

Abstract
The development of devices for the precise and controlled delivery of therapeutics has grown rapidly over the last few decades. Drug delivery materials must provide a depot with delivery profiles that satisfy pharmacodynamic and pharmacokinetic requirements resulting in clinical benefit. Therapeutic efficacy can be limited due to short half-life and poor stability. Thus, to compensate for this, frequent administration and high doses are often required to achieve therapeutic effect, which in turn increases potential side effects and systemic toxicity. This can potentially be mitigated by using materials that can deliver drugs at controlled rates, and material design principles that allow this are continuously evolving. Affinity-based release strategies incorporate a myriad of reversible interactions into a gel network, that have affinities for the therapeutic of interest. Reversible binding to the gel network impacts the release profile of the drug. Such affinity-based interactions can be modulated to control the release profile to meet pharmacokinetic benchmarks. Much work has been done developing affinity-based control in the context of polymer-based materials. However, this strategy has not been widely implemented in peptide-based hydrogels. Herein, we present recent advances in the use of affinity-controlled peptide gel release systems and their associated mechanisms for applications in drug delivery.
Graphical Abstract
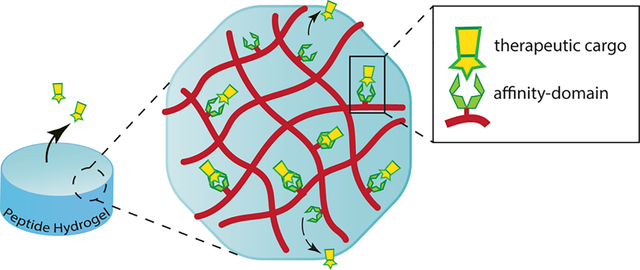
This review explores the potential of self-assembled peptide hydrogels to modulate and control the release of therapeutic cargo via reversible affinity-based interactions, along with their associated mechanisms for drug-delivery applications.
Introduction
The development of hydrogels as functional biomaterials continues to be a rapidly growing area of investigation towards applications in drug-delivery, tissue engineering, immunoengineering, biosensing and wound healing.1–11 Hydrogels are characterized by a water-swollen three-dimensional porous matrix, capable of housing a myriad of cargo including small molecules, peptides, proteins, and nucleic acids for their eventual release and delivery.12 Moreover, many gels have thixotropic properties that render them injectable at a targeted tissue, enabling local delivery of the payload while diminishing off-target toxicity.2 Although much effort has been expended to develop strategies to control the release rates of drugs from gels, it still remains a challenge. Most gels published to date2,13 are characterized by time-dependent logarithmic release with a predominant burst phase with a few reports of systems depicting a nearly linear release profile.14,15
In a classical diffusion-based delivery scheme in the absence of any engineered affinity, the overall time it takes a therapeutic to be released from a material network (trelease) is given by (L2/D), where (L) is the diffusional pathlength of the material and (D) is the diffusivity of the cargo, i.e. [trelease (s) = L2/D (m2/m2s−1)].16,17 The diffusivity characterizing the diffusion of a given therapeutic through a gel network is dependent on several factors such as the size of the molecule and mesh size of the network, Figure 1. Larger molecules diffuse slowly exhibiting long release profiles, whereas small molecules diffuse more rapidly resulting in shorter release durations. Similarly, gel networks having larger pore-sizes facilitate faster molecular diffusion and shorter release profiles, whereas smaller pore-sized networks allow for slower diffusion, accompanied by longer release profiles. Thus, one can tune the release profile by modulating the mesh size of a gel. For polymeric systems, changing the weight % and/or molecular weight of monomer, cross-linking chemistry and reaction conditions can affect changes in mesh size.18 In self-assembled peptide systems, mesh size can be modulated by altering the peptide wt % used to formulate the gel, but it is difficult to achieve a desired pre-determined mesh size using this approach.19 Further, release rates can be further impacted by hydrogel degradation or erosion over time, which often hastens delivery.6,20,21
Figure 1.
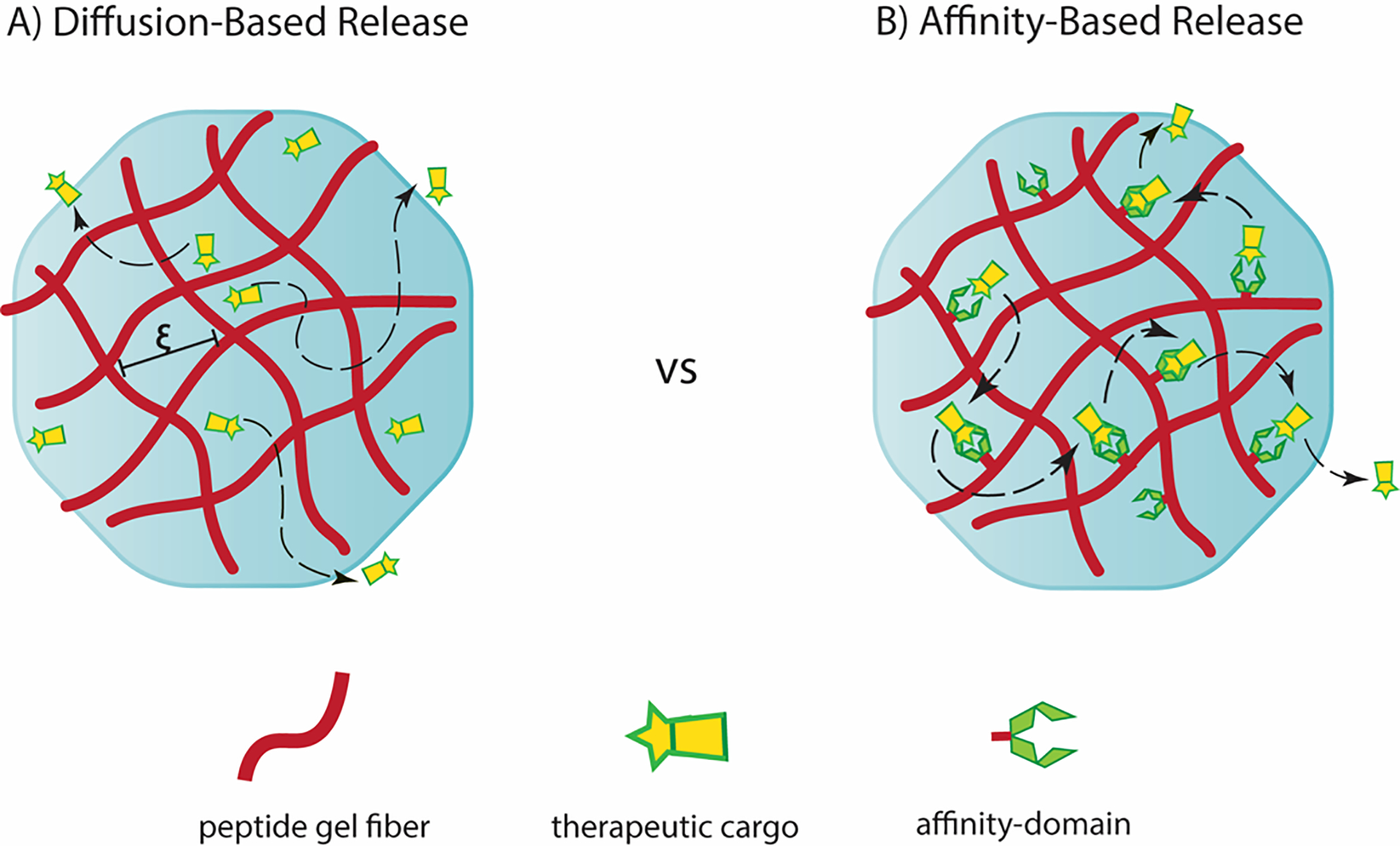
An illustration of the comparative difference between non-affinity-controlled diffusion-based and affinity-controlled hydrogel release systems (ξ represents the gel mesh size).
Affinity-controlled systems incorporate ligands or binding moieties within the gel network that form reversible binding interactions with the cargo and impact its release rate. Network-bound ligands serve as anchors that aid in prolonging the persistence time of a therapeutic within the hydrogel. A slower diffusion profile can potentially minimize early burst release of the therapeutic, facilitating a more sustained release over time and in some cases achieve linear release.16,22 Thus, these systems can provide greater control over release kinetics, which can be tuned depending on the system. Nature employs non-covalent interactions between heparin in the extracellular matrix and a myriad of growth factors (KD = 10−6 to 10−9) and has served as inspiration for the development of some of the earliest examples of affinity-controlled release systems.23,24 Heparin binding proteins can be delivered over timescales ranging from a few days to several weeks using this approach.25–32 Other complementary binding partners such as antibodies with antigens33 or albumin with small molecule therapeutics34 have also served in the design of this class of material. While affinity-controlled hydrogels have been prepared employing hyaluronic acid,25,26 gelatin26,27 and other polymer-based scaffolds,35–37 hydrogels prepared from self-assembling peptides may serve as useful alternatives that have unique attributes. The synthesis of their peptide building blocks is precise with respect to sequence, composition, length, and reproducibility as a result of advances in solid phase peptide methodology, making their self-assembled gels highly amenable to biomedical applications.38–40 In addition, there is ample scope for sequence-specific incorporation of a wide variety of chemical functionalities and non-natural motifs that can influence bulk material properties. Peptide-based gels are typically biocompatible and do not cause adverse immune response unless designed to do so. Further, they are readily biodegraded.
Although they hold promise, there are fewer reports of self-assembled peptide gels that demonstrate affinity-controlled release. This is surprising given their promised utility. However, there are enough examples to begin to formulate design principles and draw comparisons to polymer-based systems. For instance, most peptide-based affinity systems reported to date differ from conventional polymer-based systems with respect to the nature of the binding interaction between drug and material. In polymer-based systems, there are more material designs that utilize molecular recognition events that are specific in nature, for example, an antigen binding to its antibody. Here, molecular recognition/complexation is facilitated by a combination of primary interactions, such as electrostatics, π-effects, van der Waals as well as the hydrophobic effect if complexation is entropically driven. These same non-covalent interactions play a vital role in the formation and cross-linking of peptide gel networks3,41 and thus it is not surprising that they have been utilized in the early designs of affinity-controlled peptide systems to mediate drug-material complexation. However, therapeutic binding to the material network is driven predominantly by a single type of interaction as opposed to a combination which is typical of most of the specific molecular recognition events utilized in polymer-based affinity-controlled materials. As such, the binding events leading to complexation in peptide systems are often not as specific but can be of high affinity. Further, in polymer systems, ligands are typically ligated to the polymer network. In many peptide-based materials, self-assembly leads to fibril networks where the fibrils themselves can host drug binding. With that said, there are examples of peptide materials where ligands are incorporated to the fibril networks and those will also be discussed.
This review briefly outlines basic affinity-controlled release kinetics previously defined using polymer-based systems which are also applicable to peptide-based materials. We go on to explore peptide hydrogels that leverage reversible interactions for the loading and delivery of a variety of therapeutics, the various molecular mechanisms defining their release kinetics and future opportunities for developing next generation affinity-controlled peptide materials.
