


Abstract
De novo metagenome assembly is effective in assembling multiple draft genomes, including those of uncultured organisms. However, heterogeneity in the metagenome hinders assembly and introduces interspecies misassembly deleterious for downstream analysis. For this purpose, we developed a hybrid metagenome assembler, MetaPlatanus. First, as a characteristic function, it assembles the basic contigs from accurate short reads and then iteratively utilizes long-range sequence links, species-specific sequence compositions, and coverage depth. The binning information was also used to improve contiguity. Benchmarking using mock datasets consisting of known bacteria with long reads or mate pairs revealed the high contiguity MetaPlatanus with a few interspecies misassemblies. For published human gut data with nanopore reads from potable sequencers, MetaPlatanus assembled many biologically important elements, such as coding genes, gene clusters, viral sequences, and over-half bacterial genomes. In the benchmark with published human saliva data with high-throughput nanopore reads, the superiority of MetaPlatanus was considerably more evident. We found that some high-abundance bacterial genomes were assembled only by MetaPlatanus as near-complete. Furthermore, MetaPlatanus can circumvent the limitations of highly fragmented assemblies and frequent interspecies misassembles obtained by the other tools. Overall, the study demonstrates that MetaPlatanus could be an effective approach for exploring large-scale structures in metagenomes.
INTRODUCTION
The advancement of technologies has made it possible to automatically determine the genome sequence of an isolated bacterium using mate-pair libraries or single-molecule long reads with sufficient coverage (1). However, as the culture protocols for the majority of bacterial species have not yet been standardized, their isolation and purification to obtain sufficient DNA for sequencing might represent the most time-consuming and challenging step in projects targeting them. The de novo assembly of metagenome shotgun sequencing data has been demonstrated as an effective method that bypasses the isolation and culture—it provides important insights into the community biodiversity and the biological functions encoded in the genome of uncultured microbiota (2–5). For example, metagenomic data have been used to characterize the genomic information of the candidate phyla radiation (CPR), which is estimated to represent 15% of unidentified species in the bacterial domain (6), the Asgard superphylum (7,8), and phages in the human gut (9,10).
Nevertheless, the de novo assembly of environmental metagenomes presents several limitations, including uneven sequence coverage reflecting relative species abundance and the potential risk of interspecies misjoining. Fortunately, with rapid research advances, several metagenome assemblers have been developed to overcome these issues (11–14), where the uneven sequence coverage is utilized as a tool to distinguish the sequences of different species (15). However, when interspecies misjoining occurs, it leads to false hypotheses in a wide range of analyses, including phylogenetic, population genetics and pathway analyses. False identification of horizontal gene transfer can impede a proper understanding of the dynamics of environmental communities. Moreover, it can also lead to a secondary issue of misidentification. For example, once a chimeric genome is registered in a database, its use can lead to misidentification in other studies (16). Heterogeneity in metagenomes imposes a major limitation in the assembly of long sequences, as it introduces complexity to assembly graph structures. Therefore, there is an enormous need for a method that could efficiently remove false interspecies links.
Short-read sequencers, such as those obtained by Illumina sequencing, have been used to produce metagenomic data from deep sequencing; however, an entire chromosome has rarely been assembled. Moreover, though hundreds of thousands of microbial genomes have been sequenced (5,6,17–20), most of them are represented as sets of fragmented sequences (bins). Recently, long-read sequencers have been applied to metagenomic studies, dramatically improving the lengths of assemblies (contigs) (21–25). However, the numbers of complete circular bacterial genomes obtained in these studies are, for example, 20 (obtained from 13 human gut samples) (23) and 57 (obtained from 23 sludge samples) (25). These numbers are much fewer compared to the expected numbers of species in the microbiomes. Therefore, the development of robust assembly methods to improve the efficacy of microbial metagenomics is required.
In general, the throughput and fine-scale accuracy of short reads are higher than those of long reads. For example, the large long-read dataset (PacBio; 50 Gbp) obtained for the cow rumen microbiome did not cover many regions that were efficiently obtained from short reads (26). In addition, contigs produced from long reads sometimes contain much smaller insertion-deletion (indel) errors than those from short reads (27–29). These errors can affect gene predictions with false frameshifts (30) and hinder strain-level genome comparisons. Therefore, a hybrid assembly approach that combines the advantages of both long and short reads has been explored. Although long read-based assemblers have shown high performance in resolving repetitive regions (31,32), hybrid assemblers have exhibited higher accuracy and contiguity for some single genome (33,34) and metagenome assemblies (35), suggesting that the long read-based assembly is not an ultimate solution. Furthermore, when adopting the technologies of long-range links to metagenomic samples, the amount of long DNA fragments can be limited for some species because of their low abundance, extraction efficiencies, or DNA fragility. These factors consequently reduce the number of reads for such species, and therefore, an assembly algorithm able to join sequences using a small number of links while avoiding misjoins is necessary.
After the generation of contigs and scaffolds, binning (also called grouping or clustering) of sequences is often performed to obtain species draft genomes. Most binning tools utilize the composition of short k-mers (typically k: 4–6) and coverage depth of reads (36–38). Although binning can link sequences divided by regions that are hard to assemble such as repetitive ones, the combination of binning and assembly processes has not been implemented as a public tool.
Here, we report a hybrid metagenome assembler, MetaPlatanus. Its characteristic features include (i) long-range links (mate pair or long reads), (ii) species-specific sequence compositions and coverage depth to prevent interspecies misjoinings while scaffolding and (iii) binning to extend scaffolds. We performed benchmarks with short and long reads, showing that MetaPlatanus could assemble more regions with biologically important elements, such as genes, gene clusters, viral sequences, and near-complete genomes compared to the existing assemblers.
MATERIALS AND METHODS
Overview of MetaPlatanus
A schematic overview of MetaPlatanus is presented in Figure 1, illustrating the sequential utilization of read overlaps, differences in coverage depths between species, long-range sequence links, 4-mer compositions, and a binning result. The last four data types are useful when handling long sequences, and the workflow was designed in a stepwise and iterative manner to simultaneously achieve long scaffolds and few interspecies misjoinings. The functions related to these steps especially have methodological novelty, and they are described in the main text and highlighted in Figure 1. The other parts are described in Supplementary Methods.
Figure 1.

Overview of MetaPlatanus.
MetaPlatanus requires at least one short-read library and uses long reads and/or mate-pairs (jumping library) as long-range sequence links. An outline is presented below, and more details are provided in the Methods or Supplementary Methods sections.
The contig assembly was performed using short reads. The k-mers in the reads are counted, and the counts are used to estimate the coverage depth. The k and coverage cutoff (the minimum coverage depth of k-mers in the graph) are critical parameters for the contiguity and accuracy of the final metagenome assembly. MetaPlatanus uses multiple parameter values to reconstruct the graph iteratively (Supplementary Figure S1). The implementation of the graph and related functions are derived from Platanus (39). Junction data (k overlaps between contigs) are stored and used in subsequent steps.
