

Abstract
To date, the scaled-up manufacturing and efficient drug loading of exosomes are two existing challenges limiting the clinical translation of exosome-based drug delivery. Herein, we developed a facile magnetic extrusion method for preparing endosome-derived vesicles, also known as exosome mimetics (EMs), which share the same biological origin and similar morphology, composition, and biofunctions with native exosomes. The high yield and consistency of this magnetic extrusion method help to overcome the manufacturing bottleneck in exosome research. Moreover, the proposed standardized multi-step method readily facilitates the ammonium sulfate gradient approach to actively load chemodrugs such as doxorubicin into EMs. The engineered EMs developed and tested here exhibit comparable drug delivery properties as do native exosomes and potently inhibit tumor growth by delivering doxorubicin in an orthotopic breast tumor model. These findings demonstrate that EMs can be prepared in a facile and scaled-up manner as a promising biological nanomedicine for cancer drug delivery.
Keywords: Exosome mimetics, drug delivery, cancer, magnetic extrusion, iron oxide nanoparticles
Graphical Abstract
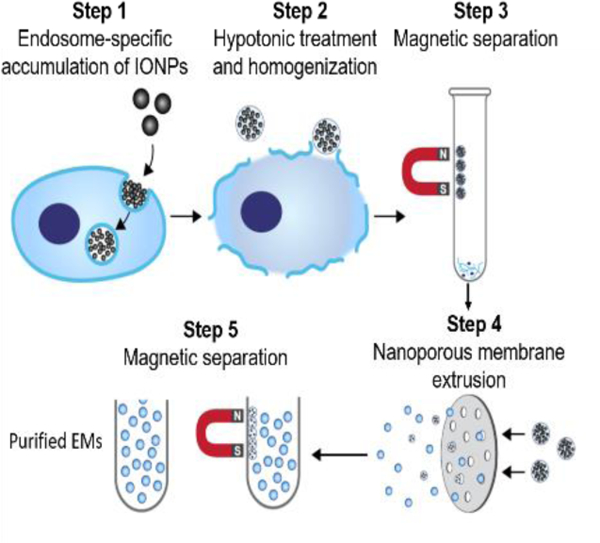
A magnetic extrusion method is developed to prepare endosome-derived vesicles, also called exosome mimetics (EMs), in a facile and scaled-up manner. The engineered EMs share the same biological origin and similar morphology, composition, and biofunctions with native exosomes, which can be utilized as a promising biological nanomedicine for cancer drug delivery.
Introduction
Exosomes are nanosized extracellular vesicles (30–150 nm) of endosomal origin secreted by most cell types[1,2]. The heterogeneous structure of exosomes features a lipid bilayer enclosing solubilized bioactive cargoes, mainly proteins and nucleic acids[3]. Upon secretion, exosomes travel long-distance and deliver their content to target cells thereby playing a key role in cell-to-cell communication and supporting both physiological (e.g., coagulation) and pathological processes (e.g., cancer metastasis)[1,2,4]. In recent years, exosomes have emerged as a novel class of versatile nanoparticles. In fact, exosomes mirror the secreting cell in composition therefore representing a novel source of diagnostic and prognostic biomarkers[1,2]. Furthermore, due to their unique structure, exosomes can be readily engineered and loaded with various therapeutic agents, constituting a promising class of biocompatible vehicles for targeted drug delivery[3]. As delivery vehicles, exosomes naturally combine unique features of biological functions such as organ tropism, breaching of biological barriers and poor immunogenicity and can outperform the majority of currently available synthetic nanoparticles (e.g., liposomes, and polymer nanoparticles) [3–5].
Despite their potential, the clinical translation of exosome-based therapeutics has been hampered by critical challenges including poorly scalable isolation methods, limited yield and low drug loading efficiency. Conventional exosome isolation methods rely on time- and labor-intensive protocols (e.g., ultracentrifugation, density gradients, size-exclusion chromatography) that require expensive instruments as well as trained personnel. Exosomes separated using conventional methods (e.g., ultracentrifugation, polymer-based precipitation, and size-exclusion chromatography) may suffer from variable degrees of particle aggregation, purity, sample loss and damage. Furthermore, the yields of conventional exosome isolation methods have been characterized as being approximately 7×107, 6×108, and 1×1010 particles/million cells for size exclusion chromatography, ultracentrifugation, and tangential flow filtration, respectively[6–8]. The yields of these exosome isolation methods have yet to meet the rapidly growing needs for exosome-based drug delivery and diagnostic applications[9,10]. Common exosome loading strategies (e.g., passive mixing, electroporation, exogenous loading) often result in low loading efficiencies (< 30%) compared to synthetic nanomedicines[11–13]. These factors, and others, represent substantial challenges to the scale-up of exosome manufacturing for drug delivery purposes which, if overcome, could advance their translation into nanomedicine.
In order to accomplish this, we have developed a novel magnetic extrusion method to generate formulations of endosome-derived vesicles in a facile and scaled-up manner. Endosomes were collected from different cell sources using iron oxide nanoparticle (IONP)-assisted magnetic separation and subsequently extruded, using a nanoporous membrane, into monodispersed endosome-derived vesicles. Given that the obtained endosome-derived vesicles share the same intracellular origin of the native exosomes, we will refer to them as exosome mimetics (EMs). We compared EM properties and in vitro and in vivo performances to those of small extracellular vesicles (EVs) isolated from conditioned media of the same cell line. Taking advantage of the controllable procedure of this method, we were able to readily use an ammonium sulfate gradient approach to actively load chemotherapeutic doxorubicin into EMs in a highly efficient manner. The engineered EMs loaded with doxorubicin (EM-Dox) were evaluated as a novel cancer nanomedicine in comparison with native small EVs and their drug delivery performance was determined and compared in vitro and in vivo.
2. Results and Discussion
Preparation and characterization of EMs.
The workflow of the magnetic extrusion method (as shown in Figure 1) takes advantage of the endosome-specific accumulation of magnetic IONPs via endocytosis-mediated cell internalization. The IONP-encapsulating endosomes are released from parental cells after hypotonic buffer treatment and homogenization and collected using magnetic separation. Purified endosomes are subsequently extruded as monodispersed endosome-derived vesicles, also called EMs. Finally, IONP-free EMs are purified from free IONPs and IONP-encapsulating EMs by magnetic separation. Compared to many other established EM synthesis protocols based on plasma membrane extrusion[14–16], the method developed in this manuscript provides the opportunity to obtain endosome-derived EMs that mirror the biological and physical properties of native exosomes.
Figure 1. Schematic illustration of the magnetic extrusion method.
