

Abstract
We report the first crystal structure of the mammalian non-heme iron enzyme cysteamine dioxygenase (ADO) at 1.9 Å resolution, which shows an Fe- and 3-histidine (3-His) active site situated at the end of a wide substrate access channel. The open approach to the active site is consistent with the recent discovery that ADO catalyzes not only the conversion of cysteamine to hypotaurine, but also the oxidation of N-terminal cysteine (Nt-Cys) peptides to their corresponding sulfinic acids as part of the eukaryotic N-degron pathway. Whole-protein models of ADO in complex with either cysteamine or an Nt-Cys peptide, generated using molecular dynamics and quantum mechanics/molecular mechanics calculations, suggest occlusion of access to the active site by peptide substrate binding. This finding highlights the importance of a small tunnel that leads from the opposite face of the enzyme into the active site, providing a path through which co-substrate O2 could access the Fe center. Intriguingly, the entrance to this tunnel is guarded by two Cys residues that may form a disulfide bond to regulate O2 delivery in response to changes in the intracellular redox potential. Notably, the Cys and tyrosine (Tyr) residues shown to be capable of forming a cross-link in human ADO reside ~7 Å from the iron center. As such, cross-link formation may not be structurally or functionally significant in ADO.
Keywords: cysteamine dioxygenase, thiol dioxygenase, O2 activation, non-heme iron enzyme
Graphical Abstract
Introduction
The metabolism of thiol-containing compounds is vital to mammalian homeostasis. Regulation of hypotaurine, taurine, and cysteine levels preserves cardiac and vascular functions and protects neural cells from excitotoxicity. Additionally, elevated cysteine levels have been associated with Alzheimer’s and Parkinson’s diseases.1 To regulate intracellular levels of these thiolate-containing metabolites, mammals use two distinct thiol dioxygenases (TDOs); i.e., cysteine dioxygenase (CDO) and cysteamine dioxygenase (ADO).2,3 The definitively established function of CDO is to catalyze the oxidation of cysteine (Cys) to cysteine sulfinic acid (CSA) via the incorporation of both oxygen atoms from molecular oxygen.4 CSA can then be catabolized into pyruvate, sulfate, and hypotaurine.5 Under normal conditions, rats or mice convert about 70–90% of CSA to hypotaurine and taurine.6 Hypotaurine has been identified as a contributor to the growth and progression of aggressive high-grade gliomas in the brain.7 Additionally, taurine has been found to serve as an osmoregulator in the heart,8 though this compound also plays a role in several essential body functions, such as regulating calcium levels, creating bile cells, balancing electrolytes in the body, and supporting the development of the nervous system.9
ADO was once believed to exclusively convert cysteamine (2-aminoethanethiol, 2-AET) to hypotaurine (Scheme 1A). However, Ratcliffe et al. proposed that the native function of ADO may instead be to catalyze the O2-dependent conversion of peptides featuring an amino-terminal cysteine (Nt-Cys, Scheme 1B) to their CSA derivatives.10 They found that mammalian ADO could replace one of the five plant thiol dioxygenases in Arabidopsis thaliana (At), termed plant cysteine oxidase 4 (PCO4), by oxidizing the plant Nt-Cys peptides both in vitro and in vivo. Oxidation of the Nt-Cys peptide substrates by PCO promotes arginylation and degradation in the N-degron pathway,11 while under anoxic conditions, the peptide persists and enhances Gα catalyzed GTP hydrolysis, leading to an attenuation of G-protein coupled signaling. Newly identified ADO substrates, regulators of G protein signaling RGS4 and RGS5, are part of the mammalian N-degron pathway and serve as negative regulators of cardiovascular function controlled by G-protein signaling.12,13
Scheme 1.
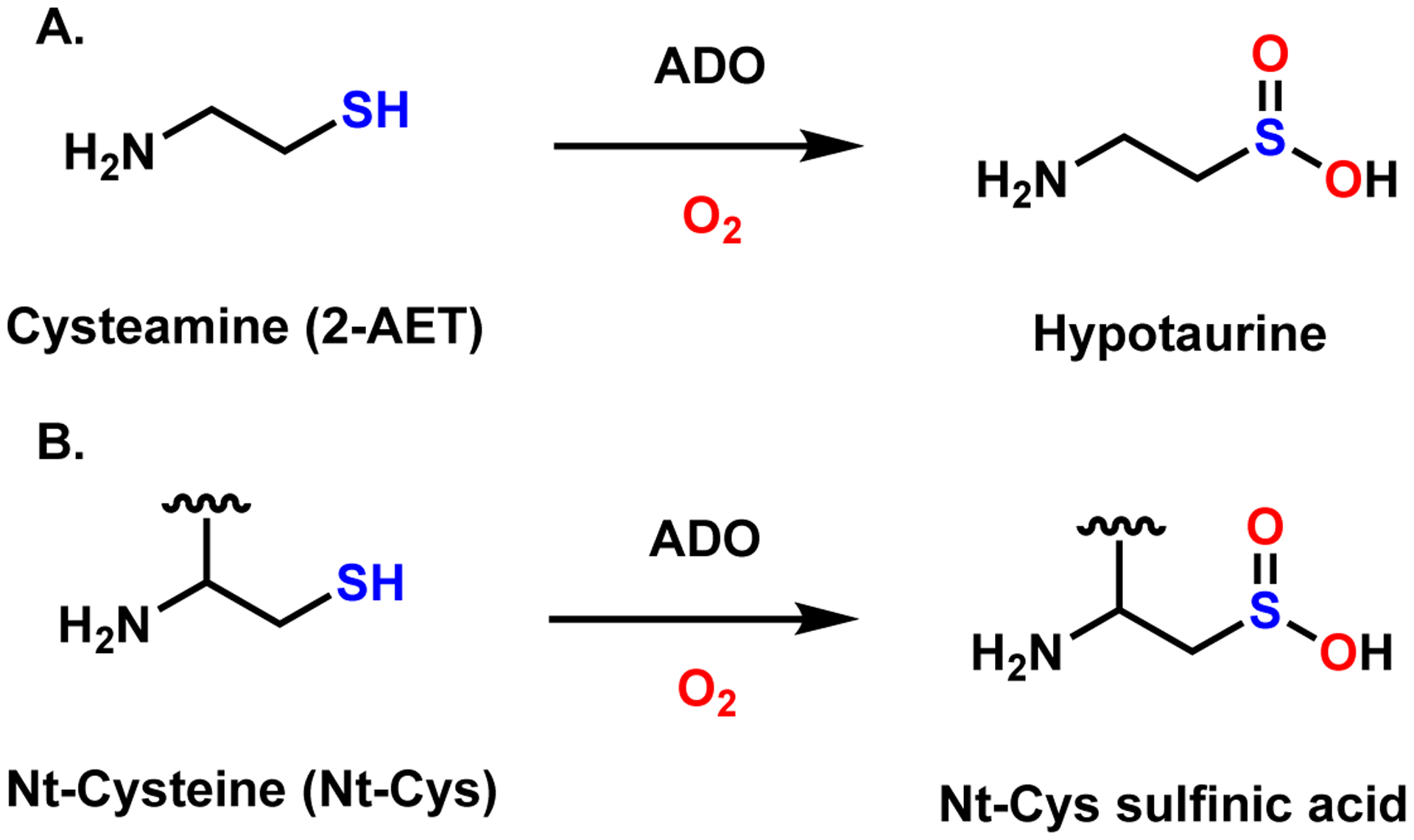
Reactions catalyzed by ADO.
