


Abstract
Mutations in genes encoding mitochondrial aminoacyl-tRNA synthetases are linked to diverse diseases. However, the precise mechanisms by which these mutations affect mitochondrial function and disease development are not fully understood. Here, we develop a Drosophila model to study the function of dFARS2, the Drosophila homologue of the mitochondrial phenylalanyl–tRNA synthetase, and further characterize human disease-associated FARS2 variants. Inactivation of dFARS2 in Drosophila leads to developmental delay and seizure. Biochemical studies reveal that dFARS2 is required for mitochondrial tRNA aminoacylation, mitochondrial protein stability, and assembly and enzyme activities of OXPHOS complexes. Interestingly, by modeling FARS2 mutations associated with human disease in Drosophila, we provide evidence that expression of two human FARS2 variants, p.G309S and p.D142Y, induces seizure behaviors and locomotion defects, respectively. Together, our results not only show the relationship between dysfunction of mitochondrial aminoacylation system and pathologies, but also illustrate the application of Drosophila model for functional analysis of human disease-causing variants.
INTRODUCTION
The aberrant aminoacylation in mitochondrial tRNAs have been linked to a wide spectrum of clinical presentations (1–6). Aminoacylation, the attachment of an amino acid to a tRNA, is catalyzed by mitochondrial aminoacyl-tRNA synthetases (mt-aaRSs) such as mitochondrial phenylalanyl–tRNA synthetase (FARS2) (7–9). In animal, 22 mitochondrial tRNAs are encoded by its own genome (mtDNA) (10,11), while all 19 mt-aaRSs are encoded by the nuclear genome, synthesized in cytosol and subsequently imported into mitochondria (12–14). Clinical presentations linked to aminoacylation deficiencies caused by these mt-aaRS defects usually display a marked bias for the central nervous system, including the encephalopathy, leukodystrophy, Perrault syndrome, sensorineural deafness and visual impairment (15–22). The clinical spectrum of FARS2 deficiency ranges from the infantile-onset phenotype, characterized by epileptic mitochondrial encephalopathy, to the later-onset phenotype, characterized by spastic paraplegia, less severe neurologic manifestations, and longer survival (23–30). In particular, patients carrying the homozygous p.Y144C and p.G309S or compound heterozygous p.G309S with p.R153G mutations in the FARS2 developed infantile-onset epileptic mitochondrial encephalopathy with seizure, developmental delay and truncal hypotonia from the birth to age of six month (23,25,27,29). The primary defects in these FARS2 mutations were the deficient aminoacylation of tRNAPhe (26,28). The aberrant tRNAPhe metabolism impaired mitochondrial translation and subsequent deficiencies of oxidative phosphorylation (25,28). However, the pathophysiology of FARS2 defiency is still poorly understood due to the lack of animal disease model.
Drosophila is a powerful model organism for understanding pathophysiology of human diseases (31). To investigate whether FARS2 deficiency caused the epileptic mitochondrial encephalopathy and spastic paraplegia in vivo, we produced the Drosophila dFARS2 knockout mutants by genome editing using the CRISPR/Cas9 system and dFARS2 knockdown model using RNAi approach. These dFARS2 knockout and knockdown fly models were characterized for the phenotypes including developmental delay and seizure. Biochemical analysis further revealed that dFARS2 deficiency led to defects in mitochondrial tRNAPhe metabolism, translation, assembly and activity of oxidative phosphorylation system (OXPHOS) complexes. Finally, we produced humanized fly models by introducing human wild type FARS2, p.G309S and p.D142Y pathogenic variants into Drosophila dFARS2 mutants and analyzed the pathologic consequence of human disease-causing FARS2 p.G309S and p.D142Y mutations.
MATERIALS AND METHODS
Drosophila stocks
All flies were reared on standard corn medium at 25°C. The following fly stocks were used: UAS-dFARS2-RNAi (Bloomington Drosophila Stock Center, BDSC64869), Daughterless-Gal4 (Da-Gal4), elav-Gal4, y1 M{vas-int.Dm}ZH-2A w*; M{3xP3-RFP.attP}ZH-86Fb (Bloomington Drosophila Stock Center, BDSC24749) and y1 M{vas-Cas9}ZH-2A w1118/FM7c (Bloomington Drosophila Stock Center, BDSC51323).
Generation of Drosophila dFARS2 mutants and transgenic flies expressing dFARS2 or human FARS2
Drosophila dFARS2KO mutant allele was generated through the CRISPR-Cas9 system (32–34). A guide RNA sequence against dFARS2 gene was designed by a CRISPR guide RNA analysis tool (http://tools.flycrispr.molbio.wisc.edu/targetFinder/). The DNA template for transcription of single-stranded guide RNA (sgRNA) was synthesized by PCR amplification according to the manufacturer's protocol with the forward primer (TAATACGACTCACTATAGGAGGAGCGTATCGCAACCAA GTTTTAGAGCTAGAAATAGC) and reverse primer (AAAAAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC). The sgRNA was transcribed from the DNA template using in vitro Transcription T7 Kit (Vazyme, R101-01) and then purified with RNeasy® Mini Kit (QIAGEN, 74104). The sgRNA was microinjected into BDSC51323 embryos. The emerging adult flies were individually crossed to If/CyO balancer flies. The progenies of these flies were used to extract the genomic DNA to identify the mutation by PCR. The sequence of the primers used for PCR are AACGTAACGCCCAAGATA and AGATGCTGCTGATTGATGTA. The PCR products were sequenced to assess the mutation in the target region.
To generate the pAttB-dFARS2 construct, a 2356 bp genomic DNA fragment containing the Drosophila FARS2 gene (CG13348) was amplified with the corresponding primers (Supplementary Table S1), and then subcloned into the pAttB vector. To generate the pUAST-FARS2-AttB construct, a human FARS2 CDS fragment (1356 bp) was amplified with the corresponding primers (Supplementary Table S1), and then subcloned into the pUAST-AttB vector. Mutation of c.G925A (p.G309S) and c.G424T (p.D142Y) were introduced through PCR based site-directed mutagenesis to give rise to pUAST-FARS2-p.G309S-AttB or pUAST-FARS2-p. D142Y-AttB construct. Primers used are listed in Supplementary Table S1. All constructs were sequenced and insered at the 86FB landing site (BDSC24749) on the third chromosome using phi-C31 integrase-mediated site-specific transgenesis.
S2 cell culture and transfection
Drosophila S2 cells were cultured in Schneider's medium (Gibco, 21720–001) with 10% FBS (Gibco, 10099-141) at 25°C. The full length dFARS2 cDNA fragment was amplified by PCR using FARS2 gene-specific primers (Forward: 5′-CCGGATCGGGGTACATGCTTCTGACGCTTC-3′ and Reverse: 5′-TCTTTGTAGTCCATGCGGCCACGTATTTGCACATT-3′), and subcloned into a pAC5.1-flag vector to generate the pAC5.1-dFARS2-Flag construct. S2 cells were transfected with pAC5.1-dFARS2-Flag plasmid using Effectene Transfection Reagent (QIAGEN, 301425).
Mitochondrial tRNA analysis
Total RNA was extracted with TRIzol (Invitrogen, 15596018) from the second instar larvae and adult fly heads. The tRNA Northern blot analysis was performed as detailed elsewhere (22,35,36). Digoxigenin (DIG) labeled Probe sequences for mitochondrial tRNAPhe, tRNALys, tRNAThr, tRNALeu(CUN), tRNAAsp, tRNAHis, tRNATyr, tRNAIle and 5S rRNA were provided in Supplementary Table S2. The hybridization and quantification of density in each band were performed as detailed previously (37).
The aminoacylation assays were carried out as detailed elsewhere (37,38). To further distinguish non-aminoacylated tRNA from aminoacylated tRNA, samples of tRNAs were deacylated with the treatment of 0.2 M NaOH after heating for 10 min at 60 oC (pH 9) and then run in parallel (37,38). Oligodeoxynucleotide probes for mitochondrial tRNAPhe, tRNALys, tRNAThr were described as above. The hybridization and quantification were carried out as described previously (37,38).
qRT-PCR and mtDNA copy number analysis
For qRT-PCR, total RNA was extracted from the second instar larvae with TRIzol (Invitrogen). Reverse transcription was performed using HiScript II 1st Strand cDNA Synthesis Kit (Vazyme, R211-02). For the measurement of mtRNA copy number, total genomic DNA was isolated from the second instar larvae using MiniBEST Universal Genomic DNA Extraction Kit (TAKARA, 9765) according to the manufacturer's protocol. Quantitative PCR analysis was carried out using an ABI 7900HT Fast Real-Time PCR System with a ChamQ SYBR qPCR Master Mix (Vazyme, Q311-02). The primers used were provided in the Supplementary Table S3.
