

Abstract
The endothelial barrier regulates interstitial fluid homeostasis by transcellular and paracellular means. Dysregulation of this semipermeable barrier may lead to vascular leakage, edema, and accumulation of pro-inflammatory cytokines, inducing microvascular hyperpermeability. Investigating the molecular pathways involved in those events will most probably provide novel therapeutic possibilities in pathologies related to endothelial barrier dysfunction. Metformin (MET) is an anti-diabetic drug, opposes malignancies, inhibits cellular transformation, and promotes cardiovascular protection. In the current study, we assess the protective effects of MET in LPS-induced lung endothelial barrier dysfunction and evaluate the role of P53 in mediating the beneficial effects of MET in the vasculature. We revealed that this biguanide (MET) suppresses the LPS-induced dysregulation in the lung microvasculature, since it suppressed the formation of the filamentous actin stress fibers, and deactivated cofilin. To investigate whether P53 is involved in those phenomena, we employed the fluorescein isothiocyanate (FITC) – dextran permeability assay, to measure paracellular permeability. Our observations suggest that P53 inhibition increases paracellular permeability, and MET prevents those effects. Our results contribute towards the understanding of the lung endothelium and reveal the significant role of P53 in the MET-induced barrier enhancement.
Keywords: Inflammation, Hyperpermeability, Vasculature, Endothelium, Vascular barrier
Introduction
Endothelial barrier dysfunction (EBD) due to endothelial hyperpermeability is associated with diabetes, sepsis, and lung injury [1]. Inflammatory factors such as lipopolysaccharides (LPS), thrombin, and reactive oxygen species (ROS) promote the contraction between endothelial cells destabilizing the cytoskeleton [2, 3]. The deeper understanding of the molecular mechanisms governing endothelial barrier homeostasis will contribute to the development of new medical countermeasures against EBD-related pathologies.
P53 devises cellular responses against extracellular and intracellular stimuli [4], and the anti-inflammatory activities of this tumor suppressor have been associated with NF-κB inhibition [5]. P53 counteracts the activation of neutrophils and macrophages, ameliorates LPS-induced acute lung injury (ALI) [6, 7], and it is induced by glucocorticoids. Those agents are widely utilized in the treatment of asthma, rheumatoid arthritis, and acute respiratory distress syndrome [8–10]. Moreover, P53 mediates AMPK activation [11, 12] and participates in the protective activities of Growth Hormone Releasing Hormone antagonists (GHRHAnt) [13, 14] and heat shock protein 90 (Hsp90) inhibitors [15] in endothelial inflammation. Those P53 inducers protect against endothelial barrier dysfunction, and exert protective activities in inflamed human tissues [16, 17]. They can also activate unfolded protein response by triggering tissue-repairing processes [18–20].
MET is the first-line treatment for type 2 diabetes and opposes gastric, pancreatic, cervical, and thyroid cancers [21–23]. It protects against lipopolysaccharide (LPS)-induced lung injury and reduces inflammation [24]. Those events are due to NF-κB inhibition [25], PI3K-Akt suppression [26], inhibition of leukocyte adhesion, and eNOS uncoupling. MET also prevents vascular aging, endothelial cell injury, and apoptosis [27–30].
In the present study, we investigated the effects of MET in LPS-induced actin cytoskeleton reorganization. Moreover, we examined the involvement of P53 in the MET-induced endothelial barrier enhancement, as measured with the Fluorescein isothiocyanate (FITC) – dextran permeability assay. Our results support the beneficial effects of MET towards barrier function, and suggest that P53 is a crucial mediator of the corresponding endothelial effects.
Materials and Methods
Reagents:
Metformin hydrochloride (TCM2009), RIPA buffer (AAJ63306-AP), anti-mouse IgG HRP linked whole antibody from sheep (95017-554), anti-rabbit IgG HRP linked whole antibody from donkey (95017-556) and nitrocellulose membranes (10063-173) were obtained from VWR (Radnor, PA).P53 (9282), phospho-Cofilin (Ser3) (3313), Cofilin (3318), phospho-myosin light chain 2 (Thr18/Ser19) (3674), and myosin light chain 2 (3672) antibodies were obtained from Cell Signaling (Danvers, MA). The β-actin antibody (A5441), Lipopolysaccharides (L4130), Corning® Transwell® cell culture inserts (CLS3470), and FITC-Dextran (46945) were purchased from Sigma-Aldrich (St Louis, MO).
