

Abstract
Tumor neoepitopes presented by major histocompatibility complex (MHC) class I are recognized by tumor-infiltrating lymphocytes (TIL) and are targeted by adoptive T-cell therapies. Identifying which mutant neoepitopes from tumor cells are capable of recognition by T cells can assist in the development of tumor-specific, cell-based therapies and can shed light on antitumor responses. Here, we generate a ranking algorithm for class I candidate neoepitopes by using next-generation sequencing data and a dataset of 185 neoepitopes that are recognized by HLA class I–restricted TIL from individuals with metastatic cancer. Random forest model analysis showed that the inclusion of multiple factors impacting epitope presentation and recognition increased output sensitivity and specificity compared to the use of predicted HLA binding alone. The ranking score output provides a set of class I candidate neoantigens that may serve as therapeutic targets and provides a tool to facilitate in vitro and in vivo studies aimed at the development of more effective immunotherapies.
Clinical studies performed in individuals with melanoma1–6, gastrointestinal cancer7,8 and breast cancer9 receiving adoptive transfer of autologous in vitro-cultured TIL indicate that T cells that recognize mutant antigens, or neoantigens, play an important role in mediating tumor regression. Further evidence that neoantigens represent important therapeutic targets has been provided by studies indicating that responses to immune checkpoint inhibitor therapies are associated with mutational load10–13. These responses were observed in tumor types bearing relatively high mutational loads, such as melanoma, non-small cell lung cancer or in mismatch repair-insufficient tumors14, which possess increased mutational burdens relative to mismatch repair-sufficient tumors that fail to respond to these therapies15.
The development of methods that use tumor DNA and RNA sequencing to identify neoantigens targeted by tumor-reactive T cells has provided insights into factors that influence antitumor responses and has served as the basis for the development of clinical treatments. These methods involve the integration of HLA binding algorithms16–18 with additional tools that include the change in binding rank of mutant and wild-type peptides19, gene expression and peptide-processing prediction algorithms20–22. In one study, machine learning was used to develop a model for neoantigen identification based on 12 features that included predicted HLA binding and peptide processing23. The training sets used in this study, however, did not include a large panel of verified cancer neoepitopes and lacked the corresponding negative candidates for these samples. In another study, an analysis of 52 known neoepitopes and a matching set of 2,760 negative candidates indicated that additional features, such as predicted peptide stability and processing efficiency, did not appear to substantially enhance a filtering method based on HLA binding predictions using an affinity threshold of 500 nM (ref.24).
The use of neoepitope prediction methods has also provided evidence for an association between increased predicted neoepitope burden and response to immune checkpoint therapy10,25. In other studies where this association was not observed, it appeared that additional factors, such as tumor mutational clonality11 and the degree of tumor infiltration by T cells26, had a significant influence on clinical responses to immune checkpoint therapy.
The current study utilizes a dataset of 185 tumor neoantigens, which were identified by screening cultured TIL from 96 individuals with metastatic cancers, and corresponding negative screening data, to develop machine learning models for candidate neoantigens. The resulting models provide an estimate of the relative influence of multiple features on MHC class I–restricted neoepitopes and provide tools that further facilitate neoantigen identification.
Results
Sample training data.
A group of 70 individuals with metastatic cancer with at least one human leukocyte antigen (HLA) class I–restricted epitope and both whole-exome and RNA-sequencing data were chosen to represent the training set (Supplementary Table 1)4,5,8,27–29. Non-synonymous single-nucleotide variants (snvs) and insertion/deletion mutations (indels) identified from these samples generated a starting dataset of 30,863 unique amino acid sequences consisting primarily of the mutant residue and the 12 residues corresponding to the upstream and downstream wild-type sequences, termed mutant nmers. The nmers included one or more flanking amino acids to facilitate the processing of class I products from the tandem minigene constructs used for screening and to aid with the detection of HLA class II neoepitopes, which are generally longer than class I epitopes but were not investigated in this study. A subset of 9,541 nmers were selected (see Methods) and evaluated for immunoreactivity by coculturing autologous antigen-presenting cells expressing mutant long peptides and tandem minigene constructs, as previously described28. Initial assay results demonstrated that 139 (1.46%) of the screened nmers, termed positive nmers, were recognized by MHC class I–restricted T cells (Supplementary Table 2). Subsequently, the 8- to 12-amino acid-long peptides containing the mutation, termed mutated minimal peptides (mmps), were screened to identify the optimal epitope and the HLA restriction element responsible for peptide presentation, resulting in the identification of 120 class I mmps and their corresponding HLA restriction elements (Supplementary Table 3).
Evaluation of binding prediction models.
Initially, the NetMHCpan4.0 binding (BA) and elution (EL) models30, MixMHCpred2.02 (ref.31), HLAthena32 and MHCflurry1.6 (ref.18) binding algorithms were evaluated to determine their performance in sorting candidate class I mmps. The peptide percentile binding ranks, representing the predicted affinity percentile of a given peptide relative to a set of random peptides, were used to help obviate biases in scoring that result from differences in the mean binding affinities of HLA class I alleles33. The models were assessed by randomly subsampling 500 known negative mmps with the 120 positive mmps and recording the area under the curve (AUC) for each model. When this process was performed 1,000 times, MHCflurry1.6 (mean AUC = 0.9947 ± 0.015) slightly outperformed NetMHCpan4.0 EL (mean AUC = 0.9938 ± 0.016) (Supplementary Table 4). All models performed well; however, HLAthena does not support alleles with a length of 12, leaving it unable to score one positive mmp, and MixMHCpred2.02 does not currently support three alleles where positive mmps were found (HLA-B*42:01, HLA-C*07:04 and HLA-A*26:08), resulting in lower AUCs. Notably, MHCflurry1.6 boosted the rankings in eight of ten positive mmps with a NetMHCpan4.0 EL percentile rank of greater than two, six of which contain cysteine residues at primary or secondary anchor positions (Supplementary Table 3 and Extended Data Fig. 1). Although the ranks of the putative cysteine anchor peptides are still among the poorest of the positive mmps, MHCflurry1.6 appeared to partially correct the underestimation of their ranks resulting from the effects of peptide oxidation on peptide elution and binding assay results34–36.
Evaluation of nmers using predicted binding rank.
The MHCflurry1.6 percentile rank was used to score mmps generated from all nmers in the training set, regardless of screening status, using the appropriate class I alleles for each individual. As the combination of differing mmp lengths and an individual’s class I composition can lead to the generation of 300 or more unique HLA–peptide combinations, a single score was given to each of the 30,863 nmers. The assigned score was the best percentile rank of all the mmps derived from an individual nmer and was then subsequently used to evaluate the sensitivity and specificity of the predicted binding rank on neoantigen selection. Analysis of the 139 mutant nmers recognized by T cells revealed that 99% of the 139 positive nmers and 95% of all nmers contained one or more mmp with a predicted affinity rank percentile of two or lower, a threshold for weak binders, and 94% of the positive nmers and 66% of all nmers contained one or more peptides that fell within a predicted rank binding threshold of 0.5 or lower, a commonly used cutoff for strong binders (Supplementary Table 5). As shown in Supplementary Table 5, similar percentages of screened and unscreened nmers remained at each evaluated cutoff, indicating that the high percentages of positives were not the result of a selection bias during nmer screening. The frequency of nmers that did not contain any predicted strong or weak binder ranged between 1% and 14% of the nmers for each of the samples (Supplementary Table 6), although the significance of this finding is unclear as the frequencies did not appear to be associated with the presence or absence of particular HLA alleles.
An mmp–HLA combination was noted as being screened if the nmer from which it was derived was tested for recognition by an individual’s TIL. An analysis of all screened mmp–HLA combinations revealed that 113 (94.2%) of the positive mmps possessed an MHCflurry1.6 percentile rank of two or lower. Of all candidate mmp–HLA combinations, 72,146 (2.6%) possessed a rank score at or below two, similar to the expected frequency at this percentile cutoff. At a more stringent cutoff of 0.5 or less, 98 (81.7%) of the positive and approximately 16,400 (0.6%) of all candidate mmp–HLA combinations would be retained (Fig. 1a and Supplementary Table 7). When assessed for their rank among all mmps derived from a given nmer, 72 of the 120 positive mmp–HLA combinations had the best predicted binding rank within that nmer, 93 were in the top two, 101 were in the top three and the remaining positives ranked between the 4th and 48th by predicted binding rank, indicating that the top predicted binder is not always the recognized peptide (Supplementary Table 8).
Fig. 1 |. Evaluating criteria for their effects on nmer discovery.
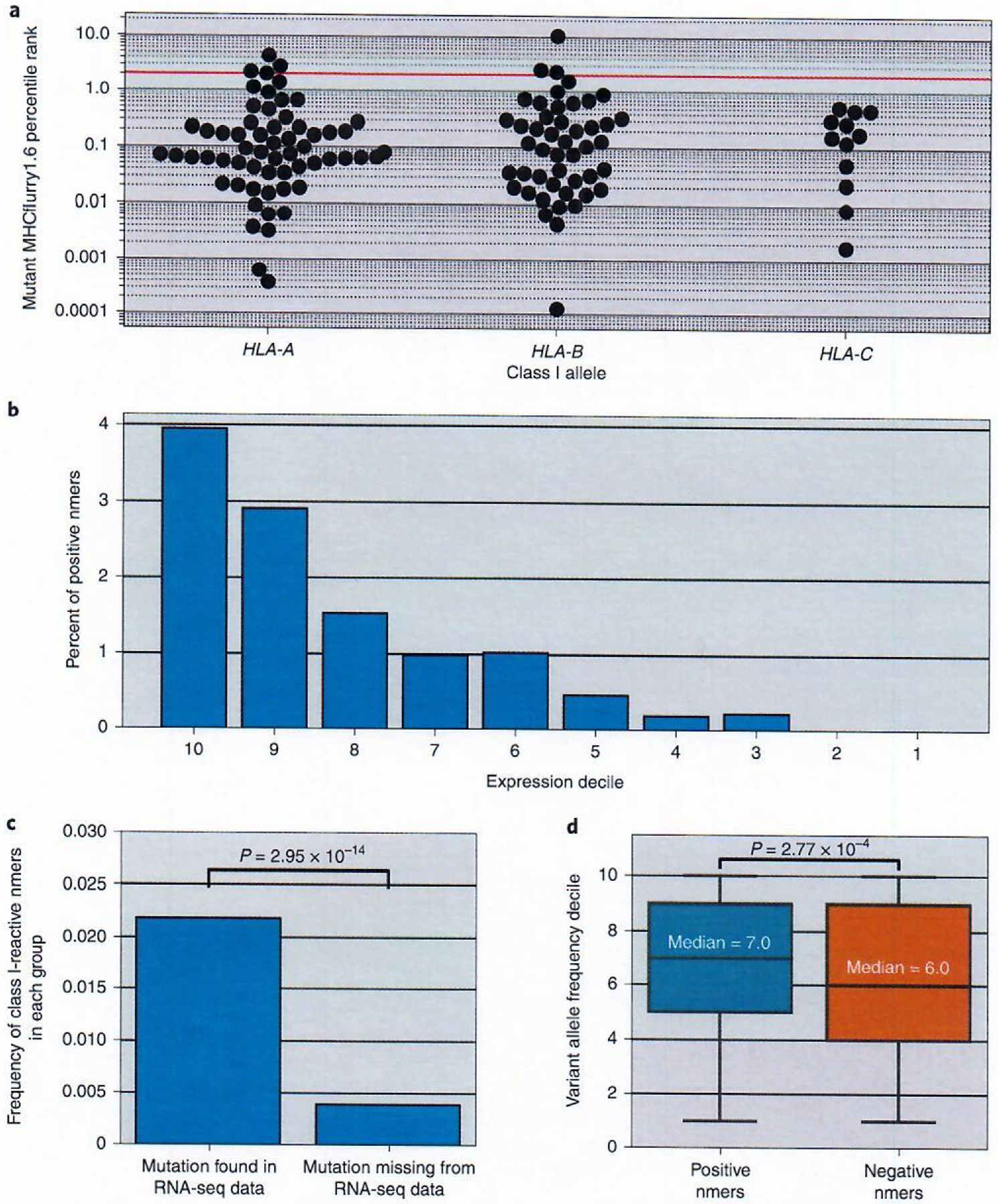
a, Known CD8+ minimal epitopes predicted MHCflurry1.6 percentile rank for each HLA; HLA-A, n = 60; HLA-B, n = 47 and HLA-C, n = 12. The red line indicates the rank two cutoff. b, Histogram plotting the percentage of positive nmers found within the screened nmers from each expression decile. N = 1,240, 1,342, 1,371, 1,317, 1,081, 870, 539, 449, 317 and 1,015 nmers for deciles 10, 9, 8, 7, 6, 5, 4, 3, 2 and 1, respectively. c, Analysis of variant presence in corresponding RNA-seq data. The frequencies of reactive class I variants seen in nmers encoded by mutations that were detected in RNA-seq data, comprising 5,686 tested nmers and 124 class I–reactive nmers were compared with those that were not seen in the RNA-seq data, comprising a total of 3,855 tested nmers and 15 class I–reactive nmers, are shown.. Significance was calculated by a two-sided Fisher’s exact test. d, Box plot comparing the VAFs of reactive (n = 139) and non-reactive (n = 9,402) nmers. The VAFs were normalized and expressed as deciles to facilitate comparisons between samples. For box plots, the box indicates quartiles two and three and interquartile range (IQR), and the median is indicated by the line in the box plot. Whiskers represent quartiles 1 and 4 ± 1.5× IQR or minimum/maximum value if within the whisker values. Significance was calculated using the Mann–Whitney U-test.
