

Abstract
Chronic lymphocytic leukemia (CLL) is characterized by disordered DNA methylation, suggesting these epigenetic changes might play a critical role in disease onset and progression. The methyltransferase DNMT3A is a key regulator of DNA methylation. Although DNMT3A somatic mutations in CLL are rare, we found that low DNMT3A expression is associated with more aggressive disease. A conditional knockout mouse model showed that homozygous depletion of Dnmt3a from B cells results in the development of CLL with 100% penetrance at a median age of onset of 5.3 months, and heterozygous Dnmt3a depletion yields a disease penetrance of 89% with a median onset at 18.5 months, confirming its role as a haploinsufficient tumor suppressor. B1a cells were confirmed as the cell-of-origin of disease in this model, and Dnmt3a depletion resulted in focal hypomethylation and activation of Notch and Myc signaling. Amplification of chromosome 15 containing the Myc gene was detected in all CLL mice tested, and infiltration of high-Myc-expressing CLL cells in the spleen was observed. Notably, hyperactivation of Notch and Myc signaling was exclusively observed in the Dnmt3a CLL mice, but not in 3 other CLL mouse models tested (Sf3b1-Atm, Ikzf3 and MDR), and Dnmt3a-depleted CLL were sensitive to pharmacologic inhibition of Notch signaling in vitro and in vivo. Consistent with these findings, human CLL samples with lower DNMT3A expression were more sensitive to Notch inhibition than those with higher DNMT3A expression. Altogether, these results suggest that Dnmt3a depletion induces CLL that is highly dependent on activation of Notch and Myc signaling.
Introduction
Recent large-scale cancer sequencing studies performed over the past decade have revealed a myriad of candidate genetic lesions that drive malignancies, however, the functional effects of these newly somatic mutations have remained largely unknown (1,2). For chronic lymphocytic leukemia (CLL), growing efforts to address this challenge have yielded the recent generation of in vivo models engineered to recapitulate the B cell-restricted expression of novel putative genetic drivers. These studies have confirmed the CLL-driving roles of alterations such as mutated SF3B1 in conjunction with ATM deletion, and hotspot mutations in IKZF3, and altogether highlight the common and diverse mechanisms underlying CLL pathogenesis (i.e., dysregulation of B-cell receptor (BCR) signaling) (3,4).
In addition to genetic lesions, recent studies have further highlighted the role of epigenetic aberrations in cancer development (5,6). DNA methylation is one of the most well-studied epigenetic abnormalities in human cancers and dysregulation of DNA methylation can lead to genome instability and the silencing of tumor suppressor genes (7,8). In line with this notion, previous studies have revealed that CLL is characterized by prominent locally disordered methylation disorder (9,10). These characteristic changes in DNA methylation suggest a critical role in the onset and progression of CLL (10–12). In mammalian cells, DNA methylation is dynamically established by the DNA methyltransferase 3 (DNMT3) family of de novo methyltransferases DNMT3A and DNMT3B and sustained by the maintenance methyltransferase DNMT1 (13). Previous studies suggested that alterations in DNA methylation/demethylation intermediates were closely linked to clinical outcomes in CLL (14).
Although mutations of DNMT3A are rarely observed in CLL, we demonstrate herein that low DNMT3A expression is associated with more aggressive disease through the analysis of two independent CLL cohorts. To explore the hypothesis that loss of DNMT3A expression functionally contributes to CLL pathogenesis, we engineered a conditional knock-out mouse model with B cell-restricted deletion of Dnmt3a through intercrossing with the CD19-Cre system. Strikingly, homozygous Dnmt3a depletion in B cells resulted in the development of CLL with 100% penetrance at a median age of onset of 5.3 months, and heterozygous Dnmt3a depletion yielded a disease penetrance of 89% with a median onset at 18.5 months, confirming its role as a haplo‐insufficient tumor suppressor. Distinct from other existing CLL models tested (i.e., Sf3b1-Atm, Ikzf3 and MDR (3,4,15)), we observed selective activation of Notch and Myc signaling, but not BCR signaling, and sensitivity to Notch inhibition, supporting this line as a Notch-driven mouse model of CLL.
Materials and Methods
Animals
All animals were housed at Dana-Farber Cancer Institute (DFCI). All animal procedures were completed in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees at DFCI (protocol#:07–068; 12–004). Dnmt3a-floxed mice were gifted from Dr. Ross Levine (MSKCC) (16). Cd19-Cre+/− mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Dnmt3a-floxed mice were intercrossed with Cd19-Cre mice to generate Cd19-Cre Dnmt3a-floxed C57BL/6J mice.
CLL patient samples
Heparinized blood samples were obtained from healthy donors and patients enrolled on clinical research protocols with informed consent. The study was conducted in accordance with the ethical guidelines approved by the Human Subjects Protection Committees of DFCI, at UCSD, by the NIH National Heart, Lung, and Blood Institute and by the Department of Internal Medicine III at Ulm University, Germany. Peripheral blood mononuclear cells (PBMC) from normal donors and CLL patients were isolated by Ficoll/Hypaque density gradient centrifugation. CD19+ B cells from normal volunteers were isolated by immunomagnetic selection (Miltenyi Biotec). PBMCs were used fresh or cryopreserved in FBS with 10% DMSO and stored in vapor-phase liquid nitrogen until the time of analysis.
Flow Cytometry
To monitor disease progression, 50µl of blood was collected from mice via submandibular bleeds into EDTA-containing tubes. Erythrocyte lysis was achieved by incubating the blood in 1 ml of ACK buffer for 5 min and then washing with PBS supplemented with 2% FCS and 2 mM EDTA. The cells were then stained with a cocktail of antibodies as described in Supplementary Materials and Methods for 20 min at 4°C. Cells were then washed and analyzed (BD LSRFortessa™ cell analyzer). For flow cytometric evaluation of specific cell surface markers, erythrocyte lysed single cell suspensions prepared from spleen, bone marrow, and peritoneal cavity were washed with PBS supplemented with 2% FCS and 2 mM EDTA and then incubated with antibodies for 20 min. Splenic marginal and follicular B cell populations were stained with a panel of antibodies as described in Supplementary Materials and Methods. All analyses of splenic and bone marrow B cells were performed using mice of 8–12 weeks old, while that of B cells from the peritoneal cavity were from mice of 4–6 weeks of age. To analyze germinal center generation, mice of 8–12 weeks of age were immunized intraperitoneally with 109 sheep red blood cells (Immunoresearch) in 150 microliters PBS. Spleens were collected 10 days after immunization and analyzed by flow cytometry to quantify germinal center B-cells using a panel of antibodies as described in Supplementary Materials and Methods. To assess proliferation of B1a cells, cells isolated from the peritoneal cavity of 8–12 weeks old Cd19-Cre+/− or Cd19-Cre+/−Dnmt3afl/fl animals, were stained with the CellTrace™ CFSE Cell Proliferation Kit (ThermoFisher Scientific) according to the manufacturer’s protocol and injected intraperitoneally into irradiated immunodeficient recipients (10-weeks old NSG mice). Eleven days following injection, recipient mice were euthanized, and peritoneal fluid was collected and a panel of antibodies as described in Supplementary Materials and Methods. CD11b+ B1a cells were analyzed for CFSE intensity by flow cytometry
Immunohistochemistry
For IHC staining, spleens were fixed in 10% buffered neutral formalin overnight followed by 70% ethanol until the tissues were processed. Spleens were then paraffin-embedded and sectioned into 20μm for staining. Bones were fixed in 10% buffered neutral formalin overnight followed by 2 hr of 70% ethanol until the tissues were processed. Hematoxylin and Eosin (H&E) staining, as well as PAX5 and CD5 immunostaining were performed on the sections using standard procedures. MYC immunohistochemistry was performed on the Leica Bond III automated staining platform using the Leica Biosystems.
LSK, B1a and CLL transfer experiments
Transfer of HSC:
Seven- to twelve-week-old female or male CD45.2+ Cd19-Cre+/−Dnmt3afl/fl were used as donors. Femurs, tibias, hips, and spines were isolated from donor mice, crushed, and ACK-lysed. Cells were enriched with anti-CD117 microbeads (Miltenyi Biotec, Bergish Gladbach, Germany) and then sorted to collect LSK (lineage– Sca-1+ c-Kit+) cells a panel of antibodies as described in Supplementary Materials and Methods.
Transfer of B1a cells:
Six- to eight-week-old female or male Cd19-Cre+/− or Cd19-Cre+/−Dnmt3afl/fl mice were used as donors. B1a cells from the peritoneal cavity were sorted and injected intraperitoneally into irradiated Cd45.1+ recipients, cells from of one donor mouse (50,000–100,000 cells) per one recipient mouse for total of five donors per genotype.
Transplantation of CLL cells:
Five million splenocytes from Cd19-Cre+/−Dnmt3afl/fl were resuspended in 100 microliters PBS and injected intravenously into 8–15-week-old NSG mice. Overall, CLL cells from 5 different donors were transplanted into recipient mice. Disease burden in the peripheral blood, spleen and bone marrow were evaluated by flow cytometry and IHC.
BCR stimulation, Notch stimulation and immunoblotting analysis
For analysis of BCR signaling transduction, purified splenic B-cells (3 × 106) or splenic CLL cells were stimulated in 1ml RPMI 1640 medium (Life Technologies, Woburn, MA) supplemented with 10% FCS, and 1% penicillin/streptomycin in the presence of 10μg/ml goat anti-mouse IgM (Southern Biotech, Birmingham, AL) for 5 or 15 min at 37°C. Cells were harvested, washed twice with cold phosphate-buffered saline (PBS), and lysed for 30 minutes on ice with RIPA buffer (Boston Bioproducts, Boston, MA) supplemented with a protease inhibitor mix (1 tablet in 10 ml RIPA buffer, Roche, San Francisco, CA) and PhosSTOP (Sigma Aldrich) according to manufacturer’s protocol. Cell debris was removed by centrifugation at 10,000×g for 15 minutes, followed by protein quantification using Pierce BCA protein assay kit (Thermo Scientific). Cell extracts were fractionated by SDS-PAGE and transferred to Immun-Blot PVDF or nitrocellulose membranes (Bio-Rad laboratories). Membranes were stained overnight at 4°C with primary antibodies and appropriate secondary antibodies, e.g., horseradish peroxidase-coupled goat antirabbit and goat anti-mouse antibodies (Rockland Immunochemicals Inc). The bands were visualized by using a Bio-Rad ChemiDoc touch imaging system. Antibodies are described in Supplementary Materials and Methods. Western-blot quantification was performed using imageJ (RRID:SCR_003070).