Cell Culture:
Bovine pulmonary artery endothelial cells (BPAEC) (PB30205) were purchased from Genlantis (San Diego, CA). Those cellswere maintained at 37°C in a humidified atmosphere of 5% CO2/95% air in Dulbecco’s modified Eagle’s medium (VWRL0101–0500) supplemented with 10% FBS (89510–186) and 1X penicillin/streptomycin (97063–708), and were used at an early passage. All reagents were purchased from VWR (Radnor, PA).
Western Blot Analysis:
Proteins were isolated from cells using RIPA buffer, and an equal amount of protein samples were separated by electrophoresis onto 12% sodium dodecyl sulfate (SDS–PAGE) Tris-HCl gels. Wet transfer was used to transfer the proteins onto nitrocellulose membranes. After blocking with 5% non-fat dry milk at room temperature, the blots were exposed to appropriate primary and secondary antibodies to detect protein signals in a ChemiDocTM Touch Imaging System from Bio-Rad (Hercules, CA). The β-actin was the loading control unless otherwise stated in the graph of densitometry.
Fluorescein isothiocyanate (FITC) - dextran permeability assay:
Paracellular influx across the BPAEC monolayer was measured using the Transwell assay system in 24-well culture plates. A total of 200,000 cells were seeded in each insert and media were changed after 24 h. Cells were treated with either vehicle (PBS) or MET (100 μM) for 24 h, and then exposed to either vehicle (0.1% DMSO) or Pifithrin (30 μM). 24 h after the addition of Pifithrin, FITC-dextran (70 kDa, 1 mg/ml) was added to the media for 30 minutes. 100 ml of basal media was removed, and fluorescence intensity was measured in a Synergy H1 Hybrid Multi-Mode Reader from Biotek (Winooski, Vermont). The excitation and emission wavelengths were 485 nm and 530 nm, respectively.
Densitometry and Statistical Analysis:
Image J software (NIH) was used to perform densitometry of the immunoblots. All values were expressed as the mean ± SEM (standard error of the mean). GraphPad Prism (version 5.01) was used to analyze the data. Student’s t-test was used to determine statistically significant differences among the groups.
RESULTS
MET induces the expression of P53 in BPAEC.
To investigate the involvement of MET in endothelial P53 expression, we employed commercially available bovine pulmonary artery endothelial cells (BPAEC). Cells were seeded on 12-well plates and were treated with either vehicle (PBS) or MET (100μM) for 12, 24, 48, and 72 h. The results shown in Fig. 1A indicate that MET induces P53 expression levels.
Figure 1. P53 mediates MET-induced endothelial integrity.
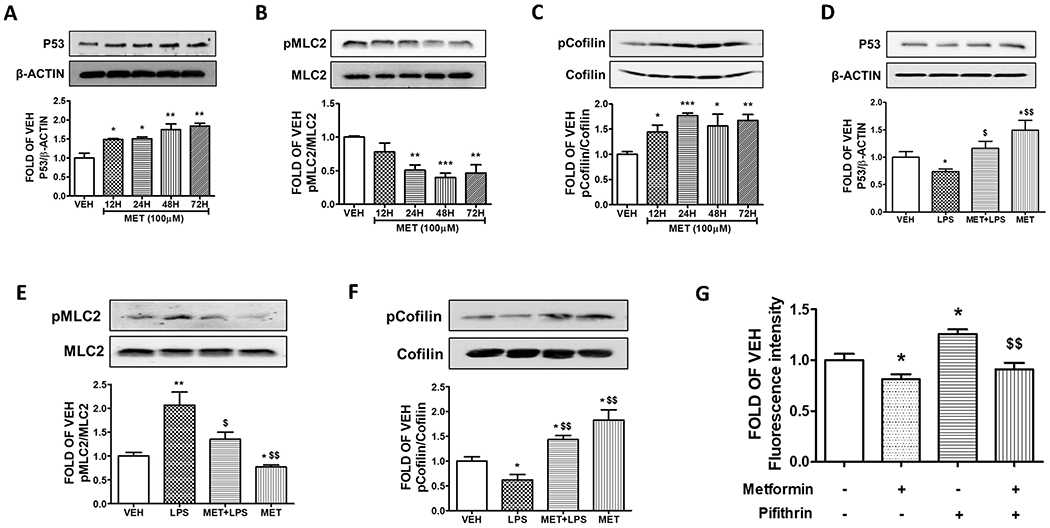
