


Abstract
DNA repair systems allow microbes to survive in diverse environments that compromise chromosomal integrity. Pathogens such as Mycobacterium tuberculosis must contend with the genotoxic host environment, which generates the mutations that underlie antibiotic resistance. Mycobacteria encode the widely distributed SOS pathway, governed by the LexA repressor, but also encode PafBC, a positive regulator of the transcriptional DNA damage response (DDR). Although the transcriptional outputs of these systems have been characterized, their full functional division of labor in survival and mutagenesis is unknown. Here, we specifically ablate the PafBC or SOS pathways, alone and in combination, and test their relative contributions to repair. We find that SOS and PafBC have both distinct and overlapping roles that depend on the type of DNA damage. Most notably, we find that quinolone antibiotics and replication fork perturbation are inducers of the PafBC pathway, and that chromosomal mutagenesis is codependent on PafBC and SOS, through shared regulation of the DnaE2/ImuA/B mutasome. These studies define the complex transcriptional regulatory network of the DDR in mycobacteria and provide new insight into the regulatory mechanisms controlling the genesis of antibiotic resistance in M. tuberculosis.
INTRODUCTION
Cells are exposed to a variety of endogenous and exogenous factors that lead to DNA damage. DNA damage is particularly relevant to an intracellular pathogen such as Mycobacterium tuberculosis as its DNA is subject to assault from a variety of host defense mechanisms, many of which are genotoxic (1–5). There are multiple types of damage that occur on DNA, the most deleterious being double-strand breaks. As DNA integrity is essential for cell survival, growth and replication, cells encode multiple genes necessary for DNA damage repair and response (6). The development of antibiotic resistance in M. tuberculosis is solely the result of chromosomal mutations that arise during replication or because of mutagenic repair and response. A major regulator of the DNA damage response (DDR) in many bacteria is the inducible SOS pathway that is activated when the LexA repressor interacts with RecA nucleoprotein filaments on single-stranded DNA and undergoes autocatalytic cleavage (7–9). Mycobacterium also encodes a second DDR pathway, the PafBC pathway, which functions as a transcriptional activator with a currently unidentified activating signal (10). Although PafBC is encoded in an operon with the ubiquitin-like pup ligase PafA, PafB and PafC do not function in the pup-proteasome system (11). Transcriptional analyses of both Mycobacterium smegmatis and M. tuberculosis lacking the PafBC pathway (ΔpafBC) or recA (ΔrecA, as a surrogate for SOS inactivation) after DNA damage have defined the transcriptional regulon controlled by each pathway (10,12,13). These studies revealed that while there are some important overlaps in the genes regulated by these pathways, the PafBC pathway controls the larger transcriptional output after DNA damage (10,13). The PafBC proteins are not DNA damage inducible and therefore their mechanism of activation is unknown.
However, there are indications that despite having the smaller transcriptional regulon, the SOS pathway has an important functional role. For instance, the SOS pathway has been implicated in the survival of persistent cells (14) as well as in the induction of adaptive mutagenesis (15) in mycobacteria. The DnaE2 polymerase, which is required for mutagenesis in M. tuberculosis and M. smegmatis, is reported to be under SOS control (15). However, prior literature has used inactivation of recA as a surrogate for SOS inactivation, due to the essential role of RecA as the LexA coprotease. Although recA null cells are clearly SOS null, RecA has pleiotropic roles in DSB repair, replication fork restart and other functions (8,16), raising the possibility that RecA inactivation may have broader effects than simply SOS inactivation (17–19). Our knowledge of the functional overlap between the SOS and PafBC pathways in the mycobacterial DDR is therefore very limited.
This work aims to elucidate how SOS and PafBC functionally contribute to the DDR after distinct types of DNA damage by specifically ablating SOS and PafBC, alone or in combination, in comparison to loss of RecA. To specifically ablate SOS and avoid confounding functions of RecA, we engineered LexA-S167A, which prevents LexA autocatalytic cleavage. Characterization of these bacterial mutants revealed that, although PafBC does control a larger gene set, the two systems are both required for repair, with SOS playing a dominant role after UV damage and PafBC more important for survival after gyrase inhibition. Studies with specific DNA damaging agents further confirmed that gyrase inhibition and replication fork perturbation specifically activate the PafBC pathway. Finally, we show that, although DnaE2, the primary mutagenic polymerase of mycobacteria, is under SOS control after UV damage, it is coregulated by SOS and PafBC after gyrase inhibition. Further, the ImuA'/B cassette, also required for mutagenesis, is under dual control and, accordingly, mutagenesis is coregulated by both pathways.
MATERIALS AND METHODS
Bacterial strains/plasmid constructions and growth conditions
Mycobacterium smegmatis strains are derivatives of mc2155 and were grown and maintained in Difco Middlebrook 7H9 media (broth) supplemented with 10% ADS (0.5% albumin, 0.085% NaCl, 0.2% dextrose) and 0.05% Tween 80 or on Difco Middlebrook 7H10 (agar) supplemented with 0.5% glycerol and 0.5% dextrose at 37°C. Gene deletions were made by homologous recombination and double negative selection (20). The point mutant of LexA was generated using the previously described oligo recombineering procedure (21). Mutant strains were confirmed by PCR using primers outside the cloned region, followed by sequencing of the amplified PCR product to confirm the strains. Mycobacterium tuberculosis strains were on the H37Rv background and were grown and maintained in 7H9 media (broth) or on 7H10 (agar) supplemented with 10% oleic acid–albumin–dextrose–catalase supplement, 0.5% glycerol and 0.01% Tyloxapol (broth only) at 37°C. For a complete strain list with relevant features, see Supplementary Table S5. Plasmids utilized in this study were generated using standard molecular techniques and along with relevant oligos are listed with their features in Supplementary Table S5.
RT-qPCR
Mycobacterium smegmatis cultures were grown to OD600 ∼ 0.5–0.6, collected by centrifugation at 3200 × g and resuspended to OD600 = 0.6. For each treatment [0.5 μg/ml ciprofloxacin, 80 ng/ml mitomycin C (MMC) or 10 ml culture exposed to 20 mJ/cm2 UV], 10 ml cultures were shaken at 37°C/150 rpm at a final OD600 = 0.3 (for UV, 5 ml treated culture in 5 ml of fresh media) for the indicated time and collected for RNA preparation, lysed by bead beating three times for 30 s and after a 24-h incubation in 500 μl RNAlater buffer RNA was isolated using the GeneJet RNA Purification Kit. Five hundred nanograms of RNA (quantified on Thermo Scientific NanoDrop 8000 spectrophotometer) was used to make cDNA using the Thermo Maxima First Strand cDNA Synthesis Kit for RT-qPCR with dsDNase. The RT-qPCR reaction was made for a TaqMan assay using Thermo Scientific DyNAmo Flash Probe qPCR Kit (10 μl of master mix, 0.1 μl of each primer, 0.05 μl of each probe, 5 μl of cDNA sample and 4.5 μl of deionized H2O per reaction) and analyzed using the Applied Biosystems 7500 Real-Time System (cycling conditions: 95°C for 7 min, 45 cycles of 95°C for 5 s and 60°C for 30 s). Primer/probe sets for target genes of dnaE2, adnA and imuB were combined with primer/probe sets for sigA as the housekeeping gene and the analysis was done by comparing the ΔΔCT for each treated strain to wild-type (WT) mc2155 untreated control. Each cDNA sample was tested in duplicate, and no RT control reactions were included in all RT-qPCR experiments to exclude spurious amplification of contaminating chromosomal DNA.
Mycobacterium tuberculosis strains were grown to OD600 ∼ 0.5–0.6, collected by centrifugation at 3200 × g and resuspended to OD600 = 0.6. For each treatment (0.5 μg/ml ciprofloxacin or 8 ml culture exposed to ∼20 mJ/cm2 UV), cultures with a final volume of 10 ml were shaken at 37°C/150 rpm at a final OD600 = 0.3 (for UV, 5 ml treated culture in 5 ml of fresh media) for 24 h, lysed in TRIzol reagent by bead beating three times for 45 s and processed using the Direct-zol Miniprep Plus Kit. RNA was treated following the rigorous DNase treatment of the TURBO DNA-free Kit. cDNA synthesis and RT-qPCR used the above-mentioned protocol for M. smegmatis cells. Primers for RT-qPCR are given in Supplementary Table S5.
Ribosomal RNA depletion and transcriptional profiling
Mycobacterium smegmatis and M. tuberculosis RNA samples for ciprofloxacin [0.5 μg/ml for 3 h (M. smegmatis) or 24 h (M. tuberculosis)] or UV [20 mJ/cm2 with a recovery period of 1 h (M. smegmatis) or 24 h (M. tuberculosis) after exposure] transcriptional profiling by RNA sequencing were isolated from cells grown and treated as described earlier. The M. smegmatis RNA samples (n = 2) were depleted for ribosomal RNA using a biotinylated oligonucleotide-based protocol (22). The M. tuberculosis RNA samples (n = 1) were depleted for ribosomal RNA using the Ilumina Ribo-Zero Plus rRNA Depletion Kit. For both rRNA depletion methods, the efficiency of rRNA depletion was variable between samples with the percentage of reads mapped to rRNA after depletion ranging from 11% to 92%. Despite this variable rRNA depletion, all samples contained >3 million nonribosomal mapping transcripts. RNA sequencing was performed as previously reported (23).
