

Abstract
High-throughput RNA sequencing (RNA-seq) has extraordinarily advanced our understanding of gene expression and disease etiology, and is a powerful tool for the identification of biomarkers in a wide range of organisms. However, most RNA-seq methods rely on retroviral reverse transcriptases (RTs), enzymes that have inherently low fidelity and processivity, to convert RNAs into cDNAs for sequencing. Here, we describe an RNA-seq protocol using Thermostable Group II Intron Reverse Transcriptases (TGIRTs), which have high fidelity, processivity, and strand-displacement activity, as well as a proficient template-switching activity that enables efficient and seamless RNA-seq adapter addition. By combining these activities, TGIRT-seq enables the simultaneous profiling of all RNA biotypes from small amounts of starting material, with superior RNA-seq metrics, and unprecedented ability to sequence structured RNAs. The TGIRT-seq protocol for Illumina sequencing consists of three steps: (i) addition of a 3' RNA-seq adapter, coupled to the initiation of cDNA synthesis at the 3' end of a target RNA, via template switching from a synthetic adapter RNA/DNA starter duplex; (ii) addition of a 5' RNA-seq adapter, by using thermostable 5' App DNA/RNA ligase to ligate an adapter oligonucleotide to the 3' end of the completed cDNA; (iii) minimal PCR amplification, to add capture sites and indices for Illumina sequencing. TGIRT-seq for the Illumina sequencing platform has been used for comprehensive profiling of coding and non-coding RNAs in ribodepleted, chemically fragmented cellular RNAs, and for the analysis of intact (non-chemically fragmented) cellular, extracellular vesicle (EV), and plasma RNAs, where it yields continuous full-length end-to-end sequences of structured small non-coding RNAs (sncRNAs), including tRNAs, snoRNAs, snRNAs, pre-miRNAs, and full-length excised linear intron (FLEXI) RNAs.
Graphic abstract:
Keywords: Group II intron reverse transcriptase, Non-LTR-retroelement, Reverse transcriptase, RNA-seq, Template switching, Transcriptomics, Illumina sequencing
Background
Most RNA-seq methods rely on a retroviral reverse transcriptase (RT) to convert target RNAs into cDNAs, which can then be sequenced on a high-throughput DNA sequencing platform ( Stark et al., 2019 ). However, retroviral RTs evolved to have inherently low fidelity and processivity, to help retrovirus evade host defenses by introducing frequent mutational variations and rapidly propagating beneficial ones by RNA recombination, which involves dissociating from one template and reinitiating on another (Onafuwa-Nuga and Telesnitsky, 2009; Hu and Hughes, 2012). Although commercial retroviral RTs have been engineered to increase their processivity and thermostability, the ability to improve these enzymes is limited by the retroviral RT structural framework, and even highly engineered retroviral RTs have difficulty reverse transcribing structured RNAs (Martín- Alonso et al., 2021 ).
TGIRT-seq is a next-generation comprehensive RNA-seq method that utilizes the beneficial biochemical properties of group II intron-encoded RTs, which are evolutionary ancestors of retroviral RTs (Belfort and Lambowitz, 2019). Group II intron RTs are largely prokaryotic enzymes that are associated with bacterial retrotransposons called mobile group II introns, and they evolved to function in retrohoming, a retrotransposition mechanism that requires reverse transcription of a highly structured group II intron RNA with high fidelity and processivity (Lambowitz and Belfort, 2015). They belong to a large subgroup of RTs encoded by non-LTR-retroelements, which include other bacterial RTs, retroplasmid RTs, and RVT RTs, as well as human LINE-1 element, insect R2 element, and other eukaryotic non-LTR-retrotransposon RTs (Xiong and Eickbush, 1990; Kojima and Kanehisa, 2008; Simon and Zimmerly, 2008; Gladyshev and Arkhipova, 2011; Zimmerly and Wu, 2015). Non-LTR-retroelement RTs differ from retroviral RTs in having a distinctive N-terminal extension (NTE) and two distinctive insertions (RT2a and RT3a) in the fingers and palm, which were shown by the crystal structure of a group II intron RT to contribute multiple additional interactions with the RNA template, and more tightly constrain the RT active site in ways that could contribute to higher fidelity and processivity (Xiong and Eickbush, 1990; Blocker et al., 2005 ; Stamos et al., 2017 ). The NTE also plays a crucial role in a proficient end-to-end template-switching activity, which enables efficient and seamless RNA-seq adapter addition ( Lentzsch et al., 2019 and 2021). TGIRTs from bacterial thermophiles combine these beneficial properties with the ability to function at high temperatures (≥60°C), which help melt out stable RNA secondary structures ( Mohr et al., 2013 ). A key to using TGIRTs and other non-LTR-retroelement RTs for RNA-seq and other applications was their fusion to a large solubility tag (e.g., maltose-binding protein), which enables them to be produced in large quantities and remain soluble and stable during storage, when freed of endogenous tightly bound nucleic acids ( Mohr et al., 2013 ; Upton et al., 2021 ). As thousands of group II intron and other non-LTR-retroelement RTs have been identified, it is likely that we have only scratched the surface in identifying optimal enzymes for RNA-seq applications.
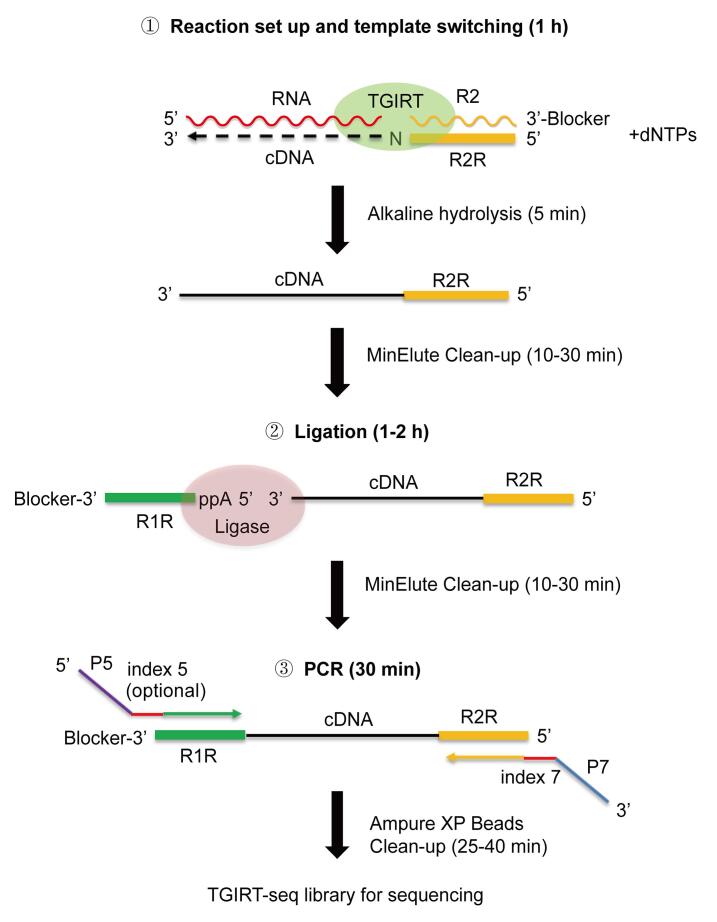
Figure 1 outlines a TGIRT-seq protocol using TGIRT-III (InGex; a proprietary version of Geobacillus stearothermophilus GsI-IIC RT; Mohr et al., 2013 ) for the simultaneous profiling of all coding and non-coding RNA biotypes in cellular, extracellular vesicle (EV), and human plasma RNA preparations without size selection (also referred to as the TGIRT Total RNA-seq method). The protocol described here is an updated version used in Yao et al. (2020) based upon earlier versions described in Qin et al. (2016) , Nottingham et al. (2016) , and Xu et al. (2019) .
