


Abstract
Cellular and virus-coded long non-coding (lnc) RNAs support multiple roles related to biological and pathological processes. Several lncRNAs sequester their 3′ termini to evade cellular degradation machinery, thereby supporting disease progression. An intramolecular triplex involving the lncRNA 3′ terminus, the element for nuclear expression (ENE), stabilizes RNA transcripts and promotes persistent function. Therefore, such ENE triplexes, as presented here in Kaposi's sarcoma-associated herpesvirus (KSHV) polyadenylated nuclear (PAN) lncRNA, represent targets for therapeutic development. Towards identifying novel ligands targeting the PAN ENE triplex, we screened a library of immobilized small molecules and identified several triplex-binding chemotypes, the tightest of which exhibits micromolar binding affinity. Combined biophysical, biochemical, and computational strategies localized ligand binding to a platform created near a dinucleotide bulge at the base of the triplex. Crystal structures of apo (3.3 Å) and ligand-soaked (2.5 Å) ENE triplexes, which include a stabilizing basal duplex, indicate significant local structural rearrangements within this dinucleotide bulge. MD simulations and a modified nucleoside analog interference technique corroborate the role of the bulge and the base of the triplex in ligand binding. Together with recently discovered small molecules that reduce nuclear MALAT1 lncRNA levels by engaging its ENE triplex, our data supports the potential of targeting RNA triplexes with small molecules.
INTRODUCTION
Long non-coding (lnc) RNAs comprise a class of transcripts that participate in normal development by regulating gene expression, and whose expression often has consequences for disease progression. Functional roles associated with lncRNAs include (i) molecular signals as markers of biological events such as stress, (ii) decoys that ‘sponge’ protein factors and chromatin modifiers, (iii) molecular guides that localize ribonucleoprotein complexes to specific chromatin targets, inducing changes in gene expression and (iv) assembling protein complexes that impact transcriptional activation or repression (1). As hallmarks of aging, lnc-RNAs are implicated in epigenetic regulation (2), proteostasis (3), intracellular communication (4), cell proliferation (5) and telomere stability (6), while their association with cancer is exemplified by MIR100HG, which promotes cell proliferation in triple negative breast cancer (7).
In consideration of their roles as promoters of gene expression, replication, assembly, virion release from infected cells and cellular transformation (8), virus-coded lncRNAs are also receiving increased attention. Kaposi's sarcoma herpesvirus (KSHV) is the etiological agent of Kaposi's sarcoma (KS), a common neoplasm in HIV-infected individuals and a major source of morbidity and mortality (9). Production and release of progeny virions from KSHV-infected cells is associated with a ∼1.1-kb viral lncRNA, designated polyadenylated nuclear (PAN) RNA, which accounts for as much as 80% of the polyadenylated RNA in a lytically-infected cell (10). Using SHAPE-mutational profiling (SHAPE-MaP), we recently probed KSHV PAN structure in its nuclear, cytoplasmic or viral environments or following cell/virion lysis and removal of proteins, therein characterizing the cis-acting Mta responsive element (MRE) and ENE (11). Although some protein binding sites were selectively localized, others were occupied in all three biological contexts. Such structural characterization of a viral lncRNA and interactions with its protein partners in discrete biological contexts, provides a broad framework for understanding the roles of PAN RNA in KSHV infection. PAN nuclear localization, abundance and stability derive from a cis-acting element, the element for nuclear expression, or ENE, that adopts a triple helix conformation by sequestering its poly(A) tail, rendering it refractory to exonuclease degradation and promoting its nuclear accumulation (12). Genetic knockdown of KSHV PAN alters nuclear export and thereby affects the availability of viral mRNAs in the lytic phase (13); however, deletion of the ENE does not preclude formation of infectious virus (14).
We (15–18) and others (19–21) have investigated the notion of targeting structured motifs of regulatory RNAs with small molecules. By exploiting RNA secondary or tertiary structure, small molecules offer the opportunity of targeting bulges, loops, junctions, pseudoknots, or higher-order structures, thereby sequestering unique RNA folds. With respect to the KSHV PAN-encoded ENE, a first step in this direction would be determining whether structural features of this triplex might constitute unique ligand binding sites. Precedent for this strategy has been established by our previous documentation of ligands that specifically recognized and down-regulated expression of the analogous ENE of metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) lncRNA (16), yet failed to perturb the homologous counterpart elements of both PAN and nuclear paraspeckle assembly transcript 1 (NEAT1) lncRNAs, the latter being frequently overexpressed in tumors (22). Herein, through a combination of biochemical analyses, nucleoside analog mutagenesis, molecular simulations and X-ray crystallography, we report a small molecule ligand that binds within a pocket formed be the PAN ENE triplex and a dinucleotide bulge at the base of the triplex.
MATERIALS AND METHODS
RNA preparation
Synthetic RNAs were either purchased from GE Dharmacon (Lafayette, CO), or generated by in vitro transcription and purified as described elsewhere (23,24). A detailed description is provided in Supplementary Table S1.
Small molecule microarray (SMM) screening
SMM screening plates were prepared as described (25). Briefly, γ-aminopropyl silane (GAPS) microscope slides (Corning) were functionalized with a short Fmoc-protected amino polyethylene glycol spacer. After piperidine deprotection, 1,6-diisocyanatohexane was coupled to the surface by urea bond formation to provide functionalized isocyanate-coated microarray slides that react with primary and secondary amines and primary alcohols to create immobilized small molecule libraries. Slides were exposed to pyridine vapor to facilitate covalent attachment, then incubated with a 1:20 polyethylene glycol:DMF (v/v) solution to quench unreacted isocyanate surface. Small molecules used in this library screen are referenced elsewhere (26).
PAN ENE core hairpin, labeled at the 5′ terminus with Cy5, and unlabeled r(A)9 were deprotected, dried according to the manufacturer's instructions and dissolved in a buffer of 50 mM sodium cacodylate, pH 6.5, 50 mM KCl, 1 mM MgCl2 and 0.1 mM EDTA (RNA Folding buffer). PAN ENE core RNA (5 μM) was mixed with an equimolar amount of r(A)9 RNA in the same buffer, heated to 95°C for 3 min, then snap-cooled on ice. Samples were assessed for triplex formation by non-denaturing polyacrylamide gel electrophoresis, including 5 mM MgCl2 in the gel and running buffer (Supplementary Figure S1).
Microarray slides were incubated with the ENE core hairpin or the triplex complex (PAN ENE core + r(A)9) (Figure 1) at a concentration of 5 μM (500 μl volume) for 2 h in the dark, then washed three times in a buffer of 12 mM NaH2PO4, pH 7.4, 137 mM NaCl, 3 mM KCl, 0.01% (v/v) Tween-20, and once with H2O. Slides were dried by centrifugation and immediately imaged for fluorescence (650 nm excitation, 670 nm emission) on a GenePix 4000a array scanner (Molecular Devices) and with a resolution of 5 or 10 μm. Scanned images were aligned with the GenePix Array List (GAL) file corresponding to the appropriate array to identify individual features. From the GPR file, JMP 9.0 (SAS) was utilized to generate the mean (μ) and standard deviation (σ) for control (DMSO-printed) spots. For each compound, duplicate spots were averaged, and a coefficient of variation (CV) was calculated. A composite Z-score was generated for each compound using the following definition:
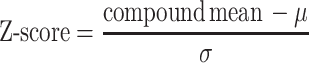 |
Figure 1.

SMM screen & compound identification. (A) Secondary structures for the ENE•r(A)9 triplex and ENE core duplex used in SMM screening. The cartoon depicts a small molecule microarray chip, upon which a library of small molecules was immobilized. Cy5-labeled RNA is passed over the microarray, and interacting ligands are identified by measuring the fluorescence signal. (B) SMM spots shown for a single small molecule performed in duplicate (i & ii) for buffer control (top lane), ENE core duplex (middle lane) and ENE•r(A)9 triplex (bottom lane). The fluorescence signal observed indicates an interaction between the RNA and the small molecule, which in this example is specific to the ENE•r(A)9 triplex. (C) Chemical structures of seven chemotypes identified by SMM screening.
Hits were determined using the following criteria: a) CV for duplicate spots of a compound <100, (b) Average Z-score for a compound >3, and (c) [(Z-score ENE•r(A)9 incubated Array) – (Z-score PAN ENE (control) Array)]/Z-score PAN ENE (control) Array) > 3. Hits were also validated by visual inspection of array images. Candidate ligands satisfying these hit criteria for the PAN ENE triplex were re-purchased from the original suppliers (Supplementary Tables S2a and S2b).
