

SUMMARY
Single-particle cryo-EM, whose full power was not realized until the advent of powerful detectors in 2012, has a unique position as a method of structure determination as it is capable of providing information not only on the structure but also on dynamical features of biomolecules. This information is of special importance in understanding virus-host interaction and explains the crucial role of cryo-EM in the efforts to find vaccinations and cures for pandemics the world has experienced in the past decade.
Graphical Abstract
Compared to the time of the viral pandemic that ravaged our planet a hundred years ago, we are now in a much better position to fight diseases – microbial or viral -- due to our detailed understanding of molecular biology and molecular genetics, and the underlying structural basis. We have learned that knowledge of molecular interactions on the atomic level is of key importance for understanding life processes in the cell and the way they are disrupted by pathogens. For this reason, structural biology employing the three major biophysical techniques of structure determination, X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy and cryogenic electron microscopy (cryo-EM) has advanced in the past 60 years to a place at the forefront of biomedical research. Development of vaccines, broad-band prophylaxis and effective drug therapeutics either rely on the accumulated knowledge base, or at least greatly benefit from it.
Single-particle cryo-EM, or the structure determination by electron microscopy from biological molecules in solution, accounts for an increasing share of biomedical structure research. Its importance as a method of biophysical visualization, next to X-ray crystallography and NMR, has been recognized by the 2017 Nobel Prize in Chemistry. It has opened up structure determination, at the atomic level, for molecules that cannot be crystallized, and is capable of providing multiple structures related to the molecule’s native states in solution, often proxies to their functional states. Particularly in the context of the research on virus-host interactions the capability of providing information on the conformational dynamics gives cryo-EM an important advantage over the other techniques. Cryo-EM data processing as it is implemented now on a number of software platforms is able to cope with conformational heterogeneity of a sample as it gives us a variety of structures sampling the entire range of states, not just one. Moreover, as pointed out before (Frank, 2018), it is even possible now to characterize the entire continuous range of conformations from a large dataset of single-particle snapshots of freely equilibrating molecules (see Dashti et al., 2020 for a recent application of this technique to the Ryanodine receptor). Here I wish to single out the role cryo-EM has played in fighting the pandemics that have sprung up in the last decade, including, and foremost in world-wide impact, COVID-19.
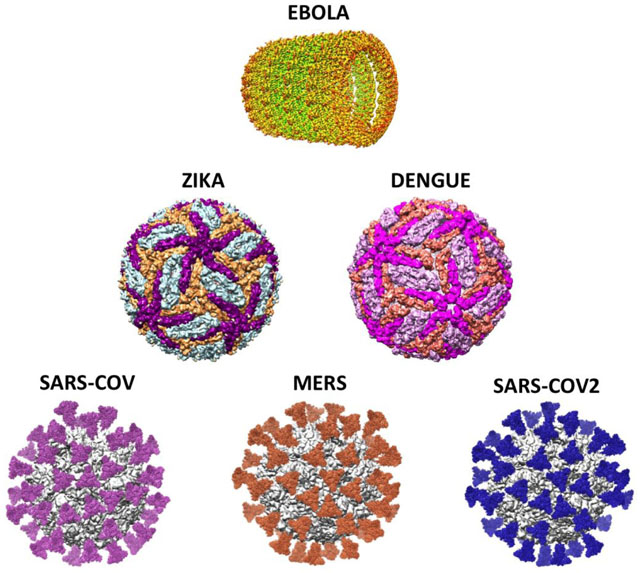
Even though its main tenets and methodology reach back more than four decades, the full power of the method could not be realized until 2012 when the new single-electron detecting cameras became commercially available. Looking at the timeline in the breakout of global diseases since that year, it is quite remarkable that the period after the “resolution revolution” coincides with the emergence of a number of menacing virus-caused pandemics: MERS (2012–2015), Ebola (2014–2016), Zika (2015–2016), Dengue (2019–2020) and now, for an uncertain period of time, COVID-19 caused by SARS-Cov2. Although it preceded this time period, SARS-Cov (2002–2004) should also be mentioned here because of its close relationship to SARS-Cov2 and MERS. In hindsight one could say the new capability became available ‘just in time’ to help us cope with an increasing number of public health crises worldwide. Here with ‘coping’ I mean the development of vaccines to prevent infection as well as effective cures, based on the knowledge gained by the determination of the structures of viruses, their components and their metabolic intermediates expressed in the host cell. In the following I will give representative examples for cryo-EM reconstructions that have played a role in the elucidation of the structures of viruses causing these pandemics. They can be combined in three groups: (1) Zika/Dengue, (2) Ebola (on its own), and (3) MERS/SARS-Cov/SARS-Cov2.