Influence of additional features on neoantigen reactivity.
Additional features that could increase the specificity and sensitivity of neoantigen detection were evaluated in the context of both nmer and mmp reactivity. To this end, the relationship between expression and the likelihood of encoding a neoantigen was evaluated using gene expression data that were transformed into expression deciles, with ten representing the highest level of expression and one representing the lowest level of expression (see Methods). Transformation was performed to normalize data across tumor samples possessing variable RNA quality. Evaluation of the effect of expression was performed using only screened nmers and mmps from our training data that included samples for which candidates were prioritized using RNA-seq expression analysis (see Methods). This analysis revealed that 3.95% of the screened nmers in the tenth decile were recognized by TIL, whereas only 0.22% of those ranked in the third decile and none in deciles two or lower were recognized (Fig. 1b and Supplementary Table 9). Given a positivity rate as low as 0.2%, screening of the more than 1,300 nmers from deciles one and two should have resulted in the detection of two or three neoantigens, indicating that the use of expression filters for screening assays did not bias these results. The number and frequency of neoantigens detected in RNA-seq libraries was also higher than variants not found in RNA-seq libraries among the screened nmers, with approximately fivefold higher frequency of positives in the subset of mutations seen in RNA-seq (Fig. 1c and Supplementary Table 10). Nevertheless, mutant transcripts could not be identified for 10% of the neoepitopes, potentially due to underrepresentation of transcripts in the RNA-seq libraries or degradation of RNA. Our data demonstrate that detection of variants in transcriptome data has a positive predictive value for candidate neoepitope ranking models.
Mutations possessing higher variant allele frequencies (VAFs) are more likely to be clonal than lower frequency variants, and their prioritization represents another strategy used during candidate neoantigen selection37. Our data validated this strategy, with positive nmers possessing significantly higher normalized VAFs (see Methods) than those encoding negative nmers, although there was a high degree of overlap between these groups (Fig. 1d and Supplementary Table 11).
Evaluation of neoepitope enrichment within a particular intra-cellular compartment was performed using WoLF Psort38 to predict the localization of proteins corresponding to the screened nmers. The result of this analysis revealed an approximately two-fold enrichment of positive nmers within the mitochondrial compartment, although the difference between positive and negative nmers was not significant when corrected for multiple comparisons (Bonferroni-corrected P value = 0.5) (Supplementary Table 12 and Extended Data Fig. 2). Microbial sequence similarity also did not appear to influence immunogenicity, as only 11 of the 120 recognized mmps aligned to a set of previously published microbial antigens (Supplementary Table 13)26.
Evaluation of candidate neoantigens.
A dataset of all mmps that could be derived from the screened nmers was used to evaluate the influence of several factors on peptide immunogenicity. As indicated in the analysis of nmers, rank binding predictions for each peptide in the context of the appropriate participant’s HLA class I alleles revealed that positive mmps possessed significantly lower MHCflurry1.6 percentile rank values than negative mmps (Fig. 2a). MHCflurry1.6 additionally offers two models that provide a score for processing and presentation. Both additional models in the MHCflurry1.6 package showed a significant difference between the scores of the positive and negative mmps (Fig. 2b,c). Evaluation of expression levels revealed that transcripts encoding positive mmps were expressed at significantly higher levels than those encoding negative mmps despite the abundance of negative mmps produced by the same transcripts (Extended Data Fig. 3). Furthermore, increased expression appeared to compensate for poor binding, as the frequency of positive mmps encoded by highly expressed genes was less impacted by binding rank than those encoded by poorly expressed genes (Fig. 2d), consistent with the hypothesis that high expression can facilitate the presentation of peptides with relatively low HLA class I binding affinities39.
Fig. 2 |. Analysis of unique minimal epitopes.
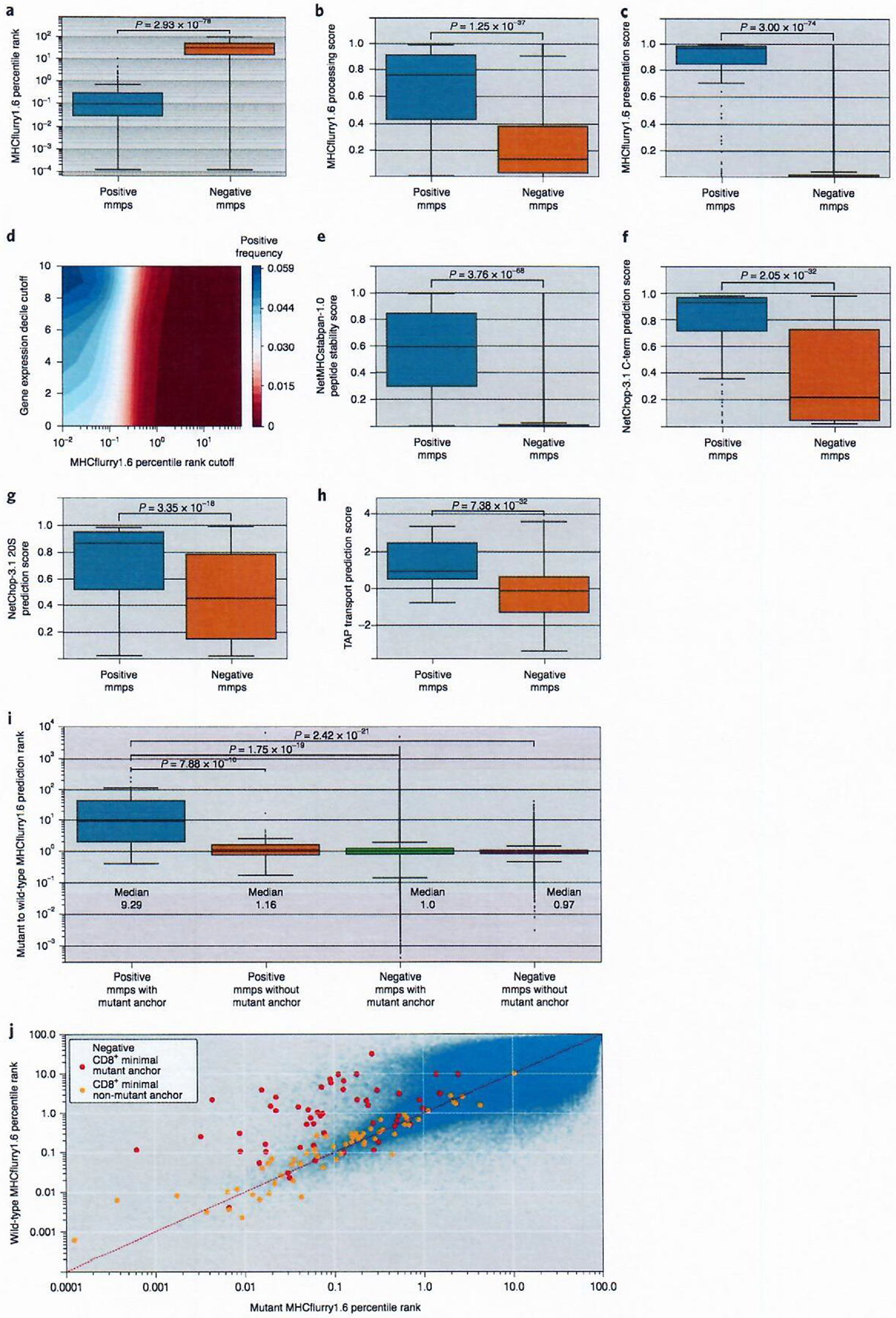
Comparison of reactive (positive mmps, n = 119) and non-reactive (negative mmps, n = 2,681,162) peptide scores for MHCflurry1.6 percentile rank binding predictions (a), MHCflurry1.6 processing score (b) and MHCflurry1.6 presentation score (c). d, Contour plot showing CD8+ frequency for minimal epitopes at different thresholds of MHCflurry1.6 percentile rank binding and expression decile. e, NetMHCstabpan-1.0 stability score for peptide–HLA class I binding complexes. Proteasomal processing score for C-terminal residues using either the NetChop-3.1 C-term (f) or 20S (g) models. h, TAP transport score. i, Box plot of MHCflurry1.6 percentile binding rank for the wild-type peptide in relation to the snv mmp (wild-type:mutant) for reactive (positive mmps, n = 54) and non-reactive (negative mmps, n = 1,083,564) mmps with mutant anchors defined as positions two, three or the C terminus and reactive (positive mmps, n = 64) and non-reactive (negative mmps, n = 1,513,799) snv mmps without mutant anchors. For box plots, the box indicates quartiles two and three and IQR, and the median is indicated by the line. Whiskers represent quartiles 1 and 4 ± 1.5× IQR or minimum/maximum value if within the whisker values. Significance was calculated by a Mann–Whitney U-test. j, Scatter plot of MHCflurry1.6 percentile binding rank for mutant mmps compared to matched wild-type mmps for snv changes. Negative mmps (n = 2,582,291) are shown in blue, positive mmps with mutations outside of anchor residue (n = 64) are shown in red and positive mmps with mutant anchors (n = 54) are shown in orange.
The contribution of peptide and HLA class I complex stability was evaluated using NetMHCstabpan-1.0 (ref.40), an artificial neural network trained to predict the half-life of peptide–HLA class I binding complexes and scores their stability. Positive mmps possessed significantly higher stability scores than negative mmps (Fig. 2e), and 73 of the 120 (61%) positive mmps possessed a stability score greater than or equal to 0.5, whereas only 0.7% of the negative mmps scored within this range (Supplementary Table 14). The combination of a MHCflurry1.6 percentile rank score of two or lower and a stability score of 0.5 or higher further reduced the negative mmps to 10,611 from 19,413 when using a 0.5 stability cutoff alone. These combined filters retained 71 positive mmps, yielding a positive frequency of 0.00667. This represents an increase in precision compared to using either a percentile binding rank of two or a stability score of 0.5 alone, where we observe a positive frequency of 0.0015 and 0.0038, respectively. These findings indicate that combining these two metrics could lead to enhanced neoepitope predictions, as previously suggested41.
