

Abstract
Angiotensin‐converting enzyme inhibitors (ACEi) are part of the indicated treatment in hypertensive African Americans. ACEi have blood pressure‐independent effects that may make them preferred for certain patients. We aimed to evaluate the impact of ACEi on anti‐fibrotic biomarkers in African American hypertensive patients with left ventricular hypertrophy (LVH). We conducted a post hoc analysis of a randomized controlled trial in which hypertensive African American patients with LVH and vitamin D deficiency were randomized to receive intensive antihypertensive therapy plus vitamin D supplementation or placebo. We selected patients who had detectable lisinopril (lisinopril group) in plasma using liquid‐chromatography/mass spectrometry analysis and compared them to subjects who did not (comparison group) at the one‐year follow‐up. The pro‐fibrotic marker type 1 procollagen C‐terminal propeptide (PICP) and the anti‐fibrotic markers matrix metalloproteinase‐1 (MMP‐1), tissue inhibitor of metalloproteinases 1 (TIMP‐1), telopeptide of collagen type I (CITP), and N‐acetyl‐seryl‐aspartyl‐lysyl‐proline (Ac‐SDKP) peptide were measured. Sixty‐six patients were included, and the mean age was 46.2 ± 8 years. No difference was observed in the number and intensity of antihypertensive medications prescribed in each group. Patients with detectable lisinopril had lower blood pressure than those in the comparison group. The anti‐fibrotic markers Ac‐SDKP, MMP‐1, and MMP‐1/TIMP‐1 ratio were higher in patients with detectable ACEi (all p < .05). In a model adjusted for systolic blood pressure, MMP‐1/TIMP‐1 (p = .02) and Ac‐SDKP (p < .001) levels were associated with lisinopril. We conclude that ACEi increase anti‐fibrotic biomarkers in hypertensive African Americans with LVH, suggesting that they may offer added benefit over other agents in such patients.
Keywords: ACE inhibitors, Ac‐SDKP, African American, collagen, left ventricular hypertrophy, MMP‐1
1. INTRODUCTION
Hypertension is the leading risk factor for cardiovascular mortality and has a particularly early incidence and high prevalence in the African American population. 1 , 2 Elevated blood pressure (BP) leads to vascular, myocardial, and renal tissue damage, which are responsible for the high burden of disease in this population. 2 With the progression of hypertension, adaptive and pathologic changes are observed in the heart and vascular beds. Some of these changes are macroscopic, such as left ventricular hypertrophy (LVH) and arterial stiffness, while other alterations are microscopic, including endothelial dysfunction, vascular rarefaction, inflammation, and fibrosis. 3 Myocardial and vascular fibrosis are of particular importance because they can lead to early cardiac dysfunction, accelerate the vascular damage by altering physiological vascular mechanics, and decrease tissue oxygenation. 3 , 4
Fibrosis is a dynamic process characterized by the formation, deposition, and late consolidation (ie, crosslinking) of fibrotic fiber, specifically collagen. 5 Simultaneous with the pro‐fibrotic process, a series of anti‐fibrotic mechanisms promote collagen release after the injury is healed and before extensive scarring (crosslinking) occurs. 6 Cardiac fibroblasts synthesize type I and III collagen fibers in the interstitium of the heart when a stressor or injury occurs, as in hypertension. During the extracellular conversion of procollagen type I into mature collagen type I, type 1 procollagen C‐terminal propeptide (PICP) is generated and released from the heart into the circulation. 6 Serum PICP levels correlate with myocardial collagen and collagen type I deposition. 7 On the other hand, matrix metalloproteinase 1 (MMP‐1), or cardiac collagenase, is the key enzyme for interstitial collagen degradation. 6 MMP‐1 releases the carboxy‐terminal telopeptide of collagen type I (CITP) into the circulation when collagen degradation occurs. 8 MMP‐1 is regulated by the tissue inhibitor of matrix metalloproteinase 1 (TIMP‐1); thus, net MMP‐1 activity can be estimated from the MMP‐1/TIMP‐1 ratio, with a higher ratio suggesting more collagen degradation or an anti‐fibrotic effect. 8 Unfortunately, after crosslinking, collagen is resistant to the MMP‐1 degradative process. The PICP/CITP ratio reflects the balance between type I collagen synthesis and degradation. 9 It is possible to predict the intensity of collagen crosslinking with the CITP/MMP‐1 ratio, where a higher value is associated with less crosslinked cardiac collagen. 10
Part of the adverse cardiac remodeling and fibrosis are explained by the renin‐angiotensin‐aldosterone system (RAAS) which includes angiotensin II (Ang II) via angiotensin receptor 1 (AT1). Ang II production has untoward effects leading to remodeling of the cardiovascular tissues, 11 including stimulation and proliferation of cardiac fibroblasts. RAAS activation stimulates collagen deposition and is involved in cardiac fibrosis. Matrix metalloproteinases (MMPs) and the tissue inhibitors of matrix metalloproteinases (TIMPs) are fundamental mediators and regulators of the cardiac fibrotic process and sensitive to the RAAS modulation. 12 Specifically, MMP‐9 and TIMP‐1 are known to be involved in models of cardiac disease. 13 Previous studies have indicated that Ang II could stimulate vascular smooth muscle cells, endothelial cells, and extracellular matrix (ECM) of cerebral vessels to secrete MMP‐9, 11 , 14 , 15 an effect that was prevented by using captopril. 14 Cardiac myocytes are known to release large amounts of MMP‐1. In addition, MMP‐1 and TIMP‐1 play a role in maintaining the ECM architecture. Imbalances in either of these proteinases reflect a disruption in the architecture of myocardial tissue. 16
Treatment with RAAS inhibitors may provide benefit independent of BP reduction 17 and are an important component of combination therapy for African American patients. 17 , 18 An important meta‐analysis indicated that angiotensin‐converting enzyme inhibitors (ACEi) have an additional protective effect over angiotensin receptor blockers (ARB) in myocardial infarction incidence, independent of the BP‐reducing effects. 19 This may be because ACEi specifically induce the release of an anti‐fibrotic and anti‐inflammatory tetra‐peptide called N‐acetyl‐seryl‐aspartyl‐lysyl‐proline (Ac‐SDKP). 20 Ac‐SDKP is degraded by the N‐terminal domain of the ACE enzyme and has been proposed as a marker of medication adherence in patients prescribed ACEi. 21 Ac‐SDKP has been shown to have strong anti‐fibrotic effects in heart, kidney, and vascular tissues in vitro and in vivo. It has also been suggested that many of the protective effects of ACEi can be ascribed to this peptide. 22 However, it has also been shown that ARBs and calcium channel blockers may have direct and indirect anti‐fibrotic effects. 23 , 24 No comparative study has yet evaluated differences in anti‐fibrotic markers in hypertensive African American patients with adaptive remodeling taking ACEi vs. antihypertensive medications from other classes. Thus, we sought to evaluate anti‐fibrotic markers in hypertensive African American patients with LVH after one year of standard intensive treatment, comparing levels in patients who were treated with the ACEi lisinopril with those who received other classes of antihypertensive medications.
