

Abstract
Alternative splicing has emerged as a fundamental mechanism for the spatiotemporal control of development. A better understanding of how this mechanism is regulated has the potential not only to elucidate fundamental biological principles, but also to decipher pathological mechanisms implicated in diseases where normal splicing networks are misregulated. Here, we took advantage of human pluripotent stem cells to decipher during human myogenesis the role of muscleblind-like (MBNL) proteins, a family of tissue-specific splicing regulators whose loss of function is associated with myotonic dystrophy type 1 (DM1), an inherited neuromuscular disease. Thanks to the CRISPR/Cas9 technology, we generated human-induced pluripotent stem cells (hiPSCs) depleted in MBNL proteins and evaluated the consequences of their losses on the generation of skeletal muscle cells. Our results suggested that MBNL proteins are required for the late myogenic maturation. In addition, loss of MBNL1 and MBNL2 recapitulated the main features of DM1 observed in hiPSC-derived skeletal muscle cells. Comparative transcriptomic analyses also revealed the muscle-related processes regulated by these proteins that are commonly misregulated in DM1. Together, our study reveals the temporal requirement of MBNL proteins in human myogenesis and should facilitate the identification of new therapeutic strategies capable to cope with the loss of function of these MBNL proteins.
Introduction
Alternative splicing has emerged in the post-genomic era as the main driver of proteome diversity. The identification that at least 95% of human genes produce multiple spliced RNA via alternative exon usage has revealed the prevalence of this additional layer of gene expression regulation (1,2). The regulation of alternative splicing varies with cell type, during development or upon cellular differentiation, thereby participating in the fine-tuning of a gene signature temporally and spatially (3). To ensure this developmental and tissue-specific regulation, alternative splicing relies on complex mechanisms involving an intricate protein–RNA network. This leads to the precise and sequential recognition of cis-acting sequences on the pre-mRNA by trans-acting RNA-binding factors. It has become apparent that these coordinated splicing networks have important physiological functions in developmental processes and that disruption of these networks negatively impacts health and contributes to human diseases.
According to some estimates, up to 50% of all pathogenic mutations may affect splicing in some way (4). Illustrating this, myotonic dystrophy type 1 (DM1) represents one paradigmatic example of genetic disease in which loss of muscleblind-like (MBNL) splicing factors is considered as the central pathological event. DM1 is an autosomal dominant neuromuscular disease caused by abnormal expansion of CTG repeats in the 3′ untranslated region of the Dystrophia myotonia protein kinase (DMPK) gene (5,6). The pathogenesis of DM1 involves an RNA gain-of-function mechanism caused by the expression of mutant RNAs containing hundreds to thousands of CUG repeats that sequester MBNL RNA-binding proteins (RBP) involved in the regulation of alternative splicing (7,8). This functional loss of MBNL proteins results in several alternative splicing misregulation events that have been associated for some of them to clinical symptoms such as myotonia, muscle weakness and insulin resistance (7,9). Three different genes encode human MBNL homologs: MBNL1, MBNL2 and MBNL3, which all share structural similarities (8,10,11). MBNL1 and MBNL2 are ubiquitously expressed, but whereas MBNL1 is found in similar levels in all tissues, MBNL2 is more abundant in the brain (10,11). In a majority of tissues MBNL1 and MBNL2 expression rises during differentiation. In contrast, MBNL3 is expressed at much lower levels, with peak expression in placenta, and its functions are poorly defined compared with its two other paralogs (11,12). According to this differential expression over time, MBNL1 and MBNL2 proteins have been shown to modulate the splicing profile of a large number of genes during fetal-to-adult transitions (13). At the functional level, knockout Mbnl1 mice display skeletal muscle defects such as myotonia or central nuclei only at an adult stage (10), whereas loss of Mbnl2 mice leads predominantly to brain phenotypes with no major defects in skeletal muscles (14). However, the exploitation of these loss-of-function models was hampered by compensatory mechanisms that result in an increased expression of Mbnl2 as observed in knockout Mbnl1 mice (15). In contrast, Mbnl1(−/−); Mbnl2(−/−) double knockouts (DKOs) are embryonic lethal, whereas Mbnl1(−/−); Mbnl2(+/−) mice are viable but develop severe phenotypes such as reduced lifespan, heart conduction block, severe myotonia and progressive skeletal muscle weakness (15,16). Concerning Mbnl3(−/−) mice, results showed decreased skeletal muscle regeneration and reduced grip strength associated with age despite low expression of Mbnl3 in adult muscles. Moreover, all the phenotypes described in Mbnl3 (−/−) mice were not associated with significant changes in alternative splicing (17,18). Altogether, these studies have highlighted the developmental function of Mbnl proteins in mice, but they also raise the question of whether MBNL proteins are similarly involved in human development as well as the precise timing at which these proteins intervene.
In this study, we focused on the role of MBNL proteins during human skeletal myogenesis. Because of their capacity to differentiate into any cell types of an organism, we based our approach on the combined use of human-induced pluripotent stem cells (hiPSCs) and CRISPR/Cas9 technology to generate hiPSCs depleted in MBNL homologs and to therefore evaluate the impact of MBNL proteins loss at the cellular and molecular level during the different developmental steps of myogenesis.
Results
CRISPR/Cas9 strategy to generate MBNL KO(s) hiPSCs
To generate MBNL knockout hiPSC lines, sgRNAs were specifically designed to target one of the first coding exons as well as the presence of an enzymatic restriction site at the PAM sequence (Fig. 1A and B) (19). Each sgRNA was complexed with SpCas9 protein to perform ribonucleoprotein transfection of dissociated hiPSCs (20). The selection of gene-edited clones was on the basis of a two-step selection process. Thanks to the use of sgRNA targeting sequences within a restriction endonuclease site (Fig. 1B; framed in purple) at the PAM level (Fig. 1B; NGG in red), restriction fragment length polymorphism was first analyzed to rapidly detect CRISPR gene editing according to total or partial loss of restriction sites (Supplementary Material, Table S1). The second selection was on the basis of Sanger DNA sequencing to select clones with INDELS that have generated STOP codons (Fig. 1C). For MBNL1, we obtained an editing efficiency of 25% with 24 pre-selected clones out of 96 clones tested (10 with a homozygous profile and 14 heterozygous). After the sequencing of these 24 clones, 3 homozygous and 2 heterozygous clones were validated (Fig. 1C and D and Supplementary Material, Fig. S1A). Concerning MBNL2, 9 clones were pre-selected out of 96 (editing efficiency of 9%) and 3 were finally validated by sequencing. We generated two heterozygous clones and obtained only one MBNL2(−/−) homozygous knockout clone (Fig. 1C and Supplementary Material, Fig. S1A). Because of the compensatory phenomena described between Mbnl2 and Mbnl1 in knockout mice (15), DKO MBNL1(−/−); MBNL2(−/−) were generated (Fig. 1C and D and Supplementary Material, Fig. S1A). The condition MBNL1(−/−); MBNL2(+/−) was also generated as Mbnl1(−/−); Mbnl2(−/−) double mice knockouts are embryonic lethal, whereas Mbnl1(−/−); Mbnl2(+/−) mice are viable but develop severe phenotypes (15,16). Finally, to decipher the contribution of MBNL3, we generated triple knockout (TKO) of MBNL1(−/−); MBNL2(−/−); MBNL3(−/−). We obtained an editing efficiency of 22% and validated five different TKO clones, two of which were considered for further analyses (Fig. 1C and D and Supplementary Material, Fig. S1A). For each generated clone, pluripotency was validated by flow cytometry of the pluripotent markers SSEA3 and TRA1-81. Genomic integrity was also verified by a karyotype analysis and SNP genotyping (Supplementary Material, Fig. S1B and C). To determine off-target activity of our gRNAs, we analyzed the most likely off-target sites, and no mutation induced by genome editing was observed (Supplementary Material, Table S2).
Figure 1.

Generation of MBNL knockout hiPSCs by CRISPR/Cas9. (A) Schematic representation of the different exons encoding human MBNL1, MBNL2 and MBNL3. Alternative exons are indicated in blue and the targeted site of each guide-RNA is indicated by an orange arrow. (B) Edited clones were first identified by RFLP. The on-target site is underlined and the PAM sequence is in red. The restriction endonuclease recognition site (BsrI in this case) is framed in purple. After locus-specific PCR and restriction digest, cleaved fragments are separated by gel electrophoresis into readily distinguishable profiles, allowing an efficient and rapid screening of single clones to further analyze by sequencing. (C) Examples of sequencing of the targeting site in human MBNL1, MBNL2 and MBNL3 genes after NHEJ-mediated knockout using the CRISPR/Cas9 system in WT hiPSCs. The MBNL1(−/−) clone harbors a deletion of 14 bp; MBNL2(−/−) a deletion of 4 bp; MBNL1(−/−); MBNL2(−/−) (DKO) a deletion of 35 bp and MBNL1(−/−); MBNL2(−/−); MBNL3(−/−) (TKO) a deletion of 7 bp. The DKO clone was generated from MBNL2(−/−) clone and the TKO clone from the DKO clone. STOP codons induced by INDELS are framed in red. (D) Schematic representation of the different gene editing experiments performed to obtain MBNL knockout hiPSCs clones. Created with Biorender.com.