BH3 profiling
BH3 profiling was performed as described previously (17). Experiments were performed using B cells from the spleen and peritoneal cavity of 2–3 month old Cd19-Cre+/− or Cd19-Cre+/−Dnmt3afl/fl, immunomagnetically isolated (B cell Isolation Kit, Miltenyi Biotec). B cells from the peritoneal cavity were pooled together to increase total cell number. Cells were suspended in MEB2 buffer (150 mM mannitol, 10 mM HEPES-KOH pH 7.5, 150 mM KCI, 1 mM EGTA, 1 mM EDTA, 0.1% BSA, 5 mM succinate). Cells were added to a 384-well plate and incubated at 25°C for 60 mins combined with different BH3 peptides in 0.001% digitonin for permeabilization. Cell were fixed with 4% formaldehyde for 10 mins at RT followed by neutralization by adding N2 buffer (1.7 M Tris base, 1.25 M glycine, pH 9.1). Cells were stained overnight at 4°C with Hoechst 33342 (H3570, Invitrogen) and anti–cytochrome c–Alexa Fluor 488 (6H2.B4/612308) and analyzed using an Intellicyt iQue flow cytometer to determine the rate of loss of cytochrome c in response to each BH3 peptide. Splenic and peritoneal cavity cells were also stained with CD19, CD11b and CD5 antibodies and gated for B2 cells (CD19+) or CD11b+ B1a cells (CD19+;CD5+;CD11b+). Assays were generally conducted in triplicate. DMSO and alamethicin were used as negative and positive controls for cytochrome c release.
RNA collection
RNA extraction was performed on sorted splenic B cells or peritoneal cavity B1a cells from young, disease-free 4–6-week-old mice and from CLL cells isolated from the spleens of diseased mice and sorted to collect B220+CD5+IgK+ cells. Total RNA was extracted using Qiagen RNAeasy kit or Qiagen RNAeasy micro kit with on-column DNase treatment (Qiagen).
In vitro Drug treatment
CLL cells from different genetic CLL mouse models (3,4,15) were resuspended in RPMI 1640 medium supplemented with 10% FBS and cultured in 96-well tissue culture plates (50,000 cells/100 μL). Cells were treated with indicated concentrations of the BCL2 inhibitor ABT-199, BTK inhibitor Ibrutinib, Notch signaling inhibitor DAPT and Wnt signaling inhibitor LGK-974. After incubation for 24 hours at 37 ℃ with 5% CO2, cell viability was measured by CellTiter-Glo® Luminescent Assay and normalized by cells treated with the DMSO vehicle control. Human primary CLL cells were plated at 2 million/mL in complete RPMI and exposed to 50mM DAPT, 1nM venetoclax or 1uM ibrutinib overnight, followed by Cell-TiterGlo assessment of cellular viability, assessed in technical triplicates. OP9 and OP9-DLL1 were kindly provided by John C. Aster (Brigham and Women’s Hospital), were seeded in 48-well plates (250K stromal cells per well) 16hrs prior to incubation with samples, and then drug treatments were performed as above.
In vivo drug treatment
Transplantation studies were performed on 8–12 weeks old immunodeficient recipient NSG mice using viably cryopreserved splenocytes from Dnmt3afl/fl CLL animals. Five million CLL cells/recipient were resuspended in 100 microliters PBS and injected intravenously into NSG. Two weeks post-transplant, CLL burden in the peripheral blood of the transplanted recipients (NSGs) was analyzed by flow cytometry and the treatment was initiated. Vehicle control (4% DMSO in corn oil) or 30 mg/kg DAPT was administered daily by intraperitoneal injections for 14 days on a 5 days on-2 days off schedule. Disease burden in the peripheral blood was evaluated by flow cytometry.
RNA library preparation, sequencing and analysis
RNA was assessed for quality and quantity (RNA 6000 Nano LabChip kit on a 2100 Bioanalyzer, both from Agilent. RNA libraries were generated using NEBNext® Single Cell/Low Input RNA Library Prep Kit for Illumina (New England BioLabs) according to manufacturer’s protocol and checked for quality and quantified using the DNA-1000 kit (Agilent) on a 2100 Bioanalyzer. Libraries were sequenced with the Illumina Sequencing SP kit on two lanes to obtain 150-base paired-end reads. RNA-seq reads were aligned to the mouse reference genome mm10 using STAR (v2.4.0.1; RRID:SCR_004463) (18) and TPM (Transcripts Per Kilobase Million) value was used to measure gene expression. Differentially expressed genes were assessed using DESeq2 (RRID:SCR_000154) (19). Transcription factor and pathway analyses were performed using Enrichr (RRID:SCR_001575) and the TRRUST transcription factor database and the MsigDB hallmark gene sets, respectively.
Quantitative PCR
Total RNA was transcribed into cDNA using the SuperscriptII reverse transcription kit (Life Technologies). The expression of mRNAs was analyzed using the TaqMan Gene Expression Assays probes as described in Supplementary Materials and Methods. Target gene expression was normalized to the mean Ct values of the housekeeping gene Gapdh.
DNA isolation, IGHV-D-J gene rearrangement analysis
Genomic DNA was isolated using the QIAGEN DNeasy Blood & Tissue Kit (Qiagen). IGHV analysis: DNA from CLL cells was subjected to PCR as previously described (20).
DNA Library Preparation, HiSeq Sequencing and data analysis
Initial DNA sample quality assessment, DNA library preparation, sequencing and bioinformatics analysis were conducted at GENEWIZ, Inc. (South Plainfield, NJ, USA). Genomic DNA sample were quantified using Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and DNA integrity was checked with 0.6% agarose gel with 50–60 ng sample loaded in each well. SureSelectXT Exome Enrichment System for Illumina Paired-End Multiplexed Sequencing Library and SureSelect Human All Exon V5 bait library were used for target enrichment DNA library preparation following the manufacturer’s recommendations (Agilent, Santa Clara, CA, USA) and the standard low-input protocol which starts with 200 ng. Briefly, the genomic DNA was fragmented by acoustic shearing with a Covaris LE200 Focused Ultra-sonicator instrument. Fragmented DNAs were cleaned up and end repaired, as well as adenylated at the 3’ends. Adapters were ligated to the DNA fragments, and adapter-ligated DNA fragments were enriched with limited cycle PCR. Adapter-ligated DNA fragments were validated using Agilent TapeStation (Agilent Technologies, Palo Alto, CA, USA), and quantified using Qubit 2.0 Fluorometer. 750 ng adapter-ligated DNA fragments were hybridized with biotinylated RNA baits at 65 C for 24 hours. The hybrid DNAs were captured by streptavidin-coated magnetic beads. After extensive wash, the captured DNAs were amplified and indexed with Illumina indexing primers. Post-captured DNA libraries were validated using Agilent TapeStation and quantified using Qubit 2.0 Fluorometer and Real-Time PCR (Applied Biosystems, Carlsbad, CA, USA). Illumina reagents and kits for DNA library sequencing cluster generation and sequencing were used for enrichment DNA sequencing. Post-captured DNA libraries were multiplexed in equal molar mass, and pooled DNA libraries were clustered on one lane of a flow cell, using the cBOT from Illumina. After clustering, the flow cell was loaded on the Illumina HiSeq instrument according to manufacturer’s instructions. The samples were sequenced using a 2 × 150 paired-end (PE) configuration. Image analysis and base calling was conducted by the HiSeq Control Software (HCS 2.0) on the HiSeq instrument. Whole exome sequencing raw FASTQ files were aligned to Genome Reference Consortium Mouse Build 38 (mm10) and the resulting BAM files were further processed by marking duplicated reads and recalibrating base qualities. To perform the copy number analysis, read coverage counts were calculated across the exome intervals for all samples. A CNV panel of normal (PoN) was created using all of the samples. The tumor read coverage data was normalized using the PoN. All of the analysis was performed following the recommendation of the GATK Best Practices Workflows using GATK4 toolkit (RRID:SCR_001876).
Reduced representation bisulfite sequencing (RRBS) and data analysis
RRBS libraries were generated from 50 ng input DNA using the Tecan Ovation RRBS Methyl-Seq System following the manufacturer’s recommendation except that we performed scaled-down half reactions from restriction digest through final repair after adapter ligation. Libraries were PCR amplified for 12 cycles. Paired-end 100-base reads were generated on Illumina HiSeq2500 rapid flow cells using custom primers (Tecan) without adding a PhiX spike-in.
Illumina sequencing reads were quality-controlled and adapter clipped using fastQC (RRID:SCR_014583) and cutadapt (21). These reads were aligned to the mouse mm9 reference genome using BSmap (22). In order to determine the methylation state of all CpGs captured and assess the bisulfite conversion rate, we used the mcall module in the MOABS software suite (RRID:SCR_012071) with standard parameter settings (23). Differentially methylated regions (DMRs) were called using metilene version 0.2–8 (24). DMRs were defined to have an absolute minimum difference in methylation of 0.2 with a maximum distance of 300 nt between CpGs within a DMR and a minimum of 5 CpGs per DMR. Missing values were allowed per group. Adjusted P-values were calculated using the -c 2 parameters, invoking Benjamini-Hochberg correction (FDR). For heatmap visualization, methylation levels of DMRs were calculated as the mean methylation of all CpGs with a DMR for each sample and plotted using the heatmap function of the R package pheatmap (RRID:SCR_016418). A DMR was associated with a gene if it overlapped or was in the promoter region (−2500nt to +500nt of TSS) of the respective gene.
Discordant read methylation was calculated from each read based on the fraction of transitions in methylation status (0 or 1) between adjacent CpGs. Paired-end reads overlapping each other were cut in the middle of the overlap to avoid interpretation of the same molecule twice and reads covering less than 3 CpGs were excluded from this analysis. For each read the methylation status of the covered CpGs was determined as unmethylated (converted base) or methylated (unconverted base). Based on this, the number of transitions in methylation status along the read were calculated and normalized to the number of possible transitions.
