

Abstract
Introduction:
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder of motor neurons. Spread of pathology to other brain areas leads to development of non-motor symptoms (NMSs). These usually remain undiagnosed because of overwhelming motor problem and are responsible for significant distress to the patient. Our objective was to explore the burden of various NMSs of patients with ALS, compare between limb-onset and bulbar-onset patients, and to correlate with severity and duration of disease.
Methods:
Fifty patients with ALS diagnosed according to revised El Escorial Criteria and 50 healthy controls were included in this study. They were assessed with NMS Questionnaire, Beck's Depression Inventory, Center for Neurologic Study-Lability Scale, Drooling Frequency and Severity Scale, Epworth Sleepiness scale, Bengali Mental State Examination, and Frontal Assessment Battery and relevant statistical analyses were carried out.
Results:
The patients with ALS had significantly increased prevalence of almost all NMSs compared to controls. There was also significant increase in depression, suicidal ideation, pseudobulbar affect, and daytime sleepiness in patients with ALS. The bulbar onset subgroup had significantly increased daytime drooling, dysphagia, nausea and vomiting, whereas the limb onset subgroup reported increased frequency of leg swelling. Executive dysfunction was detected in 24% of patients with ALS and 9.8% had mild cognitive impairment. Weight loss, frequency of falling, insomnia, unpleasant nocturnal leg sensations, difficulty having sex, depression, and cognitive impairment increased significantly with an increase in severity of the disease.
Conclusion:
NMSs were significantly more prevalent in patients with ALS. Some NMSs worsened with advancement of the disease.
Keywords: Amyotrophic lateral sclerosis, anxiety, cognition, depression, non-motor symptoms
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder of undetermined etiology of the motor neurons in motor cortex, corticospinal tracts, brainstem, and spinal cord.[1] Traditionally considered as a disease of motor system, ALS is now increasingly recognized as a “multisystem disease” with prominent non-motor symptoms (NMS).[2] It has been suggested that these NMSs may arise from pathological spread to neighboring nonmotor regions of the brain as happen in other neurodegenerative diseases.[3]
Presence of these NMSs in ALS increases morbidity and causes significant distress to the sufferer. These adversely affect the quality of life of patients with ALS and reduce their functional capacity which in turn lead to increasing caregiver burden. With non-availability of definitive or disease-modifying treatment, management of these NMSs forms one of the most important aspects in the management of patients with ALS.
NMSs are not well defined in ALS. Various series have reported these symptoms in 5%–80% of patients.[4] Such wide variation of frequencies signifies difference in methodologies employed in these series. To assess NMSs in ALS, scales developed for other diseases are usually utilized to serve the purpose.
The magnitude of burden of all NMSs in ALS is still not fully known as primary focus has always been on the motor symptoms. This is also because of possible under-reporting of these symptoms by patients and their caregivers. This knowledge gap led us to plan for this study. Our primary objectives were (a) to explore the burden of various NMSs in patients with ALS in comparison to that of age- and sex-matched healthy individuals using standard questionnaire, (b) to compare the frequency of these NMSs between limb onset and bulbar onset patients with ALS, and (c) to correlate the burden of NMSs with disease severity and duration in patients with ALS.
MATERIALS AND METHODS
This was a questionnaire based, observational, cross-sectional, comparative study conducted between March 2018 and October 2019. Consecutive patients with ALS attending the outpatient department and those admitted in the neurology inpatient were screened for enrolment in the study. Healthy volunteers were recruited from relatives of patients attending Neurology OPD for non-neurodegenerative diseases. The study was approved by Institutional Ethics Committee, and written informed consent was obtained from patients/caregivers and healthy subjects prior to recruitment.
All patients were evaluated with detailed history, general, and neurological examination. They were subjected to investigations including complete blood count, erythrocyte sedimentation rate, blood biochemical tests like thyroid function test, fasting and postprandial blood sugar, glycosylated hemoglobin, lipid profile, liver and renal function tests, skiagram of the chest, and ultrasound of the abdomen. A detailed electrophysiological test including nerve conduction and electromyography of limb, paraspinal and tongue muscles were carried out. Each patient also underwent magnetic resonance imaging study of the brain and cervical spine. Additionally, serum electrophoresis, computerized tomography of chest and abdomen, tumor markers and positron emission tomography, and so on were carried out when clinically indicated. A consensus diagnosis of ALS was made using the revised El Escorial criteria[5] by two senior neurologists. Patients with clinically definite and clinically probable ALS were included in the study. Following patients were excluded – motor neuron diseases other than ALS, having associated structural brain or spinal cord lesions; with past history of any neurological disorder, and who had any known systemic illness prior to disease onset, including diabetes.
Assessment procedure
The patients with ALS were graded according to their severity using the ALS Functional Rating Scale-Revised (ALSFRS-R).[6] This is a 12-item scale to assess the functional capacity of patients with ALS in their personal life, with maximum score of 48 and higher score suggests better functional capacity. Based on total score, patients were classified to have (i) minimal-to-mild disease (>40), (ii) mild-to-moderate disease (30–39), (iii) moderate-to-severe disease (20–29), and (iv) advanced disease (<20). NMSs were enquired using the non-motor symptoms questionnaire (NMSQuest).[7] This is a 30-item questionnaire that was developed to record various NMS of Parkinson's disease patients in previous 1 month. Although NMSQuest was not designed as a quantitative scale, a total NMS score was calculated as sum of all positive (”yes”) answers of the 30 items. Additionally, we used following scales to record specific symptoms: Beck Depression Inventory-I (BDI) (for depression),[8] Center for Neurologic Study-Lability Scale (CNS-LS) (for pseudobulbar affect),[9] Epworth's Sleepiness scale (ESS) (for excessive daytime sleepiness [EDS]),[10] Drooling Severity and Frequency Scale (DSFS) (for drooling),[11] and cognitive impairment was assessed by the Kolkata Cognitive Screening Battery (KCSB)[12] and the Frontal Assessment Battery (FAB).[13] We used these scales in the local vernacular version. All participants were subjected to rater-administered questionnaires by a single observer (AC) to reduce inter-observer variability. All data were maintained in a patient's case record form.