Western Blot analysis of (A) P53 and β-actin, (B) phosphorylated MLC2 (pMLC2) and MLC2, (C) phosphorylated Cofilin (pCofilin) and Cofilin after treatment of BPAEC with either vehicle (PBS) or 100 μM of MET for 12, 24, 48, and 72 h. The blots represent three independent experiments. The signal intensity of protein bands was analyzed by densitometry. Protein levels of P53, pMLC2, and pCofilin were normalized to β-actin, MLC2 and Cofilin respectively. *P<0.05, **P < 0.01, ***P < 0.001 vs vehicle (VEH). Means ± SEM. Western Blot analysis of (D) P53 and β-actin, (E) phosphorylated MLC2 (pMLC2) and MLC2, (F) phosphorylated Cofilin (pCofilin) and Cofilin. BPAEC were treated for 24 h with either vehicle (PBS) or MET (100μM) prior to either vehicle (PBS) or LPS (10μg/ml) exposure for 1 h. The blots shown are representative of 3 independent experiments. The signal intensity of the protein bands was analyzed by densitometry. Protein levels of P53, pMLC2, and pCofilin were normalized to β-actin, MLC2 and Cofilin respectively. *P < 0.05, **P < 0.01 vs. vehicle (VEH) and $p < 0.05, $$p < 0.01 vs. LPS. Means ± SEM. (G) BPAECs were seeded onto transwell inserts of a 24-well culture plate. After 24 h, cells were treated with either vehicle (PBS) or MET (100μM) for 24 h. Pifithrin (30μM) was then added for 24 h followed by 70 kDa FITC-dextran (1 mg/ml). At 30 minutes after FITC-dextran addition, 100 ml of basal media was removed, fluorescence intensity was measured and shown as fold changes relative to the vehicle. *P < 0.05 vs. vehicle (VEH) and $$p < 0.01 vs. Pifithrin. Means ± SEM, n = 3.
MET suppresses the activation of MLC2 in BPAEC.
To determine whether MET impacts actin cytoskeleton, bovine lung endothelial cells were treated with either vehicle (PBS) or MET (100μM) for 12, 24, 48, and 72 h. The results (Fig. 1B) indicate that MET suppresses the phosphorylation of MLC2, as compared to the vehicle (PBS)- treated cells.
MET induces the deactivation of Cofilin in BPAEC.
To assess the effects of MET in Cofilin, BPAECs were exposed to either vehicle (PBS) or MET (100μM) for 12, 24, 48, and 72 h. The Western Blot data reveal that MET deactivated (phosphorylated) Cofilin in all treatments (Fig. 1C). Cofilin severs the actin cytoskeleton, increasing permeability.
MET inhibits LPS-induced P53 suppression in BPAEC.
To induce in vitro lung endothelial inflammation, we exposed BPAECs to LPS (endotoxin from Gram-negative bacteria). The cells were pre-treated with either vehicle (PBS) or MET (100μM) for 24 h before an 1h treatment with vehicle (PBS) or LPS (10μg/ml). As shown in Fig. 1D, LPS suppresses the expression levels of P53, while MET pre-treatment counteracts the LPS-induced P53 reduction.
MET protects against LPS-induced MLC2 phosphorylation in BPAEC.
To examine whether MET inhibits the barrier disruptive effects of LPS, BPAECs were pre-treated with either vehicle (PBS) or MET (100μM) for 24 h prior to LPS (10μg/ml) exposure (1 h). LPS induces the activation of MLC2 by phosphorylation. Fig. 1E demonstrates the ability of MET to prevent the LPS-induced phosphorylation of MLC2.
MET inhibits LPS-induced activation of Cofilin in BPAEC.
Lung cells were treated with either vehicle (PBS) or MET (100μM) for 24 h prior to exposure to either vehicle (PBS) or LPS (10μg/ml) for 1 h. The data shown in Fig. 1F suggest that MET suppresses the LPS-triggered activation of cofilin.