To remove interspecies misjoinings, correction of the contig edges is performed. In this step, MetaPlatanus makes use of differences in coverage depth (abundance) between species. First, the coverage depths of all k-mers in contigs are reassigned using the result of k-mer counting for input short reads obtained in (i). Then, for each contig, edge regions with coverage depths that are largely different from the median value in the contig are divided.
To simplify the de Bruijn graph and extend contigs, MetaPlatanus untangles the cross-structures generated by repetitive sequences in a genome. This function is derived from Platanus-allee (40). The links of reads between the contigs are used. After untangling, sets of nodes without junctions are converted into contigs.
Scaffolding is executed for contigs using long-range links (paired-ends, mate-pairs, and long reads). The minimum number of reads (pairs) to join sequences is two (default) to handle low-abundant species or those whose DNA is not efficiently extracted. Interspecies misjoinings in scaffolding are excluded based on differences in coverage depths and 4-mer frequencies between species.
Paired-ends and mate-pairs are mapped to scaffolds, and then reads covering gaps and edges are collected. Then gap-closing and edge-extension are executed through the local contig assembly of the collected reads.
Steps (ii)–(v) are iterated five times. For the first three iterations, steps (iv) and (v) are omitted to obtain contigs that are long enough to be used for scaffolding. When the process returns to step (ii) for the fourth iteration, the scaffolds and local contigs are merged through the de Bruijn graph (Supplementary Figure S2).
Binning of scaffolds is performed using MetaBAT2 (38) using a 4-mer composition and coverage depth. It does not use database information, such as single-copy genes, because it is preferable to target non-prokaryotic and viral sequences. This step can accept coverage-depth data from multiple samples, including time-series ones.
Scaffolding is re-executed for each bin to further extend the sequences.
Gap-closing and edge-extension are applied to each bin. In addition, locally assembled contigs that are not used to close gaps are also added to the bins. This function is designed to include repetitive but important regions for species identification, such as rRNA genes.
Gap-closing with long reads is performed using TGS-GapCloser (41).
Polishing with short reads is performed using NextPolish (42).
The steps of (x) and (xi) are applied only when long reads are used as input.
Scaffolding in MetaPlatanus workflow
Similar to the contig assembly, the scaffolding module of MetaPlatanus was derived from Platanus, but the number of internal loops (iterations) increased with different parameters (Supplementary Figure S3). In contrast to Platanus, the function of merging heterozygous regions (bubble or branch structures) is deactivated, and the minimum number of reads (pairs) to join sequences is two. The short reads are first used, followed by long reads. The following parameters were changed and applied for each loop: sequence-length cutoff, maximum overlap length (tolerance), and minimum alignment length for long-read mapping. In the first loop for long reads, conflicting local alignments in the read mapping are excluded to avoid scaffolding of repetitive contigs.
To prevent interspecies misjoinings, the links between contigs from different species are detected utilizing coverage depths and 4-mer frequencies, as follows:
-
Estimation of the empirical distribution of coverage depth ratios between contigs from the same species:
For a pair of contigs with coverage depths c1 and c2, the coverage depth ratio is defined as min(c1, c2)/max(c1, c2). Each contig ≥2 kbp is divided equally into halves, and the coverage depth ratio is calculated for each pair. Next, the complementary cumulative distribution function (i.e. 1 − the cumulative distribution function) of the coverage depth ratio Fcoverage is empirically determined.
-
Estimation of the empirical distribution of the 4-mer frequency distances between contigs from the same species:
Considering a 4-mer from the characters {A, T, G, C} be si (i =1, 2, …, 44), and the prefix 3-mer of si be si[1, 3]. The number of occurrences of the substring s plus one is referred to as n(s), and ‘plus one’ refers to additive (Laplace) smoothing. The frequency of si is defined as fi: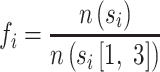
This corresponds to the posterior probability of the occurrence of si after si[1, 3]. The 4-mer frequency of a sequence (contig) is represented as the vector, and the 4-mer frequency distance between two sequences is the Euclidean distance between the pair of 4-mer frequencies. The complementary cumulative distribution function of the 4-mer frequency distances in the same species, F4-mer, is empirically determined similarly as Fcoverage (step (i)) was determined.
-
Construction of the scaffold graph:
The nodes and edges of the scaffold graph correspond to contigs and their paired-end (mate pair) linkages, respectively. Contigs are linked if the number of paired reads is ≥2.
Exclusion of links between different species.
For each pair of linked contigs, the coverage depth ratio r and the 4-mer frequency distance d are computed. The link is excluded if the following condition is satisfied:
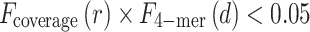 |
where the left side corresponds to the posterior probability:
P (coverage depth ratio > r and 4-mer frequency distance > d | same-species contigs).
Note that F4-mer is set to 1 for contigs <1000 bp, as these contigs are assumed to be too short for calculating their 4-mer frequencies.
The protocol for the construction of scaffolds from the graph is descrived in Supplementary Methods.
Iteration of scaffolding and gap-closing in MetaPlatanus workflow
To extend sequences, MetaPlatanus iterates scaffolding and gap-closing five times (default). For each iteration, the procedure is as follows (Supplementary Figure S3):
-
Re-calculation of coverage depths for contigs
The number of k-mers (default, k = 25) in the short reads (k-mer coverage) are stored in the contig assembly step. For each contig, the k-mer coverages are calculated for all k-mers in the contig using the stored data.
-
Correction of contig edges
To correct interspecies misjoinings in contigs, MetaPlatanus deletes the edges of the contigs when the following condition is satisfied: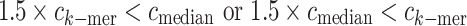
where ck-mer and cmedian are the k-mer coverage depth and the median in a contig, respectively.
-
Untangling cross-structures in a de Bruijn graph
A de Bruijn graph is reconstructed using the trimmed contigs, joining the contigs with edge overlaps of (k − 1) characters. Next, the graph is simplified as described in the ‘Untangling the cross-structures in a de Bruijn graph in MetaPlatanus workflow’ section in Supplementary Methods.
-
Scaffolding
Scaffolding is performed as described in the ‘Scaffolding in MetaPlatanus workflow’ section.
-
Gap-closing
This module is derived from Platanus (39). In addition to gap-closing, MetaPlatanus extends the edges of the scaffolds and saves the contigs that are not used in gap-closing or edge-extension for processing in the next step.
Merging of sequences using de Bruijn graphs
A de Bruijn graph (k = 1.5 × read length) is constructed from the scaffolds, and contigs are generated. The scaffolds extended in (iii) overlapped each other, and the overlapping sequences are merged through the de Bruijn graph. The resulting contigs serve as inputs for the next iteration.
