

Abstract
Background:
The mechanism responsible for initiating and controlling the immunosuppressive response after burn injury remains unknown. Interleukin-17 (IL-17) secreting Th17 (interferon [IFN]γ− IL17+) cells are a novel subset of CD4+ T cells associated with a weak, proinflammatory response that antagonizes the proinflammatory Th1 (IFNγ+ IL17−) response. Given that transforming growth factor-β and IL6 mediate Th17 cell development we hypothesized that burn injury may generate Th17 cells that could mediate postburn immunosuppression.
Methods:
After a 20% total body surface area burn in female C57BL/6 mice, wound-draining lymph nodes were harvested 3 days, 7 days, or 14 days after injury. CD4+ T cells were enriched by magnetic selection, and flow cytometry was used to identify intracellular IL17 and IFNγ in CD3+CD4+ T cells. Additional purified CD3+CD4+ T cells were cultured with Th17− polarizing IL6 and transforming growth factor-β for 4 days, and flow cytometry was again used to identify intracellular IL17 and IFNγ in CD4+ T cells.
Results:
The number and percentage of preformed Th17 cells was significantly greater in burn mice compared with sham at all time points. In addition, the ratio of Th17 cells to Th1 cells was always significantly higher in burn mice compared with sham. These differences were eliminated in Th17 polarizing conditions in vitro. CD4+ T cells never generated both IL17 and IFNγ.
Conclusion:
These results demonstrate for the first time that Th17 cells (IFNγ− IL17+) are spontaneously generated after burn injury. Given that Th17 cells (IFNγ− IL17+) are antagonistic to Th1 (IFNγ+ IL17−) cells, these results suggest a novel mechanism for initiating and controlling postburn immunosuppression that deserves further investigation.
Keywords: Burn injury, Immunology, Th17, T cells, IL-17
CD4+ T-helper (Th) cells have been classically divided into pro- (Th1) or anti-inflammatory (Th2) T cells based on the type of cytokines they produce after stimulation.1 However, a newly discovered subset of CD4+ T cells, called Th17 cells, has been shown to secrete interleukin-17 (IL-17) but not interferon-γ (IFN-γ) or IL-4.2–4 Recent studies have suggested that the main functions of IL-17 are to activate macrophages and to promote their production of inflammatory cytokines. Subsequently, these cytokines, which include tumor necrosis factor-α (TNF-α) and IL-1β recruit neutrophils to sites of infection.5 For example, IL-17 receptor deficient mice have reduced neutrophil recruitment, increased bacterial loads, and high mortality rates after challenge with Klebsiella pneumoniae.6 Furthermore, overexpression of IL-17 in the lungs results in a localized increase in TNF-α and IL-1β, as well as enhanced neutrophil accumulation and K. pneumoniae clearance.7 IL-17 receptor deficient mice are also more susceptible to infection with Candida albicans.8 Because these pathogens primarily cause infection in individuals with suppressed immune systems, this has led to the belief that Th17 cells are weak proinflammatory cells.
Naïve CD4+ T cells are regarded as being able to differentiate toward one of four lineages; Th1, Th2, Treg, or Th17 depending on many factors such as strength of antigen stimulation and cytokine microenvironment. Differentiation of naïve CD4+ T cells toward a Th17 phenotype requires the presence of transforming growth factor-β (TGF-β) and IL-6, whereas IL-23 is believed to play a role in the maintenance of Th17 effector function.9–11 This differentiation process requires a unique transcription factor, retinoic acid-related receptor (ROR)-γt. to induce transcription of the IL-17 gene.12 In addition, cells genetically deficient in the master transcription factors required for Th1 and Th2 differentiation have either undiminished or enhanced Th17 development.2 Together these findings indicate that Th17 cells are a lineage distinct from Th1 and Th2 cells. Furthermore, cytokines secreted by differentiated Th1 and Th2 cells inhibit formation of Th17 cells (Fig. 1). Conversely, when naïve CD4+ T cells are exposed to TGF-β in the absence of IL-6, they express forkhead box P3 (Foxp3), which is the master transcription factor that drives induction of Foxp3+ regulatory T cells (Treg).13 Therefore, the presence of IL-6 switches the development of naïve CD4+ T cells from a Treg pathway to a Th17 pathway.
Figure 1.
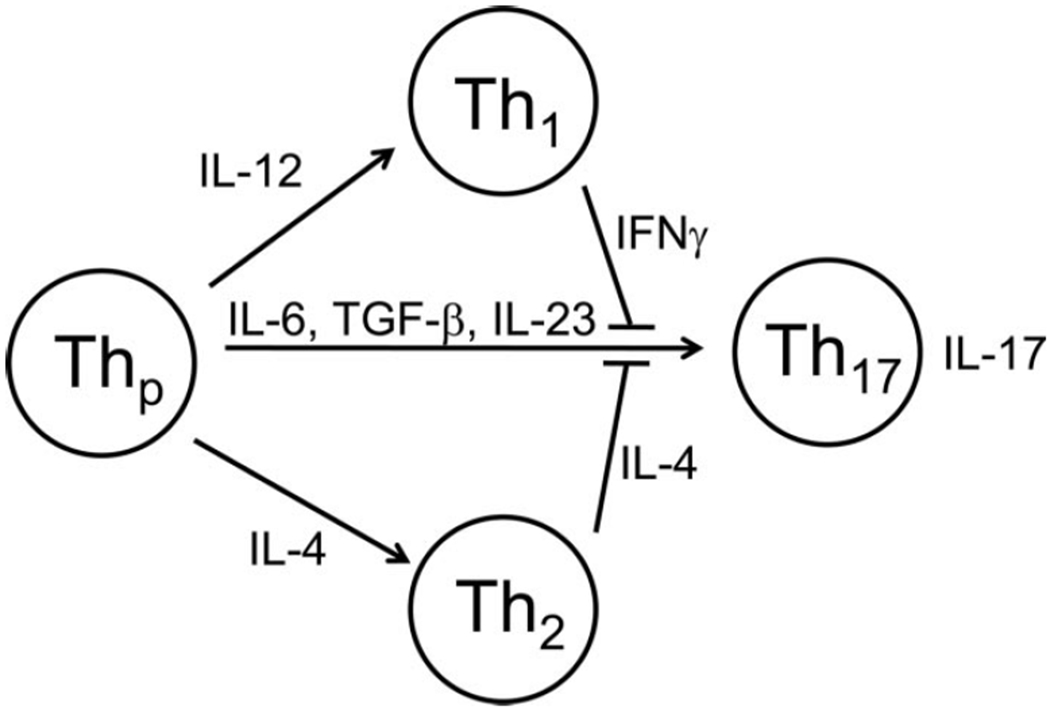
Th17 cells are a distinct lineage of CD4+ T cells. Naïve CD4+ T precursors (Thp) can differentiate toward either Th1, Th2, or Th17 phenotype. Differentiation of naïve CD4+ T cells toward a Th17 phenotype requires the presence of TGF-β and IL-6, whereas IL-23 is required for the maintenance of Th17 effector function. Unique cytokines secreted by differentiated Th1 (IFNγ) and Th2 (IL-4) cells inhibit formation of Th17 cells.
Severe burn injury causes a dangerous immune dysfunction. Much research effort has been expended into defining the immune consequence of burn in terms of lymphocyte cytokine identity.14–33 One model of burn injury suggests that there is a rapid onset of a systemic inflammatory response symptom characterized by the production of proinflammatory cytokines.23,34 If this proinflammatory state is uncontrolled, patients can experience early multiple organ dysfunction syndrome and death.35 Patients surviving this period are then thought to develop a compensatory anti-inflammatory response characterized by immune suppression and decreased resistance to infection.36
We and others have defined CD4+ and CD8+ T-cell population changes both early and late after burn in patients and animal models. After burn injury, there is an early (hours to days) proinflammatory response followed by a shift toward an anti-inflammatory phenotype in both the CD4+ and CD8+ T-cell compartments.15,22,23 More specifically, CD4+ T cells begin to produce the proinflammatory cytokines IFN-γ and IL-2 on stimulation, which are defining characteristics of Th1 cells. In addition, macrophages produce TNF-α, IL-1, and IL-6 as well as other proinflammatory cytokines.37 Furthermore, the level of TGF-β increases gradually in the burn wound throughout this phase.36,38 Following this phase after burn, the proinflammatory state of the immune system switches to an anti-inflammatory response characterized by over-production of TGF-β,15,36,38 as well as a switch in CD4+ T-cell responses from a Th1 to a Th2 (IL-4) phenotype.39 This manifests itself as decreased antigen-specific proliferation, diminished cytokine secretion, and cytotoxic T lymphocyte activity.18,22,40,41
Because most patients develop immune failure days to weeks after injury, we have been interested in both CD8+ and CD4+ T cell function during the first 2 weeks after injury. We have previously demonstrated profound alterations in the cytokine profile late after burn injury. CD8+ T cells, crucial for antipathogen and antiallograft immune responses, are impaired immediately postburn but experience increased proliferation and have a unique and dramatic altered cytokine profile later after burn (14 days).18,22,40,41 This functional enhancement is characterized by a selective peripheral T cell lymphopenia that drives a homeostatic increase in “spontaneous” memory-like CD8+ and CD4+ T cells in the periphery.14,22 Because elevated TGFβ and IL-6 levels are found after burn injury, we hypothesize that CD4+ Th17 cells are generated after burn injury in a murine model of burn.