Five classes of TDOs have been identified to date with differing native functions: namely, CDO, ADO, PCO, 3-mercaptopropionate dioxygenase (MDO), and mercaptosuccinate dioxygenase (MSDO), with CDO and ADO being the only mammalian TDOs. All TDOs belong to the cupin superfamily, which is typified by a common architecture.14 While cupin proteins tend to have low overall sequence identity, all feature a β-barrel fold and two conserved sequence motifs: G(X)5HXH(X)6G and G(X)5PXG(X)2H(X)3N. In addition to its classification as a cupin protein, ADO belongs to the PFam family PF07847 (PCO_ADO), which is distinct from the PF05995 (CDO_I) family that includes CDO and MDO. As is typical for members of the cupin superfamily, Mus musculus ADO (MmADO) shares little overall sequence identity with either Rattus norvegicus CDO (~14%, RnCDO) or PCO4 (~21%). Nevertheless, all TDOs that have previously been characterized by X-ray crystallography (CDO, MDO, and PCO) feature the same non-heme iron coordination environment, consisting of a relatively rare 3-histidine (3-His) facial triad. The impact on enzyme activity of removal of one of the coordinating His via site-directed mutagenesis and spectroscopic studies provided compelling evidence that ADO also contains the 3-His binding motif but differs with respect to secondary sphere residues that determine its substrate specificity.3,15
An alignment of the amino acid sequences of MmADO and RnCDO highlights crucial differences in the active site pocket.3 Perhaps most noticeable, the Cys93 involved in forming the Cys-Tyr cross-link of mammalian CDOs is not conserved in ADO. In RnCDO, this unusual thioether linkage increases activity by properly positioning the Cys and O2 substrates while also suppressing the coordination of a water molecule that competes with O2 binding.16,17 A Cys-Tyr cross-link motif was identified in human ADO via genetic incorporation of an unnatural amino acid, 3,5-difluoro-tyrosine, in conjunction with mass spectrometry and NMR experiments.18 While the cross-link in RnCDO forms between Cys93 and Tyr157, in human ADO the residues involved, Cys206 and Tyr208 (MmADO numbering), are separated by only one amino acid. Based on sequence alignment, the ADO cross-link motif is plausible in PCOs; however, neither X-ray crystallography nor tandem MS/MS analyses provided evidence for the formation of a Cys-Tyr thioether bond in PCO4.19 Consequently, the extent of cross-link formation in MmADO and its physiological role remain uncertain.
The lack of an X-ray crystal structure of mammalian ADO and the low overall sequence identity between ADO and other TDOs have rendered comparisons with other TDOs uncertain. While the presence of a 3-His facial triad coordinating the Fe cofactor was confirmed on the basis of spectroscopic and site-directed mutagenesis experiments, little has previously been established about ADO beyond its first coordination sphere. Thus, a structural characterization of ADO was pivotal to identifying key residues involved in substrate binding and other salient features of the protein. Here, we describe the 1.9 Å X-ray crystal structure of MmADO. As proposed on the basis of previous studies, ADO features a 3-His facial triad that coordinates the iron center. Both the X-ray crystal structure of ADO and a sequence alignment of various TDOs reveal that the structure of ADO is more similar to that of PCO than CDO. As reported for PCO,19 cross-link formation between Cys206 and Tyr208 was not observed in the structure of MmADO. Interestingly, a pair of Cys residues is identified that could form a disulfide bond, thus blocking a secondary access tunnel and controlling catalytic activity via regulating delivery of the co-substrate O2. Finally, molecular dynamics in combination with quantum mechanics/molecular mechanics calculations, validated on the basis of the X-ray crystal structure of resting ADO, were used to generate models of 2-AET and Nt-Cys bound ADO.
Materials and Methods
Preparation of Recombinant MmADO.
ADO was expressed as previously described15 with minor additions to the purification protocol. Briefly, Escherichia coli Rosetta 2(DE3) cells were transformed with a pET SUMO expression vector containing a codon-optimized Mus musculus ADO gene and protein expression was induced with the addition of isopropyl-β-D- thiogalactopyranoside to a final concentration of 0.2 mM along with ferrous ammonium sulfate to a final concentration of 100 μM. Cells were harvested 4 hrs post-induction and flash frozen.
Soluble protein was purified via immobilized metal affinity chromatography (IMAC) and size exclusion chromatography (SEC) as previously described with additional steps.15 All buffers included 5 mM tris(2-carboxyethyl)phosphine (TCEP) as a reducing agent and were degassed and bubbled with argon gas to minimize oxygen presence in buffer. Post elution from the SEC column, the ADO/SUMO fusion protein was dialyzed via centrifugation into 200 mM Tris-HCl, 150 mM NaCl, 5 mM TCEP, 5 mM imidazole, pH 8.0 IMAC buffer. Thin-layer chromatography was used to ascertain that the purified ADO protein was able to catalyze the conversion of cysteamine to hypotaurine (work is underway to develop a quantitative activity assay). The protein was then incubated overnight at room temperature with SUMO protease (ThermoFisher). ADO was separated from the His6SUMO tag via reverse IMAC. Protein aliquots to be used for crystallization were flash frozen under a stream of argon gas and stored at −80 °C.
Crystallization and structure determination.
Purified MmADO was screened for crystallization response using commercial screens, MRC SD2 microplates, and a Mosquito crystallization robot. Initial diffraction studies indicated almost no crystalline diffraction, even though the crystals seemed well-formed by light microscopy. Progressive exclusion of oxygen during protein purification and crystallization environment yielded progressively better diffraction. The exceptional crystal yielding the refined diffraction data was grown by hanging drop vapor diffusion at 293 K in a Coy anaerobic chamber. The reservoir solution was allowed to degas overnight in the anaerobic chamber, and consisted of 15% PEG 3350, 300 mM MgCl2, and 0.1 M Tris-HCl pH 8.0. The plate was withdrawn from the chamber, and the crystal was quickly cryoprotected against reservoir solution supplemented with 20% ethylene glycol and cooled by direct immersion in liquid nitrogen.
Diffraction data were collected at sectors 21 and 23 of the Advanced Photon Source, Argonne National Laboratory. The refinement data set was collected on an Eiger 9M detector at Life Sciences Collaborative Access Team beamline 21ID-D. A full 360 degree sweep of data was collected at 1.127 Å, 155 mm sample to detector distance, 0.2 degrees/frame, and 0.04 s exposure time. Data were reduced using XDS20 and autoPROC.21 Crystallographic structure solution and refinement were conducted within the Phenix suite of programs.22 The structure was solved by molecular replacement as implemented in Phaser,23 using an edited homology model from SWISS-MODEL24 derived from a PDB:6SBP template, covering residues 10–121 and 153–210 of MmADO. Molecular replacement initially produced the location and orientation of three copies of MmADO. The location and orientation of the fourth and final copy was uncovered by molecular replacement using a partially refined model based on the first three positions discovered. The structure was iteratively rebuilt in Coot25 and refined using Phenix.refine.26 Figures for all protein structures were created using PyMOL.27
Molecular Dynamics (MD) Simulations.
MD simulations were performed using the GROMACS (versions 5.1.4 and 2019.6) software package28 with the AMBER ff19SB force field.29 Initial coordinates were taken from chain D of the crystal structure reported here, keeping all crystallographic waters within 6 Å of that chain. We chose chain D for our computational studies because this is the most complete chain in the structure. Very similar results were obtained using the second most complete chain in the structure, chain B. Protonation was performed using phenix.reduce and manually adjusted, as necessary. A missing segment spanning Thr26 to Glu29 was constructed manually in PyMOL. MD parameters were generated in AmberTools,30 using the metal center parameter builder (MCPB.py) modelling tool for Fe(II) and its ligands (His100, His102, His179, Fe-bound waters, and later the Fe-bound substrates).31 The system was solvated with the SPC/E water model,32 and its total charge was neutralized by the addition of Na+ ions. Then, an energy minimization step was performed, followed by two equilibration steps: a 100 ps run under the NVT (isothermal-isochoric) ensemble and a 100 ps run under the NPT (isothermal-isobaric) ensemble. For each model, structures from the MD trajectory were clustered based on the root-mean-square deviation (RMSD) of the protein backbone. The average structures of the five most populated clusters were chosen for QM/MM optimization.
Modifications to this workflow were required for some models. In the case of the disulfide model, a bond between Cys120 and Cys169 was added to the crystal structure in silico. This system required an additional 10 ns added to its MD simulation, as the RMSD of the protein backbone was found to still be rising substantially at the end of the first 10 ns. For the substrate-bound models, starting coordinates were taken from the QM/MM-optimized ADO model, with the substrate placed into the active site using the docking software AutoDock Vina.33
Quantum Mechanics/Molecular Mechanics (QM/MM) Calculations.
Geometry optimizations were performed using the quantum mechanics/molecular mechanics (QM/MM) approach as implemented in the ONIOM method of Gaussian 16.34 The QM region was defined as the Fe ion, the side chains of its three histidine ligands (His100, His102, His179), Fe-bound water molecules, and 2-AET or the terminal cysteine of the peptide substrate if applicable. The density functional theory (DFT) portion of the calculations was performed with the unrestricted Becke, 3-parameter, Lee-Yang-Parr (UB3LYP) functional,35,36 using the triple-ζ valence plus polarization (TZVP) basis set37 for Fe and its coordinating atoms, and the 6–31G basis set for all other atoms.38 For all atoms outside of the QM region, the MM calculation was performed with the AMBER force field.39
Results
Crystallization and structure determination.