Size exclusion chromatography (SEC)
RNAs were purified by SEC as previously described (23). For experiments involving ENE triplexes, these were mixed at a 1:1.5 ratio of the ENE and its r(A)9 counterpart, respectively. All RNAs were prepared in a buffer of 1 mM MgCl2, 20 mM HEPES–KOH, pH 6.9 (Folding buffer), followed by incubation at 95°C for 3 min, snap-cooling on ice for 10 min and equilibration at room temperature for at least 1 h. Folded RNAs were fractionated through a Superdex 75 column (GE Healthcare, 24-ml bed volume) in Folding buffer, monitoring A260 of the eluate. Given their distinct elution patterns, the two-component RNA triplex was readily separated from the excess of r(A)9 or ggc(A)9.
Ribonuclease R degradation assay
Samples were prepared and run similar to as previously described (23). Briefly, RNA master mix was prepared in 1 mM MgCl2, 75 mM NaCl, and 75 mM KCl and 20 mM HEPES, pH 6.9. The sample was heated to 95°C for 3 min, snap-cooled on ice for 10 min, then incubated at room temperature for 30–60 min. After incubation, 10 μl of RNA was dispensed into each reaction tube to obtain 0.5 μg of RNA per reaction. RNase R stock solution was prepared in the same buffer as the RNA sample. To one RNA sample, 10 μl of RNase R stock solution was added to achieve a final reaction containing 3 units of enzyme per 0.5 μg of RNA. To the control sample, 10 μl of buffer was added. Next, the samples were incubated at 37°C for 5 h followed by analysis on 6% denaturing PAGE stained with ethidium bromide.
Circular dichroism (CD)
RNA was prepared at a concentration of 0.5 mg/ml in buffer of 5 mM sodium cacodylate, 50 mM KCl, and 1 mM MgCl2, pH 6.5. Triplex RNA complexes were formed by annealing at 95°C for 3 min, snap-cooling on ice for 10 min and incubating at room temperature for 1h. CD spectra were collected at room temperature on a Jasco J720 instrument with a 1 cm pathlength, using a wavelength scan from 210 to 320 nm with a resolution of 1 nm and data sampling every 2 s. CD data represent an average of five scans, baseline-subtracted, normalized and corrected to units of molar ellipticity. Thermal melting studies were performed on 0.2 mg/ml pre-formed RNA triplex in 1× PBS, in either 10 μM MgCl2 or 1 mM MgCl2, varying the temperature from 20 to 95°C with 0.1°C steps at a rate of 1°C/min. Similar changes in Tm due to ligand binding were obtained for melting experiments in 10 μM MgCl2 and 1 mM MgCl2.
Microscale thermophoresis (MST)
MST experiments were performed using a Monolith NT.115 instrument (NanoTemper Technologies, Munich, Germany), collecting each dataset in triplicate. Cy5-labeled RNA was dissolved in nuclease-free water and diluted to the required concentration in assay buffer. ENE triplex variants, compound screening and compound titration experiments were performed as follows:
ENE+r(A) 9: A 2-fold serial dilution of the unlabeled ENE core hairpin duplex was prepared in 5 mM sodium cacodylate, 50 mM KCl, 1 mM MgCl2, 0.002% (v/v) Tween20, pH 6.5 (MST buffer). 20 nM 5′-Cy5 labeled r(A)9 was added, with the final ENE concentration ranging from 25 μM to 0.048 nM. Samples were annealed at 95°C for 3 min, snap-cooled on ice for 10 min and incubated at room temperature for 1 h in the dark. Following incubation, samples were added to premium coated capillaries (NanoTemper Technologies) and MST data collected at 40% LED and 40% MST power at room temperature. Results were analyzed by MO Affinity software provided by the supplier and binding plots prepared using Prism (GraphPad). A single-site model was used to fit the binding curves and to determine the dissociation constant (Kd).
GC ENE+ggc(A) 9: A 2-fold serial dilution of the unlabeled GCENE was prepared in MST buffer with 10 nM 5′-Cy5 labeled ggc(A)9 with the final concentration of GCENE ranging from 2 μM to 0.001 nM. Experiments with GCENEΔU+ggc(A)9 were performed similarly to those performed with GCENE. Sample processing and data analysis was performed as described above. For MST studies on the A4N-, A7N- and, A10N-substituted oligos, 2-fold serial dilution of the unlabeled analog was prepared generating a concentration range from 2 μM to 0.001 nM. Experiments were prepared with 10 nM 5′-Cy5 labeled GCENE.
Compound titration with GCPAN triplex: A 2-fold serial dilution of compound 15 and 3-fold serial dilution of compounds 8, 13, 18, 19, 20, 24, 25 and compound 15 analog 15-A1 were prepared in DMSO. 19 μl of 25 nM GCPAN triplex was added to 1 μl of serially diluted compounds, with the final concentration ranging from 2.5 mM to 0.08 μM. Samples were incubated for 30 min at room temperature prior to experimentation. Sample processing and data analysis was performed as described above.
Fab-BL3-6 purification
Fab-BL3-6 purification followed the protocol by Ye et al. (27). Colonies from a freshly transformed expression plasmid were inoculated into a 2XYT/Amp100 broth starter culture and incubated at 37°C. Following overnight incubation, this was transferred into fresh 2XYT/Amp100 medium and incubated at 30°C for 24 hours. Cells were collected by centrifugation, gently resuspended in phosphate-depleted medium, and incubated for an additional 24 h at 30°C. Cells were harvested, lysed, treated with DNase I, and a clarified supernatant was obtained by centrifugation, followed by passing through a 0.45 μm filter. Clarified supernatant was loaded onto a HiTrap Protein A column (Cytiva) and eluted with 0.1N acetic acid. Pooled fractions were adjusted to neutral pH, loaded onto a HiTrap Protein G column (Cytiva) and eluted with 0.1 M glycine. Pooled fractions were adjusted to pH 5.5, loaded onto a HiTrap heparin column (Cytiva) and eluted with high salt. Fractions containing pure Fab were pooled, concentrated and buffer exchanged to 1× PBS using an Amicon 10K MWCO filtration device. Fab-BL3-6 was stored in aliquots at –80°C.
Crystallization and structure determination
For crystallization, an ENE variant was used wherein the 5′-GAAA-3′ tetraloop described by Mitton-Fry et al. (12) was replaced by Fab-BL3-6 binding motif (5′-GAAACAC-3′) (28). RNAs were fractionated by 15% denaturing polyacrylamide gel electrophoresis, followed by electroelution and ethanol precipitation. Purified GCENEFab was mixed with ggc(A)9 at a 1:1 molar ratio in RNA Folding buffer and annealed as described above. Following folding, the triplex was supplemented with 1.1 molar equivalents of purified Fab-BL3-6 and incubated on ice for 1hr (28). Fab-bound samples were concentrated to 5 mg/ml using a 3 kDa concentrator (Amicon) and complex formation confirmed by 8% native polyacrylamide gel electrophoresis. High-throughput crystallization screens were evaluated with a mosquito® crystal robot (TTP Labtech) using several commercially available screens. Large-size and best-diffracting crystals were obtained at 20°C via the hanging drop vapor diffusion method using equal volumes of the RNA and the reservoir solution (0.1 M Tris–HCl, pH 8.0, 0.2 M Li2SO4, 22% PEG 3350). Crystals appeared within 1 or 2 days and grew to full size within 2 weeks. RNA-Fab crystals were soaked in crystallization buffer containing 2 mM compound 15 in 5% DMSO for 1 min. Apo and soaked crystals were cryoprotected in 10% glycerol before flash freezing in liquid nitrogen and shipping to the Argonne National Laboratory for data collection.
Diffraction data sets were collected at 100K at the Advanced Photon Source on beamline 24-ID. All data sets were indexed, integrated and scaled in HKL3000 (29). Both the apo and soaked GCENEFab:ggc(A)9:Fab crystals were primitive monoclinic and belong to P1 space group. Cell dimensions for the apo crystals were a = 43.299 Å, b = 76.182 Å, c = 110.523 Å and α = 72.223°, β = 88.584°, and γ = 86.569° and for the soaked crystals were a = 43.128 Å, b = 76.26 Å, c = 110.664 Å and α = 71.89°, β = 88.59°, and γ = 86.579° (Supplementary Table 1). For crystals, there are two GCENE:ggc(A)9:Fab molecular complexes in the asymmetric unit. Structures were solved by molecular replacement in CCP4 suite (https://www.ccp4.ac.uk/), using the N-terminal variable domain and the C-terminal constant domain of Fab BL3-6 to search the initial density maps in Phaser (https://www.phenix-online.org/documentation/reference/phaser.html). All RNAs were subsequently built into the electron density maps using COOT (30,31). Models were refined to 3.3 Å resolution for the apo crystals and to 2.5 Å resolution for the soaked crystals in Refmac with translation/vibration and screw motions included in the final step (32). Structures were further refined using PHENIX (33). The model for the ligand was generated using the SMILES (Daylight Chemical Information Systems Inc. CA) and all figures were prepared in PyMOL (https://pymol.org/). Crystallographic coordinates and structure factors for both structures were deposited into the PDB (https://www.wwpdb.org) (apo: 6X5N; soaked: 6X5M).
