

Abstract
SARS-CoV-2, the virus that causes coronavirus disease 19 (COVID-19), is associated with a bewildering array of cardiovascular manifestations, including myocardial infarction and stroke, myocarditis and heart failure, atrial and ventricular arrhythmias, venous thromboembolism, and microvascular disease. Accumulating evidence indicates that a profound disturbance of endothelial homeostasis contributes to these conditions. Furthermore, the pulmonary infiltration and edema, and later pulmonary fibrosis, in patients with COVID-19 is promoted by endothelial alterations including the expression of endothelial adhesion molecules and chemokines, increased intercellular permeability, and endothelial-to-mesenchyme transitions. The cognitive disturbance occurring in this disease may also be due in part to an impairment of the blood-brain barrier. Venous thrombosis and pulmonary thromboembolism are most likely associated with an endothelial defect caused by circulating inflammatory cytokines and/or direct endothelial invasion by the virus. Endothelial-targeted therapies such as statins, nitric oxide donors, and antioxidants may be useful therapeutic adjuncts in COVID-19 by restoring endothelial homeostasis.
Keywords: SARS-CoV-2, endothelium, deep venous thrombosis, pulmonary embolism, myocardial infarction, cerebrovascular attack, dementia, myocarditis, nitric oxide
Introduction
The endothelium is a diaphanous film of tissue that invests the luminal surface of all blood vessels with a nonthrombogenic lining. The endothelium is critically important in vascular patency and homeostasis. It generates factors that prevent platelet adherence and aggregation, suppresses leukocyte adhesion and infiltration, relaxes the vascular smooth muscle to permit the smooth flow of blood, and inhibits abnormal growth of vascular cells to prevent intimal and medial hyperplasia. The healthy endothelium also governs the exchange of gases, nutrients, fluids, and metabolites into organs and tissues. Thus, a healthy endothelium is critical for normal cardiovascular structure and function. Viral impairment of endothelial function has been noted in multiple acute viral diseases, including coronavirus disease 19 (COVID-19, caused by the SARS-CoV-2 virus). This endothelial disruption could cause or contribute to a number of cardiovascular disorders involving thrombosis, inflammation, altered vascular permeability, contractility, and proliferation.
A Role for Endotheliopathy in Acute Cardiovascular Events
Accumulating evidence indicates that the cardiovascular manifestations of COVID-19 are likely due to a profound endothelial alteration secondary to circulating inflammatory cytokines and/or direct viral invasion of the endothelium1,2,3,4 as well as infection of supporting cells (eg, pericytes).5,6 Viral inclusion bodies have been observed by electron microscopy in tissues from patients succumbing to SARS-CoV-2, and circulating inflammatory factors in infected patients cause systemic impairment of endothelial function.7,8 A viral induction of endothelial pathology would be expected to underlie many of the cardiovascular manifestations of COVID-19. Indeed, endothelial dysfunction is a known independent risk factor for heart attack and stroke. All known risk factors for cardiovascular disease (eg, hypertension, diabetes mellitus, hypercholesterolemia) cause endothelial dysfunction long before there is any histopathological evidence of vascular disease.9,10,11 Genetic induction of endothelial dysfunction (as with endothelial nitric oxide synthase knockout mice) accelerates vascular disease.12,13
The hypothesis that an endotheliopathy underlies COVID-19 is supported by prior observations that other viral illnesses can induce endothelial dysfunction, and these viral illnesses are associated with later cardiovascular disease.14,15,16,17,18,19,20,21 For example, prior infection with influenza is known to be associated with cardiovascular disease. In this regard, it is notable that post-mortem tissues from patients succumbing to influenza or COVID-19 show greater endothelial damage from SARS-CoV-2 infection than with H1N1 infection.22 Of concern, we have shown that viral activation of cell-autonomous innate immune activation induces epigenetic alterations that facilitate nuclear reprogramming to a different cell phenotype or lineage,23,24,25,26,27 with a loss of normal cellular functions that maintain homeostasis. As discussed below, such epigenetic changes may have long-term effects and contribute to post-COVID syndrome and later cardiovascular disease.
The endotheliopathy induced by COVID-19 contributes to a hyperinflammatory response and hypercoagulability (Figure 1). Pattern recognition receptors (PRRs) on the endothelium can sense pathogen-associated molecular patterns presented by the virus as well as damage-associated molecular patterns generated by damaged cells. Activation of these endothelial PRRs stimulates an inflammatory response that causes loss of the normal vasodilatory factors such as nitric oxide (NO) and prostacyclin. Loss of these factors also facilitates platelet and leukocyte adhesion. Interaction of blood elements with the vessel wall is aggravated by the expression of endothelial adhesion molecules, chemokines, and inflammatory cytokines that precipitate a local inflammatory response. This acute endotheliopathy and cardiovascular injury is expected to be more severe in older individuals with preexisting endothelial dysfunction and cardiovascular disease.28,29,30 The increasing impairment of endothelial function with age contributes to the increased vulnerability of the elderly to SARS-CoV-2.
Figure 1.
