

Abstract
PURPOSE
The poly(ADP-ribose) polymerase (PARP) inhibitor rucaparib is approved in the US for patients with metastatic castration-resistant prostate cancer (mCRPC) and a deleterious germline and/or somatic BRCA1 or BRCA2 (BRCA) alteration. While sequencing of tumor tissue is considered the standard for identifying patients with BRCA alterations (BRCA+), plasma profiling may provide a minimally invasive option to select patients for rucaparib treatment. Here, we report clinical efficacy in BRCA+ mCRPC patients identified through central plasma, central tissue, or local genomic testing and enrolled in TRITON2.
EXPERIMENTAL DESIGN
Patients had progressed after next-generation androgen receptor–directed and taxane-based therapies for mCRPC and had BRCA alterations identified by central sequencing of plasma and/or tissue samples or local genomic testing. Concordance of plasma/tissue BRCA status and objective response rate and prostate-specific antigen (PSA) response rates were summarized.
RESULTS
TRITON2 enrolled 115 BRCA+ patients identified by central plasma (n = 34), central tissue (n = 37), or local (n = 44) testing. Plasma/tissue concordance was determined in 38 patients with paired samples and was 47% in 19 patients with a somatic BRCA alteration. No statistically significant differences were observed between objective and PSA response rates to rucaparib across the three assay groups. Patients unable to provide tissue samples and tested solely by plasma assay responded at rates no different to patients identified as BRCA+ by tissue testing.
CONCLUSION
Plasma, tissue, and local testing of mCRPC patients can be used to identify men with BRCA+ mCRPC who can benefit from treatment with the PARP inhibitor rucaparib.
Keywords: BRCA, metastatic castration-resistant prostate cancer, rucaparib, PARP inhibitor, next-generation sequencing
INTRODUCTION
The poly (ADP-ribose) polymerase (PARP) inhibitor rucaparib is approved in the United States for the treatment of patients with metastatic castration-resistant prostate cancer (mCRPC) and a deleterious BRCA1 or BRCA2 (BRCA) alteration, based on results from the phase 2 TRITON2 study (1).
Germline or somatic alterations in BRCA are observed in approximately 12% of men with advanced prostate cancer (2,3). Protein truncating alterations, such as homozygous deletions, large rearrangements, as well as frameshift, nonsense, and pathogenic missense mutations, have been identified as drivers of the disease (3,4), highlighting the need for robust approaches to select patients for treatment.
Sequencing of tumor tissue is the standard for genomic subtyping of solid cancers but has limitations in prostate cancer. While tissue samples from the prostate tumor are taken from almost all patients at diagnosis, the time to progression to mCRPC can span many years (5,6), which can impact sample quality and subsequent success of molecular profiling (7). Furthermore, tumor genomics may evolve with disease progression or development of treatment resistance (7,8), and diagnostic biopsy or radical prostatectomy samples may not reflect the genomics of advanced disease. Although DNA repair status can be accurately assessed from primary tumors in most cases (9,10), more recent and metastatic biopsies are preferred for higher NGS success rate (3); however, mCRPC largely involves skeletal spread, and many patients lack accessible soft tissue lesions (11). Biopsies from bone metastases frequently have insufficient tissue content for next-generation sequencing (NGS), and have high failure rates (3,12–16).
Blood-based assays have become a viable option for guiding treatment in tumors where tissue biopsies are difficult to obtain, such as non–small-cell lung cancer (17–19). As cancer and normal cells undergo apoptosis or necrosis, they shed cell-free DNA (cfDNA) into the bloodstream which can be isolated from peripheral blood (20–22). Patients with mCRPC are frequently found to have sufficiently high fractions of tumor-derived cfDNA (23,24), containing the DNA from primary and metastatic lesions (21) for tumor profiling through NGS. High fractions of circulating tumor DNA (ctDNA) have been reported to be associated with poor prognosis in mCRPC (25,26). Sequencing of cfDNA has shown high concordance with tissue sequencing of metastatic biopsies in several tumor types, including mCRPC (27), and is suitable to help guide patient treatment. In contrast to tissue biopsies, the collection of plasma is minimally invasive for patients, ubiquitously accessible and easily repeatable over the course of the disease.
Studies have demonstrated high concordance between BRCA alterations in the plasma and tissue of mCRPC patients (3,28), but no comparative analyses of clinical efficacy in BRCA+ patients prospectively identified through plasma or tissue testing have been reported, and skepticism towards the clinical applicability of targeted treatment in patients selected through plasma testing remains (2). Of concern are potential false-positive results from the cfDNA assay due to technical (2) or biological factors, such as low variant allele frequency (AF) and clonal hematopoiesis of indeterminate potential (CHIP) (29). Although very rarely seen in BRCA, recent analyses found CHIP mutations in non-BRCA DNA damage repair (DDR) genes frequently reported as tumor originating alterations during plasma testing of patients with advanced prostate cancer when sequencing of a matched normal sample is not performed (30). In patients with low tumor burden or less aggressive disease, somatic mutations may be missed due to insufficient amounts of ctDNA, calling into question the suitability of plasma assays to select patients for PARP inhibitor therapy (31,32). Here, we compare clinical responses to rucaparib in patients with a deleterious BRCA alteration identified by plasma, tissue, or local testing and evaluate the concordance between the selection methodologies.
METHODS
Study Description
TRITON2 (NCT02952534) is an ongoing, international, open-label, phase 2 study evaluating rucaparib in patients with mCRPC associated with DDR deficiency. Men aged ≥18 years with histologically or cytologically confirmed mCRPC, Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, and adequate organ function were enrolled. Eligible patients had a deleterious germline or somatic alteration in BRCA1, BRCA2, or 1 of 13 other DDR genes (ATM, BARD1, BRIP1, CDK12, CHEK2, FANCA, NBN, PALB2, RAD51, RAD51B, RAD51C, RAD51D, RAD54L) and disease progression following 1 to 2 lines of next-generation AR-directed therapy for prostate cancer and 1 prior taxane-based chemotherapy for castration-resistant disease. Patients treated with a PARP inhibitor, mitoxantrone, cyclophosphamide, or platinum-based chemotherapy, or with an active secondary malignancy were excluded. Patients were enrolled irrespective of measurable disease status.