DNA damage assays
Strains were grown to saturation and were diluted to an OD600 = 0.02 and grown to OD600 ∼ 0.6, collected by centrifugation at 3200 × g and resuspended to an OD600 = 0.6. For UV exposure, cells were serially diluted in phosphate-buffered saline (PBS) + 0.05% Tween 80 onto 7H10 agar plates. Agar plates were treated with the indicated doses of UV radiation with a Stratagene UV Stratalinker 1800 with 254-nm UV bulbs. Plates were wrapped in foil (to prevent potential effects of photolyase) and incubated at 37°C. For treatment with ciprofloxacin (0.5 μg/ml for 3 h) and MMC (80 ng/ml for 3 h), cultures were incubated at a final OD600 = 0.3. Cultures were washed with PBS + 0.05% Tween 80 and then were serially diluted in PBS + 0.05% Tween 80 onto 7H10 agar plates
Immunoblotting
Mycobacterium smegmatis lysates were prepared from cells exposed to ciprofloxacin (1.25 μg/ml for 1 and 3 h), UV (20 mJ/cm2 with a recovery period of 1 and 3 h after exposure), MMC (80 ng/ml for 2 and 5 h), anhydrotetracycline (ATc; 50 ng/ml for described time), cumene hydroperoxide (CHP) (50 μM for 1 h or 3 h), SKI356313 (1.9 μM for 1 or 3 h) or etoposide (24 μM for 3 h) by bead beating two times for 30 s (5-min rest on ice between each cycle). Protein quantities were normalized using the Protein A280 on the NanoDrop 8000 and normalized to an apparent concentration of 0.2 mg/ml. Blots were blocked and probed in 5% Omniblot milk in 1× PBST (PBS + 0.01% Tween 20). Equal loading was confirmed with commercially available Biolegend Escherichia coli anti-RpoB antibody (1:10 000 dilution) as a loading control. RecA antibody, raised in rabbits against purified full-length M. smegmatis RecA (8), was used at a 1:5000 dilution. For RecA-Streptag II (STII), STII (GenScript rabbit anti-NWSHPQFEK polyclonal antibody) was used at a 1:5000 dilution and the blots were blocked and probed in Strep wash buffer (150 mM NaCl, 100 mM Tris, pH 7.9/8, 1 mM EDTA). LexA antibody, raised in rabbits against purified full-length M. smegmatis LexA, was used at a 1:2000 dilution. Blots were imaged in iBright FL1000 and quantified using the iBright analysis software. Mycobacterium tuberculosis lysates were prepared from cells exposed to ciprofloxacin (1.25 μg/ml for 24 h or UV (20 mJ/cm2 for 24 h) by bead beating three times for 45 s. Blots were blocked and probed similarly to M. smegmatis samples.
UV-induced mutagenesis
Ten milliliters of each strain at OD600 = 0.6 was transferred to OmniTray single-well plates (M. smegmatis) or extra depth disposable Petri dish (M. tuberculosis) and exposed to 20 mJ/cm2 UV radiation using a Stratagene UV Stratalinker 1800. From each treated sample and its untreated control, 5 ml of culture was transferred to 5 ml of fresh media and shaken at 37°C/150 rpm for 3 h (M. smegmatis) or 24 h (M. tuberculosis). From each sample, a total of 5–9 ml of culture was cultured as 400 μl aliquots on 7H10 agar plates containing 0.5% glycerol, 0.5% dextrose and 100 μg/ml rifampicin and incubated at 37°C for 72 h (M. smegmatis) or 3 weeks (M. tuberculosis) to determine rifampicin-resistant CFU. Additional duplicates were taken from each sample and dilution plated on 7H10 agar containing no antibiotic to determine viable CFU. Resistant mutants were then normalized to viable CFU for each set of samples. Graph represents average rifampicin-resistant mutants per viable CFU.
In vitro LexA autoproteolysis
RecA-stimulated LexA autoproteolysis assays were performed as previously described (8). The LexA-S167A protein was purified as previously described (8) using the PET-SUMO system and the presence of the LexA-S167A mutation was confirmed by DNA sequencing of the expression plasmid.
Statistical analyses
Significance tests were performed in GraphPad Prism using a two-way analysis of variance (ANOVA) test on log-transformed values. All performed statistical tests were two-sided. All error bars represent standard error of the mean (SEM), unless specifically noted otherwise.
RESULTS
Genetic ablation of the DDR in M. smegmatis
To investigate the division of labor between the PafBC and SOS DDR pathways, we constructed mutants of each pathway, alone and in combination, in M. smegmatis. The PafBC pathway was ablated by deletion of the coding sequences of both proteins (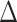 pafBC) using a previously validated homologous recombination-based knockout strategy (20). LexA is a repressor that is inactivated by RecA-stimulated autocatalytic proteolysis (24). As such, deletion of lexA results in derepression of SOS, whereas deletion of recA may be a poor surrogate for SOS ablation due to its pleiotropic roles in DNA repair. To circumvent this problem, we introduced a point mutation into the lexA chromosomal locus to direct synthesis of LexA-S167A, which renders LexA uncleavable in E. coli (25), in both the WT (mc2155) and
pafBC) using a previously validated homologous recombination-based knockout strategy (20). LexA is a repressor that is inactivated by RecA-stimulated autocatalytic proteolysis (24). As such, deletion of lexA results in derepression of SOS, whereas deletion of recA may be a poor surrogate for SOS ablation due to its pleiotropic roles in DNA repair. To circumvent this problem, we introduced a point mutation into the lexA chromosomal locus to direct synthesis of LexA-S167A, which renders LexA uncleavable in E. coli (25), in both the WT (mc2155) and 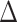 pafBC strains. These strains (WT, ΔpafBC, lexA-S167A, ΔpafBC/lexA-S167A,
pafBC strains. These strains (WT, ΔpafBC, lexA-S167A, ΔpafBC/lexA-S167A, 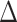 recA) were analyzed by PCR to confirm their genotypes (Figure 1A).
recA) were analyzed by PCR to confirm their genotypes (Figure 1A).
Figure 1.

Genetic ablation of the PafBC and SOS pathways in M. smegmatis. (A) Confirmation of ΔpafBC and lexA-S167A genotypes of mc2155 (WT), 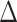 recA,
recA,  pafBC, lexA-S167A and
pafBC, lexA-S167A and  pafBC/lexA-S167A strains. The pafBC deletion allele was detected by PCR amplification with primers (OAM229 and OAM232) that amplify the genomic region upstream of pafBC yielding a product of 3059 bp in strains carrying WT pafBC and 1122 bp in strains with
pafBC/lexA-S167A strains. The pafBC deletion allele was detected by PCR amplification with primers (OAM229 and OAM232) that amplify the genomic region upstream of pafBC yielding a product of 3059 bp in strains carrying WT pafBC and 1122 bp in strains with  pafBC. The lexA-S167A mutation was detected by selective amplification using primers (OAM189 and OAM268) that anneal to the mutated LexA, yielding a product of 234 bp for lexA-S167A but no product from the WT allele. L denotes the DNA ladder. (B) LexA cleavage with DNA damage according to strain genotype. α-LexA immunoblot of mid-log-phase expression of LexA (28 kDa, bottom panel) in mc2155,
pafBC. The lexA-S167A mutation was detected by selective amplification using primers (OAM189 and OAM268) that anneal to the mutated LexA, yielding a product of 234 bp for lexA-S167A but no product from the WT allele. L denotes the DNA ladder. (B) LexA cleavage with DNA damage according to strain genotype. α-LexA immunoblot of mid-log-phase expression of LexA (28 kDa, bottom panel) in mc2155,  recA,
recA,  pafBC, lexA-S167A and
pafBC, lexA-S167A and  pafBC/lexA-S167A strains without (−) or with (+) DNA damage (20 mJ/cm2 UV). RpoB is shown as a loading control (top panel). (C) Schematic of recA-Streptag (STII) expression construct containing the PafBC (P1) and LexA (P2) binding sites in the RecA promoter. recA promoter activity was measured by α-Streptag (STII) western blot of mid-log-phase expression of RecA-STII (37 kDa) in mc2155,
pafBC/lexA-S167A strains without (−) or with (+) DNA damage (20 mJ/cm2 UV). RpoB is shown as a loading control (top panel). (C) Schematic of recA-Streptag (STII) expression construct containing the PafBC (P1) and LexA (P2) binding sites in the RecA promoter. recA promoter activity was measured by α-Streptag (STII) western blot of mid-log-phase expression of RecA-STII (37 kDa) in mc2155,  pafBC, lexA-S167A and
pafBC, lexA-S167A and  pafBC/lexA-S167A strains carrying RecA-STII driven only by the LexA repressed P2 (P1 mutated, D) or only the pafBC promoter (P2 mutated, E) with or without DNA damage (UV, 20 mJ/cm2 or ciprofloxacin, 1.25 μg/ml). RpoB is shown as a loading control for both blots in panels (D) and (E).
pafBC/lexA-S167A strains carrying RecA-STII driven only by the LexA repressed P2 (P1 mutated, D) or only the pafBC promoter (P2 mutated, E) with or without DNA damage (UV, 20 mJ/cm2 or ciprofloxacin, 1.25 μg/ml). RpoB is shown as a loading control for both blots in panels (D) and (E).