Molecular docking
Ligand docking predictions were performed using AutoDock 4.2.6 (http://autodock.scripps.edu/). Preparations of the target and ligand were performed with GUI-based AutoDockTools and Python scripts (34). Briefly, validation and partial charge calculations (Gasteiger method) of the ligand (compound 15) and target (GCPAN triplex) were saved as PDBQT files. Next, the grid files (GPF) were generated that define the search space around the RNA targets. For all target conformations tested, we manually adjusted the cuboid grid space to be centered in the middle of the ENE dinucleotide bulge at the base of the triple helix and to cover most of the target RNA. The final preparation step involved creating the docking parameter files (DPF) that specify the target and ligand parameters and the parameters for the genetic algorithm (GA) used to search for the best docking pose. GA population of 150 with the maximum number of steps per run set at 27,000 was selected. 100 GA docking runs per RNA target conformation were performed, and the ranked cluster analysis of the predicted poses was included in the post-processing step. Post-processing of the single output/log file into PDBQT, PDB and several compact output statistics text files was performed with custom Unix shell scripts, and 3D visualization of the results was performed primarily with the aid of custom scripts run in the PyMOL Molecular Graphics System V 2.3 (Schrodinger LLC).
Molecular dynamics simulations
Explicit solvent molecular dynamics simulations (MD) from 1 up-to 4 μs duration were performed using the Amber 18 package and the force field ff99OL3 (leaprc.RAN.OL3 file) for RNA (35,36). Partial atomic charges for compound 15 were computed using the Amber antechamber module. The Amber LEaP module was used to combine the two complexes in the crystallographic asymmetric unit with TIP3P waters, and monovalent ions. In the simulations of ENE triplex-compound 15 complexes, the ligand structure files were added to the LEaP input. LEaP generated the topology and coordinate files.
Explicit solvent molecular particle mesh Ewald (PME) dynamics was employed for all simulations (37,38). Cuboid solvent boxes with the minimum solute-to-box boundary distance set to 12 Å were employed. Na+ ions were first added to neutralize the net solute charge, followed by Na+/Cl– ion pairs to simulate the net system salt concentration of 0.15 M for each simulation. Simulations employed 2 fs time steps and the SHAKE algorithm to constrain all hydrogen bonds in the system. The NPT simulations used Berendsen thermostat and Berendsen algorithm (39) to maintain the system temperature at 300K and pressure at 1.0 Pa. A 9 Å non-bonded interaction cut-off distance was selected, and explicit solvent periodic boundary conditions were employed.
A 12-step equilibration protocol was used that starts with energy minimization of the solvent (and the solute, i.e. RNA or RNA-ligand complex, restrained), followed by multiple short phases of heating, dynamics and energy minimizations, during which harmonic restraints applied to the solute were slowly lowered. The equilibration protocol ends with an unrestrained heating to the target temperature of 300 K, followed by unrestrained dynamics at this temperature for a total time of 2.0 ns. After equilibration, unrestrained production MD simulations were performed for up to 4000 ns. The DSSR server was used to analyze structural aspects of the MD snapshots(40). Figures were generated with the aid of custom scripts in PyMOL. Phosphorus backbone atoms of the 51-nt bimolecular ENE and the selected internal loop nucleotides (residues G3 through C9, and G25 through C33) were used as masks in alignments of MD snapshots.
RESULTS
Ligand identification by SMM screening
Small molecule microarray (SMM) screening (Figure 1A) provides a rapid, cost-effective and high-throughput strategy to interrogate large compound libraries for their ability to bind to structured RNA motifs (26). Drug-like molecules are covalently linked to isocyanate-functionalized glass slides, then incubated with a fluorescently-labeled ENE core triplex, ENE•r(A)9, or a control duplex core (ENE) hairpin. After removal of unbound RNA, slides were imaged with a fluorescence scanner. Statistical analyses reveal any significant increases in fluorescence on the arrays correlating to specific interactions between the target RNA and ligand. Using this strategy, screening a 22,807-compound library yielded multiple chemotypes that recognized the PAN ENE•r(A)9 triplex but not its duplex counterpart (Figure 1B), and a hit rate of ∼0.16%. Based on their commercial availability and purity, compounds 8, 13, 15, 18, 19, 20 and 25 (Figure 1C) were re-purchased for further evaluation. Although screening against the ENE hairpin duplex revealed several novel chemotypes, these were not further pursued.
Enhancing ENE triplex stability
Despite crystallographic evidence of a stable PAN ENE•r(A)9 triplex (12), our initial observations suggested a surprisingly weak interaction between its oligo-A component and the core ENE duplex (Figure 2A), which we anticipated would hamper subsequent biochemical characterization of ligand binding. Specifically, a weak interaction was demonstrated by microscale thermophoresis (MST), which detects a temperature-induced change in fluorescence of a target as a function of the concentration of a non-fluorescent ligand. Using this approach, a Kd of 884 ± 15 nM was derived for the interaction between the PAN ENE core duplex and r(A)9 (Figure 2B). Furthermore, CD experiments showed no difference between the ENE core duplex in the absence or presence of r(A)9 (Figure 2C). Finally, while a PAN ENE triplex could be reconstituted and confirmed using electrophoretic mobility shift assay (Supplementary Figure S1), this dissociated when examined by size exclusion chromatography (Figure 2D). Although Mitton-Fry et al., have reported a crystal structure for the KSHV PAN ENE•r(A)9 triplex, concentrations for crystallography are several orders of magnitude higher than those used here, which likely explains these seemingly contradictory observations.
Figure 2.

Formation and stability of PAN core triplex. (A) Secondary structures of a bimolecular PAN core ENE•r(A)9 triplex (left) and the unimolecular triplex construct (right). (B) MST binding plot for ENE core titrated into 5′-Cy5 labeled r(A)9 indicates a binding affinity (Kd) of 884 ± 15 nM. (C) CD spectra of bimolecular PAN triplex and its constituents: r(A)9 (black), ENE duplex (red), and a mixture of ENE + r(A)9 (blue). (D) Silver stain gel image of fractions eluted from SEC column of the mixture of ENE + r(A)9 indicates lack of persistent triplex association. The main SEC fraction was analyzed on denaturing polyacrylamide gel electrophoresis after purification, noting the absence of r(A)9 in this fraction. Control samples were run for the ENE duplex (left lane) and r(A)9 oligo (right lane). (E) Susceptibility of the unimolecular PAN core triplex to 3′–5′ exoribonuclease R (RNaseR) digestion, evaluated by denaturing PAGE.
To identify a robust ENE triplex construct amenable to biochemical characterization, we first turned our attention to evaluate constructs whose peripheral elements (Supplementary Figure S2), which have been shown to contribute to triplex stability in homologous RNA triplexes (23), were modified. We evaluated several unimolecular ENE triplex variants to assess triplex formation using thermal melt analysis. A construct containing the wild-type apical did not exhibit a robust triplex melting profile (Supplementary Figure S2A), in contrast to those previously reported for homologous RNA triplexes (23,41). The most promising unimolecular construct, designated PAN core triplex (Figure 2A), containing a shortened apical P2 helix, generated a more noticeable triplex melting transition (Supplementary Figure S2B). However, only mild protection against 3′-5′ exonucleolytic degradation by RNase R (Figure 2E) was observed, consistent with some degree of triplex formation and 3′ end protection.
In view of the data of Figure 2 and Supplementary Figure S2, we elected to introduce minor alterations at the base of the ENE in an effort to stabilize nucleotides of the triplex-forming third strand. We introduced additional nucleotides into the unimolecular ENE core triplex (Figure 3A) to create a 3-bp ‘GC clamp’, hereafter designated GCPAN triplex, anchoring the A-rich sequence to the lower duplex through the introduction of a new P0 helix. UV melt (Supplementary Figure S2) and RNase R protection analyses (Figure 3B) both indicate a highly stabilized triplex. The bimolecular version of this construct extends the r(A)9 oligonucleotide to include -G-G-C- at its 5′ terminus (ggc(A)9), and the ENE core duplex by the complement, -G-C-C-, at its 3′ terminus (GCENE duplex) (Figure 3A). Triplex reconstitution from the two components was confirmed by CD (Figure 3C), and stable complex formation by size exclusion chromatography (Figure 3D). MST analysis (Figure 3E) determined a Kd of 12 ± 3 nM for binding of the ggc(A)9 tail to the GCENE duplex, and a parallel EMSA analysis determined a Kd of 3 ± 0.3 nM (Figure 3F and Supplementary Figure S3). Minor differences in calculated Kd are common when comparing distinct biochemical and biophysical experiences. Nonetheless, the combined data of Figure 3 thus indicate that relatively minor changes in peripheral elements of the KSHV PAN triplex sequence impart a significant increase in stability, allowing us to perform detailed biochemical investigations.
