

Abstract
Hypertrophic chondrocytes are the master regulators of endochondral ossification; however, their ultimate cell fates cells remain largely elusive due to their transient nature. Historically, hypertrophic chondrocytes have been considered as the terminal state of growth plate chondrocytes, which are destined to meet their inevitable demise at the primary spongiosa. Chondrocyte hypertrophy is accompanied by increased organelle synthesis and rapid intracellular water uptake, which serve as the major drivers of longitudinal bone growth. This process is delicately regulated by major signaling pathways and their target genes, including growth hormone (GH), insulin growth factor-1 (IGF-1), indian hedgehog (Ihh), parathyroid hormone-related protein (PTHrP), bone morphogenetic proteins (BMPs), sex determining region Y-box 9 (Sox9), runt-related transcription factors (Runx) and fibroblast growth factor receptors (FGFRs). Hypertrophic chondrocytes orchestrate endochondral ossification by regulating osteogenic-angiogenic and osteogenic-osteoclastic coupling through the production of vascular endothelial growth factor (VEGF), receptor activator of nuclear factor kappa-B ligand (RANKL) and matrix metallopeptidases-9/13 (MMP-9/13). Hypertrophic chondrocytes also indirectly regulate resorption of the cartilaginous extracellular matrix, by controlling formation of a special subtype of osteoclasts termed “chondroclasts”. Notably, hypertrophic chondrocytes may possess innate potential for plasticity, reentering the cell cycle and differentiating into osteoblasts and other types of mesenchymal cells in the marrow space. We may be able to harness this unique plasticity for therapeutic purposes, for a variety of skeletal abnormalities and injuries. In this review, we discuss the morphological and molecular properties of hypertrophic chondrocytes, which carry out important functions during skeletal growth and regeneration.
Keywords: growth plate, hypertrophy, chondrocyte, chondroclast, osteoblast, primary spongiosa, transdifferentiation, apoptosis, type X collagen, vascular endothelial growth factor, matrix metalloproteinase 9, insulin like growth factor-1, bone morphogenetic protein, SRY-Box transcription factor 9, runt-related transcription factor 2, fibroblast growth factor receptor 3
Introduction
Chondrocyte hypertrophy is a process by which cells undergo a 10 to 20-fold enlargement due to rapid volumetric increases and distinct metabolic and molecular changes. This process facilitates sustained endochondral ossification and plays an instrumental role in the explosive longitudinal bone growth observed among diverse mammalian species. Historically, hypertrophic chondrocytes have been considered as the terminal state of growth plate chondrocytes resulting in degenerative maturation, denoted by cell cycle exit, nuclear condensation and apoptosis (Bonucci et al., 2020). Yet, there is evidence that hypertrophic chondrocytes undergo “transdifferentiation” and directly become osteoblasts at the primary spongiosa (Yang et al., 2014a,b; Zhou et al., 2014; Park et al., 2015; Tsang et al., 2015; Hu et al., 2017). Thus, the “terminal” state of hypertrophic chondrocytes should be more accurately described as a “transient” state, denoted by the ability to be reprogrammed into an osteoblast-like state in response to external stimuli. Additionally, hypertrophic chondrocytes are a source of receptor activator of nuclear factor kappa-B ligand (RANKL) required to induce osteoclastogenesis and formation of the marrow space during endochondral ossification, and to maintain the balance between bone resorption and formation (Xiong et al., 2011). RANKL-mediated multinucleated “chondroclasts” are highest within the cartilaginous mineralized matrix of the hypertrophic zone (Odgren et al., 2016). Hypertrophic chondrocytes also express vascular endothelial growth factor (VEGF), a cytokine that induces angiogenesis and vascularization of the ossification center (Gerber, et al., 1999a; Harper and Klagsbrun, 1999; Zelzer et al., 2004). Thus, hypertrophic chondrocytes possess multifaceted roles to orchestrate endochondral ossification, beyond what was initially described as the terminal state of growth plate chondrocytes that are destined to apoptose. Here, we discuss the morphological properties of hypertrophic chondrocytes, as well as the molecular mechanisms underlying their diverse functions in skeletal development, growth and regeneration (Figure 1).
Figure 1. Multifactorial roles of hypertrophic chondrocytes and their molecular regulation.
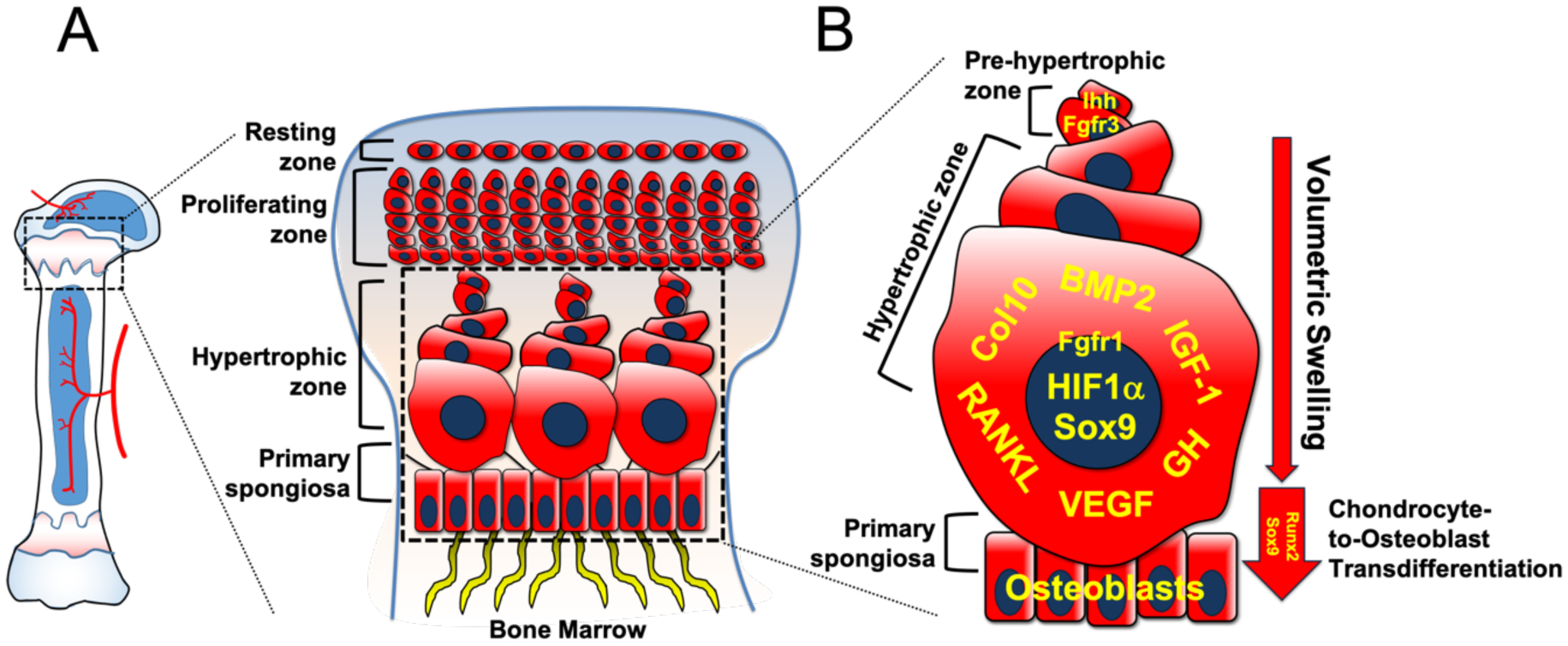
A. Magnified graphical representation of growth plate structure and morphology.
B. Enhanced cartoon of pre-hypertrophic and hypertrophic zones and primary spongiosa. Volumetric swelling due to increased synthesis of intracellular organelles and cytoplasmic water intake facilitates progressive hypertrophic chondrocyte enlargement. GH, IGF-1, Sox9, BMP2, HIF1α and FGFRs regulate chondrocyte hypertrophy, swelling, metabolism and apoptosis. Col10 is a marker for hypertrophic chondrocytes. Runx2 and Sox9 are required for transdifferentiation of hypertrophic chondrocytes into osteoblasts.
The growth plate: The fountain of bone growth
Hypertrophic chondrocytes are the descendants of chondrocytes in the resting zone of the growth plate. The growth plate is organized into three distinct layers classified by cell morphology, function and molecular signature (Hallett et al., 2019). At the top, resting chondrocytes possess stem-like properties associated with infrequent cell division and the ability to feed their daughter cells into the adjacent proliferating zone. The notion that the resting zone houses a population of stem cells was first postulated by autotransplantation experiments in rabbits (Abad et al., 2002) and subsequently by in vivo clonal analyses (Newton et al., 2019) and lineage-tracing studies in mice (Mizuhashi et al., 2018). The resting zone is maintained through the parathyroid hormone-related protein (PTHrP)–Indian Hedgehog (Ihh) feedback loop, which directs the organization and activity of the growth plate (Kronenberg, 2003). The resting zone has two functions dictating chondrocyte hypertrophy: (1) to provide a source of growth plate chondrocytes; (2) to coordinate chondrocyte differentiation into proliferative and hypertrophic cells in a non-cell autonomous manner.
Below the resting zone, proliferating chondrocytes organize vertically into columns. Once proliferative chondrocytes exhaust their mitotic capabilities, they differentiate into pre-hypertrophic chondrocytes and express Ihh. Through PTHrP–Ihh feedback regulation, IHH secreted by pre-hypertrophic cells functions in a paracrine manner to stimulate mitosis of adjacent chondrocytes in the proliferating layer, thus regulating the rate of hypertrophy (Lanske et al., 1996; Vortkamp et al., 1996). Further, pre-hypertrophic chondrocytes undergo rapid volumetric increases due to cell swelling and differentiate into hypertrophic chondrocytes.
Hypertrophic chondrocytes: morphological changes to apoptosis or transdifferentiation
Hypertrophic chondrocytes: Morphological transformation
Hypertrophic chondrocytes are the only bone cells that undergo multiple phases of volumetric increase due to hydration-induced cell swelling (Figure 2). Two classical theories for bone growth exist: (1) it is the result of mitotic activities of proliferating chondrocytes, or (2) it is the result of their cell synthetic activities, including increases in cell volume and height (Hunziker and Schenk, 1989). Early studies indicated that hypertrophic chondrocyte enlargement most significantly contributes to longitudinal bone growth (Hunziker et al., 1987), denoted by increases in absolute volume of the cellular matrix, Golgi apparatus, endoplasmic reticulum (ER) and mitochondria and an 8-fold increase in cytoplasmic water intake (Buckwalter et al., 1986). During chondrocyte hypertrophy, cell volume and height increase linearly, until the cell occupies its greatest volumetric state. This may also be the result of increased synthesis of ultrastructural components, such as hyaluronic acid and proteoglycans (Farnum et al., 1984).
Figure 2. Morphological variation of hypertrophic chondrocytes.