Isolation of mitochondria
Mitochondria were isolated from second instar larvae according to the method described by previously (39). Approximately 2 ml of second instar larvae were homogenized in 5 ml ice-cold mitochondria isolation buffer [225 mM d-mannitol, 75 mM sucrose, 30 mM Tris–HCl, pH 7.4] containing the PhosSTOP™ phosphatase inhibitor (Roche, 4906845001) and cOmplete™ protease inhibitor cocktail (Roche, 4693132001). The homogenate was centrifuged twice at 600 × g for 10 min at 4°C. After each centrifugation, the supernatant was transferred to a new tube and centrifuged at 7000 × g for 10 min at 4°C. The resulting pellet was resuspended in 1 ml ice-cold mitochondria isolation buffer. The resuspended pellet was homogenized again and then centrifuged at 9000 × g for 10 min at 4°C. The final pellet containing mitochondria was stored at −80°C.
Western blot analysis
Second instar larvae or adult fly heads were collected and homogenized in RIPA lysis buffer [50 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% SDS] containing the PhosSTOP™ phosphatase inhibitor (Roche, 4906845001) and cOmplete™ protease inhibitor cocktail (Roche, 4693132001) by grinding at 4°C for 30 min. The protein concentration was measured with BCA assay Kit (Generay, gk5012) according to the manufacturer's instructions. Samples were electrophoresed on a 10% SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked with 5% non-fat powdered milk dissolved in 1× TBST [20 mM Tris–HCl, 500 mM NaCl, 0.1% Tween 20, pH 7.4] for 1 h at room temperature. After incubation with the primary antibodies overnight at 4°C, the membrane was probed with the secondary antibodies for 1 h at RT. The primary antibodies used include anti-NDUFS3 (1:1000; Abcam, ab14711), anti-UQCRFS1 (1:1000; Abcam, ab14746), anti-CO3 (1:1000; Abcam, ab110259), anti-NDUFS1 (1:1000; Proteintech, 12444-1-AP), anti-ATP5A (1:2000; Abcam, ab14748), anti-SDHA (1:1000; Abcam, ab209986), anti-TFAM (1:1000; BOSTER, ba2827) and anti-Porin (1:2000; Abcam, ab14734). Anti-dFARS2 antibody was raised in rabbit against the C-terminal 14 amino acids ([C]VDKFKHPKTGKSSV) of the Drosophila FARS2 (1:1000; GenScript). Horseradish peroxidase conjugated Goat anti-mouse IgG (GenScript, A00160) and goat anti-rabbit IgG (GenScript, A00098) were used as secondary antibodies. Proteins were detected using the Chemi Lucent ECL detection reagents (Millipore, WBKLS0500). The blot was scanned using a gel imaging system and the image was analyzed by ImageJ software.
Biochemical assays of respiratory chain complexes and in-gel activity assays
BN-PAGE was performed on mitochondrial protein extracted from second instar larvae as detailed elsewhere (40–42). Briefly, mitochondrial proteins were isolated with solution buffer A [50 mM NaCl, 50mM imidazole, 2 mM 6-aminocaproic acid, 1 mM EDTA, 2% Triton X-100, pH 7.4] containing the PhosSTOPTM phosphatase inhibitor (Roche, 4906845001) and cOmplete™ protease inhibitor cocktail (Roche, 4693132001). 50 μg of mitochondrial protein was mixed with BN-PAGE loading buffer [final concentration: 0.5% (w/v) Coomassie Blue G-250, 50 mM 6-aminocaproic acid, 20% glycerol, pH 7.0] and separated on a 3∼11% BN-PAGE gel. Cathode buffer [50 mM Tricine, 7.5 mM imidazole, 0.02% Coomassie Blue G-250,pH 7.0] and anode buffer [25 mM imidazole, pH 7.0] were used for electrophoresis. The samples in the gel were soaked in 5% Coomassie Blue G-250 for Coomassie blue staining or transferred to a PVDF membrane. The membrane was probed with primary and secondary antibodies as described above.
For in-gel activity assays, the BN-PAGE gels were incubated in ice-cold ddH2O at 4°C for 30 min, then transferred to complex specific solutions at room temperature for 5 h. The Complex I solution was as follows: 100 mM Tris––HCl pH 7.4, 1 mg/ml Nitrotetrazolium blue chloride (NBT) and 0.1 mg/ml NADH. The Complex II solution was as follows: 5 mM Tris–HCl pH 7.4, 2.5 mg/ml NBT, 0.2 mM phenazine methasulfate and 20 mM sodium succinate (43).
Measurement of mitochondrial respiratory complex activity
Mitochondria were isolated as described above. Enzymatic activities of single respiratory chain complexes were assayed as previously described (44–46). Briefly, complex I (NADH ubiquinone oxidoreductase) activity was determined by following the oxidation of NADH with ubiquinone as the electron acceptor. The activity of Complex II was examined through the artificial electron acceptor DCPIP. Complex III (ubiquinone cytochrome c oxidoreductase) activity was measured as the reduction of cytochrome c (III) using d-ubiquinol-2 as the electron donor. The activity of complex IV (cytochrome c oxidase) was monitored by following the oxidation of cytochrome c (II). The activity of complex V was explored through the NADH oxidation via conversion of phosphoenolpyruvate to lactate by two step reactions. Citrate synthase (CS) activity was determined by catalyzing the reaction of oxaloacetic acid and acetyl-CoA with 5,5′-dithio-bis-nitrobenzoicacid as the chromogenic agent. All mitochondrial respiratory complex activities were expressed relative to citrate synthase (CS) activity.
Immunofluorescence staining
For MitoTracker staining, S2 cells were incubated for 40 min in 100 nM MitoTracker Red (Cell Signaling Technology, #9082), and then fixed with 1× PBS containing 4% paraformaldehyde (PFA) for 20 min at RT. Next, S2 cells were washed three times with 1× PBS containing 0.3% Triton-X (PBT) for 20 min at RT. The samples were blocked with 3% BSA in PBT for 1 h, and then incubated with primary antibodies overnight at 4°C. After three washes with 1× PBT, the samples were incubated with secondary antibodies for 1 h at RT. DAPI (1:1000; Invitrogen, D1306) was added for the last 10 min. The samples were washed three more times with 1× PBT and mounted in Vectershield. The primary antibodies used was rabbit anti-flag (1:1000; Sigma-Aldrich, F2555). The secondary antibodies used was goat anti-rabbit Alexa Fluor 568 (1:1000; Thermo Fisher Scientific, A-11036). Images were obtained with an Olympus FV1000 confocal microscope and processed using ImageJ and Photoshop.
Bang-sensitive behavioral testing
Bang-sensitive behavioral testing was performed as described previously (47,48). Flies were collected under CO2 at 3 days after eclosion and kept at 10 flies/vial for up to 10 days. Flies were then transferred to an empty vial 1 h before behavioral analysis. For testing, vials were mechanically stimulated by placement in a vortex mixer (Vortex-Genie 2) at the maximum speed for 20 s. The number of flies that displayed the bang-sensitive phenotype (seizure and paralysis) after the vortex were recorded. These flies were also used to analyze the recovery time. The time for each bang-sensitive fly to right itself after vortexing immediately was recorded. All flies were tested only once and the results of the same genotype were combined.
Transmission electron microscope
Fly brains were dissected and fixed with 2.5% glutaraldehyde in 1× PBS overnight and then washed three times with 1× PBS for 15 min at each step. Samples were post fixed with 1% OsO4 in 1× PBS for 1 h and washed three times with 1× PBS for 15 min at each step. The fixed samples were dehydrated by a graded series of ethanol (30%, 50%, 70%, 80%, 90% and 95%) for about 15 min at each step and absolute ethanol for 20 min, and then transferred to absolute acetone for 20 min. Samples were then infiltrated with and embedded in Spurr resin. Thin sections were cut using ultramicrotome (Leica, EM UC7) and stained with uranyl acetate and alkaline lead citrate. Afterwards, the sections were examined using a Hitachi Model H-7650 transmission electron microscope.
Survival rate analysis
For survival rates from the first instar larvae to adult, 30 first instar larvae were collected and cultured in vials with the standard medium at 25°C, and the number of pupa and eclosed adults was counted every 24 h.