The influence of peptide processing on neoepitope generation was evaluated using NetChop-3.1 to predict the efficiency of enzymatic digestion in the proteasome42. This process is responsible for generating the carboxy terminus of many HLA class I epitopes and facilitates transport through the endoplasmic reticulum by the transporter associated with antigen processing (TAP)43. Use of NetChop-3.1, independent of the MHCflurry1.6 processing tool, revealed that positive mmps possessed a significantly higher cleavage score at their C-terminal amino acid than negative mmps using either the C-term or 20S models (Fig. 2f,g). The NetChop-3.1 C-term score, which represents predicted processing by the immunoproteasome, and 20S scores, which represent predicted processing by the standard proteasome, showed similar mean scores for positive mmps (Supplementary Table 3), indicating that epitopes were not preferentially processed by either of the two dominant proteasomal subtypes. Analysis of TAP transport scores, generated by adjusting a previously reported prediction method43 to be compatible with peptides up to a length of 12 amino acids, indicated that the TAP transport efficiency scores of positive mmps were higher than those of negative mmps (Fig. 2h).
An increase in the predicted binding affinity of a mutant peptide relative to the corresponding wild-type peptide may influence neoantigen recognition by enhancing presentation of the epitope19. This has been shown in a mouse model where, for epitopes with a mutated anchor residue, the change in binding between mutant and wild-type epitopes was strongly predictive of peptide immunogenicity44. When comparing the MHCflurry1.6 ranks of mutant mmps and their corresponding wild-type peptides, positive mmps were predominantly distributed in two clusters depending on the position of the mutated residue within the mmp (Fig. 2i). An improved binding rank was observed for positive mmps containing mutations in positions two, three or the C terminus of the epitope, positions that represent primary and secondary anchor positions for the majority of HLA class I alleles. For positive mmps containing snvs in anchor residues, the median ratio of the binding ranks of the wild-type mmps to the corresponding mutant mmps was nine, whereas negative mmps with anchor residue changes possessed a median ratio of one (Fig. 2j and Supplementary Table 3). As expected, the wild-type and mutant binding ranks of positive mmps containing mutant residues at non-anchor positions and negative mmps with or without mutant anchors did not differ significantly, and the ratio observed for positive mmps with mutant anchor residues was significantly higher than that observed for each of the other three categories of mmps (Fig. 2j and Supplementary Table 3). Sixty-four of the 267,386 mmps with mutant non-anchor positions and 56 of the 178,274 mmps with mutant anchor residues were recognized with no significant difference between the frequency of positive mmps in these groups (Fisher’s exact P value = 1.37 × 10−1), indicating that both categories contributed equally to the neoepitopes in this dataset. These findings demonstrate that the difference between mutant and wild-type percentile binding rank of mmps is a relevant feature of the subset of mmps with mutant anchor residues.
Additional analyses using Immune Epitope Database (IEDB) tools45 indicated that positive mmps possessed higher immunogenicity scores and higher hydrophobicity scores in T-cell contact residues than negative mmps (Extended Data Figs. 4 and 5). These results are consistent with previous findings46, although there is overlap between the positive and negative mmps for both the IEDB and hydrophobicity analysis.
Creation of a model for mmp predictions.
The utilization of multiple features to select candidate neoepitopes has enhanced the predictive values of models as cited above; however, the relative contributions of individual features is difficult to elucidate. Compounding this further is the varying levels of correlation they have to one another (Fig. 3a). MHCflurry1.6 presentation and processing scores showed the highest levels of correlation (Spearman r = 0.88), which was expected as the processing score is used as input to generate the presentation score. The NetChop-3.1 C-term and TAP binding scores demonstrated the next highest level of correlation, with an r value of 0.64. A lower level of correlation was also seen between the NetMHCstabpan-1.0 stability score and MHCflurry1.6 binding rank. These models may not be more highly correlated because of the dependency of NetMHCstabpan-1.0 on an older binding prediction algorithm. The presence of these correlations makes it more difficult to select the features and thresholds that would result in the highest sensitivity and specificity of neoantigen detection. To address this issue, a random forest model was developed to provide an unbiased method for estimating the likelihood that an mmp would elicit a class I–restricted immune response. For this analysis, in an attempt to eliminate biases introduced in the screening selection process, screened data were randomly subsampled to generate a training set (described in Methods).
Fig. 3 |. Evaluation of mmp input features.
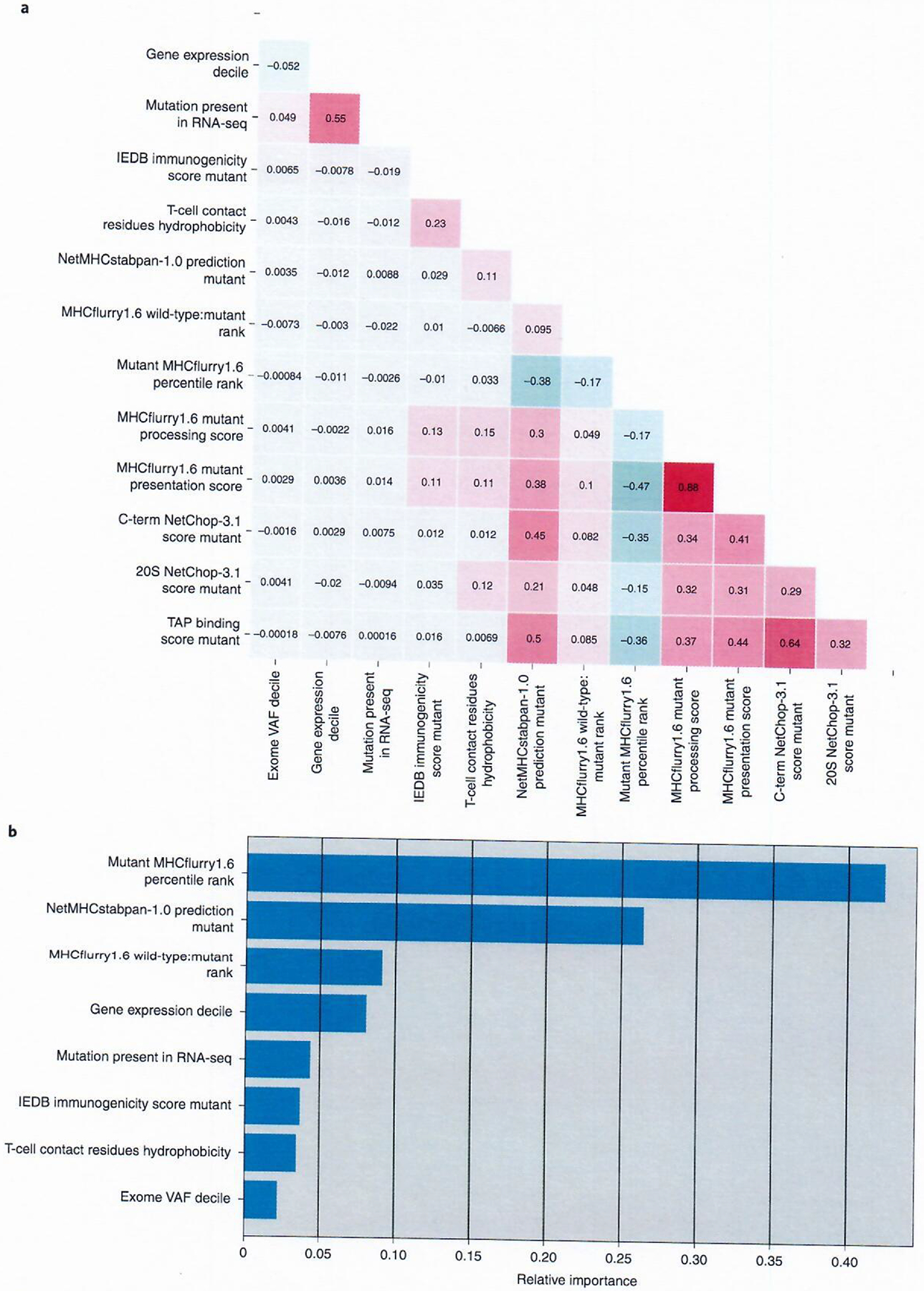
a, Spearman correlation matrix representing the associations between input features tested for the MMP model. b, The relative importance of features in the final MMP model trained with n = 120 positive mmps and n = 1,412,494 negative mmps. The relative importance represents the overall ability of a feature to purify nodes within the model.
To evaluate the ideal features for input to an mmp model, we utilized a fivefold cross validation technique where the training set was split into five equal parts; four parts were used to train the model and the fifth held-out fold was used for validation. This process was repeated until each fold was used as a validation set once. The AUC of each fold was recorded, and the average AUC and its standard deviation were used to determine how robust each model was to variations in the data. All combinations of 14 separate features were tested to determine which features should be used as input in the final mmp model. This analysis generated 16,383 individual models made up of differing input feature combinations tested across each of the five folds and produced four models with mean AUCs ≥ 0.997 (Supplementary Table 15). The input chosen for the final mmp model was gene expression decile, the mutation’s presence in RNA-seq (1 = yes, 0 = no), mutant mmp MHCflurry1.6 percentile rank, MHCflurry1.6 wild-type:mutant percentile rank ratio, NetMHCstabpan-1.0 prediction of the mutant mmp, IEDB immunogenicity score of the mutant mmp, T-cell contact residue hydrophobicity of the mutant mmp and the exome VAF decile. This set of features had the third highest mean AUC, the highest overall mean average precision score as well as both the highest sensitivity and positive predictive value at a cutoff of 0.5, the lowest cutoff score evaluated (Supplementary Table 16). For the final mmp model, termed the MMP model, MHCflurry1.6 percentile rank represented the most important feature for reducing node impurity, with a feature importance value of 0.425, followed by NetMHCstabpan-1.0 stability prediction, MHCflurry1.6 wild-type:mutant percentile rank ratio and gene expression decile (Fig. 3b). The exclusion of the TAP binding score and NetChop-3.1 cleavage scores from the final input features was likely due to the fact that they correlated with other features, such as NetMHCstabpan-1.0 stability and MHCflurry1.6 percentile rank that were included in the model. This was not unexpected as the MHCflurry1.6 model was trained using peptide elution study data, whose results would be strongly influenced by TAP transport and proteasomal cleavage. The lower feature importance value seen for gene expression level is likely due to the ability of an nmer, at any given expression level, to generate multiple negative mmps and only a single positive mmp, thus diminishing the effect of expression in reducing node impurity.
Prioritization of nmers based on analysis of mmp composition.
The identification of all positive class I mmps in our training set would require the screening of 624,276 combinations of mmps and class I alleles using binding rank alone. Due to the large false positive rate in the MMP model, identifying neoantigen-reactive T cells through the screening of nmers is a more efficient method than the use of individual mmps. Therefore, to prioritize nmers for use in T-cell screening assays, we trained a logistic regression model that takes the scores from our MMP model as input. We again performed fivefold cross validation to determine the optimal number of mmps derived from each nmer to use as input for the nmer model (see Methods). When performing each split for nmer model training, the mmps corresponding to each nmer in the training set were used to train a new MMP model for each fold evaluation using the same input features as determined in MMP model development. This ensured that the evaluation of the nmers in the validation fold was not biased due to inclusion of mmp scores used in training an MMP model. A variable number of mmp scores was tested as input for training of the NMER models, ranging from the best-scoring mmp to the top 50 mmps, as scored by the training fold mmp model. To evaluate performance, we created a second set of models using MHCflurry1.6 percentile binding rank as input again ranging from the best predicted binding mmp to the top 50. An analysis of all models revealed that those using mmp score as input outperformed models built with the MHCflurry1.6 binding score as input, confirming the enhanced performance of our MMP model score (Supplementary Table 17). The top two mmp scores were chosen as input for our final NMER model, as cross validation revealed that the models built with this input possessed the second best mean AUC (mean AUC = 0.8778 ± 0.024 versus 0.8760 ± 0.026) but the best mean average precision score (0.30 ± 0.08). The NMER model built with this input outperformed the best model built using MHCflurry1.6 rank as input, which had a mean AUC of 0.806. The NMER model also had a reduced standard deviation throughout when compared to the best model built using MHCflurry1.6 rank as input, indicating that it is a more consistent method for ranking nmers (Extended Data Fig. 6). After evaluating and determining input and parameters for the MMP and NMER models, the final models were trained using all of the data from the subsampled datasets.