The protein was produced as an ADO/SUMO fusion and crystallized under anaerobic conditions upon removal of the His6SUMO solubility tag. The structure of ADO was solved to a nominal 1.89 Å resolution (crystallographic statistics, Supporting Information Table S1). The crystallographic asymmetric unit comprises four independent copies of the protein. In all four independent monomers, a short loop of 4–8 residues between Phe21 and Pro31 was too disordered to model. In two subunits, a second 4–6 residue loop between Pro217 and Ala224 was also disordered. Results from a SuperPose analysis for the four different ADO monomers in the unit cell are presented in Table S2, and an overlay of the four independent monomers is shown in the Supporting Information Fig. S1. Coordinates and structure factors have been deposited in the Protein Data Bank (PDB) with PDB ID code 7LVZ.
Protein architecture.
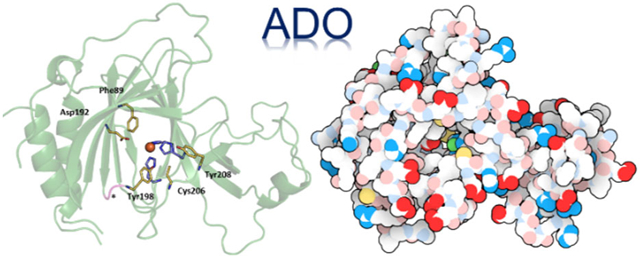
The ADO protein adopts a β-barrel structure typical of the cupin superfamily. The 3-His facial triad coordinating the catalytically relevant Fe ion lies at the center of the protein flanked by β-sheets (Figure 1A). Interestingly, in all four independent copies of the protein in the unit cell, the His130 side chain of a different ADO monomer is present in the active site (Supporting Information Fig. S2). In two cases, His130 is supplied by another protein within the asymmetric unit. In the other two cases, His130 is supplied by a symmetry related protein. In PCO,19 a His residue from the N-terminal His6 tag also protrudes into the active site. However, in contrast to what is observed for PCO, the His residue in the ADO structure does not bind directly to the Fe cofactor. Additionally, in ADO there are no obvious interactions between the His side chain and secondary sphere residues.
Figure 1.
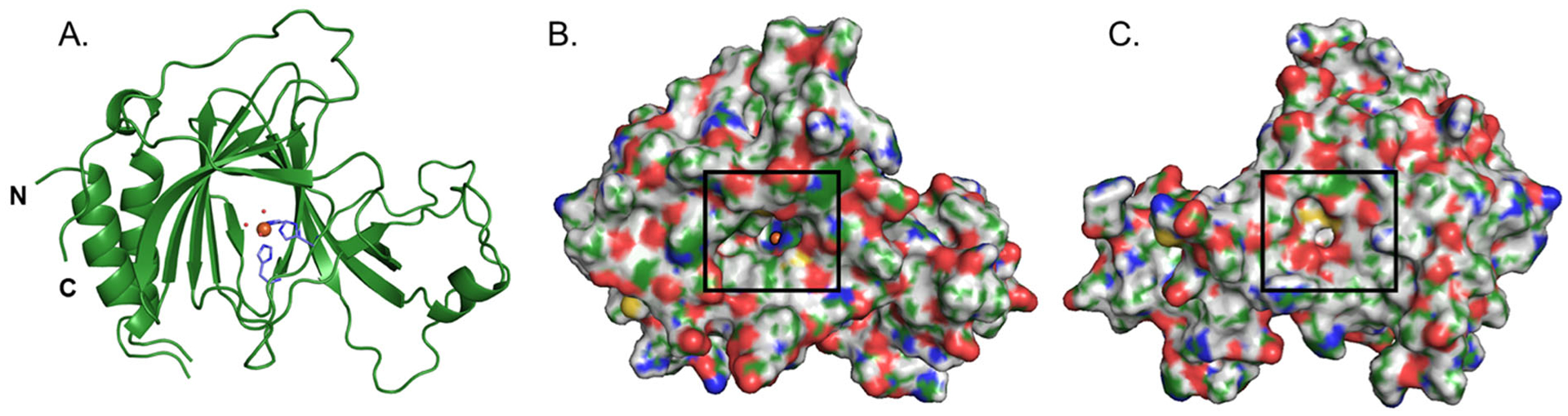
(A) Cartoon representation of the overall structure of ADO. The active site consists of an Fe ion (orange sphere) that is coordinated by a 3-His triad (purple) and 3 water molecules (red spheres). (B) Surface representation of the protein with a black box highlighting the peptide substrate access tunnel to the active site. The RBG coloring is as follows: Fe, orange; carbon, green; hydrogen white, oxygen, red; nitrogen, blue; and sulfur, yellow. (C) Same as in B but rotated 180° to reveal the entrance to the putative co-substrate (O2) tunnel, which is highlighted by a black box.
The surface representation of ADO highlights a prominent tunnel into the active site through which substrate is likely to bind (Figure 1B). Although such a tunnel also exists in CDO, the amino acids that define the CDO substrate tunnel are not conserved in ADO, and the large cavity in ADO is in a structurally distinct location as compared to CDO. Interestingly, the surface representation of the “backside” of ADO features a secondary tunnel from the protein surface to the active site (Figure 1C). It is tempting to speculate that once cysteamine or a bulky Nt-Cys substrate is bound, this much smaller tunnel could provide access for co-substrate O2 to the Fe cofactor. The entrance to this tunnel is guarded by two Cys residues pointed directly at each other with an S…S distance of ~4 Å (further discussed in Distinguishing ADO motifs).
Active site.
The Fe cofactor resides in an octahedral coordination environment composed of His100, His102, and His179 (Figure 2). Because the protein used to obtain this structure was produced and purified semi-anaerobically and crystallized under anaerobic conditions in the presence of 2-AET, the Fe center is almost certainly in the 2+ (i.e., ferrous) oxidation state. In three out of the four crystallographically independent copies in the unit cell, three well-resolved water molecules occupy the remaining coordination sites. In the fourth copy, only two coordinating water molecules are clearly resolved while additional electron density is present in two different regions, 2.18 and 3.65 Å from the Fe center. Although this electron density may seem consistent with a diatomic (e.g., O2-derived; apparent O–O distance of 1.89 Å) ligand, it is presumably associated with a third water molecule that is disordered over two positions given that ADO was crystallized under anaerobic conditions. Notably, in all four ADO monomers in the unit cell, one of the coordinated water molecules is within hydrogen-bonding distance of the hydroxyl group of Tyr198. A similar hydrogen-bonding interaction between the equivalent Tyr182 residue and a water ligand exists in PCO.19
Figure 2.
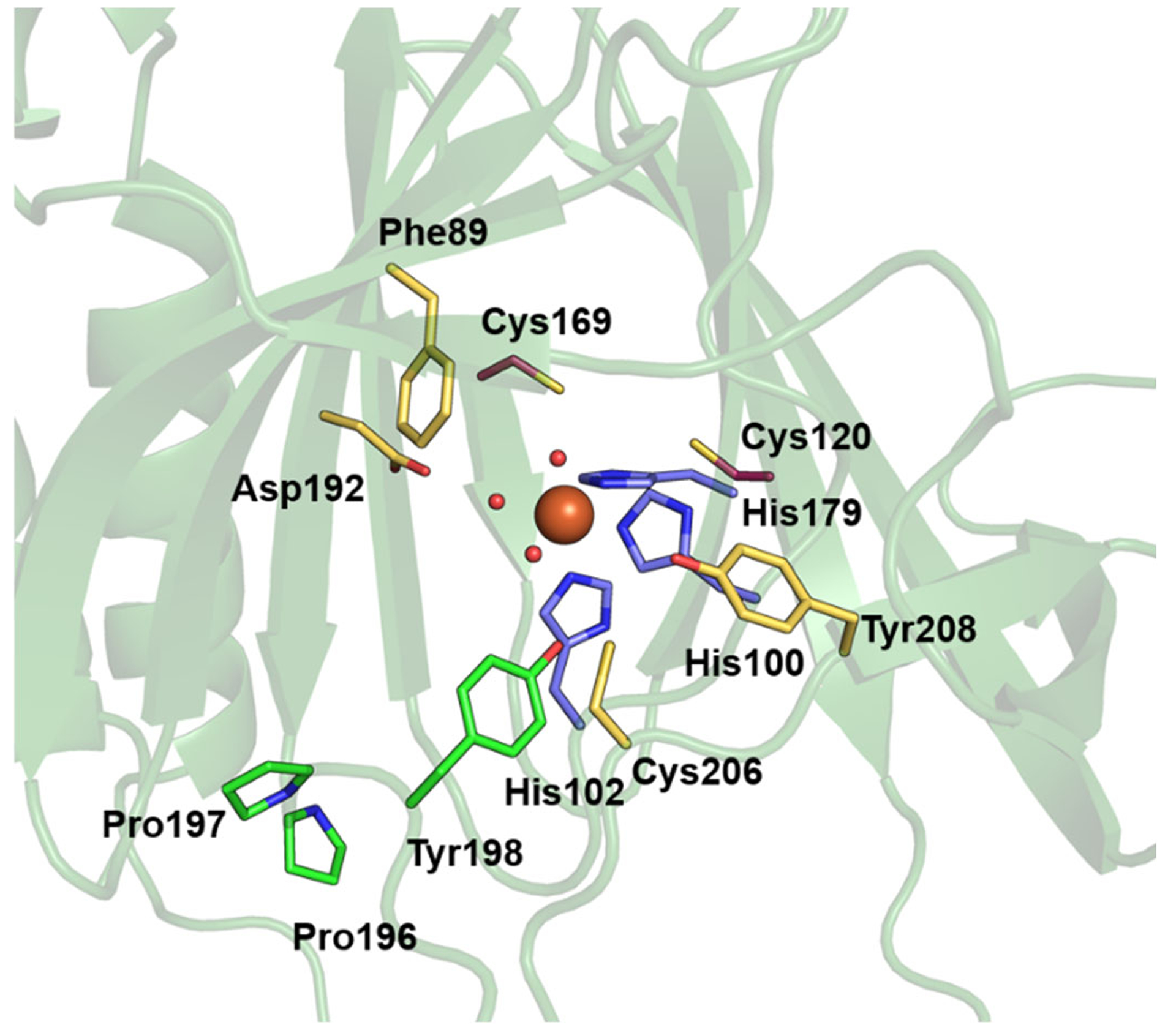
Active site region of MmADO viewed through the substrate tunnel. The Fe ion (orange sphere) is bound by His100, His102, His179 (purple), and three water molecules (red spheres). Other amino acids in the active site are highlighted in yellow (Phe89, Asp192, Cys206, and Tyr208) or green (Pro196, Pro197, and Tyr198). A stereo view of the active site is shown in the Supporting Information Fig. S3.