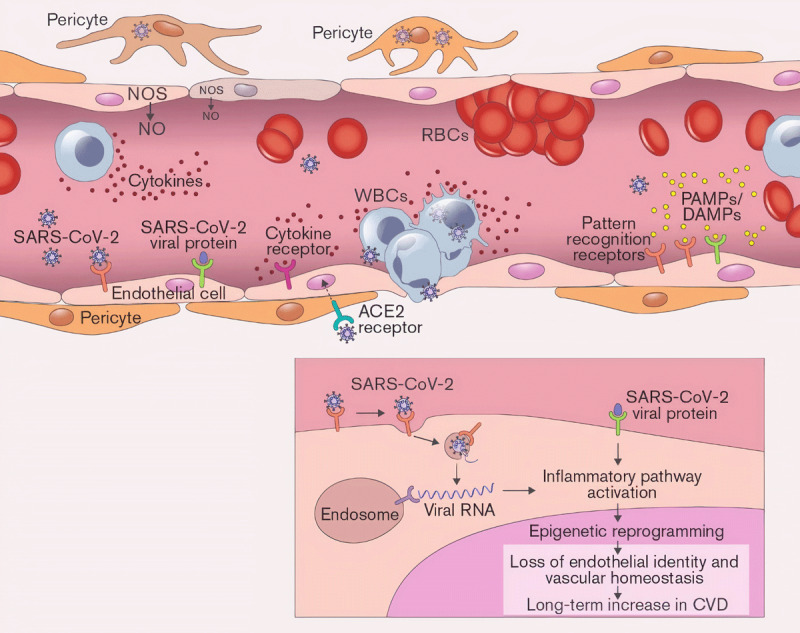
SARS-CoV-2 activates inflammatory signaling within endothelial cells (EC) that alters endothelial homeostasis and promotes inflammation, vascular permeability, and thrombosis. Epigenetic alterations contribute to the acute and chronic manifestations of COVID endotheliopathy. NOS: nitric oxide synthase; NO: nitric oxide; WBCs: white blood cells; RBCs: red blood cells; PAMP: pathogen-associated molecular patterns; DAMP: damage-associated molecular patterns; CVD: cardiovascular disease.
Depending on which vascular circulation is affected, the endothelial disturbance created by viral invasion and circulating cytokines could promote pulmonary inflammation and edema, microvascular inflammation and myocarditis, coronary thrombosis and myocardial infarction, carotid artery thrombosis and stroke, peripheral microvascular occlusion manifesting as local tissue ischemia and inflammation (eg, “COVID toes”), and venous thrombosis and pulmonary embolism. In addition to precipitating acute cardiovascular events, dysfunction of the cerebrovascular endothelium could cause or contribute to cognitive impairment.31 Notably, cardiovascular manifestations of COVID-19 can occur in the absence of respiratory symptoms.28
Venous Thrombosis as a Consequence of Covid-19 Endotheliopathy
Because the other acute cardiovascular manifestations of COVID-19 are discussed elsewhere in this issue, we highlight here the venous thrombosis that may occur in COVID-19.
Deep venous thrombosis (DVT) and its consequences (venous thromboembolism, or VTE) contributes to 100,000 deaths annually in the US.32 In contrast to arterial thrombosis, DVT research has received less attention, prompting the American Surgeon General to issue a Call to Action in 2008 to stimulate DVT research.33 Fast forward to 2020, DVT is now recognized as one of the life-threatening manifestations in patients with COVID-19 and a major determinant of mortality.34 A study of patients with severe COVID-19 in Wuhan, China, revealed that nearly 90% had lower-extremity DVT, and those with DVT had worse survival rates.34 Before the use of systemic anticoagulation in hospitalized COVID patients, the majority of those succumbing to SARS-CoV-2 had DVT, and it contributed to the demise of at least one-third of patients.35
Notably, COVID-19 is associated with endothelial activation and damage in several organs.6,36 SARS-CoV-2 invades endothelial cells in an organoid culture,37 and its viral inclusion bodies are seen in endothelial cells at autopsy. Autopsy findings of COVID-19 patients have further revealed changes in endothelial cytoarchitecture and apoptosis as well as microthrombi and larger clots in the lungs, kidneys, and mesenteric vessels—all manifestations of the virus’ vasculotropic characteristics.6 Endothelial dysfunction and venous thrombosis contribute to respiratory failure in COVID-19.38,39 These findings emphasize that endotheliopathy—and vascular thrombosis in particular—is a common aspect of severe COVID-19. Still understudied is the mechanism by which this occurs and how it might be best mitigated in ways that limit both short- and long-term consequences of this damage.