Performance of models with an independent test set.
An independent set of 26 human samples was then created to evaluate the performance of the final models (Supplementary Table 18). This test set consisted of an independent set of human samples that were being screened during the development of the MMP and NMER models. With the exception of 2 of the 26 samples in the test set with high tumor mutational burdens, the majority of nmers for each sample in the test set were screened, which allowed for the evaluation of the effectiveness of these models in identifying candidates for screening. Screening of the 3,760 nmers in the test set led to the identification of 46 positive nmers, 27 of which the reactive mmp and HLA context were identified (Supplementary Tables 19 and 20).
For 12 of the 16 samples in which the class I minimal epitope and its restriction element were known, the MMP model had a better AUC score than ranking by MHCflurry1.6 alone, although these differences were not pronounced (Supplementary Table 21). Reviewing the collective group of nmers in the test set showed that ranking by the NMER model produced an AUC of 0.911. This was an improvement when compared to ranking nmers by their best-scored mmps using any of the binding prediction models tested, where the next highest AUC of 0.781 was observed using the NetMHCpan4.0 EL model to score nmers (Fig. 4a).
Fig. 4 |. Evaluation of models in a test set.
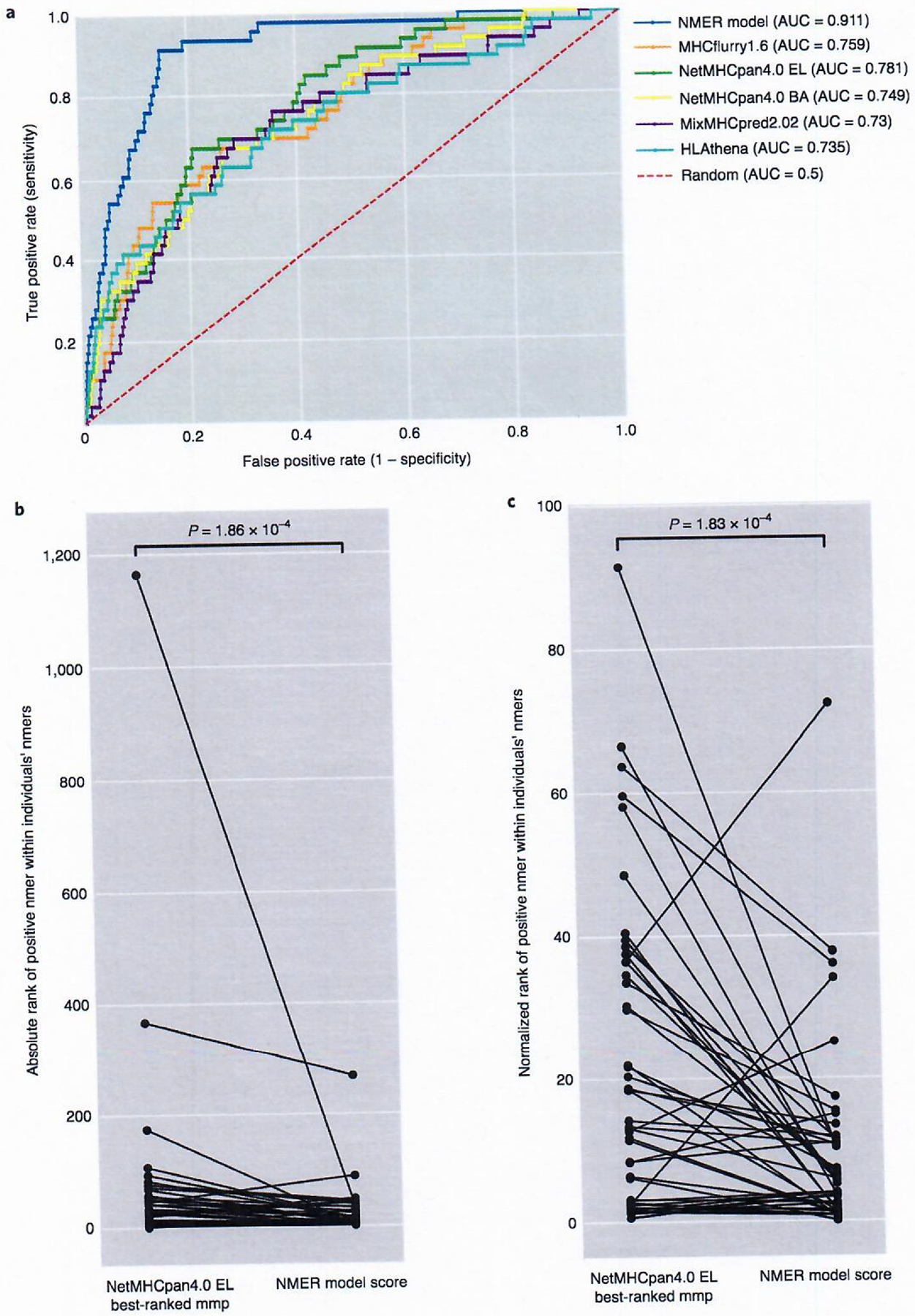
a, MMP model evaluation. Receiver operating characteristic (ROC) curve of the MMP model in a test set of n = 8,771 nmers, with n = 46 class I–reactive nmers ranked by their best-scoring mmp from five different prediction binding models. All nmers from the 26 test samples were scored and included. b, Individual rank of positive nmers (n = 46) found within an individual’s repertoire when sorted by the NMER model score or by the top NetMHCpan4.0 EL percentile binding-ranked mmp derived from each nmer. c, Percentile rank of positive nmers (n = 46) found within an individual’s repertoire when sorted by the NMER model score or by the top NetMHCpan4.0 EL percentile binding-ranked mmp derived from each nmer; P values were calculated with a two-sided Wilcoxon signed-rank test.
When evaluating all nmers on an individual sample basis, 23 of 26 samples had at least one positive nmer ranked in the top 25 using the NMER model score. In addition, 24 of 46 positive nmers were ranked in the top 10 nmers for an individual and 45 of 46 were ranked among the top 50 (Table 1). Overall, the ranks of positive nmers were significantly higher when using the NMER model than when ordered by the top-scoring NetMHCpan4.0 EL mmp within each nmer (Fig. 4b). The increase in ranking of many of the CD8+ nmers seen with the NMER model was particularly evident when the data were normalized to account for the wide variation in tumor mutational burden (Fig. 4c and Table 1). Through the use of the NMER model, all but five of the positive nmers rank in the top 20% of all nmers for a given sample.
Table 1 |.
NMER model evaluation in test set samples
| ID | Tumor type | Unique count | CD8+ nmers | Top-ranked NetMHCpan4.0 EL within nmerAUC | NMER model score AUC | Gene name | Mutant nmer | Rank within sample nmers by top NetMHCpan4.0 EL (percentile rank within sample nmers) | Rank within sample nmers by NMER model score (percentile rank within sample nmers) |
|---|---|---|---|---|---|---|---|---|---|
| 3703 | Melanoma | 289 | 4 | 0.900 | 0.911 | CCNE1 | HEVLLPQYPQQILIQIAELLDLCVL | 4 (1.38) | 2 (0.69) |
| NSDHL | FLSRILTGLNYEVPKYHIPYWVAYY | 24 (8.3) | 43 (14.88) | ||||||
| FADS3 | RHNYSRVAPLVKLLCAKHGLSYEVK | 41 (14.19) | 34 (11.76) | ||||||
| MOV10 | LDRELRGLDDIKNLKVGSVEEFQGQ | 54 (18.69) | 33 (11.42) | ||||||
| 3881 | Melanoma | 2871 | 2 | 0.767 | 0.994 | NDUFS2 | GFRGVAAQVLRPRAGVRLPIQPSRG | 175 (6.1) | 1 (0.03) |
| MELTF | GALIQRGFIRPKNCDVLTAVSEFFN | 1164 (40.54) | 35 (1.22) | ||||||
| 3942 | Rectal | 161 | 2 | 0.840 | 0.962 | NUP98 | TGLFGQTNTGFGDVGSTLFGNNKLT | 5 (3.11) | 4 (2.48) |
| KARS1 | YEIGRQFRNEGIHLTHNPEFTTCEF | 49 (30.43) | 11 (6.83) | ||||||
| 3995 | Colon | 68 | 3 | 0.774 | 0.979 | RNF213 | FVTQKRMEHFYLSFYTAEQLVYLST | 9 (13.04) | 4 (5.8) |
| TUBGCP2 | DLSIRELVHRILLVAASYSAVTRFI | 15 (21.74) | 5 (7.25) | ||||||
| KRAS | MTEYKLVVVGADGVGKSALTIQLI | 26 (37.68) | 1 (1.45) | ||||||
| 4007 | Colon | 150 | 2 | 0.990 | 0.990 | SKIV2L | PILKEIVEMLFSHGLVKVLFATETF | 2 (1.33) | 5 (3.33) |
| H3-3B | VKKPHRYRPGTVTLREIRRYQKSTE | 4 (2.67) | 1 (0.67) | ||||||
| 4014 | Lung | 254 | 2 | 0.671 | 0.901 | TGFBRAP1 | LLYHYNNQDAAAFQLWVNIVNGDVQ | 76 (29.92) | 39 (15.35) |
| USP11 | GLVEGEDYVLLPSAAWHYLVSWYGL | 93 (36.61) | 14 (5.51) | ||||||
| 4032 | Colon | 115 | 3 | 0.327 | 0.952 | API5 | DPDCVDRLLQCTQQAVPLFSKNVHS | 57 (48.72) | 6 (5.13) |
| RNF10 | RHRYLSHLPLTCKFSICELALQPPV | 68 (58.12) | 4 (3.42) | ||||||
| PHLPP1 | VNRWTRRQVILCETCLIVSSVKDSL | 107 (91.45) | 12 (10.26) | ||||||
| 4166 | Colon | 157 | 1 | 0.821 | 0.994 | NPLOC4 | VYTFSISQNPFPVENRDVLGETQDF | 29 (18.47) | 2 (1.27) |
| 4171 | Rectal | 139 | 1 | 0.609 | 0.949 | SIN3A | LGKFPELFNWFKIFLGYKESVHLET | 55 (39.57) | 8 (5.76) |
| 4202 | Melanoma | 308 | 1 | 0.987 | 0.964 | RANBP2 | VITLKKAEHLDFRHVVFGFVKDGMD | 5 (1.62) | 12 (3.9) |
| 4242 | Gastric | 189 | 1 | 0.782 | 1.000 | CDC42BPA | LRVTAGPTSLDLYVNVQRTLDNNLA | 42 (21.99) | 1 (0.52) |
| 4253 | Melanoma | 1993 | 2 | 0.903 | 0.921 | PTPN11 | KNRYKNILPFDHIRVVLHDGDPNEP | 23 (1.15) | 49 (2.46) |
| PUS1 | LGAFGRWTLRLGLRPSCSPRMAGNA | 368 (18.45) | 268 (13.43) | ||||||
| 4262 | Colon | 222 | 1 | 0.869 | 0.896 | NAMPT | WGKDHEKDAFEHTVTQFSSVPVSVV | 30 (13.33) | 24 (10.67) |
| 4264 | Esophageal | 166 | 1 | 0.879 | 0.752 | CCNYL1 | PEDLALESNPSDYPRASTIFLSKSQ | 21 (12.65) | 42 (25.3) |
| 4265 | Pancreatic | 98 | 1 | 0.670 | 0.835 | PRPF6 | TLMDMRLSQVSDTVSGQTVVDPKGY | 33 (33.67) | 17 (17.35) |
| 4268 | Colon | 95 | 2 | 0.806 | 0.973 | USP37 | PSKDAEEMRLFLYAVHQNRLPAAMK | 6 (6.32) | 2 (2.11) |
| RNF149 | PSASPAESEPQCHPSFKGDAGENTA | 33 (34.74) | 6 (6.32) | ||||||
| 4270 | Pancreatic | 82 | 1 | 0.988 | 0.667 | MCAT | GPQETLDRTVHCKPAIFVASLAAVE | 2 (2.44) | 28 (34.15) |
| 4271 | Colon | 119 | 3 | 0.566 | 0.727 | DHTKD1 | LEVSNSPLSEEAILGFEYGMSIESP | 10 (8.4) | 13 (10.92) |
| WDFY1 | LIPKEDGVITASKDRTIRVWLKRDS | 71 (59.66) | 43 (36.13) | ||||||
| CPSF6 | QFEMQSRKTTQSEQMSGEGKAGPPG | 76 (63.87) | 45 (37.82) | ||||||
| 4283 | Colon | 167 | 1 | 1.000 | 0.970 | DNMT3A | GDKNATKAGDDESEYEDGRGFGIGE | 1 (0.6) | 6 (3.57) |
| 4310 | Melanoma | 263 | 2 | 0.985 | 0.979 | COG7 | IDQVKSRMQLAAKSLQEADKWSTLS | 5 (1.9) | 4 (1.52) |
| DOCK6 | EAFTPVVYHNKSLEFYEEFKLHLPA | 6 (2.28) | 10 (3.8) | ||||||
| 4323 | Rectal | 102 | 2 | 0.940 | 0.990 | PPP2R1A | KEWAHATIIPKVSAMSGDPNYLHRM | 3 (2.94) | 4 (3.92) |
| MFSD14B | RNAESDQQGVAQVIITGIRGLCNGL | 12 (11.76) | 1 (0.98) | ||||||
| 4324 | Colon | 106 | 1 | 0.619 | 0.895 | TP53 | GNLRVEYLDDRNIFRHSVVVPYEPP | 41 (38.68) | 12 (11.32) |
| 4338 | Colon | 162 | 1 | 0.981 | 0.994 | H2AC8 | PVYLAAVLEYLTPEILELAGNAARD | 4 (2.47) | 2 (1.23) |
| 4348 | Esophageal | 126 | 1 | 0.984 | 1.000 | ACSF3 | PQQVWEKFLSSEMPRINVFMAVPTI | 2 (1.59) | 1 (0.79) |
| 4350 | Colon | 123 | 3 | 0.625 | 0.731 | GFPT1 | ACGTSYHAGVATGQVLEELTELPVM | 14 (11.38) | 1 (0.81) |
| HOOK3 | NQEGSDNEKIALFQSLLDDANLRKN | 45 (36.59) | 89 (72.36) | ||||||
| TP53 | SQKTYQGSYGFRRGFLHSGTAKSVT | 82 (66.67) | 13 (10.57) | ||||||
| 4359 | Colon | 246 | 2 | 0.891 | 0.947 | EIF2A | VLVIASTDVDKTAASYYGEQTLHYI | 5 (2.03) | 2 (0.81) |
| RPS13 | DLYHLIKKAVAVQKHLERNRKDKDA | 50 (20.33) | 26 (10.57) |
Using the NMER model score, we used varying cutoff thresholds to test the effects of filtering on the test set. This analysis revealed 93.5% of all CD8+ nmers would have been identified by screening 960 (26%) of the 3,760 screened nmers in the test set, resulting in a positive rate of 4.5% (Supplementary Table 22). The top-evaluated cutoff of 0.9 for the NMER model yielded a positive rate of 10.8%, with 26% of CD8+ nmers achieving a score of 0.9 or better. When applying cutoffs based on the top NetMHCpan4.0 EL percentile-ranked epitope, the positive rate never exceeded 8.5%, and at the cutoff for which this rate was attained, a percentile rank ≤ 0.01, only 13% of the CD8+ nmers were found (Supplementary Table 23). In addition, using NetMHCpan4.0 EL alone, a cutoff of 0.7 was required to identify 93.5% of the CD8+ nmers found in the test set, resulting in the retention of 2,324 of the 3,760 screened nmers and a positive rate of 1.9% compared to 1.2% seen in the screened test set. These findings demonstrate the enhanced predictive value of an algorithm that incorporates multiple features comprising an nmer over the use of an HLA class I binding algorithm rank alone to identify targets for active and passive immunotherapies.
Discussion
