


Abstract
Uncoordinated clashes between replication forks and transcription cause replication stress and genome instability, which are hallmarks of cancer and neurodegeneration. Here, we investigate the outcomes of head-on replication-transcription collisions, using as a model system budding yeast mutants for the helicase Sen1, the ortholog of human Senataxin. We found that RNA Polymerase II accumulates together with RNA:DNA hybrids at sites of head-on collisions. The replication fork and RNA Polymerase II are both arrested during the clash, leading to DNA damage and, in the long run, the inhibition of gene expression. The inactivation of RNA Polymerase II elongation factors, such as the HMG-like protein Spt2 and the DISF and PAF complexes, but not alterations in chromatin structure, allows replication fork progression through transcribed regions. Attenuation of RNA Polymerase II elongation rescues RNA:DNA hybrid accumulation and DNA damage sensitivity caused by the absence of Sen1, but not of RNase H proteins, suggesting that such enzymes counteract toxic RNA:DNA hybrids at different stages of the cell cycle with Sen1 mainly acting in replication. We suggest that the main obstacle to replication fork progression is the elongating RNA Polymerase II engaged in an R-loop, rather than RNA:DNA hybrids per se or hybrid-associated chromatin modifications.
Graphical Abstract
Graphical Abstract.
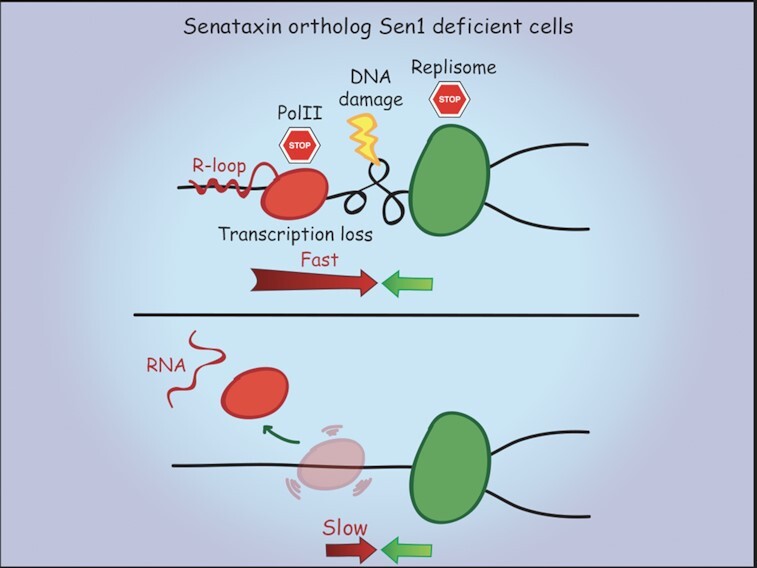
Attenuation of RNAPII elongation prevents R-loops, DNA damage and the arrest of replication and transcription at sites of head-on collisions in sen1 mutants.
INTRODUCTION
Replication stress is a source of endogenous DNA damage in both cancers and neurodegenerative disorders (1,2). Two features of replication stress include high frequency of origin firing events and enhanced fork instability at natural replication barriers (1). DNA transcription is a major cause of replication stress, especially in the case of head-on replication-transcription collisions (3). The severity of head-on collisions is linked to the accumulation of R-loops, which are structures containing an RNA:DNA hybrid and a displaced ssDNA (4–8). R-loops potentially hamper DNA replication, leading to fork arrest and collapse, thus being highly recombinogenic and mutagenic by nature (3,9). Several factors, including the tumor suppressors BRCA1 and BRCA2, limit R-loop accumulation and the consequent DNA damage (10–14). The helicase Senataxin, a physical partner of BRCA1, and the nucleases RNase H are among the best-characterized enzymes able to resolve RNA:DNA hybrids (15,16). Senataxin dysfunctions lead to multiple neurological disorders, including Ataxia (17), Amyotrophic Lateral Sclerosis (18) and Spinal Muscular Atrophy (19,20), placing this helicase as a paradigm to study the dangerous implications of R-loops in both cancer and neurodegeneration. Deficiency in Senataxin or its yeast orthologue Sen1 is associated with a number of defects caused by the alteration of R-loop homeostasis, including transcription termination failure, unscheduled gene silencing, high DNA damage sensitivity, unfaithful recombination repair events, hyper-resection at double-strand break and fork arrest at sites of replication-transcription collisions (21–30). Despite inducing DNA damage, R-loops are physiologically generated co-transcriptionally within the genome, independently of replication orientation (6,31). In fact, R-loops control gene expression by modifying the chromatin structure at sites of RNA Polymerase II (RNAPII) pausing, as promoter and termination regions (32). The role of Senataxin/Sen1 in the resolution of physiological R-loops at transcription termination sites is well established (16), while less clear is its role in the prevention of RNA:DNA hybrid-driven DNA damage. Moreover, little is known about the consequences of the collision event on the transcription side.
In Sen1-depleted cells, head-on highly transcribed RNAPII genes accumulate R-loops (24,33) and form an impenetrable barrier to replication fork (34). The activation of dormant origins near the collision sites is a mechanism to rescue stalled replication forks, while the protection of replisome prevents the nuclease-mediated processing of R-loops at the fork (34). It is currently unclear why transcription poses such a strong impediment to fork progression, considering that pervasive transcription is a general feature of both prokaryotic and eukaryotic genomes (35,36). Kuzminov proposed that is the RNAPII anchored to the DNA by an R-loop rather than the R-loop itself that arrests fork progression (37). An RNAPII unable to initiate or sustain productive elongation, because paused, arrested or backtracked, is indeed a source of R-loop-dependent DNA breaks and/or replication-transcription conflicts (12–14,38–40). R-loops have been also proposed to interfere with replication indirectly, either through the stabilization of DNA secondary structures (41,42) or alterations in the chromatin. For example, R-loops induce the formation of specific heterochromatic marks, such as phosphorylation or methylation of H3, on residues S10 (43) or K9 (44,45) respectively, that could obstruct fork progression. Increased levels of H3 acetylation, including on residue K56, also lead to R-loop-dependent DNA breaks (46,47). Finally, also the loss of heterochromatin enhances replication-transcription collisions (48) through unscheduled R-loop formation (49,50).
Here, we studied the impact of Sen1 inactivation on head-on vs co-directional replication-transcription collisions by placing the same highly expressed gene in opposite orientations to fork progression in the yeast genome. We found that head-on rather than co-directional collisions lead to R-loop accumulation and DNA damage, hindering not only replication but also transcription. To better characterize the physical nature of the transcription barrier, we took advantage of a genetic approach to identify suppressors of sen1 mutants. Also using specific point mutants in the RNAPII catalytic core, we showed that elongation attenuation, but not alterations in chromatin structure, allowed replication across transcription in sen1 mutants, preventing R-loop accumulation, the retention of inactive RNAPII on chromatin and DNA damage. Together, our data suggest that elongating RNA Polymerase II is a major contributing factor to transcription-induced replication stress.
MATERIALS AND METHODS
Yeast strains and growth conditions
The strains used in this study are isogenic derivatives of W303-1A RAD5 and listed in the Supplementary Table S1. One-step PCR-targeting method was used to delete or mutate yeast genes (51). The rpb1-N488D mutant strain, originally isolated in a different genetic background (52), was backcrossed three times to W303-1A RAD5 before being used in our experiments. The PDC1 gene, plus 830 and 320 bp upstream and downstream, respectively, was placed in either head-on (HO) or co-directional (CD) orientation to the ARS607-dependent replication on the chromosome VI using the Delitto Perfetto approach (53) in strains where the endogenous PDC1 gene on the chromosome XII was deleted. The conditional lethal strain GAL-URL-3HA-SEN1 (sen1URL) has been previously described (24). In the experiments conducted with the yeast strains carrying the GAL-URL-3HA-SEN1 system, cells were grown overnight in YPG (1% yeast extract, 2% bactopeptone, 2% galactose) and, during α-factor treatment, transferred to YPD (1% yeast extract, 2% bactopeptone, 2% glucose) to switch off Sen1 protein. Under these conditions, the Sen1 protein is depleted in 30 min (24). α-factor was used to a final concentration of 2 μg/ml and other inhibitors at concentrations indicated in the Figures.