To confirm that the LexA-S167A mutation impairs LexA cleavage in vitro, we purified the LexA-S167A protein and compared it to WT LexA in a previously described (8) LexA autoproteolysis assay catalyzed by RecA nucleoprotein filaments. These experiments confirmed that the S167A mutation abolishes autoproteolysis (Supplementary Figure S1). To confirm that LexA-S167A is resistant to cleavage in vivo, we analyzed LexA levels by immunoblotting, with or without 20 mJ/cm2 UV light (Figure 1B). In WT M. smegmatis, LexA protein was detectable at its predicted molecular weight of 28 kDa in basal conditions (Figure 1B). With UV treatment, full-length LexA became nearly undetectable, although we were unable to detect LexA proteolytic fragments (Figure 1B). In the predicted SOS-deficient strains (ΔrecA, lexA-S167A and pafBC/lexA-S167A), there was no discernible loss of LexA protein with DNA damage (Figure 1B), consistent with impaired LexA cleavage. Ablation of pafBC had no effect on LexA cleavage with damage.
To confirm that both pathways, PafBC and SOS, are functionally impaired by these mutations, we took advantage of the dual regulation of recA transcription by both pathways. The recA promoter contains binding sites for both LexA and PafBC (10,17) and is therefore subject to dual regulation (Figure 1C). We introduced mutations into the SOS box (P2) or the PafBC binding sites (P1) and used these mutated promoters to drive RecA-STII expression in WT M. smegmatis or in strains lacking PafBC, SOS or both (Figure 1D and E). When RecA is controlled only by SOS (P1 mutated, Figure 1D), RecA protein is induced with DNA damage (either UV or quinolone treatment) in WT cells. In cells lacking pafBC, RecA protein is induced by UV but not by quinolone treatment (a phenotype that will be discussed later), but RecA expression by both UV and quinolone treatment is impaired in cells lacking SOS or both pathways, indicating that the LexA-S167A strongly impairs SOS activation in vivo (Figure 1D). In contrast, when RecA is controlled only by PafBC (P2 mutated, Figure 1E), RecA protein is not induced with DNA damage in cells lacking PafBC but is inducible in WT cells as well as cells lacking SOS (Figure 1E). These experiments confirm the functional inactivation of the PafBC and SOS pathways and provide a genetic model system to dissect the relative roles of these two pathways in DNA repair and mutagenesis.
Roles of SOS and PafBC in the transcriptional DDR across DNA damaging agents
The transcriptomic profiles of both the 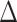 pafBC and ΔrecA strains have been assessed primarily in response to MMC in M. smegmatis and M. tuberculosis (10,12,13). However, different types of DNA damaging agents produce fundamentally different types of DNA lesions that may require unique systems for correction, the expression of which may be governed by SOS or PafBC. To investigate the possibility of DNA damage-specific responses for these pathways, we measured the transcriptional responses of the WT, ΔrecA,
pafBC and ΔrecA strains have been assessed primarily in response to MMC in M. smegmatis and M. tuberculosis (10,12,13). However, different types of DNA damaging agents produce fundamentally different types of DNA lesions that may require unique systems for correction, the expression of which may be governed by SOS or PafBC. To investigate the possibility of DNA damage-specific responses for these pathways, we measured the transcriptional responses of the WT, ΔrecA, 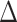 pafBC, lexA-S167A and
pafBC, lexA-S167A and 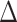 pafBC/lexA-S167A strains after exposure to UV or ciprofloxacin. In WT cells, UV and ciprofloxacin induced a common set of 185 genes (log2 fold change of ≥1.5; Supplementary Figure 1A). There were an additional 24 genes exclusively induced by ciprofloxacin, which were composed of genes predicted to be involved in replication, recombination and repair. In contrast, UV induced an additional 174 genes, most of which were genes with unknown function. To deduce whether PafBC or LexA controls different gene sets in response to different types of DNA damage, we focused on the subset of genes that were commonly induced by UV and ciprofloxacin (from our studies) as well as MMC from the literature in WT cells (10).
pafBC/lexA-S167A strains after exposure to UV or ciprofloxacin. In WT cells, UV and ciprofloxacin induced a common set of 185 genes (log2 fold change of ≥1.5; Supplementary Figure 1A). There were an additional 24 genes exclusively induced by ciprofloxacin, which were composed of genes predicted to be involved in replication, recombination and repair. In contrast, UV induced an additional 174 genes, most of which were genes with unknown function. To deduce whether PafBC or LexA controls different gene sets in response to different types of DNA damage, we focused on the subset of genes that were commonly induced by UV and ciprofloxacin (from our studies) as well as MMC from the literature in WT cells (10).
Transcriptomic profiling of the DDR pathway mutants revealed three major DDR profiles. Profile 1 consists of genes whose expression levels with DNA damage are completely dependent on the PafBC pathway, and independent of RecA, and this gene set is the largest regulon transcriptionally induced after DNA damage (Figure 2A and Supplementary Table S1), consistent with prior studies (10). Thirty-five percent of these genes are predicted to encode proteins involved in replication, recombination and repair, which has been previously noted as being overrepresented in the PafBC regulon after MMC-induced damage (10). Profile 2 consists of genes that have varying degrees of dependence on PafBC, SOS or RecA (Figure 2B and Supplementary Table S2). Of particular interest are the genes that are codependent on PafBC and SOS after ciprofloxacin damage, but are PafBC independent after UV (Figure 2B, red and blue boxes). This subset is of interest because they indicate DDR pathway clastogen-specific responses and consist of genes with important roles in DNA damage repair and response. Although UV induces a larger number of genes compared to ciprofloxacin (Supplementary Figure S2A), this pathway-specific pattern is unique to ciprofloxacin. Twenty-seven percent of the DDR pathway ciprofloxacin-specific subset has a partial or complete loss of induction in the 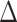 pafBC or lexA-S167A strains. Of the genes with ciprofloxacin-specific induction in the lexA-S167A strain, 82% are also either fully or partially dependent on the PafBC pathway. Profile 3 consists of genes that are DNA damage inducible but have no discernible loss of expression in any of the DDR pathway mutants (Figure 2C and Supplementary Table S3). A majority of these are genes are of unknown function. These results confirm prior results that PafBC controls a numerically larger number of DNA damage inducible genes irrespective of the type of DNA damage (Figure 2D and E), but also indicate that there are clastogen- and pathway-specific overlapping gene sets within the DDR.
pafBC or lexA-S167A strains. Of the genes with ciprofloxacin-specific induction in the lexA-S167A strain, 82% are also either fully or partially dependent on the PafBC pathway. Profile 3 consists of genes that are DNA damage inducible but have no discernible loss of expression in any of the DDR pathway mutants (Figure 2C and Supplementary Table S3). A majority of these are genes are of unknown function. These results confirm prior results that PafBC controls a numerically larger number of DNA damage inducible genes irrespective of the type of DNA damage (Figure 2D and E), but also indicate that there are clastogen- and pathway-specific overlapping gene sets within the DDR.
Figure 2.

Relative contributions of PafBC or LexA to the transcriptional DDR in mycobacteria. Gene expression heatmaps of genes that are significantly upregulated (log2 fold change ≥1.5, P-value <0.01) in WT M. smegmatis mc2155 by ciprofloxacin (0.5 μg/ml) or UV (20 mJ/cm2) from transcriptomic profiling by RNA sequencing of mc2155, ΔrecA, ΔpafBC, lexA-S167A and ΔpafBC/lexA-S167A strains. (A) The RecA-independent PafBC regulon. The heatmap displays genes in which DNA damage-induced expression is dependent on PafBC, but preserved in LexA-S167A and ΔrecA strains. The scale bar depicts normalized expression level. (B) Codependent and clastogen-specific DDR. The red box highlights genes that are RecA/SOS/PafBC dependent with ciprofloxacin, but only SOS dependent with UV. The blue boxes highlight PafBC/SOS codependent genes with ciprofloxacin stress. (C) Genes whose DNA damage-dependent expression levels are independent of either DDR pathway. Venn diagrams categorizing genes for which the DNA damage induction is abolished (WT log2 fold change is >2.5× of the compared strain) in the indicated strain backgrounds (ΔrecA, ΔpafBC, lexA-S167A and ΔpafBC/lexA-S167A) compared to mc2155 with ciprofloxacin (D) or UV (E) treatment. The genes represented in the Venn diagrams are the same genes represented in the heatmaps in panels (A)–(C).