Statistical analysis
Statistical analysis was done using the statistical package for the social sciences (SPSS) software (version 20). Mean, median and standard deviation (SD) were calculated for continuous variables. The comparison of age and sex distribution in the two study groups were made to identify age and sex matching. Comparison of means between two groups was done using unpaired t-test and for more than two groups ANOVA test was used with Tukey HSD post hoc analysis. Pearson's Chi-squared test/Fisher's exact test was used to test for any statistical association between two variables depicted in a cross-table. While computing bivariate correlations, continuity in each variable was statistically assumed. Significance of the correlation coefficient and its measurements were done by Pearson's method. P value < 0.05 was considered statistically significant for all the comparative analyses, tests of association, and correlation analysis.
RESULTS
Our study comprised 50 patients with ALS and 50 healthy control subjects. The study groups were age and sex matched. Patients with ALS had a mean age of 51.28 (±9.41) years and 80% of them were male, whereas healthy subjects had a mean age of 47.96 (±10.47) years and 74% were male. The majority (68%) of patients with ALS presented in fifth and sixth decades of their life. Out of the 50 patients with ALS, 46 (92%) had clinically definite ALS and 4 (8%) had probable ALS. The onset of symptoms as bulbar dysfunction was found in 19 (38%) patients, and onset with limb weakness in 31 (62%) patients [Table 1].
Table 1.
Demographic and clinical characteristics of the study population
| Patients with ALS | ||
|---|---|---|
| Total number | 50 | |
| Ratio of males in percentage | 80% | |
| Mean (± SD) age in years | 51.28±9.41 | |
| Mean disease duration in months | 13.92 | |
| Onset of symptom | Bulbar | 19 (38%) |
| Upper limb | 20 (40%) | |
| Lower limb | 11 (22%) | |
| Severity (ALSFRS-R score) | Minimal to mild (>40) | 5 (10%) |
| Mild to moderate (30-39) | 21 (42%) | |
| Moderate to severe (20-29) | 19 (38%) | |
| Advanced (<20) | 5 (10%) | |
ALS - Amyotrophic lateral sclerosis, ALSFRS-R - Amyotrophic lateral sclerosis functional rating scale-revised, SD - Standard deviation
The mean total NMS score was significantly higher (P < 0.001) in patients with ALS (9.3 ± 4.01) compared to healthy subjects (2.46 ± 1.80). Individual items of NMSQuest from 50 patients with ALS and 50 healthy subjects were compared and analyzed. Patients with ALS reported significantly increased daytime drooling of saliva, altered (decreased) taste/smell sensation, dysphagia, nausea or vomiting, constipation, fecal incontinence, urinary urgency, weight loss, loss of interest, difficulty concentrating, feeling sad/low, getting anxious, change in sexual behavior, falling, insomnia, vivid/frightening dreams, restless legs, and leg swelling [Table 2]. While comparing individual items of NMSQuest between bulbar and limb onset ALS subgroups [Table 2], the bulbar onset subgroup had significantly increased daytime drooling of saliva, dysphagia, nausea/vomiting and altered interest in sex, whereas the limb onset subgroup reported significantly increased frequency of leg swelling.
Table 2.
Comparison of responses of NMSQuest between patients with ALS and healthy subjects and between bulbar and limb onset ALS
| Patients with ALS (n=50) | Healthy subjects (n=50) | P | Bulbar onset (n=19) | Limb onset (n=31) | P | |
|---|---|---|---|---|---|---|
| Daytime saliva drooling | 48% | 0% | <0.001* | 78.95% | 29.03% | 0.001* |
| Altered taste/smell | 28% | 0% | <0.001* | 42.11% | 19.35% | 0.082 |
| Dysphagia/choking | 70% | 0% | <0.001* | 94.74% | 54.84% | 0.003* |
| Nausea/vomiting | 12% | 0% | 0.012* | 26.32% | 3.23% | 0.015* |
| Constipation | 40% | 6% | <0.001* | 42.11% | 38.71% | 0.812 |
| Bowel incontinence | 8% | 0% | 0.041* | 5.26% | 9.68% | 0.577 |
| Incomplete bowel evacuation | 20% | 12% | 0.275 | 26.32% | 16.13% | 0.382 |
| Urinary urgency | 22% | 4% | 0.007* | 15.79% | 25.81% | 0.407 |
| Nocturia | 12% | 6% | 0.280 | 15.79% | 9.68% | 0.519 |
| Unexplained pains | 22% | 18% | 0.617 | 26.32% | 19.35% | 0.564 |
| Unexplained weight loss | 56% | 28% | <0.001* | 68.42% | 48.39% | 0.166 |
| Forgetfulness | 8% | 4% | 0.400 | 5.26% | 9.68% | 0.577 |
| Loss of interest in what is happening around or doing things | 58% | 22% | <0.001* | 68.42% | 51.61% | 0.242 |
| Hallucinations (visual/auditory) | 0% | 0% | - | 0% | 0% | -- |
| Difficulty concentrating | 46% | 22% | 0.011* | 47.37% | 45.16% | 0.879 |
| Feeling sad/low | 92% | 46% | <0.001* | 94.74% | 90.32% | 0.577 |
| Anxious/frightened | 82% | 28% | <0.001* | 94.74% | 74.19% | 0.066 |
| Interest in sex | ||||||
| Decreased | 74% | 58% | 0.001* | 78.95% | 70.97% | 0.039* |
| No change | 14% | 42% | 0.00% | 22.58% | ||
| Increased | 12% | 0% | 21.05% | 6.45% | ||
| Difficulty in performing sex | 58% | 0% | <0.001* | 52.63% | 61.29% | 0.547 |
| Postural dizziness | 10% | 2% | 0.092 | 15.79% | 6.45% | 0.285 |
| Falling | 58% | 0% | <0.001* | 42.11% | 67.74% | 0.075 |
| Day time sleepiness | 6% | 0% | 0.079 | 5.26% | 6.45% | 0.864 |
| Insomnia | 46% | 12% | <0.001* | 52.63% | 41.94% | 0.461 |
| Vivid/frightening dreams | 42% | 12% | 0.001* | 47.37% | 38.71% | 0.547 |
| Dream enactment | 12% | 4% | 0.140 | 15.79% | 9.68% | 0.519 |
| Restless legs | 32% | 4% | <0.001* | 26.32% | 35.48% | 0.500 |
| Leg swelling | 20% | 0% | 0.001* | 5.26% | 29.03% | 0.041* |
| Excess sweating | 8% | 2% | 0.169 | 10.53% | 6.45% | 0.606 |
| Double vision | 0% | 0% | - | 0% | 0% | -- |
| Delusions | 0% | 0% | - | 0% | 0% | -- |
Using BDI, 34% of patients with ALS had moderate depression, whereas another 30% had severe depression as against 6% and 0% in healthy controls, respectively. There was a significant increase in severity of depression with increase in severity of disease (P ≤ 0.001), and it was significantly higher in moderate-severe and advanced disease compared to minimal-mild disease [Table 3]. Moreover, suicidal ideation was present in 34% of patients with ALS as compared to 10% of the healthy subjects (P = 0.004). Majority (80%) of our patients were not on antidepressant at the time of interview.