No major impairment of the early myogenic commitment in the absence of MBNL proteins
The capacity of MBNL-depleted human pluripotent stem cells to differentiate into skeletal muscle cells was evaluated by using a transgene-free protocol that can be schematically divided into three steps (21) (Fig. 2A). We first monitored the expression of the different isoforms of MBNL at the protein and mRNA level along this myogenic differentiation process. As previously reported (22), MBNL1 and MBNL2 were not found to be expressed in undifferentiated human pluripotent stem cells. Their level of expression gradually increased as differentiation progresses to reach a maximum expression after ~ 20 days of differentiation, corresponding to the beginning of the final myogenic maturation step (Fig. 2B and Supplementary Material, Fig. S2A and D).
Figure 2.

Expression profile of the three MBNL paralogs during myogenic differentiation from hiPSCs. (A) Schematic representation of the differentiation protocol used to convert hiPSCs into skeletal muscle cells. Created with Biorender.com. (B) Representative examples of western blot analysis for MBNL1 and MBNL2 at different time points of the myogenic differentiation. The detection of the two isoforms was highlighted by an arrowhead (for 40 kDa) and an arrow (for 42 kDa) as previously described (12). The graph represents the kinetics of expression of the three MBNL paralogs along the process of differentiation of WT hiPSCs. β-Actin was used as housekeeping protein for normalization. Data represent the mean ± SD values from two independent experiments. (C–D) Example of a western blot analysis for MBNL1 (C) and MBNL2 (D) proteins after 24 days of differentiation (myotube stage). The graphs on the right panel represent the quantification of MBNL proteins expression in the different conditions compared with WT. β-Actin was used as housekeeping protein for normalization. Data represent means ± SD values from three independent experiments (*P < 0.05; **P < 0.005; ***, P < 0.0005; ****P < 0.0001—ordinary one-way ANOVA, Tukey’s multiple comparisons test).
In contrast, MBNL3 expression slightly increased during the first days of differentiation followed by its decreased expression at later time points (Fig. 2B and Supplementary Material, Fig. S2A–D). As previously described in human primary myoblasts, our results also confirmed that MBNL1 is the most abundant MBNL proteins in hiPSC-derived skeletal muscle cells, although MBNL1 and MBNL2 proteins follow a similar kinetic of expression (Fig. 2B).
The analysis at the protein level demonstrated a specific lack of expression of MBNL paralogs in the different gene-edited hiPSC lines differentiated into skeletal muscle cells (Fig. 2C and D). As reported in mouse model of Mbnl loss of function (15), we also observed a compensatory increase in MBNL1 and MBNL2 expression in MBNL2(−/−) and MBNL1(−/−), respectively (Fig. 2C and D and Supplementary Material, Fig. S2E). MBNL3 level was found not be increased as a compensatory response to MBNL1 and MBNL2 loss (Supplementary Material, Fig. S2C and E).
To evaluate the functional impact of MBNL proteins loss during myogenic differentiation, the expression of stage-specific muscle markers was evaluated at different time points. As MBNL proteins are knows to be important players in DM1 pathogenesis (23,24), we used DM1 hiPSC as a reference (Fig. 3A and B). At 5 days of differentiation, ~80% of cells expressed the myogenic progenitor marker PAX3, with no significant difference between the different cell lines indicating that muscle lineage commitment is not affected by MBNL depletion (Fig. 4A and Supplementary Material, Fig. S3A). Similar observation was made at 16 days of differentiation by the quantification of cells positive for DESMIN, a muscle-specific intermediate filament. Approximatively 75% cells were DESMIN+ with no significant difference between conditions showing that the myogenic precursors developed into skeletal myoblasts in the absence of MBNL proteins (Fig. 4B and Supplementary Material, Fig. S3B).
Figure 3.
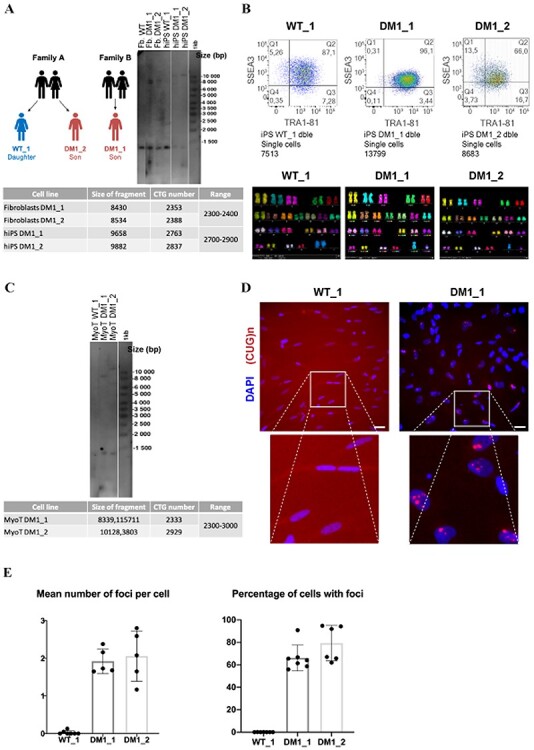
Presence of CUG foci in skeletal muscle cells derived from DM1 hiPSCs. (A) Fibroblasts from two DM1 patients and one healthy donor subject (WT; sibling of one DM1 patient) were used for reprogramming into hiPSCs. Southern blot analysis of the CTG repeats length present in the different fibroblasts and hiPSCs. (B) Characterization of WT and DM1 hiPSCs by flow cytometry for two markers of pluripotency (SSEA3 and TRA1-81). Genomic integrity of these hiPSC lines was validated by karyotyping analysis. All the MBNL KO clones were generated from the WT_1 cell line. (C) Southern blot analysis of the CTG repeat length in hiPSC-derived skeletal muscle cells (24 days after differentiation). (D) Representative images of double immunolabeling for intranuclear CUG repeat foci (red) and DAPI nuclear staining (blue) of WT and DM1 hiPSC-derived skeletal muscle cells (24 days of differentiation). The images below are higher magnifications of the inset to better visualize the presence of CUG RNA foci in DM1 nuclei. Scale bar 50 μM. (E) Quantification of the number of CUG repeat foci per cell as well as the percentage of cells with CUG RNA foci in skeletal muscle cells derived from WT and the two DM1 hiPSC lines (24 days of differentiation). The quantification of CUG repeat foci was performed as previously described (59). Data represent the mean ± SD values from at least three independent experiments in quadruplicate (*P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001—ordinary one-way ANOVA, Tukey’s multiple comparisons test).
Figure 4.
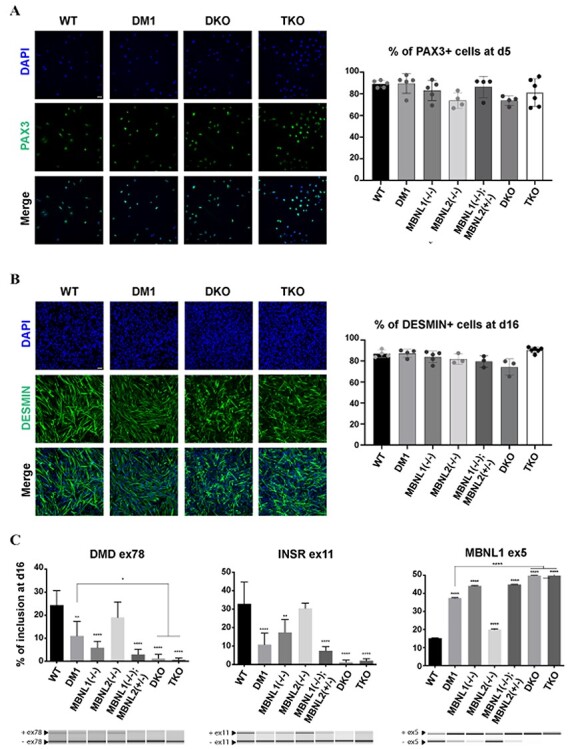
No impairment of the early myogenic commitment in the absence of the MBNL proteins. Representative images of immunolabeling for PAX3 (A) and DESMIN (B) on hiPSCs differentiated for 5 and 16 days, respectively (PAX3 and DESMIN are in green, DAPI nuclear staining is in blue). Scale bar 10 μM. The graphs on the left represent the percentage of PAX3+ or DESMIN+ cells, after automated quantification. Data represent the mean ± SD values from at least three independent experiments in quadruplicate. (C) Alternative splicing analysis for DMD exon 78, INSR exon 11 and MBNL1 exon 5 in hiPSCs-derived myoblasts (16 days of differentiation). Data represent the mean ± SD values from three independent experiments in duplicate (*P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001—ordinary one-way ANOVA, Tukey’s multiple comparisons test). Asterisks placed above individual bar correspond to the comparison with WT.