Steps (i)–(vi) are iterated five times. For the first three iterations, steps (iv) and (v) are omitted to obtain contigs that are long enough to be used for scaffolding. For the last iteration, the edge extensions in steps (v) and (vi) are not executed, and the gap-closed scaffolds represent the final output. Note that the rough concept of the iteration was derived from Platanus_B (29), which is designed for an isolated bacterial genome. However, MetaPlatanus additionally utilizes coverage depths to correct false links in the iteration, and the internal scaffolding algorithm has many differences (see ‘Scaffolding in MetaPlatanus workflow’ for details).
Scaffolding utilizing binning results in MetaPlatanus workflow
This step is executed after binning by MetaBAT2 (see Supplementary Methods). The functions related to the local difference in coverage depth and 4-mer frequencies are deactivated. In the scaffold graph, edges linking different bins are deleted. Edges between a binned sequence and an unbinned sequence are retained. Subsequently, scaffolding is performed.
Gap-closing, edge-extension and addition of contigs for each bin in MetaPlatanus workflow
After scaffolding utilizing binning, the gap-closing step is executed, and the edges of the scaffolds are then extended. Within each bin, locally assembled contigs are added if corresponding gaps or edges are not filled, short sequences that exactly match to other longer sequences are removed, and exact edge overlaps ≥32 bp are trimmed. Exact matches or edge overlaps between sequences in different bins are allowed. For each unbinned sequence, exact matches and edge overlap with the other sequences are searched, and if detected, they are excluded or trimmed. The procedures above can add repetitive sequences, such as rRNA genes, to all bins, minimizing redundancies in the entire sequence set.
Datasets for benchmarks
We downloaded the mock dataset with Illumina paired-ends (PE), Oxford Nanopore Technology (ONT) long reads, and PacBio long reads (22) (Supplementary Table S1; called ‘GIS20 mock’ hereafter) from the SRA database. The genomic DNA of 20 bacterial species was mixed with nonuniform amounts and then sequenced (Supplementary Table S2). Because the throughput of the Illumina library was relatively large, it was downsampled to the same size of each long-read library (2.44 Gbp of ONT or 5.31 Gbp of PacBio).
To generate the metagenomic mock datasets of Illumina paired-ends and mate-pairs (MP), we prepared genomic DNA samples from 20 bacteria known to inhabit the human gut (Supplementary Table S3) and mixed them in three different proportions (cases 1–3; Supplementary Tables S4 and S5; called ‘MP mock’ hereafter). The relative unevenness of constituent DNA proportions was highest in case 3 and lowest in case 1, simulating different abundance ratios. All mixtures were sequenced using Illumina HiSeq2500 and MiSeq as one paired-end (insert-size, 550 bp) and two mate-pair (insert sizes, 4 and 8 kbp) samples. We additionally prepared low-coverage datasets by downsampling (20% of case 1, 30% of case 1, and 10% of case 2).
For the benchmarks using actual environmental samples, we downloaded public metagenomic data of the human gut (22) (Supplementary Table S6) and human saliva (24) (Supplementary Table S7) samples from the SRA database. All data contained paired-end and ONT long-read libraries. For the human gut samples, we only used 18 samples with total sizes of ONT reads >1 Gbp.
RESULTS
Benchmarking with mock data
For the GIS20 mock, we ran metagenome assemblers: MetaPlatanus (version 1.3.0), metaSPAdes (version 3.15.2) (14,43), OPERA-MS (version 0.8.2) (22) and metaFlye (version 0.8.2) (44). The former three tools adopted a hybrid approach, and both short and long reads were used as input, whereas metaFlye is a long-read-based metagenome assembler. Three modes of MetaPlatanus were benchmarked: default, without TGS-GapCloser (‘w/o closing’), and without the characteristic error-correction functions (‘w/o correction’). The omitted functions in the last mode were related to coverage depths, 4-mer, binning results, and avoidance of repetitive contigs in scaffolding (see Supplementary Methods for details). OPERA-MS was executed without the assistance of reference genomes (–no-ref-clustering) to evaluate the ability for de novo assembly. The polishing tool, NextPolish (version 1.3.1) (42), was applied to the metaFlye results with short reads (‘metaFlye polished’). QUAST and metaQUAST (version 5.0.2) (45) were used to compare the reference genomes. Metrics integrating contiguity and accuracy are informative for comparing assemblers. For QUAST, NGA50 is a metric representing the NG50 value of aligned regions. NG50 is an indicator of contiguity, representing the length at which the collection of all sequences of that length or longer contains 50% of the genome size.
The GIS20 benchmark results are shown in Table 1 and Supplementary Table S8. MetaPlatanus had the highest NGA50s for both the ONT and PacBio datasets (Table 1A and B). The number of misassemblies and mean sequence identity were markedly smaller and higher, respectively, in MetaPlatanus w/o closing mode than those obtained by other tools, suggesting the high accuracy of its core regions were constructed from short reads. After applying TGS-GapCloser, MetaPlatanus had the first to third-best values for these indicators. The details of the misassemblies for each step of MetaPlatanus are shown in Supplementary Table S9. After the scaffolding step, NG50 increased >18 times, whereas the increase in the number of misassemblies was relatively low, suggesting that the scaffolding function can join many contigs without misassemblies. The number of interspecies misassemblies was counted based on the QUAST results, and it was confirmed that MetaPlatnaus had the fewest number of such misassemblies (Table 1A and B). Note that the inactivation of the correction functions of MetaPlatanus substantially increased those misassemblies and decreased the NGA50s, supporting the effectiveness of these functions. Since misassemblies can be over-estimated for draft genomes containing errors, we also evaluated the assemblies using only complete reference genomes (Table 1C and D). MetaPlatanus had the lowest and second-highest numbers for misassemblies and mean sequence identities, respectively. The NGA50 for each species was calculated using metaQUAST (45) (Supplementary Figure S4 and Supplementary Table S10), and MetaPlatanus exhibited the highest or second-highest values in most cases, regardless of coverage depth. metaFlye also had the largest values for many cases, but its performance varied widely. In addition, the mean sequence identity was substantially low for the raw result of metaFlye, suggesting the importance of short-read polishing. We additionally evaluated five binning tools (36–38,46,47) as those used in the MetaPlatanus workflow (Supplementary Table S11) and MetaBAT2’s high purity (precision) supports the validity of selecting this tool. For runtimes (Supplementary Table S12), the CPU times of MetaPlatanus were comparable to those of metaSPAdes, but MetaPlatanus' real times were the maximum when the number of threads was 24. These results provide evidence that MetaPlatanus could maximize the quality of assembled sequences at the cost of runtime. Supplementally, we prepared the datasets for which equivalent amounts of reads could be input into hybrid assemblers and metaFlye. Even in this benchmark, MetaPlatanus had NGA50s and genome fractions comparable to those of metaFlye (Supplementary Table S13).
Table 1.