Table 3.
Distribution of nonmotor, cognitive and depression scores according to severity of ALS
| Severity of ALS | P | ||||
|---|---|---|---|---|---|
|
| |||||
| Minimal to Mild | Mild to Moderate | Moderate to Severe | Advanced Disease | ||
| Total NMS score Mean (± SD) | 4.40 (± 3.36) | 8.90 (± 3.30) | 12.16 (± 4.3) | 12.40 (± 2.07) | <0.001* |
| Total BMSE score Mean (± SD) | 29.00 (± 0.71) | 27.35 (± 2.03) | 26.53 (± 1.91) | 24.00 (± 5.66) | 0.027* |
| Total FAB score Mean (± SD) | 14.20 (± 3.27) | 13.00 (± 2.74) | 13.53 (± 2.07) | 12.00 (± 1.41) | 0.664 |
| BDI score Mean (± SD) | 9.20 (± 4.27) | 19.52 (± 8.87) | 28.79 (± 9.19) | 28.00 (± 11.23) | <0.001* |
*Significant at P<0.05. ALS - Amyotrophic lateral sclerosis, BDI - Beck Depression Inventory, BMSE - Bengali Mental Status Examination, FAB - Frontal assessment battery, NMS - Non-motor symptom, SD - Standard deviation
EDS was significantly higher (P = 0.003) in patients with ALS (26%) than healthy controls (2%) using ESS. Among the patients with ALS, 24% had mild to moderate EDS, and 2% had severe EDS. Using the CNS-LS, we found pseudobulbar affect in 68% of ALS patients, and none in healthy controls (P < 0.001). There was no significant difference of pseudobulbar affect between bulbar onset (73.68%) and limb onset (64.52%) ALS (P = 0.5).
Cognitive function could not be assessed in some of the patients due to physical disability such as severe dysarthria or severe upper limb weakness. The mean total Bengali Mental State Examination (BMSE) score calculated for 41 patients with ALS was 27.05 (± 2.25), and the mean FAB score for 45 patients was 13.29 (±2.49). Although none of our patients had dementia, 11 (24.4%) had executive dysfunction (FAB score <12) and 4 (9.8%) qualified for the diagnosis of mild cognitive impairment (MCI). We did not find any difference of mean BMSE score (22.13 ± 8.32 vs 25.10 ± 6.91, P = 0.105) and FAB score (11.79 ± 3.84 vs 12.71 ± 3.84, P = 0.415) between bulbar and limb onset patients. Executive dysfunction was associated with greater dysphagia/choking (P = 0.008) and drooling (P = 0.024). Although not statistically significant, executive dysfunction was more frequent in female patients (50% vs 18.9%, P = 0.085) and those with pseudobulbar affect (40% vs 15%, P = 0.067). With increasing severity of the disease, there was deterioration of BMSE score (P = 0.027), and on post hoc analysis, there was significant difference between minimal to mild disease and advanced disease. However, FAB score did not show statistically significant difference regarding severity of disease (P = 0.664) [Table 3].
With deterioration of functional status of patients with ALS, there was a significant increase in total NMS score (P ≤ 0.001), and it was significantly higher in moderate–severe and advanced disease compared to minimal–mild disease [Table 3]. A significant negative correlation (r = ‒0.614, P < 0.001) was observed between ALSFRS-R and NMS total scores [Figure 1]. As the severity of disease increases, the ALS functional rating scale score decreases. So, the total NMS score increases with increase in disease severity. The mean total NMS scores increased with the increase in disease duration, although it was not statistically significant (P = 0.438). Comparison of individual items of NMSQuest between ALS subgroups according to severity showed that weight loss (P = 0.016), frequency of falling (P = 0.003), insomnia (P = 0.037), restless legs (P = 0.044) and difficulty having sex (P = 0.001) increased significantly with increase in severity of the disease [Figure 2]. With increasing severity of the disease, there was increase in severity of depression (P = 0.004). There was, however, no relation of drooling, pseudobulbar affect, EDS, and suicidal ideation with increasing severity of the disease.
Figure 1.