Despite the absence of phenotypes at the cellular level, we sought to evaluate the molecular consequences of MBNL depletion. We chose to focus on several gene transcripts important for skeletal muscle function and previously shown to be mis-spliced in DM1 patients’ biopsies (9,25). Although these mis-splicing events were identified in adult tissues, reverse transcriptase polymerase chain reaction (RT-PCR) analysis showed that the splicing profiles of DMD exon 78, INSR exon 11 and MBNL1 exon 5 were significantly deregulated at the myoblast stage in DM1, MBNL1(−/−), DKO and TKO compared with non-DM1 control [wild-type (WT)] (Fig. 4C and Supplementary Material, Fig. S3C). DKO showed more pronounced splicing changes than MBNL1(−/−) and DM1 conditions. A similar observation was made with the depletion of the three paralogs (Fig. 4C). In contrast, the loss of MBNL2 alone did not significantly lead to mis-splicing events when compared with non-DM1 control (WT) myoblasts illustrating the compensation of MBNL2 by MBNL1 as previously reported (23) (Fig. 4C). These observations suggested that MBNL proteins are dispensable for myogenic commitment even if MBNL loss of function led to early mis-splicing events that could interfere with later stages of myogenesis.
MBNL1 and MBNL2 depletion impacts late myogenesis and reproduces the main features associated with DM1
We next evaluated the consequences of MBNL loss of function on the capacity of hiPSC-derived myoblasts to further differentiate and fuse into myotubes (21). As a point of reference, we first characterized this late differentiation by using DM1 hiPSC-derived myoblasts. Primary or immortalized muscle cell cultures derived from muscle biopsies of DM1 patients have been shown to present altered myogenic fusion as well as DM1-associated molecular features including mutated DMPK mRNAs aggregates that sequester MBNL1 and subsequently lead to alternative splicing defects (26). Accordingly, we also observed that DM1 hiPSC-derived myoblasts under differentiation condition form smaller myotubes and present a significantly reduced fusion capacity when compared with WT hiPSC-derived myoblasts (Fig. 5A and Supplementary Material, Fig. S4A). Moreover, the main DM1-associated molecular features including mutated DMPK mRNAs aggregates, nuclear sequestration of MBNL1 and MBNL2 and presence of several mis-spliced events were detected in these cells (Figs 3C–E and 5B–C and Supplementary Material, Fig. S4B). These data demonstrate that DM1 hiPSC-derived skeletal myotubes recapitulate phenotypic and molecular traits similar to those found in skeletal muscle cell cultures isolated from DM1 patients.
Figure 5.

Depletion of MBNL1 and MBNL2 proteins impairs myogenic fusion and reproduces DM1-associated phenotypes. (A) Representative images of immunolabeling for DESMIN (green), myosin heavy chain (MF20) (red) and DAPI nuclear staining (blue) in skeletal muscle cells generated from hiPSCs after 24 days of differentiation. Scale bar 5 μM. The graph on the right represents the number of myotubes containing at least three nuclei normalized to the total number of nuclei. A minimum of 11 000 nuclei has been quantified per experiment and per cell line. Data represent mean ± SD values after normalization on the value obtained with WT hiPSC-derived skeletal muscle cells (n = at least three independent experiments). Data were analyzed with an ordinary one-way ANOVA, Tukey’s multiple comparisons test (*P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001). (B) Representative images of immunolabeling for MBNL1 and MBNL2 on hiPSC-derived skeletal muscle cells (24 days of differentiation). Scale bar 10 μM. Higher magnifications of the area in inset are provided to show the punctiform staining of MBNL1 and MBNL2 in DM1 hiPSC-derived skeletal muscle cells in comparison with WT hiPSC-derived skeletal muscle cells. Scale bar 5 μM. (C) Alternative splicing analysis of DMD exon 78, INSR exon 11 and BIN1 exon 11 were performed by RT-PCR on hiPSC-derived skeletal muscle cells (24 days of differentiation). The mean ± SD values from three independent experiments in duplicate are shown (*P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001—ordinary one-way ANOVA, Tukey’s multiple comparisons test).
In comparison, loss of either MBNL1 or MBNL2 resulted in a moderate effect on myogenic fusion, which is likely related to the compensatory phenomenon existing between the two paralogs (Fig. 5A and Supplementary Material, Fig. S4A). Immunofluorescence analysis revealed that this upregulation was mainly localized in the nucleus (Fig. 5B), in agreement with previous observations made in murine models of Mbnl loss of function (15). In contrast to single MBNL knockout lines, MBNL1(−/−); MBNL2(+/−) and DKO hiPSC-derived myotubes showed an aggravated fusion defect (Fig. 5A). Concordant with the absence of MBNL3 protein at this stage, depletion of MBNL3 in addition to the two paralogs did not exacerbate this phenotype in TKO lines when compared with DKO (Fig. 5A).
At the molecular level, and concordant with the observations made at the myoblast stage, several DM1-associated mis-splicing events, known to contribute to muscle function and maintenance such as BIN1 exon 7 and DMD exon78, were found to be mis-spliced in DM1 hiPSC-derived myotubes (Fig. 5C). For all targets examined, dramatic mis-splicing events were also observed in DKO myotubes (Fig. 5C). These data strongly suggest that MBNL1 and MBNL2 proteins regulate RNA processing essential for normal late myogenesis.
Comparative analysis of the molecular pathways misregulated between DM1, MBNL1 KO and DKO hiPSC-derived myotubes
To better evaluate the consequences of MBNL1 and MBNL2 depletions during myogenesis, we sought to conduct a comparative transcriptomic analysis between DKO and WT hiPSC-derived myotubes. We also included MBNL1(−/−) hiPSC-derived myotubes to evaluate the effect of MBNL2 compensation. For this purpose, we used AmpliSeq targeted sequencing technology allowing analysis of over 20 000 distinct human RNA targets simultaneously using a highly multiplexed amplification method. The principal component analysis (PCA) showed a distinct transcriptomic profile for each condition (Fig. 6A). The comparison between MBNL1(−/−) and WT hiPSC-derived myotubes led to the identification of 126 genes differentially expressed [false discovery rate (FDR) < 0.05; Log2FoldChange > 0.4], with 31 being upregulated and 95 downregulated in MBNL1(−/−) myotubes compared with WT (Fig. 6B). In the same order of magnitude, 122 differentially expressed genes (DEGs) were found when comparing DKO with WT hiPSC-derived myotubes, 40 upregulated and 82 downregulated in DKO myotubes. A more pronounced difference was observed between DKO and MBNL1(−/−) hiPSC-derived myotubes with 258 DEGs (Fig. 6B). All the DEGs are listed in Supplementary Material, Table S3. Strikingly, only 12 genes were commonly deregulated in MBNL1(−/−) versus WT myotubes and in DKO versus WT myotubes (Fig. 6C). Hierarchical clustering on the 122 DEGs detected in DKO compared with WT hiPSC-derived myotubes showed that MBNL1(−/−) hiPSC-derived myotubes were comparable to WT hiPSC-derived myotubes (Fig. 6D). Accordingly, gene ontology (GO) analysis of DEGs in MBNL1(−/−) versus WT myotubes revealed enrichment in multiple biological processes, but few of these are related to skeletal muscle (Fig. 6E and Supplementary Material, Table S4). These observations could be explained, at least, by the functional compensation by MBNL2 in MBNL1(−/−) myotubes.
Figure 6.
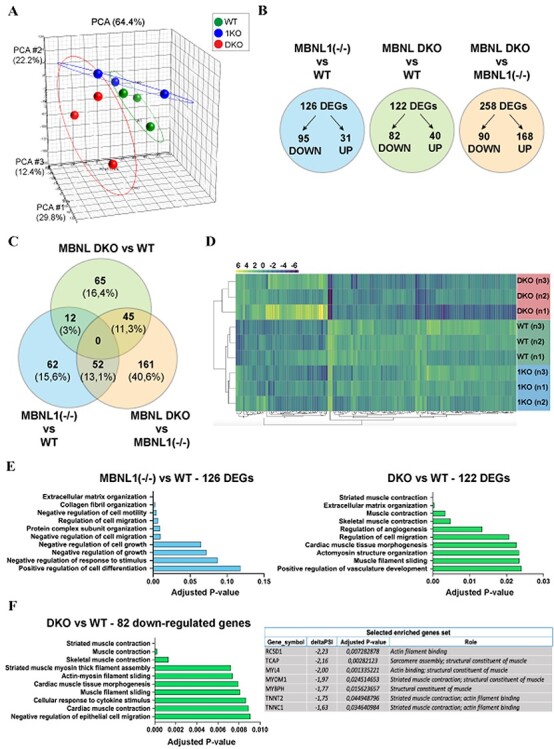
Depletion of MBNL1 and MBNL2 affects the expression of gene sets related to skeletal muscle. (A) PCA mapping indicating distinct transcriptomic profiles between WT, MBNL1(−/−) and MBNL1(−/−); MBNL2(−/−) hiPSC-derived skeletal muscle cells (24 days of differentiation). (B) Schematic representation of the number of DEGs in MBNL1 KO hiPSCs-derived myotubes compared with WT (blue), DKO myotubes compared with WT (green) and DKO compared with MBNL1 KO myotubes (orange). The detailed list DEGs for each condition is provided in Supplementary Material, Table S3. (C) Venn diagram representation of the number of DEGs commonly deregulated between the three different conditions analyzed. The proportion of each subset of DEGs relative to the total number of DEGs (396 in total) is indicated in brackets. (D) The hierarchical clustering of the 122 DEGs detected in DKO hiPSC-derived skeletal muscle compared with WT hiPSC-derived skeletal muscle cells. This analysis indicates that MBNL1 KOs are comparable to WT hiPSC-derived skeletal muscle cells regarding these DEGs. Gene expression is represented by a color code ranging from yellow for overexpressed genes to purple for underexpressed genes. (E) EnrichR-based gene set enrichment analysis of the DEGs identified between MBNL1 KO hiPSCs and WT-derived skeletal muscle cells (left), and between DKO hiPSCs and WT-derived skeletal muscle cells (right). The top 10 deregulated biological processes are classified according to their adjusted P-value. (F) Gene set enrichment analysis using EnrichR on the 82 downregulated genes in DKO hiPSC-derived skeletal muscle cells when compared with WT hiPSC-derived skeletal muscle cells. The table on the right provides a list of specific genes downexpressed in DKO hiPSC-derived skeletal muscle cells (EnrichR analysis using GO Biological Process 2018 database).