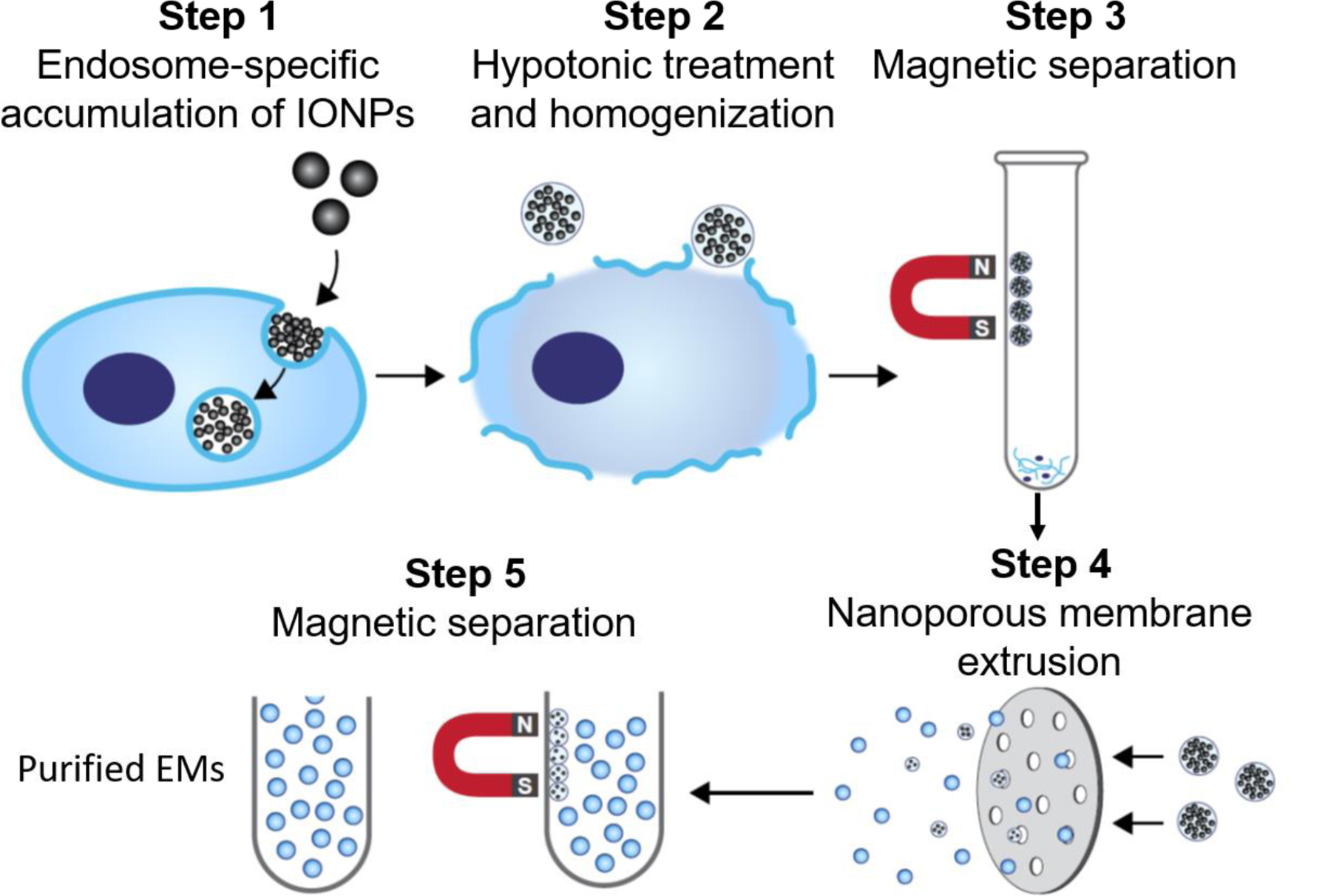
Step 1. IONPs are internalized into endosomes via endocytosis. Step 2. IONP-encapsulating endosomes are released by hypotonic treatment and homogenization. Step 3. IONP-encapsulating endosomes are isolated from other organelles and cell debris by magnetic separation. Step 4. IONP-encapsulating endosomes are extruded into monodispersed EMs via nanoporous membrane extrusion. Step 5. IONP-encapsulating EMs are eliminated from IONP-free EMs using magnetic separation.
To test our design, human breast cancer MDA-MB-231 cells were incubated with IONPs (10 nm in diameter) for 12 h, allowing IONP internalization via clathrin-mediated endocytosis pathways to occur as previously reported[17–19]. Notably, as confirmed by transmission electron microscopy (TEM) studies, IONP accumulated specifically in the endosomes and not in the other intracytoplasmatic organelles (Figure 2A). MDA-MB-231 cells were then lysed using an established hypotonic treatment followed by a homogenization step in order to release the intracytoplasmic organelles in solution[20–22]. IONP-encapsulating endosomes were separated from other organelles (e.g., nuclei, and mitochondria) and cell debris using a magnetic separator and then washed with PBS (Figure 2A). The collected IONP-encapsulating endosomes were extruded through a nanoporous membrane (200 nm in diameter) to form monodispersed EMs derived from MDA-MB-231 cells (231-EMs)[23]. The majority of free IONPs were filtered by the nanoporous membrane during the extrusion step. The remaining IONPs, together with the IONP-encapsulating EMs, were subsequently eliminated from the final EM sample by magnetic separation. As shown in Figure 2A, like native exosomes, the resulting 231-EMs are characterized by a nanosized vesicle-like structure and are delimited by a lipid bilayer.
Figure 2. Preparation and characterization of EMs.
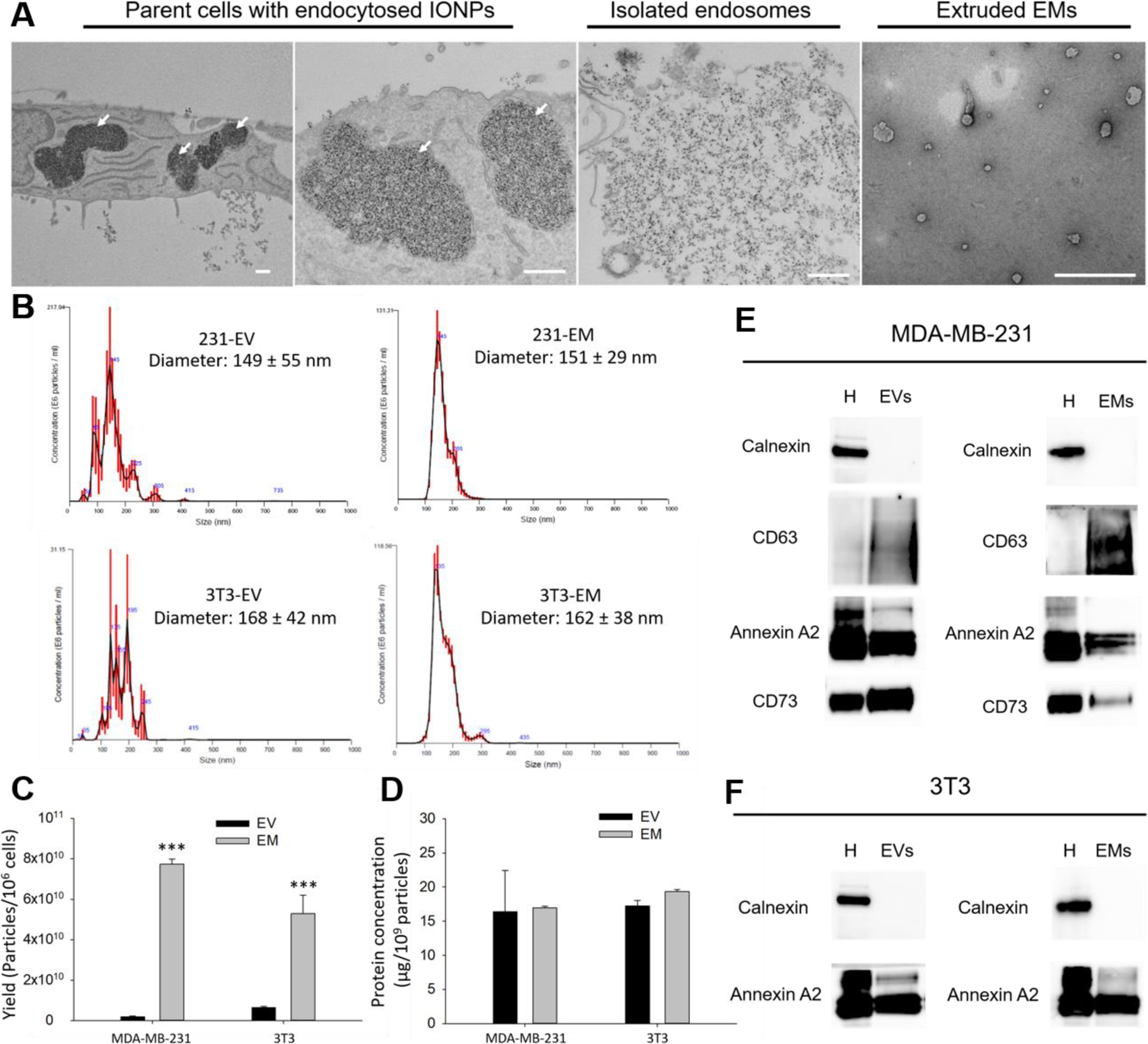
(A) The morphology and structure of parental cells with endocytosed IONPs, isolated endosomes, and extruded EMs characterized by TEM. The white arrows indicate IONP-encapsulating endosomes. The scale bars represent 500 nm. (B) Hydrodynamic diameters of 231-EM, 231-EV, 3T3-EM, and 3T3-EV characterized by NTA. (C) Yield of EMs and native EVs prepared from MDA-MB-231 and 3T3 cells. (D) Total protein concentration of EMs and native EVs prepared from MDA-MB-231 and 3T3 cells. ***P<0.001. Western blot analysis of protein marker expression in EMs, EVs, and cell homogenates (H) from MDA-MB-231 (E) and 3T3 cells (F).