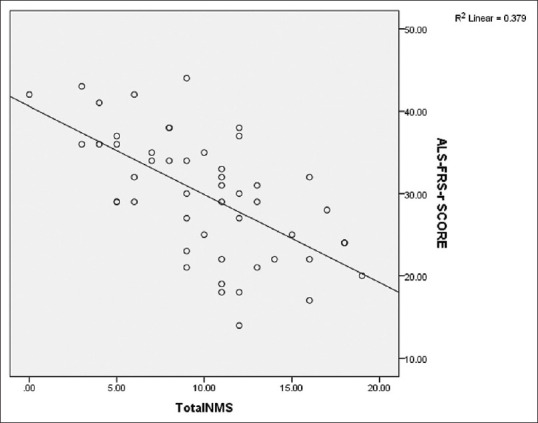
Scatter diagram showing negative correlation between total NMS and ALSFRS-R scores
Figure 2.

Frequency of various NMSs with increasing severity of the disease assessed by amyotrophic lateral sclerosis functional rating scale-revised (ALS ALSFRS-R)
Among other NMSs, fatigue was complained by 82% of patients with ALS, while 16% reported itching, and 30% complained of unexplained sensory symptoms. The sensory symptoms were in the form of tingling paresthesia in either limbs or the trunk and did not follow any definite distribution. All these patients had a normal sensory nerve conduction study, and none had pressure sores.
DISCUSSION
NMSs in ALS are not well characterized. What constitutes NMSs in a primarily motor disorder like ALS is controversial. Symptoms like drooling, dysphagia, and dyspnea are common and result from weakness of bulbar muscles. While these symptoms are considered extra-motor as they do not arise from limb weakness, their inclusion into non-motor domain is debatable.
The total non-motor score in patients with ALS was significantly higher than healthy people. A positive correlation between disease severity and non-motor scores was observed. This supports the idea that there may be a spread of disease pathology to the extra-motor areas of the brain with the progression of the disease.
Sialorrhea was complained by 52% of our patients with ALS against none in controls and most of them (40%) had either severe or profuse drooling. There was a significantly increased sialorrhea in bulbar onset patients. Sialorrhea results either from increased production or reduced clearance of saliva from the mouth. Although few studies have shown that there are alterations in saliva flow rate in ALS,[14] these alterations are usually mild. Our observation of significantly higher amount of dysphagia in bulbar onset patients strengthen this argument. However, we did not find any relation of sialorrhea and dysphagia with severity or duration of disease.
Recurrent falls were more frequent in the limb onset subgroup and increased with increase in disease severity (P = 0.003). Falls are probably multifactorial and, although primarily results from reduced strength of muscles, other factors need further probing.
Unexplained weight loss was reported by 56% of our patients with ALS and it increased with increasing severity of the disease. Studies have reported clinically severe weight loss in 15%–55% of patients with ALS in the course of the disease.[15] Higher loss of energy because of muscle fasciculations, increasing respiratory efforts, hypermetabolism, and loss of appetite are implicated for weight loss in ALS.[16] Weight loss has prognostic implication, and this has been linked with reduced survival in patients with ALS.[17]
Altered taste and smell perception was reported by 28% of patients with ALS. Various studies have also reported hyposmia in patients with ALS[18] and this has been linked to pathological TDP-43-positive inclusion, predominantly in the secondary olfactory centers especially in the hippocampus.[19] Olfactory dysfunction has also been explained by impaired respiratory function.[20]
Nausea/vomiting was reported by 12% of our patients with ALS and bulbar onset patients had a significantly increased frequency compared to limb onset subgroup. Constipation was significantly more common in our patients with ALS than healthy subjects. Delayed gastric emptying and colon transit time have been documented in patients with ALS and suggest gastrointestinal autonomic dysfunction.[21] Other factors such as decreased fluid intake due to dysphagia, inadequate fiber intake, lack of physical activity, and medications are also implicated.[21] Moreover, weakened abdominal muscles make it difficult to expel stool, even if soft.[22]
While 22% of our patients with ALS complained of urinary urgency or incontinence, only 8% reported fecal incontinence. Fecal and urinary incontinence are rare in ALS.[23] Although postural dizziness was reported by 10% of our patients with ALS, it was statistically insignificant. Autonomic symptoms such as postural dizziness and orthostatic hypotension are very rare in ALS.[24,25] The exact pathophysiology behind these autonomic symptoms is largely unknown, however, studies have demonstrated neuronal loss in the intermediolateral nucleus of the spinal cord,[26] containing autonomic neurons.
The patients with ALS complained of significantly increased neuropsychiatric problems such as loss of interest, inattention, depression, anxiety, and change in sexual behavior. Low mood and depression were complained by 92% of our patients with ALS and nearly one-third had severe depression or suicidal ideation. There was a significant increase in severity of depression with increase in severity of disease. The prevalence of depression in ALS is reported at 4%–56% depending on the assessment measure.[27,28] The presence of depression has a negative effect on the quality of life[29,30] and psychological stress is associated with a greater risk of mortality in patients with ALS.[31] Other NMSs such as poor sleep, pain, fatigue, weight loss, and psychological distress may also contribute to depression. Anxiety was reported by 82% of patients with ALS. In various studies, anxiety is reported from 0% to 30% in patients with ALS.[28] Our patients with ALS also complained of a significant alteration in sexual behavior. While 74% of patients with ALS complained of decrease in sexual interest, 12% reported an increase in sexual interest. Underlying anxiety and depression might contribute to the loss of sexual desire. Regarding the increased interest in sex, it would be interesting to consider the possibility of a frontal disinhibition component, although there was no significant difference of altered sexual interest regarding the presence of executive dysfunction.