Patients received a starting dose of 600 mg oral rucaparib twice daily. Dose reductions, in decrements of 100 mg, were permitted in case of grade ≥3 or persistent grade 2 treatment-emergent adverse events.
The study was approved by national or local institutional review boards and performed in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. Patients provided written informed consent before participation.
The primary endpoint was ORR by blinded independent radiology review (IRR) per modified Response Evaluation Criteria In Solid Tumors version 1.1 (RECIST) and Prostate Cancer Clinical Trials Working Group 3 (PCWG3) criteria in patients with measurable disease by IRR. Confirmed locally assessed prostate-specific antigen (PSA) response (≥50% decrease from baseline confirmed by a second measurement ≥3 weeks later) in all patients was a secondary endpoint. Duration of response (DOR) was defined as the time from the date of the first confirmed response to the date progression was first documented plus 1 day, and summarized using Kaplan-Meier methodology.
Efficacy analyses included BRCA+ patients enrolled by the cut-off date of 8 May 2019. The visit cutoff date was 23 December 2019; therefore, patients had a minimum of 16 weeks of follow-up, allowing for at least two radiographic tumor assessments and confirmation of response.
Patients were enrolled by three assay groups used to detect deleterious BRCA alterations: central tissue or central plasma testing or by documented evidence of a deleterious alteration from an existing test result performed locally. Patients were encouraged to submit both plasma and tissue samples for central testing. An additional blood sample for central germline testing was collected.
Central Assays
The central tissue assay was the FoundationOne® laboratory developed test (F1 LDT) for NGS of 395 cancer-related genes (including BRCA) from formalin-fixed paraffin-embedded tissue (33). The central plasma assay was the FoundationOne®Liquid laboratory developed test (F1L LDT) using circulating cfDNA isolated from plasma to sequence 70 cancer related genes (including BRCA) (20). Both central NGS assays detected germline and somatic alterations but did not distinguish between them. The Color Hereditary Cancer Test (34) was used to determine the germline status of BRCA alterations.
All assays were executed in Clinical Laboratory Improvement Amendments (CLIA)-certified and College of American Pathologists (CAP)-accredited laboratories.
RESULTS
Overview of TRITON2 Patients and Genomic Data
By the cutoff date, the TRITON2 study had enrolled 115 patients with a BRCA alteration. Clinical analyses of safety and efficacy in BRCA+ patients have been published (1).
BRCA+ patients were enrolled based on deleterious alterations in BRCA identified by central plasma (n = 34) or central tissue (n = 37) testing or by documented evidence of a deleterious alteration from local testing (n = 44; Fig 1). Patients were encouraged to submit plasma and tissue (contemporaneous or archival) samples for central testing. Several patients were BRCA+ by both sample types. In those cases, the test result used for enrollment was the result that became available first. Due to this approach, most patients had 2 or more test results: 61 patients had a BRCA+ plasma result, 59 a BRCA+ tissue result, and 38 had both. Results from central germline testing were available for all patients. An overview of BRCA+ patients and test data is shown in Figure 1.
FIG 1.
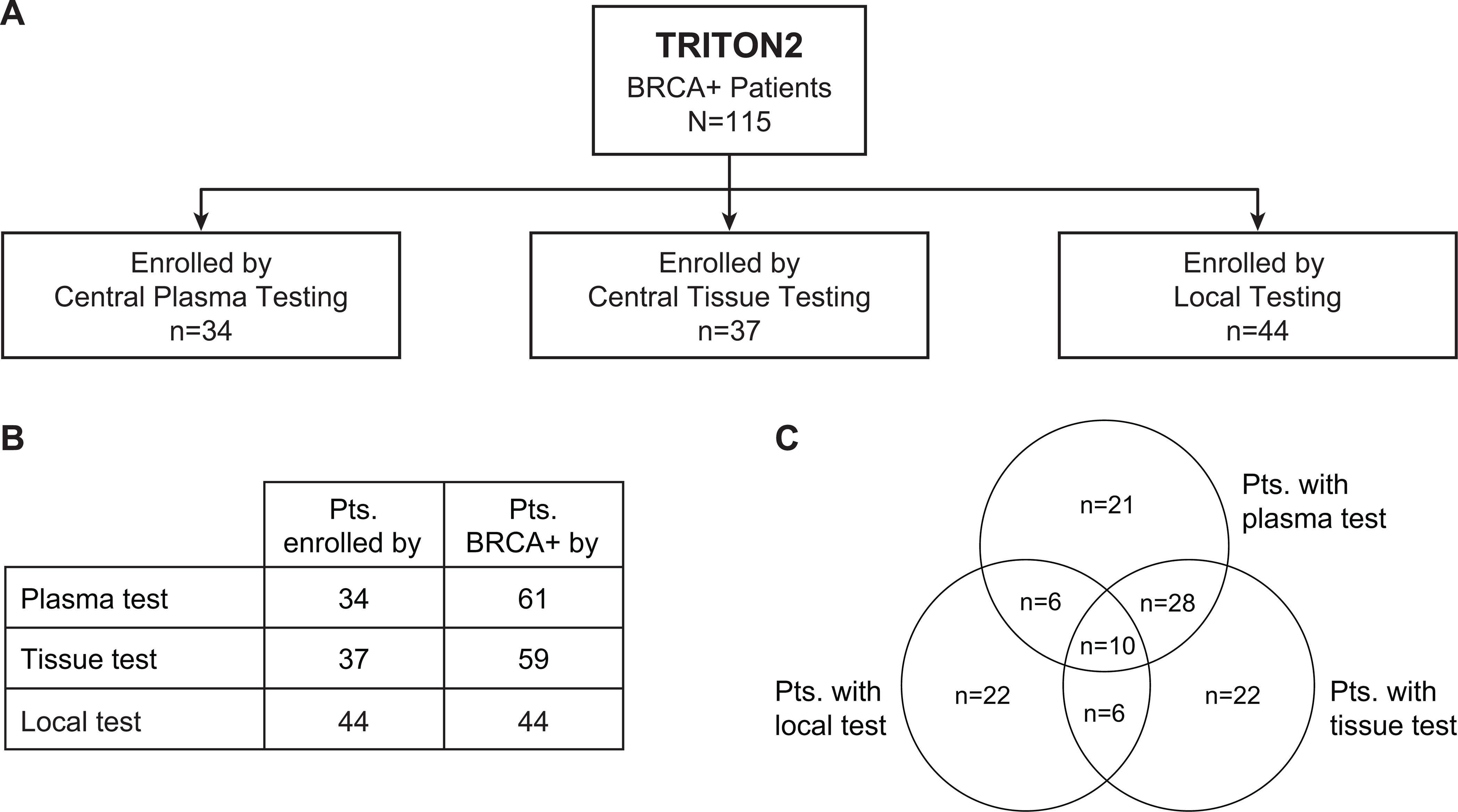
Summary of genomic testing data for patients with BRCA alterations (BRCA+) by enrollment assay type. For every BRCA+ patient enrolled in TRITON2 (n=115), 1 genomic test de novo identified an eligible deleterious BRCA1/2 alteration and was used to enroll the patient. Patients were enrolled based on central plasma testing (n=34), central tissue testing (n=34), or existing local test data (n=44) (A). Due to a comprehensive effort to obtain both, plasma and tissue samples from patients for central testing, many patients had data from multiple test types available, resulting in greater numbers of patients testing BRCA+ by plasma and tissue test, compared with the number of patients who were enrolled by each assay (B). The Venn diagram depicts the number of patients with BRCA alteration results available from 1, 2, or all 3 assay groups (C).