Dengue and Zika viruses are closely related flaviviruses transmitted by mosquitoes. A high-resolution (3.5 Å) structure of the Dengue virus type 2 was obtained by Zhang et al. (2013) in Hong Zhou’s lab at UCLA, several years before the major outbreak and actually without the help of the new cameras. This milestone study explained the role of the dynamic interplay of the virus’ E and M latch proteins with the host’s cell membrane in the infection process. The rapid determination of the high-resolution (3.8 Å) cryo-EM structure of the Zika virus (Sirohi et al., 2016) was one of the crowning achievements of the late Michael Rossmann at Purdue University. The large extent of structural similarity between the two viruses shown in the cryo-EM reconstructions of the UCLA and Purdue groups explained the observed cross-immunity conferred by infection with either virus.
To date the Ebola virus, which belongs along with the Marburg virus to the Filoviridae family, is still without known cure despite of efforts characterizing its high-resolution structure (e.g., Sugita et al., 2016). It probably has multiple paths of host entry, defying efforts to develop means to target a single one for intervention (Hoenen et al., 2019). Research has focused on structure and different modes of neutralization by antibodies. A variety of cryo-EM structures depict the binding of antibodies isolated from surviving patients (Misasi et al., 2016; Cohen-Dvashi et al., 2020).
All viruses of the third group of viruses, of the corona type, SARS Cov, MERS and SARS Cov2 are important in their relevance to the current pandemic. The reason is that structural knowledge of the closely related SARS-Cov-1 and MERS viruses preceded the advent of SARS-Cov2, and thus could be immediately exploited at the onset of the pandemic. For these three viruses, understanding the first step of infection requires a detailed knowledge of the dynamics of the molecular interaction between virus spike protein and the ACE2 receptor of the host.
The fast-paced development of vaccines for COVID-19 has put the spotlight on a novel vaccination strategy informed by this detailed knowledge. The established way of developing vaccines, which has been used for some two hundred years, goes back to the English physician Edward Jenner (1749 – 1823) and the French microbiologist Louis Pasteur (1822 – 1895). It works by weakening or inactivating the entire virus, and injecting this weakened form into the body. In that well-proven old-style approach to vaccination, antibodies are developed by the body’s immune system for a whole range of epitopes presented on the virus surface. But the recent development of the majority of vaccines for SARS-Cov2 has taken an entirely different route: through engineering a small antigenic part of the virus in the human body itself, via injection of mRNA coding for it and using the body’s own protein synthesis machinery. This de novo synthesis of the antigen from sequence information leaves opportunities open for the design of genetic modifications to promote and strengthen the immune response leading to the binding of neutralizing antibodies. The development of mRNA-based vaccines, which in all likelihood will dominate the responses to future viral outbreaks, is one of the technological breakthroughs accelerated by the urgent need to fight the COVID-19 pandemic.
Remarkably, the underlying principle of structure-based vaccine antigen design was described in a review by Graham et al. (2019) that appeared just before the onset of the COVID-19 pandemic, and can now be seen as the unwitting recipe for the massive mostly mRNA-based vaccination undertaking worldwide. That review and the original literature it cites make us understand the importance of acquiring knowledge about the dynamic features of the antigen, not just about its structure. The reason is, as cryo-EM showed for the closely related SARS Cov (Gui et al., 2017), that the spike protein is intrinsically variable, with domains in a variety of different positions, presenting a variety of different topologies to the immune system. Efficient production of immunity requires that the spike protein be stabilized in a conformation most likely encountered in the actual contact with the receptor of the host: the prefusion conformation.
We are quite fortunate (Kearns, 2020) that, because of the high similarity between SARS-Cov, MERS and SARS-Cov2 in structure and receptor binding modes, the native behavior of the spike protein and its generically engineered variants could already be characterized by cryo-EM several years before the COVID-19 outbreak (Gui et al., 2017; Pallesen et al., 2017; Yuan et al., 2017).
It is through the power of cryo-EM that we now have detailed knowledge about the structure as well as dynamical features of the spike glycoprotein alone (Wrapp et al., 2020), or bound with the ACE2 receptor (Lan et al., 2020; Yan et al., 2021), or bound with antibodies (Liu et al., 2020). In addition we have this information for different natural variants and for genetically engineered variants in stabilized prefusion forms. The intensity and scale of these focused research efforts are reflected in the March 31, 2021 release by the EM data bank (EMDB) of 136 reconstructions of the spike protein, including 36 atomic models.
Less specific than vaccinations but more broadly effective as prophylactic for a whole range of viruses is another promising strategy, the synthesis of flexible receptors designed as decoys to neutralize the attacking part of the virus. This strategy and its potential have been highlighted in a recent article in Biochemistry (Bravo et al., 2020). Virus envelope glycans that normally bind glycan-binding proteins (GBPs) of the host are intercepted by small synthetic carbohydrate receptors. Such broadband drugs may be an effective treatment option for diseases caused by several glycosylated enveloped viruses, including HIV, Zika, Dengue, influenza, MERS and SARS-CoV2. It is likely that the development of this technology to clinical trial will also benefit from the application of single-particle cryo-EM.