Affinity-Controlled Release Kinetics
Again, in a classical diffusion-based delivery scheme in the absence of any engineered affinity, the overall time it takes a therapeutic to be released from a material network (trelease) is given by (L2/D), where (L) is the diffusional pathlength of the material and (D) is the diffusivity of the cargo, i.e. [trelease (s) = L2/D (m2/m2s−1)]. In affinity-controlled systems, materials are engineered to reversibly interact with the therapeutic, which impacts the overall release time (L2/D) by a given factor depending on the exact nature of the material. Affinity-based interactions are characterized by the ability of the therapeutic to reversibly interact with its corresponding binding moiety, which is grafted to the gel network.16,42,43 The strength of the association or binding affinity is thermodynamically characterized by the dissociation constant (KD), Scheme 1. The lower the KD, the higher the binding affinity. The most straightforward method of fine-tuning release can be achieved by modulating the binding affinity (KD). However, this would be an oversimplification as there are other factors that play a role in cargo release such as the shape of the material. However, for the discussion here, we will assume a constant diffusional pathlength (L) for all shapes. A mathematical model prescribed by Vulic et al. classifies the binding and release profiles of affinity-based systems into three characteristic regimes.17 For a bimolecular affinity-based gel system, with a single receptor-cargo pair, there are typically two populations of cargo, bound and unbound. The nature of the binding equilibrium between two affinity pairs is dynamic and is kinetically characterized by association (kon) and dissociation rate (koff) constants (Scheme 1).
Scheme 1.

The dissociation of the affinity complex formed by the therapeutic cargo and the binding domain. The binding affinity (KD) is proportional to the ratio of the koff and kon rate constants.
Assuming the time it takes for the therapeutic to diffuse through the material is slower than the dissociation of the complex, two different regimes of release are possible. In regime 1, a small portion of the binding sites are occupied. As such, the release of all the cargo occurs on a single timescale of (L2/D) attenuated by (1 + [BD]T/KD), where [BD]T is the total concentration of occupied and unoccupied binding domains, Figure 2. The release rate depends on the ratio of the number of binding sites relative to KD and is independent of the amount of cargo in the gel.17
Figure 2.
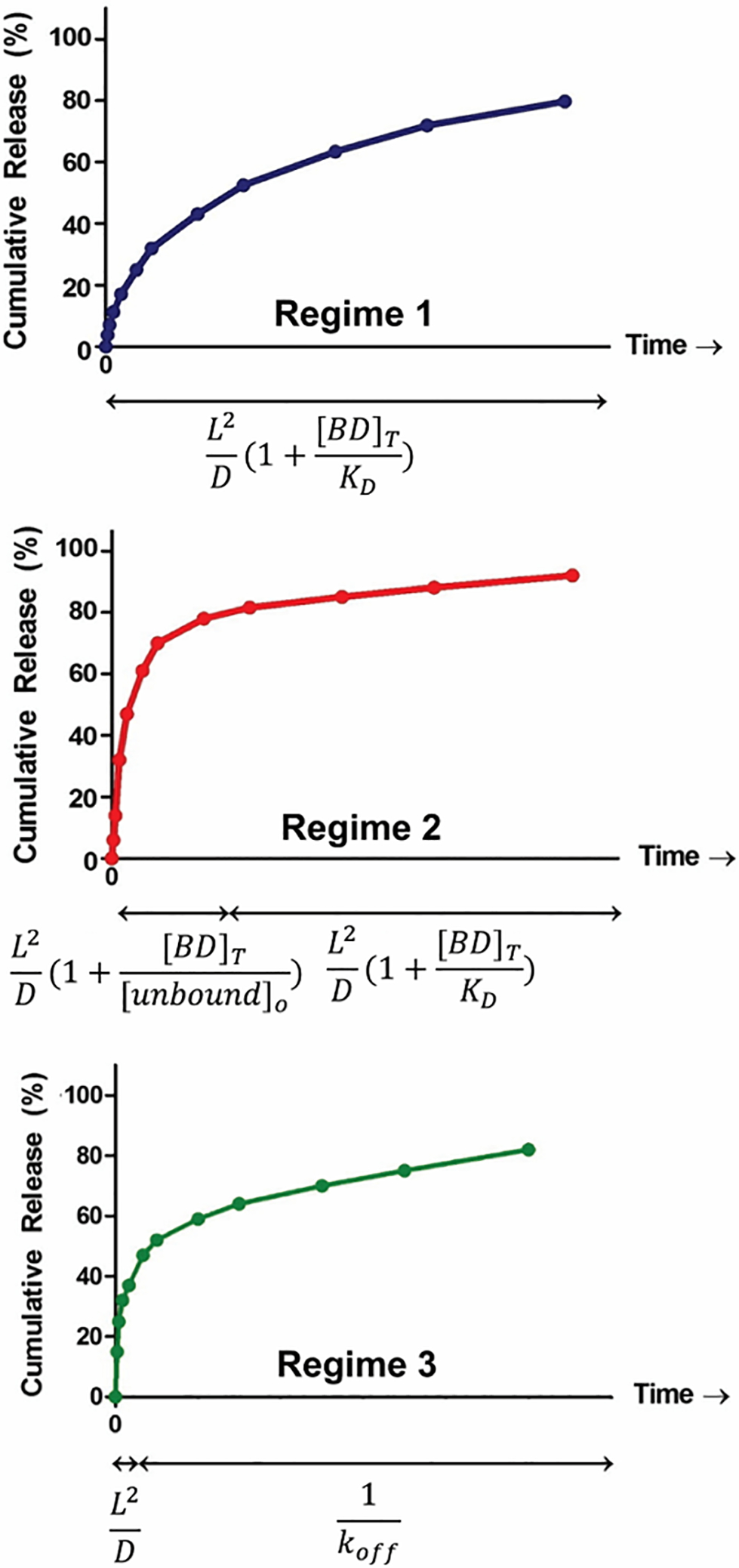
Three characteristic regimes of release of affinity-controlled systems with varying timescales for each phase of release. L and D represent the diffusion pathlength of the hydrogel and diffusivity of the cargo through the matrix, respectively. Reprinted from “Journal of Controlled Release, 197, Katarina Vulic, Malgosia M. Pakulska, Rohit Sonthalia, Arun Ramachandran, Molly S. Shoichet, Mathematical model accurately predicts protein release from an affinity-based delivery system,” Page 75, Copyright (2015), with permission from Elsevier.
Conversely, regime 2 is established when the binding sites are mostly occupied. In this case, there is an initial release that occurs over a timescale of (L2/D) attenuated by (1 + [BD]T/[unbound]o) until the cargo concentration drops to the value of KD, after which the remainder of the cargo is released over a time-scale of (L2/D) attenuated by (1 + [BD]T/KD), Figure 2. The initial release of regime 2 depends on the concentration of therapeutic in the system. The subsequent release profile of regime 2 follows the same principles outlined in regime 1.17
In regime 3, the therapeutic is released from the material more quickly than the time it takes for the dissociation of the complex. As such, release is biphasic with unbound cargo being released first in a burst mode dependent on (L2/D). This is followed by release that arises from the slower decomplexation of the bound species over a time scale proportional to 1/koff. This biphasic behavior is shown in Figure 2. The amount of cargo released during the first phase is dictated by the amount of unbound species present at equilibrium, which can be controlled by altering the KD or the initial concentrations of the unbound therapeutic and the binding domain.17
In summary, Regime 1 is characterized by a slow and steady release of cargo over a single timescale. Regimes 2 and 3 both display an early fast release followed by a subsequent slower release. For all three regimes, to achieve affinity-based release, it is imperative that the value of KD be smaller than [BD]T. When KD is larger than [BD]T, the binding domain does not appreciably form a complex with the cargo, which results in a classical diffusion-based release system with loss of affinity. The release profiles of affinity systems can thus be modulated and fine-tuned by striking a balance between diffusion and dissociation kinetics, the understanding of which can prove useful in the future design and development of affinity-controlled peptide-gel release systems.
Binding interactions of peptide-based affinity-controlled systems
Many of the peptide-based affinity-controlled systems described to date utilize predominantly one type of fundamental interaction to mediate complexation, for example electrostatics. This is most likely due to the fact that designing bimolecular interactions de novo that are specific is difficult. Further, incorporating naturally-occurring specific affinity interactions, for example hormone-receptor pairs, into a material matrix can be synthetically challenging.
Non-covalent interactions such as ion-ion and ion-dipole can be quite strong (10–90 kcal/mol). H-bonding, cation-π and π-π stacking are reported to be comparatively weaker (1–15 kcal/mol).44,45 Thus, it’s not surprising that many peptide-based affinity systems rely on electrostatics to mediate binding and controlled delivery. The strength of these interactions can be further enhanced by varying the number of interacting domains between the ligand and the therapeutic. As such, the overall affinity can be tuned depending on how this chemistry is implemented. More recently, the use of reversible covalent bonds such as imine and disulfide bonds have been introduced into the affinity moiety arsenal. With respect to peptide materials, Nature offers a range of amino acids, each distinguished by the chemical nature of their side chains, that can participate in a wide range of interactions (Table 1). Thus, for peptide-based drug-delivery systems, the chemistry of amino acids can be harnessed to engineer affinity-binding domains. Appropriate binding partners for select cargo can be identified and characterized via techniques such as isothermal titration calorimetry (ITC), surface plasmon resonance (SPR) and spectroscopic methods. Examples of peptide-based affinity-control materials are discussed and categorized below by the primary type of interaction utilized in their design. See Table 2 for a tabulation of these materials and their associated properties.