Benchmarks of GIS20 mock using QUAST
| Assembly | NG50 (bp) | NGA50 (bp) | # all-misassemblies | Mean sequence identity (%) | Genome fraction (%) | # inter-species misassemblies |
|---|---|---|---|---|---|---|
| (A) PE and ONT, all reference genomes. | ||||||
| MetaPlatanus | 757 098 | 171 795 | 201 | 99.9179 | 61.63 | 0 |
| MetaPlatanus w/o closing | 756 452 | 149 303 | 136 | 99.9879 | 59.70 | 0 |
| MetaPlatanus w/o correction | 494 840 | 148 578 | 231 | 99.9375 | 61.76 | 6 |
| metaSPAdes | 234 822 | 73 215 | 528 | 99.8405 | 63.35 | 5 |
| OPERA-MS | 168 587 | 46 454 | 512 | 99.8619 | 60.66 | 5 |
| metaFlye raw | 609 353 | 86 681 | 255 | 99.2566 | 55.28 | 1 |
| metaFlye polished | 610 750 | 90 756 | 171 | 99.9218 | 55.69 | 1 |
| (B) PE and PacBio, all reference genomes. | ||||||
| Assembly | NG50 (bp) | NGA50 (bp) | # all-misassemblies | Mean sequence identity (%) | Genome fraction (%) | # inter-species misassemblies |
| MetaPlatanus | 1 444 699 | 129 682 | 280 | 99.9390 | 69.95 | 0 |
| MetaPlatanus w/o closing | 1 444 050 | 121 228 | 163 | 99.9910 | 68.16 | 2 |
| MetaPlatanus w/o correction | 617 373 | 121 198 | 302 | 99.9507 | 69.89 | 4 |
| metaSPAdes | 374 634 | 112 139 | 219 | 99.8685 | 75.47 | 4 |
| OPERA-MS | 135 973 | 40 414 | 423 | 99.8566 | 71.51 | 13 |
| metaFlye raw | 235 065 | 11 717 | 242 | 99.7181 | 50.04 | 5 |
| metaFlye polished | 235 089 | 13 257 | 256 | 99.9221 | 50.32 | 5 |
| (C) PE and ONT, complete reference genomes only. | ||||||
| Assembly | NG50 (bp) | NGA50 (bp) | # all-misassemblies | Mean sequence identity (%) | Genome fraction (%) | |
| MetaPlatanus | 1 679 878 | 423 879 | 138 | 99.9239 | 67.07 | |
| MetaPlatanus w/o closing | 1 677 738 | 414 037 | 81 | 99.9871 | 65.23 | |
| MetaPlatanus w/o correction | 841 595 | 369 997 | 144 | 99.9477 | 67.17 | |
| metaSPAdes | 421 415 | 163 808 | 420 | 99.8632 | 68.55 | |
| OPERA-MS | 253 089 | 130 701 | 317 | 99.8962 | 66.09 | |
| metaFlye raw | 2 391 293 | 211 481 | 215 | 99.2610 | 62.97 | |
| metaFlye polished | 2 398 268 | 212 079 | 127 | 99.9178 | 63.45 | |
| (D) PE and PacBio, complete reference genomes only. | ||||||
| Assembly | NG50 (bp) | NGA50 (bp) | # all-misassemblies | Mean sequence identity (%) | Genome fraction (%) | |
| MetaPlatanus | 1 870 893 | 401 772 | 178 | 99.9355 | 75.27 | |
| MetaPlatanus w/o closing | 1 870 683 | 401 772 | 88 | 99.9890 | 73.40 | |
| MetaPlatanus w/o correction | 1 051 605 | 349 570 | 216 | 99.9465 | 75.24 | |
| metaSPAdes | 625 209 | 314 514 | 127 | 99.8649 | 81.42 | |
| OPERA-MS | 233 394 | 90 469 | 332 | 99.8426 | 77.13 | |
| metaFlye raw | 2 421 686 | 38 550 | 198 | 99.6941 | 56.46 | |
| metaFlye polished | 2 421 728 | 40 331 | 212 | 99.9126 | 56.80 | |
We additionally performed mock benchmarks (MP mock) using mate-pair libraries as another format for long-range links. The compared tools were IDBA-UD (version 1.1.3) (12), SPAdes (version 3.15.2) (48), metaSPAdes and MEGAHIT (version 1.1.3) (49). The metaSPAdes and MEGAHIT tools can only use paired-end libraries. The benchmarking demonstrated that MetaPlatanus had the largest NGA50s for all cases (Supplementary Table S14 and Supplementary Figure S5). Taken together, it is suggested that the MetaPlatanus algorithm makes use of the universal property of long-range links, not specialized for long reads.
Benchmarking with human gut samples with long reads from MinION or GridION
Potable or middle-sized nanopore sequencers such as ONT MinION and GridION are widely used and cost-effective in some cases. We used actual data of these sequencers from 18 human gut samples (‘Datasets for benchmarks’ in MATERIALS AND METHODS). The assemblers used for the GIS20 mock benchmark were tested, and contamination of human genomes was excluded (see Supplementary Methods for details). The detailed assembly statistics are shown in the Supplementary Table S15. Non-human eukaryotic sequences in assemblies were searched using MetaEuk (version 4) (50). As shown in Supplementary Table S16, the total size was less than 400 kbp (we did not discuss the performance of eukaryotic sequences).
To evaluate the contiguity of the assembly, we show the Nx-length plot, which was introduced in our previous study (15) (Supplementary Figure S6). Nx-length is defined as the length at which the collection of all sequences of that length or longer contains x bp (horizontal axis). MetaPlatanus assemblies had the largest areas under the curves in 12 out of 18 cases, suggesting that its contiguities were the highest for the majority of the samples.
Next, we counted various elements with biological functions in assemblies: protein-coding sequences (CDSs; with hits to the Swiss-Prot database (51)), biosynthetic gene clusters (≥10 kbp), and near-complete viral sequences (completeness ≥ 90%) using multiple tools (52–55) (see Supplementary Methods for details). As long-range elements, we targeted scaffolds (contigs) ≥500 kbp and bins. Considering the criteria previously published (56), we defined the categories based on the results of CheckM (version 1.0.11) (57) and Prokka (version 1.14.6) (58) as follows:
‘over-half’: completeness > 50%, contamination < 5%.
‘near-complete’: completeness > 90%, contamination < 5%, presence of 3 types of rRNAs (23S, 16S, and 5S) and at least 18 types of tRNAs.