Figure 1. Overview of the TGIRT-seq protocol for Illumina sequencing.

Major steps are: (1) Template switching from a synthetic R2 RNA/R2R DNA starter duplex with a 1-nt 3' DNA overhang (a mixture of A, C, G, and T residues, denoted N) that base pairs to the 3' nucleotide of a target RNA, and upon initiating reverse transcription by adding dNTPs, seamlessly links an R2R adapter to the 5' end of the resulting cDNA; (2) Ligation of an R1R adapter to the 3' end of the completed cDNA; and (3) Minimal PCR amplification with primers that add Illumina capture sites (P5 and P7) and barcode sequences (indices 5 and 7). The index 7 barcode is required, while the index 5 barcode is optional, to provide unique dual indices (UDIs).
In the first step used for the initiation of reverse transcription, the TGIRT enzyme template switches from a synthetic RNA template/DNA primer starter duplex that contains an RNA-seq adapter sequence directly to the 3' end of a target RNA, thereby linking the reverse complement of the adapter sequence to the 5' end of the cDNA ( Mohr et al., 2013 ; Qin et al., 2016 ). The RNA/DNA starter duplex has a 1-nt 3' DNA overhang that directs template switching with high specificity by base pairing to the 3' nucleotide of a target RNA, resulting in seamless template-switching junctions ( Lentzsch et al., 2019 and 2021). For Illumina sequencing, the starter duplex consists of a 35-nt RNA oligonucleotide that contains an Illumina Read 2 sequence (denoted R2 RNA) and has a 3’-blocking group (C3 Spacer, denoted 3SpC3) annealed to a 36-nt DNA primer containing the reverse complement of the Read 2 sequence (denoted R2R DNA), leaving the 1-nt 3’ DNA overhang. For comprehensive RNA-seq of a pool of RNAs, the 1-nt 3’ DNA overhang is a mixture of A, C, G, and T residues (denoted as N), and is added in excess to the amount of RNA template. Reverse transcription is typically carried out for 15 min at 60°C, an optimal temperature for GsI-IIC RT ( Mohr et al., 2013 ), but different times and lower or higher temperatures can be used for different applications. (e.g., Zheng et al., 2015 ; Behrens et al., 2021 ). Reverse transcription is terminated by adding NaOH, which degrades the RNA template, followed by neutralization with HCl and a MinElute cDNA clean-up step to remove unused R2R DNA.
In the next step, a second RNA-seq adapter containing the reverse complement of an Illumina Read 1 sequence (denoted R1R DNA) is attached to the 3' end of the cDNA by a single-stranded DNA ligation using thermostable 5’ App RNA/DNA ligase, and this is followed by minimal PCR amplification (no more than 12 cycles) with primers that add capture sites and indices for Illumina sequencing. Using this protocol, comprehensive TGIRT-seq libraries can be prepared from as little as 0.5 ng of human plasma RNA in approximately 5 h through completion of the PCR step.
TGIRT-seq can be done with rRNA-depleted, chemically fragmented or intact cellular RNAs, or with total RNAs from EVs or plasma. Compared to benchmark TruSeq v3 datasets of rRNA depleted, chemically fragmented Universal Human Reference RNA (UHRR), with External RNA Control Consortium (ERCC) spike-ins, TGIRT-seq: (i) better recapitulated the relative abundance of mRNAs and ERCC spike-ins; (ii) was more strand-specific; (iii) gave more uniform 5’ to 3’ gene body coverage and detected more splice junctions, particularly near the 5’ ends of genes; and (iv) eliminated sequence biases due to random hexamer priming, which are inherent in TruSeq ( Nottingham et al., 2016 ). Subsequent improvements in the TGIRT-seq method have included the use of modified RNA-seq adapter sequences that strongly decrease adapter dimer formation ( Xu et al., 2019 ), and the use of lower salt concentrations in the template-switching and reverse transcription step, to increase library yield ( Yao et al., 2020 ). If desired, a unique molecular identifier can be incorporated into the R1R adapter oligonucleotide ( Yao et al., 2020 ). Although used mainly for Illumina sequencing thus far, the RNA-seq adapter sequences could readily be reformatted for other high-throughput DNA sequencing platforms.
The TGIRT-template switching reaction for 3' RNA-seq adapter addition is the defining step of the TGIRT-seq method, while 5' RNA-seq adapter addition using thermostable 5' App DNA/RNA ligase could in principle be replaced by an alternative method. Despite relying on a single base pair between the 1-nt 3' DNA overhang of starter duplex and 3' nucleotide of the target RNA, RNA-seq adapter addition by TGIRT template switching is highly specific, yielding 97.5-99.7% precise junctions, depending upon the base pair ( Lentzsch et al., 2019 ). This high specificity reflects the fact that the 3' end of the acceptor RNA binds in a pocket formed by the NTE and RT fingertips loop, which promotes annealing of the 3' nucleotide of the acceptor RNA to the complementary 1-nt 3' DNA overhang of the starter duplex, and positions the penultimate nucleotide of the acceptor as the templating RNA base at the RT active site ( Lentzsch et al., 2021 ). A second base-pairing interaction between the templating base and a complementary incoming dNTP then results in a conformational change required for initiation of reverse transcription, an irreversible step that drives the reaction forward and ensures high specificity ( Lentzsch et al., 2021 ). The same template-switching pocket does not exist in retroviral RTs, which lack the NTE, likely leading to weaker binding of the acceptor with a greater dependence upon base-pairing interactions and propensity for artifacts resulting from template switching to alternative sites containing complementary nucleotides (e.g., Mader et al., 2001 ; Cocquet et al., 2006 ).
After reverse transcription, TGIRT enzymes can add non-templated nucleotides to the 3' end of a completed cDNA, generating 3' DNA overhangs that enable template switching to a second RNA template with a complementary 3' nucleotide. Such secondary template switches, which result in fusion reads, are suppressed in TGIRT-seq by using a relatively high salt concentration in the reaction medium (200 mM NaCl in the updated protocol and 450 mM NaCl in earlier versions; Mohr et al., 2013 ; Lentzsch et al., 2019 ; Yao et al., 2020 ). A longer-term solution may be provided by a recently described mutation that specifically inhibits non-templated addition and thus selectively inhibits secondary template switches from the 5’ end of a completed cDNA, but not from a starter duplex with a pre-formed 1-nt DNA overhang ( Lentzsch et al., 2021 ). Secondary template switches can also be used for 5' RNA-seq adapter addition ( Zhu et al., 2001 ; Picelli et al., 2013 ), and the recent detailed biochemical and structural analyses of the TGIRT template-switching reaction could be used to perfect such a method for TGIRT-seq ( Lentzsch et al., 2019 and 2021).