Table 1:
Affinity-based interactions and their associated amino acids
| Affinity-based interactions | Amino acids with participating side-chains |
|---|---|
| Non-polar interactions | Ala, Ile, Leu, Met, Phe, Pro, Trp, Val, Tyr |
| π-π/cation-π interactions | Phe, Trp, Tyr/Lys, His, Arg |
| H-bonding | Cys, Asp, Glu, His, Lys, Asn, Gln, Arg, Ser, Thr, Trp, Tyr |
| Electrostatic | Cys, Asp, Glu, His, Lys, Arg, Tyr |
| Metal ligand | Asp, Arg, Lys, Tyr, Glu, His, Cys |
Table 2:
Summary of peptide-based hydrogels that use affinity-based interactions to reversibly bind and release their cargo
| Affinity-Based Interaction | Peptide Gel Matrix | Cargo delivered | Release Duration | Ref | Notes |
|---|---|---|---|---|---|
| Electrostatic: Specific | α–helical peptide amphiphile + Heparin = β sheet nanofiber | Growth factors: FGF-2-rhodamine | 10 days | 46 | |
| Peptide-PEG conjugated gels + Heparin | Heparin binding proteins | ~ 10 days | 48 | ||
| Electrostatic: Non-specific | Heparin-mimetic peptide amphiphile | Growth factors: VEGF | 7 days | 49 | |
| β hairpin: HLT2 (cationic) | Proteins: α-lactalbumin, lactoferrin, myoglobin | 28 days | 58 | ||
| β hairpin: VEQ3 (anionic) | Proteins: α-lactalbumin, lactoferrin, myoglobin | 28 days | 58 | ||
| β sheet: RADA16 | Growth factors: β-FGF, VEGF, BDNF | > 2 days | 63 | ||
| β hairpin: STINGel MDP - K2(SL)6K2 | Cyclic dinucleotide (CDN) | > 24 hours | 64 | ||
| β hairpin: SLac MDP- (KSLSLSLRGSLSLSLKGRGDS) | Small molecule drugs: suramin | > 30 days | 66 | ||
| β hairpin: AcVES3 | Proteins: EGFP; Cytokines: IFNα | 10 days | 60 | Cargo modified with affinity tags | |
| Hydrophobic | β hairpin: MDP - K2(SL)3(SA)(SL)2K2 | Small molecule drugs: SN-38, Diflunisal, Etodolac | > 8 days | 67,68 | |
| Single amino acid hydrogel nanoparticles (HNPs) | Small molecule drugs: 5-fluorouracil | t1/2 = 3–9 hrs | 60 | ||
| MITCH gel | QK (VEGF-mimetic peptide) | > 21 days | 70 | Cargo modified with affinity tags | |
| Combination Systems: Electrostatic & Hydrophobic | Phe-modified RADA | Small molecule drugs: 5-fluorouracil | n/a | 72 | |
| Hexapeptide: NH2-WLVFFK-COOH | Small molecule drugs: 5-fluorouracil, ciprofloxacin | 72 hrs | 73 | ||
| Composite RADA gel-chitosan system with β-Cyclodextrin carriers | Small molecule drugs: dexamethasone | ~ 9 days (pH 7) > 9 days (pH 6) |
74 | ||
| Reversible Covalent: Schiff Base | Peptides with aldehyde functionality: Nap-GDFDFDpY-CHO | Small molecule drugs: Doxorubicin, Gemcitabine | > 12 hours | 80,81 | |
| Reversible Covalent: Hydrazone | Peptide amphiphile modified with hydrazine | Small molecule drugs: nabumetone | > 24 days | 82 |
Electrostatic interactions are perhaps the most widely exploited in self-assembled peptide-based gels to initiate binding between different therapeutics and a gel network. The use and application of self-assembling peptides with acidic (glutamic acid and aspartic acid) and basic amino acids (lysine, histidine and arginine) afford fibrils that can host drug binding. The ionic state of residue side chains comprising these peptides are dependent on pH and the strength of any electrostatic interaction they make with a therapeutic is dependent on ionic strength. As such, drug release can be triggered by real-time changes in pH and ionic strength.
While most of the examples discussed herein are non-specific, there are a few reports of naturally occurring affinity-binding pairs incorporated within peptide networks. Heparin-binding peptide amphiphiles, developed by the Stupp lab,46,47 were designed to specifically bind heparin sulfate-like glycosaminoglycans (HSGAG) to recruit and deliver heparin-binding proteins. Here, amphiphiles displaying the peptide LRKKLGKA, a known heparin-binding consensus sequence, forms nanostructures when heparin is added. The negatively charged heparin screens the positive charge of the peptide amphiphiles, driving self-assembly and the formation of cylindrical nanofibers where heparin chains are displayed on their surface (Figure 3A). The comparative release profiles of FGF-2-rhodamine from heparin-nucleated and Na2HPO4-nucleated (control counter-ion) hydrogels revealed a difference of over 40% in the cumulative cargo released after 10 days. The gels prepared via Na2HPO4 also resulted in a burst release of 34% within the first 10 minutes (Figure 3A).46 This demonstrates that the heparin-binding nanofibers display affinity-controlled release of FGF-2 growth factor. In separate work, a similar design was employed where composite peptide-PEG conjugated gels were formed with the addition of heparin.48 The resulting material displayed affinity-controlled release of fluorescently labelled heparin-binding peptides. In these examples, heparin coats the material networks and mediates therapy binding and release.
Figure 3.
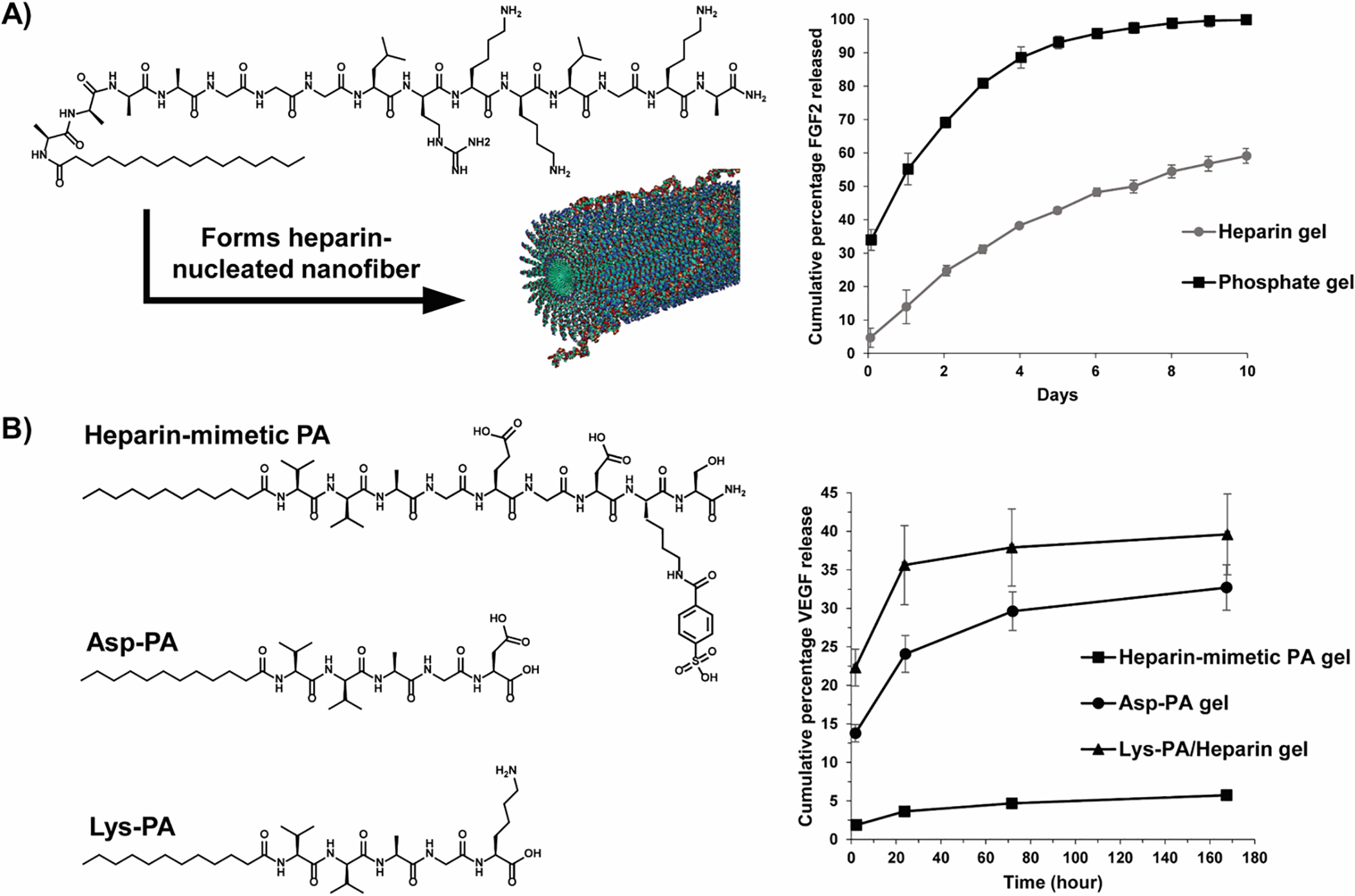
(A) A scheme outlining the formation of a heparin-nucleated fiber from a self-assembling peptide amphiphile along with its corresponding release profile. Heparin-binding enables the affinity-controlled release of FGF-2 growth factor. Adapted with permission from Chow LW, Wang L-j, Kaufman DB, Stupp SI, Self-assembling nanostructures to deliver angiogenic factors to pancreatic islets. Biomaterials. 2010; 31:6154–61 https://doi.org/10.1016/j.biomaterials.2010.04.002. Copyright 2006 American Chemical Society.
(B) Structures of heparin-mimetic peptide and control sequences (Asp-PA & Lys-PA) and their adjoining delivery profiles, depicting the release of VEGF growth factor. Adapted with permission from Mammadov R, Mammadov B, Toksoz S, et al, Heparin Mimetic Peptide Nanofibers Promote Angiogenesis. Biomacromolecules. 2011; 12:3508–19 10.1021/bm200957s. Copyright 2011 American Chemical Society.
In contrast, Mammadov et al., eliminated the need for the heparin coating by using a synthetic heparin-mimetic peptide amphiphile (PA), consisting of sulfonate, hydroxyl and carboxylic acid groups (Figure 3B).49 Gel formation was induced by the addition of Lys-terminated PA, which screens the charge of the heparin-mimetic peptide amphiphile allowing assembly leading to gel formation. The strong affinity of the VEGF growth factor with the heparin-mimetic peptide amphiphile is evident from its slow-release profile, with only 5% of the encapsulated growth factor released after 7 days (Figure 3B). Comparatively, control gels made from Asp-terminated PA and Lys-PA containing exogenous heparin, yielded burst releases of VEGF at 2 hours with 33% and 40% of the cargo released at the end of 7 days.49 This work also nicely shows that the heparin coating can be avoided simplifying material preparation without compromising affinity-controlled release.
In our lab, we have developed an extensive array of peptide-based fibrillar gels for numerous applications50–54 including delivery.55–60 These amphiphilic peptides undergo triggered self-assembly forming a fibrillar hydrogel network where fibrils are comprised of peptides that are folded into a β-hairpin secondary structure and assembled into an extended bilayered β-sheet.61 The interior of the bilayered fibrils contains hydrophobic residues and the exterior, solvent-exposed surface of the fibrils displays hydrophilic residues. Appropriately designed peptides can form fibrils displaying charged residue side chains that can be utilized for electrostatic-based affinity-controlled release of molecules. For example, when anionic peptides are used for self-assembly, negatively charged fibril networks are formed.58 Figure 4A shows schematically that when the positively charged protein lactoferrin is encapsulated, it binds avidly to the fibril network and is released slowly. Figure 4B shows the corresponding release data for lactoferrin as well as for negatively charged α-lactalbumin, whose release is governed by diffusion void of affinity control. Conversely, when gels are prepared by a cationic peptide, positively-charged fibril networks are formed that are not able to release lactoferrin in an affinity controlled manner, Figures 4 A,C. However, now α-lactalbumin can be delivered via affinity-controlled release. Neutral myoglobin is released via simple diffusion by both gels.58 A similar approach was adopted with the use of RADA16-I self-assembling peptides, a repetitious and ionic self-complementary set of beta-sheet scaffolds,62 to achieve slow and controlled delivery of differently charged cytokines of similar size – human βFGF (+), VEGF (−) and BDNF (+).63 As was the case with our system, the release of proteins from oppositely charged hydrogels was found to be slower in comparison to that from similarly charged hydrogels.
Figure 4.