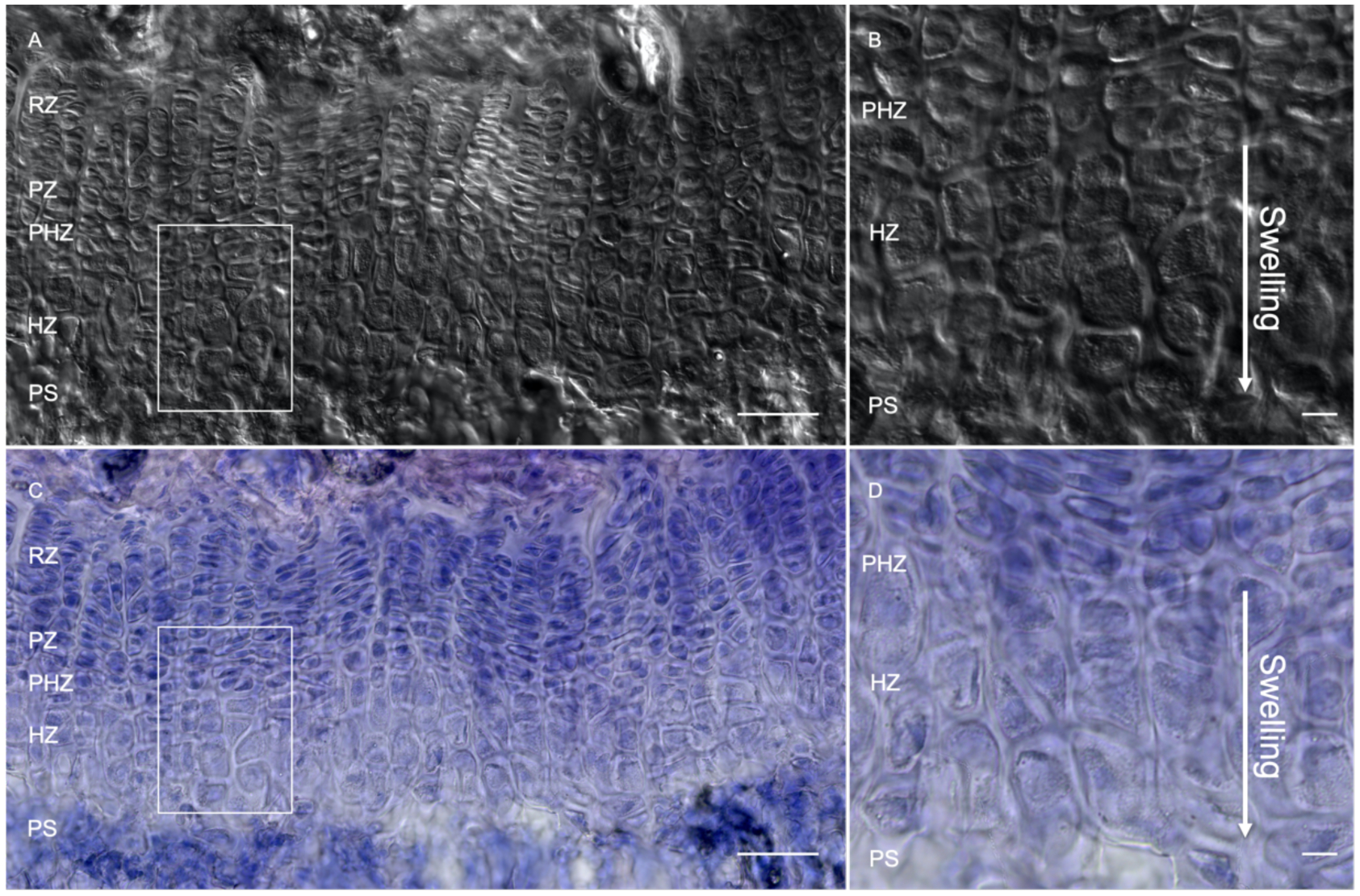
Representative differential interference contrast (DIC) (A,B) and hematoxylin and eosin staining (C,D) of the growth plate at postnatal day 36 in a C57BL/6 mouse. Magnified images (B,D) denote cellular swelling and size variation of hypertrophic chondrocytes as they move towards the primary spongiosa. RZ: resting zone, PZ: proliferating zone, PHZ: pre-hypertrophic zone, HZ: hypertrophic zone, PS: primary spongiosa. Scale bars: 100 μM.
Recently, diffraction phase microscopy was utilized to show that mammalian chondrocytes undergo three phases of volumetric increase due to swelling versus dry mass production (Cooper et al., 2013). “Dry mass” is defined as the total amount of solid substances in a cell (Ginzberg et al., 2015). During Phase 1, there is a 3-fold increase in dry mass and fluid uptake, suggesting that intracellular components of chondrocytes rapidly accumulate. Yet, during Phases 2 and 3, there are 2- and 4-fold increases in dry mass and fluid uptake, respectively, leading to stabilization of dry mass density. This was confirmed by 3D dry mass density index mapping using tomographic phase microscopy in small high-density and large low-density cells. Large chondrocytes had 60% less dry mass density in the cytoplasm. Using an independent conditional knockout study in the hindlimb, the authors demonstrated that Phase 3 entry is regulated by insulin-like growth factor 1 (IGF-1). Through Phases 1–3, hypertrophic chondrocytes increase their volume 10- to 20-fold. Therefore, hypertrophic cell size is not limited due to physical constraint but rather adaptive regulation within its environment. Thus, swelling facilitates hypertrophic cell enlargement while minimizing energetic cost. These studies shed light on the cellular characteristics enabling hypertrophic chondrocyte swelling.
Chondrocyte apoptosis: terminal differentiation followed by cell death
A group of hypertrophic chondrocytes undergoes apoptosis, as defined by physiological cell death due to sporadic or programmed cellular events leading to cytoplasmic shrinkage and maintenance of membrane integrity (Nagata, 2018). Cell cycle checkpoint proteins, p53 and Caspase proteases, play significant roles in the regulation of apoptosis (Galluzzi et al., 2018). In the articular surface, chondrocyte apoptosis is associated with degenerative musculoskeletal diseases, such as osteoarthritis (Hwang and Kim, 2015). Further, external inorganic phosphate ions are released during hydroxyapatite resorption and induce apoptosis of hypertrophic chondrocytes in vitro via nitrosative stress (Mansfield et al., 2001). Thus, hypertrophic chondrocyte apoptosis may be mediated by extrinsic factors.
Apoptosis of hypertrophic chondrocytes is also intrinsically regulated. When cultured with Caspase inhibitors, hypertrophic chondrocytes fail to undergo apoptosis, but maintain ColX synthesis (Roach et al., 2004). The morphological features of chondrocyte apoptosis differ from traditional definition, due to a lack of apoptotic bodies within the lacunae (Roach and Clarke, 2000). Observations of chick and horse terminal hypertrophic chondrocytes noted these cells are “paralyzed” or “dark”, denoted by digestions of organelles within enclosed “islands” formed by expanded or hydrated lumens of ER or vacuoles, respectively (Ahmed et al., 2007; Roach et al., 1999). Only a fraction of hypertrophic chondrocytes is labeled by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), thus, non-labeled cells with morphologically distinct DNA breaks may undergo active gene transcription (Aizawa et al., 1997; Ohyama et al., 1997). Due to the morphological and biochemical differences between classical versus chondrocyte apoptosis, “chondroptosis” has been proposed as an alternative method by which hypertrophic chondrocytes undergo combined apoptotic and autophagic processes (Roach et al., 2004; Luo et al., 2019).
A recent study has assessed morphometric parameters for classifying apoptotic hypertrophic chondrocytes, with contradictory findings (Pazzaglia et al., 2020). Using transmission electron microscopy, the authors found no evidence for expanded cytoplasm containing increased mitochondria, ribosomes, ER or Golgi apparatus in hypertrophic chondrocytes. Rather, these cells possess morphological properties similar to terminally differentiated hypertrophic cells, denoted by nuclear fragmentation and chromatin disappearance. Below the vascular invasion front, macrophages remove the degraded material produced by “hypertrophic ghosts”. These cells have been described during secondary necrosis in hypertrophic chondrocytes in response to metabolic inhibition (Pazzaglia and Congiu, 2013). Thus, there exist discrepancies in the interpretation of chondroptosis, both in terms of morphology and frequency of apoptosis-like events. Resultantly, there is a need to establish quantitative metrics and biochemical assays to accurately define hypertrophic chondrocyte state during this transition.
Hypertrophic chondrocyte transdifferentiation: the bony dilemma
Death is not the only fate of hypertrophic chondrocytes. For centuries, the idea that cells within a committed lineage can undergo alternative fates has been suggested. One example of this is transdifferentation, or the conversion of one differentiated cell type into another due to intrinsic or extrinsic factors (Merrell and Stanger, 2016).
In endochondral bones, early analyses of mouse and rat growth plates suggest that hypertrophic chondrocytes take on multiple cellular fates: apoptosis or transdifferentiation into osteoblasts (Farnum et al., 1990). Additional early investigations showed that hypertrophic chondrocytes derived from murine rib explants or bone rudiments are metabolically active, denoted by incorporation of [3H] thymidine (Crelin and Koch, 1967). EdU-labeling morphometric studies by Roach suggest similar results and postulates that terminally differentiated hypertrophic chondrocytes re-enter the cell cycle and differentiate into osteoblasts at the ossification front (Erenpreisa and Roach, 1996). Further, hypertrophic chondrocytes can undergo an osteogenic fate in response to extrinsic factors from the bone microenvironment, such as gradients of signaling molecules and high concentrations of peptides, ions and glycans (Ishizeki et al., 1996; Bianco et al., 1998; Zerega et al., 1999). These studies provided evidence that not all hypertrophic chondrocytes are destined to die and may have the potential to transdifferentiate into osteoblasts (Aghajanian and Mohan, 2018; Wolff and Hartmann, 2019; Jing et al., 2020). Yet, further investigation into the molecular and morphological changes of hypertrophic cells are required to better define chondrocyte-to-osteoblast transdifferentiation.
Hypertrophic chondrocytes as a supporter for chondroclasts
Hypertrophic chondrocytes are closely intertwined with unique matrix-resorbing cells, “chondroclasts”. Chondroclasts are a subset of osteoclasts dedicated to resorbing mineralized matrix in the hypertrophic zone (Knowles et al., 2012; Odgren et al., 2016). Chondroclasts are morphologically similar to osteoclasts, denoted by multinucleation, polarization and “ruffled bordered” membranes (Feher, 2017). Chondroclasts have been observed in the hyaline cartilage erosion area surrounding the knee joint in patients with osteoarthritis (Bromley and Woolley, 1984). Chondroclasts regulate osteogenic-angiogenic coupling by degrading extracellular matrix (ECM) in the hypertrophic zone, thus enhancing bioavailability of MMP-9 and VEGF in the ossification center (Vu et al., 1998; Gerber et al., 1999b). Chondroclasts share similar transcriptomic profiles with osteoclasts (Khan et al., 2020), but possess higher and lower levels of intracellular and extracellular Tartrate-Resistant Acid Phosphatase (TRAP), respectively (Nordahl et al., 1998). Similar genetic perturbations reduce osteoclasts and chondroclasts in the hypertrophic zone (Odgren et al., 2003). Chondroclasts regulate osteoclastic-angiogenic coupling in the ossification center, as terminal differentiation of hypertrophic chondrocytes coincides with chondroclast-mediated resorption of mineralized matrix and vascular invasion (Farnum and Wilsman, 1989; Lewinson and Silbermann, 1992). Thus, chondroclasts, a unique osteoclast subtype, resorb calcified hypertrophic cartilage, thereby maintaining balance between matrix deposition and resorption in the ossification center adjacent to the hypertrophic zone.
New insights into the molecular regulation of hypertrophic chondrocytes
Chondrocyte hypertrophy is regulated by several major signaling pathways. Here, we discuss regulatory pathways that direct chondrocyte hypertrophy, including HIF1-α, GH, IGF-1, Ihh, BMPs, Sox9, Runx2 and FGFRs (Figure 1).
Metabolic regulation of hypertrophic chondrocytes by HIF1-α signaling
Chondrocytes adapt to hypoxic environments by shifting metabolic catabolysis to anaerobic/glycolytic modes (Shapiro and Srinivas, 2007). The Crabtree effect allows cells in avascular environments with high glucose content to decrease O2 consumption through oxidative phosphorylation while maintaining low ATP production through the Pasteur effect (Hochachka and Lutz, 2001). Hypoxia inducible factor 1-alpha (HIF1-α), a transcription factor that regulates genes involved in glucose transport and the Pasteur effect in mammalian cells, is expressed by hypertrophic chondrocytes and is a survival factor for hypoxic chondrocytes by elevating expression of SRY-Box transcription factor 9 (Sox9) and glycolytic enzymes in vivo (Semenza, 2000). HIF1-α knockout mice display hypo-cellularization in the center of the hypertrophic zone associated with disorganization at the chondro-osseous junction (Pfander et al., 2003; Amarilio et al., 2007). Therefore, HIF1-α signaling uniquely regulates metabolism of hypoxic hypertrophic chondrocytes.