The active site is surrounded by a hydrophobic pocket with few residues, primarily Leu and Ile, closer than 5 Å to the Fe center. Asp192 of MmADO corresponds to the PCO4 residue Asp176, which has been shown to be critical for PCO function. Specifically, in the as-isolated Asp176Asn PCO4 variant, only 10% of the active sites contained Fe and the enzymatic activity was decreased ~10-fold even after iron supplementation.19 The next set of residues near the ADO active site are Phe89, Cys206, and Tyr208, all located ~7 Å from the cofactor. Although these residues are not close to the Fe cofactor, their locations bordering the substrate access tunnel suggest that they could aid in positioning substrate.
In human ADO, Cys220 and Tyr222 (corresponding to Cys206 and Tyr208 of MmADO) have been shown to be capable of forming a thioether cross-link.18 In the absence of a crystal structure, these two residues were presumed to be located near the Fe cofactor, as cross-link formation required the presence of 2-AET and O2. In the MmADO structure, there is no electron density supporting the existence of a cross-link between Cys206 and Tyr208. The lack of a cross-link in this structure is perhaps not surprising, because the protein used to obtain this structure was produced and purified semi-anaerobically and crystallized under anaerobic conditions in the presence of 2-AET (see Methods section). However, the fraction of cross-linked ADO purified aerobically appears to be very small based on published high-resolution mass spectra.18 Moreover, in the case of PCO4, no cross-link was observed in the X-ray crystal structure or via incubation of protein with substrate and tandem MS/MS analysis.19 Thus, the physiological relevance of the Cys-Tyr cross-link in ADO remains in question.
Distinguishing ADO motifs.
X-ray crystallographic studies revealed that the first coordination spheres of TDOs are identical, with the PCO, CDO, MDO, and, as shown here, ADO active sites all featuring an Fe center coordinated by a 3-His triad. It is the secondary sphere of the active site that boasts unique residues and sequence motifs to tailor the specific reactivity of each enzyme (e.g., the Ser153-His155-Tyr157 catalytic triad in CDO and Gln62 in MDO). The distinguishing MDO and CDO secondary sphere features are neither conserved in the ADO amino acid sequence nor replicated in the X-ray crystal structure. Contrastingly, the ADO and PCO4 active site architectures are remarkably similar, perhaps indicative of closely analogous, or even identical, functions.19 An alignment of MmADO, AtPCO4, PaMDO, MmCDO, and BsCDO was constructed (Figure 3) to guide the identification of key residues and their location in the ADO crystal structure (Figure 2).
Figure 3.
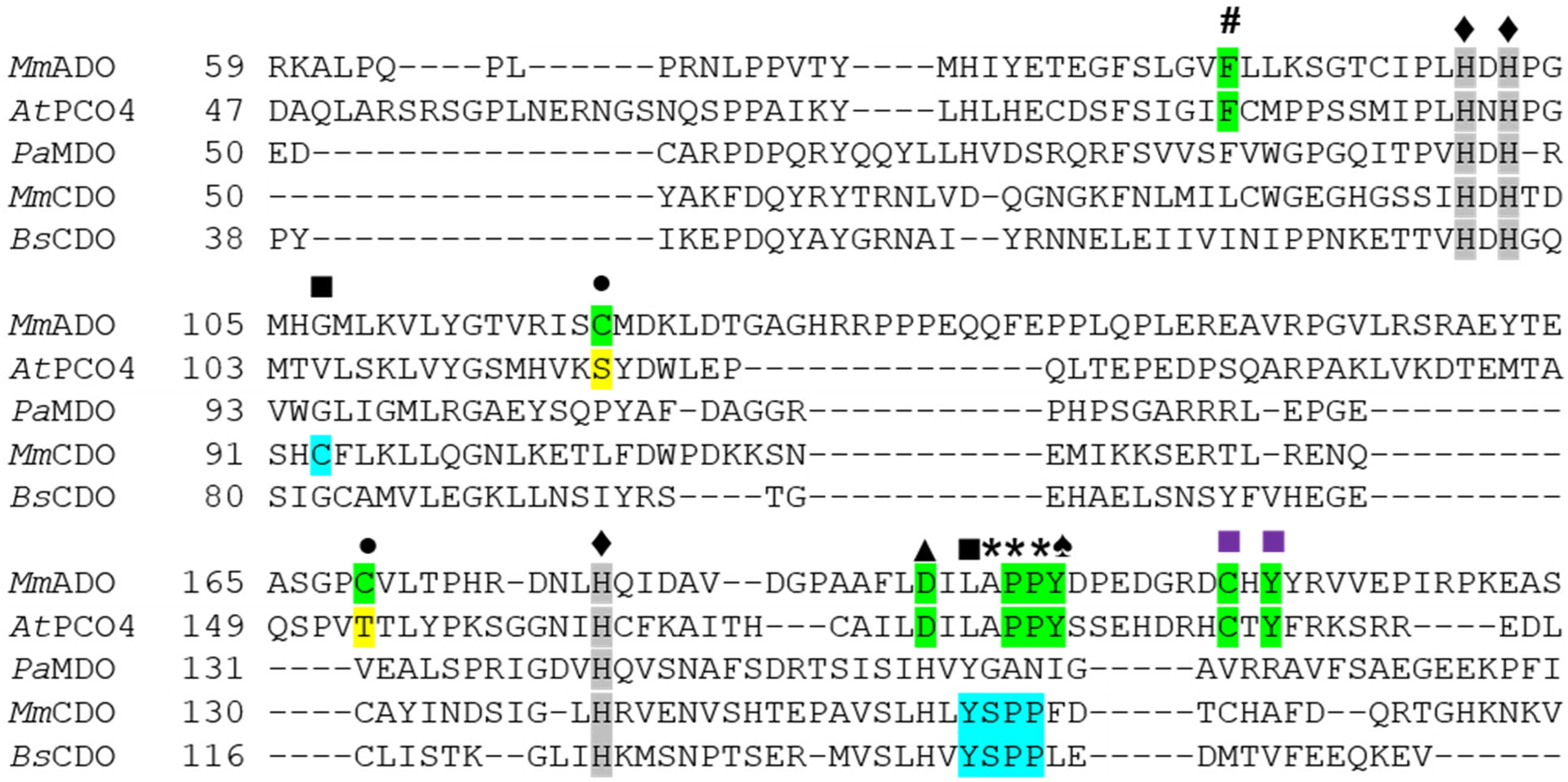
Sequence alignment showing conserved residues within TDOs. The sequences of the PCO4 from Arabidopsis thaliana (AtPCO4), the MDO from Pseudomonas aeruginosa (PaMDO), and the CDOs from Mus musculus (MmCDO) and Bacillus subtilis (BsCDO) are compared to the Mus musculus ADO (MmADO) sequence. Important ADO and PCO residues are highlighted in green, for CDO in blue, and key differences in yellow. All proteins possess a 3-His metal-binding motif marked by a ♦ and highlighted in gray. Additional motifs discussed in the text are denoted as follows: # Phe89 (MmADO numbering), ■ Cys-Tyr cross-link in MmCDO, ■ putative ADO and PCO cross-link motifs, • Cys120 and Cys169 in MmADO are replaced by Ser118 and Thr153 in PCO4, ▲ Asp192, * denotes a cis-peptide bond, and ♠ Tyr198 in MmADO and Tyr182 in AtPCO4 adjacent to the cis-peptide bond.