We extended these findings to M. tuberculosis by comparing the transcriptional response of a strain of M. tuberculosis H37Rv with a transposon insertion in pafC to both WT (H37Rv) and a pafC::tn complemented strain after exposure to both UV and ciprofloxacin. Like M. smegmatis, UV induced a larger number of genes in WT compared to ciprofloxacin (Supplementary Figure S2B). Although the numbers of genes uniquely induced by ciprofloxacin alone in both M. smegmatis and M. tuberculosis WT cells are similar (Supplementary Figure S2B), the only ciprofloxacin unique gene that was induced in both mycobacterial species was dnaQ (Rv3711c/MSMEG_6275). Mycobacterial DnaQ is a homologue of the E. coli 3′–5′ exonuclease epsilon. To confirm the PafBC and ciprofloxacin phenotypes observed in M. smegmatis, we focused on the subset of genes that were induced by both UV and ciprofloxacin (Supplementary Figure S2B). Although an M. tuberculosis LexA uncleavable strain was not available for comparison, we observed that 57% of the genes are no longer induced by either clastogen in the absence of pafC (Supplementary Table S4). We also observed a similar subset of genes that have a PafBC ciprofloxacin-dependent phenotype that includes recA (Supplementary Table S4 and Supplementary Figure S2C). These data confirm our observation of clastogen-specific gene sets within the DDR.
To confirm our RNA-seq results and gain a clearer temporal picture of the roles of the SOS and PafBC pathways in the temporal transcriptional response to different DNA damaging agents, we analyzed the mRNA encoding AdnA [a PafBC-dependent gene in our RNA-seq dataset and in the literature (10,12)] and DnaE2 [an SOS-dependent gene as defined in our RNA-seq dataset and in the literature (12)] by RT-qPCR. Analyzing the expression of adnA after treatment with UV, ciprofloxacin or MMC confirmed the results from the transcriptomic profiling that both basal and induced adnA expression are dependent on the PafBC pathway, regardless of the DNA damaging agent tested, as reflected in the significantly reduced expression of adnA with or without DNA damage in the  pafBC and
pafBC and  pafBC/lexA-S167A strains (Figure 3A–C). Impairment of the SOS pathway in both the
pafBC/lexA-S167A strains (Figure 3A–C). Impairment of the SOS pathway in both the  recA and lexA-S167A strains led to a significant increase in adnA expression in basal conditions (Figure 3A–C). However, induction of adnA in the SOS-deficient strains after DNA damage was not impaired compared to WT cells, further confirming that adnA is exclusively controlled by the PafBC- and RecA-independent pathway. The expression of adnA in the
recA and lexA-S167A strains led to a significant increase in adnA expression in basal conditions (Figure 3A–C). However, induction of adnA in the SOS-deficient strains after DNA damage was not impaired compared to WT cells, further confirming that adnA is exclusively controlled by the PafBC- and RecA-independent pathway. The expression of adnA in the  pafBC strain was rescued by expressing either M. smegmatis or M. tuberculosis PafBC (Figure 3D), confirming that these phenotypes are due to loss of PafBC function.
pafBC strain was rescued by expressing either M. smegmatis or M. tuberculosis PafBC (Figure 3D), confirming that these phenotypes are due to loss of PafBC function.
Figure 3.

Gene- and clastogen-specific requirements for PafBC and LexA in the transcriptional DDR. Normalized adnA mRNA measured by RT-qPCR in the indicated strains of M. smegmatis (black = WT, orange = ΔrecA, blue = ΔpafBC, purple = lexA-S167A and red = ΔpafBC/lexA-S167A) after exposure to (A) UV (20 mJ/cm2) (n = 3 biological replicates), (B) ciprofloxacin (0.5 μg/ml) (n = 5 biological replicates) or (C) MMC (80 ng/ml) (n = 3 biological replicates). All values are normalized to WT untreated at 1. (D) Genetic complementation. Normalized adnA mRNA measured by RT-qPCR in M. smegmatis ΔpafBC complemented with M. tuberculosis pafBC, M. smegmatis pafBC or empty vector (EV) after exposure to UV (20 mJ/cm2) (n = 2 biological replicates). All values are normalized to M. smegmatis ΔpafBC complemented with M. smegmatis pafBC. Normalized dnaE2 mRNA measured by RT-qPCR in the same M. smegmatis strains as in panels (A)–(C) after exposure to (E) UV (20 mJ/cm2) (n = 3 biological replicates), (F) ciprofloxacin (0.5 μg/ml) (n = 5 biological replicates) or (G) MMC (80 ng/ml) (n = 3 biological replicates). All values are normalized to WT untreated at 1. (H) Normalized dnaE2 mRNA in M. smegmatis ΔpafBC strains complemented with M. tuberculosis pafBC, M. smegmatis pafBC or empty vector (EV) (same strain legend as in panel D) after exposure to UV (20 mJ/cm2) (n = 2 biological replicates). All values are normalized to M. smegmatis ΔpafBC complemented with M. smegmatis pafBC untreated at 1. Normalized mRNA measured by RT-qPCR for (I)adnA or (J)dnaE2 in M. tuberculosis H37Rv, H37Rv pafC::tn and pafC::tn + pafC. All values are normalized to WT untreated at 1. Significance is calculated as *P < 0.05 or ** P< 0.01 using two-way ANOVA compared to the WT strain (mc2155 or H37Rv) at a comparable time point/condition. ‡ P < 0.05 using two-way ANOVA compared to lexA-S167A strain at a comparable time point/condition. Error bars are SEM.
dnaE2 is reported to be SOS dependent, a conclusion that is derived from its impaired expression in mycobacteria lacking recA (12). Our experiments confirm this impaired expression of dnaE2 with DNA damage in ΔrecA (Figure 3E–G). However, although loss of SOS in the lexA-S167A strain impairs dnaE2 induction to a greater degree than loss of pafBC, there is a significant impairment of dnaE2 expression in ΔpafBC, especially with ciprofloxacin and MMC. Only in the  pafBC/lexA-S167A strain does dnaE2 expression phenocopy that observed in ΔrecA (Figure 3E–G). The impaired expression of dnaE2 in the
pafBC/lexA-S167A strain does dnaE2 expression phenocopy that observed in ΔrecA (Figure 3E–G). The impaired expression of dnaE2 in the  pafBC was rescued by complementation with either M. smegmatis or M. tuberculosis pafBC (Figure 3H).
pafBC was rescued by complementation with either M. smegmatis or M. tuberculosis pafBC (Figure 3H).
We extended these findings to M. tuberculosis using the strain of M. tuberculosis H37Rv with a transposon insertion in pafC. We confirmed that adnA expression is reduced in basal conditions in cells lacking pafC, a phenotype that is complemented by the WT gene (Figure 3I). UV or ciprofloxacin induction of adnA was completely abrogated in pafC::tn (Figure 3I). In contrast, although UV-induced dnaE2 expression was independent of pafC (Figure 3J), dnaE2 induction with ciprofloxacin was substantially dependent on pafC (Figure 3J), again complemented by WT pafC (Figure 3J) as is seen in M. smegmatis. These results mirrored the transcriptional induction of both adnA and dnaE2 observed in M. tuberculosis by RNA sequencing (Supplementary Table S4). Taken together, these data confirm the PafBC dependence of adnA expression, but also implicate PafBC in dnaE2 expression under specific conditions. These data suggest that these two systems may mediate the response to different types of DNA damage and indicate that loss of recA has more severe effects on the DDR than specific ablation of the SOS pathway that more closely resembles loss of both SOS and PafBC. These data suggest a role for RecA in inducing the PafBC regulon under certain types of DNA damage.
Roles of SOS and PafBC in DNA damage survival across DNA damaging agents
Although the transcriptional output controlled by PafBC or LexA is useful to understand the relative roles of these two transcription factors, the ultimate functional outcomes of the DDR may be governed by a small number of gene products or nontranscriptional mechanisms (8). To functionally characterize the relative contributions of LexA and PafBC, we measured the survival of the mutants after treatment with the same clastogens used for transcriptional profiling. As previously described (8), loss of recA severely sensitizes M. smegmatis to all forms of DNA damage, a phenotype that could partially reflect its role in SOS induction, but also other repair roles such as DSB repair and restart of stalled replication forks (18,19). Loss of LexA cleavage severely sensitized cells to UV killing, almost to the same degree as loss of recA, with PafBC playing a more minor role, especially at low doses (Figure 4A). Only the  pafBC/lexA-S167A strain fully phenocopied loss of recA. With gyrase inhibition (ciprofloxacin), the PafBC and SOS pathways contributed nearly equally, but only loss of both pathways phenocopied loss of recA (Figure 4B). For mitomycin, loss of LexA cleavage sensitized cells to killing more than loss of PafBC (Figure 4C). The sensitivity of the
pafBC/lexA-S167A strain fully phenocopied loss of recA. With gyrase inhibition (ciprofloxacin), the PafBC and SOS pathways contributed nearly equally, but only loss of both pathways phenocopied loss of recA (Figure 4B). For mitomycin, loss of LexA cleavage sensitized cells to killing more than loss of PafBC (Figure 4C). The sensitivity of the  pafBC strain was rescued by expressing M. smegmatis or M. tuberculosis pafBC (Figure 4D). These results indicate that, despite controlling a smaller number of genes, the SOS pathway plays an important role in survival after damage. In addition, the data confirm the finding from transcriptional profiling that these two pathways respond to different types of damage, with SOS having a dominant role for UV and cross-linking and PafBC playing a more important role in the response to gyrase inhibition.
pafBC strain was rescued by expressing M. smegmatis or M. tuberculosis pafBC (Figure 4D). These results indicate that, despite controlling a smaller number of genes, the SOS pathway plays an important role in survival after damage. In addition, the data confirm the finding from transcriptional profiling that these two pathways respond to different types of damage, with SOS having a dominant role for UV and cross-linking and PafBC playing a more important role in the response to gyrase inhibition.