GH and IGF-1: Direct regulators of chondrocyte hypertrophy
Two important regulators of chondrocyte hypertrophy are growth hormone (GH) and IGF-1. Subcutaneous administration of GH and IGF-1 into rats and rabbits, respectively, stimulates [3H] thymidine incorporation into hypertrophic chondrocytes, denoting their metabolic responsiveness following GH and IGF-1 treatment (List et al., 2019). GH treatment also stimulates growth plate elongation and restores Igf1 mRNA levels in the hypertrophic zone of hypophysectomized rats, indicating that GH regulates IGF-1 expression in the growth plate (Racine and Serrat, 2020). GH-deficient mice also have decreased body length compared to controls in a sex-independent manner (Alba and Salvatori, 2004).
IGF-1 regulates endochondral bone growth by promoting chondrocyte proliferation and hypertrophy (Yakar et al., 2018). IGF-1 is one of the major hormones required for skeletal growth and is used to treat pediatric skeletal disorders, such as limb-length discrepancy and short stature (Giustina et al., 2008). Igf1 haploinsufficient mice are 10–20% smaller than controls due to decreased organ, muscle and bone mass and serum IGF-1 (Powell-Braxton et al., 1993). Igf1 knockout mice display a 35% reduction in long bone growth due to specific reductions in the linear length of hypertrophic chondrocytes, suggesting that IGF-1 regulates chondrocyte hypertrophy (Wang et al., 1999a).
IGF-1 receptor (Igf1r) deletion in mice causes delayed endochondral ossification, abnormal chondrocyte proliferation and differentiation and dwarfism (Bikle et al., 2001). Deletion of Igf1r in type II collagen alpha 1 chain (Col2a1) expressing chondrocytes caused dwarfism, expansion of the proliferating zone and increased apoptosis of hypertrophic chondrocytes (Wang et al., 2011). IGF1R signaling interacts with the PTHrP–Ihh feedback loop; in which PTHrP prolongs chondrocyte proliferation and delays their hypertrophic differentiation, thereby delaying IHH expression (Vortkamp et al., 1996). Thus, IGF-1 is necessary for skeletal growth and development due to its role as a regulator of chondrocyte hypertrophy.
Pre-hypertrophic IHH as an indirect regulator of chondrocyte hypertrophy
IHH regulates chondrocyte differentiation and skeletal morphogenesis (Lanske et al., 1996; Vortkamp et al., 1996; Chung et al., 1998; St-Jacques et al., 1999; Kobayashi et al., 2002, 2005). IHH expressed by pre-hypertrophic chondrocytes works in a concerted manner with PTHrP expressed by resting chondrocytes to maintain growth plate structure and longitudinal bone growth (Kronenberg, 2003). Ihh-deficient mice lack proper chondrocyte differentiation and mineralization due to delayed expression of type 10 collagen alpha 1 (Col10a1), a marker of hypertrophic chondrocytes (Linsenmayer et al., 1991; St-Jacques et al., 1999). Activation of Hedgehog signaling via loss of Patched-1 (PTCH1) receptor causes delayed chondrocyte hypertrophy (Mak et al., 2006). Thus, Ihh indirectly regulates chondrocyte hypertrophy through interactions with chondrocytes in the adjacent layers.
BMPs mediate chondrocyte hypertrophy via independent and complimentary mechanisms
BMPs regulate chondrocyte hypertrophy both directly and indirectly. BMP2 and BMP4 are expressed in pre-hypertrophic and hypertrophic chondrocytes (Nilsson et al., 2007) and stimulate chondrocyte hypertrophy in limb explants (de Luca et al., 2001; Hatakeyama et al., 2004). In vitro administration of BMP2 in cultured chondrocytes and limb explants targets hypertrophic chondrocytes, resulting in an increase in Rankl expression in ColX+ cells as well as Ihh and Col10a1 expression in the pre-hypertrophic and hypertrophic zones, respectively (Valcourt et al., 2002; Zhou et al., 2016). Canonical BMP signaling directly regulates chondrocyte hypertrophy, as BMP2 administration inhibits chondrocyte hypertrophy via Smad1/5/8 (Valcourt et al., 2002; Canalis et al., 2003). Col2a1-cre-specific deletion of BMP2 causes shortened long bones due to delayed formation of the hypertrophic zone (Shu et al., 2011). BMP2 induces Runx2 expression at the transcriptional and post-transcriptional levels via phosphorylation of CDK4, which inhibits chondrocyte hypertrophy via Runx2 degradation (Zhang et al., 2009). This is important, since Runx2 activation is necessary for hypertrophic chondrocyte differentiation (Ding et al., 2012) and transdifferentiation (Qin et al., 2020). Deletion of BMP signaling members, Smad6 and Bmpr1a/b, leads to chondrodysplasia due to premature hypertrophic differentiation and smaller hypertrophic zones (Yoon et al., 2005). Thus, BMP2 and members of the canonical BMP signaling pathway regulate chondrocyte hypertrophy through Runx2.
Sox9 downregulation induces hypertrophic chondrocyte transdifferentiation
Sox9 activation is required for mesenchymal condensation of the cartilaginous anlage during fetal development (Lefebvre and Smits, 2005). Sox9 is expressed in chondroprogenitor cells and becomes isolated to resting, proliferating and pre-hypertrophic chondrocytes postnatally (Zhao et al., 1997). Sox9 knockout mice have reduced chondrocyte hypertrophy due to absence of Col10a1 expression in the hypertrophic zone (Ikegami et al., 2011; Dy et al., 2012). Sox9 activates Col10a1 transcription in hypertrophic chondrocytes by binding to its promoter cooperatively with myocyte enhancer factor 2C (Mef2c) (Dy et al., 2012). Thus, Sox9-mediated Col10a1 transcription is required for chondrocyte hypertrophy. Sox9 misexpression in Col10a1+ hypertrophic chondrocytes results in reduced bone marrow formation at P0, reduced bone growth and deficiencies in Vegfa, Mmp13, Rankl and Opn expression in hypertrophic cells (Hattori et al., 2010). Further, a recent study has demonstrated that persistent Sox9 expression in the growth plate causes inhibition of chondrocyte-to-osteoblast transdifferentiation in trabecular bone associated with decreased expression of Mmp9, Mmp13, Sp7 and Col1a1 (Lui et al., 2019). Thus, downregulation of Sox9 in hypertrophic chondrocytes is necessary for vascular invasion and degradation of calcified hypertrophic cartilage in the growth plate in addition to transdifferentiation of hypertrophic chondrocytes into osteoblasts.
According to a recent study, Sox9 maintains growth plate architecture and safeguards the lineage fates of chondrocytes by preventing their dedifferentiation into mesenchymal progenitors while facilitating hypertrophic chondrocyte transdifferentiation into osteoblasts (Haseeb et al., 2021). Using an Acan-creERT2; ROSA26RtdTomato; Sox9f/f, chondrocyte-specific conditional knockout mouse, single cell RNA-sequencing analysis of chondrocytes extracted from control and mutant distal tibial and femur epiphyses discovered that transcriptomic profiles of mutant chondrocytes bypass late proliferative, pre-hypertrophic and hypertrophic stages, becoming prematurely terminally differentiated or osteoblast-like cells. These transcriptomic data were confirmed by immunohistochemical analyses, denoted by increased expression of terminal hypertrophic chondrocytes markers, Col10a1 and matrix GLA protein (Mgp) and osteoblast markers, Sp7, Col1a1 and Bglap at the transition zone of Sox9-deficient growth plates. Thus, Sox9 expression in the postnatal growth plate regulates transdifferentiation of hypertrophic chondrocytes to osteoblast-like cells.
Runx-related genes are required for chondrocyte hypertrophy
The Runx transcription factors play important roles in chondrocyte hypertrophy. During fetal development, Runx1 is expressed by early mesenchymal progenitor cells in condensations (Yamashiro et al., 2002; Smith et al., 2005). Runx2/3 are expressed in pre-hypertrophic and hypertrophic chondrocytes, suggesting direct functional roles of Runx2/3 in chondrocyte hypertrophy (Inada et al., 1999a; Kim et al., 1999; Sato et al., 2008). Runx2 regulates osteoblast differentiation in the early stages of endochondral bone formation (Komori et al., 1997; Otto et al., 1997). Genetic ablation or expression of dominant negative RUNX2 leads to reduced chondrocyte hypertrophy (Inada et al., 1999b; Ueta et al., 2001). RUNX2 transcriptionally regulates genes critical for vascular invasion and ECM synthesis, including VEGF (Zelzer et al., 2001) and MMP13, respectively (Selvamurugan et al., 2000). Runx2/Runx3 double knockout mice have loss of chondrocyte maturation due to failed formation of the hypertrophic zone and decreased Col10a1 expression (Yoshida et al., 2004). Conversely, Runx2 overexpression in chondrocytes causes premature chondrocyte hypertrophy and early induction of ColX expression in vitro (Enomoto et al., 2000) and in vivo (Takeda et al., 2001). Further, Runx2 regulates ColX transcription in hypertrophic chondrocytes (Drissi et al., 2003; Zheng et al., 2003).
A recent study shows that hypertrophic chondrocyte-specific conditional knockout of Runx2 (Col10a1-cre; Runx2f/f) causes decreased expression of Vegfa in hypertrophic chondrocytes, and Mmp13, Col1a1 in the primary spongiosa, associated with increased apoptosis and failure of chondrocyte-to-osteoblast transdifferentiation (Qin et al., 2020). Using a Col10a1-cre; Rosa26-mTFP1; Runx2f/f; 2.3Col1a1-tdTomato compound mutant mouse, the authors demonstrated that hypertrophic chondrocyte-derived trabecular and endosteal osteoblasts were significantly reduced or absent in mutants at embryonic day 17.5 (E17.5), P0 and 1-week. Primary spongiosa formation was delayed in mutants, indicated by decreased expression of bone sialoprotein 2 and Col1a1, hypertrophic chondrocyte and osteoblast markers, respectively, at E15.5. At birth, spongiosa development and trabecular bone volume were similar in wild-type and mutant mice. Thus, Runx2 is required for survival and transdifferentiation of hypertrophic chondrocytes during fetal development. Runx2, initially identified as a regulator of osteoblast formation, also plays roles in chondrocyte hypertrophy, transdifferentiation, vascular invasion and matrix deposition in the hypertrophic zone.
FGFRs play dual roles in chondrocyte hypertrophy and skeletal growth
Fibroblast growth factors receptors (Fgfr) play dual roles in promoting or inhibiting chondrocyte differentiation and endochondral bone growth. Fgfr1 and Fgfr2 are initially expressed in the embryonic perichondrium and become restricted to the hypertrophic and resting zones, respectively (Delezoide et al., 1998; Lazarus et al., 2007; Sheeba et al., 2010). Fgfr3 is expressed after the pre-condensation stage in the cartilage anlage and becomes isolated to proliferating and pre-hypertrophic chondrocytes (Ornitz and Marie, 2015). FGFR1 overactivation in humans causes appendicular skeletal deformities and dwarfism (White et al., 2005), although a similar mutation in Fgfr1 in mice had no effect on bone formation (Zhou et al., 2000). Mesoderm-specific deletion of Fgfr1 (Dermo1-cre; Fgfr1f/f) causes impaired chondrocyte hypertrophy in fetal stages (Hung et al., 2007). During postnatal development, chondrocyte-specific deletion of Fgfr1 (Col2a1-cre; Fgfr1f/f) causes hypertrophic zone expansion associated with delayed degradative maturation of hypertrophic chondrocytes (Jacob et al., 2006). Further, FGFR1 signaling delays hypertrophic differentiation of chondrocytes. Thus, Fgfr1 expression is important for regulating chondrocyte hypertrophy through unknown mechanisms. FGFR2 functions in resting and proliferating chondrocytes in a redundant manner; Dermo1-cre; Fgfr2f/f mice display normal chondrocyte proliferation and growth plate morphology (Yu et al., 2003).