Identification of genetic suppressors of sen1URL mutants
The genome-wide screen used to identify suppressors of cell lethality due to depletion of Sen1 protein was performed by crossing a query strain carrying the GAL-URL-3HA-SEN1 system with a nearly complete nonessential gene deletion library of Saccharomyces cerevisiae, according to the procedure described in (54). All the screening selection steps were carried out on YPG, to express the query construct, then sen1URL suppressors were identified for their capability to grow in YPD, as validated in independent screenings, and then confirmed in W303-1A RAD5 background.
Purification and 2D gel analysis of replication intermediates
Purification of DNA intermediates and 2D gel electrophoresis procedure were carried out as described in (55) using yeast strains synchronized in G1-phase of the cell cycle and released into 0.2 M hydroxyurea (HU). Briefly, yeast cells were exposed to in vivo psoralen cross-linking and replication intermediates were purified in the presence of CTAB. 10 μg of purified DNA were digested with different restriction enzymes, depending on the fragments to be analyzed (further information available in figure legends). DNA fragments were separated in the first dimension at 50 V for 20 h and in the second dimension at 180 V for 8 h. DNA in the gel was denatured by NaOH treatment and transferred overnight onto nylon membranes in 10× SSC buffer. Membranes were hybridized with labeled DNA probes to detect replication intermediates at the regions of interest. 2D gel images were obtained using a GE Typhoon Trio™.
DNA:RNA immuno-precipitation analysis and quantitative real-time PCR analysis
RNA:DNA hybrids were extracted and analyzed by immunoprecipitation (DRIP) using S9.6 antibodies followed by quantitative real-time PCR (qPCR) analysis, as previously described (24). Briefly, 9 μg of purified DNA was sheared by sonication to obtain 500 bp fragments, treated or not with RNase H (New England Biolabs), and incubated overnight with 7.5 μg of S9.6 monoclonal antibody bound to magnetic protein G beads (Invitrogen). For each sample, 1 μl was taken as INPUT (1%). Input and immunoprecipitated samples were treated with protease K and RNase A before purification with QIAquick PCR Purification Kit (Qiagen). Treated samples were subjected to qPCR using quantifast SYBR Green PCR Master Mix (Qiagen). Samples were run in Roche Light Cycler 480 Real-Time PCR System. RNA:DNA hybrid enrichment was determined as follows: 100 × 2(CT adjusted INPUT – CT IP S9.6). qPCR reactions were performed in triplicate.
PDC1 or SEN1 mRNA levels were measured by qPCR, as follows: the RNA was extracted using Qiagen RNeasy Kit and 1 μg of RNA was retro-transcribed with INVILO kit (Invitrogen). qPCR was done in 20 μl reaction with gene specific primers using 1 μl of cDNA diluted 1:20 using the SYBR Green (quantifast SYBR Green PCR Master Mix, Qiagen) and run in Roche Light Cycler 480 Real-Time PCR System. qPCR was also performed for ACT1 gene from each cDNA sample. PDC1 or SEN1 expression levels were calculated as follows: 2−(CT test – CT control), where test refers to the PDC1 or SEN1 gene and control refers to ACT1. All samples were run in triplicate for each independent experiment.
Chromatin immuno-precipitation analysis
Standard Chromatin Immuno-Precipitation (ChIP) was carried out as previously described in (24). Purified ‘ChIPed’ DNA was sheared to 500 bp by sonication and incubated overnight with antibodies bound to magnetic protein G beads (Invitrogen). 4 μg of anti-γ-H2AX (Cell signaling), 10 μg anti-H3K56ac (Millipore), 5 μg of anti-Rpb3 (Biolegend), 4 μg of anti-H3K4me2 (Invitrogen), 5 μg of anti-CTD-Ser2 (Abcam) or 5 μg of anti-CTD-Ser5 (Abcam) were used for ChIP. For each sample 5 μl was taken as INPUT. Input and immunoprecipitated samples were treated with protease K and RNase A before being purified with the QIAquick PCR Purification Kit (Qiagen) and then subjected to qPCR using the SYBR Green (quantifast SYBR Green PCR Master Mix, Qiagen). Samples were run in Roche Light Cycler 480 Real-Time PCR System. DNA enrichment was determined by quantitative PCR (qPCR) as follows: 100 × 2(CT adjusted INPUT – CT IP). qPCR reactions were performed in triplicate.
Chromosome spreads analysis
Yeast chromosome spreads technique was performed as described in (56) with minor modifications. Briefly, yeast strains were grown in log phase in YPG and then shifted for four hours in YPD to deplete Sen1. Cells were converted to spheroplasts and spread on glass slides in presence of SDS 20%. Slides were incubated with 0.25 μg/ml of S9.6 antibody diluted in 80 μl of blocking buffer, washed for 15 min in PBS 1X and then incubated with the secondary Alexa Fluor 488 green antibody (ThermoFisher) diluted 1:1000 in blocking buffer. In vitro RNase H treatment was achieved by incubating the slides, prior to antibody incubations, for 30 min with 2 U of enzyme (NEB) diluted in 80 μl of blocking buffer. Indirect immunofluorescence (IF) was performed using a Zeiss AxioImager M2 microscope with a 40×/NA 1.3 objective.
Transcription run-on assay by 5-bromo-UTP incorporation
Run-on experiments were performed as described in (57) with minor modifications. Briefly, 100 ml of cells in exponential phase of growth were synchronized in G1-phase of the cell cycle and released into 0.2 M HU. Cells were permeabilized using 0.6% of Sarkosyl and then transcription run-on reaction was performed in the presence of 0.75 mM of ATP, CTP, GTP and 5-bromo-UTP (BrUTP) (Sigma). Purified total RNA was extracted using Qiagen RNeasy Kit (Qiagen), 1 μg of total RNA was retro-transcribed using poly-dT primers with INVILO kit (Invitrogen) and used as control. Purified total RNA was incubated 2 h with 4 μg of anti-BrdU antibody (MBL) bound to magnetic protein G beads (Invitrogen), 1 μg of immunoprecipitated RNA was retro-transcribed using poly-dT primers with INVILO kit (Invitrogen) and analyzed by qPCR. qPCR was done in 20 μl reaction with gene specific primers using 1 μl of cDNA diluted 1:20 using the SYBR Green (quantifast SYBR Green PCR Master Mix, Qiagen) and run in Roche Light Cycler 480 Real-Time PCR System. qPCR was also performed for ACT1 gene from each cDNA sample. Nascent RNA levels were calculated as described in (58) using the ΔCq methods. All samples were run in triplicate for each independent experiment.
RESULTS
Head-on, but not co-directional, replication-transcription collisions lead to fork arrest, R-loop accumulation, DNA damage and transcription inhibition in cells lacking Sen1
We have previously shown that the inactivation of Sen1 impairs fork progression at certain R-loop-prone highly transcribed genes encountered in head-on orientation, but not co-directionally (24). To provide direct evidence that Sen1 has a specific role in preventing head-on collisions, we re-positioned the highly expressed PDC1 metabolic gene in the two opposite orientations with respect to fork progression, via a Delitto Perfetto strategy, in the proximity of the early origin of replication ARS607 on Chromosome VI (Figure 1A). The endogenous copy of the PDC1 gene was deleted in these strains. We found, by qPCR, that the expression of the manipulated copy was similar in the two opposite orientations and comparable with the expression level of PDC1 in its natural location on Chromosome XII (Supplementary Figure S1A). We then analyzed fork progression and RNA:DNA hybrid accumulation at the engineered replication-transcription conflicts, in both wild-type (WT) and Sen1-depleted cells, using 2D gel and DRIP-qPCR approaches, respectively. To deplete Sen1, which is essential for the cell growth, we took advantage of the previously described conditional lethal strain GAL-URL-3HA-SEN1 (hereafter indicated as sen1URL), in which the protein is constitutively expressed in a galactose-containing medium (YPG) and rapidly destabilized via the N-end rule pathway in glucose-containing medium (YPD) (24). Yeast strains were grown in YPG, synchronized in G1-phase of the cell cycle by α-factor treatment in YPD to deplete Sen1 and then released into HU-containing media to enrich replication intermediates for the 2D gel analysis. PDC1 transcription had no significant effects on hybrids accumulation in either orientation in WT strains, while it slowed down the progression of forks clashing in the head-on setting, as indicated by the presence of Y-shaped arcs at later time points (Figure 1A and B; Supplementary Figure S1B). In Sen1-depleted cells, forks encountering the PDC1 gene in head-on orientation were greatly impaired, leading to a persistent accumulation of replication intermediates that migrate in a big spot on the Y-shaped arc (Figure 1A). Conversely, forks moving across the PDC1 gene co-directionally were only slightly slowed down, as indicated by a transient accumulation of replication intermediates along the Y-shaped arc (Figure 1A). The strong fork pausing in Sen1-depleted cells bearing the head-on oriented PDC1 gene was also accompanied by high levels of hybrids at PDC1, while they just slightly increased when the gene was oriented co-directionally to the fork movement (Figure 1B and Supplementary Figure S1B).