Note that near-complete scaffolds/bins represent scaffolds/bins representing near-complete genomes. Consequently, MetaPlatanus had the largest number of top cases for all criteria (Figure 2 and Supplementary Table S17). In other words, it had the most sequence elements for the least seven cases compared to the other tools. These results suggest that the MetaPlatnaus assemblies have not only high contiguities but also high qualities. Note that the number of near-complete scaffolds is too small for these datasets, and we focus on this criterion in the next benchmark. For MetaPlatanus, the rates of exactly duplicated CDSs were reported to be at most 2% by CD-HIT (version 4.6) (59) (Supplementary Table S15H), and overestimations in the above results caused by duplications were expected to be limited. As expected, MetaPlatanus w/o closing had poorer results than the default mode, but the densities of CDSs were the highest in most cases (Supplementary Table S15G). The accuracy of this assembly was expected to be high, as the mock benchmarks. In contrast, the metaFlye raw assemblies had remarkably lower CDS densities and more short CDSs (Supplementary Figure S7) than the others, suggesting the importance of polishing for this tool. As an indicator of interspecies misjoinings, we calculated a minor 1k-mer rate, which is the rate of the 1k-mer assigned to the minor species within scaffolds (see Supplementary Methods for details). If this value is high, there is a possibility of many chimeric regions and interspecies misjoinings. Comparing MetaPlatanus and metaFlye, which had high performances for the Nx-length plot and long-range metrics (over-half and near-complete scaffolds), MetaPlatanus produced lower minor 1k-mer rates in 10 cases (Supplementary Table S17F and G). These results indicate that MetaPlatanus does not extend sequences at the cost of accuracy.
Figure 2.

Evaluation of the human gut assemblies using databases. (A) # CDSs with hits to Swiss-Prot. (B) # biosynthetic gene clusters (≥1 kbp). (C) # near-complete viral sequences. (D) # over-half scaffolds. See Supplementary Methods for the detection methods.
Next, we focused on cases where only MetaPlatanus could assemble long scaffolds (completeness > 90%, contamination < 5%). The relations between assemblies were determined based on top hits of alignments by Minimap2, and we extracted two cases where the maximum length in the other assemblies was less than half of the MetaPlatanus scaffold length (Supplementary Table S18). One of these scaffolds (scaffold2094 of V06-T-0501-S07) corresponds to the genome of Bifidobacterium longum (Figure 3). Here, the overall structure of the MetaPlatanus scaffold was consistent with the complete genome of B. longum strain 51A, while the other assemblies, including MetaPlatanus w/o correction, were fragmented with more than seven sequences. Notably, some edges of other sequences had high rates of minor 1k-mers (shown as red boxes in Figure 3) and were identified as the candidate regions consisting of misassemblies. In our study, metaFlye connected the sequences of B. longum and B. breve in these regions. Moreover, this scaffold displayed a high coverage depth compared to the mean (Supplementary Table S18); thus, the corresponding species was expected to be highly abundant.
Figure 3.

MetaPlatanus-specific long scaffold in the human gut assemblies. This corresponds to scaffold2094 of the sample V06-T-0501-S07 (Supplementary Table S18). The blue, red, and gray boxes indicate the assignments of 1k-mers in assemblies (see Supplementary Methods). The gray shadows are alignments between the reference genome and assemblies (tool, BLASTN; identity ≥ 80%, alignment length ≥ 500 bp, sequence length ≥ 1 kbp).
Similar to the mock datasets, the real runtime of MetaPlatanus was longer than the others (Supplementary Table S19). However, the maximum real time was 13 h (24 threads), and it took <10 h for all but one case, suggesting its practicality.
We tested various binning methods as modules in the MetaPlatanus workflow (Supplementary Table S20). Using single-sample coverage depth, MetaBAT2 had the largest number of over-half bins. The combiners, DAS Tool (version 1.1.1) (47) and MetaWRAP (version 1.3.2) (46), performed well but did not exceed MetaBAT2 for all metrics. In the benchmark using multi-sample coverage depths (18 samples), over-half and near-complete bins were increased, but the scaffolds did not. In other words, single-sample coverage information is sufficient for MetaPlatanus to construct long scaffolds.
In addition, we benchmarked the assemblers inputting all samples at once, called co-assembly (Supplementary Table S21). MetaPlatanus had the largest number of over-half and near-complete scaffolds. However, these numbers are small compared to single-sample assemblies (Supplementary Table S17), which could be attributed to the increase in heterogeneity of genomic sequences.
Benchmarking with human saliva samples with long reads from PromethION
Finally, we performed benchmarking using a high-throughput ONT sequencer. The target four samples were from human saliva (24), and Illumina paired ends (37–55 Gbp), and ONT long reads (28–75 Gbp) were downloaded (Supplementary Table S7). In addition to large total sizes, the ONT reads had N50 lengths ranging from 21 to 29 kbp, the largest values for metagenomic data, to our knowledge. De novo assembly and removal of human contaminants were performed in a manner similar to that of the human gut. The statistics of the assemblies are shown in Supplementary Table S22. The detected non-human eukaryotic sequences (Supplementary Table S23) were fewer than those in the human gut, and we do not discuss these sequences.
In the Nx-length plots (Figure 4), MetaPlatanus had a substantially larger area under the curves compared to the others, including MetaPlatanus w/o correction. The trends of CDS densities and length distributions were similar to those in the human gut benchmark, suggesting MetaPlatanus w/o closing and metaFlye raw results had high and low base-level accuracies, respectively (Supplementary Table S22G and Supplementary Figure S8). Remarkably, in the evaluation of detected elements (Figure 5 and Supplementary Table S24), MetaPlatanus achieved the highest values for all samples and metrics, except for two cases: viral sequences of SAMD00202455 (Figure 5C) and near-complete scaffolds of SAMD0020247 (Figure 5E). In these results, the superiority of MetaPlatanus is more obvious than that of the human gut benchmarks, supporting the effectiveness of its algorithm for high-throughput data. The rates of exactly duplicated CDSs were generally greater than those in the human gut benchmarks (Supplementary Table S22H), but the rates were <7% for MetaPlatanus, which were smaller than those of OPERA-MS. For minor 1k-mer rates (Supplementary Table S24F), MetaPlatanus had the smallest values in three out of four cases, and the rates increased >1.5 times for MetaPlatanus w/o correction. These results are consistent with the hypothesis that MetaPlatanus enhances the contiguity and quality of assembled sequences using error-correction functions.
Figure 4.

Nx-length plot of the human saliva assemblies. The step size of x is 10 Mbp.
Figure 5.

Evaluation of the human saliva assemblies using databases. (A) # CDSs with hits to Swiss-Prot. (B) # biosynthetic gene clusters (≥1 kbp). (C) # near-complete viral sequences. (D) # over-half scaffolds. (E) # near-complete scaffolds. See Supplementary Methods for the detection methods.
Similar to the human gut benchmark, we extracted cases in which only MetaPlatanus assembled near-complete scaffolds. There were 22 such cases (Supplementary Table S25). Notably, in the majority of cases, the coverage depths of the near-complete scaffolds were higher than the mean values. An example with the highest coverage depth (>1000 for short and long reads) is shown in Figure 6. Here, the structure of the MetaPlatanus scaffold was consistent with the reference genome of Streptococcus salivarius strain HSISS4, and the assembly of MetaPlatanus w/o correction comprised extremely short sequences. The assemblies of the other tools were also fragmented into short pieces, and the edges of some sequences indicated signs of interspecies misassemblies (red boxes in Figure 6).
Figure 6.