Virchow’s Triad and Sars-cov-2
The venous thrombosis observed in COVID-19 is due to the interaction of the three determinants of thrombosis (Virchow’s triad): vascular injury, stasis, and blood coagulability. In this case, the vascular injury is a viral-induced endotheliopathy. The stasis in the limb veins is a consequence of the immobile hospitalized patient but also the complex hemodynamics in the vicinity of the valve cusps. The increased blood coagulability is associated with immune-mediated mechanisms generated during the response to the infection.40
There is an unmet need to understand the effects of the virus and/or blood-borne inflammatory cytokines on the endothelium, understand how these effects interact with the uniquely complex venous hemodynamics (mechanotransduction), and discover endothelium-stabilizing strategies that may be cooperatively therapeutic with anticoagulants. There is a dearth of tools for investigating how these thrombi develop. Cell monolayer or simple organoid growth does not reproduce the key architecture that drives thrombosis. Most of these models forego inclusion of venous valves when it is well-known that DVT is primarily initiated at the site of venous valve pockets by endothelial disruption, blood stasis, and/or hypercoagulable blood—the three factors known as Virchow’s triad.41,42
To understand the interactions of these elements of Virchow’s triad during COVID-19 infection, three organizations—including Texas A&M University, the Houston Methodist Research Institute, and Boston University—have teamed together to bring complementary expertise in vein-chip bioengineering and innovation (Jain), molecular and computational biology of endothelial function and cell fate (Cooke), and techniques to define host response to coronaviral infection (Connor). Springing from this multidisciplinary collaboration and an urgency to address the impact of COVID-19, our objective is to understand the determinants of SARS-CoV-2–induced venous thrombosis; determine the roles of endothelial, hemodynamic, and humoral alterations; and propose therapeutic strategies. We intend to elucidate the mechanisms of COVID-19–associated venous thrombosis using our vein-on-a-chip (Figure 2).43 This vein chip is an endothelialized device incorporating the structure and unique hemodynamics proximal and distal to the venous valve cusp. We have used this chip to determine the role of major determinants of venous thrombosis—endothelium, hemodynamics, and blood components (Virchow’s triad).41,42 A salient feature of our technology is that it reveals differential endothelial phenotypes that are clinically observed but not mechanistically understood by other preclinical models.
Figure 2.
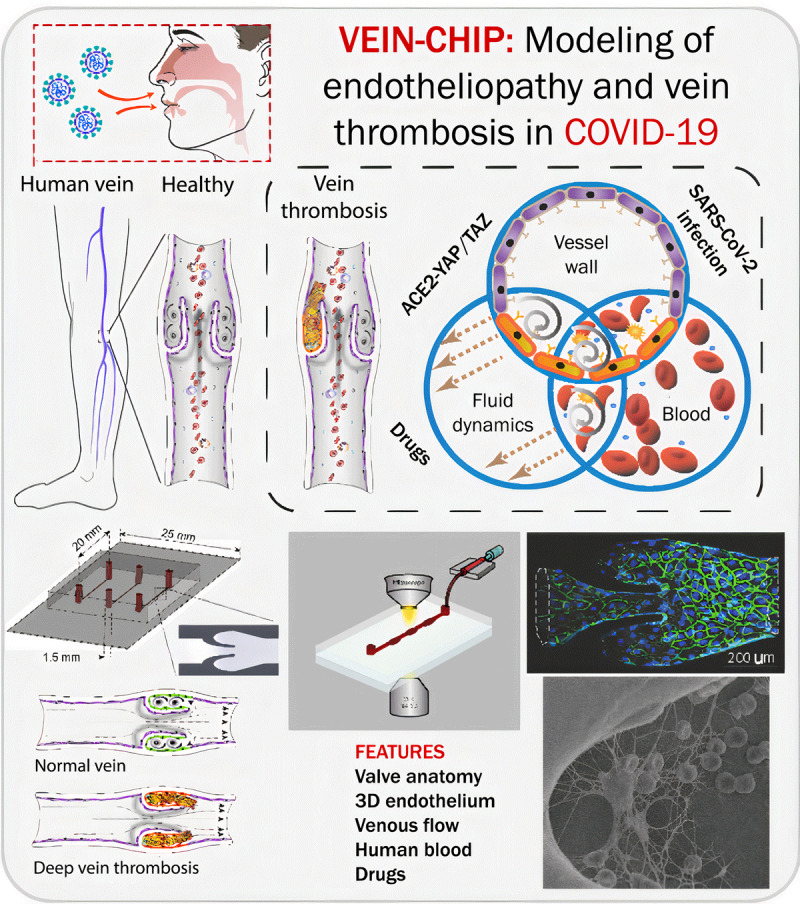
Infographic describing how the vein-chip will model endotheliopathy and thrombosis in COVID-19. Bottom fluorescence micrograph and scanning electron image taken from Rajeeva et al. Microengineered Human Vein-Chip Recreates Venous Valve Architecture and Its Contribution to Thrombosis. Small. 2020;16(49):2003401. doi: 10.1002/smll.202003401
The most common animal model of thrombosis is the mouse inferior vena cava model.44,45 Typically, venous thrombosis is induced chronically by inducing stasis or stenosis (with a ligature) or rapidly by an acute injury (eg, using chemical injury) of the inferior vena cava. While these models have decoded several key mechanisms that govern DVT, the lack of valve function (Figure 3, middle panel) and genetic differences with respect to humans can limit them in studying Virchow factors.43,46 Existing animal models of DVT are expensive and suboptimal in that they are not amenable to a reductionist approach.47 In contrast, our vein-chip microfluidic cell culture platform offers a new in vitro tool for studying how endothelial cells, humoral substances, and hemodynamics interact to influence human vein diseases or respond to therapeutics.48,49 Using such blood-vessel chips, we have elucidated mechanisms of thromboinflammation and identified potential therapeutic approaches in areas where animal models have shown limitations.44,50,51,52,53,54 With this multidisciplinary team, together with federal funding and corporate collaboration, we intend to refine therapeutic strategies for preventing and treating virally-induced venous thrombosis.