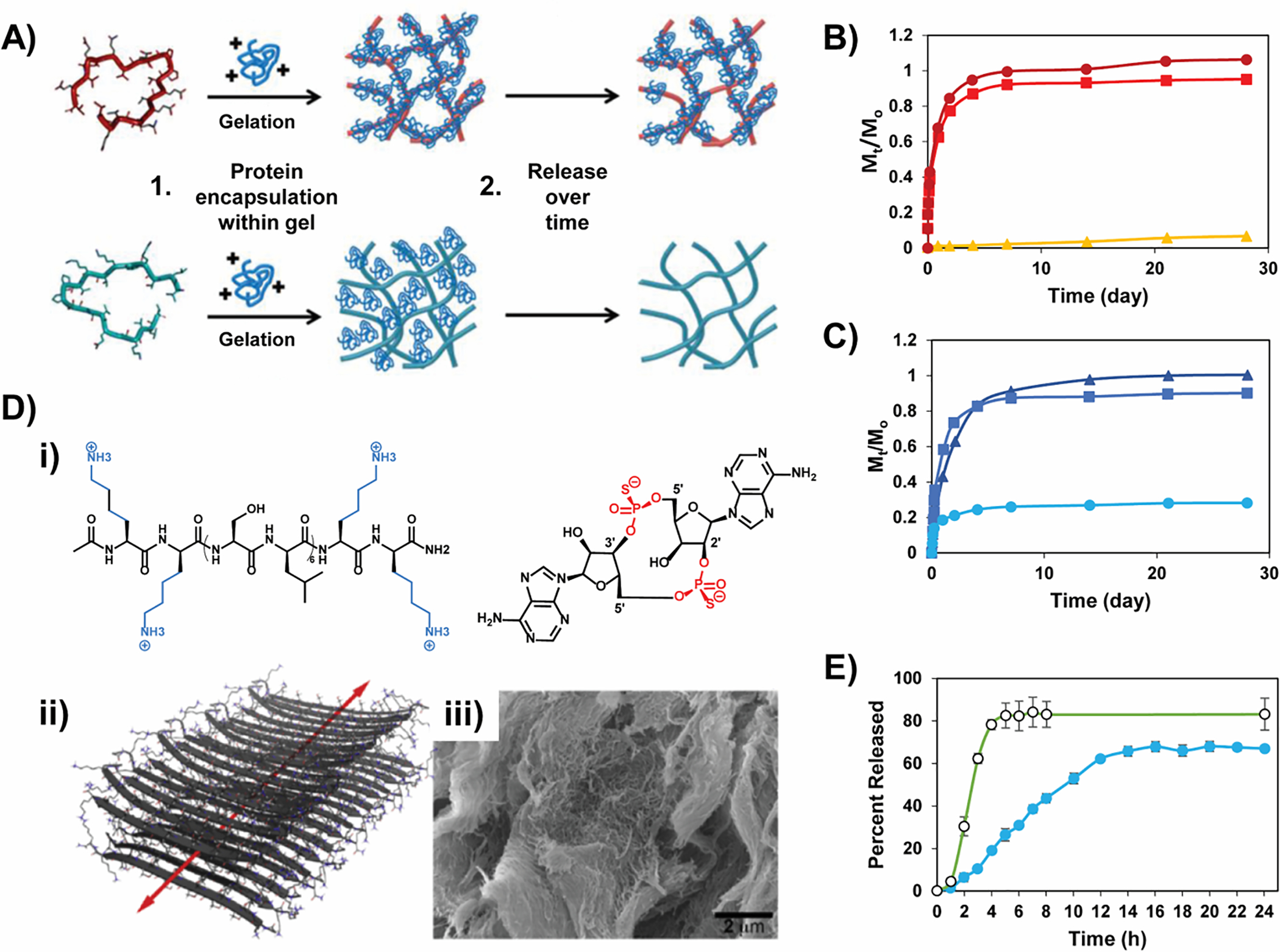
(A) A scheme contrasting the different release rates of a positively charged protein as a function of the electrostatic charge of a gel network. Relative release profiles of α-lactalbumin (●), myoglobin (◼), and lactoferrin (▴) from negatively charged (B) and positively charged gels (C). Reproduced by permission of The Royal Society of Chemistry. (D)-(i) Positively charged STINGel binds with the negatively-charged thiophoshphate groups of CDN; (ii) antiparallel beta sheet nanofiber formed by the MDP peptide, (iii) SEM micrograph of the MDP gel and, (E) Drug release profile depicting the prolonged release of CDN from MDP (blue, closed circles) relative to the neutral collagen matrix (green, open circles). Reprinted from Biomaterials, 163, David G. Leach, Neeraja Dharmaraj, Stacey L. Piotrowski, Tania L. Lopez-Silva, Yu L. Lei, Andrew G. Sikora, Simon Young, Jeffrey D. Hartgerink, STINGel: Controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy, Pages No., Copyright (2018), with permission from Elsevier.
Similar interactions were employed in the controlled delivery of a cyclic dinucleotide (CDN) from the STINGel, formed from an assembling multidomain peptide (MDP) developed by the Hartgerink lab (Figure 4D).64 This peptide forms fibrillar gels rich in antiparallel beta-sheet structure. The positively-charged network reversibly binds to the negatively charged thiophosphate moieties of CDN and extends its release. In contrast to the release rate of CDN from the STINGel, a neutral collagen gel matrix displayed an 8-fold increase in release rate for the same therapeutic, Figure 4E.64 Similarly, electrostatics was employed to affect affinity-controlled release of pro-apoptotic peptide (SDPP) from a series of peptide gels using poly-glutamate/poly-Arginine/Lysine interactions.65 A parallel study with a similar MDP sequence demonstrated the use of polyvalent drugs as ionic cross-linkers to stabilize the resulting fibril network. Negatively-charged suramin, with six sulfonate groups, allows for hydrogen bonding and electrostatic interactions with the terminal lysines of the MDP peptide. This aids in both the formation of the hydrogel network and subsequent affinity-based release of the drug over time.66
The examples above clearly show the powerful effect electrostatics can play in modulating affinity-controlled release and its utility as a design element. However, for the release of therapeutics whose conformation is important for their function, care must be taken. For example, the folded conformations of many protein therapeutics are marginally stable and binding of a charged protein directly to a material network of opposite charge can result in protein denaturation and/or aggregation. To limit direct contact between a therapeutic protein and the gel network, an interactive domain (ID) tag can be ligated onto the therapeutic protein that acts as a bridging tether between the cargo and material matrix. Such ID tags can be engineered to incorporate a suite of non-covalent interactions depending on the desired release properties. For example, we have designed a family of cationic IDs that can be appended to the N- or C-termini of proteins to facilitate their affinity-controlled release from negatively-charged hydrogel networks, Figure 5A, B.60 Figure 5C shows the effect of ligating different IDs to control the delivery of the protein EGFP. Figure 5D shows that by using combinations of ID-tagged protein, one can further tune delivery. Furthermore, our system also allows for the time-staggered delivery of different proteins, which should be beneficial in drug combination regimens.60
Figure 5.
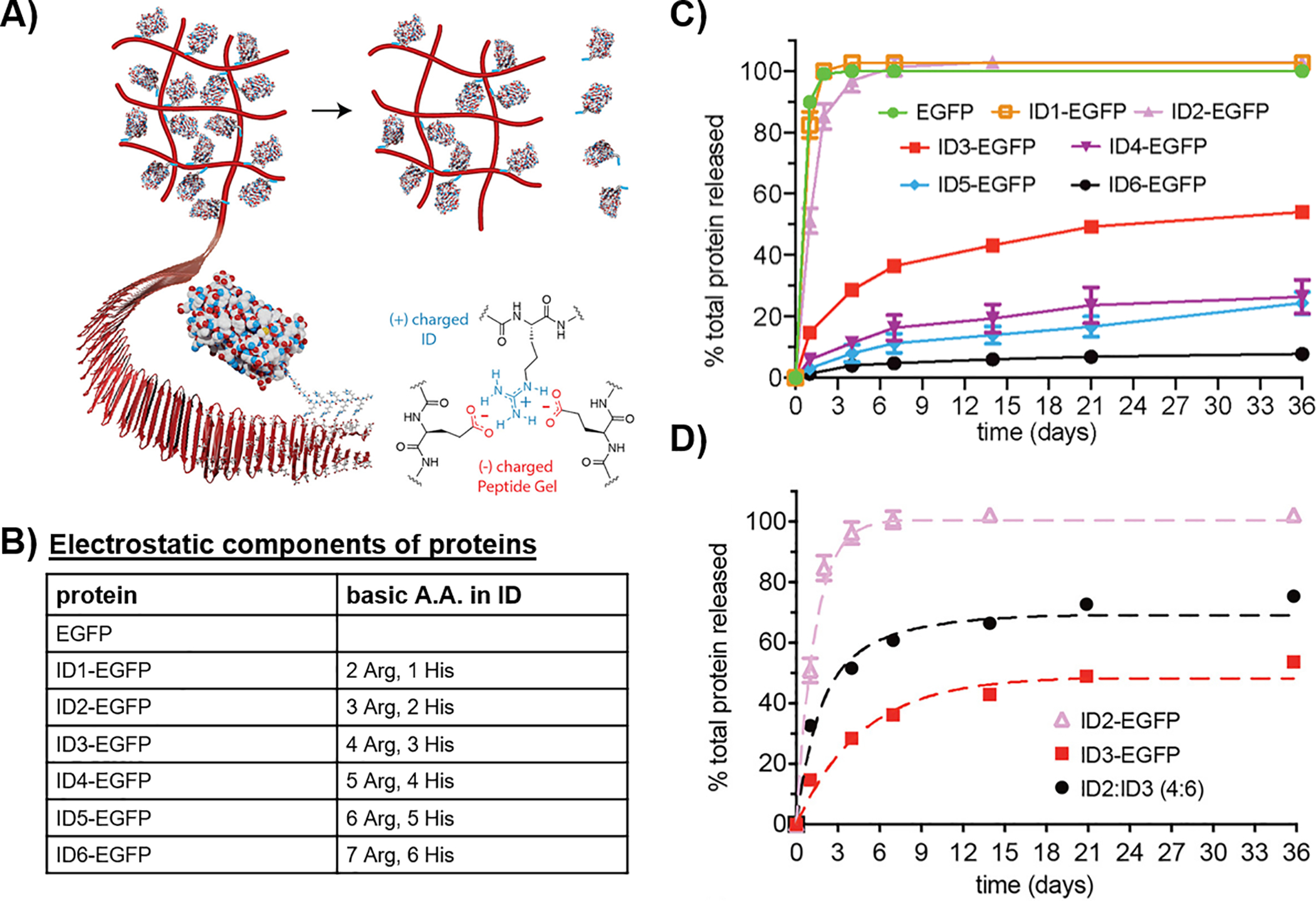
(A) Scheme depicting the affinity-controlled release of a protein from a negatively-charged peptide network via a cationic interaction domain, (B) Amino acid composition of each interaction domain (ID), (C) Comparative release profiles of EGFP tethered to different IDs and, (D) Comparative release profiles of ID-tagged IFNα. Adapted from link https://pubs.acs.org/doi/10.1021/acscentsci.9b00501. Further permissions related to the material excerpted should be directed to the ACS.