MetaPlatanus-specific long scaffold in the human saliva assemblies. This corresponds to scaffold1433 of the sample SAMD00202457 (Supplementary Table S25). The blue, red, and gray boxes indicate the assignments of 1k-mers in assemblies (see Supplementary Methods). The gray shadows are alignments between the reference genome and assemblies (tool, BLASTN; identity ≥ 80%, alignment length ≥ 500 bp, sequence length ≥ 1 kbp).
The runtime of MetaPlatanus tended to be large. However, the differences between MetaPlatanus and OPERA-MS were less than those of the former benchmarks, and MetaPlatanus real times were smaller in two cases (Supplementary Table S26). In addition, the peak memory usage of MetaPlatanus was under 256GB, while OPERA-MS exceeded this amount in two cases. However, although metaSPAdes was faster than the other hybrid assemblers, its total assembly sizes were much smaller (Supplementary Table S22A), and it might omit a substantial number of sequences in its intermediate steps.
DISCUSSION
In this study, we describe the development of the hybrid metagenome assembler, MetaPlatanus, which iteratively utilizes long-range sequence links, species-specific sequence compositions, and coverage depth. The combination of assembly and binning information is used to improve the result of each genome and yield long scaffolds without increasing misassemblies and interspecies misjoins.
In the first mock benchmark, the basic ability of these tools for sequence contiguity and accuracy was confirmed. It was also shown that MetaPlatanus could handle various types of long-range links, such as ONT, PacBio and mate-pairs. Although the metaFlye assembly exhibited high performance with large-scale accuracy and contiguity, short read-based assemblies had high fine-scale accuracy. MetaPlatanus w/o closing had the highest accuracy for all metrics, suggesting that the pre-closing results (preClose.fa) could be used to identify accurate sequences.
In the second human gut benchmark, we demonstrated that MetaPlatanus could be applied to datasets of small ONT sequencers and Illumina paired-ends. The number of samples was relatively high (18), and the N50 lengths ranged widely from 668 to 6360 bp. Thus, the robustness of the performance of MetaPlatanus was confirmed in these tests. Given the cost-effectiveness of the portable nanopore sequencer and Illumina paired-ends for pilot or small projects, MetaPlatanus could serve as a powerful tool.
Although hybrid assemblers are often reported to be advantageous for low-coverage data (33,60), in the last benchmark using high-throughput long and short reads, we demonstrated a higher efficacy of MetaPlatanus for error-correction functions than that of the other benchmarks. Here, heterogeneity in the microbiome may interfere with existing assemblers, including long read-based metaFlye. Fortunately, MetaPlatanus was found to have the potential to solve some cases by correctly removing false links. It is also remarkable that MetaPlatanus-specific near-complete scaffolds corresponded to many high-abundance bacteria. This circumstance can also be caused by heterogeneity, and these bacteria may contribute to the microbiome with important functions.
As a feature of the algorithm, OPERA-MS consists of a binning-like clustering step utilizing contig connections, coverage depths, and optionally reference genomes in the database (22). MetaPlatanus additionally uses 4-mers and binning information, and the effect of the differences was confirmed in the benchmarks above. Note that this study focuses on de novo methods, and reference information has not been well examined. Furthermore, all methods except for some tested binning tools (MaxBin2, DAS Tool and MetaWRAP) do not utilize such information, including single-copy genes.
The drawback of a scaffolding strategy is that it introduces gaps in the resulting sequences. In contrast, the long read-based assembler, metaFlye, is equipped with algorithms that assume various structures such as superbubbles and roundabout (44) in the graph to extend contigs. However, there is no comprehensive list of the types of local structures in assembly graphs, and the graph for metagenomic assembly may have other complex structures not assumed by long read-based tools. For example, a superbubble-like structure lacks some edges due to low coverage depth and a combination of inversion and large indels. The scaffolding strategy can skip the complex structures stemming from heterogeneity or repeats in metagenomes as gaps; therefore, a contig assembly strategy based on long reads is not ideal for obtaining long sequences in some cases. The cases in which MetaPlatanus succeeded in assembly but metaFlye failed despite high long-read coverage (Figure 6 and Supplementary Table S25) may reflect the advantage of the scaffolding strategy.
Although metaFlye recorded high contiguity in many cases, the fine-scale accuracies of its raw results were suggested to be low from the mock tests (Table 1) and quality assessments for actual data (Supplementary Table S17 and S24). Here, the polishing module in metaFlye was executed as the default action. In other words, metaFlye also requires additional short-read sequencing and polishing for rigorous downstream analysis. The sequencing cost per base of short-read sequencers is generally lower than that of the long-read sequencers (27,61). Considering the above, a hybrid strategy with short- and long-read sequencers is not much more costly compared to a long-read-only study design.
In conclusion, MetaPlatanus, which employs the accuracy of short reads, long-range links, species-specific features, and binning, is useful for the acquisition of metagenomic information as chromosome-scale scaffolds. We believe that the metagenome assembler developed here could help examine the contexts of sequence elements spreading over the genome.
DATA AVAILABILITY
MetaPlatanus is freely available at GitHub (https://github.com/rkajitani/MetaPlatanus) or http://platanus.bio.titech.ac.jp/metaplatanus/ under the GPLv3 license. It is also used via Bioconda (62).
Raw reads of the MP mock samples were deposited in the DDBJ Sequence Read Archive under BioProject ID PRJDB4377. Assemblies generate in this study are available at FigShare (https://doi.org/10.6084/m9.figshare.15090603.v1).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Rie Ryusui for sequencing the synthetic human gut samples.
Author contributions: T.I. and T.H. designed the study. R.K. developed de novo assembly modules. R.K. and N.H. developed a function to correct interspecies misjoinings. A.T., K.T. and Y.O. performed the sequencing of the MP mock samples. R.K., D.Y., M.O., Y.G. and Y.O. performed the benchmarks. R.K., T.I. and T.H. wrote the manuscript. All authors have read and approved the final version of the manuscript.
Contributor Information
Rei Kajitani, School of Life Science and Technology, Tokyo Institute of Technology, Meguro-ku, Tokyo 152-8550, Japan.
Hideki Noguchi, Advanced Genomics Center, National Institute of Genetics, Mishima, Shizuoka 411-8540, Japan.
Yasuhiro Gotoh, Department of Bacteriology, Graduate School of Medical Sciences, Kyushu University, Higashi-ku, Fukuoka 812-8582, Japan.
Yoshitoshi Ogura, Division of Microbiology, Department of Infectious Medicine, Kurume University School of Medicine, Asahi-machi, Kurume, Fukuoka 830-0011, Japan.
Dai Yoshimura, School of Life Science and Technology, Tokyo Institute of Technology, Meguro-ku, Tokyo 152-8550, Japan.
Miki Okuno, Division of Microbiology, Department of Infectious Medicine, Kurume University School of Medicine, Asahi-machi, Kurume, Fukuoka 830-0011, Japan.
Atsushi Toyoda, Advanced Genomics Center, National Institute of Genetics, Mishima, Shizuoka 411-8540, Japan; Comparative Genomics Laboratory, National Institute of Genetics, Mishima, Shizuoka 411-8540, Japan.