Figure 3.
A section of vein-chip. Human vein-chip more accurately models human venous architecture, flow, and thrombosis. Taken from Rajeeva et al. Microengineered Human Vein-Chip Recreates Venous Valve Architecture and Its Contribution to Thrombosis. Small. 2020;16:2003401. doi: 10.1002/smll.202003401. Image enlarged using Let’s Enhance.
Post Covid-19 Condition and Endothelial Epigenetics
Some individuals have persistent effects of SARS-CoV-2 infection long after they are serologically negative for the virus and have converted to immunopositivity. In early October 2021, the World Health Organization (WHO) classified these “long haulers” as having “post COVID-19 condition,” with symptoms of physical fatigue, tachycardia, and dyspnea with little exertion as well as cognitive impairment that interferes with activities of daily life.55,56,57 According to the WHO, the symptoms typically initiate within 3 months after probable or confirmed SARS CoV-2 infection and last at least 2 months. Although it is difficult to determine the exact number of patients who experience post COVID-19 condition, data indicate that roughly 10% to 20% continue to experience symptoms after acute infection. By comparison, recovery from influenza is complete within 2 weeks in over 90% of cases. The symptoms of those with post COVID-19 condition could be explained in part by a severe and global endothelial dysfunction that impairs pulmonary, coronary, cerebral, and skeletal muscle microvasculature.
As described above, COVID-induced endotheliopathy likely participates in the acute manifestations of the disease since it would likely exacerbate the inflammatory state, prothrombotic state, and vascular permeability—each of which may contribute to the many disease manifestations. However, what is not known, and what has not been anticipated nor adequately discussed, is the likely effect of a persistent epigenetic alteration of the endothelium (Figure 4). Specifically, epigenetic alterations in genes that regulate endothelial cell identity are likely responsible for the acute impairment or loss of normal endothelial function associated with SARS-CoV-2 infection. Furthermore, the persistence of these epigenetic alterations would be expected to cause a chronic impairment in endothelial cell function that would cause or contribute to many of the symptoms of long COVID. Chronic impairment of endothelial function would be expected to accelerate atherosclerosis and increase the risk of later arterial occlusive disease in the coronary or cerebrovascular circulations.
Figure 4.
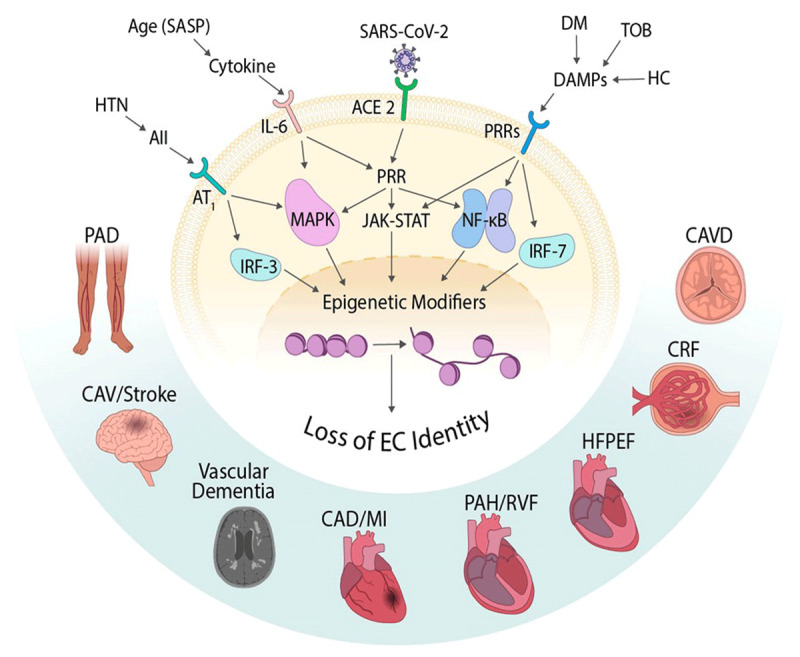
Pathways in endothelial cells (ECs) that contribute to epigenetic alterations and EC loss of function causing downstream cardiovascular disease. ACE2: angiotensin converting enzyme 2; DM: diabetes mellitus; TOB: tobacco exposure; DAMP: damage-associated molecular patterns; HC: hypercholesterolemia PRR: pattern recognition receptors; JAK/STAT: Janus kinase signal transducer and activator of transcription; NF-kB: nuclear factor kappa B; IRF-7: interferon regulatory factor 7; CAVD: calcific aortic valve disease; CRF: corticotropin-releasing factor; HFPEF: heart failure with preserved ejection fraction; PAH/RVP: pulmonary arterial hypertension/right ventricular pressure; CAD/MI: coronary artery disease/myocardial infarction; CAV: cardiac allograft vasculopathy; PAD: peripheral arterial disease; HTN: hypertension.