MATERIALS AND METHODS
Animals
Wild-type C57Bl/6 (B6) mice were purchased from Taconic Farms (Germantown, NY). All mice used in the study were maintained under specific pathogen-free conditions in the American Association of Laboratory Animal Care-accredited University of North Carolina Department of Laboratory Animal Medicine Facilities.
Mouse Burn Injury
Six to 8-week-old (15–20 g weight) female B6 mice were used as subjects in all experiments. All protocols were performed in accordance with the National Institutes of Health guidelines and approved by the University of North Carolina IACUC as previously described.15 Briefly animals were anesthetized with inhalation of isoflurane vapor (Pitman-Moore, WA Crossing, NJ) and their dorsal and flank hair clipped. A full-contact burn of ~80% total body surface area (TBSA) was produced by applying a copper rod, heated in boiling water, to the animal’s dorsum and flank for 10 seconds. Four applications of a 65 g rod (1.9 cm in diameter) were used to produce the wound, and previous biopsies of the wounds demonstrate full-thickness cutaneous burn with visible unburned muscle beneath. Mice were resuscitated with an intraperitoneal injection of Lactated Ringer’s solution (0.1 mL/gm body weight) and were given a subcutaneous injection of buprenorphine (2 mg/kg body weight) for pain control immediately after burn injury. Animals were returned to individual cages and were provided food and morphinated water ad libitum throughout the experiment. Sham controls with the 0% TBSA burn underwent all the described interventions except for the actual burn injury. There is a negligible mortality (<1%) after burn injury with this protocol. Animals were killed where indicated, and splenocytes and cells from axillary, brachial, inguinal, lumbar, and caudal peripheral lymph node (PLN) were prepared for culture or CD4+ T cell purification.
CD4+ T-Cell Purification
Cell suspensions were prepared from PLN of mice. CD4+ T cells were negatively selected by depletion of CD8+, MHC Class II+, CD11b+, and other cell types using the BD iMag Mouse CD4+ T Lymphocyte Enrichment Set according to the manufacturer’s instructions (Becton Dickinson, San Diego, CA). This method routinely provides us with >90% pure CD4+ T-cell populations.
T-Cell Stimulation for Cytokine Analysis
Splenocytes, bulk PLN cells, or purific CD4+ T cells from burn and sham mice (1 × 106 cells/mL) cells were stimulated with phorbol myristate acetate (1 μg/mL; Simga Aldrich, St. Louis, MO) and ionomycin (1 μg/mL; Sigma Aldrich) for a total of 4 hours in 1 mL of complete RPMI (10% fetal calf serum) in 24-well flat-bottom plates. Brefeldin A (3.0 μg/mL: eBioscience, San Deigo, CA) was added for the final 2 hours of culture to retain cytokines within the cell.
Th17 In Vitro Cell Polarization
Purified CD4+ T cells from burn and sham mice (1 × 106 cells/mL) cells were stimulated with plate bound antimouse CD3 antibody (1 μg/well; Becton Dickinson, San Diego, CA) and soluble antimouse CD28 antibody (5 μg/mL; BD Pharmingen, San Diego, CA) in the presence of IL-6 (50 ng/mL; BD Pharmingen, San Diego, CA) and TGF-β (1 ng/mL; PeproTech, Rocky Hill, NJ) cytokines for a total of 4 days in 0.5 mL of complete RPMI (10% fetal calf serum) in 48-well flat-bottom plates. Serum was collected from burn mice and added to cultures where indicated at a final dilution factor of 1/10.
Flow Cytometric Analysis
The panel of monoclonal antibodies used for flow cytometric analyses were anti-ROR-βt (AFKJS-9, eBioscience, San Diego, CA), anti-CD8α (53–6.7), anti-CD3ϵ (145–2C11), anti-CD4 (L3T4), anti-IFNγ (XMG-1.2) (BD Phanningen, San Diego, CA), and anti-IL-17 (eBio17B7) (eBioscience, San Diego, CA). Intracellular staining for cytokines and ROR-γt was performed using standard methods.15 Four- and five-color analysis was performed using standard methods. List mode data were collected on a FACS Cyan (Dako, Ft. Collins, CO) and analyzed using Summit software (Dako).
Statistical Analysis
Data were analyzed using Student’s t test for intracellular cytokine, absolute number, and ratio differences. Statistical significance was defined as p < 0.05 unless indicated otherwise.
RESULTS
Th17 Cells Exist in Wound-Draining Lymph Nodes But Not in Spleen After Burn Injury
We and others have previously shown a dynamic change in the CD4+ and CD8+ T-cell compartments at various time points after severe burn injury with the most dramatic changes occurring at 3 days and 14 days after burn.14–33 To examine whether preformed Th17 cells exist within peripheral immune sites at these time points, we used a model of contact burn injury in wild-type female B6 mice. Cells were harvested from spleen, inguinal, and axillary PLN at 3 days or 14 days after a 20% TBSA contact burn or sham injury. Wound-draining PLN have been shown previously to exhibit marked lymphocyte alterations after burn injury.25,42,43 T cells were stimulated with mitogen, and standard intracellular cytokine staining was employed to identity CD3+ CD4+ IL-17+ (Th17), CD3+ CD4+ IFN-γ+ (Th1), CD3+ CD8+ IL-17+ (Tc17), and CD3+ CD8+ IFN-γ+ (Tc1) cells using flow cytometry. At 3 days after burn or sham injury, we found an increased percentage of Th1 cells in the spleen (as reported previously,15) and inguinal PLN but no increase in IL-17 producing CD4+ T cells (Fig. 2). In contrast, there was a significant increase in the amount of Th17 cells found in the axillary PLN of the burn mice when compared with sham mice (Fig. 3). We did not observe any expression of IL-17 by CD8+ T cells (data not shown). At 14-day postburn and sham injury, we found a burn-dependent increase in the percentage of Th17 cells present in both the axillary and inguinal PLN but not the spleen (Fig. 3). The percentage of Th1 cells in all sites studied was significantly lower at day 14 versus day 3, corresponding to the suppressed proinflammatory state observed late after burn injury. Again, we did not observe any expression of IL-17 by CD8+ T cells at day 14 (data not shown). At all time points and in all organs studied, we did not observe at dual positive CD3+ CD4+ IL-17+ IFN-γ+ T cells, further reinforcing the notion that these are distinct Th cell lineages.
Figure 2.