The current study focused on the evaluation of neoantigens and the development of a robust model for determining their likelihood of eliciting an HLA class I–restricted response. The 185 identified neo-antigens present in the training and test sets represent a compilation of HLA class I reactivities described in a recent analysis of TIL derived from 75 individuals with metastatic gastrointestinal cancer27 and studies of additional individuals with metastatic cancers. With the exception of a single HLA-C*08:02-restricted reactivity against a KRAS p.G12D mutant peptide recognized by two TIL samples, all individuals recognized unique class I–restricted neoepitopes. The unique nature of these neoantigens highlights the need to develop strategies that facilitate the evaluation and prioritization of candidates identified from the sequencing of tumor samples. The models presented in this study attempt to balance the interplay between many of the factors involved in antigen recognition, generating a single score for each mmp and nmer. The data presented here can be used to further aid in the ongoing development of binding prediction and immunogenicity models and help to form even more accurate predictors in the future.
The set of 139 neoepitopes comprising the training set allowed for the generation of models that were evaluated using an independent testing set of 46 neoepitopes. It should be noted that positive neoantigens were identified using populations of TIL that were capable of expanding to levels needed for screening, which could be influenced by the degree of exhaustion of clonal populations and by clonal competition during in vitro expansion. In addition, these T cells were derived from tumor samples that generally ranged between 2 mm and 3 mm in diameter and thus may only provide a sampling of the total immune repertoire. These limitations could result in a significant number of immunogenic mmps and nmers being scored as negatives in our screening assays. These limitations may be reflected in the imprecision of the models developed and will hopefully be refined as more data are used to train these tools. Additionally, the imprecision is also presumably a result of our imperfect knowledge of all of the factors that influence the immunogenicity of candidate neoantigens.
Findings presented in a recent study indicating that the majority of neoantigens targeted by T cells from individuals with gastrointestinal tumors are HLA class II restricted demonstrates the need to develop similar prediction tools for class II antigens27. Of the 124 neoantigens identified from the 75 individuals evaluated in that study, there were three examples in which the same nmer was targeted by both HLA class I– and class II–restricted T cells in a single individual. Given the small number of shared HLA class I and class II neoantigens that were found, it is difficult to evaluate the significance of this finding, although it is likely that features such as gene expression also have an important influence on HLA class II neoantigen recognition.
Our models use machine learning to interrogate a large dataset of verified tumor neoantigens and a corresponding dataset of matched negative candidates from the same samples and are freely available for academic use at github.com/JaredJGartner/SB_neoantigen_Models. All datasets used in this publication are available from the National Institutes of Health (NIH) figshare repository (https://doi.org/10.35092/yhjc.c.4792338.v2) to help facilitate future studies of neoantigens. The models presented here represent a reproducible method that facilitates the identification of neoantigen targets for therapy and will help to guide future immunotherapies directed against this promising category of targets.
Methods
Sample acquisition.
Samples used in this study were from individuals enrolled in a clinical protocol and approved by the institutional review board of the National Cancer Institute. Written informed consent was obtained from all individuals included in this study. Individuals chosen for this study had at least one tumor where whole-exome and whole-transcriptome sequencing was performed with at least one HLA class I–positive nmer and includes both published1–6,8,9,27–29,36,47–54 and new data. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Next-generation sequencing.
For newly analyzed samples, whole-exome sequencing was performed at the Surgery Branch on NextSeq550 models using an Agilent SureSelectXT HS kit (5191–4029). RNA sequencing was performed at the Surgery Branch on NextSeq550 models using Illumina Truseq RNA exome kits (20020189). Data processing and variant calling were performed as described previously27.
Generation of nmers.
Following variant identification, nmers were generated by extracting the mutated amino acid and the 12 wild-type amino acids flanking the mutation where possible. For indel mutations, appropriate changes were made to the cDNA sequence, which were then translated until a stop codon or end of the transcript was encountered. The 12 amino acids before and after the non-frameshift mutations were used to generate nmers. For frameshift mutations, the 12 amino acids before the frameshift indel and all amino acids after were used for the generation of nmers.
Expression decile generation.
For an individuals’ samples, all expressed transcripts (fragments per kilobase million (FPKM) > 0) within a tumor regardless of mutation status were taken and normalized to a mean of 0 and standard deviation of 1 by subtracting the mean of the expressed transcripts and dividing by the standard deviation. The expressed transcripts were then binned into deciles, and all unexpressed transcripts were included in decile 1. For individuals with multiple RNA-seq samples, the mean for a transcript across all samples was used to bin into deciles.
VAF decile generation.
As there are varying levels of normal tissue contamination for samples, the VAF needs to be normalized to compare across samples. For an individual’s samples, all mutations were considered, and the mutated allele frequency was normalized to a mean of 0 and standard deviation of 1 by subtracting the mean of the mutant allele frequency of all mutations and dividing by the standard deviation. The normalized VAFs were then binned into deciles. For individuals with multiple sequenced tumors, the mean-normalized VAF across mutations was used to bin into deciles.
Screening of epitopes.
Neoepitope screening was performed using previously described methods28. For tumors bearing more than 200 variants, filters were generally applied to prioritize nmers for inclusion in screening assays. Filters used for these studies included the use of expression decile thresholds, the detection of variant transcripts in RNA-seq datasets and the presence of variants in multiple tumors or tumor regions. For samples 1913, 2098, 2224 and 3309, which represent the only samples for which additional criteria were used for screening, only positive nmers were included in the training set. For samples 1913 and 2098, positive neoantigens were identified using a cDNA library expression screening approach, and the sequences of negative cDNAs were indeterminant. Samples 2224 and 3309 represent the only members of the training cohort for which the NetMHCpan binding algorithm was used to identify candidate neoantigens for screening. Screening was not biased by the inclusion of the five neoepitopes from samples 2224 and 3309; however, there was no statistically significant difference between the percentage of nmers with an mmp at a given predicted binding threshold and the percentage of nmers screened at that same threshold (Supplementary Table 5). The mmps and cognate HLA class I restriction elements were determined for the six neoepitopes identified from samples 1913, 2098, 2224 and 3309, and the additional mmps derived from the positive nmers were included in the negative dataset. Filtering was not performed based on ranks generated by the models described in this manuscript or previous iterations of these models, ensuring that criteria used to select variants for screening did not bias current results. Selection of minimal peptides for screening was performed evaluating T-cell responses to one or more of the peptides representing the top mmps predicted using NetMHCpan4.0 or earlier versions of the NetMHCpan tool to autologous antigen-presenting cells or target cells transfected with gene constructs encoding participants’ HLA alleles3. For some neoepitopes, peptide titrations were performed with multiple mmps to identify the optimal minimal peptide. Given the processes that we followed for nmer and mmp selection, it is unlikely that they had a significant impact on the development of our current models.
Generation of an expression-balanced dataset.
To generate a balanced dataset, only screened nmers were included, and a random subset of 500 nmers was selected from each expression decile to reduce any bias resulting from filters used for selecting variants to perform screening assays. For deciles two and three, all nmers, 317 and 449 respectively, were selected. Random selection was performed using Python’s numpy.random.choice function.
This dataset, which included only screened nmers and has been balanced for expression, was split into five folds using the scikit-learn.StratifiedShuffleSplit function, maintaining the same ratio of positive and negative nmers in each group.