Viral invasion of endothelial cells, or exposure to inflammatory cytokines, are triggering events that activate cell-autonomous innate immune signaling in endothelial cells, which in turn may diminish or erase epigenetic determinants of endothelial identity. In the short term, this postulated endotheliopathy would be expected to cause further inflammation, vasoconstriction, and coagulation. In the long term, a persistent epigenetic alteration will promote endothelial activation manifested by endothelial adhesion molecules and chemokines, which will aggravate vascular inflammation and progression of atherosclerosis and other vascular diseases. Furthermore, in some organs this attenuation of endothelial identity will also promote endothelial-to-mesenchyme transition, leading to rarefaction of the microvasculature and disseminated fibrosis in multiple organs. Thus, a loss of endothelial identity would be expected to (1) accelerate atherosclerosis manifested as coronary, cerebrovascular, and peripheral arterial disease; (2) cause excess morbidity and mortality from strokes, vascular dementia, myocardial infarction, and critical limb ischemia; (3) generate cardiac, pulmonary, and renal fibrosis, with impaired perfusion and function of these organs; (4) increase valvular and vascular calcification; (5) increase the prevalence of hypertension and chronic renal failure; and (6) cause a persistent abnormality of the cerebral microvasculature contributing to cognitive impairment.31
Notably, endotheliopathy related to a traumatic event can persist and accelerate cardiovascular disease. Longitudinal studies have shown that individuals who were born prematurely have endothelial dysfunction as children that persists into young adulthood and a greater risk of cardiovascular disease in adulthood.58,59 These children have greater risk of infections in infancy and childhood,60 and this increase in infection exposure has been tied to later cardiovascular disease.61 Similarly, exposure to secondhand smoke in childhood is associated with increased risk of carotid plaque in adulthood.62 Recent studies reveal that activation of damage- and pathogen-associated molecular patterns cause epigenetic changes that place a cell into a state of phenotypic fluidity.23,24,25,26,27 Of course, this phenomenon can be useful in defense against a pathogen or in response to injury. However, this same inflammatory signaling can facilitate a phenotypic switch to a deleterious phenotype, depending on the context.
The hopeful news is that, despite epigenetic alterations that may persist in the absence of countervailing forces, there are potential therapeutic approaches that may reverse the aberrant epigenetic markings and restore normal endothelial cell identity and function, thereby preventing cardiovascular disease. A comprehensive characterization of COVID-19–associated endotheliopathy, and an understanding of the mechanisms of acute and chronic endothelial alterations induced by SARS-CoV-2, will lead to an improved understanding of the many manifestations of COVID-19 and a refined management approach for this and other vasculotropic viral diseases.
Key Points
Endothelial homeostasis is critical for normal cardiovascular structure and function.
Inflammatory cytokines generated during SARS-CoV-2 infection, and/or viral entry into endothelial cells, cause vascular impairment resulting in vascular inflammation, edema, and thrombosis.
An endotheliopathy likely contributes to the pulmonary and cardiovascular manifestations of COVID-19.
Some of the unusual manifestations of COVID-19 including myocarditis, cognitive disturbance, and “COVID toes” may be driven by endothelial perturbation.
Post COVID-19 condition may be due to a persisting endothelial dysfunction.
Endothelial-targeted therapies such as statins, angiotensin converting enzyme inhibitors, angiotensin receptor blockers, and nitric oxide donors may be useful in this viral disorder.
CME Credit Opportunity
Houston Methodist is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to provide continuing medical education for physicians.
Houston Methodist designates this enduring material for a maximum of .25 AMA PRA Category 1 Credit™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Click to earn CME credit: https://journal.houstonmethodist.org/articles/10.14797/mdcvj.1044/
Acknowledgements
This work was supported by NIH grants R01HL157790 (JHC, JPC, AJ), R01HL148338 (JPC), and R01HL133254 (JPC).
Funding Statement
This work was supported by NIH grants R01HL157790 (JHC, JPC, AJ), R01HL148338 (JPC), and R01HL133254 (JPC).
Competing Interests
Dr. Cooke is a consultant for and/or conducts research on behalf of HumanN, JanOne, Navitas Life Sciences, Fibralign Corp., Avita Medical, Cook Medical, and VGXI, Inc. The other authors have no disclosures.