Enrollment by Assay Group
BRCA alterations identified by central tissue or local test results included all alteration types (Fig 2). The plasma assay was not validated to detect homozygous BRCA deletions, and no patients with this alteration type were identified by the plasma assay.
FIG 2.

Germline and somatic BRCA1/2 mutation rate and frequency of deleterious BRCA1/2 alteration types identified by central plasma testing (A), central tissue testing (B), or a variety of existing data from local genomic testing (C).
Central plasma testing was used to enroll 34 of 115 (30%) BRCA+ patients, of whom 35% (12/34) had a germline and 65% (22/34) had a somatic alteration. The most common alteration types were frameshift (62%, 21/34) and nonsense (15%, 5/34) mutations. A subset of 21 patients enrolled by the plasma assay did not have a tissue sample available and was tested solely with the central plasma test.
Central tissue testing was used to enroll 37 of 115 (32%) BRCA+ patients, among them 43% (16/37) with a germline and 57% (21/37) with a somatic alteration. Homozygous BRCA deletions were detected in 41% (15/37) of patients’ tissues and frameshift mutations in 30% (11/37).
A variety of local tests were used to enroll 44 (38%) BRCA+ patients, most commonly by NGS-based gene panels (80% [35/44] patients) or germline tests (20% [9/44] patients). Of the local assays, 70% (31/44) were based on tissue profiling. Most prevalent were the FoundationOne® (33) (18% [8/44] patients) and MSK-IMPACT (35) (16% [7/44] patients) tests. Plasma-based cfDNA assays were utilized for 9% (4/44) of patients enrolled by local testing; FoundationOne Liquid (20) was used most frequently (5% [2/44] patients). Blood-based germline tests were used for 20% (9/44) of the patients enrolled by local testing, with tests from Myriad Genetics (36) (5%, 2/44) and Ambry Genetics (37) (5%, 2/44) being the most common. Local testing identified a wide range of BRCA alterations: 43% (19/44) were germline and 57% (25/44) were somatic. Patients most commonly had frameshift alterations (55%, 24/44) or homozygous BRCA loss (14%, 6/44).
Overall, of 115 BRCA+ patients in TRITON2, 13 (11%) had a BRCA1 and 102 (89%) had a BRCA2 alteration. BRCA alterations were germline in 41% (47/115) of patients and somatic in 59% (68/115).
No confounding differences were observed in baseline demographics or prognostic factors in patients across the three enrollment assay groups.
Concordance of BRCA Status by Plasma or Tissue Testing
Both plasma and tissue data were available for 38 BRCA+ patients and used to evaluate concordance of BRCA status between the assays. Tissue samples were mostly archival prostate tumor samples (84%; 32/38) with a median age of 3.5 years (range, 25 days to 19.1 years). Six (16%) patients submitted metastatic samples, of which 5 were taken within 1 year of NGS testing (range, 2.2 to 9.6 months). Plasma samples were collected prior to dosing on the first day of treatment.
In the analysis of concordance between the 2 assay types, the BRCA alterations used for enrollment were found in both tissue and plasma samples among 74% (28/38) of patients.
BRCA alterations in these patients were 50% (19/38) of germline and 50% (19/38) of somatic origin. All 19 germline alterations were detected in both tissue and plasma.
Concordant BRCA status was observed in 47% (9/19) of patients with somatic BRCA alterations. Only 1 patient in this subset had a homozygous deletion, suggesting that the discordance was predominantly due to other biological and technical challenges. For example, in 4 patients a BRCA alteration was detected in the archival tissue but not the plasma. The maximum somatic AF observed in these plasma samples was below 3%. One plasma sample was void of any alterations entirely, indicating no or very low levels of tumor DNA. One of the 4 patients in this group (25%; 95% CI, 0.6 to 80.6) had a PSA response. Another patient had measurable disease but neither a PSA nor a radiographic response.
