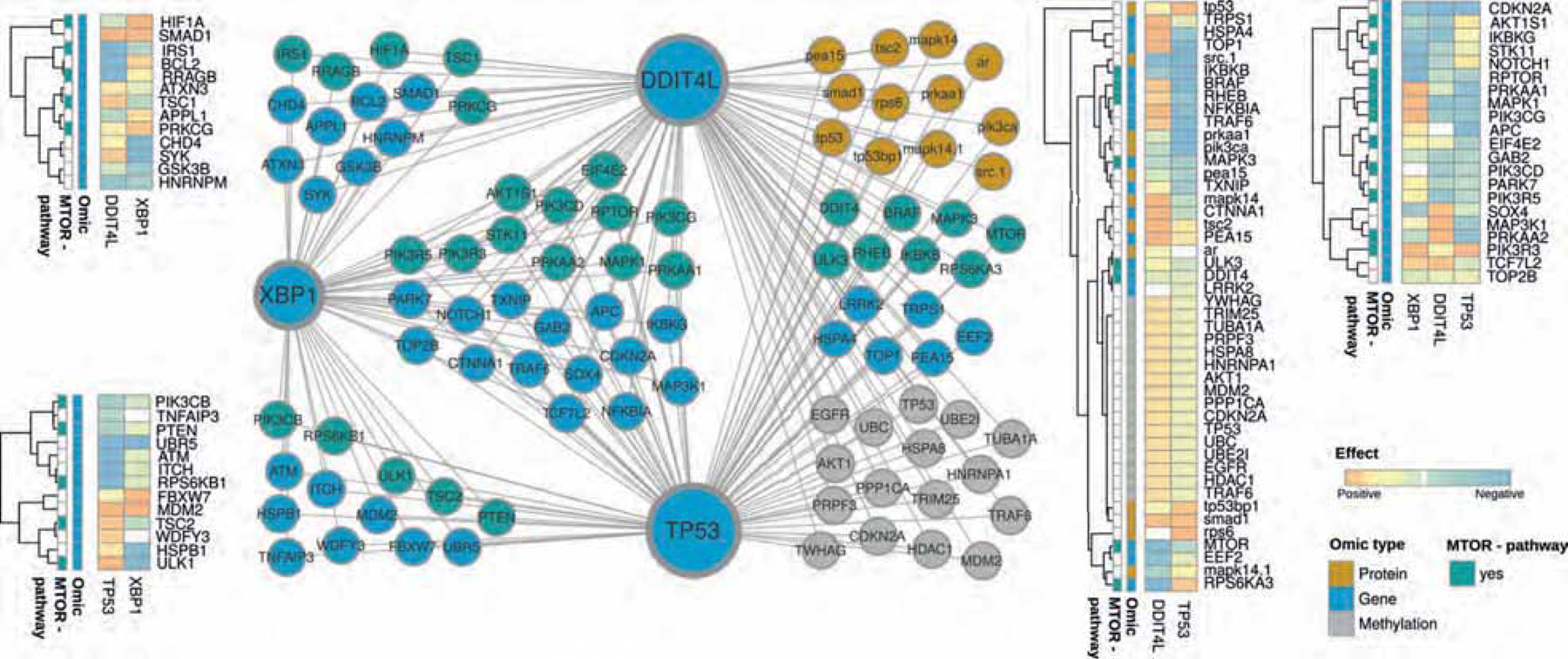
Within the network, nodes represent methylation sites (grey), proteins (orange) and genes (blue & turquoise). Connections between the nodes denote statistical identified effects obtained via KiMONo. The heatmaps on the side visualize the positive (orange) and negative (blue) effect strength between an omic feature, XBP1, TP53 and DDIT4L.