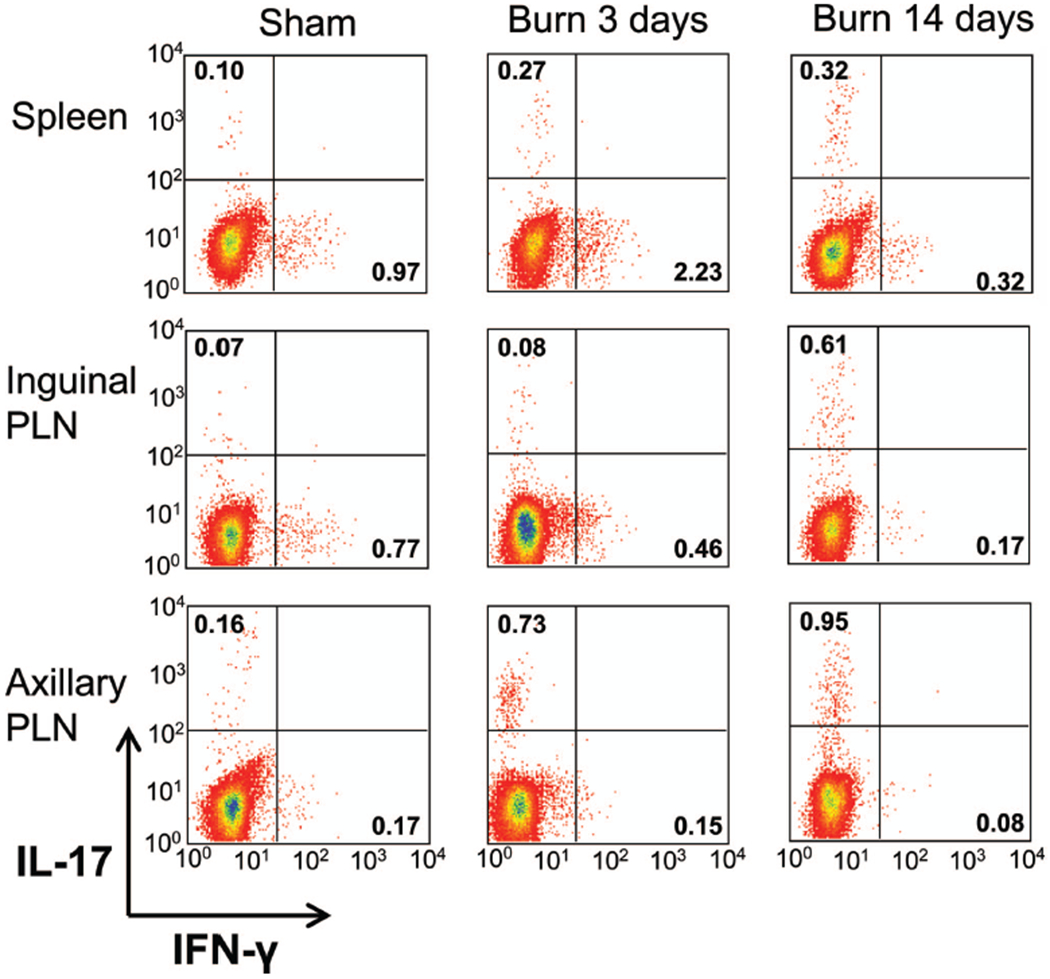
Wound-draining lymph nodes, but not the spleen, contain Th17 cells 3 days and 14 days after burn injury. Cells were harvested from spleens, as well as inguinal and axillary PLNs, 3 days or 14 days after a 20% TBSA, full-thickness burn, or sham injury. Bulk PLN cells were stimulated in vitro and intracellular cytokine staining was performed, and flow cytometry was used to measure IL-17 and IFN-γ production by CD3+ CD4+ cells. Representative examples of the flow cytometry histograms for each lymphoid organ are provided.
Figure 3.
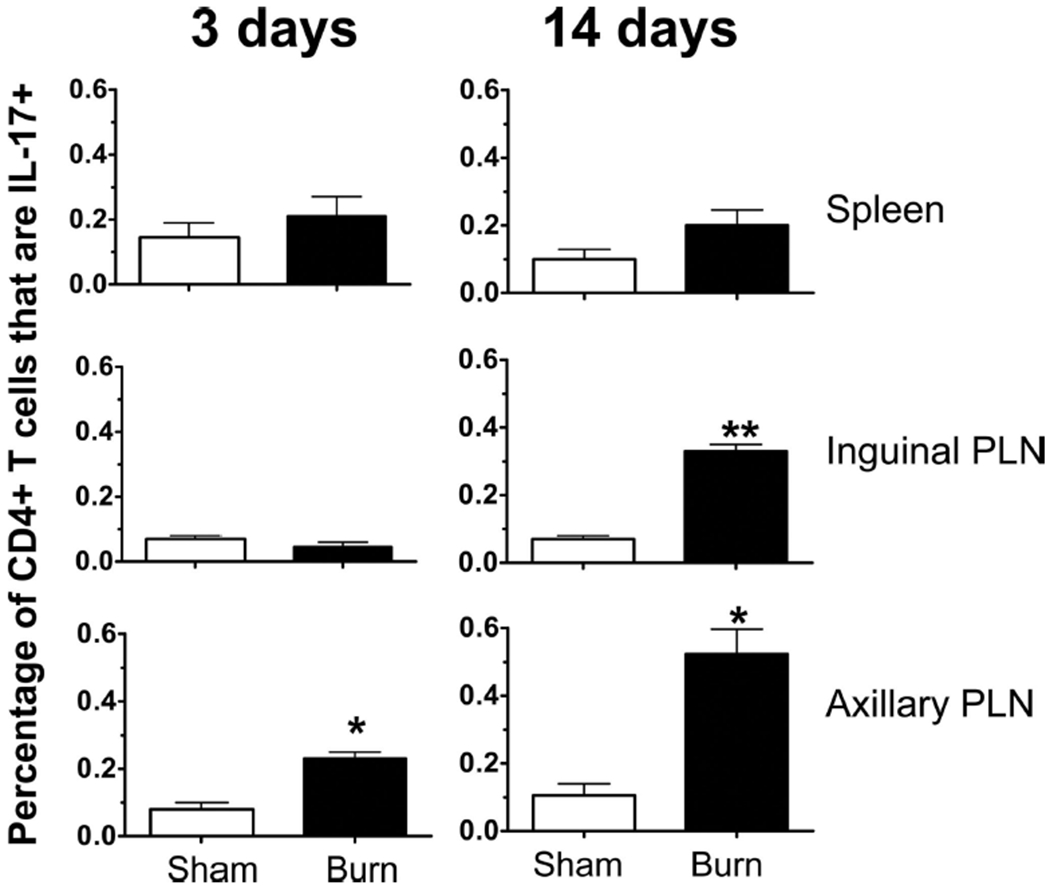
Percentage of CD4+ T cells that produce IL-17 are significantly higher in burn compared with sham. Cells were harvested from spleens, as well as inguinal and axillary PLNs, 3 days or 14 days after a 20% TBSA, full-thickness burn, or sham injury. Bulk PLN cells were stimulated in vitro and intracellular cytokine staining was performed, and flow cytometry was used to measure IL-17 and IFN-γ production by CD3+ CD4+ T cells. Data were expressed as mean ± standard error of the mean, *p ≤ 0.05; **p ≤ 0.005 compared with matched sham controls by Student’s t test (n = 4–6 mice/group).
Stimulation of CD4+ T cells by mitogen, such as phorbol myristate acetate and ionomycin, is well known to quickly and dramatically down-regulate CD4 coreceptor expression.44 To better quantify the proportion and absolute numbers of Th17 cells in the wound-draining lymph nodes, we repeated the above experiment but used a CD4+ T-cell purification step before mitogenic stimulation. At 3 days, 7 days, and 14 days after burn or sham injury, we harvested cells from various wound-draining PLN and pooled cells from each mouse to ensure enough cells were present for subsequent CD4+ T-cell purification and stimulation. At all time points, there was a statistically significant burn-dependent increase in the percentage of viable CD4+ T cells that were Th17 cells, as well as in the absolute number of Th17 cells, when compared with sham (Fig. 4).
Figure 4.
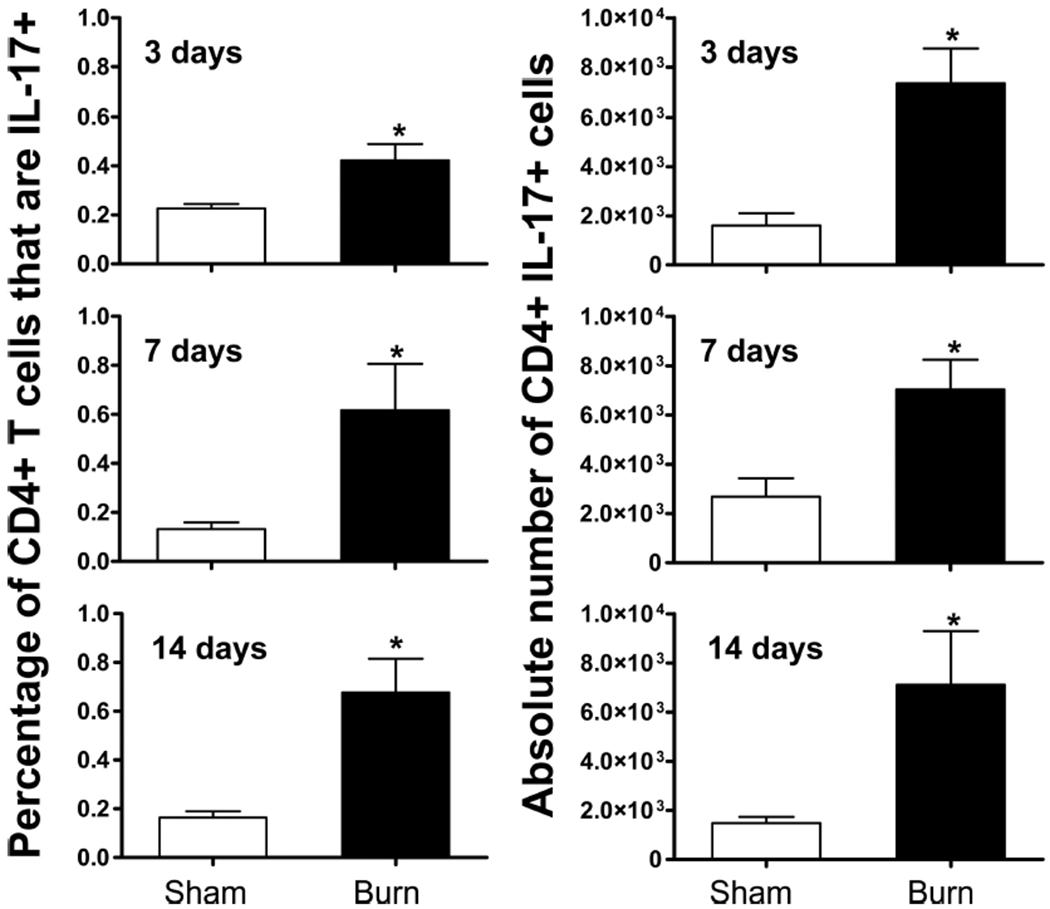
Frequency and absolute number of Th17 cells is significantly greater in the wound-draining lymph nodes of burn mice at all time points examined. Splenocytes and PLN cells were harvested at 3 days, 7 days, or 14 days after a 20% TBSA, full-thickness burn (black bar) or sham (white bar) injury. CD4+ T cells were enriched by negative magnetic selection and stimulated in vitro. Intracellular cytokine staining was performed, and flow cytometry was used to quantify IL-17+ CD4+ T cells. Data were expressed as mean ± standard error of the mean, *p ≤ 0.05 compared with matched sham controls by Student’s t test (n = 4–6 mice/group).