Hydrophobic and π-π interactions have also been used, often in combination, to affect affinity-controlled release from peptide-based gels. Typically, these interactions are used for encapsulating and delivering hydrophobic drugs, which are otherwise difficult to encapsulate within an aqueous gel reservoir. There are numerous amino acid sidechains that can be employed to facilitate hydrophobic drug-material interactions, Table 1. In an interesting case, a molecular cavity was engineered within the MDP beta-sheet scaffold discussed earlier (Figure 4D–ii) to store and subsequently release hydrophobic molecules. This was executed via manipulation of the primary sequence of the multidomain peptides, wherein the truncation of the leucine residues sandwiched within the hydrophobic layer to alanine residues resulted in a cavity suitable for the entrapment of non-polar drugs through the hydrophobic effect, Figure 6A.67 Hydrophobic drugs like diflunisal, etodolac and SN-38 were released more slowly (8 days) relative to more polar molecules such as daunorubicin and norfloxacin (4 hrs), as the latter tend to occupy sites on the exterior of the nanofibers rather than the interior. Thus, the use of the MDP peptide system can not only enable electrostatic-based affinity control, but also be tuned to facilitate hydrophobic interaction-based affinity control.
Figure 6.
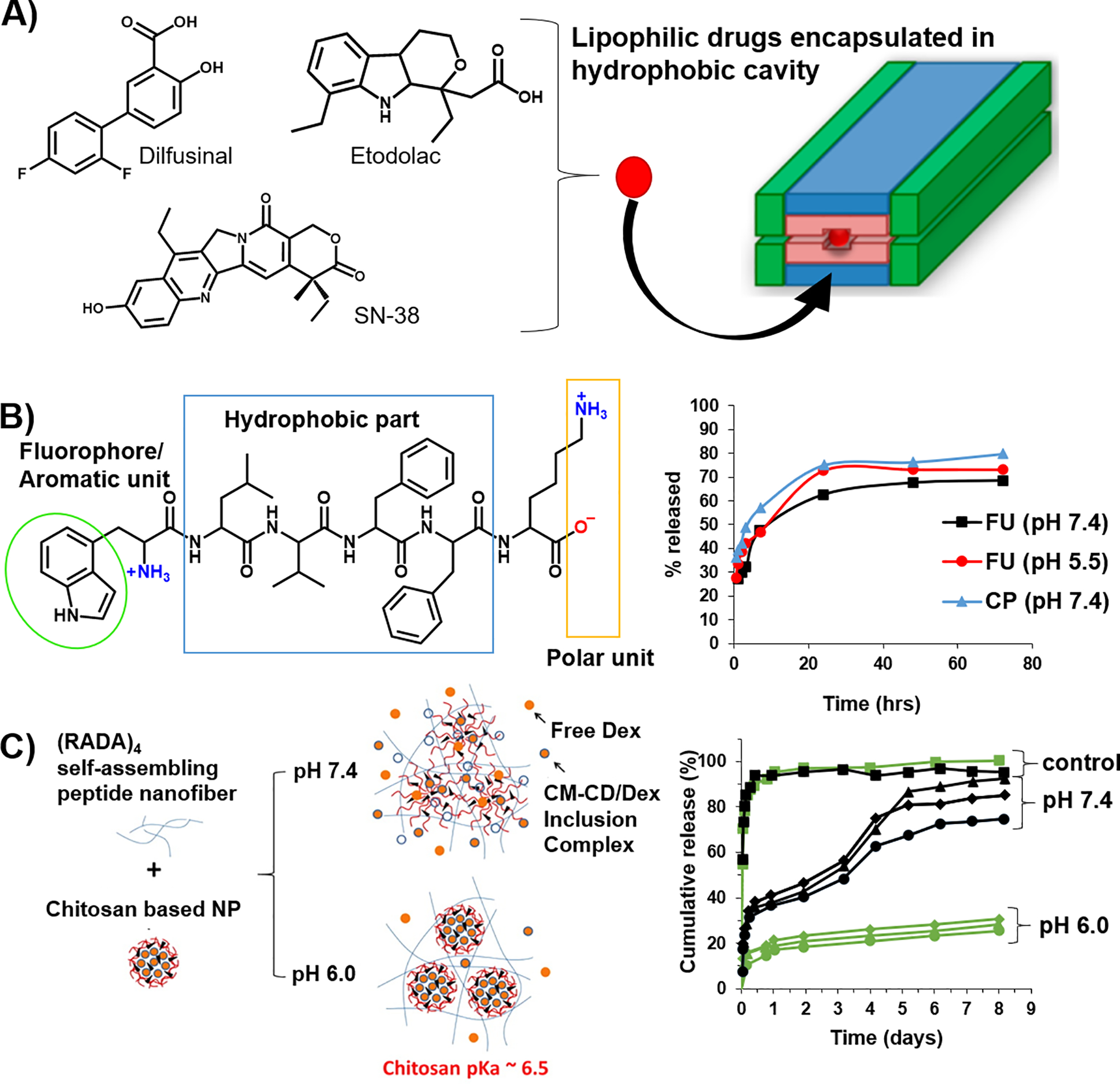
(A) MDP-based fibers are engineered with a hydrophobic cavity to host and deliver hydrophobic drugs. Adapted with permission from “Kumar VA, Shi S, Wang BK, et al, Drug-Triggered and Cross-Linked Self-Assembling Nanofibrous Hydrogels. Journal of the American Chemical Society. 2015; 137:4823–30 10.1021/jacs.5b01549.” Copyright 2016 American Chemical Society. (B) Use of an aromatic tryptophan unit within a peptide gelator for the docking of hydrophobic drugs like fluorouracil and ciprofloxacin. The release profiles display a modest faster release rate of fluorouracil under acidic conditions. Adapted with permission from “Roy K, Pandit G, Chetia M, et al, Peptide Hydrogels as Platforms for Sustained Release of Antimicrobial and Antitumor Drugs and Proteins. ACS Applied Bio Materials. 2020; 3:6251–62 10.1021/acsabm.0c00314” Copyright (2020) American Chemical Society. (C) Encapsulation of dexamethasone within cyclodextrin affords CM-β-CD/Dex inclusion complexes that are subsequently bound with chitosan to form nanoparticles that are further encapsulated into a RADA-based peptide gel. At the end of 8 days, the release of CM-β-CD/Dex inclusion complex is slower at pH 6.0 (25–31%) relative to its release at pH 7.4 (74–95%). The control group, CM-β-CD/Dex in the absence of chitosan shows a 95% cumulative release at both acidic and neutral pHs. (error bars not included for clarity). Adapted with permission from “Lu L, Unsworth LD, pH-Triggered Release of Hydrophobic Molecules from Self-Assembling Hybrid Nanoscaffolds. Biomacromolecules. 2016; 17:1425–36 10.1021/acs.biomac.6b00040.” Copyright (2016) American Chemical Society.
Tiwari et al., demonstrated affinity-controlled release using a minimalistic gel design where Fmoc-meta-aminobenzoic acid and benzoyloxy carbonyl phenylalanine were used to form hydrogel nanoparticles by self-assembly.68 The self-assembly of both molecules is mainly driven by hydrophobic collapse. The chemotherapeutic 5-fluorouracil can be incorporated into the particles via co-assembly, presumably forming π-π interactions with the self-assembling gelators.68 Subsequent, drug release over about 10 hours was observed for both gels with the Fmoc-meta-aminobenzoic acid displaying slightly longer persistence time. This is consistent with the extended π-system of the Fmoc moiety relative to the benzoyloxy group.
Similar to our discussion above on using Interaction Domain (ID) tags for electrostatic-based delivery of proteins, hydrophobic tags can also be employed. For example, a VEGF-mimetic peptide fused with different proline affinity tags can be delivered from a two-component peptide hydrogel69 demonstrating affinity-controlled release.70 The gel is formed by crosslinking tryptophan and proline-rich polypeptide domains. The VEGF mimetic is encapsulated by co-assembly where it competes for the Trp-rich domains of the gel. This design is inspired by the tryptophan WW domains of some signaling proteins, which bind to proline-rich sequences.71
Other systems use a combination of interactions to individually facilitate the encapsulation and subsequent delivery of drug. Although these following examples are not strictly considered affinity-controlled systems, they warrant mention. For example, the incorporation of phenylalanine into the sequence of self-assembling RADA-based peptides allows the encapsulation of 5-fluorouracil via the formation of π-π interactions during co-assembly.72 The drug loading efficiency was found to be 15% higher for Phe-modified RADA in comparison to a control gel void of the phenylalanine residue. Drug release results from pH-dependent destabilization of the fibers constituting the gel and as such, is not a strict example of affinity-controlled release. Roy et al. developed gels formed from a self-assembling hexapeptide (NH2-WLVFFK-COOH) that demonstrate sustained release of 5-fluorouracil and ciprofloxacin, Figure 6B.73 Efficient loading of the cargo within the gels was achieved primarily by the formation of aromatic π-π interactions between the indole moiety of the peptide’s tryptophan and the aromatic groups of the drug molecules. This system demonstrates a modest pH-dependent control over release whose molecular basis is not known. In another example, cyclodextrin is used to sequester dexamethasone and the resulting complex encapsulated with chitosan to form nanoparticles. These particles are subsequently encapsulated into a gel network formed from the self-assembly of RADA-containing peptide, Figure 6C.74 Here, the cyclodextrin is key in facilitating the loading of the hydrophobic drug. However, in this case the release of cargo was dictated by the stability of the nanoparticles in response to pH. The deprotonation of chitosan at high pH led to particle disassociation from the matrix and subsequent drug release. Although delivery is not strictly affinity-based, it is still controlled by the material’s environment.
Covalent bonds can be highly stable and can be useful for reversible capture and release of both small molecules and proteins. Ligations are a popular strategy used in the conjugation of many drug-material interfaces75 and can be tailored to respond to external stimuli. Many ligations can occur orthogonally in aqueous media under physiological conditions. Commonly employed covalent interactions that can be designed to be reversible include but are not limited to the Schiff’s base and the disulfide linkages.
Schiff’s base (imines) are dynamic covalent bonds formed via the condensation of nucleophilic amines and aldehyde groups. The scope of this reaction can be expanded to other nucleophiles such as hydrazides, acylhydrazides, and aminooxy moieties to form hydrazones, acylhydrazones and oximes,76 respectively, Figure 7A. Each of these linkages are characterized by different pH-dependent rates of hydrolysis and reformation. It is generally appreciated that hydrazones and oximes are intrinsically more stable than imines towards hydrolysis.77 Model studies using small molecules show that the hydrolytic half-life of these linkages at pH 7 range from minutes to nearly a month with the rank order of imines << alkylhydrazones ≤ acylhydrazones <<< oximes.77–79 Figure 7B plots their half-lives as a function of pH. Interestingly, imine linkages are used most frequently in the literature even though they are the most suspectable to hydrolysis at pH 7 and thus may not offer the control that hydrazone or oximes could for long delivery durations.
Figure 7.