Extrinsic mTOR inhibition reduces p-4E-BP1 levels and impedes hyperproliferation in H2b/Xbp1∆IEC mice
Based on these findings, we hypothesized that inhibition of mTOR would reconstitute suppression of intestinal epithelial proliferation in H2b/Xbp1∆IEC mice, thereby providing an actionable therapeutic intervention. Thus, we interrogated the growth properties of intestinal organoids from all four genotypes (H2b/Xbp1fl/fl, Xbp1∆IEC, H2b∆IEC and H2b/Xbp1∆IEC) using CellTiter-Glo assay, which assesses the numbers of viable cells by quantifying ATP. We first validated that despite ongoing DNA damage, H2b/Xbp1∆IEC intestinal organoids showed significantly enriched cell viability, compared to H2b∆IEC intestinal organoids (Supplementary Figure 10). We further assessed the suppressive capacity of mTOR treatment in the same experimental setting and observed that Rapamycin most strongly inhibited the growth rate of H2b/Xbp1∆IECintestinal organoids (Figure 7A,B). These findings were independently validated in H2b∆IEC and H2b/Xbp1∆IEC intestinal organoids using colony forming assays (Figure 7C). Western Blot analysis of intestinal organoids confirmed reduction of p-4E-BP1 in H2b∆IEC and H2b/Xbp1∆IEC intestinal organoids upon Rapamycin treatment (Figure 7D). To test whether mTOR inhibition might serve as a preventive mechanism to counterbalance proliferation in DNA damaged IEC, we intraperitoneally treated age- and gender-matched H2b∆IEC and H2b/Xbp1∆IEC mice (n = 3 – 4 / group) with either Rapamycin or DMSO on a daily basis for 7 days. Rapamycin treatment significantly blocked epithelial hyperproliferation (Figure 7E,F), 4E-BP1 phosphorylation (Figure 7G,H) and cell death (Figure 7I,J) in H2b/Xbp1∆IEC mice. Taken together, these data show that XBP1 restricts cellular proliferation in response to DNA damage in IEC in a mechanism involving the DDIT4l / mTOR pathway.
Figure 7: Rapamycin abolishes DDIT4L-mediated hyperproliferation by inhibiting mTOR-dependent phosphorylation of 4E-BP1.
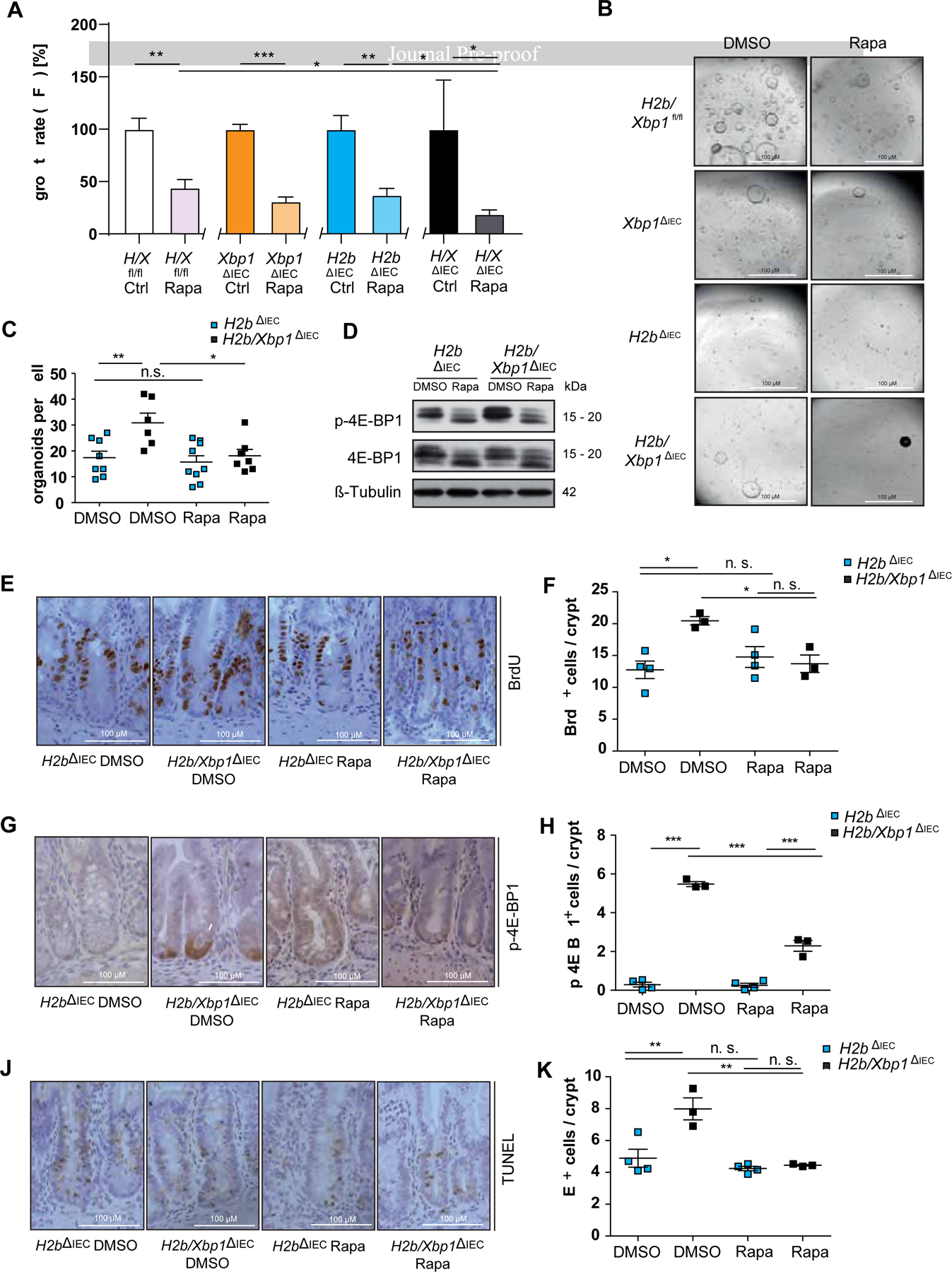
(A) CellTiter-Glo assay and representative images (B) of small intestinal organoids stimulated with either DMSO (control) or 1 μM Rapamycin for 72 hours. Representative data of n = 3 individual experiments with n = 3 technical replicates. H2b/Xbp1fl/fl/ΔIEC organoids are abbreviated as “H/Xfl/fl/ΔIEC”. (C) colony formation assay of H2bΔIEC and H2b/Xbp1ΔIEC intestinal organoids, treated with DMSO or 1 μM Rapamycin every other day over the time course of 10 days, representative