Concerning DEGs in DKO compared with WT hiPSC-derived myotubes, GO analysis revealed enrichment in several biological processes related to skeletal muscle such as ‘striated muscle contraction’ (adjusted P-value: 0.00001), ‘actomyosin structure organization’ (adjusted P-value: 0.02341) or ‘muscle filament sliding’ (adjusted P-value: 0.02348) (Fig. 6E). Strikingly, most of these muscle related-biological processes were found to be downregulated in DKO hiPSC-derived myotubes (Fig. 6F). In the same way, GO analyses of DEGs in DKO compared with MBNL1(−/−) hiPSC-derived myotubes showed similar results with multiple downregulated biological processes related to skeletal muscle (Supplementary Material, Fig. S5A and B). Among the list of downregulated genes identified in DKO hiPSC-derived myotubes, we highlighted two interesting candidates for further explorations (Fig. 6F): TCAP (also known as Telethonin) plays an essential role in the sarcomeric assembly by anchoring Titin in the Z-disk of the sarcomere (27) and is known to be involved in limb-girdle muscular dystrophy 2G (28); MYOM1 is a constituent of the sarcomeric M band and plays an important role in sarcomere assembly and integrity (29). Interestingly, MYOM1 is found mis-spliced in muscular biopsies from DM1 patients (30), and this splicing defect could be caused by the downregulation of MBNL proteins (31).
Thus, we pursued our analysis by searching for cassette exons that demonstrate a significant difference in the level of inclusion between MBNL1(−/−), DKO and WT hiPSC-derived myotubes. A total of 547 cassette exons were found to be deregulated between DKO and WT hiPSC-derived myotubes, among which 296 and 251 exhibited a significant decrease and increase in inclusion, respectively [FDR < 0.05; delta percent spliced-in (PSI) > 0.4] (Fig. 7A and Supplementary Material, Table S5). We next compared these analyses with transcriptomic data generated from DM1 hiPSC-derived myotubes (Fig. 7A). Of the 696 mis-splicing events [differentially spliced exons (DSE)] observed in DM1 hiPSC-derived myotubes, 224 (32%) appeared in MBNL1(−/−) hiPSC-derived myotubes. In the same way, 254 (36%) appeared in DKO hiPSC-derived myotubes and among these, 126 were commonly mis-spliced in DM1, MBNL1(−/−) and DKO hiPSC-derived myotubes (Fig. 7B).
Figure 7.
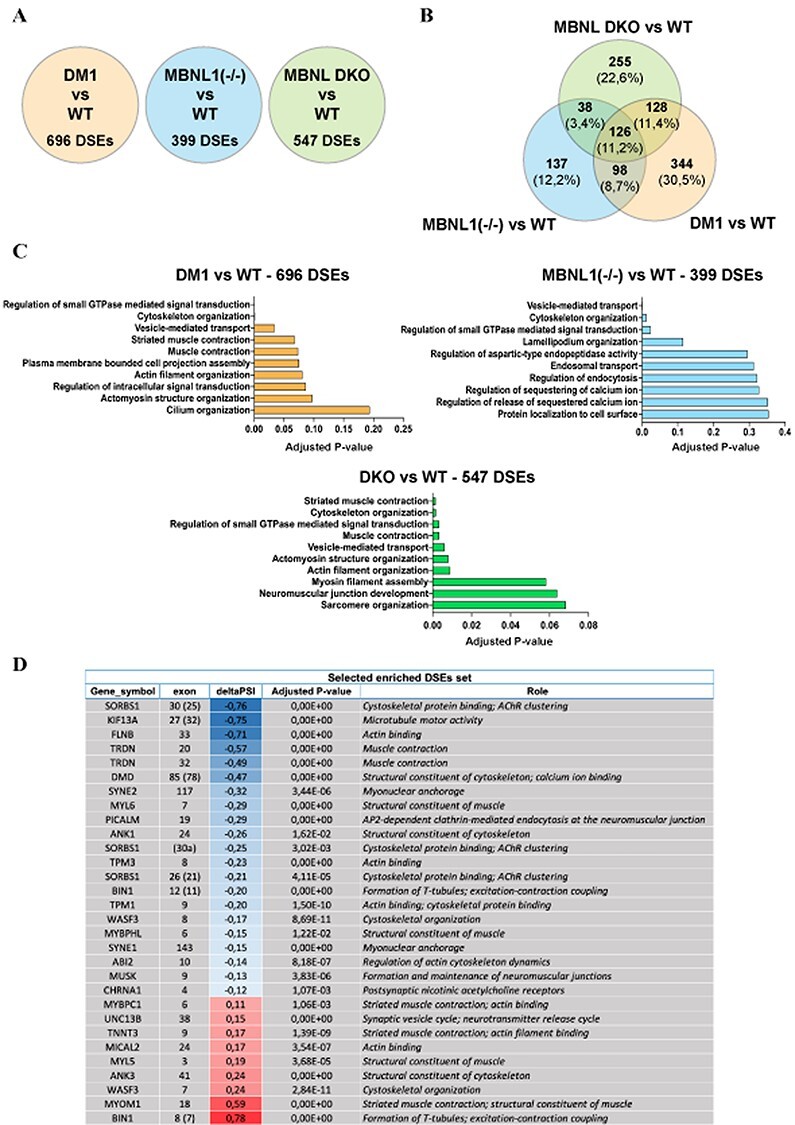
Loss of MBNL1 and MBNL2 proteins leads to a large number of alternative splicing defects related to the development of skeletal muscle. (A) Number of DSEs in DM1, MBNL1 (−/−) and in DKO hiPSCs-derived skeletal muscle cells compared with WT myotubes. (B) Venn diagram highlighting the percentage of common and different DSEs between the three conditions listed in A. All the DSEs are provided in Supplementary Material, Table S5. (C) EnrichR-based analysis of gene set enrichments using the lists of DSEs observed in DM1, MBNL1(−/−) and DKO hiPSC-derived skeletal muscle cells when compared with WT hiPSC-derived skeletal muscle cells. The top 10 most deregulated biological processes are classified according to their adjusted P-value. All the data about the enriched biological processes are detailed in Supplementary Material, Table S6. (D) List of 30 enriched DSEs covering most of the deregulated biological processes related to skeletal muscle and identified in DKO hiPSC-derived skeletal muscle cells compared with WT. For alternative splicing analysis, exons have been numbered according to FasterDB database. The numbers of the corresponding exons are indicated in brackets (EnrichR analysis using GO Biological Process 2018 database).
Functional annotation analysis of the DKO transcriptomic data revealed that the most relevant GO terms concerned skeletal muscle such as ‘sarcomere organization’ (adjusted P-value: 0.068) or ‘neuromuscular junction development’ (adjusted P-value: 0.06) (Fig. 7C and Supplementary Material, Table S6). We highlighted 30 enriched DSEs involved in most of these muscle-related biological processes deregulated in DKO myotubes (Fig. 7D) and found several already described alternative splicing defects associated with DM1 such as DMD exon 78, BIN1 exon 11 and BIN1 exon 7 (Fig. 5C and Supplementary Material, Fig. S4B) (32,33). Among these 30 DSEs, 21 were also mis-spliced in DM1 hiPSCs-derived myotubes in the same way they were in DKO myotubes (Supplementary Material, Fig. S6A). Only three DSEs (ABI2 ex10, MYL5 ex3 and TPM1 ex9) mismatched in the sense of inclusion/exclusion. Furthermore, only half of these 30 DSEs were also found deregulated in MBNL1(−/−) myotubes, which could explain why the biological processes deregulated in these cells are less related to skeletal muscle. In general, when DSEs concern both MBNL1(−/−) and DKO myotubes, the level of deregulation was worsened in DKO myotubes Supplementary Material, Fig. S6A and B). In addition to DMD exon 78, BIN1 exon 11 and BIN1 exon 7, we validated three other DSEs in our MBNL knockouts hiPSC-derived myotubes (Supplementary Material, Fig. S6B), including the aberrant inclusion of MYOM1 exon 18 in DM1 hiPSC-derived myotubes compared with WT (Supplementary Material, Fig. S6A and Supplementary Material, Table S5), which was also validated in DKO myotubes (Supplementary Material, Fig. S6B).