Figure 3.

Formation and stability of modified GCPAN triplex. (A) Secondary structures of the unimolecular GCPAN triplex (left) and bimolecular GCENE•ggc(A)9 triplex (right). (B) Susceptibility of the unimolecular GCPAN triplex to 3′–5′ exoribonuclease R (RNaseR) digestion evaluated by denaturing PAGE. (C) CD spectra for ggc(A)9 (black), GCENE (red) and GCENE+ggc(A)9 mixture (blue). Distinct spectra for each sample indicate triple association in the GCENE + ggc(A)9 mixture. (D) SEC analysis indicates enhanced stability of the GCPAN triplex. The main SEC fraction was analyzed by denaturing polyacrylamide gel electrophoresis, indicating the presence of ggc(A)9 and GCENE duplex. Bands corresponding to GCENE duplex in the control sample (left lane) and main SEC fraction (right lane) are oversaturated, resulting in a shadow effect and an appearance as a light orange band. (E) MST binding plot for GCENE titrated into 5′-Cy5 labeled ggc(A)9 indicates high affinity binding of the two RNA strands. (F) EMSA analysis of binding of GCENE duplex titrated into 32P-radiolabeled ggc(A)9 also indicates high binding affinity for these two RNA strands.
Despite these observations, a possibility we could not rule out was that, while a stable interaction of the modified GCENE core with the ggc(A)9 tail was achieved, it did not directly demonstrate triplex formation. This was confirmed by X-ray crystallography, described in the next section.
Structural confirmation of the GCPAN triple helix by X-ray crystallography
To elucidate the mode of interaction between the GCENE and ggc(A)9 tail, we determined the crystal structure of the bimolecular triplex comprising a 39nt GCENEFab duplex, containing a 7 nt Fab-binding motif, and ggc(A)9 together with Fab-BL3-6 (Figure 4A). This recently-developed strategy exploits recombinant RNA binding proteins as high-affinity crystallization chaperones that recognize a motif engineered into the target RNA (27,28,42,43). Data obtained from several crystals diffracted to 3.3 Å (Supplementary Table S3). The structure was solved by molecular replacement using Phaser (44,45) using the Fab-BL3-6 structure (PDB 6B3K) as an initial model (28). The asymmetric unit contained two complexes, and models were built using electron density that is visible for all 39 nt of GCENEFab and the 12-nt ggc(A)9. Initially, a structural model was constructed for GCENEFab and ggc(A)9 for one of the complexes. This model was then inserted into the electron density of the second complex present in the asymmetric unit and, where required, necessary bond rotations were performed. Multiple rounds of refinement were performed by Phenix (33) and the structure was solved to 3.3 Å. Superposition of the two molecules in the asymmetric unit established a backbone RMSD of 1.02 Å for the entire model (Supplementary Figure S4).
Figure 4.

GCPANFab triplex X-ray crystal structure. (A) Secondary structure of GCPAN triplex, whose apical GAAA tetra-loop was replaced by the Fab-BL3-6 binding motif (5′-GAAACAC-3′, depicted in outlined font). (B) Crystal structure of GCPAN triplex shown in stick representation and the 3.3 Å anneal-omit 2|Fo| – 2|Fc| electron density map (blue mesh) of the RNA molecule contoured at 1.5 σ level (PDB ID: 6X5N). (C) Top view of GCPAN triplex crystal structure model, depicting the P2-triplex-P1 co-axial stack. (D) Model of the five U•A–U base triples in the triplex region colored in blue (U), orange (A) and green (U). (E) A representative U•A–U base triple. (F) A representative A-minor interaction between gcc(A)9 and the P1 duplex.
In our GCPAN triplex crystal structure, the Fab-binding motif (5′-GAAACAC-3′) forms contacts with Fab-BL3-6 identical to those described in the literature (28). G14 and C20 form Watson–Crick H-bonds, while A15, A17 and C18 fold into the protein and form H-bonding interactions with Fab-BL3-6 (Supplementary Figure S5). The 3′ terminus of the A-rich polyA tail (nts 8 to 12) interacts with the U-rich stem loop of GCENE duplex (nts 4–8, Hoogsteen strand; nts 26–30, Watson–Crick strands) to form a triple helix core comprising five contiguous U•A–U triples (Figure 4D, 4E) (U4•A8–U26, U5•A9–U27, U6•A10–U28, U7•A11–U29, U8•A12–U30). All bases constituting the triple helix are within hydrogen bonding distances of 2.3–3.5 Å along the Hoogsteen and Watson–Crick faces, as observed in the previous PAN triplex core crystal structure (12), indicating that the Fab binding motif did not induce significant structural changes in GCPAN ENE triple helix.
To evaluate whether the addition of the ‘GC clamp’ impacted the structural arrangement of the triplex, we compared our GCPAN X-ray structure with the ENE•r(A)9 triplex structure (PDB ID: 3P22) published more than a decade ago (12). A superposition of the U•A-U triplex motifs from the GCPAN triplex structure with ENE•r(A)9 triplex structure resulted in an RMSD of 1.13 Å (Supplementary Figure S6A, S6B). Superposition of the triplex motif together with several surrounding base pairs above and below the triplex resulted in an RMSD of 1.4 Å (Supplementary Figure S6C). This suggests that the GC clamp did not significantly alter the triplex region or proximal residues present on either side of the triple helix region. A bent conformation was observed for the GC clamp region which resembles a similar bent conformation observed for the basal helix present in the MALAT1 RNA triplex structure (PDB ID 4PLX).
The overall rod-like structure is stabilized by co-axial stacking of the P2 apical stem and the triplex core (Figure 4C). Additional stacking interactions beneath the triplex core are presented by the P1 helix and its associated A-minor interactions; A5, A6 and A7 of ggc(A)9 form a triad of A-minor interactions with G1–C35, G2–C34, G3–C33 of the GCENE duplex (Supplementary Figure S7). Directly beneath the P1 helix, the new P0 helix is formed between the 5′ terminus of ggc(A)9 and the 3′ terminus of GCENE; formation of the A4–U36 base pair stacks immediately beneath G1–C35 and the additional G–C Watson–Crick base pairs (C3–G37, G2–C38, G1–C39) clamp the base of the structure (Supplementary Figure S8). Helix P0 deviates from the co-axial P2-triplex-P1 stack and extends away from the core of the structure.
Nearly all nucleotides within the GCPAN triplex are involved in base pairing or base triple interactions with the exception of the Fab binding motif and a dinucleotide bulge. The seven nucleotides within the Fab binding motif are involved in multiple intra- and intermolecular interactions. In contrast, A31 and U32, within the bulge at the base of the triplex, are the only nucleotides in the entire structure not directly involved in hydrogen bonding interactions. In contrast these are extruded from the helix and solvent accessible. Not surprisingly, the dinucleotide bulge is the region of weakest electron density, with only the phosphate backbone being well defined for these nucleotides (Figure 4B), suggesting it is likely flexible and highly dynamic.
Characterization of ENE-binding ligands
PAN ENE binding ligands (Figure 1C) were initially characterized by MST (Figure 5A and Supplementary Figure S9). MST is a powerful solution-based technique for the quantification of biomolecular interactions (46,47). It is based on the laser-induced, temperature-related intensity change (TRIC) and movement of the molecules along the temperature gradient (e.g. thermophoresis). Because thermophoresis is sensitive to size, charge, and solvation entropy of the molecules, MST is a highly sensitive technique to measure changes in molecular properties, including binding of small molecules to RNA (48). Kd values ranging from 128 μM to 3.2 mM were calculated, indicating relatively weak binding (Supplementary Figure S9). The exception to this was compound 15, which displayed a Kd of ∼27 μM (Figure 5A). Thermal denaturation studies suggested that compound 15 binding stabilized the GCPAN triplex, increasing the melting temperature from 46°C to 50°C (Figure 5B). Results from ITC experiments infer triplex stabilization induced by compound 15 binding, i.e. binding of GCPAN ENE to ggc(A)9 is nearly 2-fold tighter in the presence of compound 15 (Supplementary Figure S10).
Figure 5.

Compound binding to GCPAN triplex. (A) MST binding plot for compound 15 (Kd = 27 ± 7 μM) with GCPAN triplex at 37°C. (B) CD melting curves for GCPAN triplex (black) and GCPAN+compound 15 (blue) monitored at 265 nm. (C) Chemical structures of compounds 15 and 15-A1. (D) MST binding plot for compound 15-A1 (Kd = 730 ± 17 μM).