In conclusion, thanks to the development of cryo-EM as a tool of structural biology capable of capturing structure and dynamic features of molecules in their fully hydrated states, which a decade ago gained its full potential, we are now in a much better position, equipped to combat the current and future outbreaks of diseases that are life-threatening and jeopardize the very fabric of our civilization.
Acknowledgments
I would like to thank Larry Shapiro and Micah Rapp for helpful comments. This study was supported by NIGMS MIRA grant 1R35 GM139453 to J.F.
References
- Bravo MF, Lema MA, Mariaski M and Braunschweig AB (2020). Flexible synthetic carbohydrate receptors as inhibitors of viral attachment. Biochemistry 10.1021/acs.biochem.0c00732. [DOI] [PubMed] [Google Scholar]
- Cohen-Dvashi H, Zehner M, Ehrhardt S, Elad N, Klein F, Diskin R (2020). Structural basis for a convergent immune response against Ebola Virus. Cell Host and Microbe 27, 418–427. [DOI] [PubMed] [Google Scholar]
- Dashti A, Mashayekhi G, Shekhar M, Ben Hail D, Salah S, Schwander P, des Georges A, Singharoy A, Frank J and Ourmazd A (2020). Retrieving functional pathways of biomolecules from single-particle snapshots. Nat. Commun 11, 4734 (2020). 10.1038/s41467-020-18403-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J (2018). New opportunities created by single-particle cryo-EM: The mapping of conformational space. Biochemistry 57, 888–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham BS, Gilman MSA and McLellan JS (2019). Structure-based vaccine antigen design. Ann. Rev. Med 70, 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui M, Song W, Zhou H, Xu J, Chen S, Xiang Y, and Wang X (2017). Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res 27, 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenen T, Groseth A, and Feldmann H (2019). Therapeutic strategies to target the Ebola virus life cycle. Nat Rev Microbiol 17, 593–606. [DOI] [PubMed] [Google Scholar]
- Kearns S (2020) Structure of the pandemic. Structure 28, 874–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q Shi X, Wang Q, Zhang L and Wang X (2020). Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220. [DOI] [PubMed] [Google Scholar]
- Liu L, Wang P, Nair MS, Yu J, Rapp M, Wang Q, Luo Y, Chan JF-W, Sahi V, Figueroa A, Guo XV, Cerutti G, Bimela J, Gorman J, Zhou T, Chen Z, Yuen K-Y, Kwong PD, Sodroski JG, Yin MT, Sheng Z, Huang Y, Shapiro L and Ho DD (2020). Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 584, 450–456. [DOI] [PubMed] [Google Scholar]
- Misasi J, Gilman MSA, Kanekiyo M, Gui M, Cagigi A, Mulangu S, Corti D, Ledgerwood JE, Lanzavecchia A, Cunningham J, Muyembe-Tamfun JJ, Baxa U, Graham BS, Xiang Y, Sullivan NJ, and McLellan JS (2016). Structural and molecular basis for Ebola virus neutralization by protective human antibodies. Science 351, 1343–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallesen J, Wang N, Corbett KS, Wrapp D, Kirchdoerfer RD, Turner HL, Cottrell CA, Becker MM, Wang L, Shi W, Kong W-P, Andres EL, Kettenbach AN, Denison MR, Chappell JD, Graham BS, Ward AB, McLellan JS (2017). Proc. Natl. Acad. Sci. USA 114 (35) E7348–E7357; DOI: 10.1073/pnas.1707304114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG, and Kuhn RJ (2016). The 3.8 Å resolution cryo-EM structure of Zika virus. Science 352, 467–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita Y, Matsunami H, Kawaoka Y, Noda T, and Wolf M (2018). Cryo-EM structure of the Ebola virus nucleoprotein–RNA complex at 3.6 Å resolution. Nature 563, 137–140. [DOI] [PubMed] [Google Scholar]
- Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, Graham BS, McLellan JS (2020). Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R, Zhang Y, Li Y, Ye F, Guo Y, Xia L, Zhong X, Chi X, and Zhou Q (2021). Structural basis for the different states of the spike protein of SARS-CoV-2 in complex with ACE2. Cell Res. 10.1038/s41422-021-00490-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Cao D, Zhang Y, Ma J, Qi J, Wang Q, Lu G,Ying Wu Y, Yan J, Shi Y, Zhang X, and Gao GF (2017). Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun 8, 15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ge P, Yu X, Brannan JM, Bi G, Zhang Q, Schein S and Zhou ZH (2013). CryoEM structure of the mature dengue virus at 3.5-Å resolution. Nat. Struct. Mol. Biol 20, 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]