2. METHODS
We performed a post hoc analysis of a vitamin D randomized controlled trial (NCT01360476) that recruited African American men and women aged 30 to 74 who presented at the emergency departments affiliated with Wayne State University in Detroit, MI, USA, with elevated BP. Eligible individuals required a systolic BP (SBP) ≥ 160 mmHg at first measurement and at one‐hour post‐triage with evidence of LVH on cardiac magnetic resonance (CMR) imaging at outpatient follow‐up 1‐2 weeks later. Inclusion and exclusion criteria are shown in Table 1. Participants received standard antihypertensive therapy according to current guidelines for hypertensive African Americans, 17 , 18 which included initial combination therapy (diuretic plus ACEi or calcium channel antagonist) for most. Lisinopril was the ACEi prescribed in all patients receiving ACEi. All patients received a prescription for antihypertensive medications aimed at achieving intensive BP control (uniform goal SBP < 130 mmHg), utilizing an evidence‐based, standardized algorithm, for the duration of the study. 17 The number of prescribed antihypertensive medications, as well as the therapeutic intensity score (TIS), was calculated for each antihypertensive medication prescribed in each patient. Additionally, a total TIS score (all hypertensive medications combined) was also calculated. TIS is a summary measure that accounts for the number of medications and the relative doses a patient received. 25 Hypertension treatment was supplemented with vitamin D (50 000 IU) or placebo administered every other week for 52 weeks (26 total doses). Participants were followed for one year, with six antihypertensive medication titrations occurring during weeks 2, 8, 16, 28, 40, and 52.
TABLE 1.
Inclusion and Exclusion criteria
|
Inclusion Criteria:
|
Exclusion Criteria:
|
BP measurements were obtained using the BpTruTM device. BpTruTM measurements are equivalent to the daytime average of the ambulatory BP monitoring when 5 measurements without a health professional present are performed. 26 BP was measured with an appropriately sized cuff. Five seated BP measurements were taken over 5 minutes. The average was used in the analysis.
CMR imaging was performed using standard protocols. Males required a left ventricular mass index (LVMI) > 89 g/m2 and females required a LVMI > 73 g/m2 to diagnose LVH and to be included into the study.
Carotid‐femoral pulse wave velocity (PWV) was measured at screening and at 16 weeks and one year (52 weeks) post‐randomization using a commercially available, non‐invasive applanation tonometry device (SpyhgmoCor; AtCor Medical, West Ryde, Australia).
For this post hoc analysis, participants receiving antihypertensive therapy were divided into two groups: one group with detectable lisinopril in serum (lisinopril group) at 16 and 52 weeks and those in whom no lisinopril was detectable (control group). We used liquid chromatography‐mass spectrometry (LC–MS) to measure the presence and levels of lisinopril in banked serum samples obtained at the 16‐week and 52‐week (one‐year) follow‐up appointments for comparison. 27 Patients in the lisinopril group had to have detectible lisinopril at both 16‐ and 52‐week follow‐up appointments, while those in the control group had no detectable lisinopril at those time points. Patients with detectible lisinopril may not have been originally prescribed lisinopril at baseline; however, lisinopril may be added during the subsequent medication titration phase. As a result, in Table 1, there are some patients with detectible lisinopril who were not prescribed lisinopril at baseline. From the 52‐week samples, the pro‐fibrotic marker type 1 procollagen C‐terminal propeptide (PICP) and the anti‐fibrotic markers matrix metalloproteinase‐1 (MMP‐1), tissue inhibitor of MMP‐1 (TIMP‐1), the MMP‐1/TIMP‐1 ratio, telopeptide of collagen type I (CITP), and Ac‐SDKP were measured. PICP (Cat # 8003; Quidel, USA), CITP (Cat # HC0761; Neo Scientific, USA), MMP‐1 (Cat # DMP100; R&D Systems, USA), TIMP‐1 (Cat # DTM100; R&D Systems, USA), and Ac‐SDKP (Cat # A05881; SPI‐BIO, France) were measured using commercial enzyme‐linked immunosorbent assay kits according to the manufacturers’ instructions.
The study was approved by the local Institutional Review Board at Wayne State University and was in accordance with the ethical standards set forth by the Institutional Review Board for utilization of humans in research.