of n = 3 experiments with n = 3 technical replicates. (D) Western blot of H2bΔIEC and H2b/Xbp1ΔIEC intestinal organoids, treated with DMSO or 1 μM Rapamycin for 24 hours, representative of n = 3 experiments. β-Tubulin served as a loading control. Data are expressed as mean ± standard error of the mean and significance was determined using an unpaired Student’s t-test. Age-matched H2b∆IEC (n = 8, 5 females, 3 males) and H2b/Xbp1∆IEC (n = 6, 2 females, 4 males) mice received either DMSO or 1.5 mg / kg body weight Rapamycin i.p. daily for 7 days. Representative images of the SI of DMSO- vs. Rapamycin-treated H2b∆IEC and H2b/Xbp1∆IEC mice for (E) BrdU, (G) p-4E-BP1 and (I) TUNEL and accompanying statistical analysis (F) BrdU+, (H), p-4E-BP1+ and (J) TUNEL+ cells. Data are expressed as mean ± standard error of the mean and significance was determined using one-way ANOVA. * p < .05, ** p < .01, *** p < .0001.
Discussion
We demonstrate that XBP1 coordinates epithelial DDR in IEC and protects from tumorigenesis in a genetic model of chronic epithelial DNA damage. Clinical data from CRC patients support a protective role of XBP1 on overall survival (OS). Using pathway enrichment analysis, we show that low XBP1 activation is significantly associated with decreased pathway activation of the tumor suppressor p53. It must however be noted that reliable assessment of XBP1 protein is challenging in global proteome dataset and therefore this finding still warrants further validation on the protein level. Associations between XBP1 and carcinogenesis have been described in clinical datasets, albeit with contradictory effects on the clinical outcome. XBP1 function has been shown to be associated with poor OS in triple negative breast cancer and ovarian cancer31. In contrast, we here confirm and functionally delineate an epithelial-intrinsic tumor suppressive function of XBP1 in intestinal tumorigenesis. The presented findings are in line with previous data published on XBP1 and colorectal carcinogenesis, in which chemical models (AOM-DSS) and genetic models (ApcMin/+) were employed to show that epithelial Xbp1 deletion led to intestinal hyperproliferation and subsequent increased adenoma formation. However, the exact molecular mechanism on how disrupted XBP1 transforms epithelial cells into a hyperproliferative state in response to genotoxic stress remained poorly understood2. Our data now support that XBP1 crucially supports p53 activity to direct stem cell suppression as a result to DNA damage.