During fetal development, FGFR3 activates chondrocyte proliferation. By early postnatal development, FGFR3 inhibits chondrocyte proliferation and hypertrophic differentiation (Iwata et al., 2000). Activating mutations in FGFR3 in humans and mice cause impaired chondrocyte proliferation and premature hypertrophy, leading to achondroplasia (Wang et al., 1999b). Conversely, Fgfr3 deficient mice present increased hypertrophic zone linear length and prolonged endochondral ossification (Colvin et al., 1996). FGFR3-mediated inhibition of chondrocyte proliferation and hypertrophy are regulated by STAT1-p21 and MAPK-ERK signaling, respectively (Su et al., 1997; Murakami et al., 2004; Raucci et al., 2004; de Frutos et al., 2007). FGFR3-mediated suppression of Sox9 decreases pre-hypertrophic chondrocyte differentiation (Zhou et al., 2015). Mesenchymal cell-specific overactivation of FGFR3 (Prrx1-cre; Fgfr3Y637C/+) causes failure of chondrocyte-to-osteoblast transdifferentiation in a tibial fracture healing model, resulting in persistent fibrocartilages at the callus (Julien et al., 2020). In mutants, Col10a1+ cells fail to become osteoblasts, denoted by decreased vascularization and chondrocyte proliferation at the callus. The fracture defect in mutants is due to an inability for periosteal cells to differentiate into hypertrophic chondrocytes, causing an intrinsic reduction in transdifferentiation. Notably, when mutant-derived periosteal cells were transplanted to wild-type hosts, transdifferentiation occurred (Julien et al., 2020). Thus, Fgfr3 is important for chondrocyte proliferation, hypertrophy and transdifferentiation during skeletal regeneration. Yet, Fgfr3’s role during physiologic hypertrophic chondrocyte-to-osteoblast transdifferentiation remains unknown.
Hypertrophic chondrocytes regulate osteogenic-angiogenic and osteogenic-osteoclastic coupling
Hypertrophic chondrocytes as an important regulator of osteoclastogenesis
Hypertrophic chondrocytes express RANKL and regulate osteoclastogenesis. Coupling between bone-forming osteoblasts and bone-resorbing osteoclasts maintains skeletal homeostasis (Sims and Martin, 2014). RANKL is expressed by cells of the osteoblast lineage and facilitates osteoclast formation (Kong et al., 1999; Sobacchi et al., 2007). It has been known for decades that osteoblasts regulate osteoclastogenesis in vitro (Rodan and Martin, 1982; Takahashi et al., 1988). Yet, recent studies suggest that matrix-embedded osteocytes, not osteoblasts, are the primary source of RANKL (O’Brien, 2010). Ablation of osteoblasts in vivo and in vitro has no impact on Rankl expression or osteoclast number (Corral et al., 1998; Galli et al., 2009). Anabolic glucocorticoid administration in mice reduces osteoblasts and pre-osteoblasts, but not osteocytes (Weinstein et al., 1998, 2002). Conditional deletion of RANKL in limb bud mesenchyme causes significant reduction of osteoclasts below the hypertrophic zone (Xiong et al., 2011). In this study, conditional ablation of RANKL in osteoblasts (Osteocalcin-cre [Ocn-cre; Ranklf/f]; Osterix-cre [Osx-cre; Ranklf/f]) causes loss of Rankl expression in the hypertrophic zone. Further, Col10a1-cre; Ranklf/f, Osx-cre; Ranklf/f and Ocn-cre; Ranklf/f knockout mice all prevented calcified cartilage resorption by reducing RANKL expression in hypertrophic cells. Osteocytes embedded in the trabecular lacunae highly express RANKL (Nakashima et al., 2011). Osteocyte-specific deletion of RANKL (Dmp1-cre; Ranklf/f) causes decreased osteoclast number and increased trabecular bone volume, leading to osteopetrosis. Therefore, RANKL produced by hypertrophic chondrocytes and osteocytes is essential for osteoclastogenesis.
Hypertrophic chondrocytes as a central regulator of osteogenic-angiogenic coupling
Vascularization of the ossification center is an essential process to establish the marrow cavity. Capillary invasion into the cartilage template is followed by ossification. Growth factors VEGF, epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) are expressed in the growth plate and regulate vascularization (Hu and Olsen, 2016). VEGF expressed by hypertrophic chondrocytes induces vascularization of the ossification center by recruiting blood vessels (Risau, 1995; Carmeliet et al., 1996; Ferrara et al., 1996; Gerber et al., 1999a). Inhibition of VEGF protein by chimeric VEGF–IgG decreases femur length and enhances Col10a1 expression in the hypertrophic zone, associated with disorganization of metaphyseal blood vessels (Gerber et al., 1999b). VEGF-mediated metaphyseal vasculogenesis triggers apoptosis of hypertrophic chondrocytes (Gerber et al., 1999b; Harper and Klagsbrun, 1999). Col2a1-cre-specific deletion of Vegfa causes reduced cartilage formation and skeletal mineralization, delayed vascularization of the ossification center and removal of hypertrophic chondrocytes (Zelzer et al., 2004). Thus, VEGF is necessary for maintaining hypertrophic chondrocyte survival.
VEGF-mediated osteogenic-angiogenic coupling during skeletal growth has been extensively studied. Functioning cooperatively with VEGF, matrix metalloproteinase-9 (MMP-9), is expressed by hypertrophic chondrocytes and degrades cartilaginous ECM (Paiva and Granjeiro, 2017). Similar to the VEGF inhibition phenotype (Gerber et al., 1999b), Mmp-9 knockout mice have an expanded hypertrophic zone (Vu et al., 1998; Ortega et al., 2005) associated with reduced chondrocyte apoptosis, vascularization and ossification. Mmp9-deficient mice have impaired skeletal regeneration, denoted by accumulation of hypertrophic cartilage and delayed endochondral ossification during healing (Colnot et al., 2003). This was confirmed by analysis of Mmp-9-deficient growth plates, in which Mmp13 expression was elevated in the expanded hypertrophic zone (Kojima et al., 2013). Consistent with others, this suggests a role for MMP-13 to compensate for MMP-9 loss in hypertrophic cells to degrade ECM (Wu et al., 2002; Ortega et al., 2010). MMP-13-mediated ECM degradation of the hypertrophic zone also coincides with apoptosis of hypertrophic chondrocytes (Inada et al., 2004).
MMP-9-mediated ECM degradation increases bioavailability of VEGF, resulting in the recruitment of osteoclasts to the vascular front to facilitate ECM remodeling and hypertrophic chondrocyte turnover. Expansion of the hypertrophic zone and vascularization of the ossification center in Mmp9 knockout mice are partially rescued by exogenous VEGF (Ortega et al., 2010). Thus, MMP9-driven resorption of the hypertrophic zone is synergistically coupled to VEGF-mediated vasculogenesis. Apoptosis of hypertrophic chondrocytes in Mmp9-deficient mice is observed within the center of the expanded hypertrophic zone (Vu et al., 1998). Furthermore, Mmp9-deficient hypertrophic chondrocytes delay release of pro-angiogenic factors, indicating that MMP-9-driven osteogenic-angiogenic coupling in the hypertrophic zone regulates apoptosis of hypertrophic chondrocytes, ECM degradation and vasculogenesis of the metaphysis.
Alternative osteogenic cell fates of hypertrophic chondrocytes
Col10a1-mCherry+ cells are located in the metaphyseal marrow space
Type X collagen (ColX) is a short chain collagen that forms aggregates in the territorial matrix of hypertrophic chondrocytes (Schmid and Linsenmayer, 1990; Shen, 2005). Col10a1-deficient mice are viable and undergo normal bone formation (Rosati et al., 1994). Col10a1 is expressed in hypertrophic chondrocytes, according to early immunohistochemical and molecular analyses (Schmid and Linsenmayer, 1985; Iyama et al., 1991; Gu et al., 2014) and mouse reporter models (Gebhard et al., 2008; Kong et al., 1993). Analysis of Col10a1-mCherry knock-in reporter mice revealed Col10a1+ cells in the metaphyseal marrow space, in addition to in the pre-hypertrophic and hypertrophic zones (Maye et al., 2011). Yet, endogenous Col10a1 mRNA is most abundant in pre-hypertrophic and hypertrophic chondrocytes. Thus, Col10a1-mCherry+ cells in the marrow space may represent a population of apoptosis-evading chondrocytes or hypertrophic cells that have transdifferentiated.
Analysis of Col10a1-mCherry; Col3.6-Topaz; Col2.3-Emerald triple transgenic mice revealed distinct reporter activities within the growth plate and trabecular bone: Col10a1-mCherry+ cells were found in the hypertrophic zone and surrounding trabecular osteoblasts, while Col2.3-Emerald+ cells were localized to the growth plate and Col3.6-Topaz+ cells were present in the trabecular bone. Interestingly, Col10a1-mCherry+ trabecular osteoblasts do not overlap with Col3.6-Topaz+ osteoblasts. Thus, Maye et al. conclude “no evidence of chondrocyte to osteoblast transdifferentiation” (Maye et al., 2011), although their analyses were limited to late embryonic and early postnatal stages therefore not addressing the possibility that Col10a1+ cells may become osteoblast-like cells during late postnatal development. Additionally, others state that “mCherry expression fades before the onset of osteogenesis and expression of Col1a1-EGFP, and the fate of the [hypertrophic chondrocyte] cannot be traced” (Tsang et al., 2015). Could a subset of Col10a1-mCherry+ cells represent a unique osteo-chondroprogenitor population that contributes to the trabecular compartment? To address this cell fate question, advances in lineage-tracing technology have facilitated the spatiotemporal analysis of hypertrophic chondrocyte cell fates through the use of cre-loxP system (Vanhorn and Morris, 2020).
Lineage-tracing findings from pan-chondrocyte Col2a1-crER and Aggrecan-creER lines
Yang et al. demonstrated that Col2a1+ growth plate chondrocytes contributed to Col1a1+ osteoblasts in the metaphysis, using a Col2a1-creER; ROSAEYFP lineage-tracing model (Yang et al., 2014a). Analysis of Col2a1-creER; ROSAEYFP and Col2a1-creER; ROSAConfetti single and multicolor clonal lineage reporter mice demonstrated that Col2a1+ chondrocytes give rise to metaphyseal osteoblasts at low frequencies (Yang et al., 2014a). Yet, because Col2a1-creER labels all chondrocyte subtypes in the growth plate, it is unknown if Col2a1-creER-lineage-traced osteoblasts are derived from hypertrophic cells or unidentified osteo-chondroprogenitor populations at the primary spongiosa. Additionally, analysis of a “chondrocyte-specific” Aggrecan-creER (Acan-creER) lineage-tracing model (Henry et al., 2009) discovers that Acan+ cells contribute to osteoblasts at the primary spongiosa. This is also observed during skeletal regeneration, as Acan-creER+ cells contribute to 2.3Col1a1-GFP+ osteoblasts at the repair callus (Zhou et al., 2014).
Col10a1-cre: is it the right tool to study hypertrophic chondrocyte transdifferentiation?
In vivo lineage-tracing studies have demonstrated that Col10+ chondrocytes may transdifferentiate into osteoblasts and osteocytes in the trabecular and cortical bone (Yang et al., 2014a; Yang et al., 2014b; Zhou et al., 2014). In a study by Zhou et al., fetal-derived Col10a1+ hypertrophic chondrocytes expressed Col1a1 at the primary spongiosa and trabecular and endosteal surfaces during early and late postnatal development (Zhou et al., 2014). In a tandem analysis, a Col10a1int2-cre; ROSAEYFP reporter mouse shows that Col10a1+ hypertrophic chondrocytes invade into the metaphysis and trabecular bone and express osteoblast markers Col1a1, Ocn and Bsp and eventually became matrix-embedded osteocytes in the diaphysis at P20. (Yang et al., 2014b). Consistent with early reports suggesting the metabolic capability of hypertrophic chondrocytes (Crelin and Koch, 1967), the authors found that Col10a1-cre+ cells uptake BrdU in the metaphysis and are mitotically active. Further, Col10a1int2-cre; ROSAEYFP-marked cells became with perilipin+ adipocytes.