2.1. Statistics
Data were presented as frequencies (%) or mean ± standard deviation. Where normal distributions were observed, t tests and ANOVA were used to compare continuous variables between two or more categories, respectively. The Wilcoxon signed‐rank test was used when non‐normal distributions were observed. Ordinary least squares linear regression was used to determine the association between lisinopril (independent) and individual markers (dependents), controlling for week 52 SBP. Two separate sets of models were used. In one set, detectable presence of lisinopril at the week 52 visit was treated as a dichotomous indicator. In the second set, the observed lisinopril concentration at week 52 was used as a continuous predictor, to account for observed individual variation in dosage and/or adherence. Because of the order‐of‐magnitude variation observed in lisinopril levels, the reported week 52 concentrations were log‐transformed prior to inclusion in the regression models. An alpha level of 0.05 was used for all comparisons. SAS (SAS Institute, Cary, NC, USA) v9.4 was used for analyses.
3. RESULTS
Three hundred fifty‐three patients were screened, and 113 patients with CMR‐confirmed LVH were randomized. This sub‐analysis included 66 patients that had detectible antihypertensive medication serum levels at 16 and 52 weeks. The clinical characteristics of control and lisinopril groups are shown in Table 2. The average age of this cohort of hypertensive patients was 46.2 ± 8 years. The average age of patients with detectible lisinopril was 5 years older than that of patients without detectible lisinopril, with no difference in gender and body weight distribution. The number of prescribed antihypertensive medications was similar between the groups, and no difference was observed in vitamin D supplementation. TIS for each individual medication (data not shown) and the total combined TIS for all the antihypertensive medication were similar between those with detectible lisinopril and the comparison group (Table 2). The classes of antihypertensive drugs prescribed were equally distributed, except for ACEi and ARBs, which were the focus of these analyses. One patient in the lisinopril group was prescribed an ARB and an ACEi simultaneously. There were four patients who were initially prescribed ACEi in whom the drug was not detected in two consecutive screenings at 16 and 52 weeks. Because the main criterion of this analysis was the presence of detectible lisinopril, these four patients were included in the comparison group. Despite the same number of prescribed medication and TIS score, BP was lower in the lisinopril group after one year of treatment. LVMI assessed with CMR imaging was similar between the groups at the beginning of the study and numerically lower in patients with detectible lisinopril concentrations at one year of treatment (84 ± 13.6 vs 80.8 ± 12.7 g/m2); however, we did not detect a statistically significant difference in LVMI change at one year. Both groups experienced similar regression of structural heart damage after one year of intensive therapy, and arterial stiffness was similar between the groups (Table 2).
TABLE 2.
General characteristics of patients treated or not treated with ACE inhibitors
| Comparison Group | Lisinopril Group | p‐value | |
|---|---|---|---|
| N | 30 | 36 | |
| Age, years (sd) | 43.7 + 7.9 | 48.7 ± 7.5 | .01 |
| Female (%) | 60.0 | 44.0 | .21 |
| BMI (kg/m2) | 35.9 ± 10.8 | 35.1 ± 7.1 | .98 |
| Number of prescribed antihypertensive drugs | 2.3 ± 0.7 | 2.7 ± 0.8 | .07 |
| ACEi (%) | 13.0 | 94.4 | <.001 |
| ARB (%) | 56.7 | 2.8 | <.001 |
| Amlodipine (%) | 53.3 | 66.7 | .27 |
| Diuretics (%) | 86.7 | 77.8 | .35 |
| Therapeutic intensity score (sd) | 1.5(0.7) | 1.5(0.6) | .62 |
| Supp. Vitamin D (%) | 56.7 | 41.7 | .22 |
| SBP basal (mmHg) | 161.0 ± 29.3 | 162.2 ± 24.0 | .85 |
| DBP basal (mmHg) | 100.9 ± 15.3 | 102.2 ± 12.1 | .71 |
| SBP at one year (mmHg) | 139.5 ± 20.7 | 129.6 ± 13.8 | .02 |
| DBP at one year (mmHg) | 94.3 ± 12.8 | 86.3 ± 9 | <.01 |
| LVMI at one year (g/m2) | 84.0 ± 13.6 | 80.8 ± 12.7 | .33 |
| Changes in LVMI at one year—basal (g/m2) | ‐12.9 ± 15.5 | ‐17.9 ± 11.2 | .10 |
| Pulse Wave Velocity (m/s) | 6.6 ± 3.4 | 6.6 ± 3.2 | .94 |
Abbreviations: ACEi, Angiotensin‐converting enzyme inhibitors; ARB, Angiotensin II receptor blocker; BMI, body mass index; DBP, diastolic blood pressure; LVMI, left ventricular mass index; SBP, systolic blood pressure; sd, standard deviation.
The pro‐fibrotic and anti‐fibrotic markers were compared between the groups after one year of antihypertensive therapy. No difference was observed in the pro‐fibrotic marker PICP between those receiving lisinopril and the comparison group (Table 3). However, the anti‐fibrotic biomarkers Ac‐SDKP, MMP‐1, and the MMP‐1/TIMP‐1 ratio significantly increased in the group with detectible lisinopril (Table 3). In a linear regression model adjusted for SBP, the anti‐fibrotic markers Ac‐SDKP and MMP‐1/TIMP‐1 ratio were independently associated with the presence of lisinopril (Table 4) and with lisinopril concentrations (Table S1). SBP was associated with LVMI and with the presence of the pro‐fibrotic marker PICP (Table 4).
TABLE 3.
Fibrotic and anti‐fibrotic biomarkers in African Americans treated or not treated with angiotensin‐converting enzyme inhibitors
| Comparison Group | Lisinopril Group | p‐value | |
|---|---|---|---|
| PICP/CITP ratio | 41.1 ± 32.7 | 46.6 ± 44.2 | .59 |
| PICP (nM) | 84.7 ± 31.9 | 85.2 ± 28.4 | .83 |
| CITP (nM) | 3.6 ± 2.8 | 3.3 ± 2.9 | .63 |
| MMP‐1 (nM) | 3.4 ± 2.6 | 5.5 ± 4.0 | .04 |
| TIMP‐1 (nM) | 166.8 ± 36.6 | 155 ± 34.6 | .25 |
| MMP‐1/TIMP‐1 ratio | 0.02 ± 0.02 | 0.04 ± 0.03 | .04 |
| Log CITP/MMP‐1 ratio | 1.03 ± 4.10 | 0.71 ± 4.16 | .26 |
| Ac‐SDKP (nM) | 3.9 ± 2.6 | 6.3 ± 2.8 | <.001 |
Abbreviations: Ac‐SDKP, N‐acetyl‐seryl‐aspartyl‐lysyl‐proline; CITP, telopeptide of collagen type I; MMP‐1, matrix metalloproteinase‐1; PICP, type 1 procollagen C‐terminal propeptide; TIMP‐1, tissue inhibitor of metalloproteinases 1.