Figure 1.

Head-on and co-directional replication-transcription conflicts in WT and sen1 strains. (A) 2D gel analysis of replication intermediates at the PDC1 gene undergoing head-on (HO) (Left Panel) or co-directional (CD) (Right Panel) collisions with ARS607-dependent replication upon digestions with PstI (P)/BamHI (B) or NdeI (N), respectively. (B) RNA:DNA hybrid levels at PDC1 gene analyzed by DRIP-qPCR. Data represent mean ± SEM from three independent experiments. ns, not significant; **P < 0.01 (two-way ANOVA). (C) ChIP-qPCR analysis at PDC1 locus of H2A-S139 phosphorylated (γ-H2AX). Data represent mean ± SEM from three independent experiments. ****P < 0.0001 (two-way ANOVA). (D) Serial dilutions of the indicated yeast strains spotted on YPD plates with or without NaN3 grown at the semi-permissive temperature of 30°C for sen1-1.
We next wondered whether high levels of hybrids due to head-on replication-transcription collisions upon Sen1 inactivation result in DNA damage and affect PDC1 transcription. As a readout of DNA damage accumulation, we analysed the levels of H2A-S129 phosphorylation (γ-H2AX) (59) by ChIP-qPCR at PDC1 in both orientations in WT and Sen1-depleted cells treated with HU. As shown in Figure 1C, only sen1URL cells where PDC1 underwent head-on replication-transcription collisions showed high levels of γ-H2AX, compared to WT and co-directional orientation.
In budding yeast, a facultative anaerobe organism, the PDC1 gene encodes for the main isoform of pyruvate decarboxylase, a key enzyme in alcoholic fermentation and whose expression is essential under respiration-inhibitory conditions (60). We combined the strains containing the engineered head-on or co-directional replication-transcription conflict at the PDC1 gene with the thermosensitive sen1-1 allele that carries the amino-acid substitution G1747D in the helicase domain (61). We plated these mutant strains, along with their corresponding WT versions, at the semi-permissive temperature of 30°C for sen1-1 on medium containing glucose, a fermentable sugar, and Sodium azide (NaN3), an inhibitor of respiration. At 30°C, sen1-1 mutants could proliferate, while experiencing a chronic partial inactivation of Sen1 (Figure 1D). However, specifically sen1-1 strains carrying the head-on oriented PDC1 gene were unable to grow on plates containing NaN3 inhibitor (Figure 1D), demonstrating that just here, in the long run, the PDC1 transcription was impaired.
Taken together, these data indicate that, in the absence of Sen1, replication forks are significantly impaired while encountering highly expressed genes in head-on orientation, but not co-directionally, and that the fork arrest is accompanied by abnormal RNA:DNA hybrid accumulation, DNA damage and transcription hindrance.
A genomic screening to identify suppressors of sen1 mutants
Despite head-on highly transcribed genes are a strong barrier to advancing fork without Sen1, it is currently unclear whether the impediment is due to the R-loop per se, the transcribing RNA polymerase engaged in an R-loop or specific chromatin modifications. We approached this question using a high-throughput screening to identify extragenic suppressors of the Sen1’s essential role in cell growth. We crossed a query strain bearing the conditional lethal strain sen1URL with a deletion mutant library of the non-essential S. cerevisiae genes (54) with the help of an automatized robot station and we then selected double mutant strains capable to grow in YPD, that is, in the absence of Sen1. Out of 14 sen1URL suppressors isolated in our screening seven genes were confirmed in W303 background and thus considered real hits (Figure 2A). sen1URL suppressor genes include NAM7/NMD2/UPF3 of the Non-sense Mediated Decay (NMD) pathway; TMA20/TMA22 that encode factors of the RNA translation machinery; WHI3 that encodes an RNA binding protein; SPT2 that encodes a factor that assists RNAPII transcription elongation throughout nucleosomes (Figure 2A).
Figure 2.
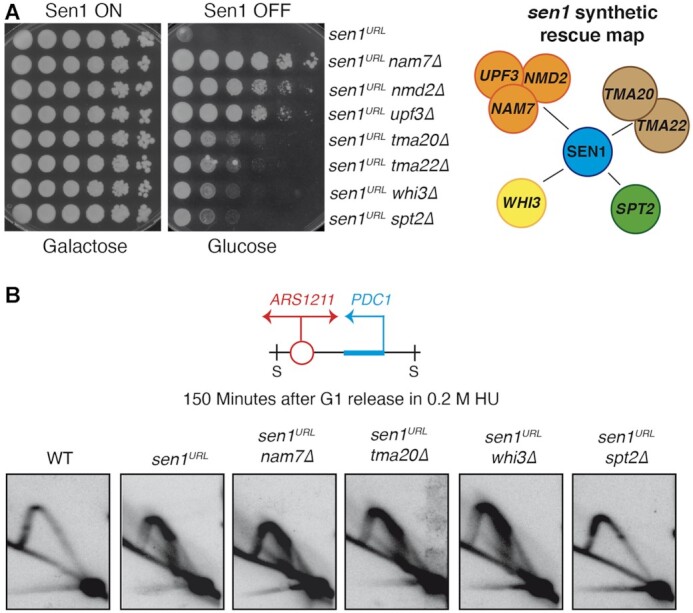
Identification of sen1URL suppressors and their contribution to replication-transcription conflicts. (A) Serial dilutions of the indicated yeast strains grown in YPG or YPD plates. A map highlighting the interactions among sen1URL suppressors is shown on the right. (B) 2D gel analysis of replication intermediates at the ARS1211-PDC1 locus upon digestion with SphI (S).
Notably, the synthetic rescue of sen1URL lethal phenotype cannot be ascribed to the restoration of normal levels of SEN1 mRNA that indeed remained low when measured by qPCR in representative suppressors of each pathway (Supplementary Figure S2A). Interestingly, the suppressors isolated in our screening also rescued DNA damage sensitivity of the thermosensitive sen1-1 allele (Supplementary Figure S2B).
Inactivation of the HMG-like factor Spt2 bypasses Sen1 requirement during replication-transcription conflicts
We wondered if sen1 suppressors were able to bypass Sen1 requirement at the fork during head-on conflicts with transcription. We focused our analysis on representative genes of each pathway isolated in our screening, namely NAM7, TMA20, WHI3 and SPT2 genes. We analyzed by the 2D gel the replication intermediates of double mutant strains, containing deletions of each of the above genes combined with the sen1URL allele, along with WT and sen1URL strains used as controls. We performed 2D gel analysis at PDC1 gene at its natural position where it is close to the ARS1211 and is a well-known hot-spot of head-on replication-transcription collisions (24,34). We observed replication forks arrested by transcription in all double mutants except for sen1URL spt2Δ, which showed a classic Y-shaped arc resembling the 2D gel pattern of the WT strain (Figure 2B). Thus, among the sen1 suppressors, specifically spt2Δ prevented the accumulation of paused forks at PDC1 locus in Sen1-depleted cells.