To demonstrate that this magnetic extrusion method enables the synthesis of EMs from different cell sources, we used the same method and experimental conditions to prepare EMs from normal murine fibroblast 3T3 cells (3T3-EMs). As shown in Figure 2B, both 231-EMs and 3T3-EMs exhibit a highly similar hydrodynamic diameter of approximately 160 nm as do native small extracellular vesicles (231-EVs and 3T3-EVs) collected from the same cell sources by ultracentrifugation. However, 231-EMs and 3T3-EMs exhibit a narrower size distribution than native small EVs, providing more consistent and reproducible biodistribution and circulation properties[24–26]. The EM yields of magnetic extrusion were determined to be 7.73 ×1010 particles/106 cells for 231-EMs and 5.29×1010 particles/106 cells for 3T3-EMs as determined via nanoparticle tracking analysis (NTA) (Figure 2C). Significantly, these yields are approximately10-fold higher than those of native small EVs (231-EVs and 3T3-EVs) prepared by the conventional ultracentrifuge method (approximately 1.97–6.51 × 109 particles/106 cells).
Compared to native EVs, these EMs showed similar protein composition and 231-EMs and 231-EVs exhibited similar total protein concentrations, 17.2 µg/109 particles and 16.4 µg/109 particles, respectively (Figure 2D). Similarly, 3T3-EMs and 3T3-EVs exhibited total protein concentrations of 19.4 µg/109 particles and 17.0 µg/109 particles, respectively. Furthermore, both 231-EM and native 231-EV samples were enriched in plasma and endosomal membrane proteins, namely cluster of differentiation (CD) 63 and CD73, both of which are EV biomarkers. Notably, EMs and EVs from both 231 and 3T3 cells exhibited a negative signal for Calnexin, an endoplasmic reticulum protein used to exclude the presence in the samples of intracytoplasmatic contaminant nanoparticles other than EVs or EMs (Figure 2E and F). Taken together, these results indicate that EMs exhibit a highly similar size, morphology and composition as do the native small EVs. We therefore hypothesized that these EMs might exhibit similar drug delivery performance as native exosomes. Given the industrial readiness of magnetic separation and nanoporous membrane extrusion[23,27], our advanced magnetic extrusion method is amenable to large scale EM manufacture that can advance EM pre-clinical and clinical translation.
Cancer cell uptake of EMs
Next, we evaluated the drug delivery potential of EMs in vitro and in vivo. We selected 3T3-EMs engineered from normal fibroblasts for our in vitro and in vivo drug delivery studies. Synthetic PEGylated liposome (PEG-LP), 3T3-EV, and 3T3-EM samples were fluorescently labeled with a lipophilic carbocyanine DiIC18 (DiR) probe that enables quantification and the ability to compare the particle uptake in vitro and the intratumor particle accumulation and organ biodistribution in vivo.
3T3-EM uptake by two cultured breast cancer cell lines, MDA-MB-231 and MDA-MB-436, was assessed using immunofluorescent staining and compared with the cellular uptake of PEG-LPs and 3T3-EVs (Figure 3A). After an 8h incubation with a fixed quantity of fluorescently labeled 3T3-EM-DiR or 3T3-EV-DiR and PEG-LP-DiR as controls, MDA-MB-231 and MDA-MB-436 were imaged by confocal microscopy. As depicted in Figure 3A, 3T3-EM-DiR, 3T3-EV-DiR, and PEG-LP-DiR were equally internalized by both breast cancer cell lines. The 3T3-EM-DiR, 3T3-EV-DiR, PEG-LP were found to be aggregated in the cytosol suggesting that these were internalized by cancer cells mainly through an endocytosis pathway. The 3T3-EM-DiR uptake from cancer cells was also determined by flow cytometry and compared with the uptake of the two controls, 3T3-EV-DiR and PEG-LP-DiR. The 3T3-EM-DiR were taken up at a similar rate as the 3T3-EV-DiR and PEG-LP-DiR by both MDA-MB-231 and MDA-MB-436 cells (Figures 3B and C).
Figure 3. Drug delivery properties of 3T3-EMs.
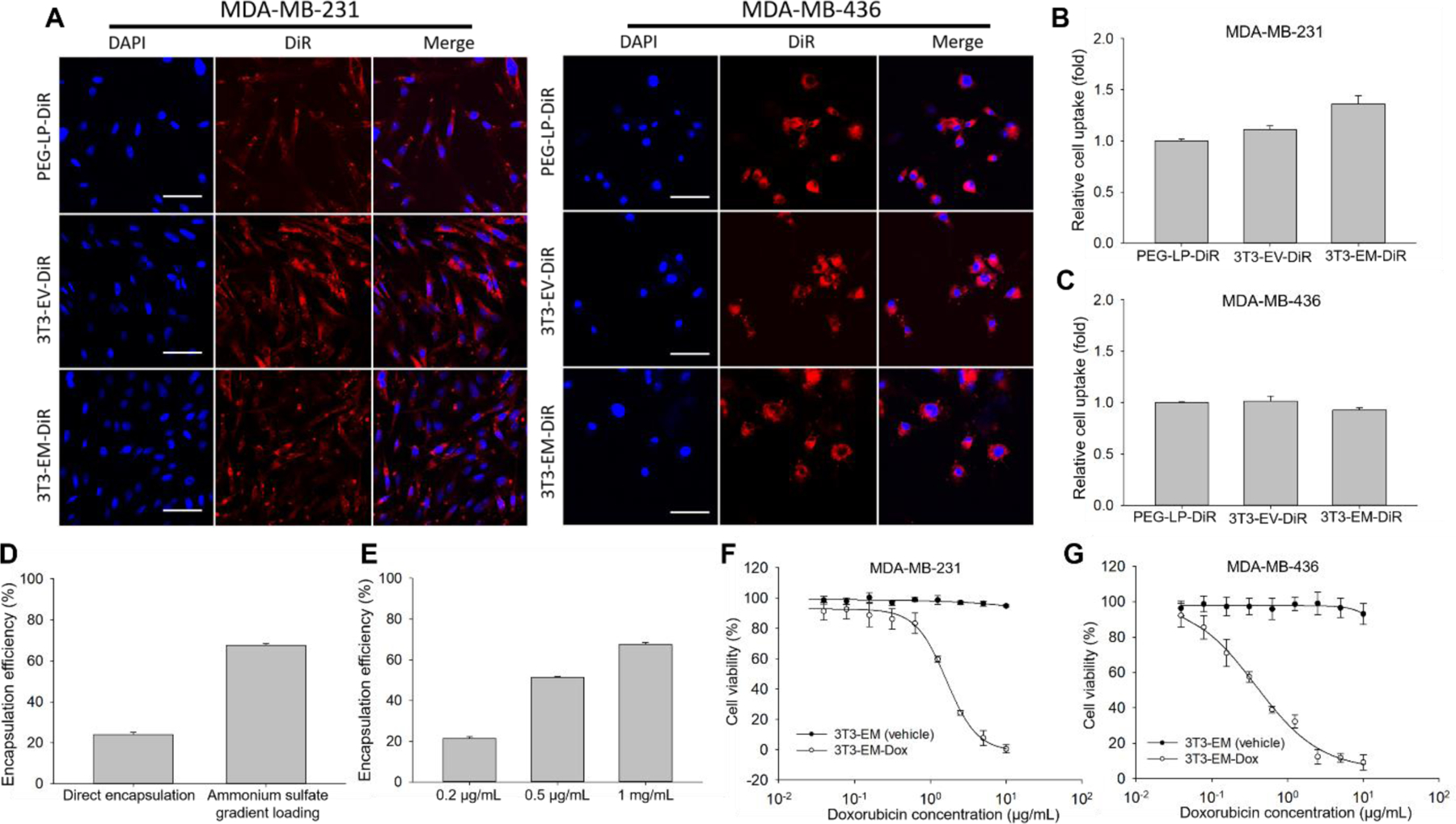
(A) Representative fluorescent microscope images of cell uptake of 3T3-EM-DiR, 3T3-EV-DiR, and PEG-LP-DiR in MDA-MB-231 and MDA-MB-436 cells. The scale bars represent 50 µm. Relative cell uptake of 3T3-EM-DiR, 3T3-EV-DiR, and PEG-LP-DiR by MDA-MB-231 (B) and MDA-MB-436 cells (C) characterized by flow cytometry. (D) Encapsulating efficiency of 3T3-EM-Dox prepared by direct encapsulation (passive loading) and ammonium sulfate gradient (active loading). (E) Encapsulating efficiency of 3T3-EM-Dox in response to different doxorubicin loading concentrations. Cytotoxic activity of 3T3-EM-Dox in treating MDA-MB-231 (F) and MDA-MB-436 (G) cells.