In the reverse analysis, 6 patients were BRCA+ by plasma but BRCA-by tissue testing. All 6 tissue samples were archival prostate samples with a median age of 2.6 years (range, 11 months to 11.5 years), and half (3/6) had low tumor purity (<25%). The majority (67%, 4/6) of BRCA alterations in the matched plasma samples were of very low (<1%) AF, suggesting subclonality. None of these 6 patients had a PSA response, and neither of the 2 patients with measurable disease had a radiographic response.
Efficacy in BRCA+ Patients by Enrollment Assay Group
Patient level genomics and responses from 113 PSA response-evaluable patients were grouped by enrollment assay and detailed in Figure 3. Two patients did not meet the follow-up criteria required to be response evaluable and were excluded from the analyses.
FIG 3.

Best percent change in prostate-specific antigen (PSA) from baseline for BRCA+ TRITON2 patients, grouped by the assay type that was used for patient enrollment. Bars were capped at 100% for visual clarity. PSA increases for the 3 leftmost patients enrolled by central plasma test were 689%, 231%, and 183%; PSA increase for the leftmost patient enrolled by local test was 133%. Additional assay, outcome, and genomics data, including BRCA1 or BRCA2 (BRCA) status from additional test results, are indicated in the tile plot. PSA response status is shown for all patients, and Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST) response status is shown for those patients who had measurable disease at baseline. The genomic data for each patient are broken into BRCA gene, germline status, alteration type, and zygosity where available. CI, confidence interval; ORR, objective response rate.
Patients enrolled by the central tissue assay had a confirmed PSA response rate of 62.2% (95% CI, 44.8 to 77.5; 23/37; Fig 4), where 15 patients with homozygous BRCA loss identified by tissue testing had higher PSA response rates (80.0%; 95% CI, 51.9 to 95.7; 12/15), compared with patients with other BRCA alteration types (50.0%; 95% CI, 28.2 to 71.8; 11/22). The median duration of PSA response was 9.0 months (range, 1.0 to 25.8+) in all patients enrolled by tissue, compared with 2.2 months (range, 1.0 to 9.0) in patients without homozygous BRCA loss and not yet reached (range, 2.7 to 25.8+ months) in those with homozygous BRCA loss.
FIG 4.

Confirmed objective radiographic (by independent radiological review [IRR]) (A) and prostate-specific antigen (PSA) response rates (B) in patients with germline or somatic BRCA alterations and in subgroups of patients identified as having BRCA alterations (BRCA+) by the 3 different assay groups used during genomic screening. RECIST, Response Evaluation Criteria in Solid Tumors version 1.1.
Patients enrolled by the central plasma assay had a response rate of 41.2% (95% CI, 24.6 to 59.3; 14/34), and a median duration of PSA response of 6.3 months (range, 3.7 to 11.7+). The response rate in patients enrolled by local assays was 59.1% (95% CI, 43.2 to 73.7; 26/44) with a median duration of PSA response of 4.6 months (range, 1.4 to 29.4+). The 95% CIs overlapped in response rates across all 3 groups. PSA response rates in all patients with germline BRCA alterations were 61.7% (95% CI, 46.4 to 75.5; 29/47) and 50.0% in patients with somatic alterations (95% CI, 37.6 to 62.4; 34/68).
Confirmed ORR by enrollment assay was evaluated in 62 patients with measurable disease: patients enrolled by central tissue testing had an ORR of 55.0% (95% CI, 31.5 to 76.9; 11/20), compared to 31.3% (95% CI, 11.0 to 58.7; 5/16) in patients enrolled by central plasma test and 42.3% (95% CI, 23.4 to 63.1; 11/26) in patients enrolled by local test, while the 95% CIs overlapped across all 3 groups (Fig 4). Patients enrolled by the tissue test and with homozygous BRCA loss had an ORR of 70.0% (95% CI, 34.8 to 93.3; 7/10), compared with 40.0% (95% CI, 12.2 to 73.8; 4/10) in those without homozygous loss. Duration of response was not yet reached (range, 2.3 to 23.0+ months) in patients enrolled by tissue testing, including patients with homozygous BRCA loss (median not yet reached; range, 4.0 to 23.0+ months) and without (6.8 months; range, 2.3 to 12.0+), compared with 6.6 months (range, 4.0 to 7.7+) in patients enrolled by plasma testing and not yet reached (range, 1.7 to 24.0+ months) in patients enrolled based on an existing local test result.
Reduction in sum of target lesions is shown in Figure S1. The ORR was 45.8% in patients with a germline BRCA alteration (95% CI, 25.6 to 67.2; 11/24) and 42.1% in patients with a somatic alteration (95% CI, 26.3 to 59.2; 16/38).
Efficacy in All Patients BRCA+ by Central Plasma and/or Tissue Testing
Assessment of efficacy by plasma and tissue assays was performed in the subgroups of all patients BRCA+ by plasma (n = 61) and/or tissue test (n = 59), regardless of their enrollment assay. Consistent with the results above, PSA reductions in the 2 groups were not different (Fig 5). Patients with homozygous BRCA deletions in tissue frequently showed significant PSA reductions and responses. PSA response rate in patients BRCA+ by the plasma assay was 47.5% (95% CI, 34.6 to 60.7; 29/61) compared to 57.6% in patients BRCA+ by tissue testing (95% CI, 44.1 to 70.4; 34/59; Fig 4). Radiographic response rates were also not different in the subsets of patients with measurable disease and BRCA+ by plasma or tissue testing.
FIG 5.