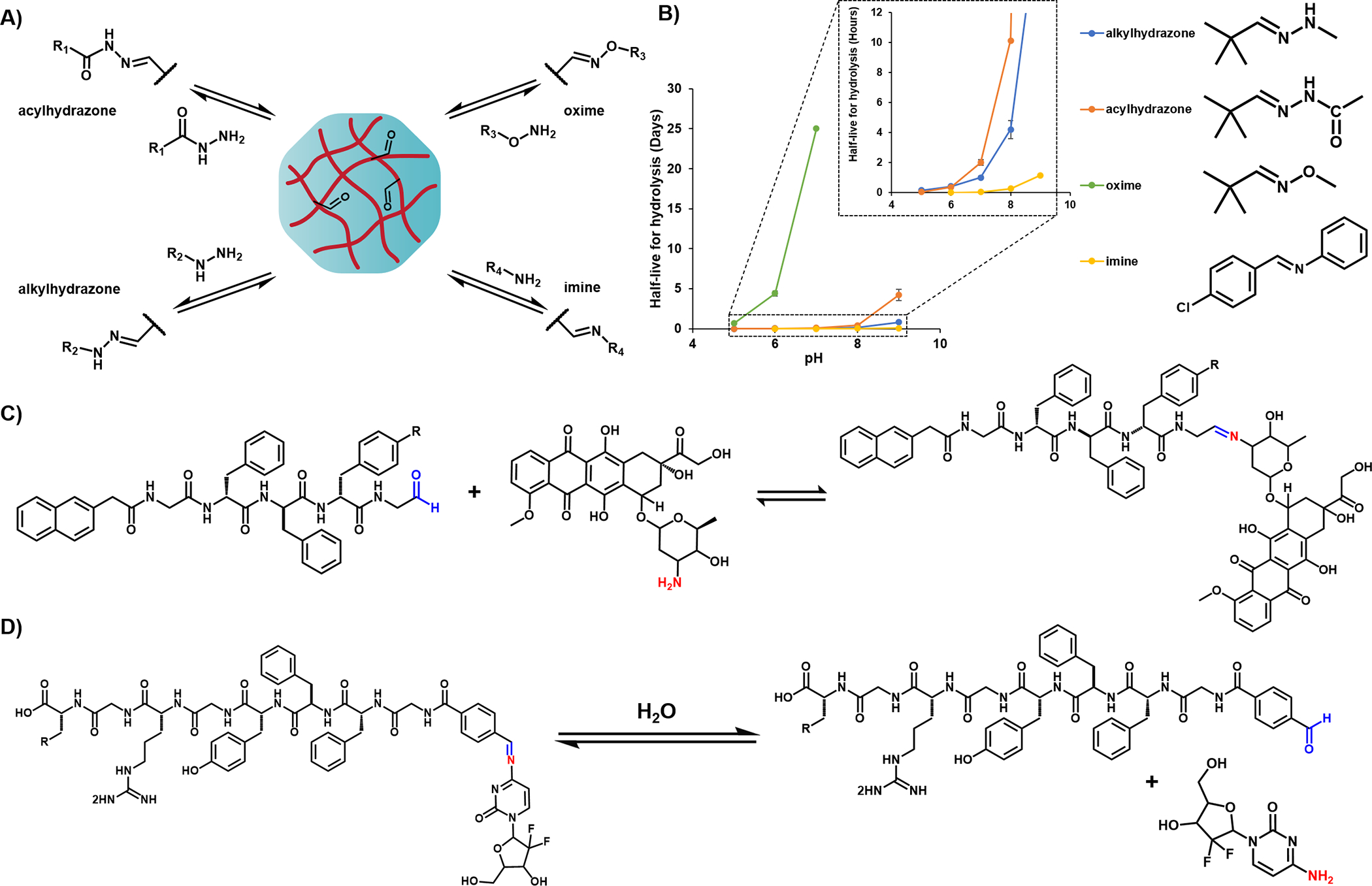
A) Scheme shows the reversible formation of imine-, alkylhydrazone-, acylhydrazone-, and oxime-based linkages between an aldehyde containing gel and amine, hydrazide, acylhydrazide, and oxyamine functional groups and. B) A plot of the relative half-life (t1/2) stabilities of imine, oxime and hydrazone linkages as a function of pH. The half-lives were estimated based on reported pseudo 1st order rate constants, where t1/2 = ln 2/k. Schemes of the aldehyde-functionalized peptide gels used to prolong the delivery of C) doxorubicin D) gemcitabine, based on hydrolytic stability of the corresponding imine complex.
The most obvious approach to the incorporation of imine linkages within peptide gel scaffolds are via the use of lysine side-chains within peptide hydrogels to release aldehyde containing molecules or vice-versa. Wang et al. reported the use of an aldehyde functionalized self-assembling peptide, Nap-GDFDFDpY-CHO that forms hydrogels capable of delivering the amine-containing chemotherapeutic, doxorubicin (Dox).80 The peptide and drug were mixed at pH 7.0 to induce imine formation (CHO-Dox gel) and induce assembly leading to gel formation, Figure 7C. The CHO-Dox gel system releases Dox more quickly at acidic pH values, correlating with Schiff base hydrolysis under those conditions. A similar strategy was used to deliver gemcitabine,81 with faster release rates realized at more acidic pH conditions. The accumulative release of gemcitabine reported after 12 hours at pH 5.0 and pH 7.4, differed by 25% indicating pH-dependence, Figure 7D. Thus, the Schiff base imine complexes can be harnessed for affinity-controlled drug release in response to acidic tumor environments. Likewise, the incorporation of hydrazides into peptide amphiphiles has been demonstrated to release a small molecule drug, nabumetone, which is tethered to the gels via a hydrazone linkage.82 Due to the higher hydrolytic stability of hydrazones relative to imines, the gels demonstrated relatively slower rates of hydrolysis at physiological pH, with a small burst release followed by a slow sustained release profile with less than 40% of the drug released after 24 days.
The thiol functionality of cysteine represents an opportunity in affinity-controlled release. Disulfide bond formation between cysteines has been extensively used to induce gel formation and modulate the mechanical properties of existing self-assembled peptide networks.83 The presence of free thiols such as glutathione in specific biological niches should permit the subsequent redox-responsive release of the bound proteins. Glutathione/glutathionedisulfide is known to be the most plentiful redox couple in vivo with differing concentration ratios at different biological sites.84,85 One can envision ligating proteins to material networks through disulfide bond formation and controlling their release in a redox-specific manner. Of course, other thiol containing molecules can also be delivered using this strategy. Surprisingly, in our literature search, we did not find examples of its use to mediate affinity-controlled delivery in peptide-based material systems. However, we did find one somewhat related example, although not strictly affinity-based. Here, a disulfide tethering strategy is used to produce prodrug hydrogelators containing pacilataxel (PTX) conjugated to a self-assembling peptide amphiphile.86 However, this system demonstrates the release of the individual peptide-drug monomer units, which occurs as a result of the dissociation of the nanofibers over time and not as a result of an affinity-based interaction. The monomer conjugates released from the gel undergo subsequent disulfide bond cleavage to release free PTX.
Future Outlook & Summary
Hydrogels play a unique role in drug-delivery, with peptide-based systems emerging as a versatile platform towards the development of precision therapeutics. Significant advances can come from unexplored binding pair combinations and other emerging reaction chemistries. For example, while there are numerous reports of the use of metal-ligand interactions in peptide gel formation,87–92 there aren’t many examples where the reversibility of metal-ligand complexation has been harnessed for drug-delivery applications. Metal-ion chelation can be highly specific and stable under a wide range of physiological conditions. Direct metal-peptide interactions with naturally occurring amino acids such as histidine-nickel complexes or other metal-binding ligands can be incorporated within the scaffold of hydrogels. For instance, drug-delivery hydrogels systems can be engineered to release metal-bound proteins fused with histidine tags, in response to biological triggers. Although an exciting possibility, attention should be given to designing systems that limit possible metal-associated toxicity.
Delivering combination therapies56 represents another opportunity. Currently, there is substantial activity in drug discovery to identify new combination therapies from existing FDA-approved drugs. Combination therapy administers multiple drugs having different mechanisms of action that together provide clinical benefit. Given the temporal resolution of biochemical pathways important to a particular disease and the different PK/PD properties of the individual drugs, it is becoming clear that the time-specific administration of combination therapies is an important factor in defining their efficacy. Thus, developing affinity-controlled systems capable of delivering multiple agents with distinct individual release kinetics would be useful.
Designing ligand/drug interactions whose KD values are sensitive to environmental cues is another exciting approach towards programmable materials development. Modulating delivery via changes in pH and ionic strength were discussed throughout the review, but other cues such as temperature, light, enzymatic action and redox potential represent material design space that is ripe for exploration. Peptide gels that are truly responsive will be far more complex than those discussed and may require the integration of multiple affinity interactions in order to respond to the cues and signals in their environment. The avenues for innovation with affinity-based release systems are limitless, allowing ample scope for customized drug-delivery solutions.
Acknowledgment
The preparation of this manuscript was supported by the Center for Cancer Research intramural research program of the National Cancer Institute, National Institutes of Health.