Climbing assay
Flies were collected as described above. For the assay, groups of 10 female flies were placed in empty vials and tapped to the bottom of the vial, and the number of flies that climbed 8 cm above the bottom within 10 s was counted.
Statistical analysis
Data shown in the figures are reported as mean ± SD. Statistical analysis was performed using the unpaired, two-tailed Student's t-test contained in the Microsoft-Excel program. Significance levels are indicated in the figure legends.
RESULTS
Drosophila FARS2 is a highly conserved mitochondrial protein
Drosophila FARS2 (CG13348, hereafter referred to as dFARS2) encodes a 453 amino acid protein with the typical mitochondrial target sequence with 27 residues at the N-terminus, predicted by MitoProt program (49). Alignment of dFARS2 with its homologs of other organisms, including Homo sapiens, Mus musculus, Danio rerio, Drosophila melanogaster and Saccharomyces cerevisiae revealed an extensive conservation of protein sequence (Supplementary Figure S1). In particular, dFARS2 shares an overall amino acid identity of 50% and 68% similarity with human FARS2, respectively (Supplementary Figure S1).
To test whether dFARS2 is localized to mitochondria, we transfected a dFARS2 construct tagged with a C-terminal FLAG into Drosophila S2 cells and examined subcellular localization by immunofluorescence analysis. Using an antibody against FLAG and Mito Tracker probes, a dye to label the mitochondrial network, we observed a complete overlap of both fluorescence signal in the transfected cells (Figure 1A). These data demonstrate the mitochondrial localization of dFARS2.
Figure 1.
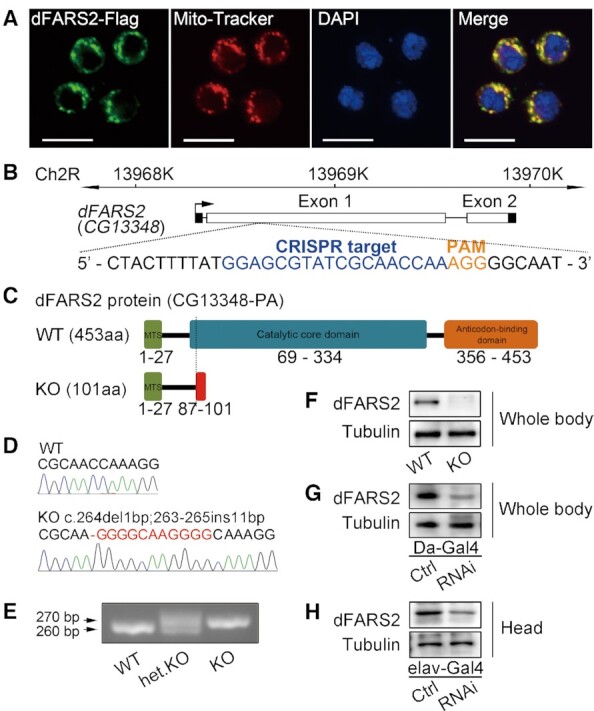
Generation of Drosophila models with dFARS2 deficiency. (A) Mitochondrial localization of dFARS2 in S2 cells. Cells were stained for Mito-Tracker (Red) and anti-Flag antibody (Green). DAPI was used to label DNA (Blue). Scale bar represents 10 μm. (B) Genome editing of dFARS2 using the CRISPR/Cas9 system. sgRNA sequence is indicated in blue and PAM sequence is indicated in orange. (C) Schematic representation of the dFARS2 wild type (WT) and dFARS2 mutant (KO) proteins. MTS indicates mitochondrial targeting sequence. (D) Partial Sanger sequencing chromatograms of the sgRNA targeting region showing for WT and dFARS2KO. A hyphen indicates a deletion and a red letter indicates an insertion. (E) PCR analysis of the sgRNA targeting region at the dFARS2 locus. (F–H) Western blot analysis of dFARS2 in protein extracts from WT and dFARS2KO second-instar larvae (F), the control and Da-Gal4 driven dFARS2 knockdown second-instar larvae (G), the control and elav-Gal4 driven dFARS2 knockdown fly heads (H).
Generation of Drosophila dFARS2 deficiency
To explore the pathological consequences of dFARS2 mutation, we first generated a Drosophila model with the loss-of-function dFARS2 mutants through the CRISPR/Cas9 system (Figure 1B) (32,50). One mutant allele, dFARS2KO, was obtained. This allele harbored a 1 bp deletion followed by an 11 bp insertion in exon 1 of dFARS2 (Homozygous and heterozygous dFARS2KO mutant flies were described as KO and het.KO respectively, while wild type flies were described as WT). In fact, this deletion caused a frame shift at codon 87 and a new stop codon at codon 101 downstream of the translation start (Figure 1C). This allele was confirmed by Sanger sequencing, PCR and Western blot analysis (Figure 1D–F). We also generated fly models with the knockdown of dFARS2 by using a UAS-dFARS2 RNAi line in combination with the GAL4/UAS binary expression system (dFARS2 knockdown flies were described as RNAi and control flies were described as Ctrl). The knockdown of dFARS2 by RNAi with a ubiquitously expressed Daughterness-Gal4 (Da-Gal4) or a pan-neuronal Gal4 (elav-Gal4) was further confirmed by the reduced level of dFARS2 protein in whole body (Figure 1G) or fly heads (Figure 1H) using western blot analysis.
dFARS2 deficiency causes developmental delay
Mutations in FARS2 are associated with several pathological states in humans, notably developmental delay and seizure (29). However, the cause-and-effect relationship has not been established in a highly tractable animal model. We took advantage of Drosophila dFARS2 deficiency models generated in this study to examine the pathological consequence.
The homozygous dFARS2KO mutants were embryonic viable, and the newly hatched larvae were able to grow and develop (Figure 2A). Notably, dFARS2KO mutant larvae showed a profound developmental delay starting on day 2 after egg laying (AEL) and some of them died during the later larval stages (Figure 2A and B). By day 5 AEL, dFARSKO mutant larvae were significantly smaller in size than control larvae (Figure 2C). The mutant larvae reached the second instar larval stage by detecting the presence of mouth hooks (Supplementary Figure S2A). Both larval lethality and size defect of dFARS2KO mutants were fully rescued with the expression of a 2.3 kb genomic DNA that covers the dFARS2 locus (Figure 2A–C). The heterozygous dFARS2KO mutants did not show developmental delay (Supplementary Figure S3A, B). Moreover, the ubiquitously knockdown of dFARS2 exhibited the same phenotypes as those in dFARS2KO mutant larvae. Da-Gal4 driven dFARS2 knockdown larvae displayed a developmental delay with markedly reduced body size during the larval stages (Figure 2D–F). Approximately 80% of these larvae were able to survive up to 10 days but arrested at the second instar as indicated by the mouth hook morphology (Supplementary Figure S2B). To further assess the effect of dFARS2 deficiency on development, we used a pan-neuron Gal4 driver elav-Gal4 to specifically knock down dFARS2 activity in the nervous system. Our analysis revealed that neuronal knockdown of dFARS2 also resulted in a significant developmental delay at both larval and pupal stages (Figure 2G–I). Despite the heavy lethality was observed during pupal development, ∼23% of elav-Gal4 driven dFARS2 knockdown animals were able to develop into adults (Figure 2I). These adult flies were further used to examine the life span. The life span was decreased in dFARS2 knockdown male and female flies compared with controls (Figure 2J). Together, these data suggest that dFARS2 inactivation results in a developmental delay.
Figure 2.