P53 mediates the MET-induced endothelial integrity.
To investigate the association of P53 with the barrier enhancing effects of MET in the context of paracellular permeability, confluent BPAECs were treated with vehicle (PBS) or MET (100μM) for 24 h in transwell inserts. Those cells were exposed to either vehicle (0.1% DMSO) or pifithrin (30μM) for 24 h. Then, FITC-dextran (70 kDa, 1 mg/ml) was added in the media for 30 min. The fluorescence intensity data of Fig. 1G demonstrates that MET enhances the endothelial barrier function, in line with our previous observations. The suppression of P53 due to pifithrin increased the paracellular permeability of the endothelial monolayer and pre-treatment with MET counteracted those events.
Discussion
Endothelial hyperpermeability is the prominent feature of inflammatory lung disease [16]. MET is a widely used hypoglycemic drug that decreases insulin resistance, inhibits hepatic glucose production, and lowers blood glucose levels [31]. Diabetes increases the progression of bacterial lung inflammation by inducing airway glucose [32]. Accumulating evidence suggests that MET reduces the progression of malignancies and improves the survival of non-small cell lung cancer patients with type 2 diabetes [33]. MET pre-treatment can also reduce airway bacterial load in hyperglycemic mice [32]; and stimulates the adenosine monophosphate-activated protein kinase (AMPK). AMPK suppresses the mammalian target of rapamycin (mTOR) [34], which is involved in obesity, cancer, type 2 diabetes, and cardiac hypertrophy [35]. Interestingly, the atypical serine/threonine kinase (mTOR) pathway is dysregulated during tumor progression through the loss of PTEN and PI3K/AKT activation, promoting invasion and metastasis [36]. AKT-mTOR signaling can also attenuate IRE1, which is a conserved unfolded protein response (UPR) transducer [37].
UPR is a signal transduction pathway that orchestrates endoplasmic reticulum (ER) proteostasis [38, 39]. Perturbation of ER protein folding induces the accumulation of dysfunctional proteins in the intracellular niche, which in turn triggers ER stress [40]. To restore ER homeostasis, cells engage UPR [41]. Three major ER membrane-embedded proteins orchestrate UPR: the inositol-requiring protein-1α (IRE1α), the activating transcription factor-6 (ATF6), and the protein kinase RNA (PKR)-like ER kinase (PERK) [42]. The ER luminal domain of those transducers is bound to the immunoglobulin heavy chain binding protein/glucose regulated protein 78 (BiP/Grp78) under unstressed conditions [43]. MET promotes the survival of cardiomyocytes by inducing BiP and stimulates the PERK-ATF4 axis to upregulate CHOP [44]. Mild induction of UPR induces the expression of P53 and enhances endothelial barrier integrity [45, 46].
P53 senses genotoxic stresses to prevent cell growth and proliferation; while MET and P53 synergize to oppose cancers [47]. P53 family proteins (p53, p63, and p73) have been associated with the metformin-mediated suppression of malignancies. MET prevents MDMX-P53 interaction to activate P53 and induce lymphoma-mediated cell death [48]. MET protected against ALI by altering mitochondria-derived ROS [25], and moderate concentrations of MET (0.1, 0.5, 1.0 mM) enhance lung endothelial barrier integrity in BPAECs [11].
Herein we investigated the effects of MET in LPS-induced barrier disruption. P53 is considered an attractive target for the development of new therapeutic possibilities against human disease [13]. We performed in vitro experiments using BPAEC to investigate the effects of MET in inflamed lung cells. Our observations suggest that P53 is involved in the beneficial effects of MET in endothelial barrier function in vitro (Fig. 1D). P53 inhibited pMLC2 (Fig. 1E) -an indicator of hyper-permeability responses- and deactivated cofilin (Fig. 1F). This is a protein that severs actin filaments to corrupt barrier integrity [49].