TABLE 4.
Regression model controlling for systolic blood pressure and detectible serum lisinopril at week 52
| R 2 | Week 52 SBP | Lisinopril | |||||
|---|---|---|---|---|---|---|---|
| Estimate | 95% Conf. Limits | p‐Value | Estimate | 95% Conf. Limits | p‐Value | ||
| LVMI (g/m2) | .1342 | 0.2636 | [0.085, 0.442] | .004 | ‐0.5463 | [−6.911, 5.818] | .864 |
| PICP/CITP | .0237 | 0.3126 | [−0.255, 0.88] | .275 | 8.603 | [−11.628, 28.834] | .399 |
| PICP | .0704 | 0.4603 | [0.039, 0.881] | .032 | 4.9843 | [−10.029, 19.998] | .509 |
| CITP | .0057 | 0.0095 | [−0.032, 0.051] | .653 | ‐0.1895 | [−1.683, 1.304] | .801 |
| MMP‐1/TIMP‐1 | .1191 | ‐0.0002 | [−0.001, 0] | .186 | 0.0139 | [0, 0.027] | .043 |
| MMP‐1 | .0893 | ‐0.0371 | [−0.089, 0.015] | .156 | 1.4337 | [−0.411, 3.278] | .125 |
| TIMP‐1 | .0274 | ‐0.0235 | [−0.54, 0.493] | .927 | ‐11.9787 | [−30.375, 6.417] | .198 |
| Ac‐SDKP | .1666 | 0.0148 | [−0.025, 0.054] | .456 | 2.4915 | [1.085, 3.898] | .001 |
Abbreviations: Ac‐SDKP, N‐acetyl‐seryl‐aspartyl‐lysyl‐proline; CITP, telopeptide of collagen type I; LVMI, Left ventricular mass‐Indexed; MMP‐1, matrix metalloproteinase‐1; PICP, type 1 procollagen C‐terminal propeptide; TIMP‐1, tissue inhibitor of metalloproteinases 1.
We observed that the majority of the patients with detectible lisinopril had a high Ac‐SDKP level, in agreement with previous reports. 21 However, six patients demonstrated low Ac‐SDKP levels despite detectible lisinopril concentrations, suggesting that these patients may not have benefited from the anti‐fibrotic effects of ACEi (Figure 1). Six (16.6%) lisinopril patients had Ac‐SDKP levels lower than 4.5 nM, 5 (13.9%) had levels between 4.5 and 5 nM, and 25 (69.4%) had levels >5 nM. We arbitrarily chose the 5 nM level based on our previous experience to separate those who are respondent (>5 nM) and non‐respondent (<5 nM). Table 5 shows the comparisons of clinical and biochemical characteristics between the respondent and non‐respondent groups. There was no difference in age, BMI, or BP between the groups. No differences were observed in LVMI and PWV. CITP and TIMP‐1 levels were higher in patients with high Ac‐SDKP levels; however, the difference did not reach statistical significance due to the low number of cases.
FIGURE 1.
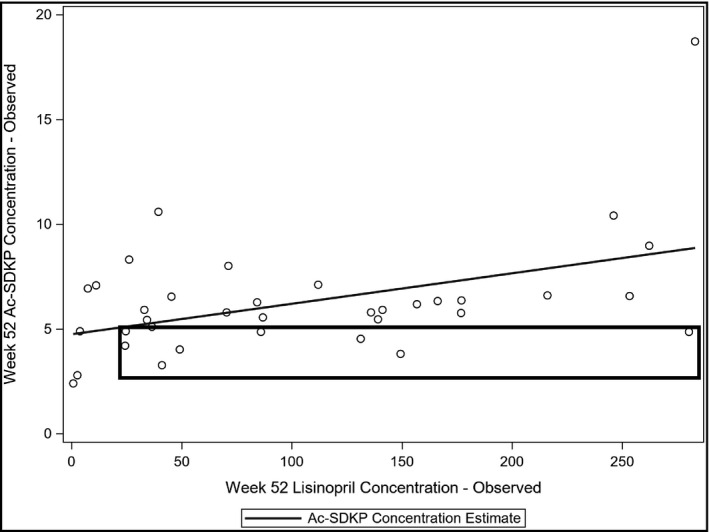
Scatter plot displaying the relationship between serum lisinopril and Ac‐SDKP. Ac‐SDKP has a moderate correlation (R 2 = .2) with lisinopril levels; however, some patients showed low Ac‐SDKP level despite the presence of lisinopril, suggesting variable Ac‐SDKP response (square area)
TABLE 5.