Data availability
The raw data described in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO) -- Accession numbers: GSE169245, GSE143711 and GSE122668.
Results
Reduced DNMT3A expression is commonly observed in CLL
We reanalyzed transcriptome (RNA-seq) data generated from 107 human CLL samples and CD19+ B cells from 8 healthy donors (HDs) (9) to evaluate the expression of DNMT3A, DNMT3B and DNMT1. We observed robust expression of DNMT3A and DNMT1 in CLLs and HDs, whereas DNMT3B was poorly expressed (Supplementary Fig. 1A). Notably, DNMT3A expression was highly variable amongst these CLL patients, and lower DNMT3A expression was associated with more aggressive disease with shorter failure-free survival (p=0.013) (Fig. 1A). We confirmed the high variability in DNMT3A expression in CLL in a separate set of 6 HDs and 17 CLLs (Supplementary Fig. 1B). We also validated the correlation between more aggressive disease and low DNMT3A expression in another independent cohort consisting of 127 treatment-naïve CLL patients (Supplementary Fig. 1C, p=0.02). Complementing this analysis, we examined DNMT3A protein expression in 25 CLLs and 6 HDs by western blot and again found a high degree of expression variability but overall lower expression of DNMT3A in CLLs (Supplementary Fig. 1D-F, Supplementary Table 1, p<0.001). Given the known key role of DNMT3A across various hematologic malignancies (25–27) and its association with more aggressive CLL that we observed, we sought to evaluate the potential impact for this lesion as a CLL driving event.
Figure 1: B cell-restricted deletion of Dnmt3a leads to CLL.
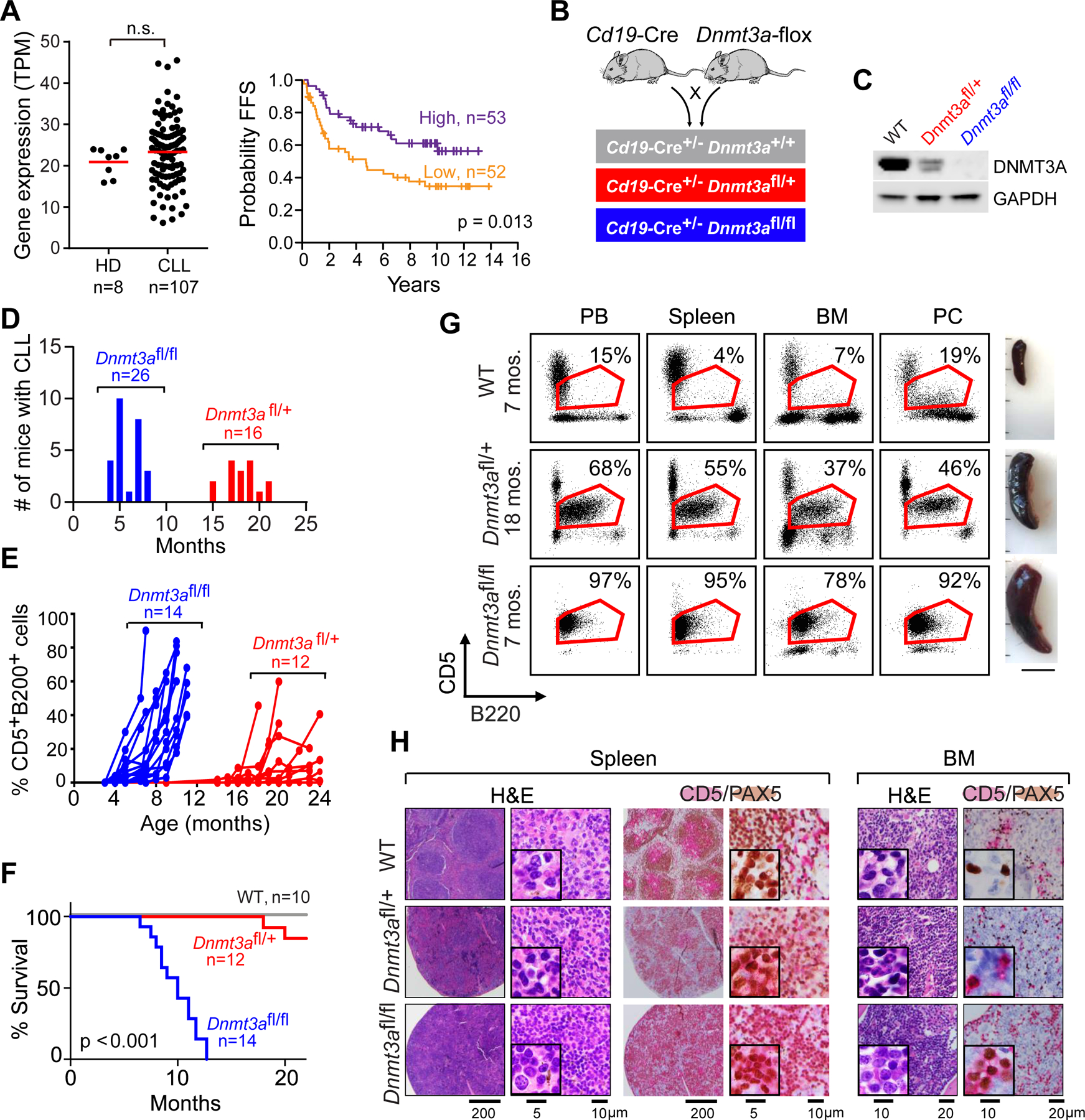
A, DNMT3A expression in human normal B cells (HD) and CLLs. Failure-free survival (FFS) from the time of sequencing sample to 1st treatment/progression or death in the DNMT3A high or low group is shown. Samples were stratified by median DNMT3A expression; there were 2 dropouts. B, Schema of the breeding strategy. C, Representative western-blots of DNMT3A expression in splenic B cells across the different genotypes (n=3). D, Age of disease onset in Dnmt3afl/+ and Dnmt3afl/fl mice. E, Detection of B220+CD5+ cells in peripheral blood over time. F, Overall survival of WT, Dnmt3afl/+ and Dnmt3afl/fl mice over time. G, Representative flow cytometry analysis of B220+ CD5+ cells within peripheral blood (PB), spleen, bone marrow (BM) and peritoneal cavity (PC) of WT, Dnmt3afl/+ and Dnmt3afl/fl CLL mice and images of the spleens. H, Representative immunohistochemical staining of CD5+(red) PAX5+ (brown) of spleen and bone marrow (BM) sections from WT and Dnmt3afl/fl CLL mice.
B cell-restricted deletion of Dnmt3a leads to generation of CLL
To investigate the role of DNMT3A depletion in vivo, we generated a B cell-restricted Dnmt3a knockout mouse model by intercrossing Cd19-Cre and Dnmt3a-LoxP mice (Fig. 1B), resulting in cohorts of wild-type (Cd19-Cre+/−, WT), heterozygous knockout (Cd19-Cre+/− Dnmt3afl/+, referred as Dnmt3afl/+) and homozygous knockout (Cd19-Cre+/−Dnmt3afl/fl, referred as Dnmt3afl/fl) mice. Successful Dnmt3a knockout was confirmed by western blot (Fig. 1C), and different groups of mice were monitored for the development of CLL through monthly flow cytometric analysis of peripheral blood for the appearance of B220+CD5+ cells up to 24 months of age. Dnmt3afl/fl mice consistently developed CLL between 4–8 months of age (median onset of 5.3, n=26) with 100% penetrance (Fig. 1D). In contrast, Dnmt3afl/+ mice developed CLL/MBL between 15–21 months of age (median of 18.5, n=16 with 89% penetrance, while none of the Cd19-Cre+/− control mice developed CLL. No malignancies other than CLL were observed in Dnmt3a knockout mice over this time period, as confirmed by IHC. A subset of mice was further monitored for disease progression and survival, showing that CLL progressed rapidly in the Dnmt3afl/fl mice (n=14), resulting in poorer survival than the Dnmt3afl/+ mice (n=12) (Fig. 1E, F).
Mice with CLL consistently displayed enlarged spleens, with infiltration of CLL cells in the peripheral blood, spleen, bone marrow and peritoneal cavity of affected mice (Fig. 1G). The presence of CLL cells in the bone marrow and spleen was confirmed by co-localization of CD5 (red) and PAX5 (brown) expression within the tissue sections (Fig. 1H). We confirmed that the CLL cells were clonal based on Igκ expression and immunoglobulin heavy chain gene variable region (IGHV) mutational status (20), which was found to be unmutated (i.e.100% homology with germline) in all five samples tested (Supplementary Table 2). We further confirmed the transplantability of the CLL cells. Following intravenous injection of CLL cells into immunodeficient mice (NSG, n=5), we consistently detected CLL cells in the peripheral blood of recipient mice 4 weeks following transplant. In each instance, upon euthanasia at 6 weeks following injection, these CLL cells were also detected in the spleen, bone marrow and peritoneal cavity by flow cytometry (Supplementary Fig. 1G). Altogether, we demonstrate that CD19-restricted deletion of Dnmt3a reliably results in CLL, confirming this lesion as a CLL driving event.
B-cell restricted loss of Dnmt3a expression alters B cell development in preleukemic animals
We examined the impact of Dnmt3a depletion on normal B cell development in 2–3 month old pre-leukemic mice using previously established flow cytometry conditions (Supplementary Fig. 2A, B) (3,4). Splenocytes from Dnmt3a knockout mice were reduced in proportion of marginal zone cells (MZ; B220+;CD23-;CD21+, p<0.001, Student’s t-test) and increased in proportion of follicular B cells (FO; B220+;CD23+;CD21-, p<0.02, Student’s t-test) compared to WT mice (Fig. 2A). In contrast, no proportion differences in bone marrow B cell subpopulations were observed (Supplementary Fig. 2C). Of note, splenic B cells from Dnmt3a knockout mice exhibited no alteration in BCR signaling activity (Supplementary Fig. 2D) or immune response to sheep red blood cells (SRBCs) (Supplementary Fig. 2E) compared to WT B cells, which differs from our previously reported CLL mouse models (3,4).