An additional study using Col10a1-cre; ROSARYFP/LacZ models demonstrated that descendants of Col10a1+ hypertrophic chondrocytes contribute to osteoblast formation at the primary spongiosa and on the trabecular and endosteal surfaces (Yang et al., 2014a). Col10a1-cre; RosaLacZ+ cells became Col1a1+ endosteal osteoblasts at P10 and at the chondro-osseous junction in cortical bone at 3 months, suggesting that Col10a1+ hypertrophic chondrocytes may commit to an osteogenic lineage in adulthood. In the same study, fetal-derived Col10a1-creERt; RosaLacZ-marked hypertrophic chondrocytes gave rise to immature Osx+ pre-osteoblasts at the primary spongiosa, Col1a1+ metaphyseal osteoblasts and Sclerostin+ osteocytes in the trabecular bone. Yet, these studies assess chondrocyte-to-osteoblast ‘transdifferentiation’ only in early postnatal time points, and therefore did not determine if conversion of Col10a1+ hypertrophic cells to osteoblasts also occur in adulthood. These studies suggest that Col10a1+ lineage traced hypertrophic chondrocytes contribute to the osteogenic pool during early postnatal endochondral bone growth.
Hypertrophic chondrocytes reenter the cell cycle and become osteoblast-like during skeletal regeneration
More recently, hypertrophic chondrocytes were confirmed to reenter the cell cycle, marked by BrdU incorporation and Ki67 expression and undergo a pro-osteogenic fate during skeletal regeneration using combinatorial histomorphometric and gene expression analyses (Hu et al., 2017). In this study, as chondrocytes in the transition zone become osteoblast-like cells, they lose expression of chondrogenic signatures, Sox9, Col2a1 and Col10a1, while beginning to express Col1a1. In addition to becoming osteoblast-like, transition zone hypertrophic chondrocytes express markers of cell pluripotency, Oct4, Sox2 and Nanog, suggesting that hypertrophic cells may revert to a pluripotent-like state during transdifferentiation into osteoblasts. These findings denote unique morphological and gene expression signatures of hypertrophic chondrocytes in response to fracture healing.
Chondrocyte-derived osteoprogenitors become osteoblasts
In a tandem analysis, these results were confirmed using bacterial artificial chromosome (BAC)-generated Col10-cre; ROSARYFP and Col10-cre; ROSALacZ reporter models (Park et al., 2015). Col10+ chondrocytes overlap with Col1a1+ and Ocn+ osteoblasts in the primary ossification center during embryonic development and later in the primary spongiosa, suggesting that these cells may originate from Col10a1+ hypertrophic chondrocytes in the growth plate. YFP+ trabecular cells isolated from the spongiosa of femoral heads of Col10-cre; RosaRYFP reporter mice were highly enriched for osteogenic markers, Ocn, Osx, Col1a1 and Runx2 at levels similar to cortical bone. According to flow cytometry analysis of cultured Col10-cre; ROSARYFP-derived endosteal osteoblasts at P7, 11% of these cells are YFP+. Thus, the authors postulate that 11% of endochondral osteoblasts are derived from hypertrophic chondrocytes that have rapidly transdifferentiated into endosteal osteoblasts. Further, a novel chondrocyte-derived osteoprogenitor (CDOP) was identified using confocal microscopy, characterized by small, condensed chondrocytes with extensive cytoplasmic vascuolization at the bottom of the hypertrophic zone. In culture, CDOPs express Col2a1, Col10, Col1a1, Osx, are enriched for the stem cell markers, Sca1, CD34, sox2 and c-myc and robustly incorporate BrdU (Park et al., 2015). These lineage tracing, morphometric and in vitro analyses suggest that Col10a1+ chondrocytes may represent “stem-like” cells that gives rise to pre-osteoblasts, osteoblasts and osteocytes at embryonic and postnatal times. We have also provided evidence of chondrocyte-to-osteoblast “transdifferentiation” based on a series of in vivo lineage-tracing experiments using a Pthrp-creER transgenic line that is specific to chondrocytes in the resting zone (Mizuhashi et al., 2018).
Lack of morphometric evidence for hypertrophic chondrocyte transdifferentiation
Recently, however, a morphometric analysis of rabbit tibial hypertrophic chondrocytes supports no evidence of chondrocyte-to-osteoblast transdifferentiation (Pazzaglia et al., 2020). The authors stipulate that in order to constitute a transdifferentiation event, hypertrophic chondrocytes must undergo: 1) a 10-fold shrinkage of size, and 2) decreases in both number and density when compared to metaphyseal osteoblasts at the vascular invasion line. The latter observation suggests the incidence of increased mitoses at the chondrocyte-to-osteoblast transdifferentiation transition zone. The authors continue to suggest that lineage-tracing analyses of transdifferentiation (Yang et al., 2014b; Zhou et al., 2014) fail to consider the possibility that “unstructured substances of apoptotic chondrocytes were still present until cleared by macrophages and that these [cells] could account for the positive fluorescent staining observed in those analyses” (Pazzaglia et al., 2020). They conclude that, “distribution and density of hypertrophic chondrocytes, macrophages and osteoblasts were consistent with a committed function for each [cell type] in the general layout of the growth plate”, based on their morphometric analyses.
Several questions remain regarding the fate of hypertrophic chondrocytes, including: (1) How often do descendants of Col10a1+ hypertrophic chondrocytes persist in adulthood and give rise to osteoblasts and osteocytes? (2) Are there unidentified osteo-chondroprogenitor populations at the interface of hypertrophic chondrocytes and newly formed bones? (3) Which molecular signals allow hypertrophic chondrocytes to alter their fate and differentiate into osteoblasts during skeletal regeneration? These outstanding questions represent future areas of investigation into the elusive nature of hypertrophic chondrocytes.
Conclusions
The ultimate cell fates of hypertrophic chondrocytes remain largely elusive due to their transient nature. Hypertrophic chondrocytes are the only skeletal cell type capable of increasing its intracellular volume through rapid water intake and increased metabolism due to accumulation of mitochondria, the Golgi apparatus and ER. Coupled with active proliferation of chondrocytes in the preceding layer, rapid enlargement of hypertrophic chondrocytes is a major driver of endochondral bone growth. Historically, hypertrophic chondrocytes have been considered as the terminal state of chondrocytes prior to apoptosis. Even this concept is debated as several varieties of “chondroptosis” denoted by “paralyzed” or “dark” cytoplasmic aggregates have been observed in hypertrophic chondrocytes. Hypertrophic chondrocytes are critical regulators of osteogenic-osteoclastic and osteogenic-angiogenic coupling activities during skeletal development, growth and regeneration. Lastly, the “terminal” state of hypertrophic chondrocytes may actually be transient; denoted by their ability to reenter the cell cycle and give rise to a newly identified, “chondrocyte-derived osteoprogenitor”-like cell, although details are not yet clear.
Chondrocyte-to-osteoblast transdifferentiation has been proposed for over a century. Early reports of this event are based on morphological characteristics, such as nuclear condensation and cellular shrinkage. More recent lineage-tracing experiments have substantially advanced our understanding of individual fates of hypertrophic chondrocytes. Hypertrophic chondrocytes represent a unique “terminally differentiated” cell type capable of giving rise to new cell types. Further investigations are required to unravel the molecular regulation of chondrocyte-to-osteoblast transdifferentiation under both physiological and pathological conditions. Resultantly, it may be possible to harness the amazingly diverse functions of hypertrophic chondrocytes in order to more effectively treat patients suffering from debilitating skeletal disorders, including skeletal abnormalities, chondrodysplasias and skeletal injuries.
Acknowledgments
We would like to thank Yuki Matsushita and Mizuki Nagata for the scientific guidance and mentorship provided throughout the drafting of this manuscript.
Funding
This research was funded by NIH/NIDCR R01DE026666, R01DE030630 (to N.O.) and NIH/NIDCR R01DE029181 (to W.O.). S.A.H. was funded by the University of Michigan Rackham Merit Fellowship supported by the University of Michigan Rackham Graduate School, the John Harvey Kellogg Memorial fund supported by the University of Michigan School of Dentistry and the T32DE007057 supported by the NIH/NIDCR.
Abbreviations
- GH
Growth Hormone
- IGF-1
Insulin-like Growth Factor-1
- IGF-1R
Insulin-like Growth Factor-1 Receptor
- Ihh
Indian Hedgehog
- PTHrP
Parathyroid Hormone related Protein
- BMP
Bone Morphogenetic Protein
- FGF
Fibroblast Growth Factor
- FGFR
Fibroblast Growth Factor Receptor
- Runx2
Runt-related Transcription Factor
- Hdac4
Histone deacetylase 4
- VEGF
Vascular Endothelial Growth Factor
- PDGF
Platelet-derived Growth Factor
- EGF
Epidermal Growth Factor
- RANKL
Receptor Activator of Nuclear Factor Kappa-B Ligand
- TRAP
Tartrate-Resistant Acid Phosphatase
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- MMP-9
Matrix Metalloproteinase-9
- MMP-13
Matrix Metalloproteinase-13
- HIF1-α
Hypoxia Inducible Factor 1-alpha
- Col2a1
Type II Collagen alpha 1 chain
- Col10a1
Type 10 Collagen alpha 1 chain
- Mef2c
Myocyte enhancer factor 2C
- Mgp
Matrix GLA protein
- ColX
Type 10 Collagen
- Acan
Aggrecan
- Adipoq
Adiponectin
- PTCH
Protein patched homolog 1
- IRX3/5
Iroquois Homeobox-containing Transcription Factors 3/5
- Col1a1
Type I Collagen alpha 1
- Ocn
Osteocalcin
- Osx
Osterix
- Sca1
Stem Cells Antigen-1
- Sox2
Sex Determining Region Y-box 2
- Sox9
Sex Determining Region Y-box 9
- CSF-1
Colony Stimulating Factor-1
- VCAM-1
Vascular Cell Adhesion Protein 1
- MALP
Marrow Adipogenic Lineage Precursor
- CDOP
Chondrocyte-derived Osteoprogenitor
- CDK
Cyclin-dependent kinases
- ER
Endoplasmic Reticulum
- ECM
Extracellular Matrix
- BAC
Bacterial artificial chromosome
Footnotes
Conflicts of Interest
The authors declare no conflict of interest. Authors declare that there are no competing financial and/or non-financial interests regarding the publication of this paper. The funders had no role in the design of the study; in the collection; analyses or interpretation of data; in writing of the manuscript; or in the decision to publish the results.