To create a dataset of minimal epitopes, all possible mmps containing the mutation were generated from the nmers in the training set. An epitope was considered negative if it was derived from a non-positive nmer or if it was a non-reactive epitope derived from a positive nmer in which the reactive minimal epitope and restriction element were known. All epitopes derived from positive nmers in which the minimal epitopes/restriction elements were unknown were excluded. For each epitope, binding predictions were calculated for the mutant and wild-type epitope for the respective participants’ class I haplotype using NetMHCpan4.0 BA and NetMHCpan4.0 EL, MixMHCpred2.02, HLAthena and MHCflurry1.6. A wild-type:mutant rank score was generated by dividing the MHCflurry1.6 percentile binding rank of the corresponding wild-type minimal peptide by that of the mmp. Proteasomal cleavage of the amino acid to generate the 8- to 12-nmer C-terminal residue were generated using NetChop version 3 on the nmer to determine the appropriate cleavage score. NetMHCstabpan version 1.0 was used to predict stability between an epitope and its respective HLA class I allele for both mutant and wild-type epitopes. IEDB immunogenicity scores for each epitope were predicted using the immunogenicity prediction tool from IEDB45. A TAP transport score was generated using a method (described in ref.55) that was adapted to accept epitopes of sizes up to 12 amino acids. T-cell contact hydrophobicity of the mutant and wild-type amino acid was determined by summing the Kyte–Doolittle hydrophobicity score for positions 4 through n − 1 of the peptide. For immunogenicity, TAP transport and hydrophobicity, all changes between the mutant and wild-type mmps were calculated by subtracting the mutant score from the wildtype. For proteasomal cleavage and peptide stability, all changes between the mutant and wild-type mmps were calculated by subtracting the wild-type score from the mutant.
mmp feature correlation.
Correlation between features was calculated using all screened mmp data (n = 2,751,320) and Python’s panda dataframe.corr tool to calculate the Spearman correlation for all features.
MMP model training.
A random forest model was trained on our balanced training mmp dataset using Python’s scikit-learn module. Features and hyperparameters were evaluated using fivefold cross validation of our training set. Training data were split into five equal parts, four of which were used to train the model, varying input features and hyperparameter settings while the remaining part was used to evaluate the trained model. Scoring of minimal epitopes was performed using the predict proba function, and the probability of an epitope being reactive was taken as the score for each epitope. The process of training a model was repeated, holding a different fold out each time, and the trained model was evaluated using the held-out portion of the data until all folds had been used as the validation set once. To identify the optimal features for use as input in the mmp model, the 14 individual features and all combinations of between 1 and 14 separate features were tested. The following features were evaluated as input: gene expression decile, mutation present in RNA-seq (binary, 1 = yes and 0 = no), mutant MHCflurry1.6 percentile rank, MHCflurry1.6 wild-type:mutant rank, C-term NetChop-3.1 score mutant, 20S NetChop-3.1 score mutant, NetMHCstabpan-1.0 prediction mutant, IEDB immunogenicity score mutant, TAP binding score mutant, T-cell contact residues hydrophobicity, T-cell contact (binary, 1 = yes and 0 = no identifying the location of the mutation), MHCflurry1.6 mutant processing score, MHCflurry1.6 mutant presentation score and exome VAF decile. This analysis generated 16,383 individual input feature combinations tested across each of the five folds, resulting in four models producing mean AUCs of ≥0.997 (Supplementary Table 12). Reviewing these four models, we chose to move forward with a model that takes as input gene expression decile, mutation present in RNA-seq (binary, 1 = yes and 0 = no), mutant MHCflurry1.6 percentile rank, MHCflurry1.6 wild-type:mutant rank (the ratio of binding rank of wild-type compared to mutant binding rank), NetMHCstabpan-1.0 prediction mutant, IEDB immunogenicity score mutant, T-cell contact residues hydrophobicity and exome VAF decile. The model parameter settings were max_features = 1, minimal_sample_leaf = 10 and number of estimators = 500. The training and validation sets were then combined, and a final model was trained on all data.
MMP model feature importance.
Feature importance for the final MMP model was generated after the final model was trained on the full balanced mmp training set (n = 1,404,674) using the feature_importances output of the scikit-learn RandomForestClassifier after the model was fit.
NMER model training.
An ensemble of 500 logistic regression models was trained using Python’s sklearm ensemble module and the nmer training dataset as input. Features and hyperparameters were evaluated using fivefold cross validation of our training set. Training data were split into five equal parts, and four fifths were used to train the model, varying input features and hyperparameter settings, while the fifth part was used to evaluate the trained model until all folds had been used as the validation set once. Feature sets tested included from the top MMP model score to the top 50 MMP model scores as well as from the top MHCflurry1.6 percentile rank score to the top 50 MHCflurry1.6 percentile rank scores. Hyperparameters evaluated were the max samples to be used when bagging, max features for the ensemble classifier and the regularization parameter for the logistic regression model being used in the ensemble. The final model features were the top two MMP model scores, and the hyperparameters were set to regularization parameter = 0.1, BaggingClassifier max_samples = 0.2, n_estimators = 500 and max features = 1. A final model was trained on all data.
Microbial sequence similarity.
Using previously published methods26, we evaluated our positive mmps for similarity to the published microbial epitope list. Briefly, all positive mmps were checked for alignment to the published microbial list using the blastp BLOSUM62 matrix with gap opening penalties of −11 and a gap extension of −1. The best match of any alignment resulting from this initial step was then scored using Biopython Bio.pairwise2 under default settings.
nmer localization predictions.
All screened nmer protein sequences were run through WoLF Psort38 to predict localization. As several different transcripts can produce the same nmer, all protein sequences that gave rise to an nmer were used. The consensus top WoLF Psort prediction for each nmer was chosen if multiple transcripts produced an nmer. In the event of two or more localization predictions in the consensus, the one with the highest WoLF Psort score was chosen to represent the localization prediction for that nmer.
Statistics and reproducibility.
No statistical method was used to determine the number of individuals considered for the training or test sets. From a pool of all individuals at the National Cancer Institute Surgery Branch, only individuals with exome and RNA-seq data from at least one tumor and at least one class I reactivity were considered. This ensured that the same data were available for all samples and that they possessed working class I machinery. Statistics were calculated using Python’s scipy.stats module. AUC values were computed using Python’s sklearn. metrics module, and Bonferroni corrections were performed using statsmodels. sandbox.stats.multicomp. Random sampling was done using Python’s numpy. random.choice, and fivefold cross validation splitting was done using scikit-learn. StratifiedShuffleSplit.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Percentile Rank Comparisons between NetMHCpan4.0 EL and MHCFlurry1.6 Percentile Rank.
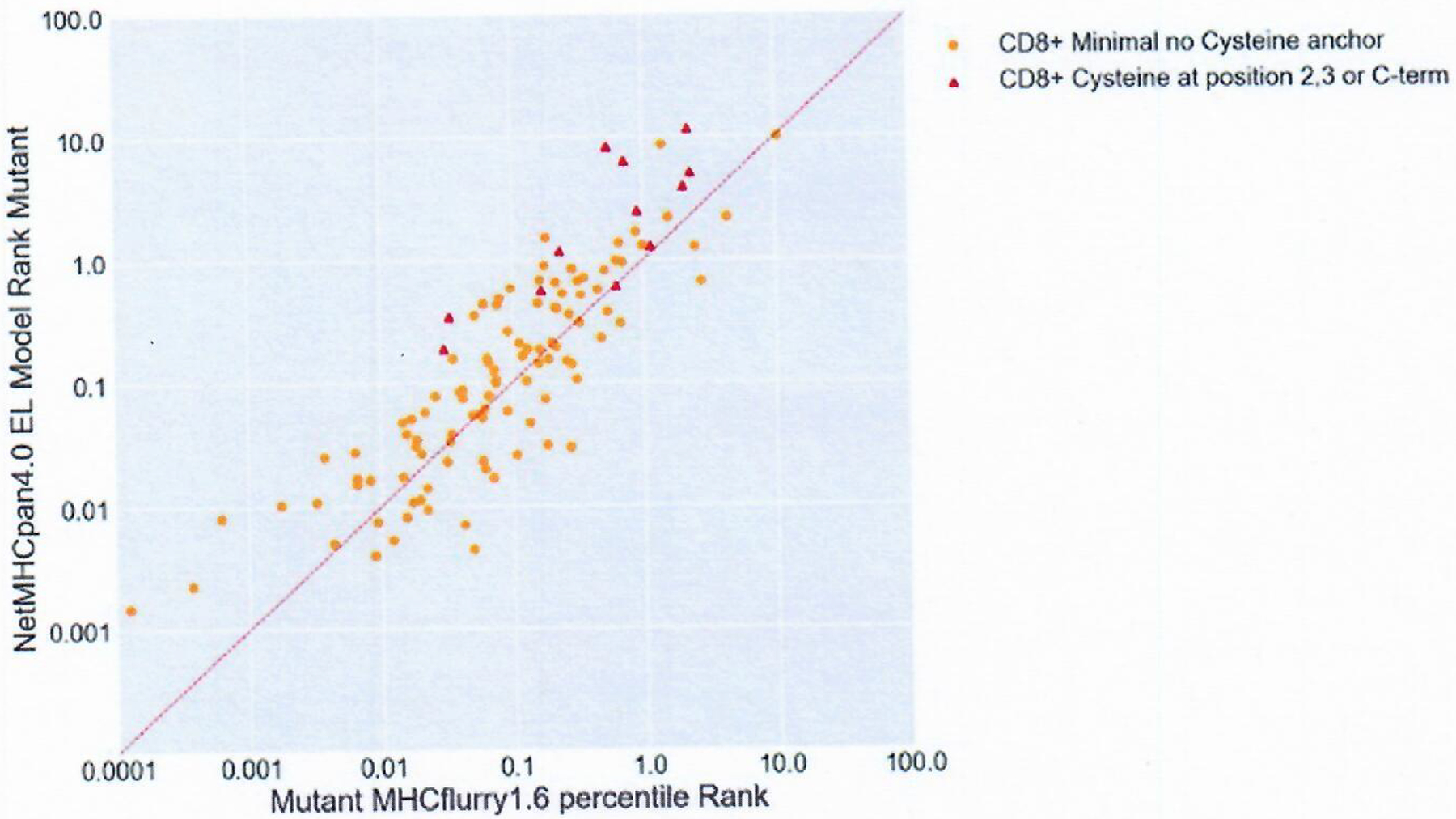
Percentile rank of positive mmps were mapped by their MHCflurry1.6 rank on the x-axis and the NetMHCpan4.0 EL model rank on the y-axis. Red Triangles correspond to mmps containing cysteine residues at positions 2,3 or C-terminus (n=12) while orange dots correspond to peptides containing cysteine residues at position 1 or between positions 3 and the C-terminus (n=107).
Extended Data Fig. 2 |. Nmer localization predictions.