We next investigated whether the disappearance of paused forks in Sen1-depleted cells upon Spt2 inactivation indicated that forks indeed progressed across the PDC1 dependent barrier. Our previous analysis showed that, in sen1URL mutants, the forks are terminally arrested at the PDC1 gene and thus the nearby dormant origin ARS1211.5 fired to rescue the replication of the locus (34). To assess whether replication forks progress through the PDC1 gene, we therefore monitored the activation status of the dormant origin ARS1211.5 in WT, sen1URL, spt2Δ and sen1URL spt2Δ strains. As shown in Figure 3A, in WT cells, forks arising from the early origin ARS1211 moved across PDC1 transcribed gene without impediments and then passively replicated the ARS1211.5 dormant origin, generating a Y-shaped arc. spt2Δ mutants showed a 2D migration pattern similar to WT cells, both at PDC1 locus and at dormant origin-containing fragment. Conversely, sen1URL mutants showed the characteristic 2D gel signal associated with fork pausing at the PDC1 locus and the consequent activation of the dormant origin ARS1211.5, as indicated by the appearance of a bubble-shaped arc. In sen1URL spt2Δ double mutants, Y-shaped arcs could be observed throughout the experiment at PDC1 locus, while barely detectable bubble signals were found at dormant origin-containing fragment. Importantly, a Y-shaped arc was clearly observed together with the bubble signal in sen1URL spt2Δ mutants, indicating that the dormant origin was also extensively passively replicated by ARS1211-dependent forks.
Figure 3.

The inactivation of Spt2 elongation factor prevents fork arrest and RNA:DNA hybrid accumulation at PDC1 gene in cells lacking Sen1. (A) 2D gel analysis of replication intermediates at the ARS1211-PDC1 and ARS1211.5 loci upon digestion with SphI (S) and BclI (B), respectively. (B) RNA:DNA hybrid and (C) PDC1 mRNA levels measured by DRIP-qPCR and qPCR, respectively. Data represent mean ± SEM from three independent experiments. ns, not significant; ****P < 0.0001 (two-way ANOVA). (D) Tetrads obtained from sporulation of diploids heterozygous for sen1-1, spt2Δ and mrc1Δ mutations grown at the semi-permissive temperature of 30°C for sen1-1.
We wondered if SPT2 deletion also suppressed the RNA:DNA hybrids at PDC1 locus in Sen1-depleted cells. As previously shown (24,34), sen1URL mutants accumulated high levels of hybrids in S-phase at the PDC1 gene, as measured by DRIP-qPCR analysis, when head-on replication-transcription conflicts take place (Figure 3B and Supplementary Figure S3). Conversely, RNA:DNA hybrids were very low in sen1URL spt2Δ double mutants, as in WT or spt2Δ single mutant controls (Figure 3B and Supplementary Figure S3). Notably, in these conditions, the decrease of the R-loop levels was not due to a pre-existent reduction in PDC1 transcription, since transcript levels were indeed not affected by SPT2 inactivation in either WT or sen1URL strains, as measured by qPCR (Figure 3C). Taking together these data suggests that, in the absence of Sen1, the further inactivation of Spt2 allows the forks to proceed across the PDC1-dependent transcription barrier, preventing R-loop formation at the same time. Importantly, in Sen1-depleted cells, we observed fork defects and accumulation of both RNA:DNA hybrids and DNA damage at other highly transcribed loci encountering replication in head-on orientation (Supplementary Figure S4). These genes include HYP2, GPM1, TPI1 and PDC5 genes, the latter known to be expressed in cells deleted for PDC1 (60). Moreover, such sen1 defects were all suppressed by SPT2 inactivation (Supplementary Figure S4). Hence, Spt2 aggravates the obstacle imposed by transcription to the forks advancing in head-on orientation when Sen1 is absent.
We have previously demonstrated that the Replication Progression Complex (RPC), composed of Mrc1/Tof1/Csm3/Ctf4, protects forks hindered by transcription from Exo1-mediated degradation in sen1 mutants (34). Deletions of any component of the RPC genes are synthetic lethal with sen1-1 allele (34). Thus, we tested if Spt2 ablation also suppressed the synthetic lethality between mutations in SEN1 andMRC1 genes. sen1-1 spt2Δ double mutants were crossed with the mrc1Δ mutants to obtain different combinations of mutations via meiosis. Representative tetrads of the 48 analyzed are shown in Figure 3D. At the semi-permissive temperature of 30°C, sen1-1 spores exhibited a slow growth phenotype, which was suppressed by spt2Δ mutation, suggesting that the interference between replication and transcription is a major cause of the spontaneous cell lethality of sen1-1 mutants. While sen1-1 mrc1Δ viable spores were never recovered, those attributable to sen1-1 spt2Δ mrc1Δ genotype developed into visible colonies in 54% of cases (Figure 3D). Thus, at least partially, SPT2 inactivation suppresses the cell lethality associated with sen1-1 mrc1 double mutants.
In conclusion, SPT2 deletion in sen1 mutants reduces replication-transcription conflicts globally, making the RPC complex largely dispensable for the stabilization of the forks.
Spt2 inactivation suppresses abnormal R-loop accumulation in cells lacking Sen1, but not RNase H enzymes
To corroborate the notion that spt2 mutants prevent RNA:DNA hybrid accumulation genome-wide in sen1 mutants, we analyzed hybrid levels on chromosome spreads by indirect immunofluorescence (IF) using the S9.6 antibody in WT, sen1URL, sen1URL spt2Δ and spt2Δ strains. In sen1URL mutants, we detected higher S9.6 fluorescence intensity signals on chromosome spreads compared to WT strain, which was significantly reduced upon the inactivation of Spt2 (Figure 4A and Supplementary Figure S5A and S5C). We also analyzed in parallel yeast strains inactivated for the RNH1 and RNH201 genes encoding the catalytic subunits of RNase H1 and H2 enzymes respectively, which, together with Sen1, are major anti-R-loop factors. By IF, rnh1Δ rnh201Δ double mutants displayed higher levels of hybrids than WT that are not reduced upon inactivation of Spt2 (Figure 4A and Supplementary Figure S5A and S5C). Moreover, while SPT2 inactivation suppressed the HU-sensitivity of sen1-1 allele, it did not rescue that of rnh1Δ rnh201Δ double mutants (Figure 4B). WT and rnh1Δ rnh201Δ double mutant strains showed a similar 2D gel migration pattern of replication intermediates at ARS1211-PDC1 locus, which differs from sen1URL (Figure 4C). Thus, unlike in cells lacking Sen1, in those lacking the RNase H enzymes, replication fork progression did not appear to be impeded by transcription.
Figure 4.
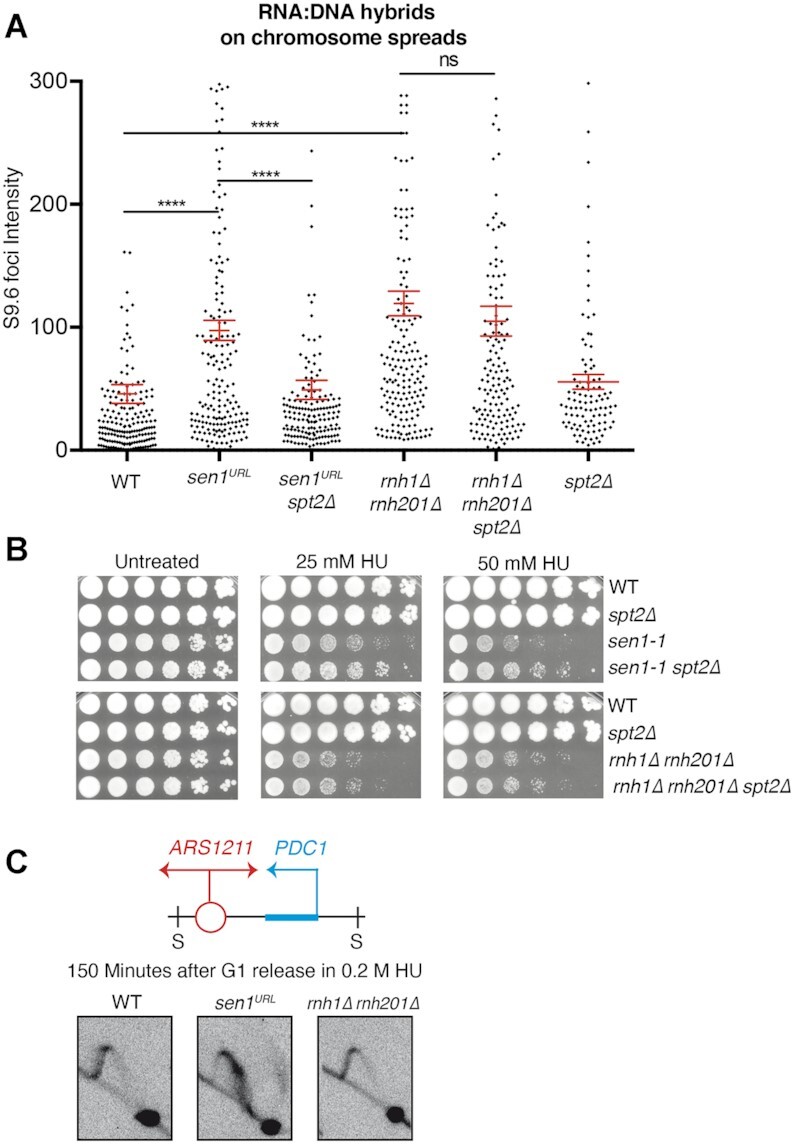
Spt2 inactivation suppresses R-loop accumulation and replication stress in cells lacking Sen1, but not RNase H enzymes. (A) S9.6 foci quantification on chromosome spreads in YPD. Data represent mean ± SEM from three independent experiments. At least 100 nuclei for each experiment were analyzed. ns, not significant; ****P < 0.0001 (unpaired two-tails t-test). (B) Serial dilutions of the indicated yeast strains spotted on YPD plates with or without HU and grown at the semi-permissive temperature of 30°C for sen1-1. (C) 2D gel analysis of replication intermediates at the ARS1211-PDC1 locus upon digestion with SphI (S).