References
- Abad V, Meyers JL, Weise M, Gafni RI, Barnes KM, Nilsson O, Bacher JD and Baron J (2002). The Role of the Resting Zone in Growth Plate Chondrogenesis. Endocrinology. 143, 1851–1857. [DOI] [PubMed] [Google Scholar]
- Aghajanian P and Mohan S (2018). The art of building bone: Emerging role of chondrocyte-to-osteoblast transdifferentiation in endochondral ossification. Bone Res. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed YA, Tatarczuch L, Pagel CN, Davies HMS, Mirams M and Mackie EJ (2007). Physiological death of hypertrophic chondrocytes. Osteoarthritis Cartilage. 15, 575–586. [DOI] [PubMed] [Google Scholar]
- Aizawa T, Kokubun S and Tanaka Y (1997). Apoptosis and proliferation of growth plate chondrocytes in rabbits. J. Bone Joint Surg B 79, 483–486. [DOI] [PubMed] [Google Scholar]
- Alba M and Salvatori R (2004). A mouse with targeted ablation of the growth hormone-releasing hormone gene: a new model of isolated growth hormone deficiency. Endocrinology. 145, 4134–4143. [DOI] [PubMed] [Google Scholar]
- Amarilio R, Viukov S. v., Sharir A, Eshkar-Oren I, Johnson RS and Zelzer E (2007). HIF1α regulation of Sox9 in necessary to maintain differentiation of hypoxic prechondrogenic cells during early skeletogenesis. Development. 134, 3917–3928. [DOI] [PubMed] [Google Scholar]
- Bianco P, Cancedda FD, Riminucci M and Cancedda R (1998). Bone formation via cartilage models: The “Borderline” chondrocyte. In Matrix Biology. 17, 185–192. [DOI] [PubMed] [Google Scholar]
- Bikle D, Majumdar S, Laib A, Powell-Braxton L, Rosen C, Beamer W, Nauman E, Leary C and Halloran B (2001). The skeletal structure of insulin-like growth factor I- deficient mice. J. Bone Min. Res 16, 2320–2332. [DOI] [PubMed] [Google Scholar]
- Bonucci E, Engfeldt B and Reinholt FP (2020). Structure and Calcification of Epiphyseal Growth Cartilage. In: Calcification in Biological Systems. 1st ed. CRC Press. [Google Scholar]
- Bromley M and Woolley DE (1984). Chondroclasts and osteoclasts at subchondral sites of erosion in the rheumatoid joint. Arthritis Rheum. 27, 968–975. [DOI] [PubMed] [Google Scholar]
- Buckwalter JA, Mower D, Ungar R, Schaeffer J and Ginsberg B (1986). Morphometric analysis of chondrocyte hypertrophy. J. Bone Joint Surg. Series A 68, 243–255. [PubMed] [Google Scholar]
- Canalis E, Economides AN and Gazzerro E (2003). Bone morphogenetic proteins, their antagonists, and the skeleton. Endo. Reviews 24, 218–235. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risaut W and Nagy A (1996). Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 380, 435–439. [DOI] [PubMed] [Google Scholar]
- Chung UL, Lanske B, Lee K, Li E and Kronenberg H (1998). The parathyroid hormone/parathyroid hormone-related peptide receptor coordinates endochondral bone development by directly controlling chondrocyte differentiation. Proc. Acad. Sci. USA 95, 13030–13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colnot C, Thompson Z, Miclau T, Werb Z and Helms JA (2003). Altered fracture repair in the abscence of MMP9. Development. 130, 4123–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin JS, Bohne BA, Harding GW, McEwen DG and Ornitz DM (1996). Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat. Genetic 12, 390–397. [DOI] [PubMed] [Google Scholar]
- Cooper KL, Oh S, Sung Y, Dasari RR, Kirschner MW and Tabin CJ (2013). Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature. 495, 375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corral DA, Amling M, Priemel M, Loyer E, Fuchs S, Ducy P, Baron R and Karsenty G (1998). Dissociation between bone resorption and bone formation in osteopenic transgenic mice. Proc. Acad. Sci. USA 95, 13835–13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crelin ES and Koch WE (1967). An autoradiographic study of chondrocyte transformation into chondroclasts and osteocytes during bone formation In vitro. Anat. Rec 158, 473–483. [DOI] [PubMed] [Google Scholar]
- de Frutos CA, Vega S, Manzanares M, Flores JM, Huertas H, Martínez-Frías ML and Nieto MA (2007). Snail1 Is a Transcriptional Effector of FGFR3 Signaling during Chondrogenesis and Achondroplasias. Dev Cell. 13, 872–883. [DOI] [PubMed] [Google Scholar]
- de Luca F, Barnes KM, Uyeda JA, De-Levi S, Abad V, Palese T, Mericq V and Baron J (2001). Regulation of growth plate chondrogenesis by bone morphogenetic protein-2. Endocrinology. 142, 430–436. [DOI] [PubMed] [Google Scholar]
- Delezoide AL, Benoist-Lasselin C, Legeai-Mallet L, le Merrer M, Munnich A, Vekemans M and Bonaventure J (1998). Spatio-temporal expression of FGFR 1, 2 and 3 genes during human embryo- fetal ossification. Mech. Develop 77, 19–30. [DOI] [PubMed] [Google Scholar]
- Ding M, Lu Y, Abbassi S, Li F, Li X, Song Y, Geoffroy V, Im HJ and Zheng Q (2012). Targeting Runx2 expression in hypertrophic chondrocytes impairs endochondral ossification during early skeletal development. J. Cell Phys 227, 3446–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drissi MH, Li X, Sheu TJ, Zuscik MJ, Schwarz EM, Puzas JE, Rosier RN and O’Keefe RJ (2003). Runx2/Cbfa1 Stimulation by Retinoic Acid Is Potentiated by BMP2 Signaling Through Interaction with Smad1 on the Collagen X Promoter in Chondrocytes. J. Cell Biochem 90, 1287–1298. [DOI] [PubMed] [Google Scholar]
- Dy P, Wang W, Bhattaram P, Wang Q, Wang L, Ballock RT and Lefebvre V (2012). Sox9 Directs Hypertrophic Maturation and Blocks Osteoblast Differentiation of Growth Plate Chondrocytes. Dev. Cell 22, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto H, Enomoto-Iwamoto M, Iwamoto M, Nomura S, Himeno M, Kitamura Y, Kishimoto T and Komori T (2000). Cbfa1 is a positive regulatory factor in chondrocyte maturation. J. Biolog. Chem 275, 8695–8702. [DOI] [PubMed] [Google Scholar]
- Erenpreisa J and Roach HI (1996) Epigenetic selection as a possible component of transdifferentiation. Further study of the commitment of hypertrophic chondrocytes to become osteocytes. Mech Ageing Dev. 87, 165–182. [DOI] [PubMed] [Google Scholar]
- Farnum CE, Wilsman NJ and Hilley HD (1984). An ultrastructural analysis of osteochondritic growth plate cartilage in growing swine. Vet. Path 21, 141–151. [DOI] [PubMed] [Google Scholar]
- Farnum CE, Turgai J and Wilsman NJ (1990). Visualization of living terminal hypertrophic chondrocytes of growth plate cartilage in situ by differential interference contrast microscopy and time- lapse cinematography. J. Ortho. Res 8, 750–763. [DOI] [PubMed] [Google Scholar]
- Farnum CE and Wilsman NJ (1989). Cellular turnover at the chondro-osseous junction of growth plate cartilage: Analysis by serial sections at the light microscopical level. J. Ortho. Res 7, 654–666. [DOI] [PubMed] [Google Scholar]
- Feher J (2017). 9.8 - Calcium and Phosphorus Homeostasis II: Target Tissues and Integrated Control (J. B. T.-Q. H. P. (Feher Second E., Ed.; pp. 933–945). Academic Press. [Google Scholar]
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, Powell-Braxton L, Hillan KJ and Moore MW (1996). Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 380, 439–442. [DOI] [PubMed] [Google Scholar]
- Galli C, Fu Q, Wang WF, Olsen BR, Manolagas SC, Jilka RL and O’Brien CA (2009). Commitment to the osteoblast lineage is not required for RANKL gene expression. J. Biolog. Chem 284, 12654–12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K and Kroemer G (2018). Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death and Diff. 25, 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhard S, Hattori T, Bauer E, Schlund B, Bösl MR, de Crombrugghe B and von der Mark K (2008). Specific expression of Cre recombinase in hypertrophic cartilage under the control of a BAC-Col10a1 promoter. Matrix Biology. 27, 693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, Wright BD, Radtke F, Aguet M and Ferrara N (1999a). VEGF is required for growth and survival in neonatal mice. Development. 126, 1149–1159. [DOI] [PubMed] [Google Scholar]
- Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z and Ferrara N (1999b). VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med 5, 623–628. [DOI] [PubMed] [Google Scholar]
- Ginzberg MB, Kafri R and Kirschner M (2015). On being the right (cell) size. Science. 348, 1245075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustina A, Mazziotti G and Canalis E (2008). Growth hormone, insulin-like growth factors, and the skeleton. Endo. Rev 29, 535–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Lu Y, Li F, Qiao L, Wang Q, Li N, Borgia JA, Deng Y, Lei G and Zheng Q (2014). Identification and characterization of the novel Col10a1 regulatory mechanism during chondrocyte hypertrophic differentiation. Cell Death and Disease. 5, e1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett SA, Ono W and Ono N (2019). Growth plate chondrocytes: Skeletal development, growth and beyond. Int. J. Mol. Sci 20, 6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J and Klagsbrun M (1999). Cartilage to bone - Angiogenesis leads the way. Nat Med. 5, 617–618 [DOI] [PubMed] [Google Scholar]
- Haseeb A, Kc R, Angelozzi M, de Charleroy C, Rux D, Tower RJ, Yao L, Pellegrino da Silva R, Pacifici M, Qin L and Lefebvre V (2021). SOX9 keeps growth plates and articular cartilage healthy by inhibiting chondrocyte dedifferentiation/osteoblastic redifferentiation. Proc. Acad. Sci. USA 118, e2019152118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama Y, Tuan RS and Shum L (2004). Distinct functions of BMP4 and GDF5 in the regulation of chondrogenesis. J. Cell Biochem 91, 1204–1217. [DOI] [PubMed] [Google Scholar]
- Hattori T, Müller C, Gebhard S, Bauer E, Pausch F, Schlund B, Bösl MR, Hess A, Surmann-Schmitt C, von der Mark H, de Crombrugghe B and von der Mark K (2010). SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development. 137, 901–911. [DOI] [PubMed] [Google Scholar]
- Henry SP, Jang CW, Deng JM, Zhang Z, Behringer RR and de Crombrugghe B (2009). Generation of aggrecan-CreERT2 knockin mice for inducible Cre activity in adult cartilage. Genesis. 47, 805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochachka PW and Lutz PL (2001). Mechanism, origin, and evolution of anoxia tolerance in animals. Comp. Biochem. Physiol. B. Biochem. Mol. Biol 130, 435–459. [DOI] [PubMed] [Google Scholar]
- Hu DP, Ferro F, Yang F, Taylor AJ, Chang W, Miclau T, Marcucio RS and Bahney CS (2017). Cartilage to bone transformation during fracture healing is coordinated by the invading vasculature and induction of the core pluripotency genes. Development. 144, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K and Olsen BR (2016). The roles of vascular endothelial growth factor in bone repair and regeneration. Bone. 91, 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung IH, Yu K, Lavine KJ and Ornitz DM (2007). FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev. Bio 307, 300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker EB and Schenk RK (1989). Physiological mechanisms adopted by chondrocytes in regulating longitudinal bone growth in rats. J. Phys 414, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker EB, Schenk RK and Cruz-Orive LM (1987). Quantitation of chondrocyte performance in growth-plate cartilage during longitudinal bone growth. J. Bone Joint Surg. Am 69, 162–173. [PubMed] [Google Scholar]
- Hwang HS and Kim HA (2015). Chondrocyte apoptosis in the pathogenesis of osteoarthritis. Int. J. Mol. Sci 16, 26035–26054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami D, Akiyama H, Suzuki A, Nakamura T, Nakano T, Yoshikawa H and Tsumaki N (2011). Sox9 sustains chondrocyte survival and hypertrophy in part through Pik3ca-Akt pathways. Development. 138, 1507–1519. [DOI] [PubMed] [Google Scholar]
- Inada M, Yasui T, Nomura S, Miyake S, Deguchi K, Himeno M, Sato M, Yamagiwa H, Kimura T, Yasui N, Ochi T, Endo N, Kitamura Y, Kishimoto T and Komori T (1999a). Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev. Dyn 214, 279–290. [DOI] [PubMed] [Google Scholar]
- Inada M, Yasui T, Nomura S, Miyake S, Deguchi K, Himeno M, Sato M, Yamagiwa H, Kimura T, Yasui N, Ochi T, Endo N, Kitamura Y, Kishimoto T and Komori T (1999b). Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev. Dyn 214, 279–290. [DOI] [PubMed] [Google Scholar]