References
- 1.Mondal S, Das S, Nandi AK, A review on recent advances in polymer and peptide hydrogels. Soft Matter. 2020; 16:1404–1454 10.1039/C9SM02127B [DOI] [PubMed] [Google Scholar]
- 2.Li J, Mooney DJ, Designing hydrogels for controlled drug delivery. Nat Rev Mater. 2016; 1:16071 10.1038/natrevmats.2016.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li J, Xing R, Bai S, Yan X, Recent advances of self-assembling peptide-based hydrogels for biomedical applications. Soft Matter. 2019; 15:1704–1715 10.1039/C8SM02573H [DOI] [PubMed] [Google Scholar]
- 4.Tesauro D, Accardo A, Diaferia C, et al. , Peptide-Based Drug-Delivery Systems in Biotechnological Applications: Recent Advances and Perspectives. Molecules. 2019; 2410.3390/molecules24020351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y, Wang F, Cui H, Peptide-based supramolecular hydrogels for delivery of biologics. Bioeng Transl Med. 2016; 1:306–322 10.1002/btm2.10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen C, Zhang Y, Hou Z, Cui X, Zhao Y, Xu H, Rational Design of Short Peptide-Based Hydrogels with MMP-2 Responsiveness for Controlled Anticancer Peptide Delivery. Biomacromolecules. 2017; 18:3563–3571 10.1021/acs.biomac.7b00911 [DOI] [PubMed] [Google Scholar]
- 7.Tang JD, Mura C, Lampe KJ, Stimuli-Responsive, Pentapeptide, Nanofiber Hydrogel for Tissue Engineering. J Am Chem Soc. 2019; 141:4886–4899 10.1021/jacs.8b13363 [DOI] [PubMed] [Google Scholar]
- 8.Dou X-Q, Feng C-L, Amino Acids and Peptide-Based Supramolecular Hydrogels for Three-Dimensional Cell Culture. Adv Mater. 2017; 29:1604062 10.1002/adma.201604062 [DOI] [PubMed] [Google Scholar]
- 9.Jain R, Roy S, Designing a bioactive scaffold from coassembled collagen–laminin short peptide hydrogels for controlling cell behaviour. RSC Adv. 2019; 9:38745–38759 10.1039/C9RA07454F [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malhotra K, Shankar S, Rai R, Singh Y, Broad-Spectrum Antibacterial Activity of Proteolytically Stable Self-Assembled αγ-Hybrid Peptide Gels. Biomacromolecules. 2018; 19:782–792 10.1021/acs.biomac.7b01582 [DOI] [PubMed] [Google Scholar]
- 11.Carrejo NC, Moore AN, Lopez Silva TL, et al. , Multidomain Peptide Hydrogel Accelerates Healing of Full-Thickness Wounds in Diabetic Mice. ACS Biomater Sci Eng. 2018; 4:1386–1396 10.1021/acsbiomaterials.8b00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dreiss CA, Hydrogel design strategies for drug delivery. Curr Opin Colloid Interface Sci. 2020; 48:1–17 10.1016/j.cocis.2020.02.001 [DOI] [Google Scholar]
- 13.Hoare TR, Kohane DS, Hydrogels in drug delivery: Progress and challenges. Polymer. 2008; 49:1993–2007 10.1016/j.polymer.2008.01.027 [DOI] [Google Scholar]
- 14.Pal P, Nguyen QC, Benton AH, Marquart ME, Janorkar AV, Drug-Loaded Elastin-Like Polypeptide–Collagen Hydrogels with High Modulus for Bone Tissue Engineering. Macromolecular Bioscience. 2019; 19:1900142 10.1002/mabi.201900142 [DOI] [PubMed] [Google Scholar]
- 15.Molina I, Li S, Martinez MB, Vert M, Protein release from physically crosslinked hydrogels of the PLA/PEO/PLA triblock copolymer-type. Biomaterials. 2001; 22:363–369 10.1016/S0142-9612(00)00192-7 [DOI] [PubMed] [Google Scholar]
- 16.Vulic K, Shoichet MS, Affinity-Based Drug Delivery Systems for Tissue Repair and Regeneration. Biomacromolecules. 2014; 15:3867–3880 10.1021/bm501084u [DOI] [PubMed] [Google Scholar]
- 17.Vulic K, Pakulska MM, Sonthalia R, Ramachandran A, Shoichet MS, Mathematical model accurately predicts protein release from an affinity-based delivery system. J Controlled Release. 2015; 197:69–77 10.1016/j.jconrel.2014.10.032 [DOI] [PubMed] [Google Scholar]
- 18.Rehmann MS, Skeens KM, Kharkar PM, et al. , Tuning and Predicting Mesh Size and Protein Release from Step Growth Hydrogels. Biomacromolecules. 2017; 18:3131–3142 10.1021/acs.biomac.7b00781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Branco MC, Pochan DJ, Wagner NJ, Schneider JP, Macromolecular diffusion and release from self-assembled β-hairpin peptide hydrogels. Biomaterials. 2009; 30:1339–1347 10.1016/j.biomaterials.2008.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashley GW, Henise J, Reid R, Santi DV, Hydrogel drug delivery system with predictable and tunable drug release and degradation rates. Proc Natl Acad Sci U S A. 2013; 110:2318 10.1073/pnas.1215498110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen W, Zhang K, Kornfield JA, Tirrell DA, Tuning the erosion rate of artificial protein hydrogels through control of network topology. Nat Mater. 2006; 5:153–158 10.1038/nmat1573 [DOI] [PubMed] [Google Scholar]
- 22.Vulic K, Shoichet MS, Tunable growth factor delivery from injectable hydrogels for tissue engineering. J Am Chem Soc. 2012; 134:882–885 10.1021/ja210638x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edelman ER, Nugent MA, Smith LT, Karnovsky MJ, Basic fibroblast growth factor enhances the coupling of intimal hyperplasia and proliferation of vasa vasorum in injured rat arteries. The Journal of Clinical Investigation. 1992; 89:465–473 10.1172/JCI115607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edelman ER, Mathiowitz E, Langer R, Klagsbrun M, Controlled and modulated release of basic fibroblast growth factor. Biomaterials. 1991; 12:619–626 10.1016/0142-9612(91)90107-L [DOI] [PubMed] [Google Scholar]
- 25.Cai S, Liu Y, Zheng Shu X, Prestwich GD, Injectable glycosaminoglycan hydrogels for controlled release of human basic fibroblast growth factor. Biomaterials. 2005; 26:6054–6067 10.1016/j.biomaterials.2005.03.012 [DOI] [PubMed] [Google Scholar]
- 26.Pike DB, Cai S, Pomraning KR, et al. , Heparin-regulated release of growth factors in vitro and angiogenic response in vivo to implanted hyaluronan hydrogels containing VEGF and bFGF. Biomaterials. 2006; 27:5242–5251 10.1016/j.biomaterials.2006.05.018 [DOI] [PubMed] [Google Scholar]
- 27.Peattie RA, Pike DB, Yu B, et al. , Effect of Gelatin on Heparin Regulation of Cytokine Release from Hyaluronan-Based Hydrogels. Drug Delivery. 2008; 15:389–397 10.1080/10717540802035442 [DOI] [PubMed] [Google Scholar]
- 28.Purcell BP, Kim IL, Chuo V, Guenin T, Dorsey SM, Burdick JA, Incorporation of sulfated hyaluronic acid macromers into degradable hydrogel scaffolds for sustained molecule delivery. Biomater Sci. 2014; 2:693–702 10.1039/C3BM60227C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung HJ, Kim HK, Yoon JJ, Park TG, Heparin immobilized porous PLGA microspheres for angiogenic growth factor delivery. Pharm Res. 2006; 23:1835–1841 10.1007/s11095-006-9039-9 [DOI] [PubMed] [Google Scholar]
- 30.Chung YI, Tae G, Hong Yuk S, A facile method to prepare heparin-functionalized nanoparticles for controlled release of growth factors. Biomaterials. 2006; 27:2621–2626 10.1016/j.biomaterials.2005.11.043 [DOI] [PubMed] [Google Scholar]
- 31.Tae G, Scatena M, Stayton PS, Hoffman AS, PEG-cross-linked heparin is an affinity hydrogel for sustained release of vascular endothelial growth factor. J Biomater Sci Polym Ed. 2006; 17:187–197 10.1163/156856206774879090 [DOI] [PubMed] [Google Scholar]
- 32.Yoon JJ, Chung HJ, Lee HJ, Park TG, Heparin-immobilized biodegradable scaffolds for local and sustained release of angiogenic growth factor. Journal of Biomedical Materials Research Part A. 2006; 79A:934–942 10.1002/jbm.a.30843 [DOI] [PubMed] [Google Scholar]
- 33.Zhao Y, Zhang J, Wang X, et al. , The osteogenic effect of bone morphogenetic protein-2 on the collagen scaffold conjugated with antibodies. J Controlled Release. 2010; 141:30–37 [DOI] [PubMed] [Google Scholar]
- 34.Lin C-C, Metters AT, Enhanced Protein Delivery from Photopolymerized Hydrogels Using a Pseudospecific Metal Chelating Ligand. Pharm Res. 2006; 23:614 10.1007/s11095-005-9395-x [DOI] [PubMed] [Google Scholar]
- 35.Wood MD, Sakiyama-Elbert SE, Release rate controls biological activity of nerve growth factor released from fibrin matrices containing affinity-based delivery systems. Journal of Biomedical Materials Research Part A. 2008; 84A:300–312 10.1002/jbm.a.31269 [DOI] [PubMed] [Google Scholar]
- 36.Sakiyama-Elbert SE, Hubbell JA, Controlled release of nerve growth factor from a heparin-containing fibrin-based cell ingrowth matrix. J Controlled Release. 2000; 69:149–158 10.1016/S0168-3659(00)00296-0 [DOI] [PubMed] [Google Scholar]
- 37.Manning CN, Schwartz AG, Liu W, et al. , Controlled delivery of mesenchymal stem cells and growth factors using a nanofiber scaffold for tendon repair. Acta Biomaterialia. 2013; 9:6905–6914 10.1016/j.actbio.2013.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uzunalli G, Guler MO, Peptide gels for controlled release of proteins. Ther Delivery. 2020; 11:193–211 10.4155/tde-2020-0011 [DOI] [PubMed] [Google Scholar]
- 39.Seow WY, Hauser CAE, Short to ultrashort peptide hydrogels for biomedical uses. Mater Today. 2014; 17:381–388 10.1016/j.mattod.2014.04.028 [DOI] [Google Scholar]
- 40.Iglesias D, Marchesan S. 2017. Chapter 9 - Short peptide self-assembled nanostructures for therapeutics innovative delivery. In Nanostructures for Novel Therapy, ed. Ficai D, Grumezescu AM:227–250: Elsevier. Number of 227–250 pp. [Google Scholar]
- 41.Gelain F, Luo Z, Zhang S, Self-Assembling Peptide EAK16 and RADA16 Nanofiber Scaffold Hydrogel. Chem Rev. 2020; 120:13434–13460 10.1021/acs.chemrev.0c00690 [DOI] [PubMed] [Google Scholar]
- 42.Pakulska MM, Miersch S, Shoichet MS, Designer protein delivery: From natural to engineered affinity-controlled release systems. Science. 2016; 351:aac4750 10.1126/science.aac4750 [DOI] [PubMed] [Google Scholar]
- 43.Delplace V, Obermeyer J, Shoichet MS, Local Affinity Release. ACS Nano. 2016; 10:6433–6436 10.1021/acsnano.6b04308 [DOI] [PubMed] [Google Scholar]
- 44.Albrecht M, Supramolecular chemistry—general principles and selected examples from anion recognition and metallosupramolecular chemistry. Naturwissenschaften. 2007; 94:951–966 10.1007/s00114-007-0282-7 [DOI] [PubMed] [Google Scholar]
- 45.Goshe AJ, Steele IM, Ceccarelli C, Rheingold AL, Bosnich B, Supramolecular recognition: On the kinetic lability of thermodynamically stable host–guest association complexes. Proc Natl Acad Sci U S A. 2002; 99:4823 10.1073/pnas.052587499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rajangam K, Arnold MS, Rocco MA, Stupp SI, Peptide amphiphile nanostructure–heparin interactions and their relationship to bioactivity. Biomaterials. 2008; 29:3298–3305 10.1016/j.biomaterials.2008.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chow LW, Wang L-j, Kaufman DB, Stupp SI, Self-assembling nanostructures to deliver angiogenic factors to pancreatic islets. Biomaterials. 2010; 31:6154–6161 10.1016/j.biomaterials.2010.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wieduwild R, Lin W, Boden A, Kretschmer K, Zhang Y, A Repertoire of Peptide Tags for Controlled Drug Release from Injectable Noncovalent Hydrogel. Biomacromolecules. 2014; 15:2058–2066 10.1021/bm500186a [DOI] [PubMed] [Google Scholar]