We also confirmed ROR-γt expression using an anti-ROR-γt antibody at these time points. ROR-γt staining was concurrent with IL-17 expression but not with IFN-γ expression at day 7 postburn, further confirming that these are Th17 cells (Fig. 5). ROR-γt was also expressed in Th17 cells at day 14, as expected, but there was also significant expression within Th1 cells. The plasticity of the Th1/Th17 lineage is an emerging concept in the current literature,12 and we are actively pursing this as a mechanism for Th17 production within burn mice.
Figure 5.
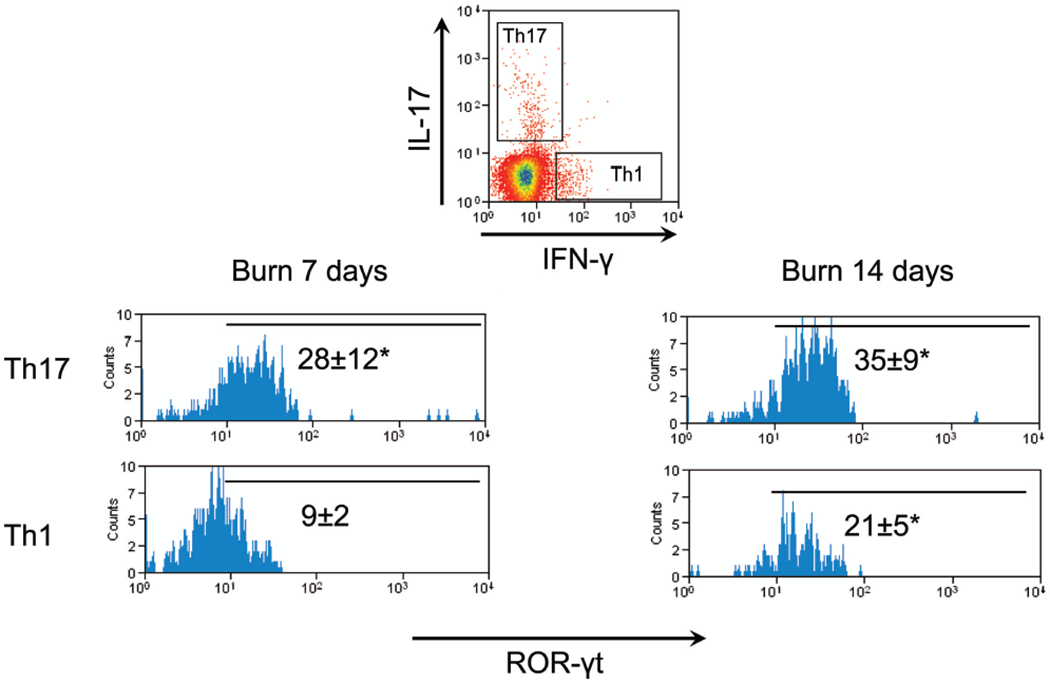
CD4+ T cells that produce IL-17 also express ROR-γt. Cells were harvested from inguinal and axillary PLN, 7 days or 14 days after a 20% TBSA, full-thickness burn injury. CD4+ T cells were enriched by negative magnetic selection and stimulated in vitro. Intracellular cytokine and ROR-γt staining was performed, and flow cytometry was used to gate Th17 (IL-17+ CD4+) and Th1 (IFN-g+ CD4+) T cells. Representative ROR-γt staining is shown in each gated population. Numbers represent mean ROR-γt fluorescent units ± standard error of the mean, *p ≤ 0.05; compared with matched isotype control by Student’s t test (n = 4–6 mice/group).
Burn Injury Induces a Dynamic Alteration in the Th1/Th17 Balance
Current studies demonstrate that strong inflammation mediated by cytokines, such as IL-6 and TGF-γ, results in Th17 differentiation of CD4+ pre-T cells (Fig. 1). To define the kinetics of Th17 to Th1 differentiation after burn injury, we calculated the ratio of the absolute number of Th17 cells (CD3+ CD4+ IL-17+) to the absolute number of Th1 cells (CD3+ CD4+ IFN-γ+) for each timepoint after sham or burn injury. For comparative purposes, we defined the sham mice having a Th17/Th1 ratio of 1.0 at each time point and normalized each burn mouse ratio to the sham. At day 3, we observed a significant shift of the proinflammatory T-cell phenotype toward a Th1 response in burn mice compared with sham, in agreement with previous studies.15,22 This shift was lost at day 7 after burn when there appeared to be equilibrium of Th17/Th1 balance similar to the sham mice. At day 14, there was a statistically significant difference between burn and sham with skewing of the proinflammatory CD4+ T-cell response to a Th17 phenotype in burn mice compared with sham (Fig. 6). These data suggest that although we observe an increase in Th17 cell number and percentage at all time points, there are increases of Th1 cells at day 3 and day 7 after burn, with day 3 having an overt burn-dependent Th1 response. We predict that day 14 represents a period after burn where the proinflammatory CD4+ T cell response is beginning to bias toward the weaker proinflammatory Th17 phenotype.
Figure 6.
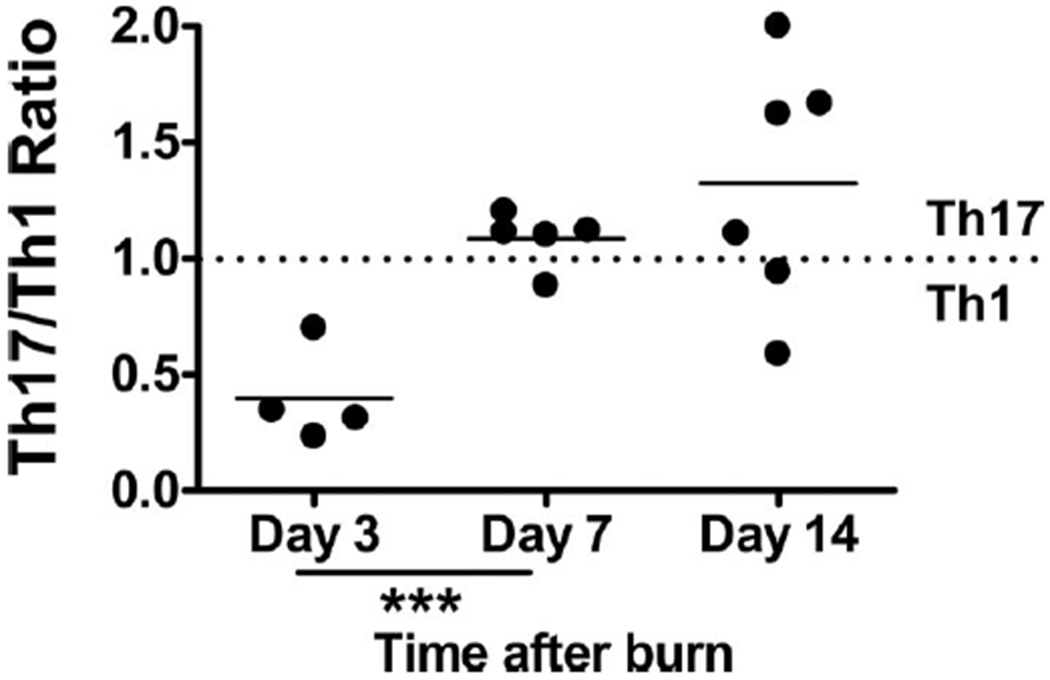
Th17/Th1 ratio is dynamic after burn injury. Cells were harvested from various PLNs at 3 days, 7 days, or 14 days after a 20% TBSA, full-thickness burn, or sham injury. Negative magnetic selection was performed to enrich the CD4+ T-cell population. CD4+ cells were stimulated in vitro before intracellular cytokine staining was performed, and flow cytometry was used to quantify IL-17+ CD4+ T cells. For each time point, the average sham Th17/Th1 ratio was set at 1.0. For each burn mouse, its Th17/Th1 ratio was normalized to the corresponding sham ratio. A ratio >1.0 represents a skew in the proinflammatory response to a Th17 phenotype, whereas a ratio below 1.0 indicates that a Th1 phenotype is dominant. ***p ≤ 0.0005 compared with matched sham controls by Student’s t test (n = 4–6 mice/group).