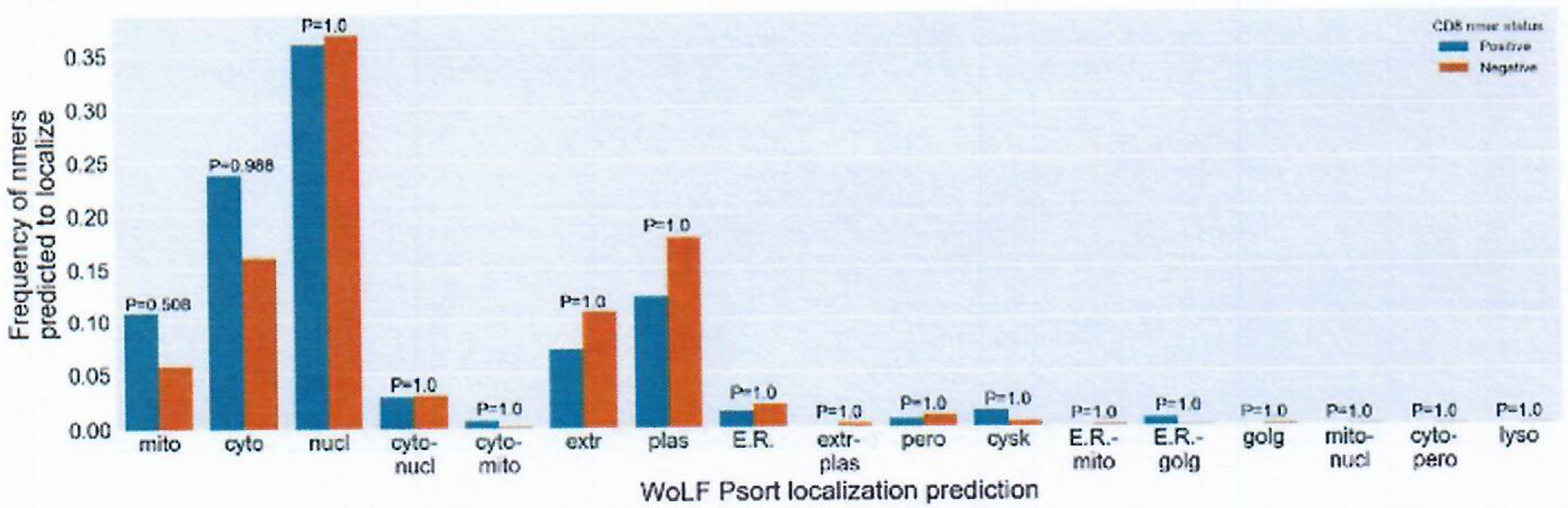
WoLF Psort algorithm was used on all nmer proteins (n=9541) to predicted for localization. Blue bars are CD8 + Positive nmers, Orange bars are negative nmers. Y-axis represents frequency of each group predicted to localize. X axis are the WoLF Psort prediction abbreviations. chlo = chloroplast, cyto = cytosol, cysk = cytoskeleton, E.R. = endoplasmic reticulum, extr = extracellular, golg = Golgi apparatus, lyso = lysosome, mito = mitochondria, nucl = nuclear, pero = peroxisome, plas = plasma membrane, vacu = vacuolar membrane . Individual totals for each groups positive and negative can be found in Supplementary Table 12. Hyphenated values denote compound prediction. P-values comparing positive to negative nmers displayed over each prediction. P-values calculated using a two-sided Fisher’s exact test and corrected using Bonferroni correction for multiple comparisons.
Extended Data Fig. 3 |. Gene expression Decile of mmps.
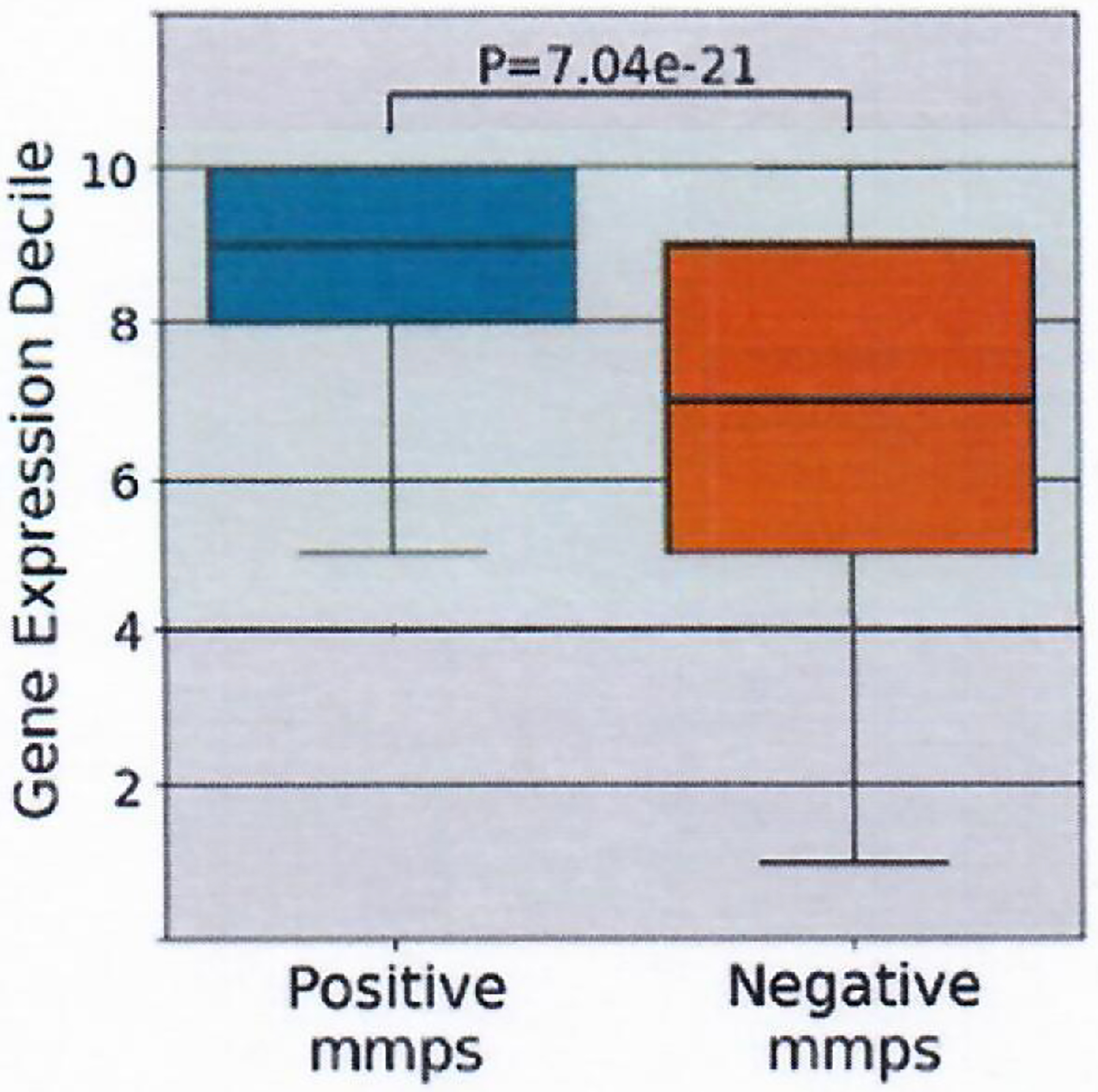
Gene expression deciles of positive (n=119) and negative mmps (n=2681162). Box indicates quartiles 2 & 3 and inter quartile range, median indicated by line in box plot, whiskers represent quartile 1 and 4 ± 1.5X IQR or minimum/maximum value if within the whisker values. Significance calculated with Mann-Whitney U test.
Extended Data Fig. 4 |. IEDB Immunogenicity scores of mmps.
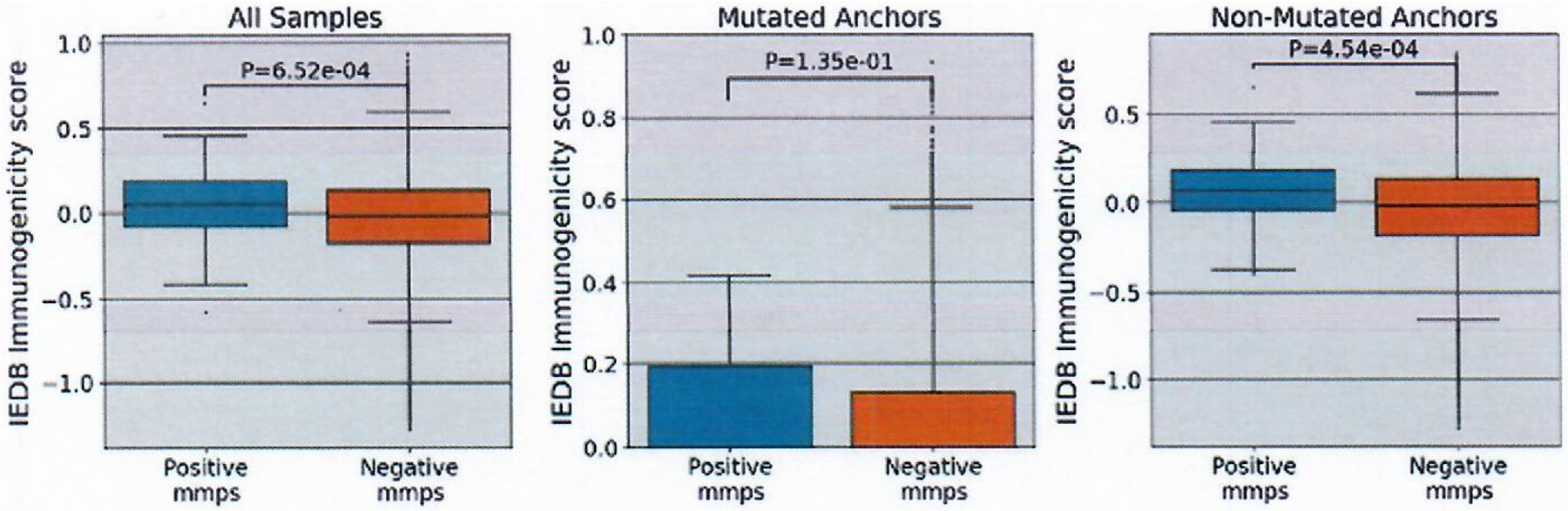
IEDB Immunogenicity scores were generated for each mmp using the IEDB immunogenicity tool. The panels are split into all mmps (positive n=119, negative n=2681162), comparison of just those with a mutation anchor in position 2,3 or C-terminus (positive n=55, negative n= 1167363) and those without mutations in position 2,3, or C-terminus (positive n= 64, negative n= 1513799). Box indicates quartiles 2 & 3 and inter quartile range, median indicated by line in box plot, whiskers represent quartile 1 and 4 ± 1.5X IQR or minimum/maximum value if within the whisker values. Significance was calculated using the Mann-Whitney U test.
Extended Data Fig. 5 |. Hydrophobicity scores of T-cell contact regions.