- Inada M, Wang Y, Byrne MH, Rahman MU, Miyaura C, López-Otín C and Krane SM (2004). Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc. Acad. Sci. USA 101, 17192–17197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizeki K, Takigawa M, Nawa T and Suzuki F (1996). Mouse Meckel’s cartilage chondrocytes evoke bone-like matrix and further transform into osteocyte-like cells in culture. Anat. Rec 245, 25–35. [DOI] [PubMed] [Google Scholar]
- Iwata T, Chen L, Li CL, Ovchinnikov DA, Behringer RR, Francomano CA and Deng CX (2000). A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Hum. Mol. Genet c9, 1603–1613. [DOI] [PubMed] [Google Scholar]
- Iyama K, Ninomiya Y, Olsen BR, Linsenmayer TF, Trelstad RL and Hayashi M (1991). Spatiotemporal pattern of type X collagen gene expression and collagen deposition in embryonic chick vertebrae undergoing endochondral ossification. Anat. Rec 229, 462–472. [DOI] [PubMed] [Google Scholar]
- Jacob AL, Smith C, Partanen J and Ornitz DM (2006). Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev. Bio 296, 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Y, Wang Z, Li H, Ma C and Feng J (2020). Chondrogenesis Defines Future Skeletal Patterns Via Cell Transdifferentiation from Chondrocytes to Bone Cells. Curr. Osteo. Rep 18, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien A, Perrin S, Duchamp de Lageneste O, Carvalho C, Bensidhoum M, Legeai-Mallet L and Colnot C (2020). FGFR3 in Periosteal Cells Drives Cartilage-to-Bone Transformation in Bone Repair. Stem Cell Rep. 15, 955–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NM, Clifton KB, Lorenzo J, Hansen MF and Drissi H (2020). Comparative transcriptomic analysis identifies distinct molecular signatures and regulatory networks of chondroclasts and osteoclasts. Arthritis Res. Ther 22, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IS, Otto F, Zabel B and Mundlos S (1999). Regulation of chondrocyte differentiation by Cbfa1. Mech. Dev 80, 159–170. [DOI] [PubMed] [Google Scholar]
- Knowles HJ, Moskovsky L, Thompson MS, Grunhen J, Cheng X, Kashima TG and Athanasou NA (2012). Chondroclasts are mature osteoclasts which are capable of cartilage matrix resorption. Virchows Archiv. 461, 205–210. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Chung UI, Schipani E, Starbuck M, Karsenty G, Katagiri T, Goad DL, Lanske B and Kronenberg HM (2002). PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development. 129, 2977–2986. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Soegiarto DW, Yang Y, Lanske B, Schipani E, McMahon AP and Kronenberg HM (2005). Indian hedgehog stimulates periarticular chondrocyte differentiation to regulate growth plate length independently of PTHrP. J. Clin. Invest 1734–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima T, Hasegawa T, de Freitas PHL, Yamamoto T, Sasaki M, Horiuchi K, Hongo H, Yamada T, Sakagami N, Saito N, Yoshizawa M, Kobayashi T, Maeda T, Saito C and Amizuka N (2013). Histochemical aspects of the vascular invasion at the erosion zone of the epiphyseal cartilage in MMP-9-deficient mice. Biomed. Res. (Japan) 34, 119–128. [DOI] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S and Kishimoto T (1997). Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 89, 755–764. [DOI] [PubMed] [Google Scholar]
- Kong RYC, Kwan KM, Lau ET, Thomas JT, Boot-hantford RP, Grant ME and Cheah KS (1993). Intron-exon structure, alternative use of promoter and expression of the mouse collagen X gene, Col10a-1. Euro J. Biochem 213, 99–111. [DOI] [PubMed] [Google Scholar]
- Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ and Penninger JM (1999). OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 397, 315–323. [DOI] [PubMed] [Google Scholar]
- Kronenberg HM (2003). Developmental regulation of the growth plate. Nature. 423, 332–336. [DOI] [PubMed] [Google Scholar]
- Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize LHK, Ho C, Mulligan RC, Abou-Samra AB, Jüppner H, Segre GV and Kronenberg HM (1996). PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 273, 663–666. [DOI] [PubMed] [Google Scholar]
- Lazarus JE, Hegde A, Andrade AC, Nilsson O and Baron J (2007). Fibroblast growth factor expression in the postnatal growth plate. Bone. 40, 577–586. [DOI] [PubMed] [Google Scholar]
- Lefebvre V and Smits P (2005). Transcriptional control of chondrocyte fate and differentiation. Birth Defects Res. C. Embryo. Today 75, 200–212. [DOI] [PubMed] [Google Scholar]
- Lewinson D and Silbermann M (1992). Chondroclasts and endothelial cells collaborate in the process of cartilage resorption. Anat. Rec 233, 504–514. [DOI] [PubMed] [Google Scholar]
- Linsenmayer TF, Chen Q, Gibney E, Gordon MK, Marchant JK, Mayne R and Schmid TM (1991). Collagen types IX and X in the developing chick tibiotarsus: Analysis of mRNAs and proteins. Development. 111, 191–196. [DOI] [PubMed] [Google Scholar]
- List EO, Berryman DE, Buchman M, Jensen EA, Funk K, Duran-Ortiz S, Qian Y, Young JA, Slyby J, McKenna S and Kopchick JJ (2019). GH Knockout Mice Have Increased Subcutaneous Adipose Tissue with Decreased Fibrosis and Enhanced Insulin Sensitivity. Endocrinology. 160, 1743–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui JC, Yue S, Lee A, Kikani B, Temnycky A, Barnes KM and Baron J (2019). Persistent Sox9 expression in hypertrophic chondrocytes suppresses transdifferentiation into osteoblasts. Bone. 125, 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo P, Gao F, Niu D, Sun X, Song Q, Guo C, Liang Y and Sun W (2019). The Role of Autophagy in Chondrocyte Metabolism and Osteoarthritis: A Comprehensive Research Review. BioMed. Res. Int e5171602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak KK, Chen MH, Day TF, Chuang PT and Yang Y (2006). Wnt/β-catenin signaling interacts differentially with Ihh signaling in controlling endochondral bone and synovial joint formation. Development. 133, 3695–3707. [DOI] [PubMed] [Google Scholar]
- Mansfield K, Teixeira CC, Adams CS and Shapiro IM (2001). Phosphate ions mediate chondrocyte apoptosis through a plasma membrane transporter mechanism. Bone. 8, 1–8. [DOI] [PubMed] [Google Scholar]
- Maye P, Fu Y, Butler DL, Chokalingam K, Liu Y, Floret J, Stover ML, Wenstrup R, Jiang X, Gooch C and Rowe D (2011). Generation and characterization of Col10a1-mcherry reporter mice. Genesis. 49, 410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrell AJ and Stanger BZ (2016). Adult cell plasticity in vivo: De-differentiation and transdifferentiation are back in style. Nat. Rev. Mol. Cell Bio 17, 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuhashi K, Ono W, Matsushita Y, Sakagami N, Takahashi A, Saunders TL, Nagasawa T, Kronenberg HM and Ono N (2018). Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature. 563, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Balmes G, McKinney S, Zhang Z, Givol D and de Crombrugghe B (2004). Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes. Dev 18, 290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata S (2018). Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol 36, 489–517. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM and Takayanagi H (2011). Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 17, 1231–1234. [DOI] [PubMed] [Google Scholar]
- Newton PT, Li L, Zhou B, Schweingruber C, Hovorakova M, Xie M, Sun X, Sandhow L, Artemov AV, Ivashkin E, Suter S, Dyachuk V, el Shahawy M, Gritli-Linde A, Bouderlique T, Petersen J, Mollbrink A, Lundeberg J, Enikolopov G and Chagin AS (2019). A radical switch in clonality reveals a stem cell niche in the epiphyseal growth plate. Nature. 567, 234238. [DOI] [PubMed] [Google Scholar]
- Nilsson O, Parker EA, Hegde A, Chau M, Barnes KM and Baron J (2007). Gradients in bone morphogenetic protein-related gene expression across the growth plate. J. Endo 193, 75–84. [DOI] [PubMed] [Google Scholar]
- Nordahl J, Andersson G and Reinholt FP (1998). Chondroclasts and osteoclasts in bones of young rats: Comparison of ultrastructural and functional features. Calcif Tissue Int. 63, 401–408. [DOI] [PubMed] [Google Scholar]
- O’Brien CA (2010). Control of RANKL gene expression. In Bone. 46, 911–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odgren PR, Philbrick WM and Gartland A (2003). Perspective. Osteoclastogenesis and Growth Plate Chondrocyte Differentiation: Emergence of Convergence. Crit. Rev. Eukaryot. Gene Expr 13, 181–193. [PubMed] [Google Scholar]
- Odgren PR, Witwicka H and Reyes-Gutierrez P (2016). The cast of clasts: catabolism and vascular invasion during bone growth, repair, and disease by osteoclasts, chondroclasts, and septoclasts. Connect. Tissue Res 57, 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama K, Farquharson C, Whitehead CC and Shapiro IM (1997). Further observations on programmed cell death in the epiphyseal growth plate: Comparison of normal and dyschondroplastic epiphyses. J. Bone Min. Res 12, 1647–1656. [DOI] [PubMed] [Google Scholar]
- Ornitz DM and Marie PJ (2015). Fibroblast growth factor signaling in skeletal development and disease. Genes. Dev 29, 1463–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega N, Behonick DJ, Colnot C, Cooper DNW and Werb Z (2005). Galectin-3 is a downstream regulator of matrix metalloproteinase-9 function during endochondral bone formation. Mol. Bio. Cell 16, 3028–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega N, Wang K, Ferrara N, Werb Z and Vu TH (2010). Complementary interplay between matrix metalloproteinase-9, vascular endothelial growth factor and osteoclast function drives endochondral bone formation. Dis. Model Mech 3, 224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RSP, Mundlos S, Olsen BR, Selby PB and Owen MJ (1997). Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 89, 765–771. [DOI] [PubMed] [Google Scholar]
- Paiva KBS and Granjeiro JM (2017). Matrix Metalloproteinases in Bone Resorption, Remodeling, and Repair. Prog. Mol. Biol. Transl. Sci 148, 203–303. [DOI] [PubMed] [Google Scholar]
- Park J, Gebhardt M, Golovchenko S, Perez-Branguli F, Hattori T, Hartmann C, Zhou X, de Crombrugghe B, Stock M, Schneider H and von der Mark K (2015). Dual pathways to endochondral osteoblasts: A novel chondrocytederived osteoprogenitor cell identified in hypertrophic cartilage. Bio. Open 4, 608–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazzaglia UE and Congiu T (2013). The cast imaging of the osteon lacunar-canalicular system and the implications with functional models of intracanalicular flow. J. Anat 222, 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazzaglia UE, Reguzzoni M, Casati L, Sibilia V, Zarattini G and Raspanti M (2020). New morphological evidence of the ‘fate’ of growth plate hypertrophic chondrocytes in the general context of endochondral ossification. J. Anat 236, 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfander D, Cramer T, Schipani E and Johnson RS (2003). HIF-1α controls extracellular matrix synthesis by epiphyseal chondrocytes. J. Cell Sci 116, 1819–1826. [DOI] [PubMed] [Google Scholar]
- Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, Gillett N and Stewart TA (1993). IGF-I is required for normal embryonic growth in mice. Genes Dev. 7, 2609–2617. [DOI] [PubMed] [Google Scholar]
- Qin X, Jiang Q, Nagano K, Moriishi T, Miyazaki T, Komori H, Ito K, von der Mar K, Sakane C, Kaneko H and Komori T (2020). Runx2 is essential for the transdifferentiation of chondrocytes into osteoblasts. PLOS Genet. 16, e1009169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine HL and Serrat MA (2020). The Actions of IGF-1 in the Growth Plate and Its Role in Postnatal Bone Elongation. Curr Oste. Rep 18, 210–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raucci A, Laplantine E, Mansukhani A and Basilico C (2004). Activation of the ERK1/2 and p38 Mitogen-activated Protein Kinase Pathways Mediates Fibroblast Growth Factor-induced Growth Arrest of Chondrocytes. J. Bio. Chem 279, 1747–1756. [DOI] [PubMed] [Google Scholar]