Taken together these data indicate that Spt2 contributes to genome-wide accumulation of RNA:DNA hybrids and replication stress caused by the absence of Sen1, but it does not contribute to the same defects caused by the inactivation of RNase H enzymes. This suggests that hybrid-driven DNA damage occurs by separable mechanisms in RNase H- and Sen1-deficient strains.
RNAPII elongation is a major obstacle during head-on replication-transcription collisions in cells lacking Sen1
Spt2 is a physical interactor of the RNAPII and assists transcription elongation by modulating chromatin structure (62). Spt2 shares homology with the human High Mobility Group B proteins (HMGB), a class of chromatin remodeling factors that destabilizes nucleosomes and promotes DNA accessibility to other proteins, including transcription factors.
We wondered if the elongating RNAPII complex or specific chromatin marks associated with transcription elongation acted as a barrier to fork progression in sen1 mutants. For this purpose, we interrogated candidate genes, which may have escaped from our genomic screening, for the capability, once deleted, to suppress the cell lethality due to the depletion of Sen1 protein (Figure 5A). We selected genes that belong to the HMGB family, NHP6A, NHP6B, HMO1, and the transcription elongation genes SPT4 and LEO1. We tested genes encoding histone H1 and H3, HHO1 and HHT1, respectively. Moreover, we analyzed BRE1, encoding the E3 ligase that promotes H2B-K123 mono-ubiquitination, a modification that favors RNAPII elongation at the early stage of transcription. Finally, we tested SET1 and JHD2 that, respectively, adds or removes H3K4 methylation, a histone mark associated with active transcription (63) and whose levels correlate inversely with those of pathological RNA:DNA hybrids (45). A specific function of Spt2, also conserved in human, is to promote the recycling of H3/H4 tetramer in the wake of RNAPII (64). Notably, in spt2 mutants, the impaired turnover of ‘old’ histones at coding genes increases the levels of newly synthetized H3 variant with acetylated K56 residue (64), a modification associated with high levels of deleterious R-loops in certain genetic contexts (47). In both WT and sen1URL strains, in either G1-phase or HU-treated cells, the inactivation of spt2 indeed resulted in high levels of H3K56 acetylation, as revealed by ChIP-qPCR analysis (Supplementary Figure S6), although in spt2 mutants high H3K56 acetylation did not correlate with hybrid accumulation (Figure 3B). To analyze the contribution of this chromatin mark in sen1 suppression, we tested RTT109 and HST3/4, encoding the enzymes that write and erase the H3K56 acetylation, respectively.
Figure 5.
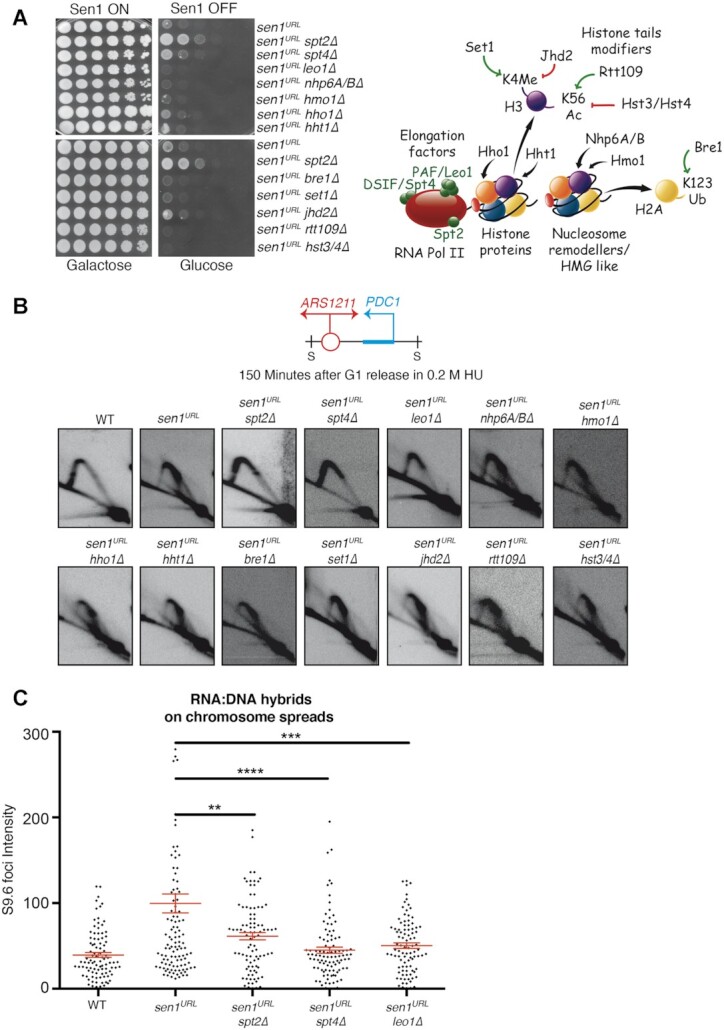
The inactivation of Spt4 or Leo1 elongation factors prevents fork arrest at PDC1 gene and the accumulation of RNA:DNA hybrids globally in cells lacking Sen1. (A) Serial dilutions of the indicated yeast strains grown in YPG or YPD plates. A schematic representation of the molecular functions of candidate genes is shown on the right. (B) 2D gel analysis of replication intermediates at the ARS1211-PDC1 locus upon digestion with SphI (S). (C) S9.6 foci quantification on chromosome spreads in YPD. Data represent mean ± SEM from three independent experiments. At least 100 nuclei for each experiment were analyzed. **P < 0.01; ***P < 0.001; ****P < 0.0001 (unpaired two-tails t-test).
We combined deletions in each of the candidate genes with the sen1URL strain and we tested the ability of double mutants to grow on YPD on plates. As shown in Figure 5A, the deletion of SPT4, but not that of the other genes tested, suppressed the cell lethality associated with the depletion of Sen1. Next, we analyzed the same candidate gene suppressors for their ability to prevent the accumulation of paused forks at ARS1211-PDC1 locus in sen1URL mutants using the 2D gel method. The deletion of SPT4 or LEO1 in Sen1-depleted cells generated a 2D gel pattern resembling that of sen1URL spt2Δ (Figure 5B). In line with this, spt4Δ or leo1Δ suppressed RNA:DNA hybrids detected by IF in cells lacking Sen1, as spt2Δ (Figure 5C and Supplementary Figure S5B and S5C).
We conclude that the alteration in the chromatin structure due to ablation of histone/non-histone proteins or the deregulation of the turnover of specific transcription-associated chromatin marks do not affect the outcome of collisions in sen1 mutants. Conversely, along with Spt2, the ablation of Spt4, the interacting partner of Spt5 in DSIF complex, or Leo1, a component of PAF complex, all proteins promoting RNAPII elongation, prevents fork arrest and R-loop accumulation due to the conflict with transcription in sen1 mutants.