Xbp1 deletion in the intestinal epithelium of mice induces a mild enteritis that turns into a fissuring, transmural inflammatory bowel disease (IBD)-like ileitis when the autophagy gene Atg16l1 is additionally deleted32. This inflammation instigates a highly regenerative process with hyperproliferative characteristics, in which functioning DNA repair and p53-mediated DNA damage responses are indispensable. It is therefore not surprising that TP53 mutations commonly occur in chronically inflamed colonic epithelia of IBD patients33–35. These initial TP53 mutations in non-neoplastic mucosae or precancerous neoplasms lead to uncontrolled cell proliferation and to a concomitant accumulation of further mutations in oncogenes and tumor suppressor genes, i.e. KRAS and APC, and may hence initiate a stepwise progression to CRC33. Interestingly, this so-called colitis-associated CRC (CAC) pathway follows a reverse mutation sequence (TP53-KRAS-APC) compared to the classical CRC route that follows the Vogelstein model (APC-KRAS-TP53)36,37. Of note, TP53 mutations are rare in the adenomatous precursors of the classical CRC pathway, whereas mutations in KRAS and APC are less prevalent in CAC38. Using the model of epithelial Rnaseh2b deficiency, we now show that H2b/Xbp1∆IEC mice present with aggressive, invasive and metastatic adenocarcinoma. Notably, we did not find a strong indication for a modulating role of the intestinal microbiome in driving the carcinogenic phenotype, as tumor incidence was comparable between single and co-housed (H2b/Xbp1∆IECvs.H2b/Xbp1fl/fl) mice.
It must be noted that 96 % of tumors arise in the small intestine and therefore conclusions on the pathogenesis of human CRC should be made with caution. Still, it needs to be pointed out that species-specific differences between mouse and human are also found e.g. in the ApcMin/+ model39, in which mice predominantly display small intestinal tumors. In line with this finding, our model shows that Xbp1 deficiency confers a site-specific vulnerability to small intestinal IEC resulting from the misled interplay of DNA damage and Xbp1 deletion, which ultimately manifests into spontaneous carcinogenesis. We further observe that orthotopically transplanted intestinal organoids from H2b/Xbp1∆IEC -derived tumors result in 100 % tumor manifestation at organoid injection site and in 66 % (liver), 100 % (lung) respective metastasis formation. These findings point to a highly aggressive tumor entity in form of metastasis formation, which has not been described in previous orthotopic tumor transplantation models yet26,27. Altogether, these data therefore provide the first in-vivo prove that impairment of ribonucleotide excision repair (RER) may indeed result into invasive and metastatic adenocarcinoma in-vivo. This finding could have potential translational impact with regard to molecular guided therapy in colorectal carcinoma, as it has been recently shown that the genome of cells with deficient RER display increased abundance of PARP-trapping lesions, which can be specifically targeted using PARP-inhibitors40.
In an attempt to understand the underlying molecular mechanism, we identified Ddit4l as a p53-dependent regulator of stem cell suppression. Importantly, DDIT4L is the paralog of DDIT4, also known as REDD1, which among other functions acts as a p53-dependent DNA damage repair gene41 and has previously been validated as prognostic marker in several malignancies42. Although our data do not directly imply a role of DDIT4L in DNA damage repair, it is conceivable that DDIT4L directs DNA damage repair modalities via mTOR, which has previously been shown e.g. in the context of CRC43 and pancreatic cancer44.
Taken together, our findings reveal a novel role of the intestinal epithelial XBP1 in coordinating epithelial DDR downstream of p53. Xbp1 deletion disrupts a fundamental feedback-mechanism which restricts epithelial proliferation via DDIT4L and 4E-BP1 and which might provide an actionable layer of cancer prevention and treatment in a subset of patients with insufficient p53 activity.
Supplementary Material
What you need to know:
BACKGROUND AND CONTEXT:
The IBD risk gene XBP1 essentially coordinates epithelial cell function and thereby contributes to intestinal mucosal homeostasis. RNaseH2 facilitates ribonucleotide excision repair and loss of epithelial RNASEH2B leads to the development of DNA damage. Whether XBP1 is involved in intestinal epithelial DNA damage responses (DDR) is not yet known.