LPS is an established inducer of inflammation which activates NF-κB via toll-like receptor 4 (TLR4) activation. Those activities induce the expression of proinflammatory cytokines (e.g. interleukin-1 beta (IL-1β), tumor necrosis factor-alpha (TNF-α), and IL-6) [50, 51], LPS phosphorylates P53 at multiple serine residues (ser6, ser15, ser33, and ser392) propelling its degradation [52, 53]. That endothelial defender (P53) can also protect the microvasculature by suppressing the transcription factor apurinic/apyrimidinic endonuclease 1/redox effector factor 1 (APE1/Ref1) [54]. We demonstrated that the expression levels of dephosphorylated cofilin in P53 null mice (P53 KO) were higher than those of the wild-type littermates, suggesting that P53 stimulates the Rac1/pCofilin pathway [55]. Those results agree with previous studies in super P53 mice, which express globally more P53 [55]. We also observed that MET induces the expression levels of P53 in BPAEC (Fig. 1A).
AMP-activated protein kinase (AMPK) is a conserved serine/threonine kinase that regulates metabolism [56]. This cellular energy sensor is activated by hypoxia, nutrient deprivation, ischemia, and heat shock, resulting in ATP depletion [12]. Metformin exerts its anti-cancer activities by stimulating AMPK pathway, which in turn attenuates the TLR4-induced neutrophil accumulation in ALI [57]. AMPK-α1 directly phosphorylates cofilin [58], stabilizing actin cytoskeleton [59]. In our studies, we observed that MET mitigated the activation of cofilin in BPAEC (Fig. 1C). Actin alignment is crucial for barrier function [45], and reorganization of actin cytoskeleton is associated with the binding of cofilin to F actin [60]. The depolymerizing activity of cofilin is abolished by its phosphorylation at serine 3 [61]. Silencing of P53 by small interfering RNA leads to the suppression of the Hsp90 inhibitor-induced phosphorylation of cofilin [55].
Lung endothelial cells are activated upon stimulation of leukocyte adhesion molecules and inflammatory cytokines. MET suppresses the LPS-stimulated secretion of TNF-α, IL-6, ICAM-1, and VCAM-1 [62, 63]. Likewise, in murine macrophages, MET weakens the LPS-induced inflammatory response via the induction of the activating transcription factor-3 (ATF-3) [64]. MET significantly inhibits the LPS-induced activation of MLC2, promoting vascular dysfunction [65]. P53 negatively regulates RhoA [39], while pifithrin potentiates the F actin formation [6].
Endothelial barrier function is maintained via transcellular and paracellular pathways regulating the tissue-fluid homeostasis [66]. Interendothelial tight and adherens junctions restrict the passage of solutes more than 3nm. The accumulation of excessive fluid into the interstitium due to the disruption of those junctions is associated with ALI. MET facilitates the assembly of epithelial tight junctions [67] and exerts beneficial effects in cystic fibrosis (CF) by blunting excessive sodium and airway surface liquid absorption. Hence, MET reduces airway inflammation [68].
There are certain limitations in our study since it was conducted in vitro. Investigations on the role of MET in LPS-induced inflammation in mice which express more [55] or less P53 [49] would further substantiate our findings, as well as experiments utilizing immunofluorescence to study the effects of MET in major cytoskeletal components (e.g. VE-Cadherin, F actin). Evans Blue would also serve as an additional approach to assess vascular leakage in vivo, and the involvement of UPR in the role of P53 towards the MET-induced endothelial barrier protection will be the subject of our future investigations.
Our study supports previous reports on the protective effects of MET towards LPS-induced endothelial barrier dysfunction. We assessed the effects of MET towards cofilin and MLC2 and investigated the effects of P53 in MET-induced endothelial barrier enhancement. Since P53 is involved in the beneficial function of MET towards the inflamed endothelium, future therapeutical interventions utilizing P53 modulators (e.g. Idasanutlin) and Metformin may deliver novel therapeutic possibilities against diseases related to endothelial barrier dysfunction.
Highlights.
Metformin (MET) protects against lipopolysaccharide (LPS)-induced lung endothelial barrier disruption.
P53 contributes to the beneficial effects of MET into the microvasculature; since it deactivates cofilin and inhibits the formation of actin stress fibers (pMLC2).
Suppression of P53 due to pifithrin increases paracellular permeability of lung endothelium, while pre-treatment with MET counteracts those events.