Efficacy in all patients with BRCA alterations (BRCA+) detected by central plasma or tissue testing, regardless of enrollment assay. Patient groups overlap but are not identical: 61 patients were identified as BRCA+ by central plasma test (A) and 59 patients were identified as BRCA+ by central tissue test (B). Bars were capped at 100% for visual clarity. PSA increases for the 3 leftmost patients in panel A were 689%, 231%, and 183%.
Patients BRCA+ Solely by Plasma Testing
Twenty-one BRCA+ patients were unable to provide a tissue sample and enrolled by plasma testing (Fig 1). These patients showed a PSA response rate of 52.4% (95% CI, 29.8 to 74.3; 11/21) and radiographic responses in 4 of 10 patients with measurable disease (40.0%; 95% CI, 12.2 to 73.8; Fig 4), with rates not different to those in patients BRCA+ by tissue testing. Patients in this group most commonly had frameshift mutations (42%; 11/21), nonsense mutations (19%; 4/21), or rearrangements (19%; 4/21; Fig S2). BRCA alterations were germline in 29% (6/21) and somatic in 71% (15/21) of patients.
DISCUSSION
The PARP inhibitor rucaparib was approved for BRCA+ mCRPC patients based on results from the TRITON2 study (1). Selection of BRCA+ patients occurred through central genomic testing of plasma or tissue and by local test result. Radiographic and PSA responses were observed independent of the assay type used to identify patients.
Both tissue and plasma assays face biological, technical, and practical challenges in the identification of BRCA+ patients. Tumor tissue testing has long been a reliable method to assess genomics in patients with mCRPC, but lack of accessible lesions or archival tissue of adequate quality is often prohibitive to assessing tumor BRCA. In addition, a single site tissue biopsy may not adequately reflect the landscape of somatic tumor heterogeneity that can develop. In contrast, patients’ plasma contains DNA from primary and metastatic lesions and can be readily collected from a minimally invasive blood draw. The data presented here provide evidence that plasma testing can detect germline and somatic driver mutations of mCRPC and identify alterations that arise through tumor heterogeneity, which in some cases may be subclonal. A technical challenge for plasma-based assays has been the detection of copy number loss, and the plasma assay used for TRITON2 enrollment did not identify patients with homozygous BRCA deletions. Patients with this alteration type have been reported to respond particularly well to rucaparib (38), and their identification has been a priority in the technical advancement of plasma-based assays. Consequentially, the plasma assay used for TRITON2 enrollment has since been updated to the FDA-approved FoundationOne®Liquid CDx (39), which detects and reports homozygous BRCA deletions.
Data from 115 BRCA+ patients enrolled in TRITON2 have shown that plasma and tissue samples from central or local testing can be used to identify mCRPC patients as BRCA+. Across these assay groups, patients’ genomic characteristics with respect to germline status and alteration type were comparable with those reported in the literature (4,7). The concordance between patients’ somatic BRCA status determined from plasma or tissue testing was 47% in 38 patients with paired samples.
In the four cases where the plasma test did not detect BRCA alterations found in tissue, there was evidence of minimal to no tumor DNA shedding in blood (estimated tumor fraction 0% to 3.7%) and lower disease burden (no visceral metastases, 2/4 with <10 bone metastases). Conversely, in six patients, the plasma assay detected a BRCA alteration which was not observed in the patient’s tissue. While there were no outstanding trends in patient characteristics or clinical factors in this discordant group, the BRCA-tissue samples were mainly archival and frequently had low tumor purity. Simultaneously, the majority of BRCA alterations detected only in plasma were of low AF and likely subclonal. These results suggest that discordance may arise from tumor heterogeneity or lack of sample purity, and larger datasets will be required to assess the impact of these factors on response to therapy.
Radiographic and PSA responses were observed in mCRPC patients with a BRCA alteration identified by central plasma, central tissue, or local testing. Although patients who were BRCA+ by plasma test were found to have numerically lower ORR and PSA response rates compared to patients who were BRCA+ by central tissue or local testing, these differences were not statistically significant, and the study was underpowered to draw definitive conclusions about differences in outcomes based on enrollment assay. A factor in this imbalance was the lack of well-performing patients with homozygous BRCA deletions combined with an enrichment of patients with subclonal BRCA alterations within the plasma selected group. A subset of patients was unable to provide tissue samples and were assessed solely with the plasma assay. Some BRCA+ patients in this subgroup experienced objective and PSA responses, suggesting that plasma testing can be a valuable tool to identify patients who may benefit from PARP inhibitor therapy. While BRCA+ patients may be identified by a variety of assays, determining the best option for any given patient may depend on many clinical and circumstantial factors and relies on clinicians’ discretion.
The potential for clonal hematopoiesis of indeterminate potential (CHIP) has raised concerns about the origin of somatic DDR gene alterations detected by plasma assays (31,32), and the dataset presented here does not include sequencing of matched whole-blood controls. While we cannot ascertain the tumoral origin of the somatic BRCA alterations, we reason that they are unlikely to be CHIP mutations. In contrast to other DDR genes, BRCA is not frequently associated with CHIP: no case of CHIP in BRCA1 and only one case in BRCA2 has been reported in advanced prostate cancer (30). Nevertheless, in cases that were identified as BRCA+ in plasma at low allele frequency but not in tissue, CHIP cannot be ruled out.
A strength of our analysis is the large amount of central genomic testing data in association with clinical outcome data. The analysis is limited because the collection of plasma and tissue samples was not concomitant, and metastatic tissue samples were rarely available. Given the expected intratumoral heterogeneity, we are unable to use the tissue NGS data as a ground truth reference for patients’ genomics to determine the positive predictive value of the plasma assay. Although TRITON2 did enroll patients with alterations in other DDR genes, the analyses presented here only include BRCA+ patients and do not allow for the calculation of the negative predictive value of the plasma assay.