Clinical and biochemical characteristics based on N‐acetyl‐seryl‐aspartyl‐lysyl‐proline level in lisinopril‐treated patients
| Ac‐SDKP < 5nM | Ac‐SDKP > 5 nM | p‐value | |
|---|---|---|---|
| N | 11 | 25 | |
| Age (years) | 47.2 ± 2.8 | 49.3 ± 7.2 | .60 |
| Female (%) | 27.3 | 52.0 | .28 |
| BMI (kg/m2) | 34.6 ± 5.2 | 35.3 ± 7.8 | .77 |
| SBP basal (mmHg) | 131.0 ± 9.2 | 129 ± 15.7 | .26 |
| DBP basal (mmHg) | 84.4 ± 7 | 87.1 ± 9.8 | .35 |
| SBP at one year (mmHg) | ‐32.1 ± 25.7 | ‐37.5 ± 23.2 | .56 |
| DBP at one year (mmHg) | ‐17.4 ± 14.5 | ‐17.3 ± 11.82 | 1.00 |
| LVMI at one year (g/m2) | 83.3 ± 9.9 | 79.7 ± 13.8 | .38 |
| Pulse Wave Velocity (m/s) | 7.34 ± 2.9 | 6.19 ± 3.3 | .34 |
| PICP/CITP ratio | 52.6 ± 38 | 43.9 ± 47.2 | .31 |
| PICP (nM) | 75.9 ± 17 | 89.3 ± 31.6 | .25 |
| CITP (nM) | 2.5 ± 2.6 | 3.6 ± 3.1 | .07 |
| MMP1/TIMP1 ratio | 0.038 ± 0.032 | 0.038 ± 0.031 | 1.00 |
| MMP‐1 (nM) | 5.4 ± 4.6 | 5.5 ± 3.8 | .90 |
| TIMP‐1 (nM) | 139.9 ± 27.1 | 161.7 ± 35.9 | .06 |
| Log CITP/MMP‐1 ratio | 0.61 ± 4.76 | 0.76 ± 4.01 | .57 |
| Ac‐SDKP (nM) | 4.05 ± 0.89 | 7.27 ± 2.79 | <0.001 |
Abbreviations: Ac‐SDKP, N‐acetyl‐seryl‐aspartyl‐lysyl‐proline; CITP, telopeptide of collagen type I; MMP‐1, matrix metalloproteinase‐1; PICP, type 1 procollagen C‐terminal propeptide; TIMP‐1, tissue inhibitor of metalloproteinases 1.
4. DISCUSSION
In this study, we found that detectable lisinopril in the serum of hypertensive African American patients with LVH on CMR was associated with an increase in the anti‐fibrotic markers MMP‐1 and Ac‐SDKP. In all patients treated with antihypertensive medication, with or without ACEi, BP decreased and LVH improved after one year of treatment with goal SBP < 130 mmHg. However, in this post hoc analysis, patients with detectable lisinopril had greater decrease in BP and improvement in LVH despite being prescribed a similar number and intensity of antihypertensive medications. We also found that the expected increase in Ac‐SDKP levels was not observed in approximately one‐third of patients with detectable levels of lisinopril, suggesting that a differential anti‐fibrotic response to ACEi might exist. This finding has potential treatment implications.
MMP‐1, secreted by fibroblasts and cardiomyocytes, is a key enzyme that participates in collagen type I degradation and the release of CITP. 28 MMP‐1 catalytic activity is inhibited by TIMP‐1, which is also produced within the fibrotic heart tissue. Thus, the ratio of MMP‐1 to TIMP‐1 reflects net MMP‐1 activity. 9 , 29 Both MMP‐1 and TIMP‐1 are released into the systemic circulation, and the plasma concentration of these proteins has been routinely used as an index of matrix remodeling in various cardiac pathologies. 9 , 10 , 30 Previous studies have indicated that the anti‐fibrotic marker MMP‐1 decreases with hypertension and in hypertensive patients, while TIMP‐1 does not increase or display any changes. 29 This is particularly true for patients with LVH, which collectively suggests that in hypertension, there is a pro‐fibrotic state favoring collagen deposition in the heart. 27 The use of a RAAS antagonist has been shown to modulate MMPs and TIMP‐1 in vitro and in animal models. 14 In our study, SBP was associated with collagen deposition (PICP levels), in agreement with this concept. Laviades et al studied a small cohort of White, Spanish hypertensive patients before and after receiving lisinopril. In agreement with our results, they found an increase in MMP‐1 activity and no changes in total TIMP‐1 after treatment with lisinopril. 30 In our study, in addition to confirming those changes in African Americans receiving lisinopril, we demonstrated that these changes are drug class‐dependent by including a comparison group of patients who received other antihypertensive medications, but not ACEi, in whom MMP‐1 values remained low. In our study, we observed no difference in total TIMP‐1 levels after lisinopril treatment. Other studies have shown that TIMP‐1 does not change after the use of ACEi; however, there are conflicting reports. 30 , 31 The discrepancy may be explained by the measurement of total vs. free TIMP‐1. One study revealed that total TIMP‐1 did not change after ACEi use, while free TIMP‐1 was diminished. 30 As we will discuss later, the level of TIMP‐1 may be affected by other regulatory metabolites that can be increased or decreased depending on the pharmacogenomic aspects of the drugs, thus blunting changes in TIMP‐1.