Our patients with ALS had significant sleep-related problems including insomnia, vivid dreams, restless legs, and EDS. Sleep-related complaints have been previously reported to occur in ALS.[32] Insomnia was reported by nearly half of our patients with ALS and this increased with the increasing severity of the disease. In a recent study from India, as many as 65% of patients with ALS were found to have insomnia.[33] The difficulty in initiating or maintaining sleep is attributable to anxiety, depression, pain, discomfort or dependency from immobilization, difficulty turning in bed, muscle cramps, restless legs, and respiratory muscle weakness.[33] The existing literature on vivid/frightening dreams, rapid eye movement sleep behavior disorders (RBD) and parasomnias in ALS is limited, although, RBD is reported in a small subset of patients with ALS.[34]
Only few studies have systematically investigated the prevalence of restless leg syndrome (RLS) in patients with ALS[35,36] and found a prevalence ranging from 14.6% to 25%. It has been suggested that progressive immobilization puts patients with ALS at a greater risk to develop RLS. Moreover, ALS being a heterogeneous disorder involving multiple systems, it might cause circadian rhythm disturbances in patients contributing to RLS.[35] Using the ESS, EDS was found in 26% of patients with ALS. Some studies showed EDS to be significantly more frequent in patients with ALS than in controls.[37] Nocturnal sleep quality and respiratory issues seem to contribute to the occurrence of EDS symptoms in patients with ALS, although the neurodegenerative process itself may be responsible.[38] Interestingly, EDS has been associated with greater cognitive and behavioral impairment.[37] Hence, it is important to assess EDS in ALS patients using established scales such as ESS, as EDS may be associated with respiratory as well as cognitive impairment.
Various studies have demonstrated the presence of frontal cognitive impairment in ALS,[39,40] including an association between ALS and frontotemporal dementia.[41] In our study, 24.4% had executive dysfunction, whereas 9.8% had MCI. Our observations are comparable with recent studies.[40,42,43] Cognitive impairment of our patients deteriorated with the advancement of the disease; however, executive dysfunction was evident in all the stages. Similar observations were made by other investigators as well.[40] This indicates that frontal dysfunction is present even in the early stages of ALS. Executive dysfunction was associated with greater dysphagia/choking (P = 0.008) and drooling (using DSFS, P = 0.024) in our patients, although there was no significant difference between bulbar and limb onset patients (P = 0.086). This suggests executive dysfunction is probably not related with the site of onset of the disease, but is correlated with the severity of bulbar symptoms. Studies have produced conflicting results regarding the association of site of onset of ALS with cognitive impairment. While some showed a greater cognitive dysfunction in bulbar onset patients,[40,44] others did not corroborate.[45] Although not statistically significant, executive dysfunction was more frequent in female patients (P = 0.085) and those with pseudobulbar affect (P = 0.067). Palmieri et al. documented greater executive dysfunction in female patients.[46] Thakore et al. found higher prevalence of pseudobulbar affect in ALS patients with cognitive dysfunction.[47]
We found 68% of patients with ALS having pseudobulbar affect, and it was more common in bulbar onset than limb onset disease. Studies have shown that pseudobulbar affect affects 20%–50% of patients with ALS, especially in patients with bulbar onset.[48,49] Pseudobulbar affect can result in significant disability, limiting social interactions, and impairing quality of life.[48]
Significant number (20%) of our patients with ALS complained of leg swelling and it was more frequent in the limb onset subgroup. The dependent edema of the hands and feet in patients with ALS results from immobility and reduced muscle pumping activity,[22] although, sympathetic hyperactivity found in ALS[49] might also contribute to edema by stimulating the renin–angiotensin–aldosterone system with resultant sodium retention.
In all, 82% of our patients with ALS complained of fatigue and 42% complained of muscle cramps. Fatigue has been reported in 44%–83% of patients with ALS and is likely multifactorial. It may develop due to sleep disruption, nocturnal cramps and nocturia, poor nutritional status, muscle weakness, and reduced vital capacity, depression, and medications including riluzole.[30,50,51] Fatigue causes reduced physical endurance and functionality, psychological distress, poor quality of life and disturbed professional and social interactions.[30,51]
Sensory symptoms were previously considered uncommon in ALS. However, patients with ALS often complain of sensory symptoms, and case series have identified objective sensory signs in 2%–10% of patients.[52] We found that 30% of patients with ALS complained of unexplained sensory symptoms with normal sensory nerve conduction study. They were in the form of tingling paresthesia in limbs or in the trunk and did not follow any definite distribution. In one of the studies, 32.5% of patients with ALS complained of sensory symptoms such as paresthesia and tingling sensation.[53] Although the occasional presence of sensory features in ALS has been a cause of diagnostic uncertainty, the coexistence of sensory symptoms does not exclude presence of ALS. Table 4 shows the comparison of frequencies of various NMSs of our ALS patients with some other studies.
Table 4.