dFARS2 deficiency leads to the developmental delay. (A) Images showing wild type (WT) and dFARS2 knockout (KO) larvae, from 1 to 5 days after egg laying (AEL). Rescue denotes the dFARS2KO carrying the genomic rescue transgene for dFARS2. Scale bars: 1 mm. (B) Graph showing the percentage of survivors for WT, dFARS2KO and rescue larvae from 1 to 5 days AEL. n = 5. (C) Graph showing the relative size of WT, dFARS2KO and rescue larvae at 5 days AEL. n = 10. (D) Images showing control and Da-Gal4 driven dFARS2 knockdown animals at various developmental stages. Scale bars: 1 mm. (E) Graph showing the percentage of survivors for control and Da-Gal4 driven dFARS2 knockdown larvae. n = 5. (F) Graph showing the relative size of control and Da-Gal4 driven dFARS2 knockdown larvae at 5 days AEL. n = 10. (G) Images showing control and elav-Gal4 driven dFARS2 knockdown animals at various developmental stages, from 1 to 15 days AEL. Scale bars: 1 mm. (H) Graph showing pupariation curves for control and elav-Gal4 driven dFARS2 knockdown larvae. n = 3. (I) Graph showing eclosion curves for control and elav-Gal4 driven dFARS2 knockdown pupae. n = 4. (J) Graph showing the survival rate of control and elav-Gal4 driven dFARS2 knockdown adult flies. n = 3. Data are presented as means ± SD. ****P< 0.0001. NS, not significant.
dFARS2 deficiency causes seizure phenotype
Seizure in the Drosophila model shares many characteristics similar to those of seizures in humans (47,50,51). Bang sensitivity assay has been widely used to measure seizure susceptibility in Drosophila (47,48). In this assay, the time required for the flies to right themselves and move after a mechanical stress was recorded to determine seizure susceptibility (47,48). The number of flies displaying bang-sensitive phenotype was also counted. In response to a strong mechanical stress, wild type flies recovered quickly while bang-sensitive mutants showed a characteristic seizure pattern (47,48). Ten-day-old elav-Gal4 driven dFARS2 knockdown and control flies were tested for bang sensitivity to assess their susceptibility to seizure. The dFARS2 knockdown female and male flies were bang sensitive and took a long time to recover from a 20-s vortex, whereas the control flies were able to right themselves immediately or within a few seconds (Figure 3A–C, supplementary video 1). Due to the larval lethality of the homozygous dFARS2KO mutants, the heterozygous dFARS2KO mutants were examined and no seizure behaviors were observed (Supplementary Figure S3C, D). Furthermore, the brain of dFARS2 knockdown adult flies appeared to be smaller than in the controls (Figure 3D). These data suggest that dFARS2 deficiency induces seizure phenotype in Drosophila.
Figure 3.

dFARS2 deficiency causes seizure phenotype in adult flies. (A) Setup for the Bang-sensitive assay. Also see supplementary video 1. (B) Graph showing the percentage of control and elav-Gal4 driven dFARS2 knockdown flies with BS paralytic phenotypes (% Bang sensitive paralysis). n = 4. (C) Graph showing the recovery time of control (female: n = 13; male: n = 11) and elav-Gal4 driven dFARS2 knockdown (female: n = 34; male: n = 36) flies after BS paralysis. (D) Images showing the brain of control and elav-Gal4 driven dFARS2 knockdown flies. Scale bars represent 100 μm. (E, F) Transmission electron microscope images showing the untrastructure of brain tissues from control and elav-Gal4 driven dFARS2 knockdown flies. Magnifications of (E) and (F) are 40 000× and 100 000× respectively. Data are presented as means ± SD. ***P< 0.001. ****P< 0.0001. Scale bars represent 0.2 μm (E) and 0.1 μm (F).
Next, we examined the ultrastructure of mitochondria in the central brain. Thin sections of the central brain regions of 10-day-old elav-Gal4 driven dFARS2 knockdown and age-matched control flies were examined using transmission electron microscopy. Mitochondria in the controls were densely packed with intact cristae. In contrast, dFARS2 knockdown flies displayed loosely packed mitochondria with disorganized cristae (Figure 3E and F). The cristae became round and gave mitochondria a honeycomb-like appearance (Figure 3E and F). The mitochondria morphological defects in the dFARS2 knockdown flies suggest that the seizure phenotypes could be derived from mitochondrial dysfunction.
Reduced aminoacylation of mitochondrial tRNAPhe
As dFARS2 is a mitochondrial phenylalanyl–tRNA synthetase, we next performed a series of biochemical assays to further confirm the validity of our dFARS2 deficiency models.
To evaluate whether the dFARS2 deficiency affected the mitochondrial tRNA aminoacylation, we measured the level of aminoacylation using Northern Blot. In order to do this, total RNA was isolated from the larvae or adult fly heads and separated on an acid urea polyacrylamide gel which allows separation of uncharged tRNA from slow migrated aminoacylated tRNA. The presence of an mt-tRNA was assayed by hybridization to the complementary oligonucleotide probes. As a control for deacylated tRNA, an aliquot of each sample was deacylated under alkaline conditions (DA) and separated on the gel along with the untreated sample (Amino). The level of aminoacylated mt-tRNAPhe was reduced in dFARS2KO mutants compared to those in wild type larvae (Figure 4A). However, the aminoacylation levels of two other mitochondrial tRNA, mt-tRNALys and mt-tRNAThr, were similar in both wild type and dFARS2KO mutant larvae (Figure 4A). These results were also confirmed in dFARS2 knockdown larvae (Figure 4B). Da-Gal4 or elav-Gal4 driven dFARS2 RNAi caused a reduction of aminoacylated mt-tRNAPhe in third instar larvae or fly heads, as compared to the control (Figure 4B).
Figure 4.

The effects of dFARS2 deficiency on the aminoacylation and steady-state levels of tRNAs. (A, B) In vivo aminoacylation of mitochondrial tRNA assays. Fifty or one hundred μg of total RNAs purified from various larvae (A) the whole body of dFARS2KO and WT second instar larvae; (B) the whole body of Da-Gal4 driven dFARS2 knockdown second instar larvae or the head of elav-Gal4 driven dFARS2 knockdown and control flies) under acid conditions were electrophoresed through an acid (pH 5.2) 9% polyacrylamide-7 M urea gel, electroblotted, and hybridized with DIG-labeled oligonucleotide probes specific for the tRNAPhe, tRNALys and tRNAThr, respectively. The charged (upper band) and uncharged (lower band) forms of different tRNAs were separated by the gel system. Samples were deacylated (DA) by heating for 10 min at 60°C at pH 9, electrophoresed, and hybridized with DIG-labeled oligonucleotide probes as described above. (C, D) Northern blot analysis of eight tRNAs. Ten μg of total RNA from various larvae were electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with DIG-labeled oligonucleotide probes specific for tRNAPhe, tRNALys, tRNAThr, tRNALeu(CUN), tRNAAsp, tRNAHis, tRNATyr, tRNAIle and 5S rRNA, respectively. (E, F) Quantification of relative tRNA levels. The content of each tRNA was normalized to that of 5S rRNA. Calculations were based on three independent experiments. Error bars indicate two standard deviations (SD) of the means. * P< 0.05, ** P< 0.01, *** P< 0.001, **** P< 0.0001. NS, not significant.
Increased steady-state levels of mitochondrial tRNAs
To examine the effect of dFARS2 deficiency on the steady-state level of mitochondrial tRNAs, total RNAs were isolated and analyzed by Northern blot. Loss of dFARS2 resulted in obvious increases in the steady-state levels of eight mitochondrial tRNAs (tRNAPhe, tRNALys, tRNAThr, tRNALeu(CUN), tRNAAsp, tRNAHis, tRNATyr, tRNAIle) analyzed (Figure 4C). For comparison, the levels of mitochondrial tRNAs were quantified by normalizing to the reference 5S rRNA. The mean values of tRNAPhe, tRNALys, tRNAThr, tRNALeu(CUN), tRNAAsp, tRNAHis, tRNATyr, tRNAIle in dFARS2KO mutant larvae were 200.9%, 166.3%, 236.6%, 194.0%, 145.1%, 137.4%, 187.9% and 193.5%, compared to those in control larvae (Figure 4E). Similarly, the mean value of tRNAPhe, tRNALys, tRNAThr, tRNALeu(CUN), tRNAAsp, tRNAHis, tRNATyr, tRNAIle in Da-Gal4 driven dFARS2 RNAi larvae were 251.3%, 210.1%, 185%, 200.2%, 161%, 135%, 146.1% and 170.8%, compared to those in control larvae (Figure 4D and F). Moreover, an increased level of mitochondrial tRNAPhe (153.5%) was also observed in elav-Gal4 driven dFARS2 knockdown fly heads, compared to that in control fly heads (Figure 4D and F). The increase in the steady-state levels of mitochondrial tRNA in our dFARS2 deficiency models suggests a potential compensatory response of mitochondrial defects.
We next investigated the effect of dFARS2 deficiency on mitochondrial transcription. The protein level of mitochondrial transcription factor TFAM was slightly elevated in both dFARS2KO mutant and RNAi larvae (Supplementary Figure S4A). In addition, the levels of mtDNA-encoded transcripts, including ND4, CYTB, ATP6 and 16S rRNA were increased in both dFARS2KO mutant and RNAi larvae, suggesting an increase of overall mitochondrial transcription (Supplementary Figure S4B and C). Furthermore, we performed qPCR to examine the level of mitochondrial DNA. Both dFARS2KO mutant and RNAi knockdown larvae exhibited increased mtDNA levels, as shown for ND4, CYTB, ATP6 and 16S rRNA genomic DNA fragments (Supplementary Figure S4D and E). These results are consistent with the notion that there is a compensatory response in dFARS2 deficiency.