In summary, the analyses of concordance and efficacy among genomic testing data from the TRITON2 study demonstrated that plasma, tissue, or local testing of mCRPC patients can be used to identify BRCA+ mCRPC patients who can benefit from treatment with the PARP inhibitor rucaparib.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
Skepticism toward the efficacy of PARP inhibitor treatment in mCRPC patients with a BRCA1/2 mutation identified by a cfDNA based plasma assay is fueled by concerns of false positives and false negatives. The comparative analyses of clinical efficacy in rucaparib-treated mCRPC patients prospectively identified as having a BRCA1/2 mutation through liquid or solid biopsies shown here address these concerns.
Objective and PSA responses to rucaparib were observed in patients with a BRCA1/2 alteration identified by plasma, tissue, or local testing, including within the subset of patients who were unable to provide a tissue sample and tested solely with the plasma assay.
Plasma, tissue, and local testing of mCRPC patients are suitable clinical tools to identify men with BRCA1/2-mutated mCRPC who can benefit from treatment with the PARP inhibitor rucaparib.
ACKNOWLEDGEMENT
Funded by Clovis Oncology; supported in part by the National Cancer Institute (NCI) Cancer Center Support Grant No. P30-CA008748, NCI Prostate Specialized Program of Research Excellence (SPORE) Grant No. P50-CA092629-16, Department of Defense Prostate Cancer Research Program Grant No. W81XWH-17-1-0124, and a Prostate Cancer Foundation Young Investigator Award (W.A.); and supported in part by a Prostate Cancer Foundation Challenge Award and NCI Prostate SPORE Grant No. P50-CA180995 (A.P.). Editorial support funded by Clovis Oncology was provided by Shelly Lim and Stephen Bublitz of Ashfield MedComms, an Ashfield Health company.
Research support for the study: This study was funded by Clovis Oncology.
Footnotes
Disclosure of Potential Conflicts of Interest
Wassim Abida has served in a consulting or advisory role for Clovis Oncology, Daiichi Sankyo, Janssen Pharmaceuticals, MORE Health, and ORIC Pharmaceuticals and has received financial support for travel and/or accommodation from Clovis Oncology and ORIC Pharmaceuticals. His institution has received research funding from Clovis Oncology, AstraZeneca, and Zenith Epigenetics. He is funded by National Cancer Institute (NCI) Cancer Center Support Grant P30 CA 008748, NCI Prostate Specialized Program of Research Excellence (SPORE) grant P50 CA092629-16, Department of Defense Prostate Cancer Research Program grant W81XWH-17-1-0124, and a Prostate Cancer Foundation Young Investigator Award.
Akash Patnaik has served in a consulting or advisory role for Exelixis, Janssen, and Jounce Therapeutics; has received honoraria from Clovis Oncology, Merck, GlaxoSmithKline, Prime Inc, and Roche; and has received research funding from Clovis Oncology, Bristol-Myers Squibb, and Progenics. He is funded by Prostate Cancer Foundation Challenge Award and NCI Prostate Specialized Program of Research Excellence (SPORE) grant (P50CA180995).
Jeremy Shapiro has served in a consulting or advisory role for Amgen, Astellas, Ipsen, Merck, and Roche and received financial support for travel from Amgen and Merck.
Alan H. Bryce has received honoraria from Astellas and Bayer.
Ray McDermott has served in a consulting or advisory role for Bayer, Janssen, and Pfizer; has received support for travel and/or accommodation from Celgene, Janssen, and Pfizer; and has received research funding related to clinical trials from Clovis Oncology, Amgen, Bayer, Bristol-Myers Squibb, and Merck.
Brieuc Sautois has served in a consulting or advisory role for Clovis Oncology, Astellas, Janssen, and Sanofi and received financial support for travel and/or accommodation from Janssen.
Nicholas J. Vogelzang has served in a consulting or advisory role for Clovis Oncology, Astellas, AstraZeneca, Bayer, Caris, Eisai, Janssen, Merck, Pfizer, and Sanofi; holds stock options from Caris; and serves as an editor for Up-To-Date.
Richard Bambury has served in a consulting or advisory role for Bayer, Janssen, Roche, and Pfizer and has received support for travel and/or accommodation from Abbvie, Ipsen, and Pfizer. He is a co-founder of Portable Medical Technology.
Jingsong Zhang has served in a consulting or advisory role and/or on speakers bureaus for Clovis Oncology, AstraZeneca, Bayer, Dendreon, Merck, Sanofi, and Seattle Genetics and received research funding from Astellas Pharma, AstraZeneca, and Bayer.
Josep M. Piulats has served in a consulting or advisory role for Clovis Oncology, Astellas, BeiGene, Bristol-Myers Squibb, Janssen, Pfizer, Merck, Novartis, Roche-Genentech, Sanofi, ImmunoCore, and VCN Biosciences; has received financial support for travel and/or accommodation from Janssen and Roche-Genentech; and has received research funding from AstraZeneca, Bristol-Myers Squibb, Incyte, Janssen, Merck, and Pfizer.
Arif Hussain has served in a consulting or advisory role for AstraZeneca, Bayer, Bristol-Myers-Squibb, Janssen, Merck, and Novartis.
Charles J. Ryan has received research support from Clovis Oncology and has served as a consultant for AstraZeneca and Bayer.
Axel S. Merseburger has served as a principal investigator or subinvestigator for clinical studies by Clovis Oncology, Astellas, Bayer, GlaxoSmithKline, Ipsen, Janssen, Novartis, Pfizer, Teva, and Wyeth; has served as a speaker for Astellas, Bayer, GlaxoSmithKline, Hexal, Ipsen, Janssen, Novartis, Sanofi Aventis, and Teva; has served on an advisory board for Clovis Oncology, Astellas, Bayer, Ipsen, Janssen, Novartis, Pfizer, and Teva; and has received research grants from Wyeth.