Comparison of frequencies of various NMSs of ALS with other studies
| Study | Present Study | Gunther et al.[20] | Martinez et al.[54] | Panda et al.[33] | Oskarsson et al.[42] |
|---|---|---|---|---|---|
| Year and place of study | 2018-2019 | 2012-2015 | 2018 | 2014-2016 | 2010 |
| Kolkata, India | Dresden, Germany | Mexico | Delhi, India | ||
| Total number of patients with ALS in study | n=50 | n=90 | n=112 | n=40 | n=16 |
| Mean total NMS score | 9.3 | - | - | - | - |
| Daytime saliva drooling | 48% | 41% | - | - | - |
| Altered taste/smell | 28% | 21% | - | - | - |
| Dysphagia/choking | 70% | 66% | - | - | - |
| Nausea/vomiting | 12% | 10% | 2% | - | - |
| Constipation | 40% | 26% | 7% | - | - |
| Bowel incontinence | 8% | 8% | 1% | - | - |
| Incomplete bowel evacuation | 20% | 27% | - | - | - |
| Urinary urgency | 22% | 51% | 2% | - | - |
| Nocturia | 12% | 50% | - | 62.5% | - |
| Unexplained pains | 22% | 18% | 26% | 22.5% | - |
| Unexplained weight loss | 56% | 21% | - | - | - |
| Forgetfulness | 8% | 13% | - | - | - |
| Loss of interest in what is happening around | 58% | 27% | 1% | - | - |
| Hallucinations (visual/auditory) | 0% | 0% | - | - | - |
| Difficulty concentrating | 46% | 23% | - | - | - |
| Feeling sad/low | 92% | 47% | 47% | - | - |
| Anxious/frightened | 82% | 23% | 18% | 57.5% | - |
| Change in sex drive | 86% | 23% | - | - | - |
| Difficulty in performing sex | 58% | 24% | - | - | - |
| Postural dizziness | 10% | 19% | 1% | - | - |
| Falling | 58% | 42% | - | - | - |
| Day time sleepiness | 6% | 10% | 1% | - | - |
| Insomnia | 46% | 49% | 1% | 65% | - |
| Vivid/frightening dreams | 42% | 12% | - | - | - |
| Dream enactment | 12% | 7% | - | - | - |
| Restless legs | 32% | 30% | 1% | 5% | - |
| Leg swelling | 20% | 28% | 4% | - | - |
| Excess sweating | 8% | 24% | 4% | - | - |
| Double vision | 0% | 0% | - | - | - |
| Delusions | 0% | 0% | - | - | - |
| Suicidal ideation | 34% | - | - | - | - |
| Pseudobulbar affect | 68% | - | 13% | - | - |
| Fatigue | 82% | - | 24% | - | - |
| Muscle cramps | 42% | - | - | 42.5% | - |
| Sensory paresthesia | 30% | - | 13% | - | - |
| Unexplained Itching | 16% | - | - | - | - |
| Executive dysfunction | 24.4% (FAB scale) | - | - | - | 50% (FAB scale) |
| Mild cognitive impairment (MMSE/BMSE) | 9.8% | - | 1% | - | - |
ALS - Amyotrophic lateral sclerosis, BMSE - Bengali Mental State examination, MMSE - Mini Mental State Examination, FAB - Frontal assessment battery
We used NMSQuest in this study due to the lack of a dedicated scale for NMSs in ALS. However, we perceived some flaws in using NMSQuest in ALS. Some of the symptoms included in the questionnaire such as dysphagia and falling could be explained by motor impairment itself; hence, we should be careful in counting them as ‘nonmotor’ symptoms. We also felt that double vision is unlikely in ALS and may be omitted. On the other hand, some symptoms should be additionally enquired about routinely in ALS. These include pseudobulbar affect, fatigue, muscle cramp, tingling/paresthesia, early satiety/postprandial fullness and unexplained itching. Moreover, cognition particularly frontal behaviour and executive function as well as depression should be evaluated in detail using comprehensive instruments.
Limitations
Lack of validated questionnaire to detect NMSs in ALS led us to use NMSQuest, a tool developed for IPD. A comprehensive dedicated tool needs to be developed. We used FAB and BMSE for assessment of cognitive impairment. Alternative tools could have been better as BMSE has its inherent limitation for frontal type of cognitive impairment. Being cross-sectional study, it does not provide whole spectrum, and a longitudinal study is required to understand the burden and to find correlation of NMSs with disease progression.
CONCLUSION
Our study comprehensively investigated various NMSs and provided information even on the infrequently present NMSs of ALS. These can be broadly categorized into neuropsychiatric, cognitive, autonomic, sleep and gastrointestinal disturbances. NMSs are common, and these are present across the disease course of ALS making it a ‘multisystem disease’. Some NMSs worsen with advancement of the disease. To the best of our knowledge, this is the first study in India which attempted to document the spectrum of NMSs of ALS.
Statement of ethics
The institutional ethics committee approved the study. Patients were recruited only after taking written informed consent from the patient and his/her caregiver or legally acceptable representative to participate in the study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4:3. doi: 10.1186/1750-1172-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van der Graaff MM, de Jong JM, Baas F, de Visser M. Upper motor neuron and extra-motor neuron involvement in amyotrophic lateral sclerosis: A clinical and brain imaging review. Neuromuscul Disord. 2009;19:53–8. doi: 10.1016/j.nmd.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol. 2010;11:301–7. doi: 10.1038/nrm2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fang T, Jozsa F, Al-Chalabi A. Nonmotor symptoms in amyotrophic lateral sclerosis: A systematic review. Int Rev Neurobiol. 2017;134:1409–41. doi: 10.1016/bs.irn.2017.04.009. [DOI] [PubMed] [Google Scholar]
- 5.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–9. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 6.Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. BDNF ALS Study Group, 1A complete listing of the BDNF Study Group.The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. J Neuro Sci. 1999;169:13–21. doi: 10.1016/s0022-510x(99)00210-5. [DOI] [PubMed] [Google Scholar]