Reductions in the levels of the subunits of OXPHOS complexes
Having shown that dFARS2 is required for mitochondrial tRNAPhe aminoacylation, we proceeded to test whether loss of dFARS2 has an effect on the levels of OXPHOS components. Western blot analysis showed that the levels of the mtDNA-encoded complex IV subunit (CO3) and the nuclear gene encoded complex III subunit (UQCRFS1), complex I subunits (NDUFS1 and NDUFS3), complex V (ATP5A) and were reduced in dFARS2KO mutant as well as dFARS2 RNAi larvae (Figure 5A). However, the level of complex II subunit (SDHA) was not altered in both dFARS2KO mutant and RNAi larvae (Figure 5A). The levels of NDUFS1, NDUFS3, SDHA, UQCRFS1, CO3 and ATP5A were 40.6%, 19.9%, 103%, 30.2%, 38.7% and 71.4% in the dFARS2KO larvae, 31.1%, 23%, 98%, 29%, 25.8% and 86.6% in the dFARS2 knockdown larvae, relative to the mean values measured in the control larvae, respectively (Figure 5B).
Figure 5.

Western blotting analysis of mitochondrial proteins. (A) Ten micrograms of total proteins from various larvae were electrophoresed through a denaturing polyacrylamide gel, electroblotted and hybridized with antibodies for 6 subunits of OXPHOS (mtDNA-encoded CO3, complex IV, nuclear-encoded UQRCFS1, complex III; NDUFS3 and NDUFS1, complex I; ATP5A, complex V; SDHA, complex II), and Porin as a loading control, respectively. (B) Quantification of relative mitochondrial protein levels. Average content of each polypeptide was normalized to the average content of Porin in each genotype. n = 3. Calculations were based on three independent experiments. Graph details and symbols are explained in the legend to Figure 4.
Defective assembly of OXPHOS complexes
We then examined the assembly of the individual OXPHOS complexes. Mitochondria were isolated from the second instar larvae and analyzed by blue native polyacrylamide gel electrophoresis (BN-PAGE) and Western blot. The levels of assembled OXPHOS complexes I and V were obviously decreased in both dFARS2KO mutant and dFARS2 RNAi larvae (Figure 6A). Consistent with the BN-PAGE results, Western blot analyses revealed that depletion of dFARS2 caused substantial reduction in OXPHOS complexes I, III, IV and V components, but not in complexes II components (Figure 6B). The levels of complex I, II, III, IV and V were 11.7%, 103.3%, 5.4%, 17.6% and 36.9% in the dFARS2KO larvae, 10.8%, 95%, 3.8%, 21.8% and 42% in the dFARS2 knockdown larvae, relative to the mean values measured in the control larvae, respectively (Figure 6C).
Figure 6.

Defective assembly of OXPHOS complexes. (A) BN-PAGE analysis of mitochondrial respiratory chain complexes in mitochondrial protein extracts from larvae with different genotypes. Left: WT and dFARS2KO larvae; Right: control and Da-Gal4 driven dFARS2 knockdown second instar larvae. Positions of specific complexes are indicated. (B) Western blot analysis of mitochondrial proteins after BN-PAGE. Mitochondria extracted from various larvae were solubilized with 20% Triton X-100, electroblotted and hybridized with antibodies specific for subunits of OXPHOS, respectively, and with Porin as a loading control. Asterisk indicates unknown complexes. (C) Quantification of the levels of complexes I, II, III, IV and V1 in various larvae. The calculations were based on three independent determinations. Graph details and symbols are explained in the legend to Figure 4.
Altered activities of OXPHOS complexes
OXPHOS assembly defects can affect respiratory chain activity. Thus, we tested the effect of dFARS2 depletion on the oxidative phosphorylation using established biochemical assays to measure the activities of individual respiratory complexes. Complex I activity was measured as the rate of NADH oxidation in the presence of ubiquinone. The nucleus-encoded Complex II activity was determined using dichlorophenolindophenol (DCPIP) as an artificial electron acceptor and succinate as the substrate. Complex III activity was measured by the rate of cytochrome c reduction using dubiquinol-2 as an electron donor. Complex IV activity was quantified by the rate of cytochrome c oxidation. Complex V activity was determined as the rate of NADH oxidation using phosphoenolpyruvate as an electron acceptor. Our results showed that the enzyme activities of OXPHOS complexes I, III, IV and V were reduced in dFARS2KO mutant and dFARS2 RNAi larvae, whereas the complex II activity was unaffected (Figure 7A and B). For comparison, the levels of mitochondrial complex activity were normalized to the average content of citrate synthase (CS).The mean values of Complex I, II, III, IV and V activities in dFARS2KO mutant were 25.6%, 101.8%, 40.4%, 68.8% and 65.7%, relative to the mean values measured in control larvae, respectively (Figure 7A). Similarly, the mean values of complexes I, II, III, IV and V activities were 45.0%, 92.6%, 32.4%, 41.5% and 49.5% in dFARS2 RNAi larvae, compared to those in control larvae, respectively (Figure 7B).
Figure 7.

Enzymatic activities of respiratory chain complexes. The activities of respiratory chain complexes were investigated by enzymatic assay on complexes I, II, III, IV and V in mitochondria isolated from WT and dFARS2KO second instar larvae (A), and control and Da-Gal4 driven dFARS2 knockdown second instar larvae (B). The calculations were based on three independent experiments. Graph details and symbols are explained in the legend to Figure 4.
Characterization of two human FARS2 pathogenic variants in the dFARS2 mutant flies
To further utilize the Drosophila dFARS2 knockout mutant model and determine the functional consequence of disease variants identified in human patients, we performed rescue experiments through expression of either wild type or mutated human FARS2 into dFARS2KO mutant flies.
We chose two human FARS2 variants, p.G309S and p.D142Y, for this analysis. p.G309S has been linked with the early-onset epileptic mitochondrial encephalopathy, whereas p.D142Y is associated with the less severe spastic paraplegia (27,29). We first generated transgenic flies with UAS-human wild type FARS2 and FARS2 bearing p.G309S or p.D142Y variant. To ensure the same expression levels, the constructs were subcloned into the same vector and integrated into the same genomic landing site in the Drosophila genome. When ubiquitously expressed under the control of Da-Gal4, wild type and two mutated versions of human FARS2 were able to partially rescue the larval growth defects and led to the eclosion of adult flies (Figure 8A). We noticed that while there was a slightly delay during pupal developmental for dFARS2KO mutants expressing human wild type FARS2, the developmental delay was much severer in dFARS2KO mutants carrying p.G309S or p.D142Y variant (Figure 8B). In order to provide further evidence for the disease-causing nature of the variants, we also tested for their seizure susceptibility in those surviving adult flies carrying wild type or mutated FARS2 transgenes. dFARS2KO mutant flies expressing wild type FARS2 showed a low level of bang sensitivity comparable to wild type controls (Figure 8C and D, supplementary video 2). Notably, there was an induced seizure phenotype in dFARS2KO mutants upon the expression of FARS2 p.G309S variant (Figure 8C and D, supplementary video 2). In contrast, there was no obvious seizure phenotype in dFARS2KO mutants carrying p.D142Y variant of human FARS2 (Figure 8C and D, supplementary video 2). Given that p.D142Y variant of human FARS2 was reported in patients with spastic paraplegia, we then performed the climbing assay to test whether these flies display locomotion defects. Flies were placed into an empty vial and tapped down to the bottom of the vial, and the number of flies able to pass an 8-cm mark successfully within 10 s was recorded to generate a climbing index. Indeed, dFARS2KO mutant flies carrying p.D142Y variant of human FARS2 showed a reduced climbing ability compared to controls (Figure 8E). A slightly reduced climbing ability was also observed for dFARS2KO mutant flies with wild type FARS2 or p.G309S variant (Figure 8E). These results suggest that our fly models could separate the disease phenotypes associated with two distinct mutations. Moreover, the efficiency of aminoacylated mt-tRNAPhe in the dFARS2KO mutant larvae carrying wild type human FARS2 was comparable with that in wild type larvae (Figure 8F and G). Notably, dFARS2KO mutant larvae carrying p.G309S mutation reduced about 15%, while the levels of aminoacylation in dFARS2KO mutants carrying p.D142Y mutation was comparable with that in wild type larvae (Figure 8F and G). The steady levels of mitochondrial tRNAPhe and tRNALys were not changed among these larvae (Figure 8H). The in-gel activity analysis revealed that introducing p.G309S or p.D142Y variant into dFARS2KO mutant background caused a slight reduction of Complex I activity, but did not affect Complex II activity (Figure 8I). These data provide evidences that human FARS2 p.G309S mutation causes developmental delay and seizure and p.D142Y mutation is associated with spastic paraplegia.