Figure 4.

Functional outputs of the DDR are codependent on LexA and PafBC. Survival of mc2155 (black), ΔrecA (orange), ΔpafBC (blue), lexA-S167A (purple) and ΔpafBC/lexA-S167A (red) after exposure to (A) UV (0, 5, 10 or 20 mJ/cm2) (n = 4 biological replicates), (B) ciprofloxacin (0.5 μg/ml) (n = 4 biological replicates) or (C) MMC (80 ng/ml) (n = 5 biological replicates). (D) Survival of ΔpafBC strains complemented with M. tuberculosis pafBC, M. smegmatis pafBC or empty vector (EV) and mc2155 complemented with EV after exposure to UV (20 mJ/cm2) (n = 2 biological replicates). Significance is calculated as *P < 0.05 using two-way ANOVA compared to the WT strain at a comparable time point/condition. ‡P < 0.05 using two-way ANOVA compared to lexA-S167A strain at a comparable time point/condition. Error bars are SEM.
Roles of SOS and PafBC in adaptive mutagenesis
DnaE2 has previously been shown to play a major role in adaptive mutagenesis after UV-induced DNA damage (15). As we observed that dnaE2 expression is controlled by both SOS and PafBC when DNA damage was induced by MMC and ciprofloxacin, but not by UV, we quantitated the functional output of adaptive mutagenesis by measuring the appearance of rifampin resistance (RifR) after UV light exposure. As expected, UV strongly induces the frequency of RifR in WT cells and loss of recA abolishes that induction (Figure 5A) (8). Surprisingly, we found that loss of either DDR pathway ( pafBC and lexA-S167A strains) impaired mutagenesis (Figure 5A), whereas loss of both pathways eliminated mutagenesis (Figure 5A). The loss of mutagenesis in the
pafBC and lexA-S167A strains) impaired mutagenesis (Figure 5A), whereas loss of both pathways eliminated mutagenesis (Figure 5A). The loss of mutagenesis in the  pafBC mutant could be rescued by complementation using M. smegmatis pafBC (Figure 5B). Extending these findings to M. tuberculosis revealed a reduction in adaptive mutagenesis in the absence of pafC, a phenotype that was complemented by the WT pafC gene (Figure 5C). The decrement of UV-induced mutagenesis in the absence of the PafBC pathway was surprising because dnaE2 expression was not significantly reduced after UV exposure in either M. smegmatis or M. tuberculosis pafBC single mutants (Figure 3E and J). Although DnaE2 activity is required for its mutagenic role in vivo (15), the mutasome also consists of the ImuA/B cassette, which are also required for mutagenesis in vivo (26). These genes are reported to be SOS regulated (12), but recent data also indicate that there is a PafBC binding site in the imuA′ (MSMEG_1620) promoter and DNA damage-induced expression of imuA'/B was impaired in M. smegmatis lacking pafBC (10). Consistent with these prior data, our transcriptomic profiling revealed that imuA'/B are strongly induced by both UV and quinolone damage and this induction is abolished in cells lacking recA, a finding previously interpreted to indicate SOS dependence. To clarify the regulation of imuA'/B, we measured the expression of imuB, which is an operon with imuA' (26), by RT-qPCR and found that imuB expression was significantly impaired by the loss of either the SOS or PafBC pathway (Figure 5D and E). We observed a partial impairment of imuB in both the lexA-S167A and ΔpafBC knockout strains, with a stronger impairment conferred after ciprofloxacin-induced DNA damage and only in the pafBC/lexA-S167A strain did we observe complete loss of imuB expression that phenocopied loss of recA (Figure 5D). In M. tuberculosis, imuB expression was induced with UV exposure, and was mildly impaired in cells lacking pafC (Figure 5E). However, imuB induction after ciprofloxacin was abolished in pafC::tn cells, a phenotype that was complemented by WT pafC (Figure 5E).
pafBC mutant could be rescued by complementation using M. smegmatis pafBC (Figure 5B). Extending these findings to M. tuberculosis revealed a reduction in adaptive mutagenesis in the absence of pafC, a phenotype that was complemented by the WT pafC gene (Figure 5C). The decrement of UV-induced mutagenesis in the absence of the PafBC pathway was surprising because dnaE2 expression was not significantly reduced after UV exposure in either M. smegmatis or M. tuberculosis pafBC single mutants (Figure 3E and J). Although DnaE2 activity is required for its mutagenic role in vivo (15), the mutasome also consists of the ImuA/B cassette, which are also required for mutagenesis in vivo (26). These genes are reported to be SOS regulated (12), but recent data also indicate that there is a PafBC binding site in the imuA′ (MSMEG_1620) promoter and DNA damage-induced expression of imuA'/B was impaired in M. smegmatis lacking pafBC (10). Consistent with these prior data, our transcriptomic profiling revealed that imuA'/B are strongly induced by both UV and quinolone damage and this induction is abolished in cells lacking recA, a finding previously interpreted to indicate SOS dependence. To clarify the regulation of imuA'/B, we measured the expression of imuB, which is an operon with imuA' (26), by RT-qPCR and found that imuB expression was significantly impaired by the loss of either the SOS or PafBC pathway (Figure 5D and E). We observed a partial impairment of imuB in both the lexA-S167A and ΔpafBC knockout strains, with a stronger impairment conferred after ciprofloxacin-induced DNA damage and only in the pafBC/lexA-S167A strain did we observe complete loss of imuB expression that phenocopied loss of recA (Figure 5D). In M. tuberculosis, imuB expression was induced with UV exposure, and was mildly impaired in cells lacking pafC (Figure 5E). However, imuB induction after ciprofloxacin was abolished in pafC::tn cells, a phenotype that was complemented by WT pafC (Figure 5E).
Figure 5.

Mycobacterial mutagenesis requires both SOS and PafBC. (A) Frequency of rifampin-resistant mutants per 108 cells in M. smegmatis mc2155, ΔrecA, ΔpafBC, lexA-S167A and ΔpafBC/lexA-S167A with or without DNA damage (20 mJ/cm2 UV) (n = 4 biological replicates). # indicates that no RifR colonies were recovered. (B) Frequency of rifampin-resistant mutants per 108 cells in WT M. smegmatis, ΔpafBC or ΔpafBC complemented with M. smegmatis pafBC after exposure to UV (20 mJ/cm2) (n = 2 biological replicates). (C) pafBC is required for mutagenesis in M. tuberculosis. Frequency of rifampin resistance per 108 cells in M. tuberculosis strains of H37Rv, transposon insertion mutant of pafC (pafC TN) and transposon insertion mutant of pafC complemented with pafC (pafC comp) with or without DNA damage (20 mJ/cm2 UV) (n = 6 biological replicates). (D) Normalized imuB mRNA measured by RT-qPCR in the indicated strains of M. smegmatis after exposure to either UV (20 mJ/cm2) (n = 3 biological replicates) or ciprofloxacin (0.5 μg/ml) (n = 3 biological replicates). All values are normalized to WT untreated at 1. (E) Normalized imuB mRNA measured by RT-qPCR in the indicated strains of M. tuberculosis after exposure to either UV (20 mJ/cm2) (n = 4 biological replicates) or ciprofloxacin (0.5 μg/ml) (n = 4 biological replicates). All values are normalized to WT untreated at 1. Significance is calculated as *P < 0.05 using two-way ANOVA compared to WT untreated. Error bars are SEM.