- 7.Chaudhuri KR, Martinez-Martin P, Schapira AH, Stocchi F, Sethi K, Odin P, et al. International multicenter pilot study of the first comprehensive self-completed nonmotor symptoms questionnaire for Parkinson's disease: The NMSQuest study. Mov Disord. 2006;21:916–23. doi: 10.1002/mds.20844. [DOI] [PubMed] [Google Scholar]
- 8.Beck AT, Ward C, Mendelson M, Mock J, Erbaugh J. Beck depression inventory (BDI) Arch Gen Psychiatry. 1961;4:561–71. doi: 10.1001/archpsyc.1961.01710120031004. [DOI] [PubMed] [Google Scholar]
- 9.Moore SR, Gresham LS, Bromberg MB, Kasarkis EJ, Smith RA. A self report measure of affective lability. J Neurol Neurosurg Psychiatry. 1997;63:89–93. doi: 10.1136/jnnp.63.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johns MW. A new method for measuring daytime sleepiness: The Epworth sleepiness scale. Sleep. 1991;14:540–5. doi: 10.1093/sleep/14.6.540. [DOI] [PubMed] [Google Scholar]
- 11.Rashnoo P, Daniel SJ. Drooling quantification: Correlation of different techniques. Int J Pediatr Otorhinolaryngol. 2015;79:1201–5. doi: 10.1016/j.ijporl.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 12.Das SK, Banerjee TK, Mukherjee CS, Bose P, Biswas A, Hazra A, et al. An urban community-based study of cognitive function among non-demented elderly population in India. Neurol Asia. 2006;11:37–48. [Google Scholar]
- 13.Dubois B, Slachevsky A, Litvan I, Pillon BF. The FAB: A frontal assessment battery at bedside. Neurology. 2000;55:1621–6. doi: 10.1212/wnl.55.11.1621. [DOI] [PubMed] [Google Scholar]
- 14.Garuti G, Rao F, Ribuffo V, Sansone VA. Sialorrhea in patients with ALS: Current treatment options. Degener Neurol Neuromuscul Dis. 2019;9:19–26. doi: 10.2147/DNND.S168353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desport JC, Preux PM, Truong CT, Courat L, Vallat JM, Couratier P. Nutritional assessment and survival in ALS patients. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:91–6. doi: 10.1080/14660820050515386. [DOI] [PubMed] [Google Scholar]
- 16.Marin B, Desport JC, Kajeu P, Jésus P, Nicolaud B, Nicol M, et al. Alteration of nutritional status at diagnosis is a prognostic factor for survival of amyotrophic lateral sclerosis patients. J Neurol Neurosurg Psychiatry. 2011;82:628–34. doi: 10.1136/jnnp.2010.211474. [DOI] [PubMed] [Google Scholar]
- 17.Moglia C, Calvo A, Grassano M, Canosa A, Manera U, D’Ovidio F, et al. Early weight loss in amyotrophic lateral sclerosis: Outcome relevance and clinical correlates in a population-based cohort. J Neurol Neurosurg Psychiatry. 2019;90:666–73. doi: 10.1136/jnnp-2018-319611. [DOI] [PubMed] [Google Scholar]
- 18.Viguera C, Wang J, Mosmiller E, Cerezo A, Maragakis NJ. Olfactory dysfunction in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2018;5:976–81. doi: 10.1002/acn3.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeda T, Iijima M, Uchihara T, Ohashi T, Seilhean D, Duyckaerts C, et al. TDP-43 pathology progression along the olfactory pathway as a possible substrate for olfactory impairment in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2015;74:547–56. doi: 10.1097/NEN.0000000000000198. [DOI] [PubMed] [Google Scholar]
- 20.Günther R, Schrempf W, Hähner A, Hummel T, Wolz M, Storch A, et al. Impairment in respiratory function contributes to olfactory impairment in amyotrophic lateral sclerosis. Front Neurol. 2018;9:79. doi: 10.3389/fneur.2018.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toepfer M, Schroeder M, Klauser A, Lochmüller H, Hirschmann M, Riepl RL, et al. Delayed colonic transit times in amyotrophic lateral sclerosis assessed with radio-opaque markers. Euro J Med Res. 1997;2:473–6. [PubMed] [Google Scholar]
- 22.Jackson CE, McVey AL, Rudnicki S, Dimachkie MM, Barohn RJ. Symptom management and end-of-life care in amyotrophic lateral sclerosis. Neurol Clin. 2015;33:889–908. doi: 10.1016/j.ncl.2015.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baltadzhieva R, Gurevich T, Korczyn AD. Autonomic impairment in amyotrophic lateral sclerosis. Curr Opin Neurol. 2005;18:487–93. doi: 10.1097/01.wco.0000183114.76056.0e. [DOI] [PubMed] [Google Scholar]
- 24.Piccione EA, Sletten DM, Staff NP, Low PA. Autonomic system and amyotrophic lateral sclerosis. Muscle Nerve. 2015;51:676–9. doi: 10.1002/mus.24457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimizu T, Hayashi H, Kato S, Hayashi M, Tanabe H, Oda M. Circulatory collapse and sudden death in respirator-dependent amyotrophic lateral sclerosis. J Neurol Sci. 1994;124:45–55. doi: 10.1016/0022-510x(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi H, Oyanagi K, Ikuta F. The intermediolateral nucleus in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 1993;86:190–2. doi: 10.1007/BF00334889. [DOI] [PubMed] [Google Scholar]
- 27.Rabkin JG, Albert SM, Del Bene ML, O'sullivan I, Tider T, Rowland LP, et al. Prevalence of depressive disorders and change over time in late-stage ALS. Neurology. 2005;65:62–7. doi: 10.1212/01.wnl.0000167187.14501.0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurt A, Nijboer F, Matuz T, Kübler A. Depression and anxiety in individuals with amyotrophic lateral sclerosis: Epidemiology and management. CNS Drugs. 2007;21:279–91. doi: 10.2165/00023210-200721040-00003. [DOI] [PubMed] [Google Scholar]
- 29.Lou JS, Reeves A, Benice T, Sexton G. Fatigue and depression are associated with poor quality of life in ALS. Neurology. 2003;60:122–3. doi: 10.1212/01.wnl.0000042781.22278.0a. [DOI] [PubMed] [Google Scholar]
- 30.Kübler A, Winter S, Ludolph AC, Hautzinger M, Birbaumer N. Severity of depressive symptoms and quality of life in patients with amyotrophic lateral sclerosis. Neurorehabil Neural Repair. 2005;19:182–93. doi: 10.1177/1545968305276583. [DOI] [PubMed] [Google Scholar]