Gedske Daugaard has served in a consulting or advisory role for Clovis Oncology, Astellas, AstraZeneca, Janssen, Merck, and Sanofi.
Axel Heidenreich has served as a principal investigator or subinvestigator for clinical studies by Clovis Oncology, Astellas, Bayer, Ipsen, Janssen, Novartis, Pfizer, Teva, and Wyeth; has served as a speaker for Astellas, Bayer, BMS, Dendreon, Hexal, Ipsen, Janssen, MSD, Novartis, Sanofi Aventis, and Takeda; has served on an advisory board for Clovis Oncology, Astellas, Bayer, Ipsen, Janssen, Novartis, Pfizer, and Teva; and has received research grants from Wyeth.
Karim Fizazi has served in a consulting and advisory role for Clovis Oncology, Amgen, Astellas, AstraZeneca, Bayer, CureVac, ESSA, Janssen, Orion Pharma, Roche-Genentech, and Sanofi.
Celestia S. Higano has served in a consulting or advisory role for Clovis Oncology, Aptevo, Asana, Astellas, Bayer, Blue Earth Diagnostics, Churchill Pharmaceuticals, Dendreon, Endocyte, Ferring, Hinova, Janssen, Myriad, Orion, and Pfizer and has received research funding from Clovis Oncology, Aptevo, Aragon Pharma, Astellas, AstraZeneca, Bayer, Dendreon, eFFECTOR Therapeutics, Emergent, Ferring, Genentech, Hoffman-LaRoche, Medivation, and Pfizer.
Laurence E. Krieger has served in a consulting and advisory role and/or on speakers bureaus for Clovis Oncology, Astellas, AstraZeneca, Bayer, Bristol-Myers Squibb, Ipsen, Janssen, Merck Sharp & Dohme, and Roche and has received financial support for travel and accommodation from Astellas, AstraZeneca, Ipsen, Janssen, and Merck Sharp & Dohme.
Cora N. Sternberg has served as a consultant or advisor for Clovis Oncology, Astellas Pharma, AstraZeneca, Bristol-Myers Squibb, Foundation Medicine, Immunomedics, Incyte, Medscape, Merck, Merck Sharp & Dohme, Pfizer, Sanofi Genzyme, Roche/Genentech, and UroToday.
Simon P. Watkins, Darrin Despain, Andrew D. Simmons, Andrea Loehr, Melanie Dowson, and Tony Golsorkhi are employees of Clovis Oncology and may own stock or have stock options in that company.
Simon Chowdhury has served in a consulting or advisory role and/or on speakers bureaus for Clovis Oncology, Astellas, Bayer, BeiGene, Janssen, and Pfizer; has received honoraria from GlaxoSmithKline; has received financial support for travel and accommodation from Clovis Oncology and Beigene; and holds stock in Curve.life..
All other authors have nothing to disclose.
REFERENCES
- 1.Abida W, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R, et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol 2020;38:3763–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stetson D, Ahmed A, Xu X, Nuttall BRB, Lubinski TJ, Johnson JH, et al. Orthogonal comparison of four plasma NGS tests with tumor suggests technical factors are a major source of assay discordance. JCO Precis Oncol 2019;3:1–9. [DOI] [PubMed] [Google Scholar]
- 3.Green F, Shapiro JD, McDermott R, Piulats JM, Reid A, Ostler P, et al. Comprehensive genomic profiling of >1000 plasma and tumor tissue samples from metastatic castration-resistant prostate cancer (mCRPC) patients gives insight into targeted treatment strategies. Cancer Res 2019;79:abstr 727. [Google Scholar]
- 4.Chung JH, Dewal N, Sokol E, Mathew P, Whitehead R, Millis SZ, et al. Prospective comprehensive genomic profiling of primary and metastatic prostate tumors. JCO Precis Oncol 2019;3:PO.18.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scher HI, Solo K, Valant J, Todd MB, Mehra M. Prevalence of prostate cancer clinical states and mortality in the United States: estimates using a dynamic progression model. PLOS ONE 2015;10:e0139440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scher HI, Heller G. Clinical states in prostate cancer: toward a dynamic model of disease progression. Urology 2000;55:323–7. [DOI] [PubMed] [Google Scholar]
- 7.Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol 2017;1:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinkamp MP, O’Mahony OA, Brogley M, Rehman H, Lapensee EW, Dhanasekaran S, et al. Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy. Cancer Res 2009;69:4434–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mateo J, Seed G, Bertan C, Rescigno P, Dolling D, Figueiredo I, et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J Clin Invest 2020;130:1743–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schweizer MT, Sivakumar S, Tukachinsky H, Coleman I, De Sarkar N, Yu EY, et al. Concordance of DNA repair gene mutations in paired primary prostate cancer samples and metastatic tissue or cell-free DNA. JAMA Oncol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong SK, Mohamad NV, Giaze TR, Chin KY, Mohamed N, Ima-Nirwana S. Prostate cancer and bone metastases: the underlying mechanisms. Int J Mol Sci 2019;20:2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ross RW, Halabi S, Ou SS, Rajeshkumar BR, Woda BA, Vogelzang NJ, et al. Predictors of prostate cancer tissue acquisition by an undirected core bone marrow biopsy in metastatic castration-resistant prostate cancer--a Cancer and Leukemia Group B study. Clin Cancer Res 2005;11:8109–13. [DOI] [PubMed] [Google Scholar]
- 13.McKay RR, Zukotynski KA, Werner L, Voznesensky O, Wu JS, Smith SE, et al. Imaging, procedural and clinical variables associated with tumor yield on bone biopsy in metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis 2014;17:325–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gandaglia G, Abdollah F, Schiffmann J, Trudeau V, Shariat SF, Kim SP, et al. Distribution of metastatic sites in patients with prostate cancer: A population-based analysis. Prostate 2014;74:210–6. [DOI] [PubMed] [Google Scholar]