Drug delivery properties of EMs
Compared to native exosomes, EMs offer an important advantage in that they can be efficiently loaded with small therapeutic molecules using active loading approaches (e.g., ammonium sulfate gradient loading[28]). In the course of the magnetic extrusion protocol, the aqueous solution inside IONP-encapsulating endosomes was replaced with a 240 mM ammonium sulfate solution that generated a concentration gradient across the lipid membrane. Coupling the concentration gradient with a dialysis step, doxorubicin molecules in solution can actively enter and accumulate inside EMs with a high loading efficiency. EM loading efficiency was compared using two different mechanisms: direct encapsulation (passive loading) and ammonium sulfate gradient loading (active loading). As shown in Figure 3D, at the same doxorubicin concentration (1 mg/mL), the ammonium sulfate gradient method achieved a doxorubicin loading efficiency of 68% in 3T3-EMs, whereas only 23% of doxorubicin was loaded into the EMs by direct encapsulation. 3T3-EM loading efficiency was also evaluated at three different doxorubicin concentrations: 0.2 mg/mL, 0.5 mg/mL, and 1 mg/mL, which are commonly used in nanomedicine preparation[29]. Interestingly, 3T3-EM loading efficiency was positively correlated with the concentration of the doxorubicin in solution (Figure 3E). This ammonium sulfate gradient protocol is a well-established method for the loading of many other types of therapeutic agents into synthetic liposomes such as, but not limited, small non-coding genetic material[30,31].
Next, we determined the anti-cancer activity of our engineered doxorubicin-encapsulating 3T3-EMs (3T3-EM-Dox). By incubating 3T3-EM-Dox with MDA-MB-231 or MDA-MB-436 cells at serial concentrations of doxorubicin from 0 mg/mL to 5 mg/mL, we found that 3T3-EM-Dox effectively ablated both MDA-MB-231 and MDA-MB-436 cells (Figures 3F and G). The half-maximum inhibitory concentrations (IC50s) of 3T3-EM-Dox were 1.48 µg/mL for MDA-MB-231 cells and 0.41 µg/mL for MDA-MB-436 cells. In comparison, empty 3T3-EMs (vehicle) show no cytotoxicity in these tested cells, suggesting the 3T3-EMs are safe and biocompatible nanovehicles for cancer drug delivery. These in vitro results indicate that 3T3-EMs can effectively deliver doxorubicin into cancer cells, supporting the further investigation of their therapeutic efficacy in animal models.
In vivo biodistribution of EMs
We next investigated the tumor accumulation and organ biodistribution of 3T3-EMs in an orthotopic, syngeneic 4T1 breast tumor model using in vivo NIR fluorescent imaging (Figure 4A). Notably, this biodistribution study was conducted in immunocompetent BALB/c mice with their homologous 4T1 tumors, featuring a complete immune system that can more faithfully recapitulate the interactions between the breast tumor microenvironment and 3T3-EMs in comparison with other immunocompromised mouse models. Mice bearing orthotopic breast tumors received an intravenous injection of 3T3-EM-DiR, 3T3-EV-DiR, and PEG-LP-DiR at an equivalent dosage of 109 particles per animal. Twenty-four hours post-injection, mice were imaged by NIR fluorescent imaging. As shown in Figures 4B and D, mice treated with 3T3-EM-DiR and 3T3-EV-DiR showed comparable intratumor particle accumulation, approximately 1.5-fold higher than PEG-LP-DiR. These results suggest that the two biogenic nanoparticles, namely 3T3-EMs and 3T3-EVs, are more efficient in tumor targeting and accumulation compared to synthetic PEG-LP-DiR. The in vivo tumor uptake data obtained from live mice precisely matched ex vivo NIR fluorescent images of excised tumors (Figure 4C).
Figure 4. In vivo biodistribution of 3T3-EMs.