CD4+ T Cells From Burn and Sham Mice Have Similar Ability to Polarize Toward a Th17 Phenotype
Various cytokines are known to polarize a proinflammatory response toward a Th17 phenotype, such as IL-6 and TGFβ. IL-23 is known to “fix” this phenotype in place. As a possible cellular mechanism for increased Th17 development, we assessed the ability of CD4+ T cells isolated from burn and sham mice to polarize in vitro toward a Th17-phenotype. We also tested whether serum collected from burn mice was able to aid in the ability to polarize. Serum was collected and CD4+ T cells were purified from PLN 3 days and 14 days after burn or sham injury. The CD4+ T cells were activated in vitro via T-Cell receptor ligation with anti-CD3 antibody and anti-CD28 costimulation in presence of IL-6 and TGF-β for 4 days, which has been previously shown to promote Th17 differentiation.2,3 Sham and burn T cells were also stimulated in the presence or absence of burn serum. Th17 and Th1 CD4+ T cells were identified after each culture condition using intracellular cytokine staining and flow cytometry. At both 3 days and 14 days, CD4+ T cells from burn mice had an equivalent ability to polarize to Th17 cells when compared with sham in the presence of know polarizing conditions (Fig. 7). We predicted that as the Th17 phenotype was maximal at day 14, the presence of IL-23 or some other unidentified serum factor would also be maximal at this time point. Addition of 14-day burn serum had no effect on Th17 polarization or stabilization of sham or burn CD4+ T cells (Fig. 8) nor was there significant difference between the response to serum alone and to any of the treatments shown in Figure 8. These data suggest that burn injury has no inherent effect on the ability of CD4+ T cells to bias toward a Th17 phenotype under in vitro polarizing cytokine conditions. For example, serum from burn mice may not contain any soluble factors, such as IL-23, which affects Th17 polarization.2,9–11
Figure 7.
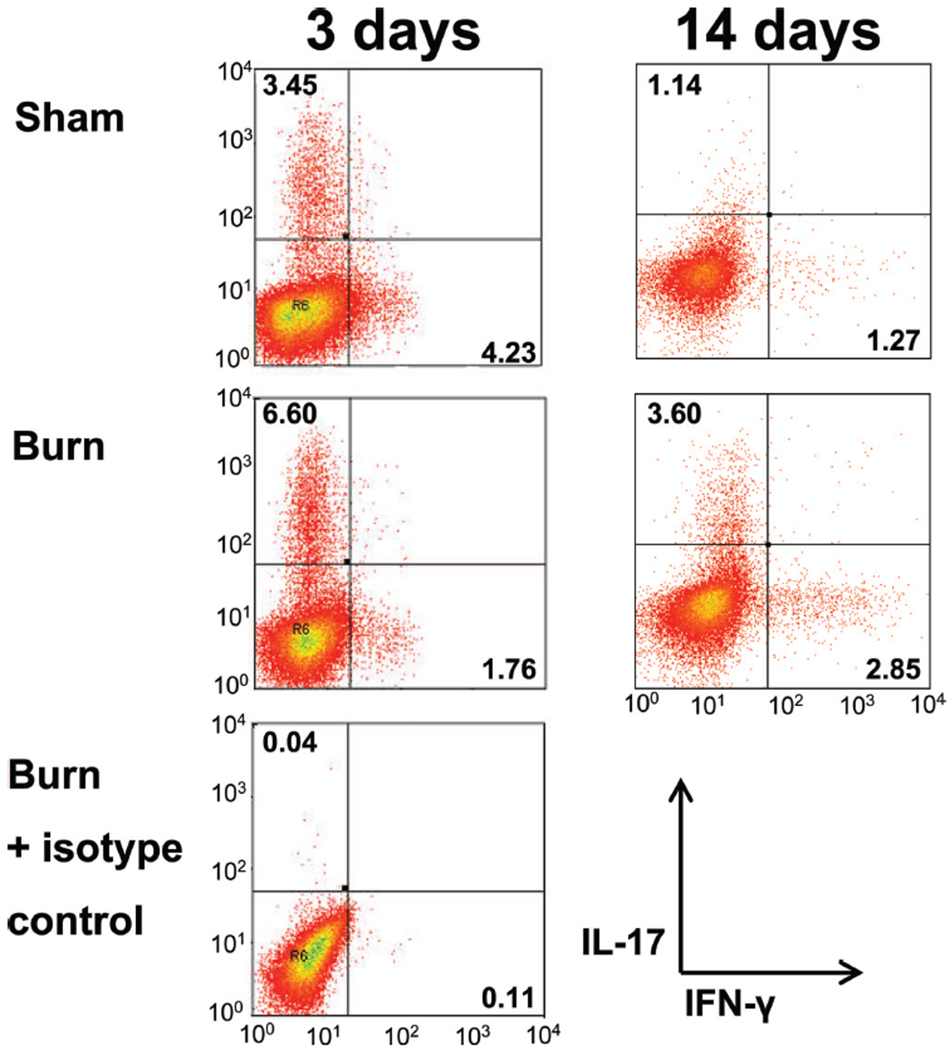
CD4+ T cells from burn and sham mice have similar abilities to polarize to Th17 cells. Cells were harvested from various PLNs at 3 days or 14 days after a 20% TBSA, full-thickness burn, or sham injury. CD4+ T cells were enriched by negative magnetic selection and cultured with plate bound anti-CD3, soluble anti-CD28, IL-6, and TGF-β for 4 days. IL-17 and IFN-γ production by CD3+ CD4+ cells was analyzed using flow cytometric staining. Representative examples of the histograms for each timepoint are provided.
Figure 8.
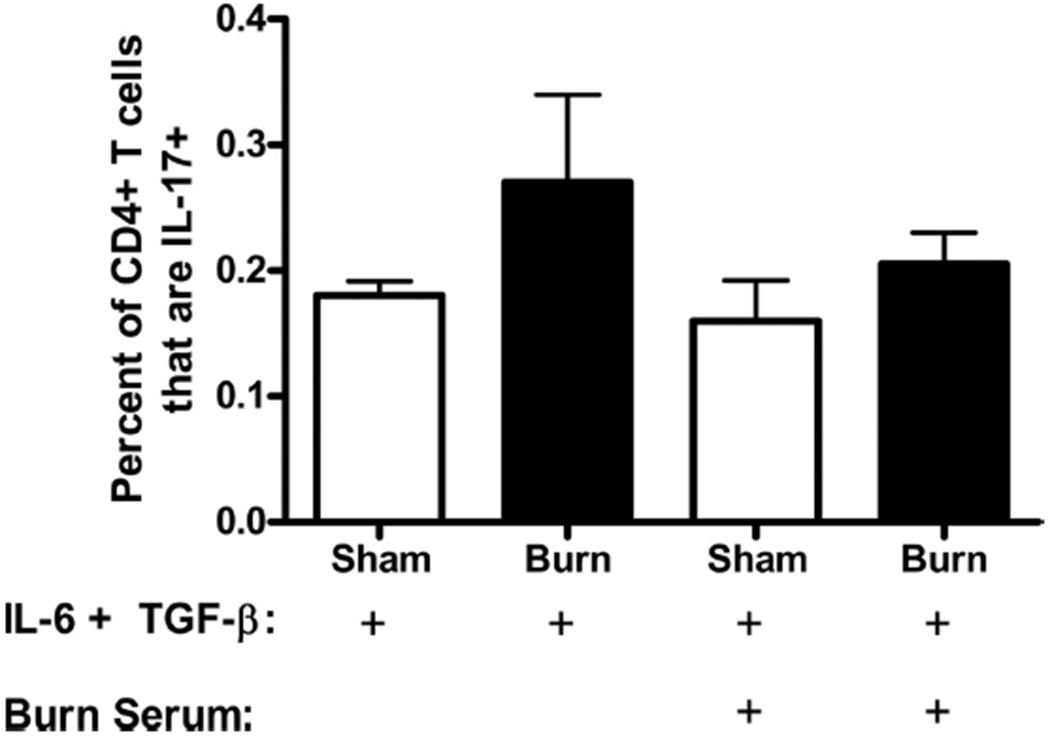
Burn serum does not contain a soluble factor that increases that ability of CD4+ T cells to polarize to a Th17 phenotype. Cells were harvested from various PLNs at 14 days after a 20% TBSA, full-thickness burn, or sham injury. Burn serum was also collected at 14 days after burn injury. CD4+ T cells were enriched by negative magnetic selection. Cells were then cultured with plate bound anti-CD3, soluble anti-CD28, IL-6, and TGF-β in the presence or absence of burn serum for 4 days. IL-17 and IFN-γ production by CD3+ CD4+ cells was analyzed using flow cytometric staining. Isotype refers to staining with isotype control antibodies. Data were expressed as mean ± standard error of the mean (n = 4–6 mice/group).