In simple terms, compound 15 can be described as comprising a three-ring aromatic unit connected to an aliphatic sidechain (Figure 5C). In order to evaluate which component mediated GCPAN triplex binding, three derivatives were synthesized, concentrating on the aromatic moiety (Figure 5C, Supplementary Figure S11). Removing the cyclopentane ring gave rise to compound 15-A1, whose affinity for GCPAN triplex was reduced ∼27-fold (730 μM versus 27 μM, Figure 5D). Additional alterations (Supplementary Figure S11) including modification of the pyrimidine ring (15-A2) or simultaneous removal of the benzene and cyclopentane rings (15-A3) resulted in complete loss of compound binding (data not shown). Collectively, these results suggest a requirement for the three-ring aromatic portion of compound 15 for stacking within the GCPAN triplex.
As a first step in identifying compound binding interactions with GCPAN triplex, we attempted to crystallize the complex by soaking apo crystals with compound 15. Although soaked crystals diffracted to higher resolution (2.5 Å soaked versus 3.3 Å apo) density corresponding to the ligand was not identified. Overall, the apo and soaked crystals overlay with 1.1 Å RMSD, demonstrating two highly organized triplexes (Figure 6A). Interestingly, soaked crystals exhibited an alternate arrangement in the vicinity of bulge nucleotides U30, A31, U32 and C33, indicating a local structural rearrangement within the crystal (Figure 6B). While the electron density for A31 and U32 is clear primarily only for the phosphate backbone in both the apo and soaked crystals (Figure 6C, D), the rearrangements of this dinucleotide between the two structures are consistent with a dynamic bulge buttressed by a highly stable triplex structure (Figure 6A). Within this region, U32 undergoes the most dramatic alteration between the apo and soaked structures. In the former, U32 is extruded from the helix and forms a stacking interaction with A31 while in the soaked crystal it rotates towards the minor groove of the Watson-Crick face of the triplex region. We therefore propose that the dinucleotide bulge may provide a ‘molecular door’ which closes upon compound 15 binding, and may explain stabilization of the triplex observed upon complex formation (Figure 5B, Supplementary Figure S10).
Figure 6.

Ligand soaking induces structural changes within the ENE dinucleotide loop. (A) Overlay of the apo structure (PDB ID: 6X5N) and the structure obtained after soaking the crystals with compound 15 (PDB ID: 6X5M). The primary region of difference between the two structures is located near the dinucleotide bulge, colored red in the apo structure and cyan in the ligand-soaked structure. (B) Key differences are between nucleotides U30, A31, U32 and C33, comprising the AU dinucleotide bulge and flanking nucleotides on either side. (C) A portion of the 3.3 Å resolution anneal-omit 2|Fo| – 2|Fc| electron density map (blue mesh) corresponding to the bulge region from the apo crystal contoured at 1.5σ with the apo structural model in red. (D) A portion of the 2.5 Å resolution anneal-omit 2|Fo| – 2|Fc| electron density map (blue mesh) corresponding to the bulge region from the ligand-soaked crystal contoured at 1.5σ level with the soaked structural model in cyan. Backbone phosphate density is clearly depicted, while nucleobase density is not well defined.
Since the precise location of compound 15 binding could not be identified within the soaked crystal, we performed several mutagenesis approaches to examine the impact on compound 15 binding. First, we applied a nucleoside analog mutagenesis strategy, whereby we replaced adenosine with purine riboside, or nebularine, (Figure 7A) at three positions within ggc(A)9 (positions 4, 7 and 10; Figure 7B), with the goal of preserving base stacking and triplex stability while assessing consequences of removing the adenine exocyclic primary amine on compound binding. Examples of purine riboside mutagenesis include studies on hammerhead ribozymes (49), structural features of the -G-N-R-A- tetraloop (50), selection of the HIV polypurine tract (51) and APOBEC3G substrate specificity (52). Second, we deleted bulge nucleotide U32, referred to as GCENEΔU (Figure 7B), due to its implied role in compound 15 binding based on the observed local rearrangement of this nucleotide in the soaked crystal (Figure 6C, D).
Figure 7.

Compound 15 binding to mutant GCPAN triplexes. (A) Chemical structures for adenosine and nebularine (N). (B) Secondary structure model for the GCPAN triplex indicating the location of individual nebularine substitutions at positions 4, 7 or 10 of ggc(A)9 (red) and deletion of U32 shown as ΔU (blue). (C–F) MST binding experiments performed at 37°C examining triplex formation between the N-modified ggc(A)9 & Cy5-labeled GGENE and for the triplex formation between GCENEΔU with Cy5-labeled ggc(A)9, respectively. (G–J) MST binding experiments performed at 37°C by titrating compound 15 into A4N-, A7N-, A10N- and ΔU-substituted GCPAN triplexes, respectively.
Prior to assessing compound 15 binding to these mutant constructs, we performed MST binding experiments to demonstrate that bimolecular triplex formation was not abrogated by each mutation. Stability of the reconstituted triplex harboring individual nebularine mutants at positions 4 and 7 indicated no destabilization relative to the unsubstituted GCPAN triplex (Figures 3E and 7C and D). A minor destabilizing effect was observed upon nebularine substitution at position 10 (Figure 7E). Similarly, the GCENEΔU reconstituted triplex (Figure 7F) indicated slight destabilization relative to the unsubstituted GCPAN triplex. Nonetheless, the changes in stability upon nebularine and GCENEΔU substitutions are not >3-fold different compared to the GCPAN triplex.
Next, we examined compound 15 binding to the modified triplexes. Figure 7G–I report the affinity of compound 15 for nebularine-substituted GCPAN triplexes. A Kd of 60 μM for the A4N-substituted triplex (Figure 7G) is only 2-fold reduced relative to the unsubstituted GCPAN triplex (27 μM, Figure 5A). For the A7N-substituted triplex, a Kd of 100 μM for compound binding (Figure 7H) would suggest its proximity to the ligand binding site. In sharp contrast, a Kd of 5 ± 1 mM was determined for compound 15 binding to the A10N substituted triplex (Figure 7I). Coupled with 3-fold reduced triplex stability and effective elimination of compound 15 binding, the results of the A10N substitution strongly argue for this region of the triplex as an important structural region involved in ligand binding. Deletion of U32 (GCPANΔU) also significantly reduced the binding affinity of compound 15 (Figure 7J, Kd = 200 μM), suggesting interactions with U32 contribute to binding. A10 is located close to the dinucleotide bulge (A31, U32), implicating this region as an important binding pocket, a notion we subsequently strengthened by molecular modeling and simulations. Binding within the region between the triplex and dinucleotide bulge would be consistent with the initial SMM screen, which showed that compound 15 failed to bind to the ENE core duplex lacking these structural features.
Molecular modeling confirms compound 15 targets the GCPAN triple helix
To further investigate the potential ligand binding site and mode of interaction between the GCPAN triplex and compound 15 we employed computational docking and molecular dynamics simulations (see Materials and Methods). Using AutoDock4, 100 unbiased docking pose predictions were calculated against the apo and the soaked crystal structures (Supplementary Figure S12). The distribution of docking poses was nearly evenly split between the major groove and minor groove side of the crystal structure and there was no overall preference for a specific docking site (Figure S12). However, most of these triplex-ligand docking poses did not persist in MD simulations for more than 100 ns of simulation time, indicating a potentially suboptimal RNA-ligand interaction.
Because local structure variations within the crystal near the dinucleotide bulge resulted from compound soaking (Figure 6), we reasoned that structural dynamics might facilitate compound 15 binding. To explore the impact of RNA dynamics on ligand binding, we first performed 1 μs MD simulations to identify potentially important structural rearrangements within putative binding regions of the GCPAN triplex (Supplementary Figure S12). We then evaluated the simulated structures and noted that the most prominent conformational rearrangements were found near the dinucleotide bulge. Specifically, the bases of the dinucleotide bulge, A31 and U32, rotated out and stacked on each other concomitant with a slight opening of the base pair between the U30 and A8 of ggc(A)9. Together, these movements created an enlarged opening suitable for ligand docking. Docking to the MD-generated RNA conformation improved the best predicted docking scores from -7.17 to -9.40 kcal/mol (Supplementary Figure S13). 14 of the 100 poses predicted for the MD conformation were placed between the dinucleotide bulge and the central U•A-U triplets, including near A10 of ggc(A)9 whose substitution with nebularine significantly disrupted compound binding (Figure 7I). Subsequent MD simulations of the new RNA-ligand complexes yielded a stable, long-lasting interaction within this binding pocket, the most stable of which remained in complex for up to 4 μs of simulation (Figure 8, Supplementary Figures S13–S14).
Figure 8.