- Risau W (1995). Differentiation of endothelium. FASEB J. 279, 1747–1756. [PubMed] [Google Scholar]
- Roach HI, Aigner T and Kouri JB (2004). Chondroptosis: A variant of apoptotic cell death in chondrocytes? Apoptosis. 9, 265–277. [DOI] [PubMed] [Google Scholar]
- Roach HI and Clarke NMP (2000). Physiological cell death of chondrocytes in vivo is not confined to apoptosis: New observations on the mammalian growth plate. J. Bone Joint Surg. B 82, 601–613. [DOI] [PubMed] [Google Scholar]
- Roach HI and Clarke NMP (1999). “Cell paralysis” as an intermediate stage in the programmed cell death of epiphyseal chondrocytes during development. J. Bone Min Res 14, 1367–1378. [DOI] [PubMed] [Google Scholar]
- Rodan GA and Martin TJ (1982). Role of osteoblasts in hormonal control of bone resorption- a hypothesis. Calc Tiss Int. 33, 349–351. [DOI] [PubMed] [Google Scholar]
- Rosati R, Horan GSB, Pinero GJ, Garofalo S, Keene DR, Horton WA, Vuorio E de Crombrugghe B and Behringer RR (1994). Normal long bone growth and development in type X collagen-null mice. Nat. Genet 8, 129–135. [DOI] [PubMed] [Google Scholar]
- Sato S, Kimura A, Ozdemir J, Asou Y, Miyazaki M, Jinno T, Ae K, Liu X, Osaki M, Takeuchi Y, Fukumoto S, Kawaguchi H, Haro H, Shinomiya KI, Karsenty G and Takeda S (2008). The distinct role of the runx proteins in chondrocyte differentiation and intervertebral disc degeneration: Findings in murine models and in human disease. Arthritis Rheum. 58, 2764–2775. [DOI] [PubMed] [Google Scholar]
- Schmid TM and Linsenmayer TF (1985). Immunohistochemical localization of short chain cartilage collagen (type X) in avian tissues. J. Cell Bio 100, 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid TM and Linsenmayer TF (1990). Immunoelectron microscopy of type X collagen: Supramolecular forms within embryonic chick cartilage. Dev. Bio 138, 53–62. [DOI] [PubMed] [Google Scholar]
- Selvamurugan N, Pulumati MR, Tyson DR and Partridge NC (2000). Parathyroid hormone regulation of the rat collagenase-3 promoter by protein kinase A-dependent transactivation of core binding factor α1. J. BioChem 275, 5037–5042. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2000). Expression of hypoxia-inducible factor 1: Mechanisms and consequences. Biochem. Pharmacol 59, 47–53. [DOI] [PubMed] [Google Scholar]
- Shapiro IM and Srinivas V (2007). Metabolic consideration of epiphyseal growth: Survival responses in a taxing environment. Bone. 59, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheeba CJ, Andrade RP, Duprez D and Palmeirim I (2010). Comprehensive analysis of fibroblast growth factor receptor expression patterns during chick forelimb development. Int. J. Dev. Bio 54, 1517–1526. [DOI] [PubMed] [Google Scholar]
- Shen G (2005). The role of type X collagen in facilitating and regulating endochondral ossification of articular cartilage. Orthod. Craniofac. Res 8, 11–7. [DOI] [PubMed] [Google Scholar]
- Shu B, Zhang M, Xie R, Wang M, Jin H, Hou W, Tang D, Harris SE, Mishina Y, O’Keefe RJ, Hilton MJ, Wang Y and Chen D (2011). BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J. Cell Sci 124, 3428–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NA and Martin TJ (2014). Coupling the activities of bone formation and resorption: a multitude of signals within the basic multicellular unit. BoneKEy Rep. 3, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith N, Dong Y, Lian JB, Pratap J, Kingsley PD, van Wijnen AJ, Stein JL, Schwarz EM, O’Keefe RJ, Stein GS and Drissi MH (2005). Overlapping expression of Runx1(Cbfa2) and Runx2(Cbfa1) transcription factors supports cooperative induction of skeletal development. J. Cell Phys 203, 133–143. [DOI] [PubMed] [Google Scholar]
- Sobacchi C, Frattini A, Guerrini MM, Abinun M, Pangrazio A, Susani L, Bredius R, Mancini G, Cant A, Bishop N, Grabowski P, del Fattore A, Messina C, Errigo G, Coxon FP, Scott DI, Teti A, Rogers MJ, Vezzoni P and Helfrich MH (2007). Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat. Genetic 39, 960–962. [DOI] [PubMed] [Google Scholar]
- St-Jacques B, Hammerschmidt M and McMahon AP (1999). Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su WCS, Kitagawa M, Xue N, Xie B, Garofalo S, Cho J, Deng C, Horton WA and Fu XY (1997). Activation of Stat1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature. 386, 288–292. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Akatsu T, Udagawa N, Sasaki T, Yamaguchi A, Moseley JM, Martin TJ and Suda T (1988). Osteoblastic cells are involved in osteoclast formation. Endocrinology. 123, 2600–2602. [DOI] [PubMed] [Google Scholar]
- Takeda S, Bonnamy JP, Owen MJ, Ducy P and Karsenty G (2001). Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 15, 467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang KY, Chan D and Cheah KSE (2015). Fate of growth plate hypertrophic chondrocytes: Death or lineage extension? Dev. Growth Differ 57, 179–192. [DOI] [PubMed] [Google Scholar]
- Ueta C, Iwamoto M, Kanatani N, Yoshida C, Liu Y, Enomoto-Iwamoto M, Ohmori T, Enomoto H, Nakata K, Takada K, Kurisu K and Komori T (2001). Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J. Cell Bio 153, 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcourt U, Gouttenoire J, Moustakas A, Herbage D and Mallein-Gerin F (2002). Functions of transforming growth factor-β family type I receptors and Smad proteins in the hypertrophic maturation and osteoblastic differentiation of chondrocytes. J. Bio Chem 277, 33545–33558. [DOI] [PubMed] [Google Scholar]
- Vanhorn S and Morris SA (2020). Next-Generation Lineage Tracing and Fate Mapping to Interrogate Development. Dev. Cell 56, 7–21. [DOI] [PubMed] [Google Scholar]
- Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM and Tabin CJ (1996). Regulation of rate of cartilage differentiation by Indian Hedgehog and PTH-related protein. Science. 273, 613–622. [DOI] [PubMed] [Google Scholar]
- Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM and Werb Z (1998). MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypetrophic chondrocytes. Cell. 93, 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhou J and Bondy CA (1999a). Igf1 promotes longitudinal bone growth by insulin-like actions augmenting chondrocyte hypertrophy. The FASEB J. 13, 1985–1990. [DOI] [PubMed] [Google Scholar]
- Wang Y, Spatz MK, Kannan K, Hayk H, Avivi A, Gorivodsky M, Pines M, Yayon A, Lonai P and Givol D (1999b). A mouse model for achondroplasia produced by targeting fibroblast growth factor receptor 3. Proc. Acad. Sci. USA 96, 4455–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Yongmei, Cheng Z, Elalieh HZ, Nakamura E, Nguyen MT, MacKem S, Clemens TL, Bikle DD and Chang W (2011). IGF-1R signaling in chondrocytes modulates growth plate development by interacting with the PTHrP/Ihh pathway. J. Bone Min. Res 26, 1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein RS, Chen JR, Powers CC, Stewart SA, Landes RD, Bellido T, Jilka RL, Michael Parfitt A and Manolagas SC (2002). Promotion of osteoclast survival and antagonism of bisphosphonate-induced osteoclast apoptosis by glucocorticoids. J. Clin. Invest 109, 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein RS, Jilka RL, Michael Parfitt A and Manolagas SC (1998). Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts end osteocytes by glucocorticoids potential mechanisms of their deleterious effects on bone. J. Clin. Invest 102, 274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, Fields J, Yu X, Shaw NJ, McLellan NJ, McKeown C, FitzPatrick D, Yu K, Ornitz DM and Econs MJ (2005). Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am. J. Hum. Genet 76, 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff LI and Hartmann C (2019). A Second Career for Chondrocytes—Transformation into Osteoblasts. Curr. Osteo. Rep 17, 129–137 [DOI] [PubMed] [Google Scholar]
- Wu CW, Tchetina EV, Mwale F, Hasty K, Pidoux I, Reiner A, Chen J, van Wart HE and Poole AR (2002). Proteolysis involving matrix metalloproteinase 13 (collagenase-3) is required for chondrocyte differentiation that is associated with matrix mineralization. J. Bone Min. Res 17, 639–651. [DOI] [PubMed] [Google Scholar]
- Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC and O’Brien CA (2011). Matrix-embedded cells control osteoclast formation. Nat. Medicine 17, 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakar S, Werner H and Rosen CJ (2018). 40 years of IGF1: Insulin-like growth factors: Actions on the skeleton. J. Mol. Endocrinol 61, T115–T137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro T, Åberg T, Levanon D, Groner Y and Thesleff I (2002). Expression of Runx1, −2 and −3 during tooth, palate and craniofacial bone development. Gene Exp. Patt [DOI] [PubMed] [Google Scholar]
- Yang G, Zhu L, Hou N, Lan Y, Wu XM, Zhou B, Teng Y and Yang X (2014a). Osteogenic fate of hypertrophic chondrocytes. In Cell Res. 24, 1266–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Tsang KY, Tang HC, Chan D and Cheah KSE (2014b). Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Acad. Sci. USA 111, 12097–12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR and Lyons KM (2005). Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc. Acad. Sci. USA 102, 5062–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida CA, Yamamoto H, Fujita T, Furuichi T, Ito K, Inoue KI, Yamana K, Zanma A, Takada K, Ito Y and Komori T (2004). Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 18, 952–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA and Ornitz DM (2003). Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 130, 3063–3074. [DOI] [PubMed] [Google Scholar]
- Zelzer E, Glotzer DJ, Hartmann C, Thomas D, Fukai N, Soker S and Olsen BR (2001). Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech. Dev 106, 97–106. [DOI] [PubMed] [Google Scholar]
- Zelzer E, Mamluk R, Ferrara N, Johnson RS, Schipani E and Olsen BR (2004). VEGFA is necessary for chondrocyte survival during bone development. Development. 131, 2161–2171. [DOI] [PubMed] [Google Scholar]
- Zerega B, Cermelli S, Bianco P, Cancedda R and Descalzi Cancedda F (1999). Parathyroid hormone [PTH(1–34)] and parathyroid hormone-related protein [PTHrP(1–34)] promote reversion of hypertrophic chondrocytes to a prehypertrophic proliferating phenotype and prevent terminal differentiation of osteoblast-like cells. J. Bone Min. Res 14, 1281–1289. [DOI] [PubMed] [Google Scholar]
- Zhang M, Xie R, Hou W, Wang B, Shen R, Wang X, Wang Q, Zhu T, Jonason JH and Chen D (2009). PTHrP prevents chondrocyte premature hypertrophy by inducing cyclin-D1-dependent Runx2 and Runx3 phosphorylation, ubiquitylation and proteasomal degradation. J. Cell Sci 22, 1382–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Eberspaecher H, Lefebvre V and de Crombrugghe B (1997). Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev. Dyn 209, 377–386. [DOI] [PubMed] [Google Scholar]
- Zheng Q, Zhou G, Morello R, Chen Y, Garcia-Rojas X and Lee B (2003). Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J. Cell Bio 162, 833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou N, Li Q, Lin X, Hu N, Liao JY, Lin LB, Zhao C, Hu ZM, Liang X, Xu W, Chen H and Huang W (2016). BMP2 induces chondrogenic differentiation, osteogenic differentiation and endochondral ossification in stem cells. Cell Tiss. Res 366, 101–111. [DOI] [PubMed] [Google Scholar]
- Zhou X, von der Mark K, Henry S, Norton W, Adams H and de Crombrugghe B (2014). Chondrocytes Transdifferentiate into Osteoblasts in Endochondral Bone during Development, Postnatal Growth and Fracture Healing in Mice. PLoS Genet. 10, e1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YX, Xu X, Chen L, Li C, Brodie SG and Deng CX (2000). A Pro250Arg substitution in mouse Fgfr1 causes increased expression of Cbfa1 and premature fusion of calvarial sutures. Hum. Mol. Genet 9, 2001–2018. [DOI] [PubMed] [Google Scholar]
- Zhou ZQ, Ota S, Deng C, Akiyama H and Hurlin PJ (2015). Mutant activated FGFR3 impairs endochondral bone growth by preventing SOX9 downregulation in differentiating chondrocytes. Hum. Mol. Genet 24, 1764–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]