DISCUSSION
Many variables contribute to the development, regulation, and effector functions of CD4+ Th1, Th17, and regulatory T cells. We hypothesized that burn injury would have a profound effect on the balance of Th1/Th17 T cells. The main reasoning behind this was that certain cytokines known to promote Th17 polarization are present after burn injury, namely IL-6 in the serum and TGF-β in the burn wound.
When examining bulk and CD4+-purified T-cell populations, we observed an increase in percentage of CD4+ cells that were expressing IL-17 (Figs. 2 and 3) in PLN draining the burn wound. However, these cells were not detected in the spleen. This localization of the Th17 cells might be due to higher concentrations of TGF-β in the draining PLN, or cells trafficking to the PLN from the wound where they were initially formed. The increase at day 14 compared with day 3 of Th17 cell number in the bulk T-cell cultures from axillary and inguinal PLN (Figs. 2 and 3) suggests that the signals generated from burn accumulate over time or increase later after burn, with a possible spread into more distal PLN (but not into the spleen). The Th17 response is observed bilaterally in the axillary and inguinal PLN. However, it should be noted that because of the way we perform our burn injury, corresponding lymph nodes are approximately equidistant to the burn wound. However, it does appear that proximity to the wound corresponds to Th17 expansion (Fig. 2). This is under further study.
To eliminate the possibility of underestimating the absolute number of Th17, due to likely downregulation of the CD4 coreceptor during T-cell stimulation, we purified the CD4+ T-cell population before culturing. To generate enough cells for this selection process, various wound-draining PLN were collected and pooled together for each mouse. With the addition of the CD4+ T-cell purification, we also saw a significant increase in the number of Th17 T cells in wound draining PLN across all time points compared with sham (Fig. 4). Although the percentage and cell number differences between burn and sham are statistically significant, the values are modest. This is likely because of the inefficiency and low sensitivity of intracellular cytokine staining at revealing polarized T-cell subsets. We identified ROR-γt staining within these cells to confirm their phenotype.
Although the proportion and absolute number of Th17 cells are important to study, the role of the Th1/Th17 balance has been postulated as being key for overall proinflammatory status in human and animal studies. Indeed, after burn, we observed a dynamic shift in this ratio. To account for variability between intracellular cytokine staining between experiments, we defined the sham as having a Th1/Th17 ratio of 1.0 at each case so that data from different experiments could be compared. In agreement with previous studies,15,22 we observed an early significant shift of the proinflammatory T-cell phenotype toward a Th1 response in burn mice compared with sham. This shift was lost at day 7 after burn, but at the day 14 time point, there was a clear skewing of the proinflammatory CD4+ T-cell response to a Th17 phenotype in burn mice compared with sham (Fig. 6). These data suggest that an increase of Th1 cells at day 3 and day 7 after burn offsets the increased Th17 differentiation we observed, meaning that Th1-promoting effects of burn over-ride the Th17-polarizing effects of IL-6 and TGF-β produced in response to burn injury.
These results suggest that day 3 represents a period of overt burn-dependent Th1 responses to antigen. Because IFN-γ is known to inhibit naïve CD4+ T cells development toward a Th17 pathway, this would likely impact the ability of the patient to recruit neutrophils to sites of infections. Similarly, IL-12 is another cytokine driving differentiation into Th1 cells. Although it has been shown that inducing a stronger Th1-response by IL-12 administration can protect against cecal ligation and puncture sepsis in mice,45 such a response has not been specifically studied with respect to the role of Th17 cells and pathogens that require rapid neutrophil clearance (e.g., Pseudomonas, Klebsiella, and Candida) mediated effectively by Th17 cells. Indeed, when burn mice are challenged with Pseudomonas aeruginosa, they experience high levels of mortality within 2 to 3 days after injury correlated with a overt proinflammatory response.46 Therefore, driving differentiation at these early time points toward a Th17 differentiation pathway could be a potential benefit for patients.
In contrast to the early proinflammatory Th1 phase, it seems that day 14 in our mouse model represents a period after burn where the proinflammatory CD4+ T-cell response is tempered, appearing to bias toward the Th17 phenotype on stimulation. This is a period of time we have previously defined as having unique and dramatic altered cytokine profile after burn injury, with exaggerated cytokine responses but no clear dominant Th1 or Th2 phenotype. Because elevated TGF-β and IL-6 levels are found after burn injury, we hypothesize that CD4+ Th17 cells are generated late after burn injury and become the dominant Th phenotype.
The mechanism for this Th17-dominance is currently unclear. The driving force for the Th1/Th2 cytokine imbalance at day 14 after burn injury seems to be intense homeostatic expansion of “spontaneous” memory-like CD8+ and CD4+ T cells as a consequence of lymphopenia early after burn injury.22 Th17 cells, also often defined as possessing a memory-like phenotype, have not yet been studied as being a consequence of homeostatic proliferation. Another potential mechanism is that Th17 polarization can be driven in vitro and in vivo by innate stimulation through Toll-like receptors. (TLR).47 We have shown altered TLR expression on innate cells (e.g., macrophages express lower TLR) and adaptive cell (T cells express higher levels of TLR) late after burn injury.48,49 Therefore, we predict that T cells are stimulated directly or indirectly via innate cells to various innate stimuli produced by burn injury, such as endogenous innate signaling molecules50–58 released from the burn wound. Although the results suggest that burn serum did not affect Th17 polarization of sham CD4+ T cells during in vitro stimulation under TGF-β and IL-6 conditions, the serum levels of such endogenous innate signaling molecules might not be high enough to detect any differences. Experiments to test the role of innate stimulation after burn in affecting adaptive T cell responses are underway.
Attempts to improve the T cell response to burn injury by manipulating cytokines has been largely been unsuccessful and are not generally used in the clinic. Our data suggest why cytokine manipulation may be unsuccessful. For example, the CD4+ T cell phenotype after burn injury is dynamic and may already be set along a certain proinflammatory phenotype when proinflammatory cytokines, such as IFN-γ or IL-2, are being administered. In addition, we have shown previously that immune dysfunction is clearly more of a late, rather than early, problem after injury as infection, sepsis, and multiple system organ failure develop weeks to months after burn injury. Thus, the majority of lymphocyte studies that focus solely on the early response to burn injury (<10 days) or have been given an additional insult, such as cecal ligation and puncture, may have missed an important characteristic of the T cell response to injury.
DISCUSSION.
Dr. Matthew Rosengart (Oakland, Pennsylvania): Doctor Peitzman, Doctor Cioffi, members and guests and specifically authors Neely and colleagues, thank you for the opportunity to discuss this intriguing and provocative study.
For decades we have acknowledged that the insult of thermal injury all too frequently leads to the development of an immunosuppressed state, characterized by an increased risk for secondary infection. In this study, authors Neely and colleagues further our understanding by identifying the presence of a Th17 population of lymphocytes in the nodal basins of thermally injured mice. Interestingly, similar events did not occur in the spleen. Distinct from Th1 and Th2 cells in both development and function, this small subpopulation is induced from naïve T cells by concomitant exposure to TGF-γ and IL-6. stabilized by IL-23, and clearly integral to a competent immune response to extracellular organisms. Furthermore, the authors characterize a temporal development, a shift from Th1 to Th17 phenotype that occurs over the span of approximately 14 days. They speculate, as the title alludes, that this population may contribute to post-burn immunosuppression. The authors should be congratulated for their work and for embarking into a relatively unexplored field.