References
- 1.Teuwen LA, Geldhof V, Pasut A, Carmeliet P. COVID-19: the vasculature unleashed. Nat Rev Immunol. 2020. Jul;20(7):389–391. doi: 10.1038/s41577-020-0343-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Libby P, Lüscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. 2020. Sep 1;41(32):3038–3044. doi: 10.1093/eurheartj/ehaa623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iba T, Connors JM, Levy JH. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflamm Res. 2020. Dec;69(12):1181–1189. doi: 10.1007/s00011-020-01401-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagashima S, Mendes MC, Camargo Martins AP, et al. Endothelial Dysfunction and Thrombosis in Patients With COVID-19-Brief Report. Arterioscler Thromb Vasc Biol. 2020. Oct;40(10):2404–2407. doi: 10.1161/ATVBAHA.120.314860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He L, Mäe MA, Muhl L, et al. Pericyte-specific vascular expression of SARS-CoV-2 receptor ACE2 – implications for microvascular inflammation and hypercoagulopathy in COVID-19. BioRxiv. 2020. May.2005.2011.088500. doi: 10.1101/2020.05.11.088500 [DOI] [Google Scholar]
- 6.Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020. May 2;395(10234):1417–1418. doi: 10.1016/S0140-6736(20)30937-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McConnell MJ, Kawaguchi N, Kondo R, et al. Liver injury in COVID-19 and IL-6 trans-signaling-induced endotheliopathy. J Hepatol. 2021. Sep;75(3):647–658. doi: 10.1016/j.jhep.2021.04.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang S, Tanaka T, Inoue H, et al. IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci U S A. 2020. Sep 8;117(36):22351–22356. doi: 10.1073/pnas.2010229117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Sun X, Carmeliet P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019. Sep 3;30(3):414–433. doi: 10.1016/j.cmet.2019.08.011 [DOI] [PubMed] [Google Scholar]
- 10.Paneni F, Beckman JA, Creager MA, Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur Heart J. 2013. Aug;34(31):2436–43. doi: 10.1093/eurheartj/eht149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease - a 30th anniversary update. Acta Physiol (Oxf). 2017. Jan;219(1):22–96. doi: 10.1111/apha.12646 [DOI] [PubMed] [Google Scholar]
- 12.Huang PL. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol Metab. 2009. Aug;20(6):295–302. doi: 10.1016/j.tem.2009.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atochin DN, Huang PL. Role of endothelial nitric oxide in cerebrovascular regulation. Curr Pharm Biotechnol. 2011. Sep;12(9):1334–42. doi: 10.2174/138920111798280974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blum A, Giladi M, Weinberg M, et al. High anti-cytomegalovirus (CMV) IgG antibody titer is associated with coronary artery disease and may predict post-coronary balloon angioplasty restenosis. Am J Cardiol. 1998. Apr 1;81(7):866–8. doi: 10.1016/s0002-9149(98)00019-8 [DOI] [PubMed] [Google Scholar]
- 15.Du Y, Zhang G, Liu Z. Human cytomegalovirus infection and coronary heart disease: a systematic review. Virol J. 2018. Feb 6;15(1):31. doi: 10.1186/s12985-018-0937-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Badawi A, Di Giuseppe G, Gupta A, Poirier A, Arora P. Bayesian network modelling study to identify factors influencing the risk of cardiovascular disease in Canadian adults with hepatitis C virus infection. BMJ Open. 2020. May 5;10(5):e035867. doi: 10.1136/bmjopen-2019-035867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hemmat N, Ebadi A, Badalzadeh R, Memar MY, Baghi HB. Viral infection and atherosclerosis. Eur J Clin Microbiol Infect Dis. 2018. Dec;37(12):2225–2233. doi: 10.1007/s10096-018-3370-z [DOI] [PubMed] [Google Scholar]
- 18.Gattone M, Iacoviello L, Colombo M, et al. Chlamydia pneumoniae and cytomegalovirus seropositivity, inflammatory markers, and the risk of myocardial infarction at a young age. Am Heart J. 2001. Oct;142(4):633–40. doi: 10.1067/mhj.2001.118118 [DOI] [PubMed] [Google Scholar]
- 19.Nieto FJ, Adam E, Sorlie P, et al. Cohort study of cytomegalovirus infection as a risk factor for carotid intimal-medial thickening, a measure of subclinical atherosclerosis. Circulation. 1996. Sep 1;94(5):922–7. doi: 10.1161/01.cir.94.5.922 [DOI] [PubMed] [Google Scholar]
- 20.Warren-Gash C, Hayward AC, Hemingway H, et al. Influenza infection and risk of acute myocardial infarction in England and Wales: a CALIBER self-controlled case series study. J Infect Dis. 2012. Dec 1;206(11):1652–9. doi: 10.1093/infdis/jis597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peretz A, Azrad M, Blum A. Influenza virus and atherosclerosis. QJM. 2019. Oct 1;112(10):749–755. doi: 10.1093/qjmed/hcy305 [DOI] [PubMed] [Google Scholar]