Altogether, these data revealed sets of disrupted biological pathways associated to the loss of MBNL1 and MBNL2 that may be particularly relevant in the pathological context of DM1.
Discussion
Cell-type-specific or developmental stage-specific alternative splicing events are largely controlled by recruiting RBPs that recognize specific regulatory sequences embedded in the pre-mRNA transcripts. In this study, we described the temporal requirement of the MBNL RBP family for defining human skeletal muscle maturation stages. Our results demonstrated that MBNL1 and MBNL2 were dispensable for the early myogenic commitment but played an important role for the late stages of myogenesis. When compared with hiPSC generated from DM1 patients, our results show that loss of MBNL1 and MBNL2 recapitulate the main cellular and molecular features associated to this neuromuscular disease. This study provides insights into the function of MBNL proteins during myogenesis. Given the technical limitations of studying embryonic and fetal human tissues, our study establishes new human cellular models to explore the role of these proteins in tissue development. Furthermore, the different hiPSC-based models developed in this study also pave the way toward the development of new therapeutic strategies capable to cope the loss of function of MBNL proteins.
Although transcriptional regulation has been demonstrated to play an important role in myogenesis, there is a growing recognition of the contributions of alternative splicing to skeletal muscle development as well as the refinement of muscle function (34). Skeletal muscle was also one of the first tissues in which broad use of alternative splicing to increase cellular complexity was documented (35). A recent study has estimated the presence of hundreds of RBPs expressed in muscle of different species such as human, rodent, flies and worms, but only a small proportion (~3%) have been studied (36). Specifically, the role of CELF (CUG-BP and ETR-3 like), MBNL and Fox families in controlling muscle-specific splice events has been largely documented in murine models. The Rbfox proteins are among the most heavily studied RBPs in the context of myogenesis. Gain- and loss-of-function experiments have demonstrated that Rbfox1 and Rbfox2 coordinately regulate splicing of the transcription factors Mef2a and Mef2d to maintain the expression and proper activity of muscle-specific isoforms (34,36). More recently, the generation of skeletal muscle-specific knockout of Rbfox1 and Rbfox2 in adult mice also revealed a role of these alternative splicing factors in the maintenance of skeletal muscle mass. This function could at least be attributable to the altered expression of calpain 3 splice isoforms that are not degraded rapidly leading to the accumulation of active calpain 3 and a defective proteostasis (37). As illustrated by this example, a major part of our knowledge has been gleaned from studies in animal models. Given the large number of RBPs expressed and with potential phenotypes in human muscle, this clearly illustrates the need for further studies examining the function of RBPs in human myogenesis.
In this context, human pluripotent stem cells represent an attractive cell source for producing myogenic cells and deciphering the function of different RBPs during this process. Until recently, the only efficient protocols allowing differentiation of human pluripotent stem cells into a reasonably mature muscle cells relied on the forced expression of transcription factors such as MyoD or Pax3/7 (38). These direct reprogramming approaches aim to bypass early developmental stages by overexpressing myogenic regulators, and the cellular events occurring during differentiation remain largely unclear (39). More recently, stepwise induction protocols on the basis of the use of small molecules and growth factors have been established and appeared as an alternative approach to better recapitulate the successive developmental stages involved in skeletal myogenesis (21,40). By using such approach, our study adds a new dimension to the role of MBNL proteins in defining human skeletal muscle maturation stages. The exact mechanisms by which MBNL proteins affect these processes are, however, unclear. In contrast to Rbfox1/2 for which depletion has been shown to inhibit myoblast fusion in vitro through the mis-splicing of Mef2D (41,42), our results indicate that depletion of MBNL proteins leads to the aberrant expression of hundreds of genes. Because several of these genes encode factors involved in sarcomere organization, actin dynamics and vesicular trafficking events required for membrane fusion (43), further experiments will be required to evaluate the exact MBNL-based mechanisms required for myogenic differentiation.
Among the three MBNL paralogs, our results confirmed that MBNL1 is the most abundant MBNL protein during in vitro myogenesis (12,14). We also observed a meticulous compensation of MBNL1 by MBNL2 because MBNL2 protein expression was increased in either MBNL1(−/−) and MBNL1(−/−); MBNL2(+/−) compared with WT and MBNL2(+/−) hiPSC-derived myotubes, respectively. Moreover, transcriptomic analyses showed that several skeletal muscle-related biological processes were downregulated in DKO myotubes compared with MBNL1(−/−) myotubes. Taking together, our results strengthened this compensation phenomenon between MBNL1 and MBNL2 proteins in myogenesis as suggested in prior studies in mice (15,16). In contrast to MBNL1 and MBNL2, the expression of MBNL3 is found to be very low and limited in duration. The role of MBNL3 during myogenesis is not well understood. Whereas constitutive expression of Mbnl3 in murine muscle precursors’ cells appears to inhibit terminal differentiation (44), it has been recently shown that Mbnl3-depleted primary murine myoblasts displayed defective in vitro differentiation (24). Our results showed that TKOs hiPSC-derived skeletal muscle cells recapitulated all the phenotypes identified in DKOs suggesting that MBNL3 might not play such important role during human myogenesis.
Sequestration of MBNL proteins is considered a major player in DM1 pathology. Comparative transcriptomic analysis between Mbnl1 knockout mice and transgenic mice expressing CUG expansion RNA have shown that the majority of changes induced by the abnormal expansions in skeletal muscle could be explained by reduced activity of Mbnl1 (23,45,46). However, it has been shown more recently that the combined loss of Mbnl1 and Mbnl2 was insufficient to reproduce the extensive gene expression changes detected in DM1, particularly in its congenital form, which is the most severe form of the disease associated with large numbers of CTG expansion (24,47,48). Accordingly, we showed here that the loss of MBNL1 as well as its combined loss with MBNL2 do not fully recapitulate the gene expression changes that we observed in skeletal muscle cells derived from DM1 hiPSC carrying large number of CTG expansions. Altogether, these results strongly suggest that additional pathways may play non-negligible roles in the disease. Among the different hypotheses that can be formed, MBNL1 has been implicated in altered miRNA biogenesis (48), RNA localization (23) and in the misregulation of alternative polyadenylation (49) in DM1, and it would be relevant to investigate the effects of MBNL loss on these regulatory pathways. In addition, the stabilization and overexpression of CUG-BP1, because of its hyperphosphorylation as a result of the alteration of the GSK3β pathway in DM1, have been shown to lead to several splicing defects (50,51). Furthermore, the splicing factor hnRNP-H found to be deregulated in DM1 may also contribute to the disease spliceopathy (52).
Finally, to this day, only a couple of studies have described the generation of DM1 hiPSC-derived skeletal muscle cells and the capacity of these cellular systems to reproduce the main molecular features associated with DM1, namely the nuclear aggregation of mutated mRNAs and some alternative splicing defects. Despite promising results, all these attempts to generate skeletal muscle cells from DM1 hiPSCs were on the basis of the forced expression of myogenic transcription factors MyoD or Pax7, which, as discussed above, are often undefined (53–55). The experimental system described here uses a transgene-free protocol to generate skeletal muscle cells from hiPSCs and opens new perspectives in term of disease modeling and drug screening. First, the different well-characterized steps of the protocol help to decipher the exact impact of the mutation on myogenic differentiation. Whereas most of the studies have focused on human primary skeletal muscle cells or immortalized skeletal muscle cells originating from DM1 patients’ biopsies (26), our results indicate that early myogenic commitment is not affected by the presence of the DM1 mutation. In addition, the transgene-free protocol described here includes a freezing step allowing the possibility to generate large cell banks. Consequently, this represents an unlimited source of material that could be used for drug screening.
In summary, our results provide important new experimental support for the MBNL loss-of-function model for DM and introduce new human cellular models to examine the tissue-specific consequences of MBNL loss.
Materials and Methods
Cell culture
hiPSCs were derived by Phenocell® from human primary fibroblasts reprogrammed using sendai virus vectors carrying OCT4, KLF4, SOX2 and c-Myc (56). Two DM1 hiPSC lines were generated from two independent patients’ fibroblasts containing ~2500 CTG repeats. DM1 hiPSC carry a mutant DMPK gene containing ~2700 CTG repeats. One control hiPSC line corresponding to the sibling matched control was also generated. Informed consents were obtained from all the patients included in this study, complying with the ethical guidelines of the institutions and with the legislation requirements of the country of origin. Experimental protocols were approved by the French minister of health (2019-A02599-48). All the hiPSC lines were used at passages around p10–p20 and were maintained in StemMACS iPS-Brew XF medium (Miltenyi Biotec®) in vitronectin (Gibco®)-coated culture dishes. Culture medium was changed every 2 days, and confluent cells were manually passaged every 5–7 days.
hiPSCs skeletal muscle differentiation experiments were performed using the commercially available Genea Biocells® skeletal muscle differentiation media following the protocol described previously (21). Briefly, hiPSCs were dissociated with StemPro Accutase Cell Dissociation Reagent (Gibco®) and plated at 3000 cells per cm2 onto collagen I-coated plates (Gibco®) and maintained for 10 days in skeletal muscle induction medium (SKM01). At day 10, cells were dissociated with trypsin–EDTA 0.05% (Gibco®), plated at 5000 cells per cm2 onto collagen I-coated plates and maintained for 6 days in skeletal myoblast medium (SKM02). At day 16, myoblasts were dissociated and cryopreserved. For terminal differentiation, cells were thawed at 10 000 cells per cm2 in SKM02 and after 2–3 days, when cells reached confluence, the medium was replaced by the myotube medium (SKM03). Cells were then maintained for 5–6 days in SKM03.
sgRNAs design
sgRNAs and primers used in this study are reported in Supplementary Material, Table S1.