Figure 8.

Functional analysis of two human FARS2 variants in dFARS2 mutants. (A) Images showing the larvae with different genotypes at 5 days AEL (left) and adult female flies at 15 days AEL (right). Scale bars represent 1 mm. (B) Graph showing pupariation curves for control, dFARS2KO larvae expressing human wild type FARS2, FARS2 p.G309S mutation or FARS2 p.D142Y mutation. n = 4. (C) Graph showing the percentage of female flies (control, KO + FARS2, KO + FARS2G309S or KO + FARS2D142Y) with BS paralytic phenotypes. n = 4. (D) Graph showing the recovery time of flies as described in (C) after BS paralysis. n = 7 for control; n = 8 for KO + FARS2; n = 17 for KO + FARS2G309S; n = 9 for KO + FARS2D142Y. (E) Graph showing the climbing index of flies as described in (C). n = 5. (F)In vivo aminoacylation of tRNAPhe assay in control and dFARS2KO third instar larvae expressing human wild type FARS2 (KO + FARS2), FARS2 p.G309S mutation (KO + FARS2G309S) or FARS2 p.D142Y mutation (KO + FARS2D142Y). (G) Quantification of aminoacylation level of tRNAPhe among larvae with different genotypes. (H) Northern blot analysis of tRNAPhe and tRNALys in various third instar larvae. (I) In-gel activity analysis from various third instar larvae. The left panel shows in gel activity of complex I, and the right panel shows in-gel activity of complex II. The calculations were based on 3–4 independent experiments. Graph details and symbols are explained in the legend to Figure 4.
DISCUSSION
In the present study, we have generated Drosophila dFARS2 knockout and knockdown models and analyzed disease related phenotypes. We found that dFARS2 mutations or ubiquitously knockdown of dFARS2 result in developmental delay and lethality at the second instar larval stages. Similar phenotypes have been reported in Drosophila SARS2 and MARS2 mutants (52,53). Furthermore, the lethality is also observed in mouse DARS2 knockout (54). These findings in model organisms are consistent with the absence of patients reported so far with an allele combination of complete loss of function of mitochondrial aminoacyl-tRNA synthase, as such mutations might be incompatible with life.
Neurological disorder is one major phenotype in patients with various mitochondrial aminoacyl-tRNA synthetase mutations (55,56). However, a proper animal model for studying these neuronal defects was not available. We specifically knocked down dFARS2 in the nervous system and observed that dFARS2 reduction leads to a developmental delay and induces seizure. These results clearly suggest that mitochondrial tRNA aminoacylation and protein translation are crucial for development as well as the maintenance of neuronal functions. The mitochondria of dFARS2 knockdown fly brains display abnormal morphology, which indicates their dysfunctions. The changes in mitochondrial morphology have previously been reported in flies depleted of SARS2 or SARS2-related gene (53,57). The phenotypes in these dFARS2 knockdown flies mirror the defects reported in patients with FARS2 mutations, indicating the potential use of these models in dissecting the molecular mechanisms of FARS2 deficiency (29). Interestingly, previous reported MARS2 knockout and SARS2 knockdown fly models have shown some features related to neurological defects (52,53). However, the seizure behaviors of MARS2 and SARS2 models were not further examined (52,53). It might be interesting to see whether seizure induction is a more general phenotype associated with ARS2 mutations in Drosophila in the future.
We have shown that mitochondrial tRNAPhe aminoacylation activity was severely reduced in dFARS2 mutant and knockdown larvae, indicating dFARS2 is essential for aminoacylation of tRNAPhe. Synthesis of cognate mitochondrial aminoacyl tRNAs by specific ARS2s is important for the fidelity in mitochondrial protein translation (8,29). In the absence of dFARS2, decreased charged tRNAPhe can impair mitochondrial protein synthesis and reduce the efficiency of protein translation. Our western blot analysis revealed that the levels of both mitochondria and nuclear encoded OXPHOS components for complex I, III, IV and V were decreased in dFARS2 deficiency larvae, indicating reduced protein synthesis and/or protein stability. Moreover, the ability of these proteins to assemble into the OXPHOS complexes was compromised in dFARS2 deficiency, as demonstrated by the Blue native gel assay. The defective assembly of OXPHOS complexes further indicates that balanced production of nuclear and mitochondrial OXPHOS polypeptides is important for respiratory complex assembly. dFARS2 deficiency specifically reduces the activity of OXPHOS complexes that contain mtDNA encoded subunits. However, the entirely nuclear encoded complex II activity was not affected. These differential effects on the OXPHOS complexes have been observed for mutations in several other mitochondrial genes. Interestingly, we found that the levels of a subset of mitochondrial mRNAs and tRNAs were increased in dFARS2 mutants. Moreover, the mitochondrial DNA levels were also upregulated. This could be explained by a compensatory response to mitochondrial dysfunction (58,59). However, the increase in the steady-state levels of these mt-tRNAs was not able to overcome the loss of dFARS2, as the OXPHOS complexes were not assembled properly and resulted in compromised mitochondria respiration. Together, these data demonstrate the biochemical characteristics observed in dFARS2 deficiency Drosophila larvae largely resemble the phenotypes in human cell lines carrying the mutations of FARS2 gene.
Moreover, an important finding of our study is that expression of human disease-causing variants in Drosophila dFARS2 mutants can partially recapitulate some features of the disease. Expression of human wild type FARS2 and FARS2 carrying the p.G309S or p.D142Y variant in the dFARS2 mutants could rescue the viability of the mutants. Compared with human wild type FARS2, expression of human FARS2 with p.G309S or p.D142Y variant in dFARS2 mutants leads to a severer developmental delay during the pupal stage. Furthermore, the adult dFARS2 mutant flies carrying human FARS2 with p.G309S variant display a seizure defect. In contrast, the adult dFARS2 mutant flies carrying human FARS2 with p.D142Y variant display a strong locomotion defect. These data together provide experimental evidence for the pathogenicity of the p.G309S and p.D142Y variants and also indicate the phenotypic difference between these two variants. Our results highlights the importance of the Drosophila in vivo model to verify the pathogenic effects of FARS2 variants with respect to the human disease.
In summary, our Drosophila dFARS2 knockout and knockdown models accurately recapitulate many phenotypic features of human disease caused by FARS2 mutations. These models will be valuable for identifying the underlying pathomechanism of ARS2 diseases and screening the suppressors of ARS2 mutations as well as drugs with a therapeutic potential.
DATA AVAILABILITY
The authors declare that [the/all other] data supporting the findings of this study are available within the article [and its supplementary information files].
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Bloomington Drosophila stock center, Vienna Drosophila Resource Center and Tsinghua fly center for fly stocks. We thank Drs Jun Ma and Feng He for their constructive suggestions.
Contributor Information
Wenlu Fan, Division of Human Reproduction and Developmental Genetics, Women's Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China; Institute of Genetics, Zhejiang University, 866 Yuhangtang Road, Hangzhou, Zhejiang 310058, China; Zhejiang Provincial Key Laboratory of Precision Diagnosis and Therapy for Major Gynecological Diseases, Women's Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310006, China.
Xiaoye Jin, Division of Human Reproduction and Developmental Genetics, Women's Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China; Institute of Genetics, Zhejiang University, 866 Yuhangtang Road, Hangzhou, Zhejiang 310058, China; Zhejiang Provincial Key Laboratory of Precision Diagnosis and Therapy for Major Gynecological Diseases, Women's Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310006, China.
Man Xu, Division of Human Reproduction and Developmental Genetics, Women's Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China; Institute of Genetics, Zhejiang University, 866 Yuhangtang Road, Hangzhou, Zhejiang 310058, China; Zhejiang Provincial Key Laboratory of Precision Diagnosis and Therapy for Major Gynecological Diseases, Women's Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310006, China.
Yongmei Xi, Division of Human Reproduction and Developmental Genetics, Women's Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China; Institute of Genetics, Zhejiang University, 866 Yuhangtang Road, Hangzhou, Zhejiang 310058, China.
Weiguo Lu, Department of Gynecologic Oncology, Women's Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310006, China; Cancer Center, Zhejiang University, Hangzhou, Zhejiang 310058, China.