Figure 2: B1a cells expansion in the peritoneal cavity of Dnmt3afl/fl mice results in CLL development.
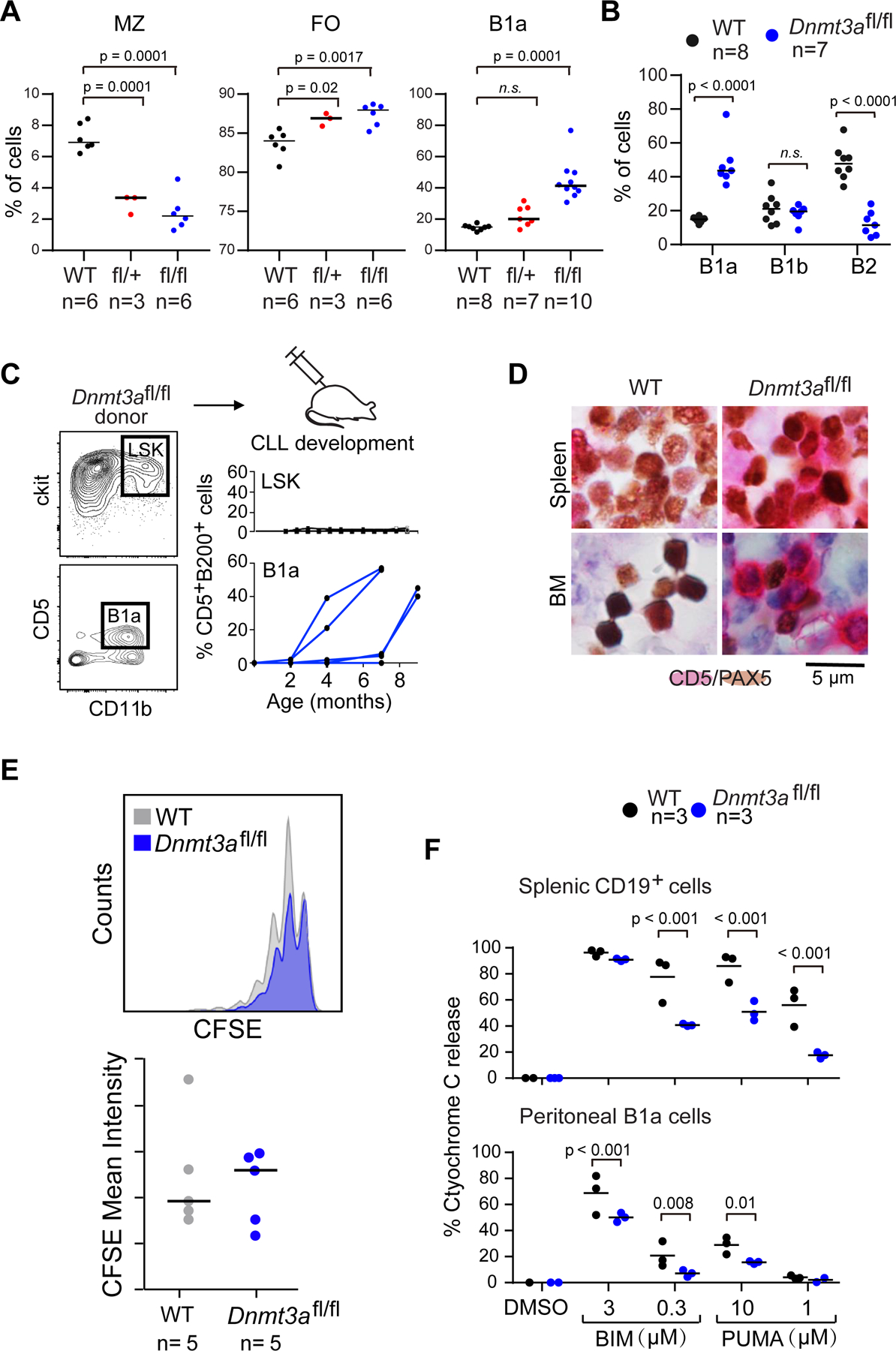
A, Analysis of the percentage of marginal zone B cells (MZ), follicular B cells (FO) from the total splenocytes suspension and the percentage of B1a from peritoneal B cells of WT, Dnmt3afl/fl mice. Data were compared using one way ANOVA followed by Dunnett multiple comparison test. B, Proportions of bone marrow B cell subpopulations. C, Transfer of Lin-;ScaI+;c-kit+ (LSK) or B1a cells from Dnmt3afl/fl or WT donor mice to CD45.1 recipient mice and flow cytometry analysis of B220+ CD5+ cells within peripheral blood over time (n=5). D, Representative immunohistochemical staining of CD5+(red) and PAX5+ (brown) of spleen and bone narrow (BM) sections from CD45.1 recipient mice following tranfer of B1a cells from WT and Dnmt3afl/fl CLL mice. E, B1a cells were isolated from WT and Dnmt3afl/fl mice, stained with CSFE and injected intraperitoneally into CD45.1 recipient mice. B1a cells were analyzed for CFSE intensity by flow cytometry 11 days post injection. F, BH3 profiles of peritoneal B1a cells and splenic CD19+ cells.
We detected a substantially increased proportion of CD11b+ B1a cells (CD19+;CD43+;CD5+;CD11b+) in the peritoneal cavity of Dnmt3afl/fl mice (Fig. 2B), suggesting B1a cells as the cell-of-origin of CLL in this model system. Testing this hypothesis, we transplanted marrow hematopoietic stem cells (LSK cells, n=5) or B1a cells (n=5) isolated from Dnmt3afl/fl mice into irradiated CD45.1+ recipient mice. While no recipient mice transplanted with LSKs developed CLL up to 12 months following transplantation, clonal CD45.2+B220+CD5+Igk+ CLL cells were detectable in peripheral blood of recipient mice transplanted with B1a cells starting 10 weeks after transplantation (Fig. 2C). IHC staining of splenic and bone marrow tissue sections confirmed the presence of CD5 and PAX5 double positive CLL cells among the engrafted cells (Fig. 2D). Infiltration of CD45.2 CLL cells was evident in the peripheral blood, spleen, bone marrow and peritoneal cavity of affected mice (Supplementary Fig. 2F-G). Consistent with the parental model, CLL cells exhibited no obvious alteration in BCR signaling activity (Supplementary Fig. 2H). Together, these data support the notion that CLL cells in this model arise from the B1a population.
To determine the mechanism of Dnmt3afl/fl B1a cell expansion, we analyzed cell division of B1a cells from WT or Dnmt3afl/fl animals using carboxyfluorescein succinimidyl ester (CFSE) staining. Peritoneal cells were isolated, labeled with CFSE and injected intraperitoneally into irradiated immunodeficient recipients (NSG, n=5). Cell division was assessed by measuring the corresponding decrease in cell fluorescence via flow cytometry. Since no difference (p=0.72) in cellular proliferation between the two groups was observed (Fig. 2E), we asked whether Dnmt3afl/fl B1a expansion was caused by reduced cell death. Indeed, both splenic CD19+ B cells and peritoneal B1a cells from Dnmt3afl/fl mice showed reduced apoptotic priming, induced by BH3 peptides (i.e, BIM and PUMA), compared to WT cells (Fig. 2F). Dnmt3a depletion thus appears to affect B1a expansion through a reduction in cell death, rather than increased proliferation.
Dnmt3a deletion alters the methylomes and transcriptomes of preleukemic B1a cells
Given the known role of Dnmt3a as a de novo methyltransferase, we evaluated the impact of Dnmt3a depletion on global DNA methylation of preleukemic B1a cells (i.e. absence of circulating CLL cells at the time of analysis) and of B2 cells (as a comparator population), using reduced representation bisulfite sequencing (RRBS). Although no difference in the average CGI methylation levels was observed between Dnmt3afl/fl versus WT B cells (n=3 per group; Fig. 3A), increased local methylation disorder (measured by percent discordant reads [PDR]) was observed in CGI shelves and background regions (i.e. CpG sites that are not in CpG islands, shores or shelves) in Dnmt3afl/fl B1a cells compared to WT B1a cells (Fig. 3B). We identified a set of differentially methylated regions (DMRs) (difference>0.2, Supplementary Table 3), mostly hypomethylated, in Dnmt3afl/fl versus WT B1a cells (Fig. 3C, D). As representative examples, hypomethylated DMRs in the promoter regions of the Cxcr7, Wnt10b and Hes5 genes are shown in Fig. 3E. In contrast, only minor changes in methylation were observed in Dnmt3afl/fl B2 cells. Notably, complete depletion of Dnmt3a was confirmed by RNA-seq in both Dnmt3afl/fl B1a and B2 cells (Supplementary Fig. 3A), suggesting that the methylation differences between the cell types could not be attributed to incomplete knockout of Dnmt3a in B2 cells. As DNMTs might have overlapping functions to regulate DNA methylation, we quantified the expression of Dnmt3a, Dnmt3b and Dnmt1 by real-time PCR in these cells. Our results revealed higher levels of Dnmt3b (p=0.005) in B2 cells compared to B1a cells (Supplementary Fig. 3B), suggesting that Dnmt3b might functionally compensate for the loss of Dnmt3a in Dnmt3afl/fl B2 cells. We further confirmed that Dnmt3b1, the active isoform of Dnmt3b gene, was stably expressed in Dnmt3afl/fl B2 cells (Supplementary Fig. 3C). Pathway analysis revealed that genes associated with those DMRs were highly enriched in Notch, TNF-α, TGF-β, and IL-2/STAT5 signaling, and apoptosis (Supplementary Table 4).
Figure 3: Changes in methylatome and transcriptome associated with Dnmt3a depletion in pre-leukemia B cells.