We also found a significant increase in the anti‐fibrotic peptide Ac‐SDKP associated with ACEi treatment. This naturally occurring peptide is derived from thymosin β4 and is degraded by ACE. The use of ACEi increases Ac‐SDKP in animal models and in humans. 22 Increased Ac‐SDKP has demonstrated a reduction in collagen synthesis, 32 a decline in collagen deposition, 33 and decreased collagen crosslinking 34 in the heart, arteries, and kidney. In the past, the presence of Ac‐SDKP has been proposed as a marker of medication adherence in patients prescribed ACEi. 21 In our study, we measured the presence of lisinopril using LC–MS and observed that approximately one‐third of the patients with detectible lisinopril did not display an increase in Ac‐SDKP levels as we had expected. Thus, there may be variability in the response to ACEi specifically related to the pleiotropic effects of these drugs, since the degree of BP reduction is similar in those with high and low levels of Ac‐SDKP. Several ACE polymorphisms have been described, including insertion/deletion (I/D) polymorphisms. The exploration of the effects of the I/D polymorphism of the gene encoding angiotensin I converting enzyme on Ac‐SDKP is limited to a single study. 35 This study indicated that the DD polymorphism only affects the timing of Ac‐SDKP metabolism, with no effects on the basal Ac‐SDKP level even in patients who received ACEi along with an exogenous Ac‐SDKP infusion. However, other polymorphisms have not been explored. Interestingly, in patients with high Ac‐SDKP, we observed a decrease in CITP and an increase in TIMP‐1, albeit without statistical power to determine whether this was simply a fortuitous finding. The decrease in CITP is in line with the anti‐crosslinking effects of Ac‐SDKP, as previously described. 34 We previously reported that Ac‐SDKP increases TIMP‐1 in myocardial tissue, preventing cardiac rupture due to exaggerated anti‐fibrotic effects. 36 Currently, it is not known exactly how TIMP‐1 is regulated in the context of ACE inhibition, but we speculate TIMP‐1 may be playing a role in preventing exaggerated extracellular matrix degradation in some patients with an extensive anti‐fibrotic response. The variable response in TIMP‐1 according to Ac‐SDKP levels may explain why some authors report no change while others report an increase in TIMP‐1 under ACEi. 30 , 31 , 37 Pharmacogenomic and metabolomic profiles are an important aspect of precision medicine, a personalized therapeutic approach to attain the best results for an individual patient. The RAAS system, particularly ACE, produces a variety of BP responses, side effects, and pharmacokinetic properties related to ACE polymorphisms and differences in drug metabolism enzymes. 38 Thus, these variable responses may help to identify patients more susceptible to the anti‐fibrotic effects of ACE inhibition. However, more studies are needed to shed light on the mechanism behind these distinct responses.
Similar regression of structural heart damage by CMR after one year between the two groups confirm the importance of BP reduction over antihypertensive class. However, anti‐fibrotic and fibrotic biomarkers have been associated with histological myocardial changes in many studies and is expected to have more sensibility to detect pre‐clinical or incipient changes than CMR. 7 , 8 , 9 Other studies have shown this dissociation between biomarkers and CMR. 39
One important aspect of this study, other than the increase of the anti‐fibrotic markers, is that we did not observe a difference in collagen metabolism markers between the groups at the one‐year follow‐up, confirming that BP control is the most important factor for improving organ fibrosis. Other drug classes, such as ARBs and calcium channel blockers, also have anti‐fibrotic effects that may explain the similar collagen metabolite results. 40 , 41 Additionally, the variability of the PICP level and the relative low number of patients may contribute to explain the lack of statistical differences between the groups despite the BP differences. Longer follow‐up would potentially provide more robust data for determining any differences in the fibrosis rate, crosslinking state, or target organ damage.
Despite the similar number and intensity of medications prescribed, patients with detectible lisinopril serum concentrations had the largest decrease in BP than patients not prescribed lisinopril or with undetectable serum lisinopril concentration. The explanation for this finding is unclear, but may include medication adherence or other factors, such as diet and physical exercise. MMP‐1 and Ac‐SDKP are associated with the presence of lisinopril (adjusted for BP), indicating a direct effect of ACEi on these variables. Although this study was not designed to compare ARB and ACEi, based on our findings and the current guidelines, our study supports the use of ACEi in African American hypertensive patients in addition to diuretics or calcium antagonists, reserving ARB as an alternative for those who suffer from adverse reactions to ACEi.
There are some aspects of this study that are important to consider. This is a post hoc analysis of a clinical trial evaluating the effect of vitamin D supplementation in hypertension; thus, it was not originally conceived to evaluate the effect of ACEi on cardiac fibrosis and its biomarkers. However, the number of patients receiving vitamin D supplementation was similar in both groups, decreasing the chance of any vitamin D‐related effect. Additionally, any non‐significance difference in biomarkers can be underpowered. The number of patients, the follow‐up period, and the age of the participants made it difficult to establish hard end points or surrogates of end organ damage. Thus, the findings cannot be extrapolated to generate any conclusion on the benefits of ACEi in this population. Moreover, because we measured lisinopril serum concentration at two discrete time points, it is possible that corresponding values represent recent intake rather than long‐term medication adherence. However, we did see relationships between increasing lisinopril serum concentrations and changes in fibrosis, BP, and LV mass, suggesting that detectable ACEi by LC–MS reflected protracted medication use.
Our study contributes to the knowledge of heart damage associated with hypertension. The cutting‐edge technology used in our study to evaluate a very specific hypertensive population with LVH under intensive BP therapy provides unique and important insights to help further our understanding of the fibrotic process in hypertension. In addition, our study generates new hypotheses to explore the contribution of ACEi to the treatment of hypertension beyond their BP‐reducing effects.
We conclude that the use of lisinopril in hypertensive African Americans with LVH is associated with an increase in anti‐fibrotic markers and a reduction in LV mass after one year of intensive antihypertensive therapy.
CONFLICT OF INTEREST
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Cesar A. Romero, MD, PhD, Benjamin Wasinski, MS, Aaron Brody, MD, Candace D. McNaughton, MD, PhD, Rafael Fridman, MD, Oscar A. Carretero, MD, and Phillip D. Levy, MD, MPH involved in study conception and design, and data collection, interpretation of data, drafting of the manuscript and revising it critically for important intellectual content, and final approval of the manuscript submitted. Shobi Mathew, MS, Michael J. Twiner, MD, PhD, and John M. Flack, MD involved in study conception and design, and interpretation of data, drafting of the manuscript and revising it critically for important intellectual content, and final approval of the manuscript submitted. Brian Reed, BS involved in analysis and interpretation of data and final approval of the manuscript submitted. Rachelle Dawood, BS involved in data collection, interpretation of data, drafting of the manuscript and revising it critically for important intellectual content, and final approval of the manuscript submitted.
Supporting information
Table S1
ACKNOWLEDGMENTS
The authors thank all students and research technicians who were involved with the data collection and retention of participants enrolled in the study.