These data indicate that both the PafBC and SOS pathways contribute to adaptive mutagenesis in mycobacteria, in part through shared regulation of the dnaE2 cofactors imuA'/B. The PafBC binding sequence in M. smegmatis has been deduced from chromatin immunoprecipitation and transcriptomic experiments and consists of a 22-nt sequence with a highly conserved 5′ TGTCGG sequence (10). This motif resembled prior analyses of the RecA-independent promoter motif present in DNA damage inducible genes in mycobacteria (27). To deduce whether PafBC-dependent, ciprofloxacin-induced transcription at the imuA'/B locus differs from UV-induced transcription, we visualized RNA sequencing reads in the putative imuA' (rv3395c) promoter. No reads mapped to imuA' in basal conditions, and we observed strong induction of imuA' transcription with UV, with reads mapping to an approximate transcription start site immediately 3′ to a consensus binding site for the LexA repressor (Supplementary Figure S3). In contrast, ciprofloxacin-induced imuA' transcription was detectable 166 nt 5′ from the LexA site at a position that overlaps the divergently transcribed gene rv3395a, which is of unknown function. Inspection of the complementary strand DNA sequence in the vicinity of these reads revealed a putative PafBC binding site, with a perfectly conserved 5′ element (TTGTCGGTG) and a conserved but slightly divergent 3′ sequence spacing (AGCT) compared to that previously reported. Consistent with our RT-qPCR data, ciprofloxacin-induced transcription from this PafBC site was abolished by the pafC transposon insertion and restored in the complemented strain (Supplementary Figure S3). UV-induced transcription beginning at the LexA site was unaffected by the pafC::tn insertion (Supplementary Figure S3). Taken together, these data strongly support that imuA'B is dual regulated by LexA and PafBC in a clastogen-specific manner (27).
Dual regulation of the RecA promoter is clastogen specific
The data presented earlier indicate that PafBC and SOS have both shared and independent roles in the DDR, which may reflect overlapping functionality of their regulons or dual regulation of specific gene products, as is the case for imuA' /B. recA is one example of a gene directly regulated by both PafBC and SOS as it contains binding sites for LexA and PafBC in its promoter (17) (Figure 1C). To further examine whether SOS and PafBC respond to distinct signals, we used RecA expression to measure the temporal output of both pathways. RecA protein expression after UV treatment was strongly induced by 1 h, and although the  pafBC strain has lower baseline RecA expression without DNA damage, RecA levels reach the same level by 3 h (Figure 6A). A similar pattern was observed in SOS-deficient cells, indicating that both pathways control UV-induced RecA expression. In contrast, ciprofloxacin treatment strongly induced RecA in WT cells, but this induction was abolished in cells lacking PafBC, and preserved in cells lacking SOS (Figure 6B). In the case of MMC, all three tested strains had similar expression levels of RecA both with and without DNA damage (Figure 6C). This ciprofloxacin-specific loss of RecA induction is rescued by complementing
pafBC strain has lower baseline RecA expression without DNA damage, RecA levels reach the same level by 3 h (Figure 6A). A similar pattern was observed in SOS-deficient cells, indicating that both pathways control UV-induced RecA expression. In contrast, ciprofloxacin treatment strongly induced RecA in WT cells, but this induction was abolished in cells lacking PafBC, and preserved in cells lacking SOS (Figure 6B). In the case of MMC, all three tested strains had similar expression levels of RecA both with and without DNA damage (Figure 6C). This ciprofloxacin-specific loss of RecA induction is rescued by complementing  pafBC with either M. smegmatis or M. tuberculosis pafBC (Figure 6D). These findings indicate that the overlapping functions of these pathway are in part governed by the type of DNA damage. Most remarkable is the finding that quinolones appear to be specific inducers of the PafBC arm of the DDR. To confirm this finding in M. tuberculosis, we treated the H37Rv pafC transposon mutant with ciprofloxacin and UV and measured RecA protein. Although UV-induced RecA expression was unaffected by loss of pafC, quinolone-induced RecA expression was completely dependent on pafC, consistent with the results in M. smegmatis (Figure 6E). These results indicate that ciprofloxacin is a specific inducer of the PafBC pathway.
pafBC with either M. smegmatis or M. tuberculosis pafBC (Figure 6D). These findings indicate that the overlapping functions of these pathway are in part governed by the type of DNA damage. Most remarkable is the finding that quinolones appear to be specific inducers of the PafBC arm of the DDR. To confirm this finding in M. tuberculosis, we treated the H37Rv pafC transposon mutant with ciprofloxacin and UV and measured RecA protein. Although UV-induced RecA expression was unaffected by loss of pafC, quinolone-induced RecA expression was completely dependent on pafC, consistent with the results in M. smegmatis (Figure 6E). These results indicate that ciprofloxacin is a specific inducer of the PafBC pathway.
Figure 6.

Ciprofloxacin is a selective inducer of the PafBC pathway. α-RecA western blot of mid-log-phase expression of RecA (37 kDa) in M. smegmatis WT, ΔpafBC and lexA-S167A after exposure to (A) UV (20 mJ/cm2 with a recovery period of 1 or 3 h after exposure), (B) ciprofloxacin (1.25 μg/ml for 1 or 3 h) or (C) MMC (80 ng/ml for 2 or 5 h). RpoB is shown as a loading control. Quantification of RecA levels normalized to RpoB levels from three biological replicates for each of the DNA damaging agents is shown in the right panels of (A)–(C). All RpoB normalized RecA levels are displayed as a percentage of WT untreated set at 100%. Significance is calculated as *P < 0.05 using two-way ANOVA compared to mc2155 at a comparable time point/condition. Error bars are SEM. (D) α-RecA immunoblot of mid-log-phase expression of RecA (37 kDa) in M. smegmatis ΔpafBC complemented with M. tuberculosis pafBC, M. smegmatis pafBC or empty vector (EV) after exposure to UV (20 mJ/cm2) or ciprofloxacin (1.25 μg/ml) after 3 h. (E) α-RecA western blot of mid-log-phase expression of RecA (37 kDa) in M. tuberculosis strains of WT H37Rv, transposon insertion mutant of pafC (pafC TN) and transposon insertion mutant of pafC complemented with pafC (pafC comp) after exposure to UV (20 mJ/cm2 with a recovery period of 24 h after exposure) or ciprofloxacin (1.25 μg/ml for 24 h). RpoB is shown as a loading control.
Damage survival and mutagenesis of SOS and PafBC are not due to impaired RecA expression
Although the above-mentioned data clearly delineate overlapping and clastogen-specific roles for SOS and PafBC in RecA expression, and RecA has multiple roles in DNA repair and mutagenesis, the contribution of impaired RecA expression to the functional defects observed in cells lacking SOS, pafBC or both is unknown. To examine this question, we placed RecA expression under the control of an ATc inducible promoter (irecA) and complemented the WT,  recA,
recA,  pafBC, lexA-S167A and
pafBC, lexA-S167A and  pafBC/lexA-S167A strains. Addition of ATc induced RecA protein accumulation at a higher level than WT cells in the absence of DNA damage and enhanced UV-induced RecA accumulation (Supplementary Figure S4A). UV treatment of the ΔrecA strain complemented with irecA revealed no inducibility of RecA, but constant RecA levels with ATc (Supplementary Figure S4A). irecA restored RecA expression in the SOS- and PafBC-deficient strains (Supplementary Figure S4B) including the doubly deficient strain, which has nearly undetectable RecA levels.
pafBC/lexA-S167A strains. Addition of ATc induced RecA protein accumulation at a higher level than WT cells in the absence of DNA damage and enhanced UV-induced RecA accumulation (Supplementary Figure S4A). UV treatment of the ΔrecA strain complemented with irecA revealed no inducibility of RecA, but constant RecA levels with ATc (Supplementary Figure S4A). irecA restored RecA expression in the SOS- and PafBC-deficient strains (Supplementary Figure S4B) including the doubly deficient strain, which has nearly undetectable RecA levels.
To test whether enforced RecA expression could reverse the DNA damage sensitivity phenotypes observed with SOS- and PafBC-deficient cells, we measured killing with UV with and without RecA expression. Although irecA expression did not change the survival profile of the WT cells, it did fully rescue the severe survival defect of the  recA strain treated with UV, indicating that irecA encodes a functional RecA protein that is expressed at levels that can rescue complete RecA deficiency (Supplementary Figure S4C). However, restoration of RecA expression had no effect on the survival of SOS-, PafBC- or doubly deficient cells (Supplementary Figure S4C). Similarly, irecA did not change the DNA damage-induced transcription of adnA in WT cells and was unable to rescue the loss of adnA transcription in either the
recA strain treated with UV, indicating that irecA encodes a functional RecA protein that is expressed at levels that can rescue complete RecA deficiency (Supplementary Figure S4C). However, restoration of RecA expression had no effect on the survival of SOS-, PafBC- or doubly deficient cells (Supplementary Figure S4C). Similarly, irecA did not change the DNA damage-induced transcription of adnA in WT cells and was unable to rescue the loss of adnA transcription in either the  pafBC or
pafBC or  pafBC/lexA S167A strains (Supplementary Figure S4D). However, irecA fully rescued dnaE2 expression in ΔrecA, and partially rescued the transcription of dnaE2 in the lexA-S167A and
pafBC/lexA S167A strains (Supplementary Figure S4D). However, irecA fully rescued dnaE2 expression in ΔrecA, and partially rescued the transcription of dnaE2 in the lexA-S167A and  pafBC/lexA-S167A strains (Supplementary Figure S4E). These results indicate that, despite the prominent coregulation of RecA expression by PafBC and SOS, the defective RecA expression conferred by loss of these pathways does not explain their damage sensitivity.
pafBC/lexA-S167A strains (Supplementary Figure S4E). These results indicate that, despite the prominent coregulation of RecA expression by PafBC and SOS, the defective RecA expression conferred by loss of these pathways does not explain their damage sensitivity.