Funding:
Our research is supported by the 1) R&D, Research Competitiveness Subprogram (RCS) of the Louisiana Board of Regents through the Board of Regents Support Fund (LEQSF(2019-22)-RD-A-26) (P.I: NB), 2) Faculty Research Support Program from the College of Pharmacy, ULM (P.I: NB) 3) Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (NIGMS) (5 P20 GM103424-20).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: None.
References
- 1.Claesson-Welsh L, Dejana E, and McDonald DM, Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol Med, 2021. 27(4): p. 314–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guillot L, et al. , Alveolar epithelial cells: master regulators of lung homeostasis. Int J Biochem Cell Biol, 2013. 45(11): p. 2568–73. [DOI] [PubMed] [Google Scholar]
- 3.Ruaro B, et al. , The History and Mystery of Alveolar Epithelial Type II Cells: Focus on Their Physiologic and Pathologic Role in Lung. Int J Mol Sci, 2021. 22(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kubra KT, et al. , P53 is Subjected to Lipoteichoic Acid-Induced Phosphorylation in the Lungs. TH Open, 2020. 4(3): p. e173–e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barabutis N, P53 in acute respiratory distress syndrome. Cell Mol Life Sci, 2020. 77(22): p. 4725–4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barabutis N, et al. , p53 protects against LPS-induced lung endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol, 2015. 308(8): p. L776–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uddin MA and Barabutis N, P53 in the impaired lungs. DNA Repair (Amst), 2020. 95: p. 102952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dejager L, et al. , Dominance of the strongest: inflammatory cytokines versus glucocorticoids. Cytokine Growth Factor Rev, 2014. 25(1): p. 21–33. [DOI] [PubMed] [Google Scholar]
- 9.Li H, et al. , Glucocorticoid receptor and sequential P53 activation by dexamethasone mediates apoptosis and cell cycle arrest of osteoblastic MC3T3-E1 cells. PLoS One, 2012. 7(6): p. e37030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meduri GU, et al. , Pharmacological principles guiding prolonged glucocorticoid treatment in ARDS. Intensive Care Med, 2020. 46(12): p. 2284–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uddin MA, et al. , Metformin in acute respiratory distress syndrome: An opinion. Exp Gerontol, 2021. 145: p. 111197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siejka A, Barabutis N, and Schally AV, GHRH antagonist MZ-5-156 increases the expression of AMPK in A549 lung cancer cells. Cell Cycle, 2011. 10(21): p. 3714–8. [DOI] [PubMed] [Google Scholar]
- 13.Barabutis N, Growth Hormone Releasing Hormone in Endothelial Barrier Function. Trends Endocrinol Metab, 2021. 32(6): p. 338–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akhter MS, et al. , Involvement of the unfolded protein response in the protective effects of growth hormone releasing hormone antagonists in the lungs. J Cell Commun Signal, 2021. 15(1): p. 125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barabutis N, Heat shock protein 90 inhibition in the inflamed lungs. Cell Stress Chaperones, 2020. 25(2): p. 195–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barabutis N, Schally AV, and Siejka A, P53, GHRH, inflammation and cancer. EBioMedicine, 2018. 37: p. 557–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akhter MS, et al. , Elucidation of the Molecular Pathways Involved in the Protective Effects of AUY-922 in LPS-Induced Inflammation in Mouse Lungs. Pharmaceuticals (Basel), 2021. 14(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barabutis N, Unfolded Protein Response in Acute Respiratory Distress Syndrome. Lung, 2019. 197(6): p. 827–828. [DOI] [PubMed] [Google Scholar]
- 19.Barabutis N, Unfolded Protein Response: A Regulator of the Endothelial Barrier. Endocr Metab Sci, 2021. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akhter MS and Barabutis N, Suppression of reactive oxygen species in endothelial cells by an antagonist of growth hormone-releasing hormone. J Biochem Mol Toxicol, 2021: p. e22879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Courtois S, et al. , Metformin targets gastric cancer stem cells. Eur J Cancer, 2017. 84: p. 193–201. [DOI] [PubMed] [Google Scholar]