Sequence biases in TGIRT-seq using TGIRT-III determined in reaction medium containing 450 mM NaCl and 60°C are limited to the first three nucleotides from the 5’ end of the RNA, corresponding to known sequence biases of the thermostable 5’ App RNA/DNA ligase ( Hafner et al., 2011 ), and the 3' nucleotide of the target RNA, reflecting a preference for, or stability of different base pairs with the 1-nt 3’ DNA overhang of the starter duplex for TGIRT template switching ( Xu et al., 2019 ). The 5’-adapter ligation step using thermostable 5’ App RNA/DNA ligase at high temperature has no major “co-folding” bias due to base pairings between the adapter and acceptor nucleic acids, and is not mitigated by using an R1R adapter with randomized nucleotides near its 5' end, as in 4 N protocols used for other ligases ( Xu et al., 2019 ). The template-switching bias for the 3' nucleotide of the target RNA can be remediated by using unequal ratios of 3'-overhang nucleotides in the starter duplex, to compensate for the base-pairing biases, and overall bias can be corrected by using a bias correction algorithm, based on a random forest regression model of all factors contributing to bias ( Mohr et al., 2013 ; Xu et al., 2019 ). Additionally, biochemical analysis showed that differences in the rates and amplitudes of TGIRT template-switching to acceptor RNAs with different 3' nucleotides are smaller at 200 mM than at 450 mM NaCl, suggesting that template-switching biases might be decreased by the lower salt concentration used in the updated protocol ( Lentzsch et al., 2019 ). TGIRT-III is more efficient than retroviral RTs in reverse transcribing through expanded GC-rich repeat sequences, such as those characteristic of myotonic dystrophy and familial amyotropic lateral sclerosis ( Carrell et al., 2018 ), but more prone to indels at homopolymer runs, particularly if followed by a stable hairpin structure ( Penno et al., 2017 ). The latter may reflect that the TGIRT enzyme is less likely to dissociate at stable RNA secondary structures than are retroviral RTs, and continues to incorporate nucleotides via slippage until it can read through such impediments. TGIRT-III does not template switch efficiently to 3' poly(A) tails of eukaryotic mRNAs ( Yao et al., 2021 ).
In ribodepleted, chemically fragmented RNA preparations, TGIRT-III gave reliable quantitation of RNA spike-ins ≥60 nt ( Nottingham et al., 2016 ; Boivin et al., 2018 ). In heterogeneously sized RNA preparations, however, TGIRT-seq under-represents RNAs <60 nt, particularly very small RNAs, such as miRNAs and short tRNA fragments (tRFs; Boivin et al., 2018 ; Yao et al., 2020 ), requiring an orthogonal method, such as RT-qPCR, or a hybridization-based assay to determine their relative abundance. This size bias occurs at the initial template-switching step used for 3' RNA-seq adapter addition and likely reflects that 5' regions of longer RNAs that extend outside the template-switching pocket can bind to additional sites on the outer surface of the protein ( Lentzsch et al., 2021 ). These additional sites likely correspond to basic surface residues that function in binding group II intron RNAs for RNA splicing and retrohoming, but are not required for reverse transcription, and thus relatively easy to remove without affecting performance in RNA-seq ( Zhao et al., 2018 ; Stamos et al., 2019 ). Additionally, this size bias appears to be significantly lower for another TGIRT enzyme (TeI4c RT; Mohr et al., 2013 ), which is not yet sold commercially.
While TGIRT-seq of ribodepleted chemically fragmented RNAs is best for mRNA quantitation, TGIRT-seq of intact (i.e., non-chemically fragmented) RNA preparations has been useful for the analysis of tRNAs and other structured sncRNAs, for which TGIRT-seq yields full-length end-to-end sequences ( Katibah et al., 2014 ; Shurtleff et al., 2017 ; Yao et al., 2020 ). An initial TGIRT-based protocol for tRNA-seq used gel purification and template switching to RNAs that have a C-terminal A residue in combination with an RNA demethylase to obtain full-length reads of mature tRNAs ( Zheng et al., 2015 ). However, the simpler TGIRT-seq total RNA-seq protocol described here also gives largely full-length end-to-end sequences of mature tRNAs and tRNA fragments without demethylase treatment ( Katibah et al., 2014 ; Qin et al., 2016 ; Shurtleff et al., 2017 ), as does a more recent TGIRT-based tRNA-seq method (mim-tRNAseq; Behrens et al., 2021 ). The TGIRT enzyme is highly processive and, given enough time, pauses at post-transcriptional modifications that affect base pairing until it can read through via distinctive patterns of misincorporation that can be used to identify the modification ( Katibah et al., 2014 ; Shen et al., 2015 ; Zheng et al., 2015 ; Qin et al., 2016 ). This read-through via misincorporation ability has also been used to map chemical modifications for RNA structure mapping in procedures such as DMS-MaPseq ( Zubradt et al., 2017 ; Wu and Bartel, 2017; Wang et al., 2018 ). The ability of TGIRT-III to give full-length tRNA sequences was key to demonstrating that mature tRNAs, rather than tRNA fragments, predominate in human plasma and EVs ( Qin et al., 2016 ; Shurtleff et al., 2017 ; Yao et al., 2020 ), and in distinguishing mature from pre-tRNAs bound by the human interferon-induced protein IFIT5 ( Katibah et al., 2014 ). TGIRT template-switching has also been used to measure levels of tRNA aminoacylation, which blocks 3’ tRNA ends for template switching ( Evans et al., 2017 ).
In addition to tRNAs, TGIRT-seq also gives full-length end-to-end sequences of other structured sncRNAs, enabling the identification of novel snoRNAs in cellular RNA preparations ( Boivin et al., 2020 ), and distinguishing pre-miRNA hairpins from mature miRNAs in human plasma ( Qin et al., 2016 ; Yao et al., 2020 ). In recent work, TGIRT-seq of human plasma and cellular RNAs revealed the presence of thousands of short full-length excised linear intron (FLEXI) RNAs, many of which have stable predicted RNA secondary structures, that would make them difficult to identify by other methods ( Yao et al., 2020 and 2021). TGIRT enzymes and variations of the TGIRT-seq method have also been used for high-throughput sequencing of protein-bound RNAs or RNA fragments by RIP-seq and irCLIP ( Katibah et al., 2014 ; Zarnegar et al., 2016 ), and for ssDNA-seq of human plasma DNA, providing information about nucleosome positioning and DNA methylation sites that can be used to identify tissues of origin (Wu and Lambowitz, 2017). Going forward, we anticipate that the current version of the TGIRT-seq method will be subject to continued improvement, by using the structural and biochemical information now available for GsI-IIC RT (TGIRT-III) to enhance methods for 5' and 3' RNA-seq adapter addition, as well as by using other natural and engineered versions of TGIRTs and other non-LTR-retroelement RTs.
Materials and Reagents
Use reagents and solutions that are RNA grade and nuclease free. Store solutions in frozen aliquots to avoid repeated freezing and thawing.