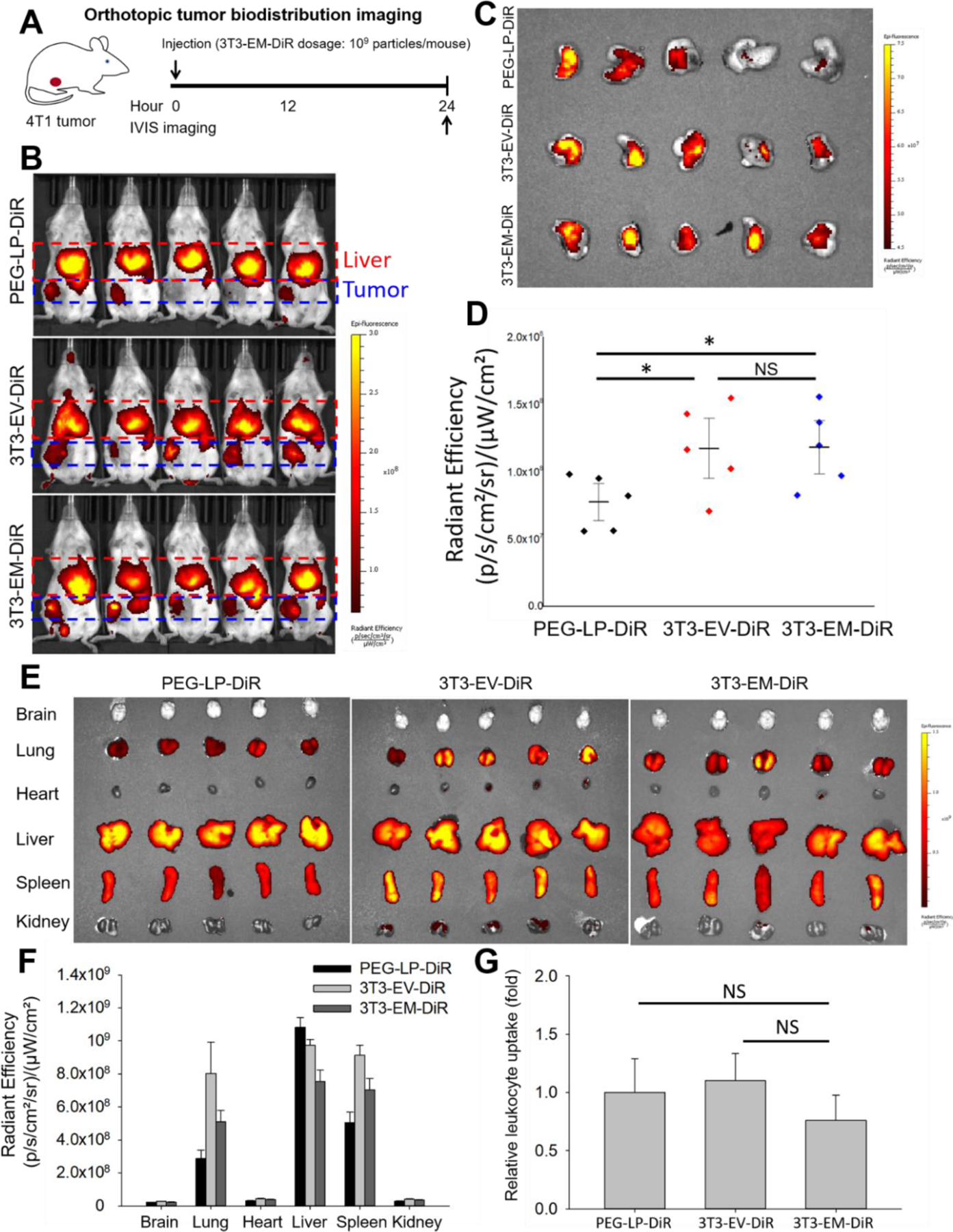
(A) Schematic design of orthotopic tumor biodistribution imaging. (B) In vivo NIR fluorescent images of BALB/c mice at 24 h after the administration of PEG-LP-DiR, 3T3-EV-DiR, and 3T3-EM-DiR (n = 5 per group). (C) Ex vivo NIR fluorescent images of 4T1 tumors treated by PEG-LP-DiR, 3T3-EV-DiR, and 3T3-EM-DiR. (D) Quantified tumor accumulation of PEG-LP-DiR, 3T3-EV-DiR, and 3T3-EM-DiR. (E) Representative ex vivo NIR fluorescent images of major organs (brain, lung, heart, liver, spleen, and kidney). (F) Quantified organ distribution of PEG-LP-DiR, 3T3-EV-DiR, and 3T3-EM-DiR. (G) Circulating leukocyte uptake of PEG-LP-DiR, 3T3-EV-DiR, and 3T3-EM-DiR quantified by flow cytometry (n = 4 to 5 per group). * P<0.05, NS-not significant; unpaired student t-test.
The accumulation of 3T3-EM-DiR, 3T3-EV-DiR, and PEG-LP-DiR in 6 major organs, namely brain, lung, heart, liver, spleen, and kidney (Figures 4E and F) was analyzed. Results showed that, like synthetic PEG-LP-DiR, the off-tumor accumulation of 3T3-EM-DiR and 3T3-EV-DIR is in the liver, spleen, and lung. The interaction of the mouse immune system with the injected nanoparticles was investigated measuring the circulating leukocyte uptake of 3T3-EM-DiR, 3T3-EV-DiR, and PEG-LP-DiR. Mouse blood was collected 24h after the injection of 3T3-EM-DiR, 3T3-EV-DiR, and PEG-LP-DiR. Circulating leukocytes were isolated from the blood by red blood cell lysis followed by a centrifugation step. Leukocyte uptake was quantified using flow cytometry. As shown in Figure 4G, 3T3-EM-DiR, 3T3-EV-DiR, and PEG-LP-DiR showed comparable uptake from circulating leukocytes. These in vivo biodistribution results suggest that EMs have a highly similar drug delivery performance compared to the native EVs collected from the same cell source and are significantly more efficient in reaching the tumor in comparison to liposomes.
In vivo therapeutic activity of EMs.
Next, using the established syngeneic 4T1 orthotopic breast tumor model, we investigated the in vivo antitumor activity of 3T3-EM-Dox (Figure 5A). 4T1 tumor-bearing mice were randomly divided into four groups and received intravenous injections of PBS (sham), 3T3-EMs (vehicle), free doxorubicin (free Dox), or 3T3-EM-Dox, at an equivalent doxorubicin dosage of 2.5 mg/kg via tail-vein injection. Dox-encapsulating native 3T3-EVs (3T3-EV-Dox) were not able to be evaluated in this therapy study due to the inherent inability to prepare them with the same 3T3-EM-Dox parameters. As has been reported, the Dox loading efficiency of EVs has been reported to be only 10% using the ammonium sulfate gradient loading method[32], while the Dox loading efficiency for EMs is 68%. As shown in Figures 5B and C, after four treatments, 3T3-EM-Dox exhibited the highest inhibitory effect on 4T1 tumor growth among all tested groups. The quantified tumor volume and mass revealed that 3T3-EM-Dox significantly reduced 4T1 tumor growth by 41%, approximately 1.34-fold more efficient than Free Dox (Figure 5D). During the treatment, we observed no weight loss (Figure 5E). These in vivo efficacy results indicate that 3T3-EM-Dox represent safe and efficient nanotherapeutics for the treatment of breast cancer.
Figure 5. Therapeutic efficacy of 3T3-EM-Dox.