SpCas9 target sequences within the first coding exon of each MBNL paralog were determined by CRISPOR (http://crispor.tefor.net/). sgRNAs with the highest predicted efficiency, the lowest number of potential off-targets and having an enzymatic restriction site at the PAM sequence were selected. crRNA and tracrRNA were synthesized by Integrated DNA Technologies®.
RNP transfection of hiPSCs
Control hiPSCs were dissociated with StemPro Accutase Cell Dissociation Reagent (Gibco®), plated in 24-well plates at 40 000 cells per cm2 and transfected the next day with a mixed of 30 nM of sgRNA (by duplexing RNA oligos: crRNA and tracrRNA), 30 nM of SpCas9 purified protein (gift from Jean-Paul Concordet, MNHN–CNRS UMR 7196/INSERM U1154) and Lipofectamine Stem Transfection Reagent (ThermoFisher Scientific®), according to the manufacturer’s protocol.
Genomic DNA extraction and PCR
Genomic DNA was extracted from transfected hiPSCs either with QIAmp DNA Micro and Mini Kit (Qiagen®) or with QuickExtract DNA extraction solution (Lucigen®), according to the manufacturer’s instructions. PCR were performed using Q5 High-Fidelity DNA Polymerase (NEB®) with 30 ng gDNA, 38 cycles of 10″ at 98°C, 15″ at 60°C and 45″ at 72°C, with a final 10′ extension and with primers framing the target area according to each guide used (Supplementary Material, Table S1).
RFLP assays to identify edited clones
Restriction fragment length polymorphism (RFLP) presents the advantage to easily and rapidly estimate the presence of gene editing in a mixed population or in single clones. Briefly, 2 μl PCR products were digested by 5 units of restriction enzyme (NEB®): BsrI for MBNL1 and MBNL2 clones, and SmlI for MBNL3 clones. Cleaved fragments were then separated by gel electrophoresis. If CRISPR-mediated gene editing was successful, NHEJ-mediated INDELS abolished the restriction endonuclease recognition site. After restriction digest, clones that had lost their restriction site were selected and corresponding PCR products were sequenced by Sanger DNA sequencing (Eurofins®).
To ensure the purity of each clone selected, we used TOPO-TA cloning Kits for Sequencing (Invitrogen®) and One Shot TOP10 chemically competent Escherichia coli (Invitrogen®) according to manufacturer’s instructions. In order to determine the presence of sublcones, at least 30 TA clones for each CRISPR cell line generated were analyzed by Sanger DNA sequencing (Eurofins®).
Ion AmpliSeq transcriptome human gene expression kit
For each sample, 50 ng of total RNA was reverse transcribed using the Ion AmpliSeq Transcriptome Human Gene Expression kit following the protocol of the manufacturer (Thermofisher Scientific®). The cDNA libraries were amplified and barcoded using Ion AmpliSeq Transcriptome Human Gene Expression core panel and Ion Xpress Barcode Adapter (Thermofisher Scientific®). The amplicons were quantified using Agilent High Sensitivity DNA kit before the samples were pooled in sets of eight. Emulsion PCR and enrichment was performed on the Ion OT2 system instrument using the Ion PI Hi-Q OT2 200 kit (Thermofisher Scientific®). Samples were loaded on an Ion PI v3 Chip and sequenced on the Ion Proton System using Ion PI Hi-Q sequencing 200 kit chemistry (200 bp read length; Thermofisher Scientific®). The quality control of the sequencing data was evaluated as described previously (57). The DEG between conditions was calculated with (v1.18.1 using R v3.4.1). Genes were considered differentially expressed when their adjusted P-value was <0.05 and their|log2FoldChange|was >0.4 (options: lfcThreshold = 0.4, altHypothesis = ‘greaterAbs’). Data are available on GEO database (GSE161029).
RNA-Seq library preparation and sequencing (TrueSeq polyA)
For each sample, 100 ng of total RNA was used. mRNAs were purified using poly-T oligo from TruSeq Stranded kit (Illumina®) and were fragmented and reverse transcribed using the TruSeq Stranded kit following the protocol of the manufacturer. Final library was quantified using Agilent high-sensitivity DNA kit before the samples were pooled in sets of six for sequencing. A total of 2 nM of pooled libraries were denatured, and the quantity of 1.8 pM was used for cluster generation before paired-end sequencing on an Illumina Nextseq550 (high output 2 × 150 bp run). Samples were sequenced with an average of 89 532 366 reads (paired-end) per sample.
The quality control of the sequencing data was evaluated using FastQC (v0.11.2). The reads were trimmed using Prinseq (v0.20.4) (--trim-right 20) and filtered by average quality score (--trim-qual 20) and cutadapt (v1.16) (58).
For the alternative splicing analysis, the raw reads were annotated on GRCh37.75 EnsEMBL reference annotation by Tophat2 (v2.0.13) and analyzed using only reads on exonics junctions with FaRLine pipeline and FasterDB database (59). Exons were considered differentially spliced when their adjusted P-value was <0.05 and their DeltaPSI value was >0.1. Data are available on GEO database (GSE161897).
RT-PCR and Agilent DNA chips analysis for alternative splicing
Total RNA was extracted using the RNeasy Micro/Mini kit (Qiagen®) and reverse transcribed using random hexamers and Superscript III Reverse Transcriptase kit (Invitrogen®) according to the manufacturer’s protocol. For splicing analysis, PCR amplification were carried out with recombinant Taq DNA polymerase (Invitrogen®) and primers listed in Supplementary Material, Table S1. The amplification was performed using a first step at 95°C for 3′ followed by 35 cycles of 45″ at 95°C, 30″ at 60°C, 1′ at 72°C, and a final 10′ extension at 72°C. The PCR products were analyzed using Agilent® DNA chips and quantified with the BioAnalyzer 2100.
Fluorescent in situ hybridization
Cell preparation was performed as previously described (22). Briefly, cells were fixed with phosphate-buffered solution (PBS) buffer solution containing 4% paraformaldehyde (PFA) for 8′ at room temperature and incubated overnight at 4°C with 70% ethanol (VWR®). After washes, cells were rehydrated with a solution of 5 mM of MgCl2 (Sigma-Aldrich®) for 10′ at room temperature and then sequentially incubated with prehybridization buffer for 10′ and hybridization buffer containing the probe overnight at 37°C under agitation. Washing steps consisted in pre-warmed washing buffer for 1 h at 37°C. Finally, cells were washed twice in PBS buffer and incubated 5′ in a Hoechst 33528 solution (Sigma-Aldrich®).
Nuclear foci detection was analyzed using the Cellomics CX7 high-content imaging system (Thermofisher Scientific®) with the 20× objective and an automatically focus cell preparation. Images were acquired in high-resolution camera mode on two channels and quantification was performed as previously described with the software attached. Each image was taken with z-stack and application of maximum projection.
Immunocytochemistry
After fixation with 4% PFA for 10′ at room temperature, cells were incubated over night at 4°C with the following primary antibodies: DESMIN (R&D System®, 1:100), PAX3 (DSHB®, 1:30), α-ACTININ (Sigma®, 1:250), MF20 (DSHB®, 1:200), MBNL1–MB1a(4A8) (DSHB®, 1:100), MBNL2–MB2a(3B4) (DSHB®, 1:100). Appropriated Alexa fluorescent secondary antibodies (Invitrogen®, 1:1000) and Hoechst (5 μg/ml) were added for 1 h. Image acquisitions were performed on the ImageXpress micro imaging system (Molecular Devices®) with the 20× objective and with three channels. Images were analyzed using MetaXpress software by quantifying the percentage of cells stained for PAX3 or DESMIN compared with the total number of cells.
Quantification of in vitro myoblasts fusion was performed counting the number of myotubes with at least three nuclei (manually counted) normalized to the total number of nuclei (at least 11 000 nuclei counted per experiment and per cell line).
Protein extraction and western blot analysis
Western blots analyses were performed as previously described (59). Briefly, cells were lyzed in RIPA 1× buffer (Sigma®) containing protease inhibitors (Sigma®) and phosphatase inhibitors (Roche®). Proteins were quantified by Pierce BCA Protein Assay kit (Pierce®). Protein extracts (15–20 μg) were loaded on a 4–12% sodium dodecyl sulphate–polyacrylamide gel electrophoresis gradient (NuPage Bis–Tris gels, Invitrogen®) and transferred onto Gel Transfer Stacks Nitrocellulose membranes (Invitrogen®) using the iBlot2 Dry Blotting System (Invitrogen®). Membranes were then incubated overnight at 4°C with the following primary antibodies: MBNL1–MB1a(4A8) (DSHB®, 1:1000), MBNL2–MB2a(3B4) (DSHB®, 1:1000), MBNL3–5A11(sc-136 168) (Santa Cruz, 1:100). After hybridization of the peroxidase-conjugated secondary antibody (1:10000), immunoreactive bands were revealed by using Amersham ECL Select Western Blotting Detection Reagents (GE Healthcare®). Equal protein loading was verified by the detection of β-actin using the A3854 Monoclonal Anti-β-Actin−Peroxidase antibody (Sigma®, 1:10000).