1.5 ml DNA LoBind microcentrifuge tubes (Eppendorf, catalog number: 022431021)
2 ml DNA LoBind microcentrifuge tubes (Eppendorf, catalog number: 022431048)
Ep Dualfilter T.I.P.S LoRetention 0.1-10 μl (Eppendorf, catalog number: 0030078632)
Ep Dualfilter T.I.P.S LoRetention 2-100 μl (Eppendorf, catalog number: 0030078659)
Ep Dualfilter T.I.P.S LoRetention 20-300 μl (Eppendorf, catalog number: 0030078675)
Ep Dualfilter T.I.P.S LoRetention 50-1,000 μl (Eppendorf, catalog number: 0030078683)
8 × 0.2 ml PCR reaction tube strip with attached flat cap (Simport, catalog number: T3202N)
Dithiothreitol (DTT), 1 M (Thermo Fisher Scientific, catalog number: P2325)
dNTPs mix, 25 mM each (Thermo Fisher Scientific, catalog number: R1122). Dilute to 20 mM each with RNase-free water before use
-
TGIRT-III enzyme (InGex, catalog number: TGIRT50)
Note: Store TGIRT-III enzyme received from a supplier at -80°C until ready for use. Store opened tubes at -20°C. TGIRT-III may lose activity after 3 months of storage at -20°C (Behrens et al., 2021).
MinElute Reaction Cleanup kit (Qiagen, catalog number: 28204 or 28206) or MinElute PCR Purification kit (QIAGEN, catalog number: 28004 or 28006)
5’ DNA adenylation kit (New England Biolabs, catalog number: E2610S/L)
Oligo Clean & Concentrator kit (Zymo Research, catalog number: D4060/4061)
Thermostable 5’ app DNA/RNA ligase (New England Biolabs, catalog number: M0319S/L)
Phusion high-fidelity PCR master mix with HF buffer (Thermo Fisher Scientific, catalog number: F531S/L)
AMPure XP (Beckman Coulter, catalog number: A63881)
High sensitivity DNA kit (Agilent, catalog number: 5067-4626)
(Optional) RNA 6000 Pico kit (Agilent, catalog number: 5067-1513)
(Optional) Small RNA kit (Agilent, catalog number: 5067-1548)
-
Oligonucleotides: All oligonucleotides should be HPLC-purified RNase-free grade
-
R2 RNA
5’-rArArG rArUrC rGrGrA rArGrA rGrCrA rCrArC rGrUrC rUrGrA rArCrU rCrCrA rGrUrC rArC/3SpC3/-3’
Note: Other blockers such as 3’ Amino Modifier C6 dT(3AmMC6T) from IDT can also be used.
-
R2R DNA
5’-GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG ATC TTN-3’ (N = equimolar A, T, G, C)
Note: The R2R DNA used in the current TGIRT-seq protocol differs from that used in earlier versions. It has a single nucleotide change (insertion of the underlined T residue at the -3 position from the 3' end) that strongly decreases formation of R1R-R2R adapter-dimers during the ligation step (Xu et al., 2019). A complementary A residue is inserted at the corresponding position in R2 RNA (underlined A; see above).
-
R1R DNA
5’-/5Phos/GAT CGT CGG ACT GTA GAA CTC TGA ACG TGT AG/3SpC3/-3’
Note: The Read 1 (R1) sequence corresponds to the small RNA sequencing primer site used in the NEBNEXT Small RNA Library Prep Set for Illumina sequencing.
-
6N unique molecular identifier (UMI) R1R DNA
5’-/5Phos/NNN NNN GAT CGT CGG ACT GTA GAA CTC TGA ACG TGT AG/3SpC3/-3’
Note: UMI nucleotides (machine-mixed equimolar A, C, G, and T residues, denoted N) are added at the 5’ end of the R1R sequence. The number of N nucleotides can be changed to suit the complexity of the samples being sequenced and the number of duplicates expected after PCR.
-
Illumina barcode PCR primer (P5)
5’-AAT GAT ACG GCG ACC ACC GAG AT BARCODE C TAC ACG TTC AGA GTT CTA CAG TCC GAC GAT C-3’
Note: The barcode sequence in the primer is the sense strand (e.g., ATCACG in the primer for TruSeq Barcode 01 (TSBC01) on the Illumina website). This barcode is optional but recommended to provide unique dual indices (UDI) for libraries sequenced on a NovaSeq instrument.
-
Illumina barcode PCR primer (P7)
5’-CAA GCA GAA GAC GGC ATA CGA GAT BARCODE GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG ATC T-3’
Note: The barcode sequence in the primer should be the reverse complement of the barcode listed on the Illumina website (e.g., CGTGAT in the primer for TSBC01).
-
UltraPure DNase/RNase-free distilled water (Thermo Fisher Scientific, catalog number: 10977015) or equivalent from other companies or in house sources
Trizma hydrochloride solution (Tris-HCl), pH 7.5, 2 M (Sigma-Aldrich, catalog number: T2944)
EDTA, 0.5 M, pH 8.0, RNase-free (Thermo Fisher Scientific, catalog number: AM9260G)
Sodium chloride solution, 5 M, RNase-free (Thermo Fisher Scientific, catalog number: AM9760G)
Magnesium chloride solution, BioUltra for molecular biology, 2 M (Sigma-Aldrich, catalog number: 68475-100ML-F)
RNA CenturyTM-Plus Markers (e.g., Thermo Fisher Scientific, catalog number: AM7145)
AMPure XP beads (Beckman Coulter, catalog number: A63880)
mirVana miRNA Isolation kit (Thermo Fisher Scientific, catalog number: AM1560)
Total Exosome RNA and Protein Isolation kit (Thermo Fisher Scientific, catalog number: 4478545)
TRIzol LS Reagent (Thermo Fisher Scientific, catalog number: 10296010)
Turbo DNase (Thermo Fisher Scientific, catalog number: AM2238)
DNase I (Zymo Research, catalog number: E1010)
Exonuclease I (Lucigen, catalog number: X40520K)
Illumina Ribo-Zero Plus rRNA Depletion kit (Illumina, catalog number: 20040526)
NEBNext Magnesium RNA Fragmentation Module (New England Biolabs, catalog number: E6150S)
RNA Clean & Concentrator-5 (Zymo Research, catalog number: R1013)
Equipment
-20°C freezer
T100 96-well PCR thermal cycler (Bio-Rad, catalog number: 1861096), Veriti 9044 60-well thermal cycler (Applied Biosystems, catalog number: 4384638), or equivalent thermal cycler with a heated lid and stable temperature control
Microcentrifuge (Eppendorf, catalog number: 2231000768)
DynaMag-2 Magnet (Thermo Fisher Scientific, catalog number: 12321D)
2100 Bioanalyzer instrument (Agilent, catalog number: G2939BA)
Chip priming station (Agilent, catalog number: 5065-4401)
Software
-
2100 Expert software/upgrade (Agilent, catalog number: G2946CA)
Note: The Bioanalyzer software should be bundled with the instrument. If not, an upgraded version is purchasable from Agilent.