TDOs are relatively small proteins with sequences containing 200 to 300 amino acids. MmADO, composed of 256 amino acids, boasts a remarkable 33 Pro residues, compared to 23 and 7 found in AtPCO4 and RnCDO, respectively. Interestingly, the ADO structure shows a cis-peptide bond between Pro196 and Pro197. Jameson et al. established that the presence or absence of a cis-peptide bond between Ser158 and Pro159 results in a different positioning of the key Tyr157 residue (RnCDO numbering) involved in the thioether cross-link of CDO.40,41 Although in CDO the Tyr157 residue precedes this cis-peptide motif, in ADO a Tyr residue (Tyr198) is present directly after Pro196 and Pro197; thus, the orientation of Tyr198 is also controlled by a cis-peptide motif. Intriguingly, Tyr198 and Tyr208 reside equidistant and in a similar position from Cys206 (Figure 2, yellow). This Tyr198-Cys206-Tyr208 sandwich borders the substrate tunnel near the protein-solvent barrier. An analogous loop is conserved in PCO4, where it was postulated to play a role in peptide substrate recognition and binding.19 On the basis of these observations, it is tempting to speculate that the Tyr198 residue in ADO plays a larger role in substrate binding and enzyme function than does Tyr208.
The backside tunnel to the active site (Figure 1C) is lined by two surface-exposed Cys residues, Cys120 and Cys169 (Figure 2, yellow). While not bonded in this crystal structure, the residues are 4.8 Å apart and gate a potential secondary access tunnel to the active site. We hypothesize that under proper oxidizing conditions, these residues could form a disulfide bond so as to control O2 access to the active site, and thus enzyme activity. Computational studies aimed at assessing the feasibility of this hypothesis, as well as the generation of faithful models of substrate-bound ADO are presented next.
Computational analysis of resting and substrate-bound ADO.
Repeated attempts were made to crystallize ADO with substrate 2-AET bound. As these attempts were unsuccessful, computational methods were used to generate whole-protein models of ADO complexed with 2-AET and the CKGL tetramer, representative of RGS5. Initially, a computational model of ADO in the absence of substrate was constructed starting from the crystal structure reported in this study to assess the feasibility of the computational approach chosen. The conformational landscape was sampled by first performing an unrestrained molecular dynamics (MD) simulation in GROMACS,28 after which a clustering algorithm was used to select five representative ADO structures that encompassed the majority of conformations accessed during the simulation. The geometries of these structures were optimized using a quantum mechanics/molecular mechanics (QM/MM) approach. The optimized model with the lowest energy was used for further analysis and as the foundation for subsequent calculations of substrate-bound ADO. Importantly, the QM/MM optimized lowest-energy ADO model shows minor deviations from the crystal structure, with the root mean square deviation (RMSD) of the backbone N and C atomic positions being 1.29 Å (for 614 atoms, using the default outlier cut-off value in PyMOL of 2 Å) even though a single ADO monomer was used in our calculations (Supporting Information Fig. S4). Additionally, both the prominent substrate channel and smaller secondary channel are preserved in the computational model, as are the bond lengths and angles of the active site.
In the QM/MM optimized ADO model, the Cys120 and Cys169 residues remain in close proximity, with an S…S distance of 3.5 Å (compared to ~4.8 Å in the crystal structure), hinting at the possibility of disulfide bond formation under oxic conditions. To further evaluate this possibility, a model of ADO containing a disulfide bond between these two residues was generated by altering the crystal structure in silico to install an S–S bond and using the computational workflow described above. Interestingly, the optimized ADO models with and without Cys120…Cys169 disulfide display only minor structural differences, both overall and in the active site region (Supporting Information Figs. S5–8). However, a significant narrowing of the backside substrate channel occurs in response to disulfide bond formation, which lends credence to the possibility of a gating mechanism that modulates O2 access based on cellular oxidative conditions.
Initial coordinates for substrate-bound ADO models were generated using the docking software AutoDock Vina42 to place substrate into the active site of the QM/MM-optimized ADO model lacking the Cys120…Cys169 disulfide bond. Both 2-AET and the CKGL peptide were bound to Fe in a monodentate fashion with thiol-only coordination, as stipulated by previous spectroscopic studies of substrate-bound ADO.15,43 Despite monodentate substrate binding, two water ligands were removed from the Fe center, one trans to His179 to allow for sulfur coordination and one trans to His100 to generate an open coordination site that would be required for O2 binding and enzymatic turnover. These substrate-bound models were then subjected to MD simulations and QM/MM optimizations, as described above for resting ADO. In the case of 2-AET-bound ADO, the large size of the substrate cavity allowed for substantial conformational changes of this small substrate during the MD simulation, with the amine group pointing toward Asp192, Cys206, or nearby water molecules at different time points. In the lowest-energy QM/MM optimized model (Figure 4A), the amine group of 2-AET is positioned roughly equidistant from Tyr198 and Cys206. Notably, upon 2-AET binding to ADO, the putative secondary substrate access channel becomes more defined, now extending from the surface to the active site so that it connects to the larger substrate channel.
Figure 4.
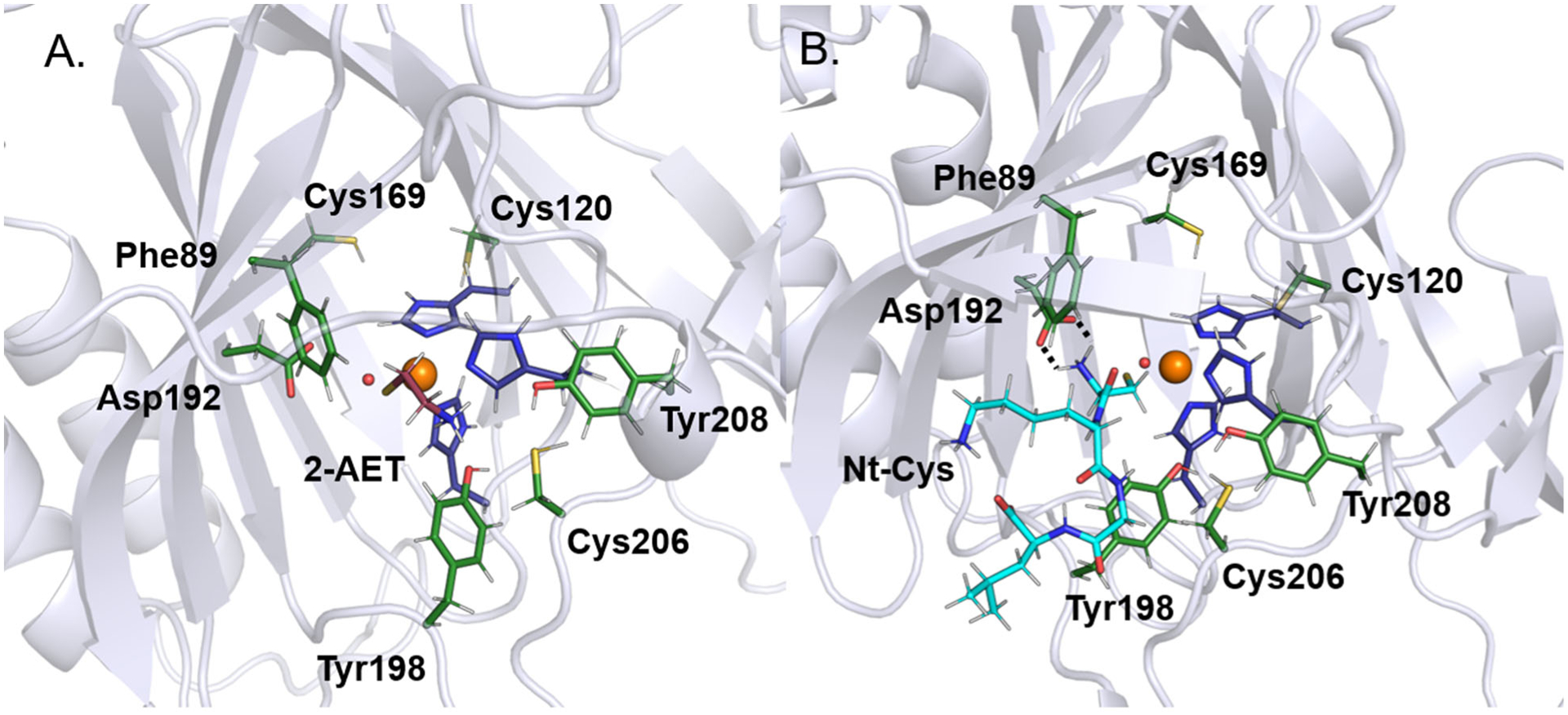
Active site regions of the QM/MM-optimized structures of ADO complexed with (A) 2-AET (burgundy sticks) and (B) the CKGL peptide (cyan sticks), representative of RGS5. The hydrogen-bonding interaction between the amine group of the peptide and Asp192 is indicated by broken lines.