Based on these results, we expected that the RNAPII was retained on chromatin during the collision in Sen1 absence, thus contributing to the replication fork barrier. To test our hypothesis, we set up a ChIP-qPCR experiment to analyze RNAPII binding at PDC1 gene in WT and sen1URL strains. sen1URL mutants combined with deletions in SPT2, SPT4 or LEO1 genes were also analyzed in the same experimental conditions, together with a strain carrying a deletion of the PDC1 gene promoter (no PDC1 transcription), as a negative control for RNAPII loading (Figure 6A). Yeast strains were synchronized in G1-phase of the cell cycle in YPD and then released into S-phase in the presence of HU. Samples were collected at 150 min in HU and the levels of Rpb3, a subunit of the RNAPII, were measured by ChIP -qPCR. Rpb3 was indeed found at PDC1 gene and even at higher levels in sen1URL mutants compared to WT strain, while it returned to normal levels when SPT2, SPT4 or LEO1 were also deleted (Figure 6A). Notably, such Rpb3 accumulation on chromatin in sen1URL mutants was not observed in G1-phase (Supplementary Figure S7A). However, we observed elevated levels of Rpb3 on chromatin during S-phase in sen1URL mutants and the suppression of this phenotype by spt2 mutation at other highly transcribed genes undergoing head-on collisions with replication (Supplementary Figure S4D).
Figure 6.
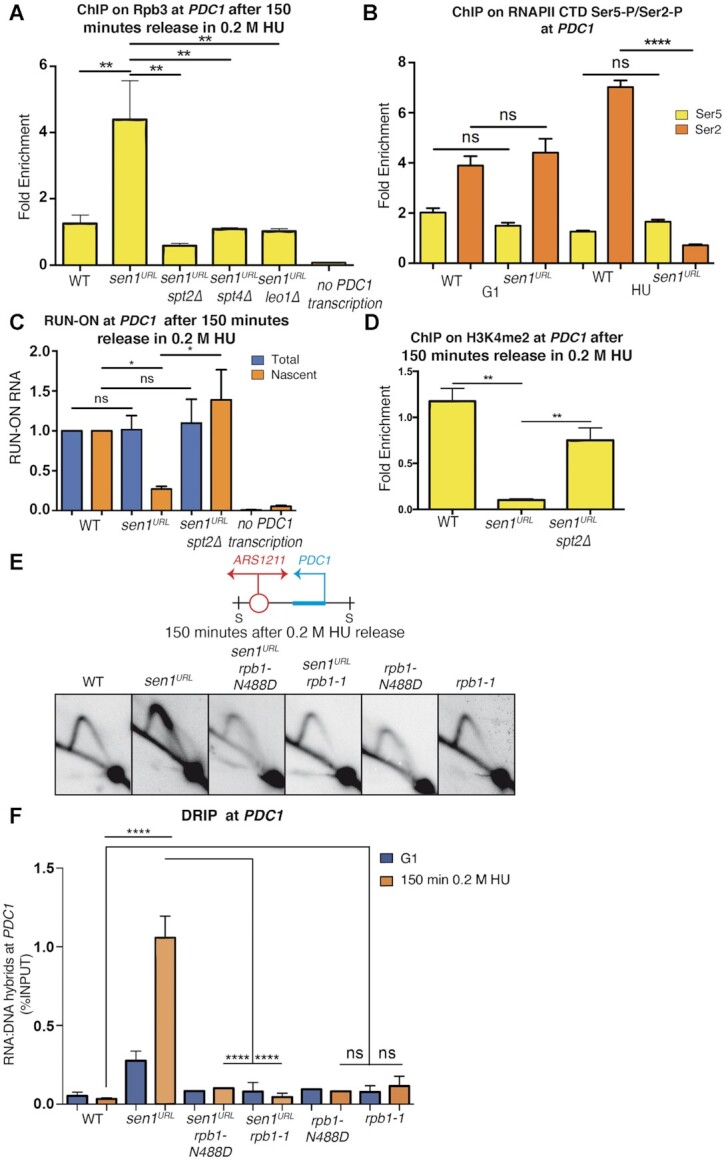
Elongating RNAPII contributes to R-loop-dependent fork barrier in Sen1-depleted cells. (A) ChIP-qPCR analysis at PDC1 locus of the Rpb3 subunit of the RNAPII complex. Data represent mean ± SEM from three independent experiments. **P < 0.01 (two-way ANOVA). (B) ChIP-qPCR analysis at PDC1 locus of phospho-CTD RNAPII isoforms in cells collected in G1 or after 150 minutes in 0.2M HU. Signals originating from CTD-Ser2 or CTD-Ser5 were normalized to total RNAPII levels. Data represent mean ± SEM from three independent experiments. ns, not significant; ****P < 0.0001 (two-way ANOVA). (C) Analysis of BrUTP-labeled nascent PDC1 transcripts and total PDC1 levels compared to ACT1 mRNA and normalized to WT levels. ns, not significant; *P < 0.05 (two-way ANOVA) (D) ChIP-qPCR analysis at PDC1 locus of histone H3 lysine 4-dimethylated (H3K4me2). Data represent mean ± SEM from three independent experiments. **P < 0.01 (two-way ANOVA) (E) 2D gel analysis of replication intermediates at the ARS1211-PDC1 locus upon digestion with SphI (S). (F) RNA:DNA hybrid levels at PDC1 gene analyzed by DRIP-qPCR. Data represent mean ± SEM from three independent experiments. ns, not significant; ****P < 0.0001 (two-way ANOVA).
We next wondered whether the RNAPII accumulating at PDC1 in sen1URL mutants was arrested by the advancing fork and thus incapable to sustain elongation. To test this, we monitored by ChIP-qPCR the levels of RNAPII phosphorylated within the carboxy terminal domain at serine 5 (CTD-Ser5P) or serine 2 (CTD-Ser2P) residues, which are hallmarks of early transcription or active elongation, respectively (65). The analysis was conducted on WT and sen1URL strains arrested in either HU or G1-phase, that is, when replication-transcription collisions took place or not, respectively. As shown in Figure 6B, while the levels of CTD-Ser5P were similar in WT and sen1URL strains, those of CTD-Ser2P were reduced in the absence of Sen1, but specifically in HU-treated cells. Taken together these data suggest that, in the absence of Sen1, RNAPII is elongation-proficient, but upon clashing with the fork it becomes unable to sustain transcription. To investigate more directly this aspect, we set up a transcription run-on assay to analyze nascent PDC1 transcript by BrUTP incorporation in S-phase of the cell cycle. WT, sen1URL, sen1URL spt2 and no PDC1 transcription strains were collected upon HU-treatment and incubated with BrUTP to label nascent RNA. Total PDC1 transcript levels were similar in WT and mutant strains, as also previously observed (Figure 3C), with the obvious exception of the strain not expressing PDC1 (Figure 6C). Consistent with the analysis of the dynamic of CTD-Ser5/2 phosphorylation, we recovered low levels of nascent PDC1 transcripts in Sen1-depleted cells than WT (Figure 6C), suggesting that RNAPII is arrested upon unscheduled collisions with the fork. Such transcription arrest in the absence of Sen1 was suppressed by spt2 mutation (Figure 6C). As a further proof that RNAPII was in fact not transcribing after the collision with the fork in sen1URL mutants, we tested by ChIP-qPCR the accumulation of H3K4 di-methylated (H3K4me2), a marker of active transcription (63). As shown in Figure 6D, H3K4me2 levels decreased in HU-treated sen1URL mutants relative to WT, while the inactivation of Spt2 partially rescued such sen1-dependent defect.
Taken together, our findings suggest that, in the absence of Sen1, elongating RNAPII and replication fork arrest each other when clashing in head-on orientation. Attenuation of elongation, by slowing down RNAPII progression, prevents fork stalling and the concomitant accumulation of RNA:DNA hybrids and DNA damage, which preclude gene expression in the long run (Figure 1D).
To further support this model, we took advantage of a point mutation allele that affects the RNAPII catalytic core (rpb1-N488D), specifically impairing transcription elongation rate both in vivo and in vitro (52,66). As shown in Figure 6E, F and Supplementary Figure S7B, rpb1-N488D suppressed both replication fork defects and RNA:DNA hybrid accumulation at the PDC1 gene in Sen1-depleted cells. Thus, although the inhibition of RNAPII elongation causes per se a delay in fork progression (67), it compensates for the stronger R-loop-dependent replication pausing due to Sen1 inactivation.