Procedure
-
Preparation of 10× Starter Duplex (Table 1)
-
Prepare 10 µM R2R DNA containing an equimolar ratio of A, C, G, and T residues at the 1-nt DNA overhang position by hand-mixing equal volumes of four separate R2R DNA oligonucleotides with 3' A, C, G, or T residues (10 µM each) in a 1.5-ml Eppendorf Lobind microcentrifuge tube or equivalent and vortex. Aliquot the stock solution and store at -20°C until use. A volume of 50 μl of 10× Starter Duplex is sufficient for 25 reactions (2 μl per reaction).
Note: R2R DNA with unequal ratios of 1-nt 3’ DNA overhang nucleotides can be used to mitigate template-switching biases in TGIRT-seq (Mohr et al., 2013; Xu et al., 2019) ,
Set up the above reaction components in a sterile PCR tube for annealing of oligonucleotides to form the Starter Duplex.
Incubate the mixed 10× Starter Duplex components at 82°C for 2 min in a pre-heated thermocycler.
-
Cool to 25°C with a 10% ramp or at a rate of 0.1°C/s.
Note: The annealed R2 RNA/R2R DNA Starter Duplex should be prepared freshly each time before the template-switching reaction. Discard any left over after use.
-
-
Template-switching reverse transcription reaction
-
Set up the following reaction components in a sterile PCR tube adding the TGIRT-III enzyme last (Table 2).
Notes:
Because TGIRT enzymes can also template switch to DNA, RNA samples should be DNase treated prior to TGIRT-seq. Minimum RNA Inputs for DNase-treated RNA samples are: 2 ng rRNA-depleted, chemically fragmented cellular RNA; 20-50 ng rRNA-depleted, unfragmented cellular RNAs; 500 pg plasma RNA; 2 ng RNA extracted from highly purified extracellular vesicles. Measure low RNA concentrations with a Qubit or Bioanalyzer. See Notes 1-4 at the end of the text for further details regarding preparation of RNA samples for TGIRT-seq.
We use 200 mM NaCl in this protocol instead of 450 mM NaCl used in earlier versions to increase the efficiency of template switching without prohibitively increasing multiple sequential template switches, which result in artifactual fusion reads. See also Note 5 below.
Do a pilot experiment to determine the optimal enzyme concentration for your samples using an Agilent Bioanalyzer or equivalent instrument to monitor library quality and quantity.
A template-switching reverse transcription reaction using the TGIRT-III enzyme to a commercial RNA ladder (e.g., Thermo Fisher Scientific, catalog number: AM7145) can be carried through the procedure as a positive control.
Pre-incubate at room temperature for 30 min, then initiate template switching and reverse transcription by adding 1 μl of 20 mM dNTPs (an equimolar mixture of 20 mM each dATP, dCTP, dGTP, and dTTP).
Incubate at 60°C for 15 min for whole cell, EV, or plasma RNAs, 5-10 min for mature miRNAs, or up to 60 min for long or heavily modified RNAs. The optimal incubation time should be determined experimentally for different RNA templates.
Add 1 μl of 5 M NaOH and incubate at 95°C for 3 min.
-
Cool to room temperature and neutralize with 1 μl of 5 M HCl.
Note: Steps B4 and B5 are needed because TGIRT-III binds very tightly to nucleic acids. Alkaline hydrolysis degrades the RNA template and enzyme but not the cDNA products.
Add 78 μl of nuclease-free water to bring to a final volume of 100 μl.
-
Clean up the cDNAs with a MinElute Reaction Cleanup kit (QIAGEN, catalog number: 28204) or a MinElute PCR Purification kit (QIAGEN, catalog number: 28004) and elute in 10 μl of QIAGEN elution buffer.
Note: Incubate the column with the elution buffer at room temperature before centrifugation to maximize recovery.
The procedure can be interrupted here, with the cDNAs stored at -20°C.
Proceed with R1R adenylation, thermostable ligation, and Phusion PCR amplification.
-
-
R1R 5' DNA adenylation using the New England Biolabs kit (New England Biolabs, catalog number: E2610S/L)
Note: If the adenylated oligonucleotide is purchased from a commercial supplier, proceed to the ligation step.
Set up the following reaction components in a sterile PCR tube (Table 3):
Incubate at 65°C for 1 h.
Incubate at 85°C for 5 min to inactivate the enzyme.
-
Clean up with an Oligo Clean & Concentrator kit (Zymo Research, catalog number: D4060) and elute in 10 μl of nuclease-free water for a final concentration of 10 μM 5’-end adenylated R1R DNA.
Note: If required for a large number of TGIRT-seq libraries, we recommend scaling up the adenylation reaction by doing multiple (e.g., 6× to 8×) 20-μl reactions with the same amounts of enzyme and oligonucleotide, and then combining products for clean-up, as higher elution volume helps with efficient recovery of adenylated oligonucleotides. After clean-up, recovered material should be 80-100 ng/μl for R1R and 100-120 ng/μl for 6N UMI R1R, measured with a Nanodrop spectrophotometer using the ssDNA setting. Adenylation can be monitored using an Agilent Bioanalyzer with a Small RNA kit. The adenylated oligonucleotide can be stored at -20°C for up to two weeks.
-
Thermostable ligation of a 5’ adenylated adapter (New England Biolabs, catalog number: M0319S/L)
Set up the following reaction components in a sterile PCR tube (Table 4):
-
Incubate at 65°C for 1-2 h.
Note: We recommend 2 h ligation. However, 1 h ligation can be used if the starting material is abundant.
Add 80 μl of nuclease-free water for a final volume of 100 μl.
-
Clean up the ligated cDNAs with a MinElute Reaction Cleanup kit (QIAGEN, catalog number: 28204) and elute in 23 μl of QIAGEN elution buffer (incubate the column with elution buffer at room temperature before centrifugation to maximize recovery.
Note: The procedure can be interrupted here with the ligated cDNAs stored at -20°C.
Proceed with Phusion PCR amplification.
-
PCR amplification (Thermo Fisher Scientific, catalog number: F531S/L)
Set up the following reaction components in a sterile PCR tube (Table 5):
-
PCR cycles:
Denaturation 98°C for 5 s.
-
Up to 12 cycles of 98°C for 5 s, 60°C for 10 s, 72°C for 15-30 s/kb, hold at 4°C.
Notes:
Minimizing the number of cycles of PCR amplification decreases bias and duplicate reads. Twelve PCR cycles is generally satisfactory for TGIRT-seq with RNA inputs indicated in Section B. The number of cycles can be increased or decreased for different RNA inputs and library complexities.
The procedure can be interrupted here and the PCR products stored at -20°C.
Use AMPure XP beads (Beckman Coulter, catalog number: A63880) to deplete adapter-dimers and enrich for desired DNA sizes in the library. The default ratio is 1.4× v/v (70 μl of beads/50 μl of PCR reaction). The ratio of beads to sample volume can be adjusted depending on the size profile of RNAs being sequenced.
-
Check library quality by analyzing 1 μl on a Bioanalyzer with a High Sensitivity DNA Analysis kit (Figure 2).
Notes:
If needed, an additional round of the 1.4× AMPure XP beads clean-up can be performed to further deplete levels of adapter dimers. More extensive AMPure XP beads clean-up can result in loss of library products.
The final TGIRT-seq library can be stored at -20°C until submitted for sequencing.