Xiaohang Yang, Division of Human Reproduction and Developmental Genetics, Women's Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China; Institute of Genetics, Zhejiang University, 866 Yuhangtang Road, Hangzhou, Zhejiang 310058, China; Zhejiang Provincial Key Laboratory of Genetic and Developmental Disorders, Zhejiang University, Hangzhou, Zhejiang 310058, China.
Min-Xin Guan, Institute of Genetics, Zhejiang University, 866 Yuhangtang Road, Hangzhou, Zhejiang 310058, China; Zhejiang Provincial Key Laboratory of Genetic and Developmental Disorders, Zhejiang University, Hangzhou, Zhejiang 310058, China.
Wanzhong Ge, Division of Human Reproduction and Developmental Genetics, Women's Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China; Institute of Genetics, Zhejiang University, 866 Yuhangtang Road, Hangzhou, Zhejiang 310058, China; Zhejiang Provincial Key Laboratory of Precision Diagnosis and Therapy for Major Gynecological Diseases, Women's Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310006, China; Cancer Center, Zhejiang University, Hangzhou, Zhejiang 310058, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Program of China [2018YFC1003200 to W.G., 2021YFC2700902 to M.X.G.], and National Natural Science Foundation of China [31970668 to W.G., 82030028 to M.X.G.]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Florentz C., Sohm B., Tryoen-Toth P., Putz J., Sissler M.. Human mitochondrial tRNAs in health and disease. Cell Mol. Life Sci. 2003; 60:1356–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Suzuki T., Nagao A., Suzuki T.. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 2011; 45:299–329. [DOI] [PubMed] [Google Scholar]
- 3. Zheng J., Ji Y.C., Guan M.X.. Mitochondrial tRNA mutations associated with deafness. Mitochondrion. 2012; 12:406–413. [DOI] [PubMed] [Google Scholar]
- 4. Tyynismaa H., Schon E.A.. Mixing and matching mitochondrial aminoacyl synthetases and their tRNAs: a new way to treat respiratory chain disorders?. Embo Mol. Med. 2014; 6:155–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boczonadi V., Ricci G., Horvath R.. Mitochondrial DNA transcription and translation: clinical syndromes. Essays Biochem. 2018; 62:321–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wallace D.C. Mitochondrial genetic medicine. Nat. Genet. 2018; 50:1642–1649. [DOI] [PubMed] [Google Scholar]
- 7. Bullard J.N., Cai Y.C., Demeler B., Spremulli L.L.. Expression and characterization of a human mitochondrial phenylalanyl-tRNA synthetase. J. Mol. Biol. 1999; 288:567–577. [DOI] [PubMed] [Google Scholar]
- 8. Ibba M., Soll D.. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000; 69:617–650. [DOI] [PubMed] [Google Scholar]
- 9. Sissler M., Gonzalez-Serrano L.E., Westhof E.. Recent advances in mitochondrial aminoacyl-tRNA synthetases and disease. Trends Mol. Med. 2017; 23:693–708. [DOI] [PubMed] [Google Scholar]
- 10. Garesse R. Drosophila melanogaster mitochondrial-DNA - gene organization and evolutionary considerations. Genetics. 1988; 118:649–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Andrews R.M., Kubacka I., Chinnery P.F., Lightowlers R.N., Turnbull D.M., Howell N.. Reanalysis and revision of the cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999; 23:147–147. [DOI] [PubMed] [Google Scholar]
- 12. Wallace D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 2005; 39:359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meyer-Schuman R., Antonellis A.. Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Hum. Mol. Genet. 2017; 26:R114–R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gonzalez-Serrano L.E., Chihade J.W., Sissler M.. When a common biological role does not imply common disease outcomes: disparate pathology linked to human mitochondrial aminoacyl-tRNA synthetases. J. Biol. Chem. 2019; 294:5309–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pierce S.B., Gersak K., Michaelson-Cohen R., Walsh T., Lee M.K., Malach D., Klevit R.E., King M.C., Levy-Lahad E.. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in perrault syndrome. Am. J. Hum. Genet. 2013; 92:614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Diodato D., Melchionda L., Haack T.B., Dallabona C., Baruffini E., Donnini C., Granata T., Ragona F., Balestri P., Margollicci M.et al.. VARS2 and TARS2 mutations in patients with mitochondrial encephalomyopathies. Hum. Mutat. 2014; 35:983–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coughlin C.R., Scharer G.H., Friederich M.W., Yu H.C., Geiger E.A., Creadon-Swindell G., Collins A.E., Vanlander A.V., Van Coster R., Powell C.A.et al.. Mutations in the mitochondrial cysteinyl-tRNA synthase gene, CARS2, lead to a severe epileptic encephalopathy and complex movement disorder. J. Med. Genet. 2015; 52:532–540. [DOI] [PubMed] [Google Scholar]
- 18. McMillan H.J., Humphreys P., Smith A., Schwartzentruber J., Chakraborty P., Bulman D.E., Beaulieu C.L., Majewski J., Boycott K.M., Geraghty M.T.et al.. Congenital visual impairment and progressive microcephaly due to lysyl-transfer ribonucleic acid (RNA) Synthetase (KARS) Mutations: the expanding phenotype of aminoacyl-transfer RNA synthetase mutations in human disease. J. Child. Neurol. 2015; 30:1037–1043. [DOI] [PubMed] [Google Scholar]
- 19. Simon M., Richard E.M., Wang X.J., Shahzad M., Huang V.H., Qaiser T.A., Potluri P., Mahl S.E., Davila A., Nazli S.et al.. Mutations of human NARS2, encoding the mitochondrial asparaginyl-trna synthetase, cause nonsyndromic deafness and leigh syndrome. Plos. Genet. 2015; 11:e1005097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang P.P., Jin X.F., Peng Y.Y., Wang M., Liu H., Liu X.L., Zhang Z.J., Ji Y.C., Zhang J.J., Liang M.et al.. The exome sequencing identified the mutation in YARS2 encoding the mitochondrial tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic manifestation of Leber's hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum. Mol. Genet. 2016; 25:584–596. [DOI] [PubMed] [Google Scholar]
- 21. Finsteree J., Zarrouk-Mahjoub S.. Phenotypic spectrum of DARS2 mutations. J. Neurol. Sci. 2017; 376:117–118. [DOI] [PubMed] [Google Scholar]
- 22. Fan W.L., Zheng J., Kong W.Z., Cui L.M., Aishanjiang M., Yi Q.Z., Wang M., Cang X.H., Tang X.W., Chen Y.et al.. Contribution of a mitochondrial tyrosyl-tRNA synthetase mutation to the phenotypic expression of the deafness-associated tRNASer(UCN) 7511A >G mutation. J. Biol. Chem. 2019; 294:19292–19305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Elo J.M., Yadavalli S.S., Euro L., Isohanni P., Gotz A., Carroll C.J., Valanne L., Alkuraya F.S., Uusimaa J., Paetau A.et al.. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum. Mol. Genet. 2012; 21:4521–4529. [DOI] [PubMed] [Google Scholar]
- 24. Shamseldin H.E., Alshammari M., Al-Sheddi T., Salih M.A., Alkhalidi H., Kentab A., Repetto G.M., Hashem M., Alkuraya F.S.. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 2012; 49:234–241. [DOI] [PubMed] [Google Scholar]
- 25. Almalki A., Alston C.L., Parker A., Simonic I., Mehta S.G., He L.P., Reza M., Oliveira J.M.A., Lightowlers R.N., McFarland R.et al.. Mutation of the human mitochondrial phenylalanine-tRNA synthetase causes infantile-onset epilepsy and cytochrome c oxidase deficiency. BBA-Mol. Basis. Dis. 2014; 1842:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang Y., Liu W., Fang Z.P., Shi J., Che F.Y., He C.X., Yao L.B., Wang E.D., Wu Y.M.. A newly identified missense mutation in FARS2 causes autosomal-recessive spastic paraplegia. Hum. Mutat. 2016; 37:165–169. [DOI] [PubMed] [Google Scholar]
- 27. Cho J.S., Kim S.H., Kim H.Y., Chung T., Kim D., Jang S., Lee S.B., Yoo S.K., Shin J., Kim J.I.et al.. FARS2 mutation and epilepsy: possible link with early-onset epileptic encephalopathy. Epilepsy. Res. 2017; 129:118–124. [DOI] [PubMed] [Google Scholar]