Tomomi Kuwahara, Department of Molecular Microbiology, Faculty of Medicine, Kagawa University, Miki-cho, Kita-gun, Kagawa 761-0793, Japan.
Tetsuya Hayashi, Department of Bacteriology, Graduate School of Medical Sciences, Kyushu University, Higashi-ku, Fukuoka 812-8582, Japan.
Takehiko Itoh, School of Life Science and Technology, Tokyo Institute of Technology, Meguro-ku, Tokyo 152-8550, Japan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
JSPS KAKENHI [JP16H06279 (PAGS), JP15H05979, JP18K19286, JP19H03206, JP20K15769, JP21K19211] from the Ministry of Education, Culture, Sports, Science and Technology of Japan; AMED [JP16gm6010003] from the Japan Agency for Medical Research and Development.
Conflict of interest statement. None declared.
REFERENCES
- 1. Chin C.-S., Alexander D.H., Marks P., Klammer A.A., Drake J., Heiner C., Clum A., Copeland A., Huddleston J., Eichler E.E.et al.. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods. 2013; 10:563–569. [DOI] [PubMed] [Google Scholar]
- 2. Tyson G.W., Chapman J., Hugenholtz P., Allen E.E., Ram R.J., Richardson P.M., Solovyev V.V., Rubin E.M., Rokhsar D.S., Banfield J.F.. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004; 428:37–43. [DOI] [PubMed] [Google Scholar]
- 3. Venter J.C., Remington K., Heidelberg J.F., Halpern A.L., Rusch D., Eisen J.A., Wu D., Paulsen I., Nelson K.E., Nelson W.et al.. Environmental genome shotgun sequencing of the Sargasso sea. Science. 2004; 304:66–74. [DOI] [PubMed] [Google Scholar]
- 4. Qin J., Li R., Raes J., Arumugam M., Burgdorf K.S., Manichanh C., Nielsen T., Pons N., Levenez F., Yamada T.et al.. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010; 464:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xie F., Jin W., Si H., Yuan Y., Tao Y., Liu J., Wang X., Yang C., Li Q., Yan X.et al.. An integrated gene catalog and over 10,000 metagenome-assembled genomes from the gastrointestinal microbiome of ruminants. Microbiome. 2021; 9:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brown C.T., Hug L.A., Thomas B.C., Sharon I., Castelle C.J., Singh A., Wilkins M.J., Wrighton K.C., Williams K.H., Banfield J.F.. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature. 2015; 523:208–211. [DOI] [PubMed] [Google Scholar]
- 7. Spang A., Saw J.H., Jørgensen S.L., Zaremba-Niedzwiedzka K., Martijn J., Lind A.E., van Eijk R., Schleper C., Guy L., Ettema T.J.G.. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature. 2015; 521:173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zaremba-Niedzwiedzka K., Caceres E.F., Saw J.H., Bäckström D., Juzokaite L., Vancaester E., Seitz K.W., Anantharaman K., Starnawski P., Kjeldsen K.U.et al.. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature. 2017; 541:353–358. [DOI] [PubMed] [Google Scholar]
- 9. Dutilh B.E., Cassman N., McNair K., Sanchez S.E., Silva G.G.Z., Boling L., Barr J.J., Speth D.R., Seguritan V., Aziz R.K.et al.. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014; 5:4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Devoto A.E., Santini J.M., Olm M.R., Anantharaman K., Munk P., Tung J., Archie E.A., Turnbaugh P.J., Seed K.D., Blekhman R.et al.. Megaphages infect Prevotella and variants are widespread in gut microbiomes. Nat. Microbiol. 2019; 4:693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boisvert S., Raymond F., Godzaridis E., Laviolette F., Corbeil J.. Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol. 2012; 13:R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peng Y., Leung H.C.M., Yiu S.M., Chin F.Y.L.. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012; 28:1420–1428. [DOI] [PubMed] [Google Scholar]
- 13. Haider B., Ahn T.-H., Bushnell B., Chai J., Copeland A., Pan C.. Omega: an overlap-graph de novo assembler for metagenomics. Bioinformatics. 2014; 30:2717–2722. [DOI] [PubMed] [Google Scholar]
- 14. Nurk S., Meleshko D., Korobeynikov A., Pevzner P.A.. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017; 27:824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Namiki T., Hachiya T., Tanaka H., Sakakibara Y.. MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res. 2012; 40:e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shaiber A., Eren A.M. Composite metagenome-assembled genomes reduce the quality of public genome repositories. mBio. 2019; 10:e00725-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stewart R.D., Auffret M.D., Warr A., Wiser A.H., Press M.O., Langford K.W., Liachko I., Snelling T.J., Dewhurst R.J., Walker A.W.et al.. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 2018; 9:870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Parks D.H., Rinke C., Chuvochina M., Chaumeil P.-A., Woodcroft B.J., Evans P.N., Hugenholtz P., Tyson G.W.. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017; 2:1533–1542. [DOI] [PubMed] [Google Scholar]
- 19. Pasolli E., Asnicar F., Manara S., Zolfo M., Karcher N., Armanini F., Beghini F., Manghi P., Tett A., Ghensi P.et al.. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell. 2019; 176:649–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Almeida A., Mitchell A.L., Boland M., Forster S.C., Gloor G.B., Tarkowska A., Lawley T.D., Finn R.D.. A new genomic blueprint of the human gut microbiota. Nature. 2019; 568:499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stewart R.D., Auffret M.D., Warr A., Walker A.W., Roehe R., Watson M.. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 2019; 37:953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bertrand D., Shaw J., Kalathiyappan M., Ng A.H.Q., Kumar M.S., Li C., Dvornicic M., Soldo J.P., Koh J.Y., Tong C.et al.. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nat. Biotechnol. 2019; 37:937–944. [DOI] [PubMed] [Google Scholar]
- 23. Moss E.L., Maghini D.G., Bhatt A.S.. Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nat. Biotechnol. 2020; 38:701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yahara K., Suzuki M., Hirabayashi A., Suda W., Hattori M., Suzuki Y., Okazaki Y.. Long-read metagenomics using PromethION uncovers oral bacteriophages and their interaction with host bacteria. Nat. Commun. 2021; 12:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Singleton C.M., Petriglieri F., Kristensen J.M., Kirkegaard R.H., Michaelsen T.Y., Andersen M.H., Kondrotaite Z., Karst S.M., Dueholm M.S., Nielsen P.H.et al.. Connecting structure to function with the recovery of over 1000 high-quality metagenome-assembled genomes from activated sludge using long-read sequencing. Nat. Commun. 2021; 12:2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bickhart D.M., Watson M., Koren S., Panke-Buisse K., Cersosimo L.M., Press M.O., Van Tassell C.P., Van Kessel J.A.S., Haley B.J., Kim S.W.et al.. Assignment of virus and antimicrobial resistance genes to microbial hosts in a complex microbial community by combined long-read assembly and proximity ligation. Genome Biol. 2019; 20:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Maio N., Shaw L.P., Hubbard A., George S., Sanderson N.D., Swann J., Wick R., AbuOun M., Stubberfield E., Hoosdally S.J.et al.. Comparison of long-read sequencing technologies in the hybrid assembly of complex bacterial genomes. Microbial Genomics. 2019; 5:e000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Giordano F., Aigrain L., Quail M.A., Coupland P., Bonfield J.K., Davies R.M., Tischler G., Jackson D.K., Keane T.M., Li J.et al.. De novo yeast genome assemblies from MinION, PacBio and MiSeq platforms. Sci. Rep. 2017; 7:3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kajitani R., Yoshimura D., Ogura Y., Gotoh Y., Hayashi T., Itoh T.. Platanus_B: an accurate de novo assembler for bacterial genomes using an iterative error-removal process. DNA Res. 2020; 27:dsaa014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Watson M., Warr A.. Errors in long-read assemblies can critically affect protein prediction. Nat. Biotechnol. 2019; 37:124–126. [DOI] [PubMed] [Google Scholar]