We also tested another mutant of the RNAPII core, the rpb1-1 thermosensitive allele (68) that causes R-loop-independent RNAPII retention on chromatin, slowing down replication (67). Accordingly, we found that rpb1-1, as rpb1-N488D, did not show per se RNA:DNA hybrid accumulation at PDC1, while it rescued both hybrids and the strong fork pausing in sen1 mutants (Figure 6E, F and Supplementary Figure S7B). We conclude that a major impediment to a fork advancing in a head-on orientation is the RNAPII retained on chromatin with RNA:DNA hybrids.
DISCUSSION
In this study, we provide further evidence that highly transcribed genes are a major impediment to a replication fork encountered in head-on orientation in yeast cells lacking Sen1. We observed RNA:DNA hybrid hyper-accumulation during this type of collisions in sen1 mutants and a minor increase of these structures in co-directional clashes. These results are consistent with the previously described dynamic of hybrid accumulation in sen1 mutants at some highly-expressed genes (24) and genome-wide (33). While our findings are not in contrast with the notion that R-loops tend to accumulate in both types of collisions (6,31), they do suggest that R-loops contribute to fork arrest only in head-on clashes. We also found that, in sen1 mutants, fork impairment and R-loop accumulation in head-on collisions lead to DNA damage and transcription hindrance. In this regard, we observed that not only RNAPII is arrested while clashing with the fork, but also that gene expression is turned off in the long run. One possibility is that R-loops might directly or indirectly damage the transcribing DNA template, thus inducing transcription loss over time, even if we cannot exclude that other mechanisms may contribute to gene silencing. However, the stalling of both replication fork and RNAPII, R-loop accumulation, DNA damage and the inhibition of gene expression are all correlated, likely consequent, outcomes of head-on and not co-directional collisions.
According to in vitro studies, Sen1 helicase binds to 5′ overhangs of either an RNA or DNA filament and, by translocating in 5′ to 3′ direction, efficiently dismantles both RNA:DNA hybrids and DNA duplexes (69–71). Thus, Sen1 could act at two levels in limiting R-loop accumulation during head-on replication-transcription collisions: i) Sen1 could chase the RNAPII by moving on RNA, thus preventing RNA:DNA hybrid formation behind the RNAPII, promoting transcription termination (16); its physical interaction with the Spt5/4 elongation factors travelling with the RNAPII (72) could be instrumental for this role; ii) as a component of the replisome (24,73), Sen1 could translocate on ssDNA and remove an R-loop-engaged RNAPII, possibly promoted by the excess of positive torsional stress generated while replication and transcription converge (37) (Figure 7A). Such topological constraints, acting as an obstacle to RNAPII forward movement, could also induce its backtracking (74), eventually triggering the formation of stable RNA:DNA hybrids ahead of the RNAPII (38). Such anterior R-loops, involving the 3′ end of nascent RNA, could be targeted by Sen1 as it translocates on ssDNA (Figure 7A). While the displacement of paused/backtracked RNAPII by Sen1 might quickly resume replication and transcription, works in humans (75) and bacteria (76) suggests that co-transcriptional R-loops can also trigger fork reversal. The processing of reversed forks by recombination or a cycle of fork cleavage-religation is then required to restart both replication and transcription. The removal of RNAPII/R-loop by Sen1/Senataxin could be crucial to reactivate transcription also in this context. In both scenarios, while counteracting R-loop formation, Sen1 also removes RNAPII from the DNA template. Indeed, our findings indicate that RNAPII is an integral part of the transcription barrier in sen1 mutants since it is retained on chromatin with RNA:DNA hybrids during the head-on collision with the replication fork. Thus, R-loops alone are not likely the obstacle to replication in sen1 mutants. The finding that a mutation in the RNAPII core, rpb1-1, causing RNAPII retention on DNA in a RNA:DNA hybrid-independent manner (67), suppresses fork pausing in sen1, infers that RNAPII alone is not a main replication barrier. Thus, we propose that a major impediment is the elongating RNAPII trapped to the DNA template via the formation of a stable R-loop (Figure 7B). In line with this, we found that either a specific mutation in the RNAPII core enzyme affecting elongation rate, rpb1-N488D, or the inactivation of proteins that assist RNAPII elongation relieves the head-on transcription-dependent fork barrier and RNA:DNA hybrid accumulation in sen1 mutants. RNAPII elongation factors include Spt2, Spt4, which together with Spt5, forms the DSIF complex and Leo1, a component of the PAF complex. All these factors facilitate RNAPII elongation through chromatin. While Spt2 was shown to be important for the proper reconstitution of chromatin behind the transcribing RNAPII (64), structural studies in mammals suggest that DSIF and PAF complexes may promote DNA rewinding at the upstream edge of the transcription bubble (77). Through different mechanisms, these factors play a role in stabilising RNAPII on DNA template, controlling the elongation processivity, defined as the ability of the RNAPII to travel the entire length of the gene without dissociating from DNA (78). Neither the inhibition of chromatin marks associated with elongation (H2B-K123 mono-ubiquitination, H3K4 methylation and H3K56 acetylation) nor the disruption of chromatin structure due to depletion of histone or non-histone proteins, is sufficient per se to relieve the transcription barrier in sen1 mutants. Since RNAPII can be engaged in a nucleosome-free DNA template, our data suggest that a stable RNAPII, regardless of the chromatin context, is evidently a major obstacle to fork progression in sen1 mutants.
Figure 7.
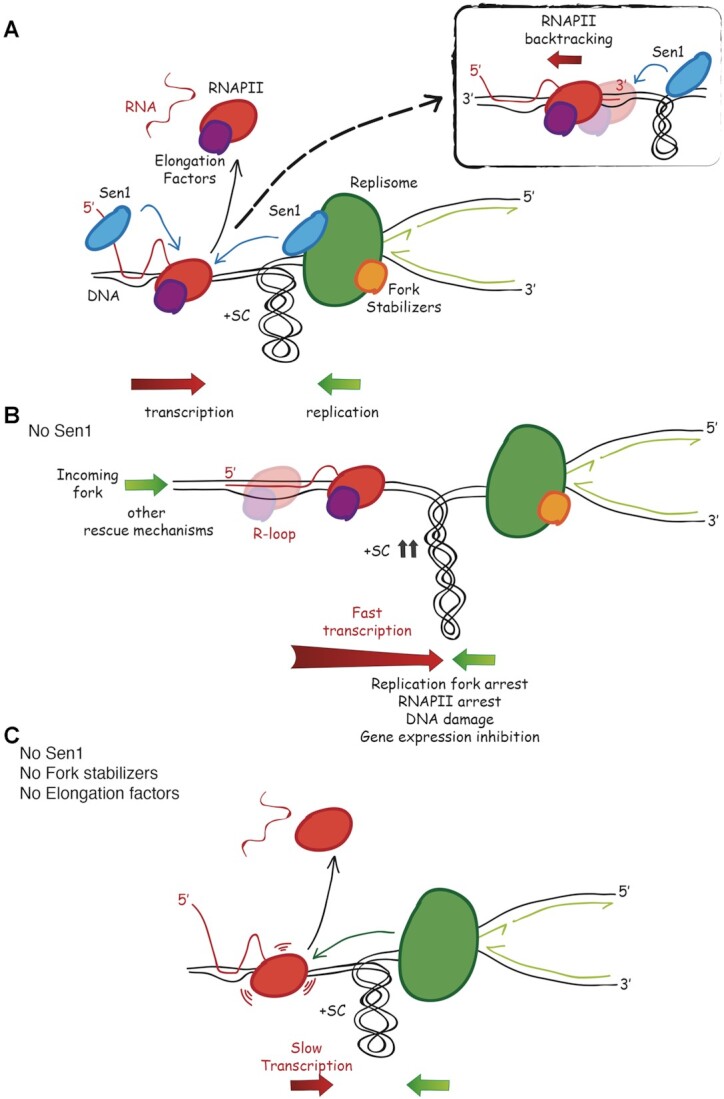
Model for the interplay between Sen1 and RNAPII elongation in head-on replication-transcription conflicts. (A) Sen1, while translocates in 5′ to 3′ direction along nascent RNA or DNA at the fork, removes elongating RNAPII together with RNA:DNA hybrids, facilitating replisome progression past transcription. Upon building up of positive supercoils at the site of head-on replication-transcription collision, paused RNAPII may undergo backtracking, generating an R-loop that involves the exposed 3′ end of nascent RNA (Box). Such anterior R-loop may be also targeted by Sen1. (B) In the absence of Sen1, elongating RNAPII, upon clashing with the replication fork, engages in a stable R-loop. Both RNAPII and replication forks are arrested during the collision, leading to DNA damage and, in the long run, to transcription inhibition. Mechanisms alternative to Sen1 can eventually rescue the replication block. (C) Attenuation of elongation processivity prevents R-loop-mediated anchoring of RNAPII to DNA template, allowing the replisome to displace it, even in the absence of Sen1 and fork stabilizer factors.