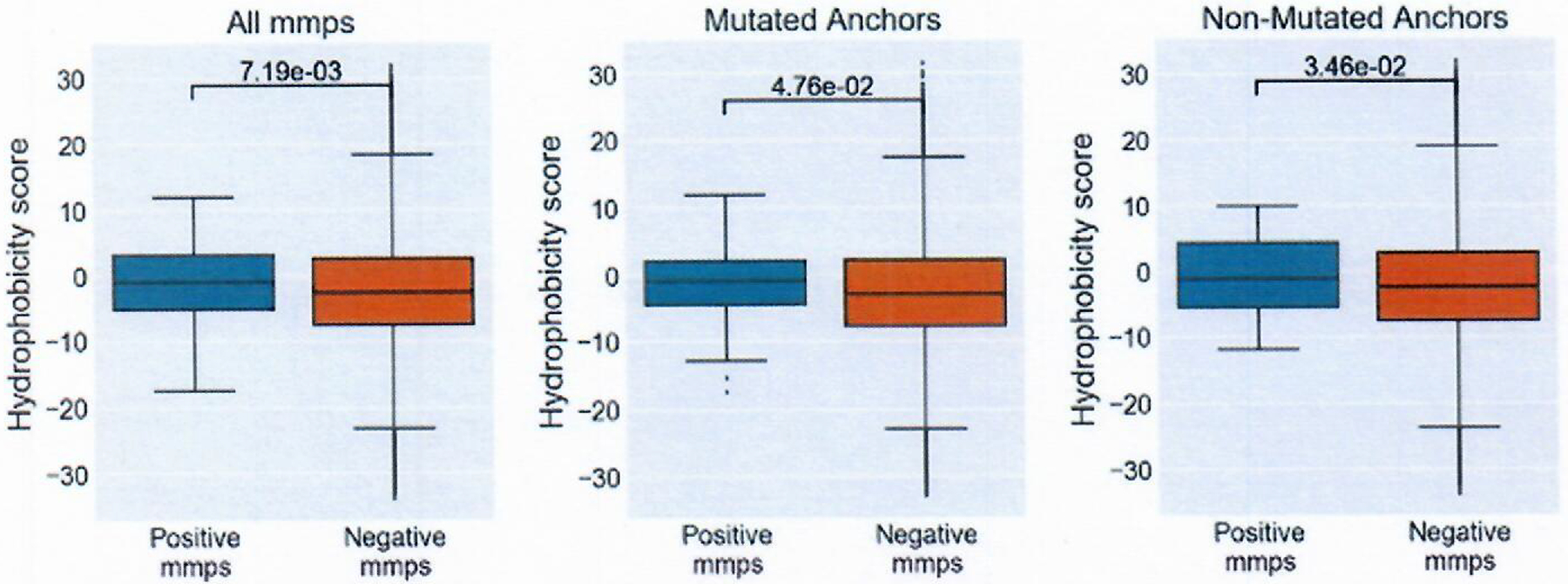
Hydrophobicity scores were calculated summing the Kyte-Doolittle hydrophobicity score of positions 4 through n-1. The panels are split into all mmps (positive n=119, negative n=2681162), comparison of just those with a anchor in position 2,3 or C-terminus (positive n=55, negative n= 1167363) and those without mutations in position 2,3, or C-terminus (positive n= 64, negative n= 1513799). Box indicates quartiles 2 & 3 and inter quartile range, median indicated by line in box plot, whiskers represent quartile 1 and 4 ± 1.5X IQR or minimum/maximum value if within the whisker values. Significance calculated with Mann-Whitney U test.
Extended Data Fig. 6 |. Top NMER models using either MMP score of MHCflurry Score as input.
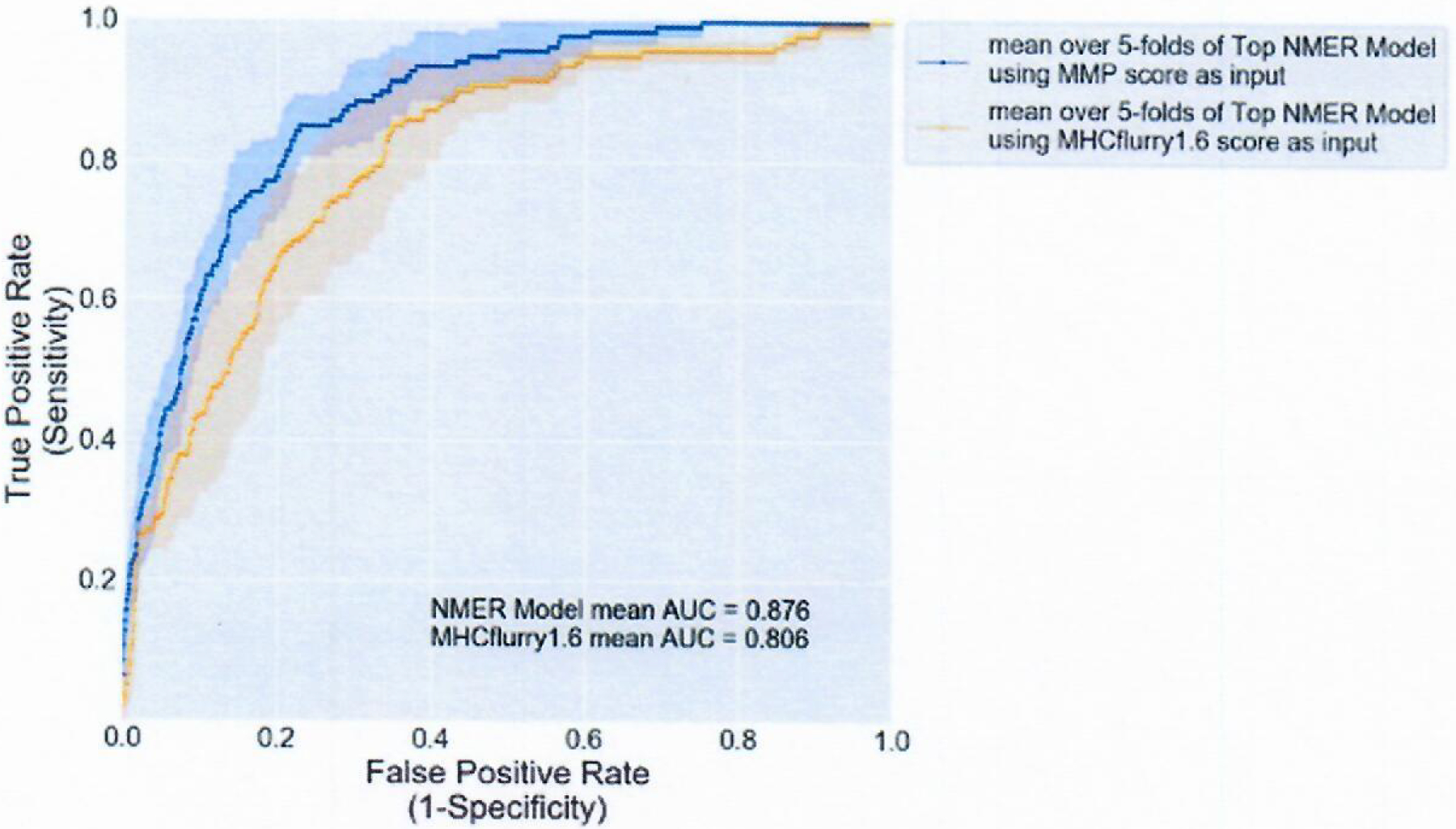
ROC curve showing the mean performance of the top models using either MMP model scores or MHCflurry scores as input. Solid line represents mean for each model across n=5 folds, shaded area is the standard deviation at each point along the x-axis.
Supplementary Material
Acknowledgements
We thank members of the NIH High Performance Computing (HPC) group for all of their support, assistance and technical advice. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). We also thank all members of the tissue procurement team for all of their efforts in acquiring and maintaining the specimens used in this study.
Footnotes
Code availability
The models developed and presented in this paper are available at https://github.com/JaredJGartner/SB_neoantigen_Models.
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s43018-021-00197-6.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s43018-021-00197-6.
Data availability
All next-generation sequencing data are available on dbGap under accession number phs001003.v1.p1. Source data are available from the NIH figshare repository at https://doi.org/10.35092/yhjc.c.4792338.v2 (ref.56).
References
- 1.Huang J et al. T cells associated with tumor regression recognize frameshifted products of the CDKN2A tumor suppressor gene locus and a mutated HLA class I gene product. J. Immunol 172, 6057–6064 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou J, Dudley ME, Rosenberg SA & Robbins PF Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J. Immunother 28, 53–62 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robbins PF et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med 19, 747–752 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu YC et al. Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. J. Immunol 190, 6034–6042 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu YC et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res 20, 3401–3410 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prickett TD et al. Durable complete response from metastatic melanoma after transfer of autologous T cells recognizing 10 mutated tumor antigens. Cancer Immunol. Res 4, 669–678 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tran E et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tran E et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med 375, 2255–2262 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zacharakis N et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med 24, 724–730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizvi NA et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGranahan N et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hellmann MD et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell 33, 843–852 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le DT et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le DT et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med 372, 2509–2520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peltomaki P DNA mismatch repair and cancer. Mutat. Res 488, 77–85 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Peters B & Sette A Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinf. 6, 132 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alvarez B et al. NNAlign_MA; MHC peptidome deconvolution for accurate MHC binding motif characterization and improved T-cell epitope predictions. Mol. Cell. Proteomics 18, 2459–2477 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Donnell TJ et al. MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst. 7, 129–132 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Duan F et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J. Exp. Med 211, 2231–2248 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bulik-Sullivan B et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol 37, 55–63 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Hundal J et al. pVACtools: a computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol. Res 8, 409–420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bjerregaard AM, Nielsen M, Hadrup SR, Szallasi Z & Eklund AC MuPeXI: prediction of neo-epitopes from tumor sequencing data. Cancer Immunol. Immunother 66, 1123–1130 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim S et al. Neopepsee: accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann. Oncol 29, 1030–1036 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Kosaloglu-Yalcin Z et al. Predicting T cell recognition of MHC class I restricted neoepitopes. Oncoimmunology 7, e1492508 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown SD et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 24, 743–750 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balachandran VP et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551, 512–516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parkhurst MR et al. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discov. 9, 1022–1035 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tran E et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 350, 1387–1390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lo W et al. Immunologic recognition of a shared p53 mutated neoantigen in a patient with metastatic colorectal cancer. Cancer Immunol. Res 7, 534–543 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jurtz V et al. NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol 199, 3360–3368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gfeller D et al. The length distribution and multiple specificity of naturally presented HLA-I ligands. J. Immunol 201, 3705–3716 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Sarkizova S et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol 38, 199–209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paul S et al. HLA class I alleles are associated with peptide-binding repertoires of different size, affinity, and immunogenicity. J. Immunol 191, 5831–5839 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen W, Yewdell JW, Levine RL & Bennink JR Modification of cysteine residues in vitro and in vivo affects the immunogenicity and antigenicity of major histocompatibility complex class I-restricted viral determinants. J. Exp. Med 189, 1757–1764 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen JL et al. Structural and kinetic basis for heightened immunogenicity of T cell vaccines. J. Exp. Med 201, 1243–1255 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sachs A, et al. Impact of cysteine residues on MHC binding predictions and recognition by tumor-reactive T cells. J. Immunol 205, 539–549 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahin U et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Horton P et al. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abelin JG et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity 46, 315–326 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rasmussen M et al. Pan-specific prediction of peptide–MHC class I complex stability, a correlate of T cell immunogenicity. J. Immunol 197, 1517–1524 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jorgensen KW, Rasmussen M, Buus S & Nielsen M NetMHCstab—predicting stability of peptide–MHC-I complexes; impacts for cytotoxic T lymphocyte epitope discovery. Immunology 141, 18–26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Groettrup M, Kirk CJ & Basler M Proteasomes in immune cells: more than peptide producers? Nat. Rev. Immunol 10, 73–78 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Larsen MV et al. An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur. J. Immunol 35, 2295–2303 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Capietto AH et al. Mutation position is an important determinant for predicting cancer neoantigens. J. Exp. Med 217, e20190179 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Calis JJ et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol 9, e1003266 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chowell D et al. TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proc. Natl Acad. Sci. USA 112, E1754–E1762 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cohen CJ et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J. Clin. Invest 125, 3981–3991 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gros A et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Invest 124, 2246–2259 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gros A et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med 22, 433–438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parkhurst M et al. Isolation of T-cell receptors specifically reactive with mutated tumor-associated antigens from tumor-infiltrating lymphocytes based on CD137 expression. Clin. Cancer Res 23, 2491–2505 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stevanovic S et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 356, 200–205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deniger DC et al. T-cell responses to TP53 “Hotspot” mutations and unique neoantigens expressed by human ovarian cancers. Clin. Cancer Res 24, 5562–5573 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yossef R et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 3, e122467 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gros A et al. Recognition of human gastrointestinal cancer neoantigens by circulating PD-1+ lymphocytes. J. Clin. Invest 129, 4992–5004 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Larsen MV et al. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinf. 8, 424 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gartner J Datasets for ‘Development of a model for ranking candidate HLA class I neoantigens based upon datasets of known neoepitopes’. figshare 10.35092/yhjc.c.4792338.v2 (2020). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All next-generation sequencing data are available on dbGap under accession number phs001003.v1.p1. Source data are available from the NIH figshare repository at https://doi.org/10.35092/yhjc.c.4792338.v2 (ref.56).