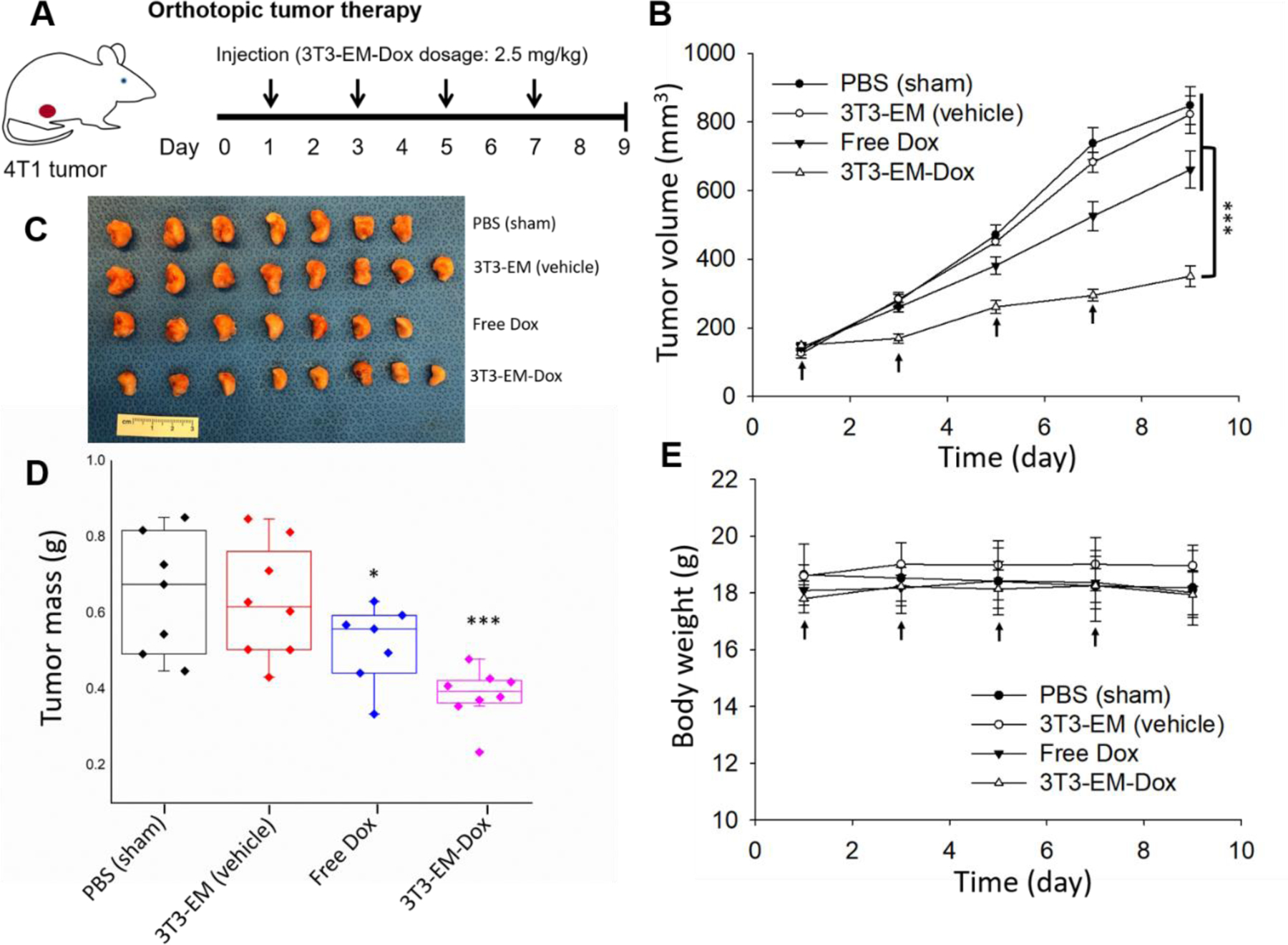
(A) Schematic design of orthotopic tumor therapy model. (B) Tumor progression was closely monitored by tumor volume measurement using a caliper. (C) Image of excised orthotopic TNBC tumors from mice treated with PBS (sham), 3T3-EM (vehicle), Free Dox, 3T3-EM-Dox. (n = 7 to 8 per group). (D) Tumor mass at end point (day 9) was quantified by weight. (E) Mouse body weight during the treatment of PBS (sham), 3T3-EM (vehicle), Free Dox, 3T3-EM-Dox. *P < 0.05; ***P < 0.001.
3. Conclusion
In summary, we have developed a novel and scalable magnetic extrusion method for the production of endosome-derived EMs that can be used as drug delivery vehicles for cancer treatment. The therapeutic payloads can be effectively loaded into EMs using an ammonium sulfate gradient method that is not limited to chemodrugs but is also expected to apply to other types of therapeutics (e.g., nucleic acids and proteins). These engineered EMs have broad drug delivery applications not limited to cancer that include other life-threatening diseases. Going forward, we intend to genetically modify the parental cell source of EMs in order to express tumor-targeting fusion proteins on the endosome membranes of cells thereby enabling us to develop active targeting EMs to optimize EM tumor specificity further and to reduce off-target adverse effects.
4. Experimental Section
Materials
Gibco® Dulbecco’s Modified Eagle Medium (DMEM), Roswell Park Memorial Institute 1640 Medium (RPMI-1640), Dulbecco’s phosphate-buffered saline (PBS), 4′,6-diamidino-2-phenylindole (DAPI), Triton X-100, 0.25% trypsin/2.6 mM ethylenediaminetetraacetic acid (EDTA) solution were purchased from Invitrogen (Carlsbad, CA, USA). Iron oxide nanoparticles (10 nm in diameter) were purchased from Ocean Nanotech (San Diego, CA, USA) and were coated with oligosaccharides to improve their water solubility as previously reported[33]. Ammonium sulfate, glutaraldehyde, paraformaldehyde, picric acid, sodium cacodylate, uranyl formate, doxorubicin (Dox) were purchased from Sigma-Aldrich (St. Louis, MO, USA). 1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindotricarbocyanine Iodide (DiR), Pierce BCA Protein Assay Kit, polycarbonate track-etched (PCTE) membrane (200 nm in diameter), Lab-Tek II Chamber Slide System, and Slide-A-Lyzer dialysis cassette (MWCO20 KD) were purchased from Thermo Fisher Scientific (Pittsburgh, PA, USA). Red blood cell (RBC) lysis buffer (10X) was purchased from BioLegend (San Diego, CA, USA). 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-distearoyl-sn-glycero-3 phosphoethanolamine-N-poly(ethylene glycol)-2000 (DSPE-PEG) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). FLOAT-A-LYZER G2 dialysis tubing (MWCO1,000 kDa) was purchased from Spectrum Laboratories (Rancho Dominguez, CA, USA). The Dojindo cell counting kit CCK-8 was purchased from Dojindo Molecular Technologies (Rockville, MD, USA). Formvar/carbon-coated grid was purchased from Electron Microscopy Sciences (Hatfield, PA, USA).
Cell culture
Two human breast cancer cell lines (MDA-MB-231 and MDA-MB-436), one murine normal embryonic fibroblast cell line (3T3), and one murine breast cancer cell line (4T1) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). MDA-MB-231, MDA-MB-436, and 3T3 cells were cultured in DMEM, 4T1 cells in RPMI-1640, with all recommended supplements. All cells were maintained at 37°C in a humidified incubator with 5% CO2.
EM preparation and isolation
EMs were prepared using a magnetic extrusion method developed by us. Briefly, 5×105 MDA-MB-231 or 3T3 cells were seeded into each well of 6-well plate and iron oxide nanoparticles (IONPs, 10 in diameter) were added to cells at a final concentration of 50–200 µg/mL. After 12 h incubation, non-internalized and cell surface-bound IONPs were removed by PBS (pH 7.4) washing. Cells taking up IONPs were collected by trypsinization and washed with PBS. The collected IONP-uptaken cell pellets were resuspended in a hypotonic buffer for 15 min followed by homogenization with 10 strokes of a Dounce homogenizer. After cell lysis, the IONP-encapsulating endosomes were isolated from cell debris and other cellular organelles by magnetic separation for 1 h. The purified IONP-encapsulating endosomes were washed by PBS and resuspended in 240 mM ammonium sulfate solution and were subsequently extruded into monodisperse nanoscale endosome-derived vesicles, also known as exosome mimetics (EMs) using a Lipex Extruder (Transferra Nanosciences, Burnaby, Canada) with a double-decked 200 nm polycarbonate track-etched nanoporous membrane. After extrusion, IONP-free EMs were further purified from free IONPs and IONP-encapsulating EMs by magnetic separation and were dialyzed in PBS (pH 7.4) using a Slide-A-Lyzer dialysis cassette (MWCO 20 kDa) overnight at room temperature (RT). Doxorubicin was then added to 1010 particles/mL EM solution to a final concentration of 0.2–1 mg/mL and incubated for 6 h to facilitate active loading. The resulting doxorubicin-encapsulating EM (EM-Dox) solution was dialyzed in PBS (pH 7.4) to remove free doxorubicin using a Slide-A-Lyzer dialysis cassette (MWCO 20 kDa) overnight at RT. Blank EMs were prepared using the same protocol save for replacing the ammonium sulfate solution with PBS (pH 7.4) without the active loading steps.
Native exosomes and polyethylene-glycol modified liposomes (PEG-LPs) were also prepared as controls. MDA-MB-231 or 3T3-derived small EVs (231-EVs or 3T3-EVs) were isolated using an established ultracentrifugation method previously reported by us[34,35]. Synthetic PEG-LPs were prepared by a lipid mixture of DOPC and DSPE-PEG(2000) at a molar ratio of 95:5 via an established nanoporous membrane extrusion method[36–39].