Romero CA, Mathew S, Wasinski B, et al. Angiotensin‐converting enzyme inhibitors increase anti‐fibrotic biomarkers in African Americans with left ventricular hypertrophy. J Clin Hypertens. 2021;23:1008–1016. 10.1111/jch.14206
NCT01360476 https://clinicaltrials.gov/ct2/show/NCT01360476
Funding information
This work was partially funded by the National Heart, Lung, and Blood Institute Grant 5‐P01‐HL‐028982 (O. Carretero) and K23LH125670 (C. McNaughton), by the Veterans Administration Tennessee Valley Healthcare System Geriatric Research Education Clinical Center and VA Office of Rural Health (ORH‐10808), and by 1R01MD005849‐01A1 (P. Levy).
Contributor Information
Cesar A. Romero, Email: CROMEROCBA@HOTMAIL.COM.
Phillip D. Levy, Email: plevy@med.wayne.edu.
REFERENCES
- 1. Collaborators GBDRF . Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990‐2017: A systematic analysis for the global burden of disease study 2017. Lancet. 2018;392:1923‐1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carnethon MR, Pu J, Howard G, et al. American Heart Association Council on E, Prevention, Council on Cardiovascular Disease in the Y, Council on C, Stroke N, Council on Clinical C, Council on Functional G, Translational B, Stroke C. Cardiovascular health in african americans: A scientific statement from the american heart association. Circulation. 2017;136:e393‐e423. [DOI] [PubMed] [Google Scholar]
- 3. Cortese F, Cecere A, Maria Cortese A, et al. Vascular, cardiac and renal target organ damage associated to arterial hypertension: Which noninvasive tools for detection? J Hum Hypertens. 2020;34(6):420‐431. [DOI] [PubMed] [Google Scholar]
- 4. Muiesan ML, Paini A, Aggiusti C, Bertacchini F, Rosei CA, Salvetti M. Hypertension and organ damage in women. High Blood Press Cardiovasc Prev. 2018;25:245‐252. [DOI] [PubMed] [Google Scholar]
- 5. Hinderer S, Schenke‐Layland K. Cardiac fibrosis ‐ a short review of causes and therapeutic strategies. Adv Drug Deliv Rev. 2019;146:77‐82. [DOI] [PubMed] [Google Scholar]
- 6. Gonzalez A, Lopez B, Ravassa S, San Jose G, Diez J. Reprint of "the complex dynamics of myocardial interstitial fibrosis in heart failure. Focus on collagen cross‐linking". Biochim Biophys Acta Mol Cell Res. 2020;1867:118521. [DOI] [PubMed] [Google Scholar]
- 7. Querejeta R, Lopez B, Gonzalez A, et al. Increased collagen type i synthesis in patients with heart failure of hypertensive origin: Relation to myocardial fibrosis. Circulation. 2004;110:1263‐1268. [DOI] [PubMed] [Google Scholar]
- 8. Ravassa S, Trippel T, Bach D, et al. Biomarker‐based phenotyping of myocardial fibrosis identifies patients with heart failure with preserved ejection fraction resistant to the beneficial effects of spironolactone: Results from the aldo‐dhf trial. Eur J Heart Fail. 2018;20:1290‐1299. [DOI] [PubMed] [Google Scholar]
- 9. Ravassa S, Ballesteros G, Lopez B, et al. Combination of circulating type i collagen‐related biomarkers is associated with atrial fibrillation. J Am Coll Cardiol. 2019;73:1398‐1410. [DOI] [PubMed] [Google Scholar]
- 10. Aguiar P, Azevedo O, Pinto R, et al. Biomarkers of myocardial fibrosis: Revealing the natural history of fibrogenesis in fabry disease cardiomyopathy. J Am Heart Assoc. 2018;7:e007124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo YS, Wu ZG, Yang JK, Chen XJ. Impact of losartan and angiotensin ii on the expression of matrix metalloproteinase‐9 and tissue inhibitor of metalloproteinase‐1 in rat vascular smooth muscle cells. Mol Med Rep. 2015;11:1587‐1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ren M, Hao S, Yang C, et al. Angiotensin ii regulates collagen metabolism through modulating tissue inhibitor of metalloproteinase‐1 in diabetic skin tissues. Diab Vasc Dis Res. 2013;10:426‐435. [DOI] [PubMed] [Google Scholar]
- 13. Sangaralingham SJ, Wang BH, Huang L, et al. Cardiorenal fibrosis and dysfunction in aging: Imbalance in mediators and regulators of collagen. Peptides. 2016;76:108‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Su YY, Li HM, Yan ZX, et al. Renin‐angiotensin system activation and imbalance of matrix metalloproteinase‐9/tissue inhibitor of matrix metalloproteinase‐1 in cold‐induced stroke. Life Sci. 2019;231:116563. [DOI] [PubMed] [Google Scholar]
- 15. Wakisaka Y, Chu Y, Miller JD, Rosenberg GA, Heistad DD. Spontaneous intracerebral hemorrhage during acute and chronic hypertension in mice. J Cereb Blood Flow Metab. 2010;30:56‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mihailovici AR, Deliu RC, Margaritescu C, et al. Collagen i and iii, mmp‐1 and timp‐1 immunoexpression in dilated cardiomyopathy. Rom J Morphol Embryol. 2017;58:777‐781. [PubMed] [Google Scholar]
- 17. Flack JM, Sica DA, Bakris G, et al. International Society on Hypertension in B. Management of high blood pressure in blacks: An update of the international society on hypertension in blacks consensus statement. Hypertension. 2010;56:780‐800. [DOI] [PubMed] [Google Scholar]
- 18. Whelton PK, Carey RM, Aronow WS, et al. 2017 acc/aha/aapa/abc/acpm/ags/apha/ash/aspc/nma/pcna guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: Executive summary: A report of the american college of cardiology/american heart association task force on clinical practice guidelines. Circulation. 2018;138:e426‐–e483. [DOI] [PubMed] [Google Scholar]