Molecular docking and simulations of triplex-ligand complexes. (A) Predicted binding pose of compound 15 to the GCPAN triplex conformation after 340 ns of molecular dynamics simulation. Space-filling model of the triplex−ligand complex with GCENE in light gray and the dinucleotide bulge shown in the shades dark gray and labeled; ggc(A)9 shown in orange, with A10 highlighted in light orange; compound 15, shown in cyan with predicted binding pose in the minor grove (−7.60 kcal/mol). (B) The stable RNA−ligand complex conformation at 4000 ns shows stacking of compound 15 on U32, partial interaction with A31 and within the vicinity if A10 of ggc(A)9. Overall, this complex achieves a generally tighter fit of the ligand into the binding pocket. U30 is rotated into the major groove and is not visible in this view. (C) Enlarged view of the binding pocket depicting interactions of compound 15 near the bulge loop from the MD conformation at the midpoint of the simulation (2000 ns). Compound 15 stacks with U32 and partly with A31. The aliphatic sidechain of compound 15 passes to the major groove side. In addition to stacking interactions, compound 15 is stabilized by hydrogen bonds throughout the MD simulations, generally involving U32 and A9 of ggc(A)9. Shown here are three hydrogen bond interactions with U32 and one with A9 of ggc(A)9 (dashed red lines). Details of these interactions are listed below the structure.
Within this pocket on the minor groove side of the triplex, U32 presents a binding platform, supported by stacking with A31, on which the aromatic rings of compound 15 stack. Compound 15 forms hydrogen bonds with U32 and also with A9 of ggc(A)9 (Figure 8C). The aliphatic sidechain of compound 15 threads through an opening to the major groove side where it is intermittently enclosed by U30 (Supplementary Figure S14). In the most stable docking pose the pentameric ring of the aromatic unit is oriented toward U32, where it remains throughout 3 independent simulations of >3 μs. Interestingly, in MD simulations of complexes wherein the pentameric ring is oriented toward A31 (not shown) compound 15 dissociates much more rapidly, further supporting placement of compound 15 in proximity to U32. The strength of the stacking between U32 and compound 15 was further supported through coordinated movements during the simulation wherein the distance between the nucleobase of U32 and compound 15 was maintained at 3.5–4.0 Å. In this way, the MD simulations explain the experimentally observed loss in compound 15 binding affinity upon deletion of U32 (Figure 7J).
Our MD simulations of RNA-ligand complexes also corroborate an indirect mechanism of ligand binding destabilization upon introducing the A10N substitution (Figure 7I). Specifically, there are only 3 base triples which are consistently maintained throughout the simulation—those involving A9, A10 and A11 of the ggc(A)9 oligo. Removal of the exocyclic amine in the A10N substitution may increase the local dynamics within this region, resulting in disruption of the binding pocket and abrogation of the interaction between A9 and compound 15.
DISCUSSION
Nuclear KSHV PAN RNA accumulation (53) is associated with a triplex structure near its 3′ terminus that sequesters and protects the poly(A) tail from cellular mRNA decay machinery. The KSHV ENE triplex has counterparts in MALAT1, an important cancer-related lncRNA, which is likewise retained in the nucleus (54); telomerase-associated RNA TER, which forms the central component of a dynamic RNA regulatory element (55,56); transposable element RNAs of plants and fungi (57) and various viral genomes and non-coding RNAs (58). The increasing recognition of the role of RNA triple helix structures thus suggests their dissection at the molecular level might provide novel therapeutic modalities. Proof-of-principle for targeting RNA triplex structures has been provided by Bhowmik et al., who have demonstrated binding of the natural alkaloid berberine and synthetic analogs to a model -U-A-U- triplex (59), and by our recent discovery of ligands that recognize the MALAT1 ENE triplex and reduce its nuclear expression (16).
Small molecule binders targeting the KSHV ENE might be adapted to antagonize a viral RNA component as opposed to currently-used activators and inhibitors of cellular enzymes (60,61). We therefore adopted an SMM strategy to identify structurally-distinct chemotypes that selectively recognized the ENE triplex. Although initial screening used the bimolecular PAN construct described by Mitton-Fry et al. (12), the ease with which it dissociated, shown here by a combination of CD, size exclusion chromatography and MST, presented a challenge to detailed chemotype characterization. This technical problem was alleviated by introducing a 3 base-pair G-C ‘clamp’ at the base of the lower stem, increasing affinity for the modified r(A)9 triplex-forming strand ∼70-fold while preserving the structural integrity of the triple helix (Supplementary Figure 6). This relatively minor adjustment supported thorough biochemical characterization of compound 15 as a triplex-binding ligand with moderate affinity.
Soaking of compound 15 into the ENE apo crystal improved the overall diffraction resolution and led to structural rearrangements within the dinucleotide bulge, but failed to provide reliable density for the ligand. Our inability to define the ligand binding site necessitated an indirect approach of ‘atomic mutagenesis’, involving replacing adenosine with the ‘shape mimic’ nebularine at different positions in the ggc(A)9 tail of the GCPAN triplex. This strategy, which has emerged as a powerful tool to unravel specific interactions in complex RNA molecules, demonstrated that a nebularine substitution in the central U•A–U triplet of the ENE triplex severely compromised compound 15 binding affinity. Similarly, removal of U32 (GCENEΔU), one of two nucleotides comprising the single stranded bulge, decreased the binding affinity of compound 15 to the triplex. While data from the nebularine substitutions and the GCENEΔU deletion mutant (Figure 7) cannot distinguish between a direct or indirect effect of the resulting destabilization of compound 15 binding, MD simulations likewise supported the notion that the compound 15 binding site was located in close proximity to the dinucleotide bulge. RNA–ligand complex stabilities in MD simulations suggest the binding mode encompasses strong stacking interactions and multiple hydrogen bonding interactions. Furthermore, rather than dock to a well-defined pre-formed pocket, compound binding to the PAN triplex may be dynamic, with a ligand-induced fit mechanism. Limited medicinal chemistry has also shown that perturbing the three-ring composition of compound 15 severely compromises its binding affinity, highlighting the importance of stacking interactions.
Although only a limited number of RNA triplexes have been documented, the specificity of compound 15 binding is an important consideration. As indirect evidence for specificity, triple helix-binding ligands we recently identified as targeting the ENE triplex of lncRNA MALAT1 (16) were not selected in the current SMM screen. More importantly, saturation‐transfer difference (STD) NMR, a ligand‐based screening technique that builds on the Nuclear Overhauser Effect (62), has provided direct evidence that MALAT1-binding ligands failed to recognize both the structurally-related ENE elements of nuclear paraspeckle assembly transcript 1 (NEAT1) and KSHV PAN lncRNAs (16). A more comprehensive understanding of the structural basis for ENE triplex specificity will require further chemical or nuclease probing, but it is worthwhile noting that in contrast to the MALAT1 and NEAT1 ENEs, which comprise two triple helical units separated by a G-C base pair, the PAN ENE is a single unit of five U•A–U base triples. Moreover, although there are minor differences between the MALAT1 and NEAT1 and PAN ENE triplexes, differing in the number of base triples, the single-stranded ‘bulge’ at the base of the triple helix differs between the three ENE triplexes. Such subtle differences in length and sequence composition of the bulge nucleotides may contribute towards selective ENE triplex targeting. The presence of single-stranded bulge nucleotides in many ENE triplexes (57) implicates this as a potentially ubiquitous pocket for small molecule targeting. Additional investigation will be required to elucidate the molecular interactions guiding small molecule specificity to the putative binding pockets formed by these varying bulge nucleotides.
Finally, although we have not determined the biological consequences of compound 15 binding to the ENE triplex, Rosetto and Pari showed that the minor capsid protein, ORF26, possessed RNA binding properties, proposing that it might provide a ‘tether’ that sequestered PAN lncRNA into the budding virion (63). Our chemoenzymatic probing studies of virion-associated PAN have also implicated the ENE triplex as a major ORF26 binding site (11). Such observations suggest that the ORF26-ENE interaction might be disrupted by compound 15 binding, with consequences for KSHV pathogenesis, providing a novel strategy that targets a viral lncRNA as opposed to a cellular factor.
DATA AVAILABILITY
Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 6X5M (soaked), 6X5N (apo).