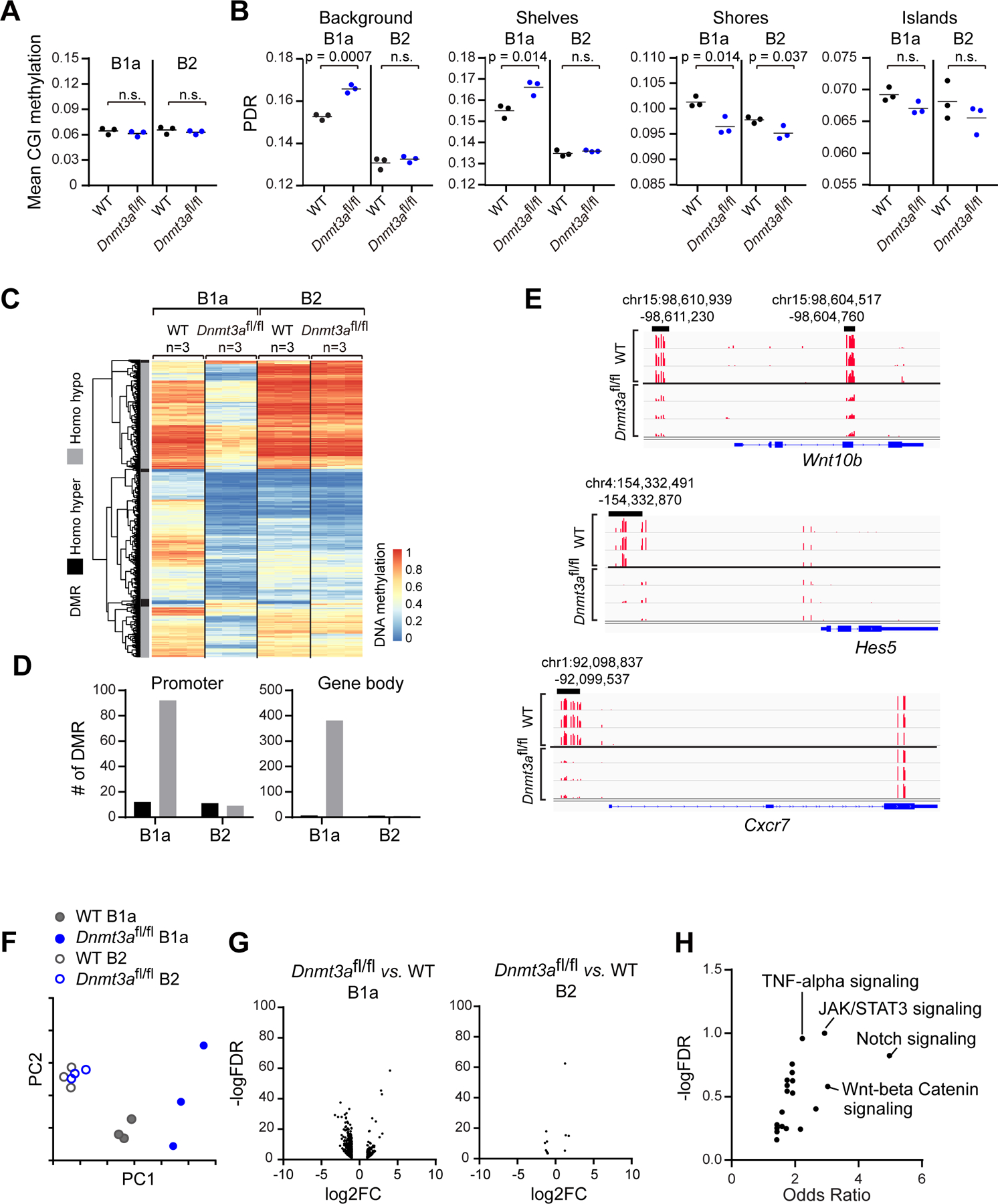
A, Mean CpG island (CGI) methylation in WT (n=3) and Dnmt3afl/fl (n=3) B1a or B2 cells. B, Percent discordant reads (PDR) associated with different DNA regions in different cells. CpG islands (CGIs) were downloaded from the UCSC genome browser (defined as regions > 500 bp, > 55% GC and expected/observed CpG ratio of >0.65), CGI shores +/−2Kb from islands, CpG shelves +−2Kb from CGI shores. All the other regions were defined as background. (n=3) C, Heatmap of differentially methylated regions (DMRs) between WT and Dnmt3afl/fl B1a or B2 cells (n=3 each). D, Number of DMRs between Dnmt3afl/fl and WT B1a cells associated with promoter regions or gene body. E, Examples of DMRs in Dnmt3afl/fl B1a cells versus WT B1a cells. F, PCA analysis of transcriptome data of WT and Dnmt3afl/fl B1a or B2 cells. G, Volcano plot of differentially expressed genes between Dnmt3afl/fl vs. WT B1a or B2 cells. H, Signaling pathways enriched for differentially expressed genes in Dnmt3afl/fl B1a cells versus WT B1a cells.
Given the prominent hypomethylation changes observed in Dnmt3a depleted B1a cells, we investigated whether these would lead to altered gene transcript expression. Indeed, PCA analysis of bulk transcriptomes generated from Dnmt3afl/fl and WT B1a, and B2 cells (n=3 per group) revealed clear differences between Dnmt3afl/fl and WT B1a cells, but not between Dnmt3afl/fl and WT B2 cells, in line with the methylation changes (Fig. 3F). However, in Dnmt3a depleted B1a cells, hypomethylation events were not necessarily associated with upregulation of gene expression (Supplementary Fig. 3D), suggesting that the methylation status per se was insufficient to determine gene expression, in line with prior findings (28). Nevertheless, we detected 460 downregulated and 168 upregulated genes in the Dnmt3afl/fl B1a cells compared to WT B1a cells (Fig. 3G-left, FDR<0.05, fold change >2; Supplementary Table 5), while the changes in B2 cells were negligible (Fig. 3G-right). Differentially expressed genes in Dnmt3afl/fl B1a were highly enriched for several oncogenic signaling pathways, including Notch, TNF-α, JAK/STAT3 and Wnt-beta catenin signaling (Fig. 3H, Supplementary Table 6).
Changes in methylome and transcriptome associated with CLL development
Upon investigating the methylomes of the CLL cells arising in Dnmt3afl/fl animals compared to WT and Dnmt3afl/fl B1a cells, we observed a marked increase in CGI methylation rate in CLL cells (Fig. 4A). This was accompanied by increased PDR levels in background regions and CpG shelves, and decreased PDR levels in CpG islands (Fig. 4B). Notably, we identified multiple DMRs in CLL cells compared to either WT or Dnmt3afl/fl B1a cells (difference>0.2; Fig. 4C-E, Supplementary Tables 7-8), a large fraction of which were hypermethylated. In the promoter regions of the CLL cells, we identified 1514 hypermethylated and 247 hypomethylated DMRs compared to WT B1a cells, and 1574 hypermethylated and 103 hypomethylated DMRs compared to Dnmt3afl/fl B1a cells. In gene body regions, we identified 855 hypermethylated and 1088 hypomethylated DMRs compared to WT B1a cells, and 892 hypermethylated and 503 hypomethylated DMRs compared to Dnmt3afl/fl B1a cells (examples of the differentially methylated DMRs in the promoters shown in Fig. 4F). Of the 466 hypomethylated DMRs observed in the pre-leukemia Dnmt3afl/fl B1a cells (Supplementary Table 3), only 45 remained hypomethylated in Dnmt3afl/fl CLL cells compared to WT B1a (including Hes5), and the others remained unchanged (n=383) or even hypermethylated (n=38), suggesting a dramatic reprogramming of the methylation profile associated with progression to CLL. Genes with promoter and/or gene body DMRs in CLL compared to WT B1a cells were highly enriched for several oncogenic signaling pathways including Notch and Hedgehog signaling, epithelial mesenchymal transition and Wnt signaling (Supplementary Table 9).
Figure 4: Changes in methylome and transcriptome associated with Dnmt3a-depleted CLL cells.