- 19. Blood Pressure Lowering Treatment Trialists C , Turnbull F, Neal B, et al. Blood pressure‐dependent and independent effects of agents that inhibit the renin‐angiotensin system. J Hypertens. 2007;25:951‐958. [DOI] [PubMed] [Google Scholar]
- 20. Rasoul S, Carretero OA, Peng H, et al. Antifibrotic effect of ac‐sdkp and angiotensin‐converting enzyme inhibition in hypertension. J Hypertens. 2004;22:593‐–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Azizi M, Ezan E, Nicolet L, Grognet JM, Menard J. High plasma level of n‐acetyl‐seryl‐aspartyl‐lysyl‐proline: A new marker of chronic angiotensin‐converting enzyme inhibition. Hypertension. 1997;30:1015‐1019. [DOI] [PubMed] [Google Scholar]
- 22. Peng H, Carretero OA, Vuljaj N, et al. Angiotensin‐converting enzyme inhibitors: A new mechanism of action. Circulation. 2005;112:2436‐2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Romero CA, Orias M, Weir MR. Novel raas agonists and antagonists: Clinical applications and controversies. Nat Rev Endocrinol. 2015;11:242‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamazaki T, Komuro I, Zou Y, et al. Efficient inhibition of the development of cardiac remodeling by a long‐acting calcium antagonist amlodipine. Hypertension. 1998;31:32‐38. [DOI] [PubMed] [Google Scholar]
- 25. Levy PD, Willock RJ, Burla M, et al. Total antihypertensive therapeutic intensity score and its relationship to blood pressure reduction. J Am Soc Hypertens. 2016;10:906‐916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Beckett L, Godwin M. The bptru automatic blood pressure monitor compared to 24 hour ambulatory blood pressure monitoring in the assessment of blood pressure in patients with hypertension. BMC Cardiovasc Disord. 2005;5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dias E, Hachey B, McNaughton C, et al. An lc‐ms assay for the screening of cardiovascular medications in human samples. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;937:44‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chalikias GK, Tziakas DN. Biomarkers of the extracellular matrix and of collagen fragments. Clin Chim Acta. 2015;443:39‐47. [DOI] [PubMed] [Google Scholar]
- 29. Lopez B, Gonzalez A, Diez J. Role of matrix metalloproteinases in hypertension‐associated cardiac fibrosis. Curr Opin Nephrol Hypertens. 2004;13:197‐204. [DOI] [PubMed] [Google Scholar]
- 30. Laviades C, Varo N, Fernandez J, et al. Abnormalities of the extracellular degradation of collagen type i in essential hypertension. Circulation. 1998;98:535‐540. [DOI] [PubMed] [Google Scholar]
- 31. Onal IK, Altun B, Onal ED, Kirkpantur A, Gul Oz S, Turgan C. Serum levels of mmp‐9 and timp‐1 in primary hypertension and effect of antihypertensive treatment. Eur J Intern Med. 2009;20:369‐372. [DOI] [PubMed] [Google Scholar]
- 32. Rhaleb NE, Peng H, Harding P, Tayeh M, LaPointe MC, Carretero OA. Effect of n‐acetyl‐seryl‐aspartyl‐lysyl‐proline on DNA and collagen synthesis in rat cardiac fibroblasts. Hypertension. 2001;37:827‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang F, Yang XP, Liu YH, et al. Ac‐sdkp reverses inflammation and fibrosis in rats with heart failure after myocardial infarction. Hypertension. 2004;43:229‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gonzalez GE, Rhaleb NE, Nakagawa P, et al. N‐acetyl‐seryl‐aspartyl‐lysyl‐proline reduces cardiac collagen cross‐linking and inflammation in angiotensin ii‐induced hypertensive rats. Clin Sci (Lond). 2014;126:85‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Azizi M, Junot C, Ezan E, Hallouin MC, Guyene T, Menard J. Functional consequences of angiotensin‐converting enzyme gene polymorphism on n‐acetyl‐ser‐asp‐lys‐pro degradation and angiotensin ii production. J Mol Med (Berl). 2002;80:492‐498. [DOI] [PubMed] [Google Scholar]
- 36. Nakagawa P, Romero CA, Jiang X, et al. Ac‐sdkp decreases mortality and cardiac rupture after acute myocardial infarction. PLoS One. 2018;13:e0190300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li‐Saw‐Hee FL, Edmunds E, Blann AD, Beevers DG, Lip GY. Matrix metalloproteinase‐9 and tissue inhibitor metalloproteinase‐1 levels in essential hypertension. Relationship to left ventricular mass and anti‐hypertensive therapy. Int J Cardiol. 2000;75:43‐47. [DOI] [PubMed] [Google Scholar]
- 38. Flaten HK, Monte AA. The pharmacogenomic and metabolomic predictors of ace inhibitor and angiotensin ii receptor blocker effectiveness and safety. Cardiovasc Drugs Ther. 2017;31:471‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pichler G, Redon J, Martinez F, et al. Cardiac magnetic resonance‐derived fibrosis, strain and molecular biomarkers of fibrosis in hypertensive heart disease. J Hypertens. 2020;38(10):2036‐2042. [DOI] [PubMed] [Google Scholar]
- 40. Ishimitsu T, Kobayashi T, Honda T, et al. Protective effects of an angiotensin ii receptor blocker and a long‐acting calcium channel blocker against cardiovascular organ injuries in hypertensive patients. Hypertens Res. 2005;28:351‐359. [DOI] [PubMed] [Google Scholar]
- 41. Sevilla MA, Voces F, Carron R, et al. Amlodipine decreases fibrosis and cardiac hypertrophy in spontaneously hypertensive rats: Persistent effects after withdrawal. Life Sci. 2004;75:881‐891. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1