- 22.Yamana H, et al. , Metformin Inhibits Proliferation and Tumor Growth of QGP-1 Pancreatic Neuroendocrine Tumor Cells by Inducing Cell Cycle Arrest and Apoptosis. Anticancer Res, 2020. 40(1): p. 121–132. [DOI] [PubMed] [Google Scholar]
- 23.Yudhani RD, et al. , Metformin Modulates Cyclin D1 and P53 Expression to Inhibit Cell Proliferation and to Induce Apoptosis in Cervical Cancer Cell Lines. Asian Pac J Cancer Prev, 2019. 20(6): p. 1667–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saisho Y, Metformin and Inflammation: Its Potential Beyond Glucose-lowering Effect. Endocr Metab Immune Disord Drug Targets, 2015. 15(3): p. 196–205. [DOI] [PubMed] [Google Scholar]
- 25.Zmijewski JW, et al. , Mitochondrial respiratory complex I regulates neutrophil activation and severity of lung injury. Am J Respir Crit Care Med, 2008. 178(2): p. 168–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isoda K, et al. , Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol, 2006. 26(3): p. 611–7. [DOI] [PubMed] [Google Scholar]
- 27.Arunachalam G, et al. , Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br J Pharmacol, 2014. 171(2): p. 523–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis BJ, et al. , Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes, 2006. 55(2): p. 496–505. [DOI] [PubMed] [Google Scholar]
- 29.Hattori Y, et al. , Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension, 2006. 47(6): p. 1183–8. [DOI] [PubMed] [Google Scholar]
- 30.Sena CM, et al. , Metformin restores endothelial function in aorta of diabetic rats. Br J Pharmacol, 2011. 163(2): p. 424–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rena G, Hardie DG, and Pearson ER, The mechanisms of action of metformin. Diabetologia, 2017. 60(9): p. 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gill SK, et al. , Increased airway glucose increases airway bacterial load in hyperglycaemia. Sci Rep, 2016. 6: p. 27636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen H, et al. , Synergistic effects of metformin in combination with EGFR-TKI in the treatment of patients with advanced non-small cell lung cancer and type 2 diabetes. Cancer Lett, 2015. 369(1): p. 97–102. [DOI] [PubMed] [Google Scholar]
- 34.Buzzai M, et al. , Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res, 2007. 67(14): p. 6745–52. [DOI] [PubMed] [Google Scholar]
- 35.Waise TMZ, et al. , Inhibition of upper small intestinal mTOR lowers plasma glucose levels by inhibiting glucose production. Nat Commun, 2019. 10(1): p. 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mossmann D, Park S, and Hall MN, mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer, 2018. 18(12): p. 744–757. [DOI] [PubMed] [Google Scholar]
- 37.Sanchez-Alvarez M, Del Pozo MA, and Bakal C, Publisher Correction: AKT-mTOR signaling modulates the dynamics of IRE1 RNAse activity by regulating ER-mitochondria contacts. Sci Rep, 2018. 8(1): p. 6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kubra KT, et al. , Unfolded protein response in cardiovascular disease. Cell Signal, 2020. 73: p. 109699. [DOI] [PubMed] [Google Scholar]
- 39.Kubra KT, et al. , Luminespib counteracts the Kifunensine-induced lung endothelial barrier dysfunction. Curr Res Toxicol, 2020. 1: p. 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barabutis N, Unfolded Protein Response supports endothelial barrier function. Biochimie, 2019. 165: p. 206–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barabutis N, Unfolded Protein Response in Lung Health and Disease. Front Med (Lausanne), 2020. 7: p. 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uddin MA, et al. , Effects of Heat Shock Protein 90 Inhibition In the Lungs. Med Drug Discov, 2020. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kubra KT and Barabutis N, Brefeldin A and Kifunensine modulate the LPS-induced lung endothelial hyperpermeability in human and bovine cells. Am J Physiol Cell Physiol, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quentin T, et al. , Metformin differentially activates ER stress signaling pathways without inducing apoptosis. Dis Model Mech, 2012. 5(2): p. 259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akhter MS, et al. , Kifunensine compromises lung endothelial barrier function. Microvasc Res, 2020. 132: p. 104051. [DOI] [PubMed] [Google Scholar]