- 28. Vantroys E., Larson A., Friederich M., Knight K., Swanson M.A., Powell C.A., Smet J., Vergult S., De Paepe B., Seneca S.et al.. New insights into the phenotype of FARS2 deficiency. Mol. Genet. Metab. 2017; 122:172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Almannai M., Wang J.L., Dai H.Z., El-Hattab A.W., Faqeih E.A., Saleh M.A., Al Asmari A., Alwadei A.H., Aljadhai Y.I., AlHashem A.et al.. FARS2 deficiency; new cases, review of clinical, biochemical, and molecular spectra, and variants interpretation based on structural, functional, and evolutionary significance. Mol. Genet. Metab. 2018; 125:281–291. [DOI] [PubMed] [Google Scholar]
- 30. Sahai S.K., Steiner R.E., Au M.G., Graham J.M., Salamon N., Ibba M., Pierson T.M.. FARS2 mutations presenting with pure spastic paraplegia and lesions of the dentate nuclei. Ann. Clin. Transl. Neur. 2018; 5:1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wangler M.F., Yamamoto S., Chao H.T., Posey J.E., Westerfield M., Postlethwait J., Hieter P., Boycott K.M., Campeau P.M., Bellen H.J.et al.. Model organisms facilitate rare disease diagnosis and therapeutic research. Genetics. 2017; 207:9–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gratz S.J., Cummings A.M., Nguyen J.N., Hamm D.C., Donohue L.K., Harrison M.M., Wildonger J., O’Connor-Giles K.M.. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 2013; 194:1029–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Housden B.E., Lin S.L., Perrimon N.. Cas9-based genome editing in Drosophila. Method Enzymol. 2014; 546:415–439. [DOI] [PubMed] [Google Scholar]
- 34. Yu Z.S., Ren M.D., Wang Z.X., Zhang B., Rong Y.K.S., Jiao R.J., Gao G.J.. Highly efficient genome modifications mediated by CRISPR/Cas9 in Drosophila. Genetics. 2013; 195:289–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen D.N., Zhang Z.M., Chen C., Yao S.H., Yang Q.X., Li F., He X., Ai C., Wang M., Guan M.X.. Deletion of Gtpbp3 in zebrafish revealed the hypertrophic cardiomyopathy manifested by aberrant mitochondrial tRNA metabolism. Nucleic. Acids. Res. 2019; 47:5341–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiao Y., Wang M., He Q.F., Xu L., Zhang Q.H., Meng F.L., Jia Z.D., Zhang F.G., Wang H.B., Guan M.X.. Asymmetrical effects of deafness-associated mitochondrial DNA 7516delA mutation on the processing of RNAs in the H-strand and L-strand polycistronic transcripts. Nucleic. Acids. Res. 2020; 48:11113–11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou M., Xue L., Chen Y.R., Li H.Y., He Q.F., Wang B.B., Meng F.L., Wang M., Guan M.X.. A hypertension-associated mitochondrial DNA mutation introduces an m1G37 modification into tRNAMet, altering its structure and function. J. Biol. Chem. 2018; 293:1425–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Enriquez J.A., Attardi G.. Analysis of aminoacylation of human mitochondrial tRNAs. Methods Enzymol. 1996; 264:183–196. [DOI] [PubMed] [Google Scholar]
- 39. Baggio F., Bratic A., Mourier A., Kauppila T.E., Tain L.S., Kukat C., Habermann B., Partridge L., Larsson N.G.. Drosophila melanogaster LRPPRC2 is involved in coordination of mitochondrial translation. Nucleic. Acids. Res. 2014; 42:13920–13938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jha P., Wang X., Auwerx J.. Analysis of mitochondrial respiratory chain supercomplexes using blue native polyacrylamide gel electrophoresis (BN-PAGE). Curr. Protoc. Mouse Biol. 2016; 6:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ji Y.C., Zhang J.J., Lu Y.Y., Yi Q.Z., Chen M.Q., Xie S.P., Mao X.T., Xiao Y., Meng F.L., Zhang M.L.et al.. Complex I mutations synergize to worsen the phenotypic expression of Leber's hereditary optic neuropathy. J. Biol. Chem. 2020; 295:13224–13238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Meng F.L., Zhou M., Xiao Y., Mao X.T., Zheng J., Lin J.X., Lin T.X., Ye Z.Z., Cang X.H., Fu Y.et al.. A deafness-associated tRNA mutation caused pleiotropic effects on the m1G37 modification, processing, stability and aminoacylation of tRNAIIe and mitochondrial translation. Nucleic. Acids. Res. 2021; 49:1075–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jin X.F., Zhang Z.M., Nie Z.P., Wang C.H., Meng F.L., Yi Q.Z., Chen M.Q., Sun J.J., Zou J., Jiang P.P.et al.. An animal model for mitochondrial tyrosyl-tRNA synthetase deficiency reveals links between oxidative phosphorylation and retinal function. J. Biol. Chem. 2021; 296:100437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bourgeron T., Rustin P., Chretien D., Birch-Machin M., Bourgeois M., Viegas-Pequignot E., Munnich A., Rotig A.. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat. Genet. 1995; 11:144–149. [DOI] [PubMed] [Google Scholar]
- 45. Thorburn D.R., Chow C.W., Kirby D.M.. Respiratory chain enzyme analysis in muscle and liver. Mitochondrion. 2004; 4:363–375. [DOI] [PubMed] [Google Scholar]
- 46. Zhang Q.H., He X., Yao S.A., Lin T.X., Zhang L.W., Chen D.N., Chen C., Yang Q.X., Li F., Zhu Y.M.et al.. Ablation of Mto1 in zebrafish exhibited hypertrophic cardiomyopathy manifested by mitochondrion RNA maturation deficiency. Nucleic. Acids. Res. 2021; 49:4689–4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Parker L., Padilla M., Du Y., Dong K., Tanouye M.A.. Drosophila as a model for epilepsy: bss is a gain-of-function mutation in the para sodium channel gene that leads to seizures. Genetics. 2011; 187:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kroll J.R., Wong K.G., Siddiqui F.M., Tanouye M.A.. Disruption of endocytosis with the dynamin mutant shibirets1 suppresses seizures in Drosophila. Genetics. 2015; 201:1087–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Claros M.G., Vincens P.. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996; 241:779–786. [DOI] [PubMed] [Google Scholar]
- 50. Song J., Tanouye M.A.. From bench to drug: human seizure modeling using Drosophila. Prog. Neurobiol. 2008; 84:182–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Parker L., Howlett I.C., Rusan Z.M., Tanouye M.A.. Seizure and epilepsy: studies of seizure disorders in Drosophila. Int. Rev. Neurobiol. 2011; 99:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bayat V., Thiffault I., Jaiswal M., Tetreault M., Donti T., Sasarman F., Bernard G., Demers-Lamarche J., Dicaire M.J., Mathieu J.et al.. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 2012; 10:e1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guitart T., Picchioni D., Pineyro D., Ribas de Pouplana L.. Human mitochondrial disease-like symptoms caused by a reduced tRNA aminoacylation activity in flies. Nucleic. Acids. Res. 2013; 41:6595–6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dogan S.A., Pujol C., Maiti P., Kukat A., Wang S., Hermans S., Senft K., Wibom R., Rugarli E.I., Trifunovic A.. Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 2014; 19:458–469. [DOI] [PubMed] [Google Scholar]
- 55. Ognjenovic J., Simonovic M.. Human aminoacyl-tRNA synthetases in diseases of the nervous system. RNA Biol. 2018; 15:623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schaffer A.E., Pinkard O., Coller J.M.. tRNA metabolism and neurodevelopmental disorders. Annu. Rev. Genomics Hum. Genet. 2019; 20:359–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Guitart T., Bernardo T.L., Sagales J., Stratmann T., Bernues J., de Pouplana L.R.. New aminoacyl-tRNA synthetase-like protein in insecta with an essential mitochondrial function. J. Biol. Chem. 2010; 285:38157–38166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Meiklejohn C.D., Holmbeck M.A., Siddiq M.A., Abt D.N., Rand D.M., Montooth K.L.. An incompatibility between a mitochondrial tRNA and its nuclear-encoded tRNA synthetase compromises development and fitness in Drosophila. PLos Genet. 2013; 9:e1003238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wredenberg A., Lagouge M., Bratic A., Metodiev M.D., Spahr H., Mourier A., Freyer C., Ruzzenente B., Tain L., Gronke S.et al.. MTERF3 regulates mitochondrial ribosome biogenesis in invertebrates and mammals. PLoS Genet. 2013; 9:e1003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that [the/all other] data supporting the findings of this study are available within the article [and its supplementary information files].