- 31. Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M.. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017; 27:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kolmogorov M., Yuan J., Lin Y., Pevzner P.A.. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019; 37:540–546. [DOI] [PubMed] [Google Scholar]
- 33. Di Genova A., Buena-Atienza E., Ossowski S., Sagot M.-F.. Efficient hybrid de novo assembly of human genomes with WENGAN. Nat. Biotechnol. 2020; 39:422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haghshenas E., Asghari H., Stoye J., Chauve C., Hach F.. HASLR: fast hybrid assembly of long reads. iScience. 2020; 23:101389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brown C.L., Keenum I.M., Dai D., Zhang L., Vikesland P.J., Pruden A.. Critical evaluation of short, long, and hybrid assembly for contextual analysis of antibiotic resistance genes in complex environmental metagenomes. Sci. Rep. 2021; 11:3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alneberg J., Bjarnason B.S., de Bruijn I., Schirmer M., Quick J., Ijaz U.Z., Lahti L., Loman N.J., Andersson A.F., Quince C.. Binning metagenomic contigs by coverage and composition. Nat. Methods. 2014; 11:1144–1146. [DOI] [PubMed] [Google Scholar]
- 37. Wu Y.-W., Simmons B.A., Singer S.W.. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2016; 32:605–607. [DOI] [PubMed] [Google Scholar]
- 38. Kang D.D., Li F., Kirton E., Thomas A., Egan R., An H., Wang Z.. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019; 7:e7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kajitani R., Toshimoto K., Noguchi H., Toyoda A., Ogura Y., Okuno M., Yabana M., Harada M., Nagayasu E., Maruyama H.et al.. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014; 24:1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kajitani R., Yoshimura D., Okuno M., Minakuchi Y., Kagoshima H., Fujiyama A., Kubokawa K., Kohara Y., Toyoda A., Itoh T.. Platanus-allee is a de novo haplotype assembler enabling a comprehensive access to divergent heterozygous regions. Nat. Commun. 2019; 10:1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu M., Guo L., Gu S., Wang O., Zhang R., Peters B.A., Fan G., Liu X., Xu X., Deng L.et al.. TGS-GapCloser: a fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience. 2020; 9:giaa094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hu J., Fan J., Sun Z., Liu S.. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 2020; 36:2253–2255. [DOI] [PubMed] [Google Scholar]
- 43. Antipov D., Korobeynikov A., McLean J.S., Pevzner P.A.. hybridSPAdes: an algorithm for hybrid assembly of short and long reads. Bioinformatics. 2016; 32:1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kolmogorov M., Bickhart D.M., Behsaz B., Gurevich A., Rayko M., Shin S.B., Kuhn K., Yuan J., Polevikov E., Smith T.P.L.et al.. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat. Methods. 2020; 17:1103–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mikheenko A., Saveliev V., Gurevich A.. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics. 2016; 32:1088–1090. [DOI] [PubMed] [Google Scholar]
- 46. Uritskiy G.V., DiRuggiero J., Taylor J.. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018; 6:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sieber C.M.K., Probst A.J., Sharrar A., Thomas B.C., Hess M., Tringe S.G., Banfield J.F.. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018; 3:836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D.et al.. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012; 19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li D., Liu C.-M., Luo R., Sadakane K., Lam T.-W.. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015; 31:1674–1676. [DOI] [PubMed] [Google Scholar]
- 50. Levy Karin E., Mirdita M., Söding J.. MetaEuk—sensitive, high-throughput gene discovery, and annotation for large-scale eukaryotic metagenomics. Microbiome. 2020; 8:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. The UniProt Consortium UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021; 49:D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hyatt D., Chen G.-L., LoCascio P.F., Land M.L., Larimer F.W., Hauser L.J.. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010; 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Buchfink B., Xie C., Huson D.H.. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015; 12:59–60. [DOI] [PubMed] [Google Scholar]
- 54. Blin K., Shaw S., Steinke K., Villebro R., Ziemert N., Lee S.Y., Medema M.H., Weber T.. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019; 47:W81–W87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nayfach S., Camargo A.P., Schulz F., Eloe-Fadrosh E., Roux S., Kyrpides N.C.. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021; 39:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bowers R.M., Kyrpides N.C., Stepanauskas R., Harmon-Smith M., Doud D., Reddy T.B.K., Schulz F., Jarett J., Rivers A.R., Eloe-Fadrosh E.A.et al.. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017; 35:725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Parks D.H., Imelfort M., Skennerton C.T., Hugenholtz P., Tyson G.W.. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015; 25:1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014; 30:2068–2069. [DOI] [PubMed] [Google Scholar]
- 59. Fu L., Niu B., Zhu Z., Wu S., Li W.. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012; 28:3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ye C., Hill C.M., Wu S., Ruan J., Ma Z.. DBG2OLC: Efficient Assembly of Large Genomes Using Long Erroneous Reads of the Third Generation Sequencing Technologies. Sci. Rep. 2016; 6:31900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jeon S.A., Park J.L., Kim J.-H., Kim J.H., Kim Y.S., Kim J.C., Kim S.-Y.. Comparison of the MGISEQ-2000 and Illumina HiSeq 4000 sequencing platforms for RNA sequencing. Genomics Inform. 2019; 17:e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Grüning B., Dale R., Sjödin A., Chapman B.A., Rowe J., Tomkins-Tinch C.H., Valieris R., Köster J.. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat. Methods. 2018; 15:475–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MetaPlatanus is freely available at GitHub (https://github.com/rkajitani/MetaPlatanus) or http://platanus.bio.titech.ac.jp/metaplatanus/ under the GPLv3 license. It is also used via Bioconda (62).
Raw reads of the MP mock samples were deposited in the DDBJ Sequence Read Archive under BioProject ID PRJDB4377. Assemblies generate in this study are available at FigShare (https://doi.org/10.6084/m9.figshare.15090603.v1).