EM characterization
The morphology of IONP-internalized MDA-MB-231 cells, IONP-encapsulating endosomes and extruded EMs was determined by transmission electron microscopy (TEM). These biological samples were first fixed with a mixed solution of 2.5% glutaraldehyde, 1.25% paraformaldehyde and 0.03% picric acid in 0.1 M sodium cacodylate buffer (pH 7.4) for 2 h at RT followed by adsorption onto a Formvar/carbon-coated grid and staining with uranyl formate. The grids were imaged using a JEOL 1200EX transmission electron microscope (JEOL, Peabody, MA, USA) and images were taken with an AMT 2k CCD camera. The size and concentration of EMs, native EVs, and PEG-LPs were measured using nanoparticle tracking analysis (NanoSight NS300, Malvern Instruments, Malvern, UK). The protein concentration of EMs and native EVs were measured using a Pierce BCA protein assay.
Western Blot Analyses
Samples were mixed with lithium dodecyl sulfate (LDS)-sample buffer 4X reducing (Thermo Fisher Scientific) and boiled (5 min at 95 °C). MDA-MB-231 and 3T3 cell homogenates were used as controls for EM and EV sample analysis. Cell homogenates were obtained by adding Cell Lysis Buffer 10X (Cell signaling) to the culture plates. Cells were kept on ice for five minutes, scraped, and transferred into low protein binding microtubes for centrifugation (12,000 rcf for 5 min). Supernatants were collected for analysis. The protein content was measured with a Bradford assay (Bio-Rad) and the same amount of total protein (20 μg for cell homogenates, and 5 μg for EV and EM samples) was loaded, electrophoresed on a polyacrylamide gel (4–12%), and analyzed by Western blot. The following antibodies were used: rabbit polyclonal anti-Annexin A2 (1:500 dilution; clone 8235S; Cell Signaling), rabbit polyclonal anti-calnexin (1:1000 dilution; clone GTX112886; GeneTex), rabbit monoclonal anti-CD73 (1:500 dilution; clone D7F9A; Cell Signaling), mouse monoclonal anti-CD63 (1:500 dilution; clone TS63; abcam), anti-rabbit immunoglobulin G (IgG) secondary horseradish peroxidase (HRP)-linked antibody (1:3000; Cell Signaling Technology, USA), and anti-mouse IgG secondary HRP-linked antibody (1:3000; Thermo Fisher Scientific, USA). The WesternSure Pre-stained Chemiluminescent Protein Ladder (LI-COR, USA) was used to confirm the molecular weights of the target proteins. Immunoreactive bands were identified using the SuperSignal West Femto (Thermo Fisher Scientific, USA).
Fluorescent labeling of EMs
For cell uptake and biodistribution studies, EMs, native EVs, and PEG-LPs were fluorescently labeled with a lipophilic carbocyanine DiIC18 (DiR) dye. Briefly, 1010 particles/mL EM solution was incubated with 40 µg/mL DiR on a rotator for 30 min at RT. The resulting DiR-labeled EMs (EM-DiR) were dialyzed in PBS to remove free DiR using a FLOAT-A-LYZER G2 dialysis tubing (MWCO 1,000 kDa) overnight. DiR labeled EVs (EV-DiR), and PEG-LPs (PEG-LP-DiR) were also prepared using the same conditions as controls.
Cell uptake of EMs
100,000 cells (MDA-MB-231 or MDA-MB-436) were seeded in 8-well Lab-Tek II Chamber Slide System and allowed to attach overnight. Cells were incubated with EM-DiR, EV-DiR or PEG-LP-DiR at a concentration of 103 particles/cell in DMEM with 10% FBS for 8 h at 37 oC. DAPI was used to stain cell nuclei. After an 8 h incubation, cells which took up the EM were washed with PBS (pH7.4) and fixed with 4% Formalin solution and dried overnight in the dark. Fluorescent images of stained cells were obtained using a Leica TCS SP5 confocal fluorescent microscope (Leica Microsystems, Buffalo Grove, USA).
Doxorubicin encapsulation efficiency measurement
The doxorubicin encapsulating efficiency of EM-Dox and control liposomes was determined using a colorimetric assay. A standard calibration curve was first generated from serially diluted free doxorubicin solutions and appropriate backgrounds measured at an absorbance wavelength of 490 nm using a FilterMax F3 Multi-Mode Microplate Reader (Molecular Devices, San Jose, USA). Then, 500 μL EM-Dox or PEG-LP-Dox solution was added to 500 μL of 0.5% Triton X-100 in a microcentrifuge tube and vortexed for 1 min to disrupt the structure of the lipid bilayer. This microcentrifuge tube was incubated at 37oC for 1h. The released doxorubicin was directly measured and calculated using its standard calibration curve.
In vitro therapeutic efficacy of EM-Dox
The in vitro therapeutic efficacy of EM-Dox was determined using a Dojindo cell viability assay. Briefly, 104 cells (MDA-MB-231 or MDA-MB-436) were seeded in each well of a 96 well plate and incubated for 24 h. Cells were then treated with PBS, EM (vehicle), and EM-Dox at Dox concentrations ranging from 0 to 50 µg/mL for 8 h. Cells were rinsed with PBS and grown for 48 h. Cell viability was determined using a Dojindo cell counting kit according to the protocol provided by the manufacturer.
In vivo biodistribution and antitumor efficacy of EM-Dox
Animal studies were performed according to the protocols approved by the Institutional Animal Care and Use Committees of Boston Children’s Hospital. For in vivo biodistribution studies, orthotopic, syngeneic 4T1 breast tumors were established by orthotopically injecting 2×106 4T1 cells into the fourth right mammary fat pad of female BALB/c mice (EnvigoRMS, Indianapolis, IN, USA) as previously described by us[37,38,40,41]. When tumors volumes reached approximately 500 mm3, 4T1 tumor-bearing mice were randomized into three treatment groups (n=5 per group) and received intravenous administration of 3T3-EM-DiR, 3T3-EV-DiR, and PEG-LP-DiR at an equivalent nanoparticle concentration of 109 particles in 100 µL PBS (pH 7.4) per mouse via tail-vein injection. At 24h after the injection, in vivo NIR fluorescence imaging was performed using an IVIS Lumina II system (Caliper, Hopkinton, MA, USA). All mice were then sacrificed and the ex vivo NIR fluorescence intensity of various organs (brain, heart, liver, lung, kidney, and spleen) and excised tumors was measured using IVIS Lumina II. Leukocytes were isolated from the whole blood of the mice via centrifugation at 1,200 rpm for 15 min followed by removal of the red blood cells using RBC lysis buffer. The fluorescence intensity of leukocytes treated with 3T3-EM-DiR, 3T3-EV-DiR, and PEG-LP-DiR was quantified using a BD FACSCalibur Flow Cytometer (BD Biosciences, San Jose, USA).
For the antitumor efficacy experiments, when orthotopic 4T1 breast tumors reached 100 mm3 in volume, mice were randomly divided into four different groups (n=7–8 per group) and received intravenous administration of PBS (sham), 3T3-EM without Dox (vehicle), free doxorubicin (Free Dox), and 3T3-EM-Dox at a Dox dose of 2.5 mg/kg at every two days using tail-vein injection. Tumor growth was monitored using a caliper and mouse body weights were measured every two days. Nine days after treatment, orthotopic tumors were excised to measure their mass.
Statistical analysis
All of the experimental data were measured in triplicate at least and are presented as mean ± standard deviation unless otherwise mentioned. Statistical variance of two comparison groups was performed at a significance level of P < 0.05 based on a paired Student’s t-test. One and two-way ANOVA with Bonferroni post hoc tests were used to analyze statistical variance when making multiple comparisons. All statistical analyses were performed using SigmaPlot 12.0 software.