- 22.Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med. 2020. Jul 9;383(2):120–128. doi: 10.1056/NEJMoa2015432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J, Sayed N, Hunter A, et al. Activation of innate immunity is required for efficient nuclear reprogramming. Cell. 2012. Oct 26;151(3):547–58. doi: 10.1016/j.cell.2012.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sayed N, Wong WT, Ospino F, et al. Transdifferentiation of human fibroblasts to endothelial cells: role of innate immunity. Circulation. 2015. Jan 20;131(3):300–9. doi: 10.1161/CIRCULATIONAHA.113.007394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng S, Zhou G, Gu Q, Chanda PK, Ospino F, Cooke JP. Transdifferentiation Requires iNOS Activation: Role of RING1A S-Nitrosylation. Circ Res. 2016. Oct 14;119(9):e129–e138. doi: 10.1161/CIRCRESAHA.116.308263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai L, Reineke E, Hamilton DJ, Cooke JP. Glycolytic Switch Is Required for Transdifferentiation to Endothelial Lineage. Circulation. 2019. Jan 2;139(1):119–133. doi: 10.1161/CIRCULATIONAHA.118.035741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dal-Pra S, Hodgkinson CP, Dzau VJ. Induced cardiomyocyte maturation: Cardiac transcription factors are necessary but not sufficient. PLoS One. 2019. Oct 17;14(10):e0223842. doi: 10.1371/journal.pone.0223842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inciardi RM, Lupi L, Zaccone G, et al. Cardiac Involvement in a Patient With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020. Jul 1;5(7):819–824. doi: 10.1001/jamacardio.2020.1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo T, Fan Y, Chen M, et al. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020. Jul 1;5(7):811–818. doi: 10.1001/jamacardio.2020.1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi S, Qin M, Shen B, et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. 2020. Jul 1;5(7):802–810. doi: 10.1001/jamacardio.2020.0950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stephan BCM, Harrison SL, Keage HAD, Babateen A, Robinson L, Siervo M. Cardiovascular Disease, the Nitric Oxide Pathway and Risk of Cognitive Impairment and Dementia. Curr Cardiol Rep. 2017. Aug 11;19(9):87. doi: 10.1007/s11886-017-0898-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stone J, Hangge P, Albadawi H, et al. Deep vein thrombosis: pathogenesis, diagnosis, and medical management. Cardiovasc Diagn Ther. 2017. Dec;7(Suppl 3):S276–S284. doi: 10.21037/cdt.2017.09.01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.National Center for Biotechnology Information [Internet]. Bethesda, MD: US National Library of Medicine; c2021. The Surgeon General’s Call to Action to Prevent Deep Vein Thrombosis and Pulmonary Embolism; 2008. [cited 2021 Oct 25]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK44178/ [Google Scholar]
- 34.Ren B, Yan F, Deng Z, et al. Extremely High Incidence of Lower Extremity Deep Venous Thrombosis in 48 Patients With Severe COVID-19 in Wuhan. Circulation. 2020. Jul 14;142(2):181–183. doi: 10.1161/CIRCULATIONAHA.120.047407 [DOI] [PubMed] [Google Scholar]
- 35.Wichmann D, Sperhake JP, Lütgehetmann M, et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Ann Intern Med. 2020. Aug 18;173(4):268–277. doi: 10.7326/M20-2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Escher R, Breakey N, Lämmle B. Severe COVID-19 infection associated with endothelial activation. Thromb Res. 2020. Jun;190:62. doi: 10.1016/j.thromres.2020.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monteil V, Kwon H, Prado P, et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell. 2020. May 14;181(4):905–913.e7. doi: 10.1016/j.cell.2020.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teuwen LA, Geldhof V, Pasut A, Carmeliet P. COVID-19: the vasculature unleashed. Nat Rev Immunol. 2020. Jul;20(7):389–391. doi: 10.1038/s41577-020-0343-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mortus JR, Manek SE, Brubaker LS, et al. Thromboelastographic Results and Hypercoagulability Syndrome in Patients With Coronavirus Disease 2019 Who Are Critically Ill. JAMA Netw Open. 2020. Jun 1;3(6):e2011192. doi: 10.1001/jamanetworkopen.2020.11192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanff TC, Mohareb AM, Giri J, Cohen JB, Chirinos JA. Thrombosis in COVID-19. Am J Hematol. 2020. Dec;95(12):1578–1589. doi: 10.1002/ajh.25982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lowe GDO. Virchow’s triad revisited: abnormal flow. Pathophysiol Haemost Thromb. 2003. Sep-2004 Dec;33(5–6):455–7. doi: 10.1159/000083845 [DOI] [PubMed] [Google Scholar]
- 42.National Center for Biotechnology Information [Internet]. Bethesda, MD: US National Library of Medicine; c2021. Rumbaut RE, Arterial Thiagarajan P., Venous, and Microvascular Hemostasis/Thrombosis; 2008. [cited 2021 Oct 25]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK53453/ [Google Scholar]