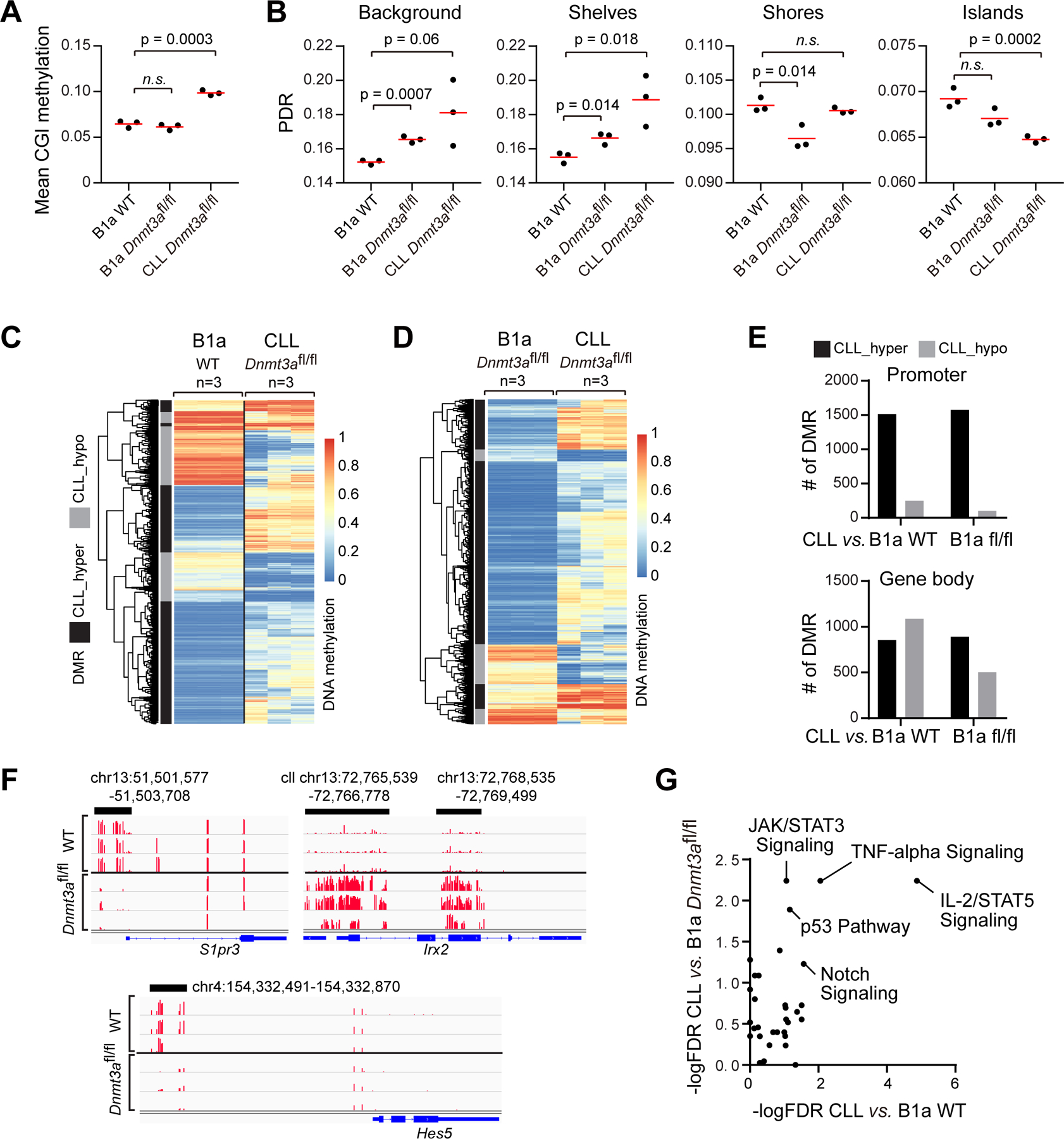
A, Mean CGI methylation in WT and Dnmt3afl/fl B1a or CLL cells (n=3). B, PDR associated with different DNA regions in different cells. CpG islands (CGIs) were downloaded from the UCSC genome browser (defined as regions > 500 bp, > 55% GC and expected/observed CpG ratio of >0.65), CGI shores +/−2Kb from islands, CpG shelves +−2Kb from CGI shores. (n=3) C, Heatmap of DMRs between Dnmt3afl/fl CLL cells and WT B1a. D, Heatmap of DMRs between Dnmt3afl/fl B1a and Dnmt3afl/fl CLL cells (n=3 each). E, Number of DMRs in Dnmt3afl/fl CLLs versus Dnmt3afl/fl or WT B1a cells associated with promoter regions or gene body. F, Examples of DMRs in Dnmt3afl/fl CLL cells versus WT B1a cells. G, Signaling pathways enriched for differentially expressed genes between Dnmt3afl/fl CLL cells versus WT B1a cells, as well as between Dnmt3afl/fl CLL cells versus Dnmt3afl/fl B1a cells.
RNA-seq analysis of Dnmt3afl/+ and Dnmt3afl/fl CLLs (n=3 each) revealed that expression changes in Dnmt3afl/+ and Dnmt3afl/fl CLLs compared to WT B1a cells were highly concordant (Supplementary Fig. 3E, R=0.72, p<0.0001), suggesting a consistent transcriptional reprogramming during CLL development. We thus combined the two groups to identify commonly altered genes in CLL cells. We identified more upregulated (n=2801) than downregulated (n=1244) genes in CLL cells compared to WT B1a cells (FDR<0.05, FC>2; Supplementary Table 10). Moreover, we identified 3030 upregulated and 1652 downregulated genes in CLL versus Dnmt3afl/fl B1a cells (Supplementary Table 11). To determine the signaling pathways affected in CLL cells, we performed an unbiased pathway enrichment analysis of these differentially expressed genes using MSigDB hallmark gene sets. This allowed us to identify several pathways that were commonly enriched in both comparisons; again, we observed enrichment of Notch signaling genes (Fig. 4G, Supplementary Table 12), pointing to its putative important role in CLL development.
Compared to methylome data of human CLLs (11), we identified 20 hypermethylated genes and 210 hypomethylated genes that are shared between human and mouse CLLs. The 444 DMRs of these genes in human CLLs are shown in Supplementary Table 13. These genes were highly enriched for TGF-beta signaling, UV response and mitotic spindle (all FDR<0.005), supporting a role of DNA damage repair pathway in CLL development. To determine the gene signatures associated with poor DNMT3A expression in human CLL, we reanalyzed transcriptome profiles of 8 samples with the lowest DNMT3A expression in the cohort (Fig. 1A). Compared to normal B cells (n=8), altogether there were 715 upregulated and 428 downregulated genes in these CLLs (FC>2, FDR<0.05). Of these genes, we identified 104 upregulated and 18 downregulated genes that overlapped with the Dnmt3a mouse CLL model (Supplementary Table 14). Pathway analysis revealed that these genes were highly enriched for apical junction, IL-2/STAT5 signaling and complement (all FDR<0.05).
Hyperactivation of Notch and Myc signaling in Dnmt3afl/fl CLL
Closer inspection of the role of the Notch signaling pathway in the Dnmt3a-depleted CLL models revealed a general upregulation of Notch signaling genes in CLL cells compared with either WT or Dnmt3afl/fl B1a cells (Fig. 5A). To investigate whether upregulation of Notch signaling genes was associated with abnormal activation of the signaling pathway, we examined expression changes of known Notch targets in these CLL cells (29). As a comparison, we also examined Notch target expression in 3 other CLL genetically-engineered mouse models, namely those driven by MDR, Ikzf3 or Sf3b1-Atm lesions (3,4,15) (Fig. 5B). The distinct features of these mouse models are summarized in Supplementary Fig. 3F. Indeed, we observed high variability of Notch target expression across the different CLL models, with selective upregulation of Notch targets exclusively observed in the Dnmt3a-depleted CLL model but not in others (Fig. 5C). The results remained robust even after we separated Dnmt3afl/+ CLLs from Dnmt3afl/fl CLLs, supporting a unique role of Notch activation in this model (Supplementary Fig. 3G). We were able to validate upregulation of several well-established Notch targets (i.e., Hes5, Dtx1 and St3gal6) in CLL cells versus normal B cells (Fig. 5D).
Figure 5: Activation of Notch-Myc signaling in Dnmt3afl/fl CLLs.
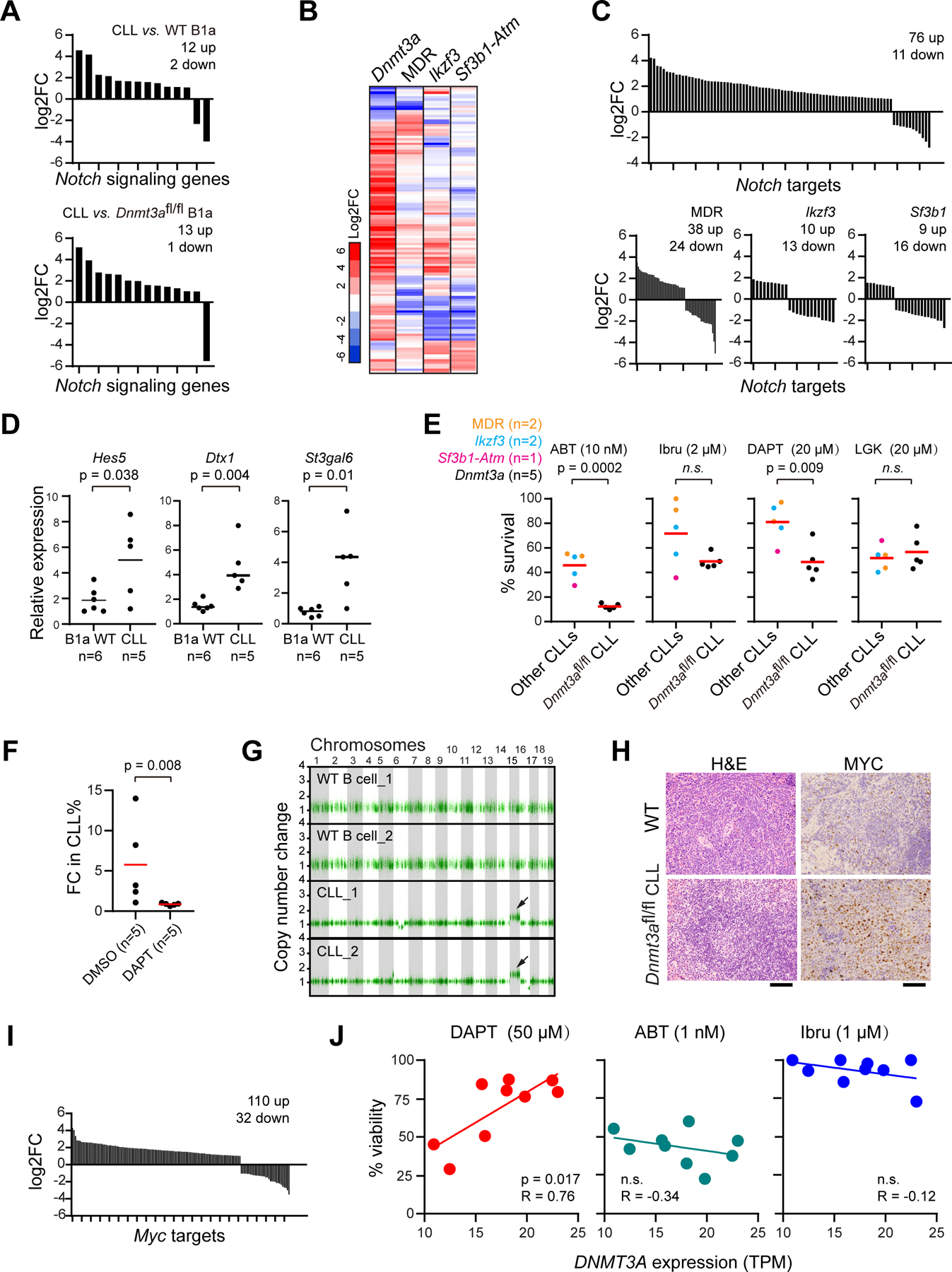
A, Changes in expression of significantly altered Notch signaling genes in Dnmt3afl/fl CLLs versus WT B1a cells. B, Heatmap of change in expression of Notch signaling genes in different mouse models. C, Changes in significantly altered Notch target genes in different CLL mouse models. D, qPCR analysis of expression of well-known Notch target genes in different cells. E, Sensitivity of different CLL cells with various driver mutations to different drugs, including the BCL2 inhibitor ABT-199 (venetoclax, ABT), BTK inhibitor Ibrutinib (Ibru), the Notch signaling inhibitor DAPT, and Wnt signaling inhibitor LGK-974 (LGK). F, Fold-change (day 14 vs. day 0) of the percentage of circulating CLL cells of transplanted mice treated with DMSO or 30mg/kg DAPT. G, Copy number analysis of WT B cells or Dnmt3afl/fl CLL (n=2). Arrows: Amplification of Chr 15. H, IHC analysis of Myc in spleen sections of Dnmt3afl/fl CLLs versus WT B1a cells. I, Change in expression of Myc targets in Dnmt3afl/fl CLLs versus WT B1a cells. J, The correlation between DNMT3A expression and response to different drugs in human primary CLLs (n=9) in vivo, measured by CellTiterGlo.