-
Sequencing and data analysis
TGIRT-seq libraries can be sequenced on any Illumina sequencing instrument to obtain single-end (SE) or paired-end (PE) reads. Read length and read depth are dependent upon the needs of the experiment. We have used PE75 on a NextSeq 500 with 330 million reads output for analysis of cellular, EV, and plasma RNAs. We have also used PE150 on a NovaSeq with at least 700 million reads output for large projects or samples requiring high-depth analysis.
Because TGIRT-seq simultaneously profiles coding and non-coding RNAs, read mapping pipelines for TGIRT-seq typically use a sequential mapping strategy with specialized databases to first map reads with rRNA and tRNA sequences encoded at multiple loci, and sncRNAs embedded within introns in protein-coding genes, before mapping the remaining reads to a human genome reference sequence. In addition to end-to-end alignment, a local alignment step is typically included to more efficiently capture reads with non-templated nucleotides added to the 3' end of cDNAs by TGIRT enzymes. Detailed protocols for read processing and mapping, including UMI deconvolution of duplicate reads, and peak calling for the identification of protein-protected mRNA fragments, structured excised intron RNAs, and intron RNA fragments as potential biomarkers in human plasma can be found in Yao et al. (2020) .
Additional TGIRT-seq read mapping protocols used for different sample types and applications, including statistical tests and details of replicates for different applications, can be found in the following references: plasma RNA ( Qin et al., 2016 ); total cellular RNA, including spike-ins ( Nottingham et al., 2016 ); plasma DNA, including nucleosome positioning and mapping of DNA methylation sites (Wu and Lambowitz, 2017); sncRNAs ( Boivin et al., 2018 ); total cellular RNAs, including detailed analysis and remediation of sequencing biases ( Xu et al., 2019 ).
Table 1. Components needed for 10× (1 μM) Starter Duplex.
| Components | Volume (final concentration) |
|---|---|
| 10 μM R2 RNA | 5 μl (1 μM) |
| 10 μM R2R hand-mixed DNA | 5 μl (1 μM) |
| 10× TE buffer (100 mM Tris-HCl, pH 7.5, 10 mM EDTA) | 5 μl (10 mM Tris-HCl, pH 7.5, 1 mM EDTA) |
| Nuclease-free H2O | to 50 μl |
Table 2. Components needed for template-switching reverse transcription reaction.
| Components | Volume (final concentration) |
|---|---|
| RNA sample | 0.5-50 ng or <100 nM |
| 5× Reaction Buffer (1 M NaCl, 25 mM MgCl2, 100 mM Tris-HCl, pH 7.5) | 4 μl (200 mM NaCl, 5 mM MgCl2, 20 mM Tris-HCl, pH 7.5) |
| 10× DTT (100 mM; avoid excessive freezing and thawing) | 2 μl (10 mM) |
| 10× Starter Duplex (after annealing) | 2 μl (100 nM final) |
| TGIRT-III enzyme (10 μM) | 1 or 2 μl (500 nM or 1 μM final) |
| Nuclease-free H2O | to 19 μl |
Table 3. Components needed for 5' DNA adenylation.
| Components | Volume |
|---|---|
| 10× reaction buffer (DNA Adenylation Buffer) | 2 μl |
| 1 mM ATP | 2 μl |
| 100 μM 5’phos/3’SpC3 R1R DNA | 1 μl |
| Mth RNA Ligase | 2 μl |
| Nuclease-free water | to 20 μl |
Note: The New England Biolabs kit includes 10× DNA Adenylation buffer, 1 mM ATP, and Mth RNA ligase.
Table 4. Components needed for thermostable ligation of 5’ adapter.
| Components | Volume |
|---|---|
| 10× reaction buffer (NEBuffer 1) | 2 μl |
| 50 mM MnCl2 | 2 μl |
| cDNA from template-switching reaction | Up to 10 μl |
| Thermostable 5’ AppDNA/RNA Ligase | 2 μl |
| 10 μM 5’-end adenylated R1R DNA | 4 μl |
| Nuclease-free water | To 20 μl if using less than 10 μl cDNA |
Table 5. Components needed for PCR amplification.
| Components | Volume (final concentration) |
|---|---|
| 2× Phusion High-Fidelity PCR Master Mix in HF buffer | 25 μl |
| 10 μM Illumina P5 primer | 1 μl (200 nM) |
| 10 μM Illumina P7 Primer | 1 μl (200 nM) |
| cDNA from thermostable ligation | Up to 23 μl |
| Nuclease-free water | To 50 μl, if using less than 23 μl cDNA |
Figure 2. Bioanalyzer traces of representative TGIRT-seq libraries obtained by using an Agilent Bioanalyzer with a High Sensitivity DNA kit.

A. Ribo-depleted and chemically fragmented HeLa S3 cellular RNA (20 ng starting material after ribodepletion). Library products for cellular RNAs that were fragmented to 70-90 nt run at 190-210 bp. B. Plasma RNA (0.5 ng of starting material). Library products run at 140-240 bp, with the peak at ~200 bp corresponding to library products from tRNAs. Residual adapter dimers (AD), which are present at low concentrations in the final libraries, run at ~120 bp for R1R-R2R, and slightly larger with the UMI R1R adapter or Unique Dual Index (UDI) PCR primer. Peaks smaller than 100 bp correspond to unused PCR and adapter oligonucleotides (P+A). The lower (35 bp) and upper (10,380 bp) markers are internal markers provided with the High Sensitivity DNA kit. DNA lengths (bp) are determined by the software program based on the ladder provided with the kit.
Notes
We have used various commercial kits to isolate RNAs for TGIRT-seq. The commercial kits differ significantly in RNA yield and can bias for different RNA biotypes. We have used the mirVana miRNA Isolation kit (Thermo Fisher Scientific, catalog number: AM1560) with good results for TGIRT-seq of human cellular RNA. For EVs, we obtained good results by using the Total Exosome RNA and Protein Isolation kit (Thermo Fisher Scientific, catalog number: 4478545). For isolating human plasma RNAs, we have tried a number of different extraction reagents and kits, including TRIzol LS Reagent (Thermo Fisher Scientific, catalog number: 10296010) and the Direct-zol RNA Miniprep (Zymo Research, catalog number: R2051); QIAamp ccfDNA/RNA Extraction kit (QIAGEN, catalog number: 55184); and miRNeasy Serum/Plasma Advanced kit (QIAGEN, catalog number: 217204). Each kit has advantages and disadvantages for input amount, ease of use, and RNA yield. In our hands, the QIAamp ccfDNA/RNA kit was more efficient for extracting DNA than RNA ( Yao et al., 2020 ).
TGIRT enzymes can template switch from the R2 RNA/R2R DNA starter duplex to the 3' end of DNA fragments, and this activity has been used for sequencing single-stranded DNA for analysis of nucleosome positioning and mapping of DNA methylation sites (Wu and Lambowitz, 2017). For RNA-seq, it is important to remove as much DNA as possible to minimize DNA reads. We have used Turbo DNase (Thermo Fisher Scientific, catalog number: AM2238) and a combination of DNase I (Zymo Research, catalog number: E1010) and Exonuclease I (Lucigen, catalog number: X40520K) ( Yao et al., 2020 ). We clean up DNase-treated RNAs using an RNA Clean & Concentrator-5 kit (Zymo Research, catalog number: R1013) with a modified 8× V/V (ethanol/input RNA) protocol.