- 15.Sailer V, Schiffman MH, Kossai M, Cyrta J, Beg S, Sullivan B, et al. Bone biopsy protocol for advanced prostate cancer in the era of precision medicine. Cancer 2018;124:1008–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lorente D, Omlin A, Zafeiriou Z, Nava-Rodrigues D, Perez-Lopez R, Pezaro C, et al. Castration-resistant prostate cancer tissue acquisition from bone metastases for molecular analyses. Clin Genitourin Cancer 2016;14:485–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malapelle U, Sirera R, Jantus-Lewintre E, Reclusa P, Calabuig-Farinas S, Blasco A, et al. Profile of the Roche cobas(R) EGFR mutation test v2 for non-small cell lung cancer. Expert Rev Mol Diagn 2017;17:209–15. [DOI] [PubMed] [Google Scholar]
- 18.Hsiue EH, Lee JH, Lin CC, Yang JC. Profile of the therascreen(R) EGFR RGQ PCR kit as a companion diagnostic for gefitinib in non-small cell lung cancer. Expert Rev Mol Diagn 2016;16:1251–7. [DOI] [PubMed] [Google Scholar]
- 19.Satouchi M, Tanaka H, Yoshioka H, Shimokawaji T, Mizuno K, Takeda K, et al. Detection of epidermal growth factor receptor gene T790M mutation in cytology samples using the cobas((R)) EGFR mutation test. Lung Cancer 2017;111:190–4. [DOI] [PubMed] [Google Scholar]
- 20.Clark TA, Chung JH, Kennedy M, Hughes JD, Chennagiri N, Lieber DS, et al. Analytical validation of a hybrid capture–based next-generation sequencing clinical assay for genomic profiling of cell-free circulating tumor DNA. J Mol Diagn 2018;20:686–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oliveira KCS, Ramos IB, Silva JMC, Barra WF, Riggins GJ, Palande V, et al. Current perspectives on circulating tumor DNA, precision medicine, and personalized clinical management of cancer. Mol Cancer Res 2020;18:517–28. [DOI] [PubMed] [Google Scholar]
- 22.Husain H, Velculescu VE. Cancer DNA in the circulation: the liquid biopsy. JAMA 2017;318:1272–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wyatt AW, Azad AA, Volik SV, Annala M, Beja K, McConeghy B, et al. Genomic alterations in cell-free DNA and enzalutamide resistance in castration-resistant prostate cancer. JAMA Oncol 2016;2:1598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Volik S, Alcaide M, Morin RD, Collins C. Cell-free DNA (cfDNA): clinical significance and utility in cancer shaped by emerging technologies. Mol Cancer Res 2016;14:898–908. [DOI] [PubMed] [Google Scholar]
- 25.Annala M, Fu S, Bacon JVW, Sipola J, Iqbal N, Ferrario C, et al. Cabazitaxel versus abiraterone or enzalutamide in poor prognosis metastatic castration-resistant prostate cancer: a multicentre, randomised, open-label, phase II trial. Ann Oncol 2021;32:896–905. [DOI] [PubMed] [Google Scholar]
- 26.Maurice-Dror C, Fonseca N, Herberts C, Fan W, Wyatt AW, Chi KN. Circulating tumor DNA fraction (ctDNA%) to independently predict for clinical outcomes in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol 2021;39 (suppl 15):5049. [Google Scholar]
- 27.Adalsteinsson VA, Ha G, Freeman SS, Choudhury AD, Stover DG, Parsons HA, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun 2017;8:1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wyatt AW, Annala M, Aggarwal R, Beja K, Feng F, Youngren J, et al. Concordance of circulating tumor DNA and matched metastatic tissue biopsy in prostate cancer. J Natl Cancer Inst 2017;109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bowman RL, Busque L, Levine RL. Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell 2018;22:157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jensen K, Konnick EQ, Schweizer MT, Sokolova AO, Grivas P, Cheng HH, et al. Association of clonal hematopoiesis in DNA repair genes with prostate cancer plasma cell-free DNA testing interference. JAMA Oncol 2021;7:107–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beware liquid biopsies to guide PARP blockade. Cancer Discov 2021;11:6. [DOI] [PubMed] [Google Scholar]
- 32.Reichert ZR, Jones MA, Alumkal JJ. A CHIP in the armor of cell-free DNA-based predictive biomarkers for prostate cancer. JAMA Oncol 2021;7:111–2. [DOI] [PubMed] [Google Scholar]
- 33.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crawford B, Adams SB, Sittler T, van den Akker J, Chan S, Leitner O, et al. Multi-gene panel testing for hereditary cancer predisposition in unsolved high-risk breast and ovarian cancer patients. Breast cancer research and treatment 2017;163:383–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 2015;17:251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reid R, DiGiovanni M, Bernhisel R, Brown K, Saam J, Lancaster J. Inherited germline mutations in men with prostate cancer. J Clin Oncol 2018;36 (suppl 6S):abstr 357. [Google Scholar]
- 37.Pritzlaff M, Tian Y, Reineke P, Stuenkel AJ, Allen K, Gutierrez S, et al. Diagnosing hereditary cancer predisposition in men with prostate cancer. Genet Med 2020;22:1517–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abida W, Campbell D, Patnaik A, Sautois B, Shapiro J, Vogelzang N, et al. Genomic characteristics associated with clinical activity of rucaparib in patients (pts) with BRCA1 or BRCA2 (BRCA)-mutated metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol 2020;38:abstr 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.FoundationOne® Liquid CDx. Technical Information. Foundation Medicine, Inc; 2020. [September 22, 2020]. Available from: https://assets.ctfassets.net/w98cd481qyp0/wVEm7VtICYR0sT5C1VbU7/cc6ac2109785d70fe6d91903b241006f/FoundationOne_Liquid_CDx_Technical_Specifications.pdf. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.