I have a few questions:
IL-17 carries a predominantly pro-inflammatory effect, activating macrophages to induce the release of inflammatory cytokines, such as TNF-α and IL-1. These cytokines induce neutrophil recruitment, and acute inflammation in which neutrophils are prominent is almost hallmark of Th17-driven inflammation. Furthermore, the elaboration of IL-17 appears integral to the host response to the challenge of extracellular organisms, as mice deficient in IL-17 have increased susceptibility to and complications from Klebsiella sp., Candida sp.. In the context of your title, can you provide further insight into how this small, specialized subpopulation of Th17 cells might underlie the immunsuppression of thermal injury as so stated? Have you conducted studies in which you modulate the induction of this population (through TGF-β or TGF-βR blockade) or its effector function (i.e. IL-17 inhibition or IL-17R−/− mice) and assessed immunocompetency after thermal insult? By contrast, does adoptive transfer of these lymphocytes alter immunocompentency?
Th17-mediated immunity is particularly important at epithelial and mucosal surfaces. In fact, Th17 cells are causally implicated in psoriasis and inflammatory bowel disease, and antibodies against IL-17 or the IL-17R have been developed and are under evaluation in psoriasis and Crohn’s disease. In the context of the known loss of barrier and immune function at the integument and gastrointestinal system, have you characterized the Th17 response at these regions, in draining nodal basins, in your burn model. I would be further interested in how modulating IL-17 may facilitate restoration of the immune barrier function at these sites.
A number of pathogens induce mainly a Th17 response, including the aforementioned for which IL-17 deficient mice are more susceptible. Do you have data on the colonization of the wounds with these specific organisms and could colonization explain the underlying induction of Th17. We typically treat burns with silvadene to minimize colonization. Do you think that this might alter your results, though be more clinically representative? This might also explain why nodal basins draining these wounds would host a significant Th17 response, by contrast to the spleen.
I was intrigued by the nodal predilection of Th17 development, yet the bland response in the spleen. In light of the known mechanism by which naïve T cells are induced to differentiate into Th17 phenotype, specifically the concomitant presence of TGF-B and IL-6, can you provide any mechanistic data underlying this apparent nodal predilection? Specifically is the local cytokine mileau (i.e. TGF-β and IL-6 concentrations) different within the spleen than within the lymph node. Several other necessary events could also explain this disparate differentiation, including the activation of the transcription factor ROR-γt, the expression of IL-23, the IL-23R, and IL-21. Do you have any data on whether these processes occur differently within different lymphatic organs. I can’t help but reflect on Kevin Trancey’s cholinergic anti-inflammatory pathways conducted through splenic vagal efferent’s. Perhaps you could comment on this work in the context of the disparate response you noted in lymph nodes and the spleen.
In closing I applaud the authors for their work and am grateful for the kind provision of a copy of the manuscript for review. I thank you for your time and the society for the priveledge of the podium.
Dr. Crystal J. Neely (Chapel Hill, North Carolina): Thank you, Doctor Rosengart for reading the manuscript ahead of time and providing your interesting comments and provocative questions.
To begin, although we believe that TH17 cells are weak pro-inflammatory cells, we do believe that they are involved in immunosuppression so it is true that the emergence of T17 cells after burn injury may seem counterintuitive but we know that the relative T cell lymphopenia 14 days after – we know that there is a relative T cell lymphopenia 14 days after burn injury and that unique T cell populations arise by this time and modulate the response.
For example, we have previously shown that there is a hyper-responsive CD8 positive T cell emergence late after burn injury and that they can mediate allogenic skin graft rejection in the face of overall immune suppression.
The emergency of this TH17 population also seems to make sense because of their principle function in signaling and recruitment of neutrophils. Whether the response is effective or beneficial has not been yet – has not been assessed yet and requires further study.
As for the questions regarding the TGF blockade as well as the adoptive transfer of these cells, this has not been performed yet and this is something that we are very interested in doing in the near future.
Your question regarding TH17 cells and the gut, skin, and mesenteric lymph nodes are important and require further investigation. We are not sure why TH17 cells are not elevated in the spleen.
But we believe that it is interesting that we have found in the local environment where IL-6 and TGF-beta are elevated that there are TH17 cells.
The relative contribution of TH17 cells to the overall immune response is not known but it is also not surprising. Other relatively small T cell subsets are known to be substantially, have substantial regulatory effects.
Suffice it to say no one is quite sure what controls the T cell responses to burn injury and what we can do about it.
To address your questions about the infection models, we have been performing the burn model without infectious complications in the lab for over 20 years.
I have been performing recent studies in the lab using pseudomonas challenge following burn injury. The controls for those experiments we’ve never seen infection or colonization out so therefore we don’t expect that the arisal of T17 cells is due to unexpected infections.
And thank you for the opportunity to present here today.
Acknowledgments
Supported by NIGMS Grant R01 GM076250 (to B.A.C.) and the North Carolina Jaycee Burn Center.
Footnotes
Presented at the 68th Annual Meeting of the American Association for the Surgery of Trauma, October 1–3, 2009, Pittsburgh, Pennsylvania.
REFERENCES
- 1.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 2.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. [DOI] [PubMed] [Google Scholar]
- 3.Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361:888–898. [DOI] [PubMed] [Google Scholar]
- 5.Dragon S, Saffar AS, Shan L, Gounni AS. IL-17 attenuates the anti-apoptotic effects of GM-CSF in human neutrophils. Mol Immunol. 2008;45:160–168. [DOI] [PubMed] [Google Scholar]
- 6.Ye P, Rodriguez FH, Kanaly S, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye P, Garvey PB, Zhang P, et al. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Cell Mol Biol. 2001;25:335–340. [DOI] [PubMed] [Google Scholar]
- 8.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of inter-leukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis. 2004;190:624–631. [DOI] [PubMed] [Google Scholar]
- 9.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. [DOI] [PubMed] [Google Scholar]
- 10.Mangan PR, Harrington LE, O’Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. [DOI] [PubMed] [Google Scholar]
- 11.Veldhoen M, Hocking RJ, Atkins CJ, et al. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. [DOI] [PubMed] [Google Scholar]
- 12.Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. [DOI] [PubMed] [Google Scholar]
- 13.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. [DOI] [PubMed] [Google Scholar]
- 14.Buchanan I, Maile R, Frelinger J, Fair JH, Meyer AA, Cairns BA. The effect of burn injury on CD8+ and CD4+ T cells in an irradiation model of homeostatic proliferation. J Trauma. 2006;61:1062–1068. [DOI] [PubMed] [Google Scholar]
- 15.Cairns BA, Maile R, Buchanan I, et al. CD8 T cells express a T-helper 1-like phenotype after burn injury. Surgery. 2001;130:210–216. [DOI] [PubMed] [Google Scholar]
- 16.Cho K, Adamson LK, Park J, Greenhalgh DG. Burn injury-mediated alterations in cell cycle progression in lymphoid organs of mice. Shock. 2003;19:138–143. [DOI] [PubMed] [Google Scholar]
- 17.Guo Z, Kavanagh E, Zang Y, et al. Burn injury promotes antigen-driven Th2-type responses in vivo. J Immunol. 2003;171:3983–3990. [DOI] [PubMed] [Google Scholar]
- 18.Hultman CS, Cairns BA, deSerres S, Frelinger JA, Meyer AA. Burn injury impairs second-set rejection and CTL reactivity in mice primed by cultured keratinocyte allografts. Transplantation. 1995;60:584–589. [DOI] [PubMed] [Google Scholar]