Cellular RNAs are rRNA-depleted to minimize reads mapping to rRNAs. As rRNA removal procedures are not 100% specific for rRNAs, different rRNA depletion kits can also affect reads corresponding to other RNA biotypes, including RNAs of interest. Thus, the same rRNA depletion kit should be used for all samples in a study. The Illumina Ribo-Zero Plus rRNA Depletion kit (Illumina, catalog number: 20040526) with the manufacturer's protocol gives satisfactory results for TGIRT-seq [5-10% rRNA reads in the final library, although in our hands it is not as effective at removing rRNA as the original (now discontinued) version of that kit (RiboZero Gold, <1-2% rRNA reads)].
For chemical fragmentation of cellular RNAs, we use the NEBNext Magnesium RNA Fragmentation Module (New England Biolabs, catalog number: E6150S). We typically use 50 ng rRNA-depleted total cellular RNA as input and heat at 94°C for 5-7 min for fragmentation. The RNA input and fragmentation time should be optimized by the researcher according to the RNAs of interest and desired read length for Illumina sequencing. After fragmentation, we clean up the RNA by using an RNA Clean & Concentrator-5 kit (Zymo Research, catalog number: R1013) with a modified 8× V/V (ethanol/input RNA) protocol. Fragmented cellular RNAs generated with divalent cations should be treated with T4 polynucleotide kinase with 3’-phosphatase activity to remove 3' phosphates and 2’,3’-cyclic phosphates, which impede TGIRT template-switching ( Mohr et al., 2013 ; Nottingham et al., 2016 ). This inhibition is less pronounced at 200 mM than at 450 mM NaCl ( Lentzsch et al., 2019 ; Yao et al., 2020 ).
The high salt concentration (450 mM NaCl) used in initial TGIRT-seq protocols to suppress multiple end-to-end template switches by the TGIRT enzyme also decreases the efficiency of the initial template-switching reaction from the R2 RNA/R2R DNA Starter Duplex ( Lentzsch et al., 2019 ). Multiple template switches are particularly problematic for miRNA sequencing. The updated protocol using 200 mM NaCl to increase the efficiency of initial template-switching reaction from the R2 RNA/R2R DNA starter duplex gave comprehensive libraries of coding and non-coding RNAs in human plasma with acceptable levels of fusion reads (0.5-4%), which include multiple template switches ( Yao et al., 2020 ).
With low RNA inputs (e.g., for human plasma and EV RNAs), Bioanalyzer traces of TGIRT-seq libraries may show an adapter dimer peak but no software-called library product peaks. In those cases, 1 μl of the library can be amplified with another 12 cycles of PCR to detect library products. If library products can be detected after this further PCR amplification, the original library without further amplification should be okay for sequencing. After the additional amplification, adapter dimer levels will be even higher (as the short products amplify preferentially), but the user should expect to observe desired products of the predicted sizes. If library products are still not detected, it likely means that library preparation failed due to an RNA input problem (quality or quantity).
A lack of both a product peak and an adapter dimer peak (120-125 bp) in Bioanalyzer traces of the final TGIRT-seq library indicates that the ligation reaction was unsuccessful, most likely due to failure of the R1R adenylation step or ligation.
Acknowledgments
The development of TGIRT-seq methods has been supported by NIH grant R35 GM136216 and Welch Foundation Grant F-1607. This protocol has been adapted from Yao et al. (2020).
Competing interests
Thermostable group II intron reverse transcriptase enzymes and methods for their use are the subject of patents and patent applications that have been licensed by the University of Texas and East Tennessee State University to InGex, LLC. A.M.L, some former and present members of the Lambowitz laboratory, and the University of Texas are minority equity holders in InGex, LLC, and receive royalty payments from the sale of TGIRT enzymes and kits employing TGIRT template-switching activity and from the sublicensing of intellectual property to other companies.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1. Behrens A., Rodschinka G. and Nedialkova D. D.(2021). High-resolution quantitative profiling of tRNA abundance and modification status in eukaryotes by mim-tRNAseq. Mol Cell 81(8): 1802–1815..e1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Belfort M. and Lambowitz A. M.(2019). Group II Intron RNPs and Reverse Transcriptases: From Retroelements to Research Tools. Cold Spring Harb Perspect Biol 11(4): a032375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blocker F. J., Mohr G., Conlan L. H., Qi L., Belfort M. and Lambowitz A. M.(2005). Domain structure and three-dimensional model of a group II intron-encoded reverse transcriptase. RNA 11(1): 14-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boivin V., Deschamps-Francoeur G., Couture S., Nottingham R. M., Bouchard-Bourelle P., Lambowitz A. M., Scott M. S. and Abou-Elela S.(2018). Simultaneous sequencing of coding and noncoding RNA reveals a human transcriptome dominated by a small number of highly expressed noncoding genes. RNA 24(7): 950-965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boivin V., Reulet G., Boisvert O., Couture S., Elela S. A. and Scott M. S.(2020). Reducing the structure bias of RNA-Seq reveals a large number of non-annotated non-coding RNA. Nucleic Acids Res 48(5): 2271-2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carrell S. T., Tang Z., Mohr S., Lambowitz A. M. and Thornton C. A.(2018). Detection of expanded RNA repeats using thermostable group II intron reverse transcriptase. Nucleic Acids Res 46(1): e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cocquet J., Chong A., Zhang G. and Veitia R. A.(2006). Reverse transcriptase template switching and false alternative transcripts. Genomics 88(1): 127-131. [DOI] [PubMed] [Google Scholar]
- 8. Evans M. E., Clark W. C., Zheng G. and Pan T.(2017). Determination of tRNA aminoacylation levels by high-throughput sequencing. Nucleic Acids Res 45(14): e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gladyshev E. A. and Arkhipova I. R.(2011). A widespread class of reverse transcriptase-related cellular genes. Proc Natl Acad Sci U S A 108(51): 20311-20316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hafner M., Renwick N., Brown M., Mihailović A., Holoch D., Lin C., Pena J. T., Nusbaum J. D., Morozov P., Ludwig J., et al.(2011). RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA 17(9): 1697-1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hu W. S. and Hughes S. H.(2012). HIV-1 reverse transcription. Cold Spring Harb Perspect Med 2(10): a006882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Katibah G. E., Qin Y., Sidote D. J., Yao J., Lambowitz A. M. and Collins K.(2014). Broad and adaptable RNA structure recognition by the human interferon-induced tetratricopeptide repeat protein IFIT5. Proc Natl Acad Sci U S A 111(33): 12025-12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kojima K. K. and Kanehisa M.(2008). Systematic survey for novel types of prokaryotic retroelements based on gene neighborhood and protein architecture. Mol Biol Evol 25(7): 1395-1404. [DOI] [PubMed] [Google Scholar]