Statistical analysis
All data were processed using Prism 8®. Values are represented as mean ± standard deviation (SD). For comparisons of more than two groups, statistical analyses were performed with by ordinary one-way analysis of variance (ANOVA) using Tukey’s multiple comparisons test. Values of P < 0.05 were considered significant (*P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001).
Supplementary Material
Acknowledgements
I-Stem is part of the Biotherapies Institute for Rare Diseases (BIRD) supported by the Association Française contre les Myopathies (AFM-Téléthon). We thank Geneviève Gourdon’s team for the southern blot analyses of the CTG repeats length. We gratefully acknowledge support from the PSMN (Pôle Scientifique de Modélisation Numérique) of the ENS of Lyon for computing resource. We thank Lina El-Kassar and Karine Giraud-Triboult for karyotyping the cell lines.
Conflict of Interest statement. None declared.
Contributor Information
Antoine Mérien, INSERM/UEPS UMR 861, Paris Saclay University, I-STEM, 91100 Corbeil-Essonnes, France.
Julie Tahraoui-Bories, INSERM/UEPS UMR 861, Paris Saclay University, I-STEM, 91100 Corbeil-Essonnes, France.
Michel Cailleret, INSERM/UEPS UMR 861, Paris Saclay University, I-STEM, 91100 Corbeil-Essonnes, France.
Jean-Baptiste Dupont, INSERM/UEPS UMR 861, Paris Saclay University, I-STEM, 91100 Corbeil-Essonnes, France.
Céline Leteur, CECS/AFM, I-STEM, 91100 Corbeil-Essonnes, France.
Jérôme Polentes, CECS/AFM, I-STEM, 91100 Corbeil-Essonnes, France.
Alexandre Carteron, CECS/AFM, I-STEM, 91100 Corbeil-Essonnes, France.
Hélène Polvèche, CECS/AFM, I-STEM, 91100 Corbeil-Essonnes, France.
Jean-Paul Concordet, INSERM U1154/CNRS UMR7196, MNHN TACGENE, 75005 Paris, France.
Christian Pinset, CECS/AFM, I-STEM, 91100 Corbeil-Essonnes, France.
Margot Jarrige, CECS/AFM, I-STEM, 91100 Corbeil-Essonnes, France.
Denis Furling, Sorbonne Université, INSERM, Association Institut de Myologie, Centre de recherche en myologie, 75013 Paris, France.
Cécile Martinat, INSERM/UEPS UMR 861, Paris Saclay University, I-STEM, 91100 Corbeil-Essonnes, France.
Funding
Laboratoire d’Excellence Revive, Investissement d’Avenir (grant ANR-10-LABX-73); French National research Agency (grant ANR-16-CE17-0018); doctoral school—Innovation Thérapeutique, du fondamental à l’appliqué (ED 569) from Paris Saclay University.
Author Contributions
A.M., M.C. and C.M.: conception and design. A.M., J.T. and C.M.: investigation and data collection. A.M., J.T., J.B., H.P., M.J., J.P., C.L. and C.M.: formal analysis and interpretation. D.F. and C.M.: provision of different cellular resources. C.M.: financial support. A.M. and C.M.: manuscript writing.
References
- 1. Wang, E.T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., Kingsmore, S.F., Schroth, G.P. and Burge, C.B. (2008) Alternative isoform regulation in human tissue transcriptomes. Nature, 456, 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pan, Q., Shai, O., Lee, L.J., Frey, B.J. and Blencowe, B.J. (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet., 40, 1413–1415. [DOI] [PubMed] [Google Scholar]
- 3. Merkin, J., Russell, C., Chen, P. and Burge, C.B. (2012) Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science, 338, 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cooper, T.A., Wan, L. and Dreyfuss, G. (2009) RNA and disease. Cell, 136, 777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brook, J.D., McCurrach, M.E., Harley, H.G., Buckler, A.J., Church, D., Aburatani, H., Hunter, K., Stanton, V.P., Thirion, J.P., Hudson, T. et al. (1992) Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell, 69, 385. [DOI] [PubMed] [Google Scholar]
- 6. Mahadevan, M., Tsilfidis, C., Sabourin, L., Shutler, G., Amemiya, C., Jansen, G., Neville, C., Narang, M., Barcelo, J., O'Hoy, K. et al. (1992) Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science, 255, 1253–1255. [DOI] [PubMed] [Google Scholar]
- 7. Miller, J.W., Urbinati, C.R., Teng-Umnuay, P., Stenberg, M.G., Byrne, B.J., Thornton, C.A. and Swanson, M.S. (2000) Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J., 19, 4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fardaei, M., Rogers, M.T., Thorpe, H.M., Larkin, K., Hamshere, M.G., Harper, P.S. and Brook, J.D. (2002) Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet., 11, 805–814. [DOI] [PubMed] [Google Scholar]
- 9. Nakamori, M., Sobczak, K., Puwanant, A., Welle, S., Eichinger, K., Pandya, S., Dekdebrun, J., Heatwole, C.R., McDermott, M.P., Chen, T. et al. (2013) Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol., 74, 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kanadia, R.N., Johnstone, K.A., Mankodi, A., Lungu, C., Thornton, C.A., Esson, D., Timmers, A.M., Hauswirth, W.W. and Swanson, M.S. (2003) A muscleblind knockout model for myotonic dystrophy. Science, 302, 1978–1980. [DOI] [PubMed] [Google Scholar]
- 11. Konieczny, P., Stepniak-Konieczna, E. and Sobczak, K. (2014) MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res., 42, 10873–10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holt, I., Jacquemin, V., Fardaei, M., Sewry, C.A., Butler-Browne, G.S., Furling, D., Brook, J.D. and Morris, G.E. (2009) Muscleblind-like proteins: similarities and differences in normal and myotonic dystrophy muscle. Am. J. Pathol., 174, 216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin, X., Miller, J.W., Mankodi, A., Kanadia, R.N., Yuan, Y., Moxley, R.T., Swanson, M.S. and Thornton, C.A. (2006) Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet., 15, 2087–2097. [DOI] [PubMed] [Google Scholar]
- 14. Charizanis, K., Lee, K.Y., Batra, R., Goodwin, M., Zhang, C., Yuan, Y., Shiue, L., Cline, M., Scotti, M.M., Xia, G. et al. (2012) Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron, 75, 437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee, K.Y., Li, M., Manchanda, M., Batra, R., Charizanis, K., Mohan, A., Warren, S.A., Chamberlain, C.M., Finn, D., Hong, H. et al. (2013) Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med., 5, 1887–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goodwin, M., Mohan, A., Batra, R., Lee, K.Y., Charizanis, K., Fernandez Gomez, F.J., Eddarkaoui, S., Sergeant, N., Buee, L., Kimura, T. et al. (2015) MBNL sequestration by toxic RNAs and RNA misprocessing in the myotonic dystrophy brain. Cell Rep., 12, 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Poulos, M.G., Batra, R., Li, M., Yuan, Y., Zhang, C., Darnell, R.B. and Swanson, M.S. (2013) Progressive impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice. Hum. Mol. Genet., 22, 3547–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choi, J., Dixon, D.M., Dansithong, W., Abdallah, W.F., Roos, K.P., Jordan, M.C., Trac, B., Lee, H.S., Comai, L. and Reddy, S. (2016) Muscleblind-like 3 deficit results in a spectrum of age-associated pathologies observed in myotonic dystrophy. Sci. Rep., 6, 30999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Concordet, J.P. and Haeussler, M. (2018) CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res., 46, W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ramakrishna, S., Kwaku Dad, A.B., Beloor, J., Gopalappa, R., Lee, S.K. and Kim, H. (2014) Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res., 24, 1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Caron, L., Kher, D., Lee, K.L., McKernan, R., Dumevska, B., Hidalgo, A., Li, J., Yang, H., Main, H., Ferri, G. et al. (2016) A human pluripotent stem cell model of facioscapulohumeral muscular dystrophy-affected skeletal muscles. Stem Cells Transl. Med., 5, 1145–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Venables, J.P., Lapasset, L., Gadea, G., Fort, P., Klinck, R., Irimia, M., Vignal, E., Thibault, P., Prinos, P., Chabot, B. et al. (2013) MBNL1 and RBFOX2 cooperate to establish a splicing programme involved in pluripotent stem cell differentiation. Nat. Commun., 4, 2480. [DOI] [PubMed] [Google Scholar]
- 23. Wang, E.T., Cody, N.A., Jog, S., Biancolella, M., Wang, T.T., Treacy, D.J., Luo, S., Schroth, G.P., Housman, D.E., Reddy, S. et al. (2012) Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell, 150, 710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thomas, J.D., Sznajder, L.J., Bardhi, O., Aslam, F.N., Anastasiadis, Z.P., Scotti, M.M., Nishino, I., Nakamori, M., Wang, E.T. and Swanson, M.S. (2017) Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev., 31, 1122–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakamori, M., Kimura, T., Fujimura, H., Takahashi, M.P. and Sakoda, S. (2007) Altered mRNA splicing of dystrophin in type 1 myotonic dystrophy. Muscle Nerve, 36, 251–257. [DOI] [PubMed] [Google Scholar]