To examine the therapeutic vulnerabilities of the diverse genetic CLL models to various drugs, we exposed CLL cells to the BCL2 inhibitor ABT-199 (venetoclax, ABT), BTK inhibitor Ibrutinib (Ibru), the Notch signaling inhibitor daptomycin (DAPT), the Wnt signaling inhibitor LGK-974 (LGK) and DMSO. The optimal working concentration of different drugs had been determined by treating tumor cells with multiple doses (Supplementary Fig. 4A). CLL cells from the 4 distinct CLL models were treated with two concentrations of each drug for 24 hours, and percent survival was calculated as the ratio between the cellular viability following test drug exposure and DMSO. Dnmt3a-depleted CLLs were more sensitive to ABT than the other CLLs, consistent with the BH3 results suggesting that Dnmt3a-depleted CLLs were more dependent on BCL2 for survival (Supplementary Fig. 4B). Moreover, Dnmt3a-depleted CLL cells were highly sensitive to Notch inhibition by DAPT (Fig. 5E, Supplementary Fig. 4C), supporting hyperactivation of Notch signaling in these CLL cells and high dependency on this pathway for survival. In vivo, NSG mice transplanted with Dnmt3afl/fl CLLs and subsequently treated with DAPT (30mg/kg; n=5) or DMSO (n=5) for 14 days likewise showed highly sensitivity to treatment with Notch inhibition (Fig. 5F, p=0.008).
To determine if Dnmt3afl/fl CLLs had a distinct genetic profile, we performed whole-genome sequencing of the CLL cells. Notably, our analysis revealed amplification of chromosome 15 in these CLL cells (Fig. 5G), consistent with our other genetically-engineered CLL models (3,4), and thereby suggesting a common oncogenic mechanism contributing to CLL generation. A putative oncogenic driver localized in chromosome 15 is Myc, a direct downstream target activated by Notch signaling. Consistent with Myc copy number gain, immunohistochemical (IHC) staining of spleen sections also revealed strong upregulation of MYC protein expression in Dnmt3afl/fl CLLs compared to WT B cells (Fig. 5H). In line with this notion, we observed that a large fraction of known Myc targets in B cells (30) were preferentially upregulated in CLL cells versus normal B cells (Fig. 5I). In contrast, very few somatic mutations (26 and 32, respectively) were identified in these CLL samples (Supplementary Table 15).
To determine if the aforementioned insights identified in our mouse model were reflected in human CLL, we evaluated the relationship between baseline expression of DNMT3A and sensitivity to Notch inhibition in primary untreated CLL samples (Supplementary Table 16). Nine samples with differing levels of DNMT3A were exposed to 50 μM DAPT, 1 nM ABT-100 or 1 μM ibrutinib overnight followed by Cell-TiterGlo assessment of cellular viability (31–33). Primary CLLs with lower expression of DNMT3A were more sensitive to DAPT, but not to the other two drugs, supporting a specific dependence on Notch signaling for survival (Fig. 5J). Moreover, activation of Notch signaling through co-culturing with Notch ligand-expressing OP9 stroma could sensitize DNMT3A-high CLLs (n=5) to DAPT (Supplementary Fig. 4D), again supporting a direct link between Notch activity and the observed drug response.
Discussion
DNMT3A is essential for de novo DNA methylation and its inactivation in embryos results in lethally of mice at 4 weeks of age (34). In blood malignancies, dysregulation of Dnmt3a, whether by mutation or altered expression, is increasingly appreciated to be crucial to the pathogenesis of both myeloid and lymphoid malignancies. Dnmt3a is essential for hematopoietic stem cells (HSC) differentiation, and hematopoietic lineage skewing was observed in its absence (35,36). Previous murine model studies have only occasionally identified CLL resulting from disruption of Dnmt3a expression, while developing aggressive myeloid or lymphoid neoplasms with early (<12 month) onset (Supplementary Table 17). Conditional knock-out of Dnmt3a in Mx1-Cre;Dnmt3afl/fl mice by pIpC treatment, followed by transplantation of whole BM cells predominantly resulted in myeloid neoplasms, including myelodysplastic syndromes (MDS, 76%), acute myelogenous leukemia (AML, 8%) and MDS/myeloproliferative neoplasms (MPN) (16%) (37). Interestingly, deletion of Dnmt3a together with expression of mutant c-Kit lead to the development of B-ALL in 25% of the mice, suggesting cooperation between the two mutations is necessary for acute B cell lymphomagenesis (37). In parallel studies, irradiated mice transplanted with Mx1-Cre;Dnmt3afl/fl HSCs develop wide spectrum of blood malignancies, including T-cell and B-cell acute lymphocytic leukemia, myelodysplastic syndrome, acute myeloid leukemia and primary myelofibrosis (16,38,39). In contrast, when homozygous Dnmt3a knock-out in HSCs through the EmSR-tTA;Teto-Cre system, peripheral T cell lymphomas (PTCL) and CLL were observed, whereas heterozygous deletion of Dnmt3a resulted in MBL/CLL (65% of the cases) and myeloproliferative disease (MPD, 15%) (40–42). In another study, deletion of Dnmt3a specifically in B cells resulted in expansion of B1a cells and development of monoclonal leukemia (43). Finally, deletion of Dnmt3a using the Vav-Cre;Dnmt3afl/fl model resulted in MDS (53%), AML (7%), MPN (7%) and mixed phenotype acute leukemia (13%) (44). The heterogeneity of disease phenotypes observed in these studies may be explained by the distinct conditional strategies, as Mx1-cre and Vav1-cre are known to impact transgene expression also in non-HSCs populations, including endothelial and bone marrow mesenchymal cells (45). Overall, all these studies support the role of Dnmt3a as a tumor suppressor across cell lineages.
In the current study, by restricting Dnmt3a loss to B cells through intercrossing with CD19-Cre mice, we detected only the generation of CLL (without occurrence of myeloid disease or PTCL). Alterations in methylation produced by lack of DNMT3A present within the distinct cellular context and transcriptional regulatory milieu of B1a cells consistently led to disease. Notably, we identified that DNMT3A depletion resulted in the activation of the key CLL-associated pathway of Notch signaling as well as Myc overexpression. Thus, distinct from other existing genetically engineered mouse models of CLL which prominently feature dysregulation of BCR signaling, this DNMT3A-depleted model most critically dysregulates Notch. As NOTCH mutations and altered expression have been highly linked to CLL generation, our results provide a novel mechanism linking epigenetic alterations in CLL to NOTCH dysregulation.
Our analysis of the changes in the methylation profiles of our heterozygous and homozygous Dnmt3a knockout mouse models and the mechanism of Notch activation within these models provided us with insight into the process by which Dnmt3a depletion exerts its pro-oncogenic activity. To determine the influence of Dnmt3a depletion on methylation we performed RRBS analysis. The principle of RRBS is to focus coverage on CpG dense regions of the genome, but not the whole genome, in order to reduce sequencing requirements and cost. Using RRBS, about 50% of covered CpGs are located in CpG islands (CGIs) and the remaining CpGs span other CpG dense regions including gene bodies and repeats. Of note, CGIs are often located close to gene promoters, and methylation of CGIs is one of the major mechanisms of gene expression regulation (46). We observed two levels of change in the DNA methylation profiles within our models. In the pre-leukemic setting, we found that loss of Dnmt3a induced limited but significant hypomethylation whose impact was reduced apoptotic priming and facilitated accumulation of B1a cells. Following CLL transformation, we observed the CLL cells to become hypermethylated. This process was seemingly independent of Dnmt3a function as this is also observed in other CLL mouse models with WT Dnmt3a, suggesting that the hypermethylation is a response to, rather than a cause of, CLL progression, in line with prior findings (28). In combination with Dnmt3a-associated methylation changes, these result in transcriptional dysregulation of several oncogenic pathways that are important for CLL development, particularly Notch signaling.
Altogether, we identified an association between low DNMT3A expression and poor failure-free survival in human datasets, and have confirmed a causal role of Dnmt3a depletion in CLL generation in mice. Our results support the interaction between Dnmt3a-dependent methylation change and activation of Notch and Myc signaling as a mechanism by which Dnmt3a depletion induces CLL. Moreover, the Dnmt3a models provide a unique opportunity for the study of non-mutational Notch activation, and a useful platform for the study of Notch-signaling targeted therapeutics.
Supplementary Material
Statement of significance:
Loss of DNMT3A expression is a driving event in CLL and is associated with aggressive disease, activation of Notch and Myc signaling, and enhanced sensitivity to Notch inhibition.
Acknowledgements
The authors thank the members of the Wu lab, in particular Drs. Erin Parry and Satyen Gohil for their valuable feedback and critical insights. The authors are also grateful for the Dana-Farber Cancer Institute Animal Research Facility technical team, and Flow Cytometry Core for truly excellent technical support and the Dana-Farber/Harvard Cancer Center in Boston, MA, for the use of the Specialized Histopathology Core. Dana-Farber/Harvard Cancer Center is supported in part by an NCI Cancer Center Support Grant (NIH 5 P30 CA06516). The authors are also grateful to Binyamin Knisbacher, Cynthia Hahn, John Aster, Oriol Olive, Adrian Wiestner and Tuan Tran for their generous support. This study was supported by grants from the NIH/National Cancer Institute (NIH/NCI) (P01 CA206978, P01-CA081534, R01CA216273, UG1CA233338). This work was further supported in part by the Lymphoma Research Foundation (A.B) and the Leukemia Lymphoma Society (A.B). A.B is an Awardee of the Weizmann Institute of Science – Israel National Postdoctoral Award Program for Advancing Women in Science. E.t.H. is a Scholar of the American Society of Hematology. A.M. is supported by the Max Planck Society). C.S is supported by the Intramural Research Program of the NHLBI, NIH. E.T. and S.S. received research support by DFG SFB1074 subproject B1 and B2. J.R.B is supported by the NIH R01 CA 213442, P01 CA206978 and P01-CA081534.