- 46.Akhter MS, Uddin MA, and Barabutis N, Unfolded protein response regulates P53 expression in the pulmonary endothelium. J Biochem Mol Toxicol, 2019. 33(10): p. e22380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yi Y, et al. , Role of p53 Family Proteins in Metformin Anti-Cancer Activities. J Cancer, 2019. 10(11): p. 2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gu J, et al. , Metformin Induces p53-Dependent Mitochondrial Stress in Therapy-Sensitive and - Resistant Lymphoma Pre-Clinical Model and Primary Patients Sample with B-Cell Non-Hodgkin Lymphoma (NHL). Blood, 2015. 126: p. 4008–4008. [Google Scholar]
- 49.Uddin MA, et al. , P53 deficiency potentiates LPS-Induced acute lung injury in vivo. Curr Res Physiol, 2020. 3: p. 30–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barabutis N, Verin A, and Catravas JD, Regulation of pulmonary endothelial barrier function by kinases. Am J Physiol Lung Cell Mol Physiol, 2016. 311(5): p. L832–L845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uddin MA, et al. , Induction of the NEK family of kinases in the lungs of mice subjected to cecal ligation and puncture model of sepsis. Tissue Barriers, 2021: p. 1929787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barabutis N, Uddin MA, and Catravas JD, Hsp90 inhibitors suppress P53 phosphorylation in LPS - induced endothelial inflammation. Cytokine, 2019. 113: p. 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia Y, et al. , Phosphorylation of p53 by IkappaB kinase 2 promotes its degradation by beta-TrCP. Proc Natl Acad Sci U S A, 2009. 106(8): p. 2629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uddin MA, et al. , P53 supports endothelial barrier function via APE1/Ref1 suppression. Immunobiology, 2019. 224(4): p. 532–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barabutis N, et al. , Wild-type p53 enhances endothelial barrier function by mediating RAC1 signalling and RhoA inhibition. J Cell Mol Med, 2018. 22(3): p. 1792–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mayer A, et al. , AMP-activated protein kinase regulates lymphocyte responses to metabolic stress but is largely dispensable for immune cell development and function. Eur J Immunol, 2008. 38(4): p. 948–56. [DOI] [PubMed] [Google Scholar]
- 57.Zhao X, et al. , Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol, 2008. 295(3): p. L497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y, et al. , AMP-activated protein kinase/myocardin-related transcription factor-A signaling regulates fibroblast activation and renal fibrosis. Kidney Int, 2018. 93(1): p. 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suurna MV, et al. , Cofilin mediates ATP depletion-induced endothelial cell actin alterations. Am J Physiol Renal Physiol, 2006. 290(6): p. F1398–407. [DOI] [PubMed] [Google Scholar]
- 60.McGough A, et al. , Cofilin changes the twist of F-actin: implications for actin filament dynamics and cellular function. J Cell Biol, 1997. 138(4): p. 771–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Komarova YA, et al. , Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ Res, 2017. 120(1): p. 179–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun J, et al. , Protective effects of metformin on lipopolysaccharideinduced airway epithelial cell injury via NFkappaB signaling inhibition. Mol Med Rep, 2019. 19(3): p. 1817–1823. [DOI] [PubMed] [Google Scholar]
- 63.Millar FR, et al. , The pulmonary endothelium in acute respiratory distress syndrome: insights and therapeutic opportunities. Thorax, 2016. 71(5): p. 462–73. [DOI] [PubMed] [Google Scholar]
- 64.Kim J, et al. , Metformin suppresses lipopolysaccharide (LPS)-induced inflammatory response in murine macrophages via activating transcription factor-3 (ATF-3) induction. J Biol Chem, 2014. 289(33): p. 23246–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Waschke J, et al. , Requirement of Rac activity for maintenance of capillary endothelial barrier properties. Am J Physiol Heart Circ Physiol, 2004. 286(1): p. H394–401. [DOI] [PubMed] [Google Scholar]
- 66.Komarova Y and Malik AB, Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol, 2010. 72: p. 463–93. [DOI] [PubMed] [Google Scholar]
- 67.Peng L, et al. , Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr, 2009. 139(9): p. 1619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Myerburg MM, et al. , AMPK agonists ameliorate sodium and fluid transport and inflammation in cystic fibrosis airway epithelial cells. Am J Respir Cell Mol Biol, 2010. 42(6): p. 676–84. [DOI] [PMC free article] [PubMed] [Google Scholar]