- 49.Mammadov R, Mammadov B, Toksoz S, et al. , Heparin Mimetic Peptide Nanofibers Promote Angiogenesis. Biomacromolecules. 2011; 12:3508–3519 10.1021/bm200957s [DOI] [PubMed] [Google Scholar]
- 50.Medina SH, Li S, Howard OMZ, et al. , Enhanced immunostimulatory effects of DNA-encapsulated peptide hydrogels. Biomaterials. 2015; 53:545–553 10.1016/j.biomaterials.2015.02.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith DJ, Brat GA, Medina SH, et al. , A multiphase transitioning peptide hydrogel for suturing ultrasmall vessels. Nat Nanotechnol. 2016; 11:95–102 10.1038/nnano.2015.238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamada Y, Patel NL, Kalen JD, Schneider JP, Design of a Peptide-Based Electronegative Hydrogel for the Direct Encapsulation, 3D Culturing, in Vivo Syringe-Based Delivery, and Long-Term Tissue Engraftment of Cells. ACS Appl Mater Interfaces. 2019; 11:34688–34697 10.1021/acsami.9b12152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamada Y, Fichman G, Schneider JP, Serum Protein Adsorption Modulates the Toxicity of Highly Positively Charged Hydrogel Surfaces. ACS Appl Mater Interfaces. 2021; 13:8006–8014 10.1021/acsami.0c21596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fichman G, Andrews C, Patel NL, Schneider JP, Antibacterial Gel Coatings Inspired by the Cryptic Function of a Mussel Byssal Peptide. Adv Mater. 2021; n/a:2103677 10.1002/adma.202103677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun JEP, Stewart B, Litan A, et al. , Sustained release of active chemotherapeutics from injectable-solid β-hairpin peptide hydrogel. Biomater Sci. 2016; 4:839–848 10.1039/C5BM00538H [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Majumder P, Baxa U, Walsh STR, Schneider JP, Design of a Multicompartment Hydrogel that Facilitates Time-Resolved Delivery of Combination Therapy and Synergized Killing of Glioblastoma. Angew Chem, Int Ed. 2018; 57:15040–15044 10.1002/anie.201806483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Majumder P, Zhang Y, Iglesias M, et al. , Multiphase Assembly of Small Molecule Microcrystalline Peptide Hydrogel Allows Immunomodulatory Combination Therapy for Long-Term Heart Transplant Survival. Small. 2020; 16:2002791 10.1002/smll.202002791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nagy-Smith K, Yamada Y, Schneider JP, Protein release from highly charged peptide hydrogel networks. Journal of Materials Chemistry B. 2016; 4:1999–2007 10.1039/C5TB02137E [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamada Y, Chowdhury A, Schneider JP, Stetler-Stevenson WG, Macromolecule-Network Electrostatics Controlling Delivery of the Biotherapeutic Cell Modulator TIMP-2. Biomacromolecules. 2018; 19:1285–1293 10.1021/acs.biomac.8b00107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller SE, Yamada Y, Patel N, et al. , Electrostatically Driven Guanidinium Interaction Domains that Control Hydrogel-Mediated Protein Delivery In Vivo. ACS Cent Sci. 2019; 5:1750–1759 10.1021/acscentsci.9b00501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nagy-Smith K, Moore E, Schneider J, Tycko R, Molecular structure of monomorphic peptide fibrils within a kinetically trapped hydrogel network. Proc Natl Acad Sci U S A. 2015; 112:9816 10.1073/pnas.1509313112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang J, Ma PX, Cyclodextrin-based supramolecular systems for drug delivery: Recent progress and future perspective. Adv Drug Delivery Rev. 2013; 65:1215–1233 10.1016/j.addr.2013.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gelain F, Unsworth LD, Zhang S, Slow and sustained release of active cytokines from self-assembling peptide scaffolds. J Controlled Release. 2010; 145:231–239 10.1016/j.jconrel.2010.04.026 [DOI] [PubMed] [Google Scholar]
- 64.Leach DG, Dharmaraj N, Piotrowski SL, et al. , STINGel: Controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy. Biomaterials. 2018; 163:67–75 10.1016/j.biomaterials.2018.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yishay-Safranchik E, Golan M, David A, Controlled release of doxorubicin and Smac-derived pro-apoptotic peptide from self-assembled KLD-based peptide hydrogels. Polym Adv Technol. 2014; 25:539–544 10.1002/pat.3300 [DOI] [Google Scholar]
- 66.Kumar VA, Shi S, Wang BK, et al. , Drug-Triggered and Cross-Linked Self-Assembling Nanofibrous Hydrogels. J Am Chem Soc. 2015; 137:4823–4830 10.1021/jacs.5b01549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li IC, Moore AN, Hartgerink JD, “Missing Tooth” Multidomain Peptide Nanofibers for Delivery of Small Molecule Drugs. Biomacromolecules. 2016; 17:2087–2095 10.1021/acs.biomac.6b00309 [DOI] [PubMed] [Google Scholar]
- 68.Tiwari P, Rajagopalan R, Moin M, Soni R, Trivedi P, DuttKonar A, Can self-assembled hydrogels composed of aromatic amino acid derivatives function as drug delivery carriers? New J Chem. 2017; 41:308–315 10.1039/C6NJ02125E [DOI] [Google Scholar]
- 69.Wong Po Foo CTS, Lee JS, Mulyasasmita W, Parisi-Amon A, Heilshorn SC, Two-component protein-engineered physical hydrogels for cell encapsulation. Proc Natl Acad Sci U S A. 2009; 106:22067 10.1073/pnas.0904851106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mulyasasmita W, Cai L, Hori Y, Heilshorn SC, Avidity-controlled delivery of angiogenic peptides from injectable molecular-recognition hydrogels. Tissue Eng, Part A. 2014; 20:2102–2114 10.1089/ten.tea.2013.0357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Macias MJ, Wiesner S, Sudol M, WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 2002; 513:30–37 10.1016/S0014-5793(01)03290-2 [DOI] [PubMed] [Google Scholar]
- 72.Ashwanikumar N, Kumar NA, Saneesh Babu PS, et al. , Self-assembling peptide nanofibers containing phenylalanine for the controlled release of 5-fluorouracil. Int J Nanomed. 2016; 11:5583–5594 10.2147/IJN.S104707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roy K, Pandit G, Chetia M, et al. , Peptide Hydrogels as Platforms for Sustained Release of Antimicrobial and Antitumor Drugs and Proteins. ACS Appl Bio Mater. 2020; 3:6251–6262 10.1021/acsabm.0c00314 [DOI] [PubMed] [Google Scholar]
- 74.Lu L, Unsworth LD, pH-Triggered Release of Hydrophobic Molecules from Self-Assembling Hybrid Nanoscaffolds. Biomacromolecules. 2016; 17:1425–1436 10.1021/acs.biomac.6b00040 [DOI] [PubMed] [Google Scholar]
- 75.Gopinathan J, Noh I, Click Chemistry-Based Injectable Hydrogels and Bioprinting Inks for Tissue Engineering Applications. Tissue Eng Regener Med. 2018; 15:531–546 10.1007/s13770-018-0152-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu J, Liu Y, Hsu SH, Hydrogels Based on Schiff Base Linkages for Biomedical Applications. Molecules. 2019; 2410.3390/molecules24163005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kalia J, Raines RT, Hydrolytic stability of hydrazones and oximes. Angew Chem, Int Ed Engl. 2008; 47:7523–7526 10.1002/anie.200802651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cordes EH, Jencks WP, On the Mechanism of Schiff Base Formation and Hydrolysis. J Am Chem Soc. 1962; 84:832–837 10.1021/ja00864a031 [DOI] [Google Scholar]
- 79.Cordes EH, Jencks WP, The Mechanism of Hydrolysis of Schiff Bases Derived from Aliphatic Amines. J Am Chem Soc. 1963; 85:2843–2848 10.1021/ja00901a037 [DOI] [Google Scholar]
- 80.Wang Y, Zhang Y, Li X, Li C, Yang Z, Wang L, A Peptide-Based Supramolecular Hydrogel for Controlled Delivery of Amine Drugs. Chem - Asian J. 2018; 13:3460–3463 10.1002/asia.201800708 [DOI] [PubMed] [Google Scholar]
- 81.Ren C, Xu C, Li D, Ren H, Hao J, Yang Z, Gemcitabine induced supramolecular hydrogelations of aldehyde-containing short peptides. RSC Adv. 2014; 4:34729–34732 10.1039/C4RA05808A [DOI] [Google Scholar]
- 82.Matson JB, Stupp SI, Drug release from hydrazone-containing peptide amphiphiles. Chem Commun. 2011; 47:7962–7964 10.1039/C1CC12570B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Seow WY, Salgado G, Lane EB, Hauser CAE, Transparent crosslinked ultrashort peptide hydrogel dressing with high shape-fidelity accelerates healing of full-thickness excision wounds. SCIENTIFIC REPORTS. 2016; 610.1038/srep32670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tu Y, Peng F, White PB, Wilson DA, Redox-Sensitive Stomatocyte Nanomotors: Destruction and Drug Release in the Presence of Glutathione. Angew Chem, Int Ed. 2017; 56:7620–7624 10.1002/anie.201703276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kuppusamy P, Li H, Ilangovan G, et al. , Noninvasive Imaging of Tumor Redox Status and Its Modification by Tissue Glutathione Levels. Cancer Res. 2002; 62:307. [PubMed] [Google Scholar]
- 86.Chakroun RW, Wang F, Lin R, et al. , Fine-Tuning the Linear Release Rate of Paclitaxel-Bearing Supramolecular Filament Hydrogels through Molecular Engineering. ACS Nano. 2019; 13:7780–7790 10.1021/acsnano.9b01689 [DOI] [PubMed] [Google Scholar]
- 87.Knerr PJ, Branco MC, Nagarkar R, Pochan DJ, Schneider JP, Heavy metal ion hydrogelation of a self-assembling peptideviacysteinyl chelation. J Mater Chem. 2012; 22:1352–1357 10.1039/C1JM14418A [DOI] [Google Scholar]
- 88.Micklitsch CM, Knerr PJ, Branco MC, Nagarkar R, Pochan DJ, Schneider JP, Zinc-Triggered Hydrogelation of a Self-Assembling β-Hairpin Peptide. Angew Chem, Int Ed. 2011; 50:1577–1579 10.1002/anie.201006652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mei J, Zhang X, Zhu M, Wang J, Wang L, Wang L, Barium-triggered β-sheet formation and hydrogelation of a short peptide derivative. RSC Adv. 2014; 4:1193–1196 10.1039/C3RA45023F [DOI] [Google Scholar]
- 90.McEwen H, Du EY, Mata JP, Thordarson P, Martin AD, Tuning hydrogels through metal-based gelation triggers. Journal of Materials Chemistry B. 2017; 5:9412–9417 10.1039/C7TB02140B [DOI] [PubMed] [Google Scholar]
- 91.Abul-Haija YM, Scott GG, Sahoo JK, Tuttle T, Ulijn RV, Cooperative, ion-sensitive co-assembly of tripeptide hydrogels. Chem Commun. 2017; 53:9562–9565 10.1039/C7CC04796G [DOI] [PubMed] [Google Scholar]
- 92.Ji W, Yuan C, Zilberzwige-Tal S, et al. , Metal-Ion Modulated Structural Transformation of Amyloid-Like Dipeptide Supramolecular Self-Assembly. ACS Nano. 2019; 13:7300–7309 10.1021/acsnano.9b03444 [DOI] [PMC free article] [PubMed] [Google Scholar]