- 31.McDonald ER, Wiedenfeld SA, Hillel AL, Carpenter CL, Walter RA. Survival in amyotrophic lateral sclerosis: The role of psychological factors. Arch Neurol. 1994;51:17–23. doi: 10.1001/archneur.1994.00540130027010. [DOI] [PubMed] [Google Scholar]
- 32.Hetta J, Jansson I. Sleep in patients with amyotrophic lateral sclerosis. J Neurol. 1997;244:S7–9. doi: 10.1007/BF03160565. [DOI] [PubMed] [Google Scholar]
- 33.Panda S, Gourie-Devi M, Sharma A. Sleep disorders in amyotrophic lateral sclerosis: A questionnaire-based study from India. Neurol India. 2018;66:700–8. doi: 10.4103/0028-3886.232327. [DOI] [PubMed] [Google Scholar]
- 34.Boentert M. Sleep disturbances in patients with amyotrophic lateral sclerosis: Current perspectives. Nat Sci Sleep. 2019;11:97–111. doi: 10.2147/NSS.S183504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Limousin N, Blasco H, Corcia P, Arnulf I, Praline J. The high frequency of restless legs syndrome in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12:303–6. doi: 10.3109/17482968.2011.557736. [DOI] [PubMed] [Google Scholar]
- 36.Lo Coco D, Piccoli F, La Bella V. Restless legs syndrome in patients with amyotrophic lateral sclerosis. Mov Disord. 2010;25:2658–61. doi: 10.1002/mds.23261. [DOI] [PubMed] [Google Scholar]
- 37.Liu S, Huang Y, Tai H, Zhang K, Wang Z, Shen D, et al. Excessive daytime sleepiness in Chinese patients with sporadic amyotrophic lateral sclerosis and its association with cognitive and behavioural impairments. J Neurol Neurosurg Psychiatry. 2018;89:1038–43. doi: 10.1136/jnnp-2018-318810. [DOI] [PubMed] [Google Scholar]
- 38.Ahmed RM, Newcombe RE, Piper AJ, Lewis SJ, Yee BJ, Kiernan MC, et al. Sleep disorders and respiratory function in amyotrophic lateral sclerosis. Sleep Med Rev. 2016;26:33–42. doi: 10.1016/j.smrv.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 39.Goldstein LH, Abrahams S. Changes in cognition and behaviour in amyotrophic lateral sclerosis: Nature of impairment and implications for assessment. Lancet Neurol. 2013;12:368–80. doi: 10.1016/S1474-4422(13)70026-7. [DOI] [PubMed] [Google Scholar]
- 40.Crockford C, Newton J, Lonergan K, Chiwera T, Booth T, Chandran S, et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology. 2018;91:e1370–80. doi: 10.1212/WNL.0000000000006317. doi: 10.1212/WNL.0000000000006317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang JL, Lomen-Hoerth C, Murphy J, Henry RG, Kramer JH, Miller BL, et al. A voxel-based morphometry study of patterns of brain atrophy in ALS and ALS/FTLD. Neurology. 2005;65:75–80. doi: 10.1212/01.wnl.0000167602.38643.29. [DOI] [PubMed] [Google Scholar]
- 42.Oskarsson B, Quan D, Rollins YD, Neville HE, Ringel SP, Arciniegas DB. Using the frontal assessment battery to identify executive function impairments in amyotrophic lateral sclerosis: A preliminary experience. Amyotroph Lateral Scler. 2010;11:244–7. doi: 10.3109/17482960903059588. [DOI] [PubMed] [Google Scholar]
- 43.Terada T, Miyata J, Obi T, Kubota M, Yoshizumi M, Yamazaki K, et al. Frontal assessment battery and frontal atrophy in amyotrophic lateral sclerosis. Brain Behav. 2017;7:e00707. doi: 10.1002/brb3.707. doi: 10.1002/brb3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schreiber H, Gaigalat T, Wiedemuth-Catrinescu U, Graf M, Uttner I, Muche R, et al. Cognitive function in bulbar- and spinal-onset amyotrophic lateral sclerosis. A longitudinal study in 52 patients. J Neurol. 2005;252:772–81. doi: 10.1007/s00415-005-0739-6. [DOI] [PubMed] [Google Scholar]
- 45.Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65:586–90. doi: 10.1212/01.wnl.0000172911.39167.b6. [DOI] [PubMed] [Google Scholar]
- 46.Palmieri A, Mento G, Calvo V, Querin G, D’Ascenzo C, Volpato C, et al. Female gender doubles executive dysfunction risk in ALS: A case-control study in 165 patients. J Neurol Neurosurg Psychiatry. 2015;86:574–9. doi: 10.1136/jnnp-2014-307654. [DOI] [PubMed] [Google Scholar]
- 47.Thakore NJ, Pioro EP. Laughter, crying and sadness in ALS. J Neurol Neurosurg Psychiatry. 2017;88:825–31. doi: 10.1136/jnnp-2017-315622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCullagh S, Moore M, Gawel M, Feinstein A. Pathological laughing and crying in amyotrophic lateral sclerosis: An association with prefrontal cognitive dysfunction. J Neurol Sci. 1999;169:43–8. doi: 10.1016/s0022-510x(99)00214-2. [DOI] [PubMed] [Google Scholar]
- 49.Oey PL, Vos PE, Wieneke GH, Wokke JH, Blankestijn PJ, Karemaker JM. Subtle involvement of the sympathetic nervous system in amyotrophic lateral sclerosis. Muscle Nerve. 2002;25:402–8. doi: 10.1002/mus.10049. [DOI] [PubMed] [Google Scholar]
- 50.Lo Coco D, La Bella V. Fatigue, sleep, and nocturnal complaints in patients with amyotrophic lateral sclerosis. Eur J Neurol. 2012;19:760–3. doi: 10.1111/j.1468-1331.2011.03637.x. [DOI] [PubMed] [Google Scholar]
- 51.Lou JS. Approaching fatigue in neuromuscular diseases. Phys Med Rehabil Clin N Am. 2005;16:1063–79, xi. doi: 10.1016/j.pmr.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 52.Li TM, Alberman E, Swash MI. Comparison of sporadic and familial disease amongst 580 cases of motor neuron disease. J Neurol Neurosurg Psychiatry. 1988;51:778–84. doi: 10.1136/jnnp.51.6.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cho JH, Nam HW, Yon BW, Jeon BS, Roh JK, Lee SB, et al. Sensory symptoms in amyotrophic lateral sclerosis. J Korean Neurol Assoc. 1996;14:789–95. [Google Scholar]
- 54.Martínez HR, Escamilla-Oca˜nas CE, Hernández-Torre M. Síntomas neurológicos extra-motores en pacientes con esclerosis lateral amiotrófica. Neurología. 2018;33:474–6. doi: 10.1016/j.nrl.2016.05.002. [DOI] [PubMed] [Google Scholar]