- 26. Arandel, L., Polay Espinoza, M., Matloka, M., Bazinet, A., De Dea Diniz, D., Naouar, N., Rau, F., Jollet, A., Edom-Vovard, F., Mamchaoui, K. et al. (2017) Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis. Model. Mech., 10, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bertz, M., Wilmanns, M. and Rief, M. (2009) The titin-telethonin complex is a directed, superstable molecular bond in the muscle Z-disk. Proc. Natl. Acad. Sci. U. S. A., 106, 13307–133310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Markert, C.D., Meaney, M.P., Voelker, K.A., Grange, R.W., Dalley, H.W., Cann, J.K., Ahmed, M., Bishwokarma, B., Walker, S.J., Yu, S.X. et al. (2010) Functional muscle analysis of the Tcap knockout mouse. Hum. Mol. Genet., 19, 2268–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Prill, K., Carlisle, C., Stannard, M., Windsor Reid, P.J. and Pilgrim, D.B. (2019) Myomesin is part of an integrity pathway that responds to sarcomere damage and disease. PLoS One, 14, e0224206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakamori, M., Hamanaka, K., Thomas, J.D., Wang, E.T., Hayashi, Y.K., Takahashi, M.P., Swanson, M.S., Nishino, I. and Mochizuki, H. (2017) Aberrant myokine signaling in congenital myotonic dystrophy. Cell Rep., 21, 1240–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Koebis, M., Ohsawa, N., Kino, Y., Sasagawa, N., Nishino, I. and Ishiura, S. (2011) Alternative splicing of myomesin 1 gene is aberrantly regulated in myotonic dystrophy type 1. Genes Cells, 16, 961–972. [DOI] [PubMed] [Google Scholar]
- 32. Fugier, C., Klein, A.F., Hammer, C., Vassilopoulos, S., Ivarsson, Y., Toussaint, A., Tosch, V., Vignaud, A., Ferry, A., Messaddeq, N. et al. (2011) Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med., 17, 720–725. [DOI] [PubMed] [Google Scholar]
- 33. Rau, F., Laine, J., Ramanoudjame, L., Ferry, A., Arandel, L., Delalande, O., Jollet, A., Dingli, F., Lee, K.Y., Peccate, C. et al. (2015) Abnormal splicing switch of DMD's penultimate exon compromises muscle fibre maintenance in myotonic dystrophy. Nat. Commun., 6, 7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nikonova, E., Kao, S.Y. and Spletter, M.L. (2020) Contributions of alternative splicing to muscle type development and function. Semin. Cell Dev. Biol., 104, 65–80. [DOI] [PubMed] [Google Scholar]
- 35. Llorian, M. and Smith, C.W. (2011) Decoding muscle alternative splicing. Curr. Opin. Genet. Dev., 21, 380–387. [DOI] [PubMed] [Google Scholar]
- 36. Nikonova, E., Kao, S.Y., Ravichandran, K., Wittner, A. and Spletter, M.L. (2019) Conserved functions of RNA-binding proteins in muscle. Int. J. Biochem. Cell Biol., 110, 29–49. [DOI] [PubMed] [Google Scholar]
- 37. Singh, R.K., Kolonin, A.M., Fiorotto, M.L. and Cooper, T.A. (2018) Rbfox-splicing factors maintain skeletal muscle mass by regulating calpain3 and proteostasis. Cell Rep., 24, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sato, T. (2020) Induction of skeletal muscle progenitors and stem cells from human induced pluripotent stem cells. J. Neuromuscul. Dis., 7, 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chal, J. and Pourquie, O. (2017) Making muscle: skeletal myogenesis in vivo and in vitro. Development, 144, 2104–2122. [DOI] [PubMed] [Google Scholar]
- 40. Chal, J., Oginuma, M., Al Tanoury, Z., Gobert, B., Sumara, O., Hick, A., Bousson, F., Zidouni, Y., Mursch, C., Moncuquet, P. et al. (2015) Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat. Biotechnol., 33, 962–969. [DOI] [PubMed] [Google Scholar]
- 41. Pedrotti, S., Giudice, J., Dagnino-Acosta, A., Knoblauch, M., Singh, R.K., Hanna, A., Mo, Q., Hicks, J., Hamilton, S. and Cooper, T.A. (2015) The RNA-binding protein Rbfox1 regulates splicing required for skeletal muscle structure and function. Hum. Mol. Genet., 24, 2360–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Runfola, V., Sebastian, S., Dilworth, F.J. and Gabellini, D. (2015) Rbfox proteins regulate tissue-specific alternative splicing of Mef2D required for muscle differentiation. J. Cell Sci., 128, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sampath, S.C., Sampath, S.C. and Millay, D.P. (2018) Myoblast fusion confusion: the resolution begins. Skelet. Muscle, 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee, K.S., Smith, K., Amieux, P.S. and Wang, E.H. (2008) MBNL3/CHCR prevents myogenic differentiation by inhibiting MyoD-dependent gene transcription. Differentiation, 76, 299–309. [DOI] [PubMed] [Google Scholar]
- 45. Du, H., Cline, M.S., Osborne, R.J., Tuttle, D.L., Clark, T.A., Donohue, J.P., Hall, M.P., Shiue, L., Swanson, M.S., Thornton, C.A. et al. (2010) Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol., 17, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Osborne, R.J., Lin, X., Welle, S., Sobczak, K., O'Rourke, J.R., Swanson, M.S. and Thornton, C.A. (2009) Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum. Mol. Genet., 18, 1471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang, M., Weng, W.C., Stock, L., Lindquist, D., Martinez, A., Gourdon, G., Timchenko, N., Snape, M. and Timchenko, L. (2019) Correction of glycogen synthase kinase 3beta in myotonic dystrophy 1 reduces the mutant RNA and improves postnatal survival of DMSXL mice. Mol. Cell. Biol., 39, e00155–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang, E.T., Treacy, D., Eichinger, K., Struck, A., Estabrook, J., Olafson, H., Wang, T.T., Bhatt, K., Westbrook, T., Sedehizadeh, S. et al. (2019) Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Hum. Mol. Genet., 28, 1312–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Batra, R., Charizanis, K., Manchanda, M., Mohan, A., Li, M., Finn, D.J., Goodwin, M., Zhang, C., Sobczak, K., Thornton, C.A. et al. (2014) Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell, 56, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kalsotra, A., Xiao, X., Ward, A.J., Castle, J.C., Johnson, J.M., Burge, C.B. and Cooper, T.A. (2008) A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. U. S. A., 105, 20333–20338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang, E.T., Ward, A.J., Cherone, J.M., Giudice, J., Wang, T.T., Treacy, D.J., Lambert, N.J., Freese, P., Saxena, T., Cooper, T.A. et al. (2015) Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res., 25, 858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Paul, S., Dansithong, W., Kim, D., Rossi, J., Webster, N.J., Comai, L. and Reddy, S. (2006) Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J., 25, 4271–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mondragon-Gonzalez, R. and Perlingeiro, R.C.R. (2018) Recapitulating muscle disease phenotypes with myotonic dystrophy 1 induced pluripotent stem cells: a tool for disease modeling and drug discovery. Dis. Model. Mech., 11, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ueki, J., Nakamori, M., Nakamura, M., Nishikawa, M., Yoshida, Y., Tanaka, A., Morizane, A., Kamon, M., Araki, T., Takahashi, M.P. et al. (2017) Myotonic dystrophy type 1 patient-derived iPSCs for the investigation of CTG repeat instability. Sci. Rep., 7, 42522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang, Y., Wang, Z., Sun, H., Shi, C., Yang, J., Liu, Y., Liu, H., Zhang, S., Zhang, L., Xu, Y. et al. (2018) Generation of induced pluripotent stem cell line(ZZUi006-A)from a patient with myotonic dystrophy type 1. Stem Cell Res., 32, 61–64. [DOI] [PubMed] [Google Scholar]
- 56. Barrault, L., Gide, J., Qing, T., Lesueur, L., Tost, J., Denis, J.A., Cailleret, M., Aubry, L., Peschanski, M., Martinat, C. et al. (2019) Expression of miRNAs from the imprinted DLK1/DIO3 locus signals the osteogenic potential of human pluripotent stem cells. Cell, 8, 1523–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jarrige, M., Polèche, H., Carteron, A., Janczarski, S., Peschanski, M., Auboeuf, D. and Martinat, C. (2021) SISTEMA: a large and standardized collection of transcriptome datasets for human pluripotent stem cell research. IScience, 24, 102767–102788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schmieder, R. and Edwards, R. (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics, 27, 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Maury, Y., Poydenot, P., Brinon, B., Lesueur, L., Gide, J., Roqueviere, S., Come, J., Polveche, H., Auboeuf, D., Alexandre Denis, J. et al. (2019) Pluripotent stem cell-based drug screening reveals cardiac glycosides as modulators of myotonic dystrophy type 1. iScience, 11, 258–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.