Replisome perturbation induces the PafBC pathway
An essential question about the PafBC pathway is its mechanism of activation. The PafBC proteins are not themselves damage inducible (10), suggesting that a signal generated by DNA damage results in PafBC activation. Whether this occurs at the level of induced DNA binding, dimerization or some other mechanism is unknown (28). The above-mentioned data indicate that quinolones may be a specific activating signal for the PafBC pathway (Figure 6). Quinolones act by inhibiting DNA gyrase and thereby affect DNA replication and transcription, in addition to inducing protein-linked DNA double-strand breaks. To understand the induction of the PafBC pathway by quinolones, we tested other stresses that may impact DNA replication or transcription. Although oxidative stress induction by CHP (Figure 7A) and inhibiting DNA replication using the antimicrobial SKI356313 (29) (Figure 7B) both induced RecA expression in WT cells, this induction was not dependent on pafBC. In contrast, the topoisomerase inhibitor etoposide (30) induced RecA in WT cells, and this induction was substantially dependent on PafBC, but not SOS (Figure 7C). To more precisely perturb DNA replication, we expressed the alternative lesion bypass polymerase DinB1 from an ATc inducible promoter. Mycobacterial DinB1 interacts with the DNA replication machinery beta clamp (31) and therefore may stall the replication fork when overexpressed. Consistent with this prediction, DinB1 expression strongly induced RecA (Figure 7D), but this induction was abolished when DinB1Δ356–360 was expressed, which carries a mutation in the beta-clamp interacting motif (Figure 7E). RecA induction by DinB1 overexpression was lost in ΔpafBC but preserved in the SOS-deficient strain (Figure 7D), consistent with the hypothesis that perturbation of the replication fork is an inducer of the DDR pathway. Polymerase inactive DinB1 (dinB1 D113A) strongly induced RecA, but this induction was no longer pafBC dependent (Figure 7F), suggesting that this inactive polymerase activated an alternative form of DNA damage, possibly by inhibiting access of repair factors to the fork.
Figure 7.

Inhibition of DNA replisome function selectively induces the PafBC pathway. α-RecA immunoblot of mid-log-phase expression of RecA (37kb) in mc2155, ΔpafBC and lexA-S167A strains after exposure to (A) CHP (50 μM for 1 or 3 h), (B) SKI356313 (1.9 μM for 1 or 3 h), (C) etoposide (24 μM for 3 h), (D) ATc-induced overexpression of DinB1 for 4 or 6 h, (E) ATc-induced overexpression of DinB1 missing the beta-clamp interaction domain (DinB1Δ356–360) for 4 or 6 h, or (F) ATc-induced overexpression of polymerase-dead DinB1 (dinB1-D113A) for 4 or 6 h.
DISCUSSION
In this study, we have taken a comprehensive approach to examine the transcriptional and functional contributions of both the PafBC and SOS pathways in the mycobacterial DDR. Our findings reveal the requirement for both pathways to mount a full and effective response to DNA damage inflicted by various DNA damaging conditions. Our analysis of the transcriptional response of mycobacteria to distinct types of DNA damage reveals that, although the PafBC pathway controls the larger transcriptional response irrespective of the type of DNA damage, both arms of the DDR are required for survival after damage and for mutagenesis. In addition, we reveal important differences between the pathways that in part depend on the type of DNA damaging agent. Most prominently, our data indicate that quinolone antibiotics, which inhibit DNA gyrase, or replication fork perturbation, specifically induce the PafBC arm of the DDR.
Despite encoding a larger transcriptional regulon, our findings reveal that both the SOS and PafBC pathways play major roles in surviving the effects of DNA damage and in mutagenesis that results from the repair of this damage. In mycobacteria, the mutasome consists of a complex of the DnaE2 polymerase (15), along with ImuB–ImuA (26), all of which are required for the generation of rifampin resistance mutations. ImuB interacts directly with the beta clamp of the replication apparatus and DnaE2 (26). Our results indicate that a partial loss of imuA'/B expression in the absence of the PafBC pathway is accompanied by impaired mutagenesis in cells lacking pafBC. Although imuA' /B expression is partially SOS dependent, M. smegmatis or M. tuberculosis lacking pafBC or pafC, respectively, display impaired dnaE2 or imuB expression after DNA damage. This pafC dependence is particularly dramatic after gyrase inhibition, during which dnaE2 or imuB expression is completely pafBC dependent.
This quinolone-specific, pafBC-dependent response to DNA damage has potential clinical relevance. Although the in vitro activity of quinolones against M. tuberculosis has long been recognized, and they have been part of second-line therapy for multidrug-resistant tuberculosis, recent clinical data indicate a role for the quinolone moxifloxacin in 4-month rifapentine regimens to treat drug-sensitive tuberculosis (32). Coupled with our finding that there is a role for PafBC in supporting mutagenesis, our findings provide a molecular basis for the possibility that widespread use of quinolones for tuberculosis treatment may promote mutations that could enhance resistance to other antibiotics. In E. coli and other bacteria, studies have shown a requirement for the SOS pathway in inducing mutagenesis after quinolone treatment (33–35).
Our data provide perspective on prior efforts to understand the division of labor between the classical SOS pathway and the PafBC pathway. Based on the validated role for RecA as the LexA coprotease, and before the identification of PafBC, groundbreaking work demonstrated that the mycobacterial DDR had ‘RecA-dependent’ and ‘RecA-independent’ arms (12,36,37). The RecA-dependent arm was thought to represent the SOS arm of the pathway. However, the data presented here reveal a more complex relationship between RecA and SOS. Although RecA null cells are clearly SOS null, they also are clearly more severely deficient for DNA repair and mutagenesis than cells specifically ablated for SOS. Only when SOS and PafBC are both ablated do the DNA damage phenotypes resemble loss of RecA.
The activation signal for the PafBC pathway is unknown. The PafBC heterodimer is constitutively expressed and therefore one model of its activation, in part based on structural studies (28), is that a ligand generated during DNA damage binds directly to PafBC and induces a conformational change that results in DNA binding. Alternative models are possible, including sequestration of the heterodimer in basal conditions and/or induced heterodimerization. Our study does not identify the specific inducing signal for the PafBC pathway, but our data do support a model in which this signal is generated by replication fork perturbation or arrest. Given the role of RecA in replication fork restart (19), our data may suggest that in certain circumstances the activating signal for PafBC requires RecA and replication fork perturbation, a hypothesis that can be pursued by future studies.
NOTE ADDED IN PROOF
During the review process of this paper, Chengalroyen et al. posted a preprint (doi: https://doi.org/10.1101/2021.10.27.466036) reporting that nargenicin is an inhibitor of DNA replication via mycobacterial DnaE1. Transcriptomic profiling of nargenicin treated Mtb revealed that adnAB and lhr, PafBC dependent genes, are among the most strongly upregulated transcripts with nargenicin treatment.
DATA AVAILABILITY
RNA sequencing data have been deposited into the SRA as BioProject # PRJNA746693.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Heran Darwin for providing the H37Rv pafC Tn and complemented strains.
Contributor Information
Oyindamola O Adefisayo, Immunology and Microbial Pathogenesis Graduate Program, Weill Cornell Graduate School, 1300 York Avenue, New York, NY 10065, USA; Immunology Program, Sloan Kettering Institute, 1275 York Avenue, New York, NY 10025, USA.
Pierre Dupuy, Immunology Program, Sloan Kettering Institute, 1275 York Avenue, New York, NY 10025, USA.
Astha Nautiyal, Immunology Program, Sloan Kettering Institute, 1275 York Avenue, New York, NY 10025, USA.
James M Bean, Immunology Program, Sloan Kettering Institute, 1275 York Avenue, New York, NY 10025, USA.
Michael S Glickman, Immunology and Microbial Pathogenesis Graduate Program, Weill Cornell Graduate School, 1300 York Avenue, New York, NY 10065, USA; Immunology Program, Sloan Kettering Institute, 1275 York Avenue, New York, NY 10025, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [AI 064693, P30 CA008748]. Funding for open access charge: National Institutes of Health [AI 064693].
Conflict of interest statement. M.S.G. serves as a SAB member and holds equity in Vedanta Biosciences, is on the SAB of PRL-NYC, and is a consultant for Fimbrion Therapeutics.