- 14. Lambowitz A. M. and Belfort M.(2015). Mobile Bacterial Group II Introns at the Crux of Eukaryotic Evolution. Microbiol Spectr 3(1): Mdna3-0050-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lentzsch A. M., Yao J., Russell R. and Lambowitz A. M.(2019). Template-switching mechanism of a group II intron-encoded reverse transcriptase and its implications for biological function and RNA-Seq. J Biol Chem 294: 19764-19784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lentzsch A. M., Stamos J. L., Yao J., Russell R. and Lambowitz A. M.(2021). Structural basis for template switching by a group II intron-encoded non-LTR-retroelement reverse transcriptase. J Biol Chem 297(2): 100971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mader R. M., Schmidt W. M., Sedivy R., Rizovski B., Braun J., Kalipciyan M., Exner M., Steger G. G. and Mueller M. W.(2001). Reverse transcriptase template switching during reverse transcriptase-polymerase chain reaction: artificial generation of deletions in ribonucleotide reductase mRNA. J Lab Clin Med 137(6): 422-428. [DOI] [PubMed] [Google Scholar]
- 18. Martín-Alonso S., Frutos-Beltrán E. and Menéndez-Arias L.(2021). Reverse Transcriptase: From Transcriptomics to Genome Editing. Trends Biotechnol 39(2): 194-210. [DOI] [PubMed] [Google Scholar]
- 19. Mohr S., Ghanem E., Smith W., Sheeter D., Qin Y., King O., Polioudakis D., Iyer V. R., Hunicke-Smith S., Swamy S., et al.(2013). Thermostable group II intron reverse transcriptase fusion proteins and their use in cDNA synthesis and next-generation RNA sequencing. RNA 19(7): 958-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nottingham R. M., Wu D. C., Qin Y., Yao J., Hunicke-Smith S. and Lambowitz A. M.(2016). RNA-seq of human reference RNA samples using a thermostable group II intron reverse transcriptase. RNA 22(4): 597-613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Onafuwa-Nuga A. and Telesnitsky A.(2009). The remarkable frequency of human immunodeficiency virus type 1 genetic recombination. Microbiol Mol Biol Rev 73(3): 451–480., Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Penno C., Kumari R., Baranov P. V., van Sinderen D. and Atkins J. F.(2017). Stimulation of reverse transcriptase generated cDNAs with specific indels by template RNA structure: retrotransposon, dNTP balance, RT-reagent usage. Nucleic Acids Res 45(17): 10143-10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Picelli S., Björklund Å K., Faridani O. R., Sagasser S., Winberg G. and Sandberg R.(2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 10(11): 1096-1098. [DOI] [PubMed] [Google Scholar]
- 24. Qin Y., Yao J., Wu D. C., Nottingham R. M., Mohr S., Hunicke-Smith S. and Lambowitz A. M.(2016). High-throughput sequencing of human plasma RNA by using thermostable group II intron reverse transcriptases. RNA 22(1): 111-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shen P. S., Park J., Qin Y., Li X., Parsawar K., Larson M. H., Cox J., Cheng Y., Lambowitz A. M., Weissman J. S., et al.(2015). Protein synthesis. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains. Science(New York, NY) 347(6217): 75-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shurtleff M. J., Yao J., Qin Y., Nottingham R. M., Temoche-Diaz M. M., Schekman R. and Lambowitz A. M.(2017). Broad role for YBX1 in defining the small noncoding RNA composition of exosomes. Proc Natl Acad Sci U S A 114(43): E8987-E8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simon D. M. and Zimmerly S.(2008). A diversity of uncharacterized reverse transcriptases in bacteria. Nucleic Acids Res 36(22): 7219-7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stamos J. L., Lentzsch A. M. and Lambowitz A. M.(2017). Structure of a Thermostable Group II Intron Reverse Transcriptase with Template-Primer and Its Functional and Evolutionary Implications. Mol Cell 68(5): 926–939..e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stamos J. L., Lentzsch A. M., Park S. K., Mohr G., Lambowitz A. M.(2019). Non-LTR retroelement reverse transcriptase and uses thereof. PCT/US2018/054147.
- 30. Stark R., Grzelak M. and Hadfield J.(2019). RNA sequencing: the teenage years. Nat Rev Genet 20(11): 631-656. [DOI] [PubMed] [Google Scholar]
- 31. Upton H. E, Ferguson L, Temoche-Diaz M. M, Liu X, Pimentel S. C, Ingolia N. T, Schekman R and Collins K.(2021). Low-bias ncRNA libraries using ordered two-template relay: serial template jumping by a modified retroelement reverse transcriptase. BioRxiv doi: 10.1101/2021.04.30.442027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Z., Ma Z., Castillo-González C., Sun D., Li Y., Yu B., Zhao B., Li P. and Zhang X.(2018). SWI2/SNF2 ATPase CHR2 remodels pri-miRNAs via Serrate to impede miRNA production. Nature 557(7706): 516-521. [DOI] [PubMed] [Google Scholar]
- 33. Wu D. C. and Lambowitz A. M.(2017). Facile single-stranded DNA sequencing of human plasma DNA via thermostable group II intron reverse transcriptase template switching. Sci Rep 7(1): 8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu X. and Bartel D. P.(2017). Widespread Influence of 3'-End Structures on Mammalian mRNA Processing and Stability. Cell 169(5): 905–917..e911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xiong Y. and Eickbush T. H.(1990). Origin and evolution of retroelements based upon their reverse transcriptase sequences. Embo J 9(10): 3353-3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu H., Yao J., Wu D. C. and Lambowitz A. M.(2019). Improved TGIRT-seq methods for comprehensive transcriptome profiling with decreased adapter dimer formation and bias correction. Sci Rep 9(1): 7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yao J., Wu D. C., Nottingham R. M. and Lambowitz A. M.(2020). Identification of protein-protected mRNA fragments and structured excised intron RNAs in human plasma by TGIRT-seq peak calling. Elife 9: e60743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yao J., Xu H., Shelby W., Wu D. C., Ares M. and Lambowitz A. M.(2021). Human cells contain myriad excised linear introns with potential functions in gene regulation and as RNA biomarkers. BioRxiv doi: 10.1101/2020.09.07.285114. [DOI] [Google Scholar]
- 39. Zarnegar B. J., Flynn R. A., Shen Y., Do B. T., Chang H. Y., and Khavari P. A.(2016). irCLIP platform for efficient characterization of protein-RNA interactions. Nat Methods 13(6): 489-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao C., Liu F. and Pyle A. M.(2018). An ultraprocessive, accurate reverse transcriptase encoded by a metazoan group II intron. RNA 24(2): 183-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zheng G., Qin Y., Clark W. C., Dai Q., Yi C., He C., Lambowitz A. M. and Pan T.(2015). Efficient and quantitative high-throughput tRNA sequencing. Nat Methods 12(9): 835-837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu Y. Y., Machleder E. M., Chenchik A., Li R. and Siebert P. D.(2001). Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. Biotechniques 30(4): 892-897. [DOI] [PubMed] [Google Scholar]
- 43. Zimmerly S. and Wu L.(2015). An Unexplored Diversity of Reverse Transcriptases in Bacteria. Microbiol Spectr 3(2): Mdna3-0058-2014. [DOI] [PubMed] [Google Scholar]
- 44. Zubradt M., Gupta P., Persad S., Lambowitz A. M., Weissman J. S. and Rouskin S.(2017). DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo . Nat Methods 14(1): 75-82. [DOI] [PMC free article] [PubMed] [Google Scholar]