Visualization of the MD trajectory for the ADO model complexed with the relevant portion of the RGS5 peptide (CKGL) makes it apparent that important differences exist in how 2-AET and peptide substrates interact with the secondary sphere of ADO. Compared to 2-AET, the peptide interacts with a much larger number of residues lining the active site cavity. After an initial equilibration period during the MD simulation, the orientation of the terminal cysteine remained largely unchanged, with the amine group always pointing toward Asp192. In the lowest energy QM/MM optimized structure (Figure 4B), the distance between these groups is 1.98 Å, suggesting that Asp192 engages in a hydrogen bonding interaction to properly situate the peptide substrate for catalysis. Another notable residue in the model of peptide-bound ADO is Tyr198, the residue situated next to the Pro196 and Pro197 cis-peptide motif. This residue likely participates in stabilizing interactions with substrate, as it is positioned closer than 4 Å from several side chains of the peptide. Finally, while the small backside channel lined by Cys120 and Cys169 appears more restricted than in the 2-AET-bound model, with a small occlusion to the protein’s surface, it does not disappear entirely.
Discussion
Oxidation of 2-AET and Nt-Cys peptides are important biological transformations; yet, little is known about the enzyme, ADO, that catalyzes these reactions. The geometric and electronic structures of substrate-bound ADO have been spectroscopically investigated and both 2-AET and Nt-Cys have been found to coordinate to the Fe via the terminal thiolate moiety.15,43 While kinetic studies demonstrated that ADO is capable of turning over both 2-AET and Nt-Cys peptides,3,10 the substrate scope and specificity remain to be established. This work confirms that ADO adopts both the canonical cupin architecture and 3-His facial triad typical of TDOs. However, unique structural motifs and secondary sphere elements are observed in the X-ray crystal structure of ADO that are not present in CDO or MDO. In addition, this structure provides the necessary foundation for future investigations of the mechanisms of dioxygen activation and peptide substrate oxidation employed by this vital mammalian TDO.
As ADO could not be crystallized with substrate bound, MD and QM/MM computations were employed to investigate the interactions between secondary sphere residues and Fe-bound substrate. Upon binding of CKGL (representative of the RGS5 peptide) to Fe(II)ADO, the amine group of the peptide adopts a position close enough to hydrogen bond with Asp192. Interestingly, the CKGL peptide is positioned closer to Tyr198 than Tyr208, the latter of which has been proposed to form a cross-link with Cys206. The small size of 2-AET allows for greater conformational flexibility within the active site, such that the amine does not interact with the same residues as the representative RGS5 peptide model. In particular, Tyr208 is too far away from 2-AET to affect the Fe–S bonding interaction in 2-AET-bound Fe(III)ADO. In support of this computational prediction, electron paramagnetic resonance spectra obtained for the cyanide/2-AET adducts of WT Fe(III)ADO and its Tyr208Phe variant are superimposable.44
Computational modeling of peptide-bound ADO shows that substrate RGS5 would likely fill the larger active site tunnel and force co-substrate O2 delivery through a separate channel. To explore this possibility, MOLE2.5 was used to identify potential tunnels, cavities, and pores.45–47 Upon lowering the tunnel radius from the default value of 1.25 to 0.49 Å (and using an internal threshold of 0.9 Å and a cutoff ratio of 0.5 while keeping all other parameters at their default values), a tunnel was identified that starts at residues Cys120 and Cys169 and ends at the Fe atom (Supporting Information Fig. S9). Thus, our MOLE2.5 analysis corroborates the proposal that this tunnel could selectively allow O2 to access the substrate-bound active site. Furthermore, QM/MM optimized models of ADO with Cys120 and Cys169 in their dithiol and disulfide forms display minor global structural differences, implying that these residues could form a disulfide bond under oxidizing conditions. As such, Cys120 and Cys169 could serve as a redox sensor to regulate O2 access to the active site in response to changes in the intracellular redox potential. As ADO is capable of turning over a variety of Cys-containing substrates (though free L-cysteine inhibits ADO catalyzed turnover with cysteamine), it lacks the high substrate specificity characteristic of other TDOs, like CDO. The oxidation of Nt-Cys by ADO marks RGS5 for arginylation by ATE1, a poorly understood post-translational modification.48 RGS5 is up-regulated in ADO and ATE1 deficient cells, establishing that ADO acts upstream of ATE1.10 The presence of easily oxidizable residues such as Cys120/Cys169 and the use of O2 as a co-substrate could connect the redox potential of the cell to enzyme activity, coupling arginylation to oxidative signaling and precluding ADO from depleting cells of Nt-Cys substrates, as well as Cys and 2-AET, in the presence of high O2 levels.
Comparison to other thiol dioxygenases.
A sequence alignment of relevant TDOs (Figure 3) demonstrates that ADO is most closely related to AtPCO4 (21% sequence identity). Comparison of the MmADO and the AtPCO4 X-ray crystal structures demonstrates that these enzymes share similar secondary structures (the RMSD of the backbone atomic positions between chain D of MmADO and AtPCO4 is 0.90 Å for 472 atoms, using the default outlier cut-off value in PyMOL of 2 Å), as well as identical active site residues (Figure 5A and B). Intriguingly, ADO has been shown to complement the function of AtPCO4 both in vivo and in vitro, although sequence similarity analyses have established that a greater than 70% sequence identity is typically needed to extrapolate conservation of function.49,50
Figure 5.
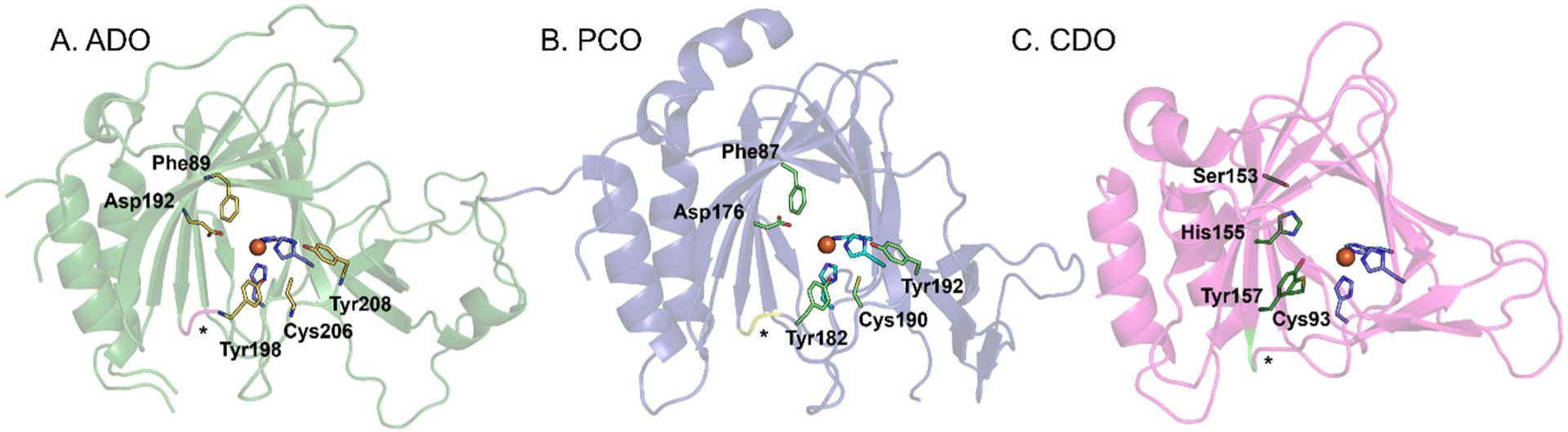
Comparison of the protein folds and active site regions of (A) MmADO (PDB: 7LVZ), (B) AtPCO4 (PDB:6S7E), and (C) RnCDO (PDB: 4JTO). The unusual cis-peptide bond that is preserved in all three enzymes is denoted by an asterisk (*).