REFERENCES
- 1. Nathan C., Shiloh M.U.. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl Acad. Sci. U.S.A. 2000; 97:8841–8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Warner D.F., Mizrahi V.. Tuberculosis chemotherapy: the influence of bacillary stress and damage response pathways on drug efficacy. Clin. Microbiol. Rev. 2006; 19:558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Darwin K.H., Nathan C.F.. Role for nucleotide excision repair in virulence of Mycobacterium tuberculosis. Infect. Immun. 2005; 73:4581–4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Houghton J., Townsend C., Williams A.R., Rodgers A., Rand L., Walker K.B., Bottger E.C., Springer B., Davis E.O.. Important role for Mycobacterium tuberculosis UvrD1 in pathogenesis and persistence apart from its function in nucleotide excision repair. J. Bacteriol. 2012; 194:2916–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Naz S., Dabral S., Nagarajan S.N., Arora D., Singh L.V., Kumar P., Singh Y., Kumar D., Varshney U., Nandicoori V.K.. Compromised base excision repair pathway in Mycobacterium tuberculosis imparts superior adaptability in the host. PLoS Pathog. 2021; 17:e1009452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dos Vultos T., Mestre O., Tonjum T., Gicquel B.. DNA repair in Mycobacterium tuberculosis revisited. FEMS Microbiol. Rev. 2009; 33:471–487. [DOI] [PubMed] [Google Scholar]
- 7. Yu X., Egelman E.H.. The LexA repressor binds within the deep helical groove of the activated RecA filament. J. Mol. Biol. 1993; 231:29–40. [DOI] [PubMed] [Google Scholar]
- 8. Wipperman M.F., Heaton B.E., Nautiyal A., Adefisayo O., Evans H., Gupta R., van Ditmarsch D., Soni R., Hendrickson R., Johnson J.et al.. Mycobacterial mutagenesis and drug resistance are controlled by phosphorylation- and cardiolipin-mediated inhibition of the RecA coprotease. Mol. Cell. 2018; 72:152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nautiyal A., Patil K.N., Muniyappa K.. Suramin is a potent and selective inhibitor of Mycobacterium tuberculosis RecA protein and the SOS response: RecA as a potential target for antibacterial drug discovery. J. Antimicrob. Chemother. 2014; 69:1834–1843. [DOI] [PubMed] [Google Scholar]
- 10. Müller A.U., Imkamp F., Weber-Ban E.. The mycobacterial LexA/RecA-independent DNA damage response is controlled by PafBC and the Pup-proteasome system. Cell Rep. 2018; 23:3551–3564. [DOI] [PubMed] [Google Scholar]
- 11. Festa R.A., Pearce M.J., Darwin K.H.. Characterization of the proteasome accessory factor (paf) operon in Mycobacterium tuberculosis. J. Bacteriol. 2007; 189:3044–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rand L., Hinds J., Springer B., Sander P., Buxton R.S., Davis E.O.. The majority of inducible DNA repair genes in Mycobacterium tuberculosis are induced independently of RecA. Mol. Microbiol. 2003; 50:1031–1042. [DOI] [PubMed] [Google Scholar]
- 13. Brzostek A., Płociński P., Minias A., Ciszewska A., Gąsior F., Pawełczyk J., Dziadek B., Słomka M., Dziadek J.. Dissecting the RecA-(in)dependent response to mitomycin C in Mycobacterium tuberculosis using transcriptional profiling and proteomics analyses. Cells. 2021; 10:1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Podlesek Z., Žgur Bertok D.. The DNA damage inducible SOS response is a key player in the generation of bacterial persister cells and population wide tolerance. Front. Microbiol. 2020; 11:1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boshoff H.I., Reed M.B., Barry C.E., Mizrahi V.. DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell. 2003; 113:183–193. [DOI] [PubMed] [Google Scholar]
- 16. Cox M.M. Regulation of bacterial RecA protein function. Crit. Rev. Biochem. Mol. Biol. 2007; 42:41–63. [DOI] [PubMed] [Google Scholar]
- 17. Gopaul K.K., Brooks P.C., Prost J.F., Davis E.O.. Characterization of the two Mycobacterium tuberculosis recA promoters. J. Bacteriol. 2003; 185:6005–6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gupta R., Barkan D., Redelman-Sidi G., Shuman S., Glickman M.S.. Mycobacteria exploit three genetically distinct DNA double-strand break repair pathways. Mol. Microbiol. 2011; 79:316–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Michel B., Sinha A.K., Leach D.R.F.. Replication fork breakage and restart in Escherichia coli. Microbiol. Mol. Biol. Rev. 2018; 82:e00013-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barkan D., Stallings C.L., Glickman M.S.. An improved counterselectable marker system for mycobacterial recombination using galK and 2-deoxy-galactose. Gene. 2011; 470:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gupta R., Unciuleac M.C., Shuman S., Glickman M.S.. Homologous recombination mediated by the mycobacterial AdnAB helicase without end resection by the AdnAB nucleases. Nucleic Acids Res. 2017; 45:762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Culviner P.H., Guegler C.K., Laub M.T.. A simple, cost-effective, and robust method for rRNA depletion in RNA-sequencing studies. mBio. 2020; 11:e00010-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hubin E.A., Fay A., Xu C., Bean J.M., Saecker R.M., Glickman M.S., Darst S.A., Campbell E.A.. Structure and function of the mycobacterial transcription initiation complex with the essential regulator RbpA. eLife. 2017; 6:e22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bell J.C., Kowalczykowski S.C.. RecA: regulation and mechanism of a molecular search engine (Trends in Biochemical Sciences, June 2016, Vol. 41, No. 6, 491–507). Trends Biochem. Sci. 2016; 41:646. [DOI] [PubMed] [Google Scholar]
- 25. Slilaty S.N., Little J.W.. Lysine-156 and serine-119 are required for LexA repressor cleavage: a possible mechanism. Proc. Natl Acad. Sci. U.S.A. 1987; 84:3987–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Warner D.F., Ndwandwe D.E., Abrahams G.L., Kana B.D., Machowski E.E., Venclovas C., Mizrahi V.. Essential roles for imuA′- and imuB-encoded accessory factors in DnaE2-dependent mutagenesis in Mycobacterium tuberculosis. Proc. Natl Acad. Sci. U.S.A. 2010; 107:13093–13098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gamulin V., Cetkovic H., Ahel I.. Identification of a promoter motif regulating the major DNA damage response mechanism of Mycobacterium tuberculosis. FEMS Microbiol. Lett. 2004; 238:57–63. [DOI] [PubMed] [Google Scholar]
- 28. Müller A.U., Leibundgut M., Ban N., Weber-Ban E.. Structure and functional implications of WYL domain-containing bacterial DNA damage response regulator PafBC. Nat. Commun. 2019; 10:4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harris K.K., Fay A., Yan H.G., Kunwar P., Socci N.D., Pottabathini N., Juventhala R.R., Djaballah H., Glickman M.S.. Novel imidazoline antimicrobial scaffold that inhibits DNA replication with activity against mycobacteria and drug resistant Gram-positive cocci. ACS Chem. Biol. 2014; 9:2572–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chatterji M., Unniraman S., Mahadevan S., Nagaraja V.. Effect of different classes of inhibitors on DNA gyrase from Mycobacterium smegmatis. J. Antimicrob. Chemother. 2001; 48:479–485. [DOI] [PubMed] [Google Scholar]
- 31. Kana B.D., Abrahams G.L., Sung N., Warner D.F., Gordhan B.G., Machowski E.E., Tsenova L., Sacchettini J.C., Stoker N.G., Kaplan G.et al.. Role of the DinB homologs Rv1537 and Rv3056 in Mycobacterium tuberculosis. J. Bacteriol. 2010; 192:2220–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dorman S.E., Nahid P., Kurbatova E.V., Phillips P.P.J., Bryant K., Dooley K.E., Engle M., Goldberg S.V., Phan H.T.T., Hakim J.et al.. Four-month rifapentine regimens with or without moxifloxacin for tuberculosis. N. Engl. J. Med. 2021; 384:1705–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thi T.D., López E., Rodríguez-Rojas A., Rodríguez-Beltrán J., Couce A., Guelfo J.R., Castañeda-García A., Blázquez J.. Effect of recA inactivation on mutagenesis of Escherichia coli exposed to sublethal concentrations of antimicrobials. J. Antimicrob. Chemother. 2011; 66:531–538. [DOI] [PubMed] [Google Scholar]
- 34. Castro R.A.D., Borrell S., Gagneux S.. The within-host evolution of antimicrobial resistance in Mycobacterium tuberculosis. FEMS Microbiol. Rev. 2021; 45:fuaa071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gillespie S.H., Basu S., Dickens A.L., O'Sullivan D.M., McHugh T.D.. Effect of subinhibitory concentrations of ciprofloxacin on Mycobacterium fortuitum mutation rates. J. Antimicrob. Chemother. 2005; 56:344–348. [DOI] [PubMed] [Google Scholar]
- 36. Dawson L.F., Dillury J., Davis E.O.. RecA-independent DNA damage induction of Mycobacterium tuberculosis ruvC despite an appropriately located SOS box. J. Bacteriol. 2010; 192:599–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davis E.O., Springer B., Gopaul K.K., Papavinasasundaram K.G., Sander P., Bottger E.C.. DNA damage induction of recA in Mycobacterium tuberculosis independently of RecA and LexA. Mol. Microbiol. 2002; 46:791–800. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data have been deposited into the SRA as BioProject # PRJNA746693.