Supplementary Material
ACKNOWLEDGEMENTS
J.T.M. and S.F.J.L.G.are grateful to Anna Lewicka and Huw Rees, University of Chicago, for technical assistance in Fab purification. M.S., W.K.K., M.L., J.T.M., J.S., J.S.S., A.W., B.A.S. and S.F.J.L.G. were supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. J.S.S. was funded by National Institutes of Health grant 1-ZIA-BC011585-07. This work has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract no. HHSN75N91019D00024 (M.S., M.L., W.K.K.). D.K. and J.P. were funded by National Institutes of Health grant R01GM102489 and the Chicago Biomedical Consortium, with support from the Searle Funds at The Chicago Community. A.A.A. and N.J.B. were funded by National Institutes of Health grant K22-HL121113A and University of the Sciences Start-up funds. A.A.A. was the recipient of a graduate fellowship from the Saudi Arabian Cultural Mission and Jazan University. R.M. and A.F. were funded by National Institutes of Health grant SC1GM111158. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Author contributions: M.S. performed X-ray crystallography, MST and CD experiments, and assisted in manuscript preparation. A.A. performed SEC, UV melts, ITC and RNase R protection experiments, and assisted in manuscript preparation. W.K.K. performed docking and MD analysis and assisted in manuscript preparation. M.L. and A.W. assisted in interpretation of X-ray crystallography data. J.T.M. performed EMSA analysis and prepared recombinant Fab. J.S.S. performed small molecule screening. J.S. assisted in interpretation of SMM data. D.K. and J.P. assisted in Fab purification and provided purified Fab. R.P.M. and A.J.F. assisted in preparation of compound 15 derivatives. B.A.S., N.J.B. and S.F.J.L.G. designed experiments for PAN variants and assisted in manuscript writing.
Notes
Present address: Deepak Koirala, Department of Chemistry and Biochemistry, University of Maryland Baltimore County, Baltimore, MD 21250, USA.
Contributor Information
Monalisa Swain, Basic Research Laboratory, National Cancer Institute, Frederick, MD 21702, USA; Basic Science Program, Frederick National Laboratory for Cancer Research, Frederick, MD 21702, USA.
Abeer A Ageeli, Department of Chemistry & Biochemistry, University of the Sciences, Philadelphia, PA 19104, USA; Chemistry Department, Faculty of Science (Female Section), Jazan University, Jazan 82621, Saudi Arabia.
Wojciech K Kasprzak, Basic Science Program, Frederick National Laboratory for Cancer Research, Frederick, MD 21702, USA.
Mi Li, Basic Science Program, Frederick National Laboratory for Cancer Research, Frederick, MD 21702, USA; Center for Structural Biology, National Cancer Institute, Frederick, MD 21702, USA.
Jennifer T Miller, Basic Research Laboratory, National Cancer Institute, Frederick, MD 21702, USA.
Joanna Sztuba-Solinska, Department of Biological Sciences, Auburn University, Auburn, AL 36849, USA.
John S Schneekloth, Chemical Biology Laboratory, National Cancer Institute, Frederick, MD 21702, USA.
Deepak Koirala, Department of Biochemistry and Molecular Biology, University of Chicago, Chicago, IL 60637, USA.
Joseph Piccirili, Department of Biochemistry and Molecular Biology, University of Chicago, Chicago, IL 60637, USA; Department of Chemistry, University of Chicago, Chicago, IL 60637, USA.
Americo J Fraboni, Department of Chemistry, Brooklyn College, City University of New York, Brooklyn, NY, 11210, USA.
Ryan P Murelli, Department of Chemistry, Brooklyn College, City University of New York, Brooklyn, NY, 11210, USA; PhD Program in Chemistry, The Graduate Center, City University of New York, New York, NY 10016, USA; PhD Program in Biochemistry, The Graduate Center, City University of New York, New York, NY 10016, USA.
Alexander Wlodawer, Center for Structural Biology, National Cancer Institute, Frederick, MD 21702, USA.
Bruce A Shapiro, RNA Biology Laboratory, National Cancer Institute, Frederick, MD 21702, USA.
Nathan Baird, Department of Chemistry & Biochemistry, University of the Sciences, Philadelphia, PA 19104, USA.
Stuart F J Le Grice, Basic Research Laboratory, National Cancer Institute, Frederick, MD 21702, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Searle Funds at The Chicago Community; National Cancer Institute (Intramural Research Program, ZIA-BC011585-07); Chicago Biomedical Consortium; National Heart, Lung, and Blood Institute [K22-HL121113A]; National Institute of General Medical Sciences [R01GM102489, SC1GM111158]; Frederick National Laboratory for Cancer Research [HHSN75N91019D00024]; University of the Sciences; Saudi Arabian Cultural Mission and Jazan University. Funding for open access charge: University of the Sciences (Start-up funds to N. Baird).
Conflict of interest statement. None declared.
REFERENCES
- 1. Marchese F.P., Raimondi I., Huarte M.. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017; 18:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kotake Y., Nakagawa T., Kitagawa K., Suzuki S., Liu N., Kitagawa M., Xiong Y.. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene. 2011; 30:1956–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weidle U.H., Birzele F., Kollmorgen G., Ruger R.. Long non-coding RNAs and their role in metastasis. Cancer Genomics Proteomics. 2017; 14:143–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li Z., Chao T.C., Chang K.Y., Lin N., Patil V.S., Shimizu C., Head S.R., Burns J.C., Rana T.M.. The long noncoding RNA THRIL regulates TNFalpha expression through its interaction with hnRNPL. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:1002–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abdelmohsen K., Gorospe M.. Noncoding RNA control of cellular senescence. Wiley Interdiscip. Rev. RNA. 2015; 6:615–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deng Z., Campbell A.E., Lieberman P.M.. TERRA, CpG methylation and telomere heterochromatin: lessons from ICF syndrome cells. Cell Cycle. 2010; 9:69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang S., Ke H., Zhang H., Ma Y., Ao L., Zou L., Yang Q., Zhu H., Nie J., Wu C.et al.. LncRNA MIR100HG promotes cell proliferation in triple-negative breast cancer through triplex formation with p27 loci. Cell Death. Dis. 2018; 9:805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu W., Ding C.. Roles of LncRNAs in viral infections. Front. Cell Infect. Microbiol. 2017; 7:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ganem D. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J. Clin. Invest. 2010; 120:939–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhong W., Ganem D. Characterization of ribonucleoprotein complexes containing an abundant polyadenylated nuclear RNA encoded by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8). J. Virol. 1997; 71:1207–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sztuba-Solinska J., Rausch J.W., Smith R., Miller J.T., Whitby D., Le Grice S.F.J.. Kaposi's sarcoma-associated herpesvirus polyadenylated nuclear RNA: a structural scaffold for nuclear, cytoplasmic and viral proteins. Nucleic Acids Res. 2017; 45:6805–6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mitton-Fry R.M., DeGregorio S.J., Wang J., Steitz T.A., Steitz J.A.. Poly(A) tail recognition by a viral RNA element through assembly of a triple helix. Science. 2010; 330:1244–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Withers J.B., Li E.S., Vallery T.K., Yario T.A., Steitz J.A.. Two herpesviral noncoding PAN RNAs are functionally homologous but do not associate with common chromatin loci. PLoS Pathog. 2018; 14:e1007389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gutierrez I.V., Dayton J., Harger S., Rossetto C.C.. The expression and nuclear retention element of polyadenylated nuclear RNA is not required for productive lytic replication of Kaposi's Sarcoma-Associated Herpesvirus. J. Virol. 2021; 95:e0009621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abulwerdi F.A., Shortridge M.D., Sztuba-Solinska J., Wilson R., Le Grice S.F., Varani G., Schneekloth J.S. Jr. Development of small molecules with a noncanonical binding mode to HIV-1 trans activation response (TAR) RNA. J. Med. Chem. 2016; 59:11148–11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abulwerdi F.A., Xu W., Ageeli A.A., Yonkunas M.J., Arun G., Nam H., Schneekloth J.S. Jr, Dayie T.K., Spector D., Baird Net al.. Selective small-molecule targeting of a triple helix encoded by the long noncoding RNA, MALAT1. ACS Chem. Biol. 2019; 14:223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Calabrese D.R., Zlotkowski K., Alden S., Hewitt W.M., Connelly C.M., Wilson R.M., Gaikwad S., Chen L., Guha R., Thomas C.J.et al.. Characterization of clinically used oral antiseptics as quadruplex-binding ligands. Nucleic Acids Res. 2018; 46:2722–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sztuba-Solinska J., Shenoy S.R., Gareiss P., Krumpe L.R., Le Grice S.F., O’Keefe B.R., Schneekloth J.S. Jr. Identification of biologically active, HIV TAR RNA-binding small molecules using small molecule microarrays. J. Am. Chem. Soc. 2014; 136:8402–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Disney M.D., Winkelsas A.M., Velagapudi S.P., Southern M., Fallahi M., Childs-Disney J.L.. Inforna 2.0: a platform for the sequence-based design of small molecules targeting structured RNAs. ACS Chem. Biol. 2016; 11:1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rzuczek S.G., Colgan L.A., Nakai Y., Cameron M.D., Furling D., Yasuda R., Disney M.D.. Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol. 2017; 13:188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Velagapudi S.P., Cameron M.D., Haga C.L., Rosenberg L.H., Lafitte M., Duckett D.R., Phinney D.G., Disney M.D.. Design of a small molecule against an oncogenic noncoding RNA. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:5898–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dong P., Xiong Y., Yue J., Hanley S.J.B., Kobayashi N., Todo Y., Watari H.. Long Non-coding RNA NEAT1: A Novel Target for Diagnosis and Therapy in Human Tumors. Front Genet. 2018; 9:471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ageeli A.A., McGovern-Gooch K.R., Kaminska M.M., Baird N.J.. Finely tuned conformational dynamics regulate the protective function of the lncRNA MALAT1 triple helix. Nucleic Acids Res. 2019; 47:1468–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sherpa C., Rausch J.W., Le Grice S.F., Hammarskjold M.L., Rekosh D. The HIV-1 Rev response element (RRE) adopts alternative conformations that promote different rates of virus replication. Nucleic Acids Res. 2015; 43:4676–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bradner J.E., McPherson O.M., Mazitschek R., Barnes-Seeman D., Shen J.P., Dhaliwal J., Stevenson K.E., Duffner J.L., Park S.B., Neuberg D.S.et al.. A robust small-molecule microarray platform for screening cell lysates. Chem. Biol. 2006; 13:493–504. [DOI] [PubMed] [Google Scholar]
- 26. Connelly C.M., Abulwerdi F.A., Schneekloth J.S. Jr. Discovery of RNA binding small molecules using small molecule microarrays. Methods Mol. Biol. 2017; 1518:157–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ye J.D., Tereshko V., Frederiksen J.K., Koide A., Fellouse F.A., Sidhu S.S., Koide S., Kossiakoff A.A., Piccirilli J.A.. Synthetic antibodies for specific recognition and crystallization of structured RNA. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Koirala D., Shelke S.A., Dupont M., Ruiz S., DasGupta S., Bailey L.J., Benner S.A., Piccirilli J.A.. Affinity maturation of a portable Fab-RNA module for chaperone-assisted RNA crystallography. Nucleic Acids Res. 2018; 46:2624–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Minor W., Cymborowski M., Otwinowski Z., Chruszcz M.. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D. Biol. Crystallogr. 2006; 62:859–866. [DOI] [PubMed] [Google Scholar]
- 30. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 31. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Winn M.D., Murshudov G.N., Papiz M.Z.. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003; 374:300–321. [DOI] [PubMed] [Google Scholar]
- 33. Liebschner D., Afonine P.V., Baker M.L., Bunkoczi G., Chen V.B., Croll T.I., Hintze B., Hung L.W., Jain S., McCoy A.J.et al.. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol. 2019; 75:861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morris G.M., Huey R., Olson A.J.. Using AutoDock for ligand-receptor docking. Curr Protoc Bioinform. 2008; 24:8.14.11–18.14.40. [DOI] [PubMed] [Google Scholar]
- 35. Case D.A., Cheatham T.E., Darden T., Gohlke H., Luo R., Merz K.M., Onufriev A., Simmerling C., Wang B., Woods R.J.. The Amber biomolecular simulation programs. J. Comput. Chem. 2005; 26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zgarbova M., Otyepka M., Sponer J., Mladek A., Banas P., Cheatham T.E., Jurecka P.. Refinement of the Cornell et al. Nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J. Chem. Theory Comput. 2011; 7:2886–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheatham T.E., Miller J.L., Fox T., Darden T.A., Kollman P.A.. Molecular-dynamics simulations on solvated biomolecular systems - the particle mesh Ewald method leads to stable trajectories of DNA, RNA, and proteins. J. Am. Chem. Soc. 1995; 117:4193–4194. [Google Scholar]
- 38. Essmann U., Perera L., Berkowitz M.L., Darden T.A., Lee H., Pedersen L.G.. A smooth particle mesh Ewald method. J. Chem. Phys. 1995; 103:8577–8593. [Google Scholar]
- 39. Berendsen H.J.C., Postma J.P.M., Vangunsteren W.F., Dinola A., Haak J.R.. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984; 81:3684–3690. [Google Scholar]
- 40. Zheng G., Lu X.J., Olson W.K.. Web 3DNA–a web server for the analysis, reconstruction, and visualization of three-dimensional nucleic-acid structures. Nucleic Acids Res. 2009; 37:W240–W246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brown J.A., Valenstein M.L., Yario T.A., Tycowski K.T., Steitz J.A.. Formation of triple-helical structures by the 3′-end sequences of MALAT1 and MENbeta noncoding RNAs. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:19202–19207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ferre-D’Amare A.R., Doudna J.A.. Crystallization and structure determination of a hepatitis delta virus ribozyme: use of the RNA-binding protein U1A as a crystallization module. J. Mol. Biol. 2000; 295:541–556. [DOI] [PubMed] [Google Scholar]
- 43. Huang L., Lilley D.M.. The molecular recognition of kink-turn structure by the L7Ae class of proteins. RNA. 2013; 19:1703–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McCoy A.J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D. Biol. Crystallogr. 2007; 63:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baaske P., Wienken C.J., Reineck P., Duhr S., Braun D. Optical thermophoresis for quantifying the buffer dependence of aptamer binding. Angew. Chem. Int. Ed. Engl. 2010; 49:2238–2241. [DOI] [PubMed] [Google Scholar]
- 47. Seidel S.A., Dijkman P.M., Lea W.A., van den Bogaart G., Jerabek-Willemsen M., Lazic A., Joseph J.S., Srinivasan P., Baaske P., Simeonov A.et al.. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods. 2013; 59:301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moon M.H., Hilimire T.A., Sanders A.M., Schneekloth J.S. Jr. Measuring RNA-ligand interactions with microscale thermophoresis. Biochemistry. 2018; 57:4638–4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fu D.J., Rajur S.B., McLaughlin L.W.. Importance of specific guanosine N7-nitrogens and purine amino groups for efficient cleavage by a hammerhead ribozyme. Biochemistry. 1993; 32:10629–10637. [DOI] [PubMed] [Google Scholar]
- 50. Worner K., Strube T., Engels J.W.. Synthesis and stability of GNRA-loop analogs. Helv. Chem. Acta. 1999; 82:2094–2104. [Google Scholar]
- 51. Rausch J.W., Le Grice S.F. Purine analog substitution of the HIV-1 polypurine tract primer defines regions controlling initiation of plus-strand DNA synthesis. Nucleic Acids Res. 2007; 35:256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rausch J.W., Chelico L., Goodman M.F., Le Grice S.F. Dissecting APOBEC3G substrate specificity by nucleoside analog interference. J. Biol. Chem. 2009; 284:7047–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhong W., Wang H., Herndier B., Ganem D. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:6641–6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brown J.A., Bulkley D., Wang J., Valenstein M.L., Yario T.A., Steitz T.A., Steitz J.A.. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat. Struct. Mol. Biol. 2014; 21:633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim N.K., Zhang Q., Zhou J., Theimer C.A., Peterson R.D., Feigon J.. Solution structure and dynamics of the wild-type pseudoknot of human telomerase RNA. J. Mol. Biol. 2008; 384:1249–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Theimer C.A., Blois C.A., Feigon J.. Structure of the human telomerase RNA pseudoknot reveals conserved tertiary interactions essential for function. Mol. Cell. 2005; 17:671–682. [DOI] [PubMed] [Google Scholar]
- 57. Tycowski K.T., Shu M.D., Steitz J.A.. Myriad triple-helix-forming structures in the transposable element RNAs of plants and fungi. Cell Rep. 2016; 15:1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tycowski K.T., Shu M.D., Borah S., Shi M., Steitz J.A.. Conservation of a triple-helix-forming RNA stability element in noncoding and genomic RNAs of diverse viruses. Cell Rep. 2012; 2:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bhowmik D., Das S., Hossain M., Haq L., Suresh Kumar G.. Biophysical characterization of the strong stabilization of the RNA triplex poly(U)*poly(A)*poly(U) by 9-O-(omega-amino) alkyl ether berberine analogs. PLoS One. 2012; 7:e37939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang S.E., Wu F.Y., Chen H., Shamay M., Zheng Q., Hayward G.S.. Early activation of the Kaposi's sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J. Virol. 2004; 78:4248–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ye J., Shedd D., Miller G.. An Sp1 response element in the Kaposi's sarcoma-associated herpesvirus open reading frame 50 promoter mediates lytic cycle induction by butyrate. J. Virol. 2005; 79:1397–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Becker W., Bhattiprolu K.C., Gubensak N., Zangger K.. Investigating protein-ligand interactions by solution nuclear magnetic resonance spectroscopy. ChemPhysChem. 2018; 19:895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rossetto C.C., Pari G.S.. Kaposi's sarcoma-associated herpesvirus noncoding polyadenylated nuclear RNA interacts with virus- and host cell-encoded proteins and suppresses expression of genes involved in immune modulation. J. Virol. 2011; 85:13290–13297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 6X5M (soaked), 6X5N (apo).