- 43.Rajeeva NK, Walther BK, Suresh R, Cooke JP, Jain A. Microengineered Human Vein-Chip Recreates Venous Valve Architecture and Its Contribution to Thrombosis. Small. 2020;16(49):2003401. doi: 10.1002/smll.202003401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pandian, NKR, Mannino RG, Lam WA, Jain A. Thrombosis-on-a-chip: Prospective impact of microphysiological models of vascular thrombosis. Curr Opin Biomed Eng. 2018. Mar;5:29–34. doi: 10.1016/j.cobme.2017.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diaz JA, Obi AT, Myers DD, et al. Critical review of mouse models of venous thrombosis. Arterioscler Thromb Vasc Biol. 2012. Mar;32(3):556–62. doi: 10.1161/ATVBAHA.111.244608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bazigou E, Makinen T. Flow control in our vessels: vascular valves make sure there is no way back. Cell Mol Life Sci. 2013. Mar;70(6):1055–66. doi: 10.1007/s00018-012-1110-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diaz JA, Saha P, Cooley B, et al. Choosing a Mouse Model of Venous Thrombosis. Arterioscler Thromb Vasc Biol. 2019. Mar;39(3):311–318. doi: 10.1161/ATVBAHA.118.311818 [DOI] [PubMed] [Google Scholar]
- 48.Ingber DE. Reverse Engineering Human Pathophysiology with Organs-on-Chips. Cell. 2016. Mar 10;164(6):1105–1109. doi: 10.1016/j.cell.2016.02.049 [DOI] [PubMed] [Google Scholar]
- 49.Benam KH, Dauth S, Hassell B, et al. Engineered in vitro disease models. Annu Rev Pathol. 2015;10:195–262. doi: 10.1146/annurev-pathol-012414-040418 [DOI] [PubMed] [Google Scholar]
- 50.Jain A, Graveline A, Waterhouse A, Vernet A, Flaumenhaft R, Ingber DE. A shear gradient-activated microfluidic device for automated monitoring of whole blood haemostasis and platelet function. Nat Commun. 2016. Jan 6;7:10176. doi: 10.1038/ncomms10176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Ceunynck K, Peters CG, Jain A, et al. PAR1 agonists stimulate APC-like endothelial cytoprotection and confer resistance to thromboinflammatory injury. Proc Natl Acad Sci U S A. 2018. Jan 30;115(5):E982–E991. doi: 10.1073/pnas.1718600115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jain A, Barrile R, van der Meer AD, et al. Primary Human Lung Alveolus-on-a-chip Model of Intravascular Thrombosis for Assessment of Therapeutics. Clin Pharmacol Ther. 2018. Feb;103(2):332–340. doi: 10.1002/cpt.742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mathur T, Singh KA, Pandian NKR, et al. Organ-on-chips made of blood: endothelial progenitor cells from blood reconstitute vascular thromboinflammation in vessel-chips. Lab Chip. 2019. Jul 23;19(15):2500–2511. doi: 10.1039/c9lc00469f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luna DJ, Pandian NKR, Mathur T, et al. Tortuosity-powered microfluidic device for assessment of thrombosis and antithrombotic therapy in whole blood. Sci Rep 2020. Apr 1;10(1):5742. doi: 10.1038/s41598-020-62768-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rubin R. As Their Numbers Grow, COVID-19 “Long Haulers” Stump Experts. JAMA. 2020. Oct 13;324(14):1381–1383. doi: 10.1001/jama.2020.17709 [DOI] [PubMed] [Google Scholar]
- 56.Marshall M. The lasting misery of coronavirus long-haulers. Nature. 2020. Sep;585(7825):339–341. doi: 10.1038/d41586-020-02598-6 [DOI] [PubMed] [Google Scholar]
- 57.WHO.int [Internet]. Geneva, Switzerland: World Health Organization; c2021. A clinical case definition of post COVID-19 condition by a Delphi consensus; 2021. Oct 6 [cited 2021 Nov 10]. Available from: http://apps.who.int/iris/bitstream/handle/10665/345824/WHO-2019-nCoV-Post-COVID-19-condition-Clinical-case-definition-2021.1-eng.pdf. [Google Scholar]
- 58.Mehta JL, Bavineni M. Premature birth, infections, and atherosclerotic cardiovascular disease. Eur Heart J. 2019. Oct 14;40(39):3275. doi: 10.1093/eurheartj/ehz441 [DOI] [PubMed] [Google Scholar]
- 59.Bavineni M, Wassenaar TM, Agnihotri K, Ussery DW, Lüscher TF, Mehta JL. Mechanisms linking preterm birth to onset of cardiovascular disease later in adulthood. Eur Heart J. 2019. Apr 7;40(14):1107–1112. doi: 10.1093/eurheartj/ehz025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller JE, Hammond GC, Strunk T, et al. Association of gestational age and growth measures at birth with infection-related admissions to hospital throughout childhood: a population-based, data-linkage study from Western Australia. Lancet Infect Dis. 2016. Aug;16(8):952–61. doi: 10.1016/S1473-3099(16)00150-X [DOI] [PubMed] [Google Scholar]
- 61.Bekkering S, Miller JE, Burgner DP. Childhood infection may mediate the relationship between suboptimal intrauterine growth, preterm birth, and adult cardiovascular disease. Eur Heart J. 2019. Oct 14;40(39):3273–3274. doi: 10.1093/eurheartj/ehz438 [DOI] [PubMed] [Google Scholar]
- 62.West HW, Juonala M, Gall SL, et al. Exposure to parental smoking in childhood is associated with increased risk of carotid atherosclerotic plaque in adulthood: the Cardiovascular Risk in Young Finns Study. Circulation. 2015. Apr 7;131(14):1239–46. doi: 10.1161/CIRCULATIONAHA.114.013485 [DOI] [PubMed] [Google Scholar]