Closer inspection of the MmADO and AtPCO4 structures discloses distinct substrate and co-substrate access tunnels. While ADO contains one larger and one smaller access tunnel, PCO4 features a single large tunnel that extends from one side of the protein all the way to the other side and past the active site. Additionally, residues Cys120 and Cys169, which line the smaller access channel in ADO, are replaced by Ser118 and Thr153 in PCO4. Thus, the coupling of enzymatic activity to the intracellular redox potential that we propose to be important for the mammalian enzyme ADO is unlikely to occur in the plant enzyme PCO4. Considering that intracellular O2 levels in mammalian cells are highly variable among different tissues, an extra layer of regulation of enzyme activity may be necessary.
The ADO metallocofactor resides in a hydrophobic pocket with one polar residue, Asp192, positioned within hydrogen bonding distance of an Fe-bound water (Figure 5A). To assess the functional role of the corresponding residue in PCO4, Asp176 (Figure 5B), the D176N variant was produced and kinetically characterized. The variant had 10% Fe incorporation and showed minimal activity even upon the addition of exogenous iron in the activity assay.19 CDO also features a polar residue in the active site, Arg60 (Figure 5C), which has been shown to promote substrate binding via the formation of a salt bridge to the Cys carboxylate tail.51 It is likely that the negatively-charged Asp192 in ADO engages in a stabilizing interaction with a positively-charged amine group of Nt-Cys substrate.
Eukaryotic CDO features a cross-link between residues Cys93 and Tyr157 (Figure 5C). The formation of this cross-link enhances enzymatic activity by repositioning Cys93 and Tyr157 so as to allow for more favorable interactions with the Cys and O2 substrates, while also preserving an open coordination site for O2 binding. Although a thioether cross-link involving Cys206 and Tyr208 (MmADO numbering) has been identified in human ADO by Liu and coworkers,18 in the crystal structure of MmADO these two residues reside ~7 Å from the Fe cofactor and no thioether bond is observed (as expected given the largely anaerobic conditions under which the enzyme was produced, purified, and crystallized). While the large separation of Cys206 and Tyr208 from the active site suggests that cross-link formation plays a minor structural or functional role in ADO, small differences between the mouse and human ADO sequences (85% sequence identity) could result in more pronounced differences between these two enzymes than one would expect. Intriguingly, as highlighted in Figure 5, the unusual cis-peptide bond that aids in the proper positioning of Tyr157 in CDO (Ser158 and Pro159) is also present in ADO (Pro196 and Pro197). In the QM/MM optimized structure of CKGL-bound ADO, Tyr198 is positioned close to the peptide substrate. The absence of a Cys206…Tyr208 cross-link in the crystal structure of MmADO, along with the lack of a direct interaction between Tyr208 and Nt-Cys or 2-AET in the computational models of substrate-bound ADO indicate that Tyr198 may be functionally more relevant than Tyr208.
In conclusion, the crystallographic and computational data of ADO presented here have revealed residues that could influence small molecule and peptide positioning within the active site. Although a thioether cross-link is key to increased turnover in mammalian CDO, a comparable contribution remains elusive in the MmADO as the Cys206 and Tyr208 residues implicated in this cross-link reside ~7 Å from the Fe cofactor. Of note, computational modeling of Nt-Cys binding to the ADO indicates that Tyr198 may play a key role in positioning a peptide substrate in the active site. Importantly, a backside tunnel is identified that could serve to deliver O2 to the peptide-bound ADO active site. This tunnel is lined by Cys120 and Cys169, with a distance and orientation compatible with redox-dependent control of O2 access through disulfide bond formation. The unique combination of separate peptide substrate and O2 binding channels, along with the potential for redox-sensitive control of O2 access to the Fe- and 3-His active site represent a new model for understanding the function of mammalian thiol dioxygenases.
Supplementary Material
Acknowledgement
The authors thank Nicholas Juntunen for performing a preliminary sequence similarity network analysis and Prof. Martha H. Stipanuk for sharing the original mouse ADO plasmid used to begin this work.
Funding
The authors are grateful for financial support from the National Institute of General Medical Sciences of the National Institutes of Health (Grant GM117120 to T.C.B.). This research was conducted in part while RLF was supported by the National Institute of General Medical Sciences of the National Institutes of Health (Grant T32GM008505). We thank Dr. Desiree Bates and Jon Ellis for their helpful suggestions regarding the computations.
Crystallization, data collection, and structure solution and refinement were conducted by the Department of Biochemistry Collaborative Crystallography Core at the University of Wisconsin, which is supported by user fees and the department. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). GM/CA@APS has been funded by the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006, P30GM138396). The Eiger 16M detector at GM/CA-XSD was funded by NIH grant S10 OD012289.
Footnotes
Supporting Information
The Supporting Information (in PDF format) is available free of charge on the ACS Publications website.
Data collection and refinement statistics; results from a SuperPose analysis for the four different ADO monomers in the unit cell; overlay of the four independent ADO monomers in the unit cell; figure showing that His130 from a neighboring molecule intrudes into the active site in each of the four crystallographically unique copies of MmADO; stereo view of the active site region of ADO; comparison of the ADO X-ray crystal structure with the QM/MM optimized structure of ADO; front and back of the QM/MM optimized structure of ADO; surface representation of the backside of QM/MM optimized structure of ADO highlighting the putative co-substrate tunnel; front and back of the QM/MM optimized structure of a model of oxidized ADO featuring a Cys120–Cys169 disulfide bond; surface representation of the backside of QM/MM optimized structure of oxidized ADO; and tunnels calculated by MOLE2.5 into the active site of the QM/MM optimized model of peptide-bound ADO.
Added in Proof While this paper was under review, the crystal structure of a human ADO variant at a resolution of 1.78 Å with nickel bound to the active site was reported (PDB entry 7REI): Wang, Y.; Shin, I.; Li, J.; Liu, A. Crystal structure of human cysteamine dioxygenase provides a structural rationale for its function as an oxygen sensor. J. Biol. Chem. 2021, 297, 101176.
The authors declare no competing financial interest.