Acknowledgments
M.A.M. acknowledges the support of NIH NCI R21CA253051 and the Breast Cancer Research Foundation. P.G. acknowledges the support of the OFD/BTREC/CTREC Faculty Career Development Fellowship of Boston Children’s Hospital. The authors thank Kristin Johnson of the Vascular Biology Program at Boston Children’s Hospital for assistance with the schematic illustrations.
Footnotes
Conflict of interest
P.G., J.H., G.M., and M.A.M. are co-inventors of a patent application filed by the Boston Children’s Hospital (filed on 16 August 2019). All other authors declare no competing interests.
References
- [1].Abels ER, Breakefield XO, Cell. Mol. Neurobiol 2016, 36, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Xu R, Rai A, Chen M, Suwakulsiri W, Greening DW, Simpson RJ, Nat. Rev. Clin. Oncol 2018, 15, 617. [DOI] [PubMed] [Google Scholar]
- [3].Wiklander OPB, Brennan MÁ, Lötvall J, Breakefield XO, EL Andaloussi S, Sci. Transl. Med 2019, 11, eaav8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Morad G, Moses MA, J. Extracell. Vesicles 2019, 8, 1627164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Busatto S, Pham A, Suh A, Shapiro S, Wolfram J, Biomed. Microdevices 2019, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Takov K, Yellon DM, Davidson SM, J. Extracell. Vesicles 2019, 8, 1560809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Haraszti RA, Miller R, Stoppato M, Sere YY, Coles A, Didiot M-C, Wollacott R, Sapp E, Dubuke ML, Li X, Shaffer SA, DiFiglia M, Wang Y, Aronin N, Khvorova A, Mol. Ther 2018, 26, 2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Busatto S, Vilanilam G, Ticer T, Lin W-L, Dickson D, Shapiro S, Bergese P, Wolfram J, Cells 2018, 7, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Colao IL, Corteling R, Bracewell D, Wall I, Trends Mol. Med 2018, 24, 242. [DOI] [PubMed] [Google Scholar]
- [10].Guo P, Huang J, Moses MA, Trends Pharmacol. Sci 2020, 41, 730. [DOI] [PubMed] [Google Scholar]
- [11].Kooijmans SAA, Stremersch S, Braeckmans K, de Smedt SC, Hendrix A, Wood MJA, Schiffelers RM, Raemdonck K, Vader P, J. Controlled Release 2013, 172, 229. [DOI] [PubMed] [Google Scholar]
- [12].Lamichhane TN, Raiker RS, Jay SM, Mol. Pharm 2015, 12, 3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Walker S, Busatto S, Pham A, Tian M, Suh A, Carson K, Quintero A, Lafrence M, Malik H, Santana MX, Wolfram J, Theranostics 2019, 9, 8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yang Z, Shi J, Xie J, Wang Y, Sun J, Liu T, Zhao Y, Zhao X, Wang X, Ma Y, Malkoc V, Chiang C, Deng W, Chen Y, Fu Y, Kwak KJ, Fan Y, Kang C, Yin C, Rhee J, Bertani P, Otero J, Lu W, Yun K, Lee AS, Jiang W, Teng L, Kim BYS, Lee LJ, Nat. Biomed. Eng 2020, 4, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lunavat TR, Jang SC, Nilsson L, Park HT, Repiska G, Lässer C, Nilsson JA, Gho YS, Lötvall J, Biomaterials 2016, 102, 231. [DOI] [PubMed] [Google Scholar]
- [16].Jang SC, Kim OY, Yoon CM, Choi D-S, Roh T-Y, Park J, Nilsson J, Lötvall J, Kim Y-K, Gho YS, ACS Nano 2013, 7, 7698. [DOI] [PubMed] [Google Scholar]
- [17].Rueda-Gensini L, Cifuentes J, Castellanos MC, Puentes PR, Serna JA, Muñoz-Camargo C, Cruz JC, Nanomaterials 2020, 10, 1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Popescu RC, Savu D, Dorobantu I, Vasile BS, Hosser H, Boldeiu A, Temelie M, Straticiuc M, Iancu DA, Andronescu E, Wenz F, Giordano FA, Herskind C, Veldwijk MR, Sci. Rep 2020, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Feng Q, Liu Y, Huang J, Chen K, Huang J, Xiao K, Sci. Rep 2018, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Storrie B, Amadden E, In Methods in Enzymology, Elsevier, 1990, pp. 203–225. [DOI] [PubMed] [Google Scholar]
- [21].Graham JM, Rickwood D, Eds., Subcellular fractionation: a practical approach, IRL Press at Oxford University Press, Oxford ; New York, 1997. [Google Scholar]
- [22].Pryor PR, Subcellular fractionation: a laboratory manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 2015. [Google Scholar]
- [23].Wagner A, Vorauer-Uhl K, J. Drug Deliv 2011, 2011, 591325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hoshyar N, Gray S, Han H, Bao G, Nanomed 2016, 11, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Blanco E, Shen H, Ferrari M, Nat. Biotechnol 2015, 33, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Danaei M, Dehghankhold M, Ataei S, Hasanzadeh Davarani F, Javanmard R, Dokhani A, Khorasani S, Mozafari MR, Pharmaceutics 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Iranmanesh M, Hulliger J, Chem. Soc. Rev 2017, 46, 5925. [DOI] [PubMed] [Google Scholar]
- [28].Haran G, Cohen R, Bar LK, Barenholz Y, Biochim. Biophys. Acta BBA - Biomembr 1993, 1151, 201. [DOI] [PubMed] [Google Scholar]
- [29].Haghiralsadat F, Amoabediny G, Sheikhha MH, Forouzanfar T, Helder MN, Zandieh-Doulabi B, Cell J 2017, 19, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lamichhane TN, Jeyaram A, Patel DB, Parajuli B, Livingston NK, Arumugasaamy N, Schardt JS, Jay SM, Cell. Mol. Bioeng 2016, 9, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jeyaram A, Lamichhane TN, Wang S, Zou L, Dahal E, Kronstadt SM, Levy D, Parajuli B, Knudsen DR, Chao W, Jay SM, Mol. Ther 2020, 28, 975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Antimisiaris S, Mourtas S, Marazioti A, Pharmaceutics 2018, 10, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Huang J, Wang L, Zhong X, Li Y, Yang L, Mao H, J Mater Chem B 2014, 2, 5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Morad G, Carman CV, Hagedorn EJ, Perlin JR, Zon LI, Mustafaoglu N, Park T-E, Ingber DE, Daisy CC, Moses MA, ACS Nano 2019, 13, 13853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Morad G, Daisy C, Otu H, Libermann T, Dillon S, Moses M, Int. J. Mol. Sci 2020, 21, 3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Guo P, You J-O, Yang J, Moses MA, Auguste DT, Biomaterials 2012, 33, 8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Guo P, Wang B, Liu D, Yang J, Subramanyam K, McCarthy CR, Hebert J, Moses† MA, Auguste† DT, Nano Lett 2018, 18, 2254. † contributed equally. [DOI] [PubMed] [Google Scholar]
- [38].Guo P, Yang† J, Liu D, Huang L, Fell G, Huang J, Moses‡ MA, Auguste‡ DT, Sci. Adv 2019, 5, eaav5010. †contributed equally. ‡ contributed equally. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Guo† P, Moses-Gardner† A, Huang J, Smith‡ ER, Moses‡ MA, Sci. Rep 2019, 9‡6195. †contributed equally. † contributed equally. ‡ contributed equally. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Guo P, Liu D, Subramanyam K, Wang B, Yang J, Huang J, Auguste† DT, Moses† MA, Nat. Commun 2018, 9:130. † contributed equally. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Guo P, Yang J, Huang J, Auguste† DT, Moses† MA, Proc. Natl. Acad. Sci 2019, 116, 18295. † contributed equally. [DOI] [PMC free article] [PubMed] [Google Scholar]