NEW FINDINGS:
Simultaneous deficiency of epithelial Xbp1 and Rnaseh2b leads to the generation of aggressive metastatic intestinal adenocarcinoma in mice. Mechanistically, Xbp1 restrains intestinal stem cell proliferation and consecutive carcinogenesis via p53-Ddit41-mediated mTOR inhibition.
LIMITATIONS:
The exact molecular mechanism of how XBP1 licences DNA damage responses needs to be elucidated.
IMPACT:
This study puts forward a novel concept in which chronic intestinal ER-Stress and DNA damage converge into a layer susceptibility for intestinal carcinogenesis.
Grant support
This work was supported by the DFG Research Training Group 1743 (P.R.), the BMBF iTREAT project (P.R.), the DFG Cluster of excellence “Precision medicine in chronic inflammation” RTF III and TI-1, the DFG CRC 1371 P11 (Project-ID 395357507; M.T.), the EKFS research grant #2019_A09 (K.A.), the Wilhelm Sander-Stiftung #2019.046.1 (K.A.), the BMBF (eMED Juniorverbund “Try-IBD” 01ZX1915A), the DFG RU5042 (P.R., K.A.), the National Institutes of Health (grants DK044319, DK051362, DK053056 and DK088199; R.S.B.), DFG CRC877 A13 and Z2 (Project-ID 125440785; P.A.), the Austrian Academy of Sciences (C.P.), the European Research Council (grant 786295; Z.T.), and grant to the Harvard Digestive Diseases Center DK034854 (R.S.B). Sequencing was performed with help of the DFG CCGA sequencing center at CAU Kiel.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
The authors have no conflict of interest.
References:
- 1.Kaser A, Lee A-H, Franke A, et al. XBP1 Links ER Stress to Intestinal Inflammation and Confers Genetic Risk for Human Inflammatory Bowel Disease. Cell 2008;134:743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niederreiter L, Fritz TMJ, Adolph TE, et al. ER stress transcription factor Xbp1 suppresses intestinal tumorigenesis and directs intestinal stem cells. J Exp Med 2013;210:2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmitt M, Greten FR. The inflammatory pathogenesis of colorectal cancer. Nat Rev Immunol 2021. [DOI] [PubMed] [Google Scholar]
- 4.Rad R, Cadiñanos J, Rad L, et al. A genetic progression model of Braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell 2013;24:15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennecke M, Kriegl L, Bajbouj M, et al. Ink4a/Arf and Oncogene-Induced Senescence Prevent Tumor Progression during Alternative Colorectal Tumorigenesis. Cancer Cell 2010;18:135–146. [DOI] [PubMed] [Google Scholar]
- 6.Reijns MAM, Rabe B, Rigby RE, et al. Enzymatic Removal of Ribonucleotides from DNA Is Essential for Mammalian Genome Integrity and Development. Cell 2012;149:1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aden K, Bartsch K, Dahl J, et al. Epithelial RNase H2 Maintains Genome Integrity and Prevents Intestinal Tumorigenesis in Mice. Gastroenterology 2019;156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Argemí J, Kress TR, Chang HCY, et al. X-box Binding Protein 1 Regulates Unfolded Protein, Acute-Phase, and DNA Damage Responses During Regeneration of Mouse Liver. Gastroenterology 2017;152:1203–1216.e15. [DOI] [PubMed] [Google Scholar]
- 9.Yamamori T, Meike S, Nagane M, et al. ER stress suppresses DNA double-strand break repair and sensitizes tumor cells to ionizing radiation by stimulating proteasomal degradation of Rad51. FEBS Lett 2013;587:3348–3353. [DOI] [PubMed] [Google Scholar]
- 10.Tao R, Chen H, Gao C, et al. Xbp1-mediated histone H4 deacetylation contributes to DNA double-strand break repair in yeast. Cell Res 2011;21:1619–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dufey E, Bravo-San Pedro JM, Eggers C, et al. Genotoxic stress triggers the activation of IRE1α-dependent RNA decay to modulate the DNA damage response. Nat Commun 2020;11:2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hiller B, Achleitner M, Glage S, et al. Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J Exp Med 2012;209:1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aden K, Tran F, Ito G, et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J Exp Med 2018;215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roper J, Tammela T, Akkad A, et al. Colonoscopy-based colorectal cancer modeling in mice with CRISPR–Cas9 genome editing and organoid transplantation. Nat Protoc 2018;13:217–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal; Vol 17, No 1 Next Gener Seq Data Anal 2011. [Google Scholar]