References
- (1).Heafield MT, Fearn S, Steventon GB, Waring RH, Williams AC, and Sturman SG (1990) Plasma cysteine and sulphate levels in patients with motor neurone, Parkinson’s and Alzheimer’s disease. Neurosci. Lett 110, 216–220. [DOI] [PubMed] [Google Scholar]
- (2).Simmons CR, Hirschberger LL, Machi MS, and Stipanuk MH (2006) Expression, purification, and kinetic characterization of recombinant rat cysteine dioxygenase, a non-heme metalloenzyme necessary for regulation of cellular cysteine levels. Protein Expr. Purif 47, 74–81. [DOI] [PubMed] [Google Scholar]
- (3).Dominy JE, Simmons CR, Hirschberger LL, Hwang J, Coloso RM, and Stipanuk MH (2007) Discovery and characterization of a second mammalian thiol dioxygenase, cysteamine dioxygenase. J. Biol. Chem 282, 25189–25198. [DOI] [PubMed] [Google Scholar]
- (4).Stipanuk MH, Dominy JE, Ueki I, and Hirschberger LL (2008) Measurement of cysteine dioxygenase activity and protein abundance. Curr. Protoc. Toxicol [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Stipanuk MH, Dominy JE, Lee J-I, and Coloso RM (2006) Mammalian cysteine metabolism: new insights into regulation of cysteine metabolism. J. Nutr 136, 1652S–1659S. [DOI] [PubMed] [Google Scholar]
- (6).Stipanuk MH (1986) Metabolism of sulfur-containing amino acids. Annu. Rev. Nutr 6, 179–209. [DOI] [PubMed] [Google Scholar]
- (7).Shen D, Tian L, Yang F, Li J, Li X, Yao Y, Lam EWF, Gao P, Jin B, and Wang R (2021) ADO/hypotaurine: a novel metabolic pathway contributing to glioblastoma development. Cell Death Discov. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Schaffer SW, Ju Jong C, KC R, and Azuma J (2010) Physiological roles of taurine in heart and muscle. J. Biomed. Sci 17, 101–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ripps H, and Shen W (2012) Review: Taurine: A “very essential” amino acid. Mol. Vis 18, 2673–2686. [PMC free article] [PubMed] [Google Scholar]
- (10).Masson N, Keeley TP, Giuntoli B, White MD, Lavilla Puerta M, Perata P, Hopkinson RJ, Flashman E, Licausi F, and Ratcliffe PJ (2019) Conserved N-terminal cysteine dioxygenases transduce responses to hypoxia in animals and plants. Science (80-. ). 365, 65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).White MD, Klecker M, Hopkinson RJ, Weits DA, Mueller C, Naumann C, O’Neill R, Wickens J, Yang J, Brooks-Bartlett JC, Garman EF, Grossmann TN, Dissmeyer N, and Flashman E (2017) Plant cysteine oxidases are dioxygenases that directly enable arginyl transferase-catalysed arginylation of N-end rule targets. Nat. Commun 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lee MJ, Tasaki T, Moroi K, An JY, Kimura S, Davydov IV, and Kwon YT (2005) RGS4 and RGS5 are in vivo of the N-end rule pathway. Proc. Natl. Acad. Sci. U. S. A 102, 15030–15035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tamirisa P, Blumer KJ, and Muslin AJ (1999) RGS4 inhibits G-protein signaling in cardiomyocytes. Circulation 99, 441–447. [DOI] [PubMed] [Google Scholar]
- (14).Stipanuk MH, Simmons CR, Karplus PA, and Dominy JE (2011) Thiol dioxygenases: Unique families of cupin proteins. Amino Acids 41, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Fernandez RL, Dillon SL, Stipanuk MH, Fox BG, and Brunold TC (2020) Spectroscopic Investigation of Cysteamine Dioxygenase. Biochemistry 59, 2450–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Driggers CM, Kean KM, Hirschberger LL, Cooley RB, Stipanuk MH, and Karplus PA (2016) Structure-Based Insights into the Role of the Cys–Tyr Crosslink and Inhibitor Recognition by Mammalian Cysteine Dioxygenase. J. Mol. Biol 428, 3999–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Davies CG, Fellner M, Tchesnokov EP, Wilbanks SM, and Jameson GNL (2014) The Cys-Tyr cross-link of cysteine dioxygenase changes the optimal ph of the reaction without a structural change. Biochemistry 53, 7961–7968. [DOI] [PubMed] [Google Scholar]
- (18).Wang Y, Griffith WP, Li J, Koto T, Wherritt DJ, Fritz E, and Liu A (2018) Cofactor Biogenesis in Cysteamine Dioxygenase: C−F Bond Cleavage with Genetically Incorporated Unnatural Tyrosine. Angew. Chemie Int. Ed 57, 8149–8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).White MD, Carbonare LD, Puerta ML, Iacopino S, Edwards M, Dunne K, Pires E, Levy C, McDonough MA, Licausi F, and Flashman E (2020) Structures of Arabidopsis thaliana oxygen-sensing plant cysteine oxidases 4 and 5 enable targeted manipulation of their activity. Proc. Natl. Acad. Sci. U. S. A 117, 23140–23147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kabsch W (2010) XDS. Acta Crystallogr. Sect. D Biol. Crystallogr D66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T, and Bricogne G (2011) Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. Sect. D Biol. Crystallogr 67, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, De Beer TAP, Rempfer C, Bordoli L, Lepore R, and Schwede T (2018) SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D Biol. Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).The PyMOL Molecular Graphics System, Version 2.0 Schrodinger, LLC. [Google Scholar]
- (28).Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindah E (2015) Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. [Google Scholar]
- (29).Tian C, Kasavajhala K, Belfon KAA, Raguette L, Huang H, Migues AN, Bickel J, Wang Y, Pincay J, Wu Q, and Simmerling C (2020) Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput 16, 528–552. [DOI] [PubMed] [Google Scholar]
- (30).Case DA, Belfon K, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham TE, Cruzeiro I, Darden VWD, Duke TA, Giambasu RE, Gilson G, Gohlke MK, Goetz H, Harris AW, Izadi R, Jhala S, Kovalenko KK, Krasny A, Kurtzman R, Lee T, LeGrand TS, Li S, Lin P, Liu C, Luchko J, Luo T, Man R, Merz V, Miao KM, Mikhailovskii Y, Monard O, Nguyen G, Onufriev H, Pan A, Pantano F, Qi S, Roe R, Roitberg DR, Sagui A, Schott-Verdugo C, Shen S, Simmerling J, Skrynnikov CL, Smith N, Swails J, Walker J, Wang RC, Wilson J, Wolf L, Wu RM, York X, M. D, and Kollman PA (2020) Amber 2020. University of California, San Francisco. [Google Scholar]
- (31).Li P, and Merz KM (2016) MCPB.py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Model 56, 599–604. [DOI] [PubMed] [Google Scholar]
- (32).Berendsen HJC, Grigera JR, and Straatsma TP (1987) The missing term in effective pair potentials. J. Phys. Chem 91, 6269–6271. [Google Scholar]
- (33).Trott O, and Olson AJ (2009) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 31, NA–NA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Frisch MJ, Trucks GW, Schlegel HE, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomberts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA, Peralta J, Ogliaro JE, Bearpark F, Heyd MJ, Brothers JJ, Kudin EN, Staroverov KN, Keith VN, Kobayashi TA, Normand R, Raghavachari J, Rendell K, Burant AP, Iyengar JC, Tomasi SS, Cossi J, Millam M, Klene JM, Adamo M, Cammi C, Ochterski R, Martin JW, Morokuma RL, Farkas K, Foresman O, B. J, and Fox JD (2016) Gaussian 16, Revision C.01. Gaussian, Inc., Wallingford CT,. [Google Scholar]
- (35).Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 98, 5648. [Google Scholar]
- (36).Lee C, Yang W, and Parr RG (1988) Development of the Colle-Salvetti correlation- energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789. [DOI] [PubMed] [Google Scholar]
- (37).Schäfer A, Huber C, and Ahlrichs R (1994) Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys 100, 5829–5835. [Google Scholar]
- (38).Hehre WJ, Ditchfield K, and Pople JA (1972) Self-consistent molecular orbital methods. XII. Further extensions of gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys 56, 2257–2261. [Google Scholar]
- (39).Bayly CI, Merz KM, Ferguson DM, Cornell WD, Fox T, Caldwell JW, Kollman PA, Cieplak P, Gould IR, and Spellmeyer DC (1995) A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc 117, 5179–5197. [Google Scholar]
- (40).Tchesnokov EP, Fellner M, Siakkou E, Kleffmann T, Martin LW, Aloi S, Lamont IL, Wilbanks SM, and Jameson GNL (2015) The cysteine dioxygenase homologue from Pseudomonas aeruginosa is a 3-mercaptopropionate dioxygenase. J. Biol. Chem 290, 24424–24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Aloi S, Davies CG, Karplus PA, Wilbanks SM, and Jameson GNL (2019) Substrate Specificity in Thiol Dioxygenases. Biochemistry 2398–2407. [DOI] [PubMed] [Google Scholar]
- (42).Trott O, and Olson AJ (2009) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 31, NA–NA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wang Y, Davis I, Chan Y, Naik SG, Griffith WP, and Liu A (2020) Characterization of the nonheme iron center of cysteamine dioxygenase and its interaction with substrates. J. Biol. Chem 295, 11789–11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Fernandez RL, Juntunen ND, Fox BG, and Brunold TC Spectroscopic Investigation of Iron (III) Cysteamine Dioxygenase in the Presence of Substrate (Analogues): Implications for the Nature of Substrate-Bound Reaction Intermediates. J. Biol. Inorg. Chem 2021, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Sehnal D, Vařeková RS, Berka K, Pravda L, Navrátilová V, Banáš P, Ionescu CM, Otyepka M, and Koča J (2013) MOLE 2.0: Advanced approach for analysis of biomacromolecular channels. J. Cheminform 5, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Jones JC, Banerjee R, Shi K, Aihara H, and Lipscomb JD (2020) Structural Studies of the Methylosinus trichosporium OB3b Soluble Methane Monooxygenase Hydroxylase and Regulatory Component Complex Reveal a Transient Substrate Tunnel. Biochemistry 59, 2946–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Banerjee R, and Lipscomb JD (2021) Small-Molecule Tunnels in Metalloenzymes Viewed as Extensions of the Active Site. Acc. Chem. Res 54, 2185–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Van V, and Smith AT (2020) ATE1-Mediated Post-Translational Arginylation Is an Essential Regulator of Eukaryotic Cellular Homeostasis. ACS Chem. Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Tian W, and Skolnick J (2003) How well is enzyme function conserved as a function of pairwise sequence identity? J. Mol. Biol 333, 863–882. [DOI] [PubMed] [Google Scholar]
- (50).Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, and Whalen KL (2015) Enzyme function initiative-enzyme similarity tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta - Proteins Proteomics 1854, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Dominy JE, Hwang J, Guo S, Hirschberger LL, Zhang S, and Stipanuk MH (2008) Synthesis of amino acid cofactor in cysteine dioxygenase is regulated by substrate and represents a novel post-translational regulation of activity. J. Biol. Chem 283, 12188–12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.