- 19.Kavanagh EG, Kelly JL, Lyons A, Soberg CC, Mannick JA, Lederer JA. Burn injury primes naive CD4+ T cells for an augmented T-helper 1 response. Surgery. 1998;124:269–276; discussion 276–267. [PubMed] [Google Scholar]
- 20.Kell MR, Kavanaugh EG, Goebel A, Soberg CC, Lederer JA. Injury primes the immune system for an enhanced and lethal T-cell response against bacterial superantigen. Shock. 1999;12:139–144. [DOI] [PubMed] [Google Scholar]
- 21.Maekawa T, Kajihara H, Okabayashi K, Otani M, Yuge O. Impairment of splenic B and T lymphocytes in the early period after severe thermal injury: immunohistochemical and electron microscopic analysis. Burns. 2002;28:329–339. [DOI] [PubMed] [Google Scholar]
- 22.Maile R, Barnes CM, Nielsen AI, Meyer AA, Frelinger JA, Cairns BA. Lymphopenia-induced homeostatic proliferation of CD8+ T cells is a mechanism for effective allogeneic skin graft rejection following burn injury. J Immunol. 2006;176:6717–6726. [DOI] [PubMed] [Google Scholar]
- 23.Mannick JA, Rodrick ML, Lederer JA. The immunologic response to injury. J Am Coll Surg. 2001;193:237–244. [DOI] [PubMed] [Google Scholar]
- 24.Murphy TJ, Ni Choileain N, Zang Y, Mannick JA, Lederer JA. CD4+CD25+ regulatory T cells control innate immune reactivity after injury. J Immunol. 2005;174:2957–2963. [DOI] [PubMed] [Google Scholar]
- 25.Organ BC, Antonacci AC, Chiao J, et al. Changes in lymphocyte number and phenotype in seven lymphoid compartments after thermal injury. Ann Surg. 1989;210:78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Sullivan ST, Lederer JA, Horgan AF, Chin DH, Mannick JA, Rodrick ML. Major injury leads to predominance of the T helper-2 lymphocyte phenotype and diminished interleukin-12 production associated with decreased resistance to infection. Ann Surg. 1995;222:482–490; discussion 490–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patenaude J, D’Elia M, Hamelin C, Garrel D, Bernier J. Burn injury induces a change in T cell homeostasis affecting preferentially CD4+ T cells. J Leukoc Biol. 2005;77:141–150. [DOI] [PubMed] [Google Scholar]
- 28.Schwacha MG. Macrophages and post-burn immune dysfunction. Burns. 2003;29:1–14. [DOI] [PubMed] [Google Scholar]
- 29.Teodorczyk-Injeyan JA, Cembrzynska-Nowak M, Lalani S, Peters WJ, Mills GB. Immune deficiency following thermal trauma is associated with apoptotic cell death. J Clin Immunol. 1995;15:318–328. [DOI] [PubMed] [Google Scholar]
- 30.Teodorczyk-Injeyan JA, Sparkes BG, Mills GB, Peters WJ. Immunosuppression follows systemic T lymphocyte activation in the burn patient. Clin Exp Immunol. 1991;85:515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zang Y, Dolan SM, Choileain NN, et al. Burn injury initiates a shift in superantigen-induced T cell responses and host survival. J Immunol. 2004;172:4883–4892. [DOI] [PubMed] [Google Scholar]
- 32.Zedler S, Bone RC, Baue AE, von Donnersmarck GH, Faist E. T-cell reactivity and its predictive role in immunosuppression after burns. Crit Care Med. 1999;27:66–72. [DOI] [PubMed] [Google Scholar]
- 33.Zedler S, Faist E, Ostenneier B, von Donnersmarck GH, Schildberg FW. Postburn constitutional changes in T-cell reactivity occur in CD8+ rather than in CD4+ cells. J Trauma. 1997;42:872–880; discussion 880–881. [DOI] [PubMed] [Google Scholar]
- 34.Kelly JL, O’Sullivan C, O’Riordain M, et al. Is circulating endotoxin the trigger for the systemic inflammatory response syndrome seen after injury? Ann Surg. 1997;225:530–541; discussion 541–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deitch EA. Multiple organ failure. Pathophysiology and potential future therapy. Ann Surg. 1992;216:117–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Qattan MM. “Late’ multiorgan failure in major burns: a “three-event” construct rather than a “two-event” construct. Burns. 2007;33:268–270. [DOI] [PubMed] [Google Scholar]
- 37.Ogle CK, Mao JX, Wu JZ, Ogle JD, Alexander JW. The 1994 Lindberg Award. The production of tumor necrosis factor, interleukin-1, interleukin-6, and prostaglandin E2 by isolated enterocytes and gut macrophages: effect of lipopolysaccharide and thermal injury. J Burn Care Rehabil. 1994;15:470–477. [PubMed] [Google Scholar]
- 38.Wei D, Ge S, Chen Y, Dai F, Su B. Expression of endogenous transforming growth factor-beta and its type I and type II receptors in rat burn wounds. Wound Repair Regen. 1997;5:229–234. [DOI] [PubMed] [Google Scholar]
- 39.Utsunomiya T, Kobayashi M, Herndon DN, Pollard RB, Suzuki F. A relationship between the generation of burn-associated type 2 T cells and their antagonistic cells in thermally injured mice. Burns. 1997:23:281–287. [DOI] [PubMed] [Google Scholar]
- 40.Hultman CS, Cairns BA, deSerres S, Frelinger JA, Meyer AA. Early, complete burn wound excision partially restores cytotoxic T lymphocyte function. Surgery. 1995;118:421–429; discussion 429–430. [DOI] [PubMed] [Google Scholar]
- 41.Hultman CS, Cairns BA, Yamamoto H, deSerres S, Frelinger JA, Meyer AA. The 1995 Moyer Award. The effect of burn injury on allograft rejection, alloantigen processing, and cytotoxic T-lymphocyte sensitization. J Burn Care Rehabil. 1995;16:573–580. [PubMed] [Google Scholar]
- 42.Ni Choileain N, Macconmara M, Zang Y, Murphy TJ, Mannick JA, Lederer JA. Enhanced regulatory T cell activity is an element of the host response to injury. J Immunol. 2006;176:225–236. [DOI] [PubMed] [Google Scholar]
- 43.Kataranovski M, Kucuk J, Colic M, et al. Post-traumatic activation of draining lymph node cells. II. Proliferative and phenotypic characteristics. Burns. 1994;20:403–408. [DOI] [PubMed] [Google Scholar]
- 44.Richie ER, McEntire B, Phillips J, Allison JP. Altered expression of lymphocyte differentiation antigens on phorbol ester-activated CD4+8+ T cells. J Immunol. 1988; 140:4115–4122. [PubMed] [Google Scholar]
- 45.O’Suilleabhain C, O’Sullivan ST, Kelly JL, Lederer J, Mannick JA, Rodrick ML. Interleukin-12 treatment restores normal resistance to bacterial challenge after burn injury. Surgery. 1996;120:290–296. [DOI] [PubMed] [Google Scholar]
- 46.Murphey ED, Sherwood ER. Bacterial clearance and mortality are not improved by a combination of IL-10 neutralization and IFN-gamma administration in a murine model of post-CLP immunosuppression. Shock. 2006;26:417–424. [DOI] [PubMed] [Google Scholar]
- 47.Jyonouchi H, Geng L, Cushing-Ruby A, Monteiro IM. Aberrant responses to TLR agonists in pediatric IBD patients; the possible association with increased production of Th1/Th17 cytokines in response to Candida, a luminal antigen. Pediatr Allergy Immunol. 2010:21(4 Pt 2):e747–e755. [DOI] [PubMed] [Google Scholar]
- 48.Cairns B, Maile R, Barnes CM, Frelinger JA, Meyer AA. Increased toll-like receptor 4 expression on T cells may be a mechanism for enhanced T cell response late after burn injury. J Trauma. 2006;61:293–298; discussion 298–299. [DOI] [PubMed] [Google Scholar]
- 49.Cairns BA, Barnes CM, Mlot S, Meyer AA, Maile R. Toll-like receptor 2 and 4 ligation results in complex altered cytokine profiles early and late after burn injury. J Trauma. 2008;64:1069–1077; discussion 1077–1068. [DOI] [PubMed] [Google Scholar]
- 50.Anders HJ, Banas B, Schlondorff D. Signaling danger: toll-like receptors and their potential roles in kidney disease. J Am Soc Nephrol. 2004;15:854–867. [DOI] [PubMed] [Google Scholar]
- 51.Dinarello CA. Infection, fever, and exogenous and endogenous pyrogens: some concepts have changed. J Endotoxin Res. 2004;10:201–222. [DOI] [PubMed] [Google Scholar]
- 52.Guillot L, Balloy V, McCormack FX, et al. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J Immunol. 2002;168:5989–5992. [DOI] [PubMed] [Google Scholar]
- 53.Iliev AI, Stringaris AK, Nau R, Neumann H. Neuronal injury mediated via stimulation of microglial toll-like receptor-9 (TLR9). FASEB J. 2004;18:412–414. [DOI] [PubMed] [Google Scholar]
- 54.Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 2002;168:5233–5239. [DOI] [PubMed] [Google Scholar]
- 55.Johnson GB, Brunn GJ, Platt JL. Cutting edge: an endogenous pathway to systemic inflammatory response syndrome (SIRS)-like reactions through Toll-like receptor 4. J Immunol. 2004;172:20–24. [DOI] [PubMed] [Google Scholar]
- 56.Kirschning CJ, Schumann RR. TLR2: cellular sensor for microbial and endogenous molecular patterns. Curr Top Microbiol Immunol. 2002;270:121–144. [DOI] [PubMed] [Google Scholar]
- 57.Marshak-Rothstein A, Busconi L, Rifkin IR, Viglianti GA. The stimulation of Toll-like receptors by nuclear antigens: a link between apoptosis and autoimmunity. Rheum Dis Clin North Am. 2004;30:559–574, ix. [DOI] [PubMed] [Google Scholar]
- 58.Paterson HM, Murphy TJ, Purcell EJ, et al. Injury primes the innate immune system for enhanced Toll-like receptor reactivity. J Immunol. 2003;171:1473–1483. [DOI] [PubMed] [Google Scholar]