- 16.Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012;7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 2014;31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breuer K, Foroushani AK, Laird MR, et al. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res 2013;41:D1228–D1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muzny DM, Bainbridge MN, Chang K, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogris C, Hu Y, Arloth J, et al. Versatile knowledge guided network inference method for prioritizing key regulatory factors in multi-omics data. Sci Rep 2021;11:6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schubert M, Klinger B, Klünemann M, et al. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat Commun 2018;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grant S Ara-C: cellular and molecular pharmacology. Adv Cancer Res 1998;72:197–233. [DOI] [PubMed] [Google Scholar]
- 24.Wang Q, Zou Y, Nowotschin S, et al. The p53 Family Coordinates Wnt and Nodal Inputs in Mesendodermal Differentiation of Embryonic Stem Cells. Cell Stem Cell 2017;20:70–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwak B, Kim DU, Kim TO, et al. MicroRNA-552 links Wnt signaling to p53 tumor suppressor in colorectal cancer. Int J Oncol 2018;53:1800–1808. [DOI] [PubMed] [Google Scholar]
- 26.Roper J, Tammela T, Cetinbas NM, et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat Biotechnol 2017;35:569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lannagan TRM, Lee YK, Wang T, et al. Genetic editing of colonic organoids provides a molecularly distinct and orthotopic preclinical model of serrated carcinogenesis. Gut 2019;68:684 LP–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tschurtschenthaler M, Adolph TE, Ashcroft JW, et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease–like ileitis. J Exp Med 2017;214:401–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simonson B, Subramanya V, Chan MC, et al. DDiT4L promotes autophagy and inhibits pathological cardiac hypertrophy in response to stress. Sci Signal 2017;10:eaaf5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corradetti MN, Inoki K, Guan K-L. The Stress-inducted Proteins RTP801 and RTP801L Are Negative Regulators of the Mammalian Target of Rapamycin Pathway. J Biol Chem 2005;280:9769–9772. [DOI] [PubMed] [Google Scholar]
- 31.Chen X, Iliopoulos D, Zhang Q, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014;508:103–107. A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adolph TE, Tomczak MF, Niederreiter L, et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013;503:272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baker A-M, Cross W, Curtius K, et al. Evolutionary history of human colitis-associated colorectal cancer. Gut 2019;68:985 LP–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brentnall TA, Crispin DA, Rabinovitch PS, et al. Mutations in the p53 gene: An early marker of neoplastic progression in ulcerative colitis. Gastroenterology 1994;107:369–378. [DOI] [PubMed] [Google Scholar]
- 35.Yin J, Harpaz N, Tong Y, et al. p53 Point mutations in dysplastic and cancerous ulcerative colitis lesions. Gastroenterology 1993;104:1633–1639. [DOI] [PubMed] [Google Scholar]
- 36.Itzkowitz SH, Yio X. Inflammation and Cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Liver Physiol 2004;287:G7–G17. [DOI] [PubMed] [Google Scholar]
- 37.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990;61:759–767. [DOI] [PubMed] [Google Scholar]
- 38.Hao XP, Frayling IM, Sgouros JG, et al. The spectrum of p53 mutations in colorectal adenomas differs from that in colorectal carcinomas. Gut 2002;50:834 LP–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su LK, Kinzler KW, Vogelstein B, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science (80- ) 1992;256:668 LP–670. [DOI] [PubMed] [Google Scholar]
- 40.Zimmermann M, Murina O, Reijns MAM, et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 2018;559:285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janic A, Valente LJ, Wakefield MJ, et al. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat Med 2018;24:947–953. [DOI] [PubMed] [Google Scholar]
- 42.Pinto JA, Rolfo C, Raez LE, et al. In silico evaluation of DNA Damage Inducible Transcript 4 gene (DDIT4) as prognostic biomarker in several malignancies. Sci Rep 2017;7:1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsieh H-J, Zhang W, Lin S-H, et al. Systems biology approach reveals a link between mTORC1 and G2/M DNA damage checkpoint recovery. Nat Commun 2018;9:3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Müller D, Shin S, Goullet de Rugy T, et al. eIF4A inhibition circumvents uncontrolled DNA replication mediated by 4E-BP1 loss in pancreatic cancer. JCI insight 2019;4:e121951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.