Footnotes
Conflict of interest statement
C.J.W. is an equity holder of BioNtech, Inc. and C.J.W. and D.N. receive research funding from Pharmacyclics. D.N. has been a consultant for H3 Biomedicine and received research funding from Celgene. A.L serves as an equity holding SAB member for Zentalis Pharmaceuticals, Flash Therapeutics, and Dialectic Therapeutics. His laboratory receives research support from Novartis. T.J.K. has received research funding and/or has served as an advisor to Ascerta/AstraZeneca, Celgene, Genentech/Roche, Gilead, Janssen, Loxo Oncology, Octernal Therapeutics, Pharmacyclics/AbbVie, TG Therapeutics, VelosBio, and Verastem. Cirmtuzumab was developed by T.J.K. and licensed by the University of California to Oncternal Therapeutics, Inc., which has provided stock/options to the university and T.J.K. C.S received research funding from Genmab. J.R.B has served as a consultant for Abbvie, Acerta/Astra-Zeneca, Beigene, Bristol-Myers Squibb/Juno/Celgene, Catapult, Eli Lilly, Genentech/Roche, Janssen, MEI Pharma, Morphosys AG, Nextcea, Novartis, Pfizer, Rigel; received research funding from Gilead, Loxo/Lilly, SecuraBio, Sun, TG Therapeutics; and served on the data safety monitoring committee for Invectys. All other authors do not have any relevant conflict of interest.
References
- 1.Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015;526:525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and Other Novel Cancer Genes in Chronic Lymphocytic Leukemia. New England Journal of Medicine 2011;365:2497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin S, Gambe RG, Sun J, Martinez AZ, Cartun ZJ, Regis FFD, et al. A Murine Model of Chronic Lymphocytic Leukemia Based on B Cell-Restricted Expression of Sf3b1 Mutation and Atm Deletion. Cancer Cell 2019;35:283–296.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lazarian G, Yin S, Ten Hacken E, Sewastianik T, Uduman M, Font-Tello A, et al. A hotspot mutation in transcription factor IKZF3 drives B cell neoplasia via transcriptional dysregulation. Cancer Cell 2021;39:380–393.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.You JS, Jones PA. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell 2012;22:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaiti F, Chaligne R, Gu H, Brand RM, Kothen-Hill S, Schulman RC, et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature 2019;569:576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kulis M, Esteller M. DNA methylation and cancer. Adv Genet 2010;70:27–56. [DOI] [PubMed] [Google Scholar]
- 8.Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene Nature Publishing Group; 2002;21:5400–13. [DOI] [PubMed] [Google Scholar]
- 9.Landau DA, Clement K, Ziller MJ, Boyle P, Fan J, Gu H, et al. Locally Disordered Methylation Forms the Basis of Intratumor Methylome Variation in Chronic Lymphocytic Leukemia. Cancer Cell 2014;26:813–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kulis M, Heath S, Bibikova M, Queirós AC, Navarro A, Clot G, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet 2012;44:1236–42. [DOI] [PubMed] [Google Scholar]
- 11.Kretzmer H, Biran A, Purroy N, Lemvigh CK, Clement K, Gruber M, et al. Preneoplastic Alterations Define CLL DNA Methylome and Persist through Disease Progression and Therapy. Blood Cancer Discov 2021;2:54–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pei L, Choi J-H, Liu J, Lee E-J, McCarthy B, Wilson JM, et al. Genome-wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics 2012;7:567–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nature Reviews Genetics Nature Publishing Group; 2018;19:81–92. [DOI] [PubMed] [Google Scholar]
- 14.Bagacean C, Tempescul A, Le Dantec C, Bordron A, Mohr A, Saad H, et al. Alterations in DNA methylation/demethylation intermediates predict clinical outcome in chronic lymphocytic leukemia. Oncotarget 2017;8:65699–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, et al. The DLEU2/miR-15a/16–1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 2010;17:28–40. [DOI] [PubMed] [Google Scholar]
- 16.Guryanova OA, Lieu YK, Garrett-Bakelman FE, Spitzer B, Glass JL, Shank K, et al. Dnmt3a Regulates Myeloproliferation and Liver-Specific Expansion of Hematopoietic Stem and Progenitor Cells. Leukemia 2016;30:1133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryan JA, Brunelle JK, Letai A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. PNAS National Academy of Sciences; 2010;107:12895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scielzo C, Bertilaccio MTS, Simonetti G, Dagklis A, ten Hacken E, Fazi C, et al. HS1 has a central role in the trafficking and homing of leukemic B cells. Blood 2010;116:3537–46. [DOI] [PubMed] [Google Scholar]
- 21.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011;17:10–2. [Google Scholar]
- 22.Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 2009;10:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun D, Xi Y, Rodriguez B, Park HJ, Tong P, Meong M, et al. MOABS: model based analysis of bisulfite sequencing data. Genome Biology 2014;15:R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jühling F, Kretzmer H, Bernhart SH, Otto C, Stadler PF, Hoffmann S. metilene: fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Res 2016;26:256–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bond J, Touzart A, Leprêtre S, Graux C, Bargetzi M, Lhermitte L, et al. DNMT3A mutation is associated with increased age and adverse outcome in adult T-cell acute lymphoblastic leukemia. 1 2019;104:1617–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin M-E, Hou H-A, Tsai C-H, Wu S-J, Kuo Y-Y, Tseng M-H, et al. Dynamics of DNMT3A mutation and prognostic relevance in patients with primary myelodysplastic syndrome. Clinical Epigenetics 2018;10:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bagacean C, Le Dantec C, Berthou C, Tempescul A, Saad H, Bordron A, et al. Combining cytogenetic and epigenetic approaches in chronic lymphocytic leukemia improves prognosis prediction for patients with isolated 13q deletion. Clinical Epigenetics 2017;9:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spencer DH, Russler-Germain DA, Ketkar S, Helton NM, Lamprecht TL, Fulton RS, et al. CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression. Cell 2017;168:801–816.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan RJH, Petrovic J, Rausch DM, Zhou Y, Lareau CA, Kluk MJ, et al. A B Cell Regulome Links Notch to Downstream Oncogenic Pathways in Small B Cell Lymphomas. Cell Rep 2017;21:784–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tesi A, de Pretis S, Furlan M, Filipuzzi M, Morelli MJ, Andronache A, et al. An early Myc‐dependent transcriptional program orchestrates cell growth during B‐cell activation. EMBO Rep [Internet] 2019. [cited 2021 Mar 20];20. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6726900/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson MA, Deng J, Seymour JF, Tam C, Kim SY, Fein J, et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood 2016;127:3215–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ponader S, Chen S-S, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood 2012;119:1182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mangolini M, Götte F, Moore A, Ammon T, Oelsner M, Lutzny-Geier G, et al. Notch2 controls non-autonomous Wnt-signalling in chronic lymphocytic leukaemia. Nat Commun 2018;9:3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okano M, Bell DW, Haber DA, Li E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell Elsevier; 1999;99:247–57. [DOI] [PubMed] [Google Scholar]
- 35.Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 2011;44:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Izzo F, Lee SC, Poran A, Chaligne R, Gaiti F, Gross B, et al. DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat Genet 2020;52:378–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Celik H, Mallaney C, Kothari A, Ostrander EL, Eultgen E, Martens A, et al. Enforced differentiation of Dnmt3a-null bone marrow leads to failure with c-Kit mutations driving leukemic transformation. Blood 2015;125:619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CV, et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 2015;125:629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kramer AC, Kothari A, Wilson WC, Celik H, Nikitas J, Mallaney C, et al. Dnmt3a Regulates T-cell Development and Suppresses T-ALL Transformation. Leukemia 2017;31:2479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haney SL, Upchurch GM, Opavska J, Klinkebiel D, Hlady RA, Suresh A, et al. Promoter Hypomethylation and Expression Is Conserved in Mouse Chronic Lymphocytic Leukemia Induced by Decreased or Inactivated Dnmt3a. Cell Rep 2016;15:1190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haney SL, Upchurch GM, Opavska J, Klinkebiel D, Appiah AK, Smith LM, et al. Loss of Dnmt3a induces CLL and PTCL with distinct methylomes and transcriptomes in mice. Sci Rep 2016;6:34222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peters SL, Hlady RA, Opavska J, Klinkebiel D, Pirruccello SJ, Talmon GA, et al. Tumor suppressor functions of Dnmt3a and Dnmt3b in the prevention of malignant mouse lymphopoiesis. Leukemia 2014;28:1138–42. [DOI] [PubMed] [Google Scholar]
- 43.Mahajan VS, Mattoo H, Sun N, Viswanadham V, Yuen GJ, Allard-Chamard H, et al. B1a and B2 cells are characterized by distinct CpG modification states at DNMT3A-maintained enhancers. Nat Commun 2021;12:2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ostrander EL, Kramer AC, Mallaney C, Celik H, Koh WK, Fairchild J, et al. Divergent Effects of Dnmt3a and Tet2 Mutations on Hematopoietic Progenitor Cell Fitness. Stem Cell Reports Elsevier; 2020;14:551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Joseph C, Quach JM, Walkley CR, Lane SW, Lo Celso C, Purton LE. Deciphering Hematopoietic Stem Cells in Their Niches: A Critical Appraisal of Genetic Models, Lineage Tracing, and Imaging Strategies. Cell Stem Cell 2013;13:520–33. [DOI] [PubMed] [Google Scholar]
- 46.Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 2005;33:5868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data described in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO) -- Accession numbers: GSE169245, GSE143711 and GSE122668.