A replication fork advancing in head-on orientation across a highly transcribed DNA template will likely encounter the first RNAPII in the proximity of the 3′ end. In sen1 mutants, elongating RNAPII cannot be efficiently removed neither in transcription termination nor during replication, resulting in an impenetrable barrier to the fork moving in the head-on orientation (Figure 7B). Alternative mechanisms could displace such barrier, including a fork coming from the opposite direction (34) and/or checkpoint-dependent pathways (79). The inhibition of elongation processivity would reduce RNAPII stability on DNA template due to its reduced ability to engage in a stable R-loop formation, thus resulting in a barrier easier to be displaced by the replisome advancing in a head-on orientation, even without the fork stabilizer RPC (Figure 7C).
Our findings are consistent with the observation that, in thermosensitive sen1 mutants, transcription readthrough at short noncoding RNAs is respectively suppressed or exacerbated by RNAPII point mutants that reduce or increase the transcription elongation rate (80). The role of Sen1 in transcription termination has been extensively studied on non-polyadenylated RNA species, as part of the complex with Nrd1 and Nab3 proteins (16,81), though R-loops accumulate also at coding genes, especially over polyA sites in sen1 mutants (22,31,82). While Sen1 requirement in transcription termination of mRNA is not currently well characterized, multiple evidence suggests that Sen1 moves with the replication forks and suppresses hyper-recombination independently from the termination complex subunit Nrd1 (22,24,73,83). Thus, according to the current literature, R-loop-dependent replication defects observed upon inactivation of Sen1 could arise from the sum of the transcription termination defects and the absence of the Sen1 activity at the fork. In a reconstituted bacterial DNA replication systems the stalled transcription elongation complexes block replication forks (84), while replisomes easily bypass R-loops alone on either lagging or leading DNA strands (85). In vivo evidence suggests that R-loops that are anchored to transcription elongation complexes strongly impair replication in UV-irradiated E.coli mutants for the RNase H enzymes (86). Moreover, faster transcription elongation caused by the depletion of RECQL5 in mammalian cells increases transcription-associated genome instability due to replication-transcription conflicts (87). Thus, not only in sen1 mutants but more in general, RNAPII engaged in elongation is a critical obstacle to fork progression, because it is more tightly anchored to the DNA template and/or inclined to undergo backtracking.
It has been reported that stimulation of RNAPII elongation prevents R-loop formation and the associated DNA damage, which seems at odds with our findings. For example, in BRCA1 or BRCA2 mutated cells, mutations in the anti-elongator NELF complex (12) or the overexpression of PAF1 (13), respectively, suppress abnormal R-loop accumulation in the proximity of the transcription start sites, by favoring RNAPII elongation. However, we note that stimulation or inhibition of RNAPII elongation should have the same outcome at promoter proximal pausing sites or in sen1 mutants, that is, to prevent unscheduled RNAPII stalling and R-loop accumulation. Thus, importantly, these studies together with our findings, suggest that RNAPII elongation either promotes or prevents R-loop accumulation, depending on the context. However, our data support the model that RNAPII in elongation mode represents the major obstacle for a converging replication fork.
We also found that, while Spt2 inactivation suppresses RNA:DNA hybrid accumulation and replication stress in sen1 mutants, it does not rescue the same defects in cells lacking RNase H proteins, whose functions are essential when Sen1 cannot be recruited at the forks (73). The genetic separation of functions we describe here may suggest that while Sen1 processes R-loops forming at the time of collisions between replication and RNAPII-dependent transcription, RNase H provides a backup for R-loop clearance in S-phase. This is consistent with recent findings showing that RNase H2 removes R-loops in a post-replicative stage (88) and RNase H1 mainly processes R-loops accumulated at high levels, such as in sen1 mutants (89).
In this study, we have also identified other suppressors of Sen1’s role in DNA damage, which differently from Spt2 or Spt4, do not bypass the requirement of the helicase in the removal of RNA:DNA hybrids in replication-transcription collisions. Other sen1 suppressors are genes of the NMD pathway, which controls the stability of diverse classes of mRNA, including those containing premature stop codons or short upstream (u) ORFs (90); the ribosome associated Tma20/22 complex, MCT1/DENR in human, that controls translation re-initiation of mRNA containing uORFs (91); Whi3, a PolyQ protein that binds and inhibits the translation of many mRNAs and is also prone to form aggregates potentially toxic for the cells (92). One possibility is that the inhibition of these pathways causes resilience to R-loop-driven DNA damage. Intriguingly, persistent DNA damage inhibits the NMD pathway in non-cycling human cells (93) and its inactivation in yeast increases DNA damage resistance by potentiating homologous recombination repair (94). The understanding of whether the inhibition of these sen1 suppressor pathways converges to modulate the same factors (for example both the NMD pathway and the DENR-MCT1 complex act on mRNA containing uORFs) and/or by-passes another Sen1 function not related to R-loop metabolism will require further investigations.
It is interesting to note that most of the sen1 suppressors identified in this study are conserved in humans. If the inactivation of those genes also bypassed Senataxin functions in DNA damage response, they could represent potential druggable targets for the cure of Senataxin neurological disorders. In this regard, it is interesting to note that the inhibition of Spt4 or Leo1 proteins in human and/or model organisms were shown to prevent the deleterious consequences of transcription across repetitive elements (95–97), whose expansion is a cause of ALS and Huntington's disease and is linked to aberrant R-loop accumulation (98).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Marco Foiani, Arianna Colosio, Elena Chiroli and Daniele Piccini for helpful discussions and advice. We are grateful to Mikhail Kashlev for kindly providing the mutant rpb1-N488D yeast strain.
Notes
Present address: Eleonora Nardini, Medical University of Vienna, Center for Pathobiochemistry and Genetics, Institute of Medical Chemistry, Vienna 1090, Austria.
Present address: Alessandra Brambati, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA.
Contributor Information
Luca Zardoni, Istituto di Genetica Molecolare “Luigi Luca Cavalli-Sforza”, CNR, 27100 Pavia, Italy; Scuola Universitaria Superiore IUSS, 27100 Pavia, Italy.
Eleonora Nardini, Istituto di Genetica Molecolare “Luigi Luca Cavalli-Sforza”, CNR, 27100 Pavia, Italy.
Alessandra Brambati, Istituto di Genetica Molecolare “Luigi Luca Cavalli-Sforza”, CNR, 27100 Pavia, Italy.
Chiara Lucca, IFOM Foundation, 20139 Milan, Italy.
Ramveer Choudhary, IFOM Foundation, 20139 Milan, Italy.
Federica Loperfido, Istituto di Genetica Molecolare “Luigi Luca Cavalli-Sforza”, CNR, 27100 Pavia, Italy.
Simone Sabbioneda, Istituto di Genetica Molecolare “Luigi Luca Cavalli-Sforza”, CNR, 27100 Pavia, Italy.
Giordano Liberi, Istituto di Genetica Molecolare “Luigi Luca Cavalli-Sforza”, CNR, 27100 Pavia, Italy; IFOM Foundation, 20139 Milan, Italy.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Associazione Italiana per la Ricerca sul Cancro [IG17714 to G.L. and IG24316 to S.S.]; MIUR [PRIN 2017KSZZJW to S.S.]; L.Z. was recipient of a ‘Arturo Falaschi’ fellowship from Adriano Buzzati-Traverso Foundation. Funding for open access charge: Associazione Italiana per la Ricerca sul Cancro [IG17714].
Conflict of interest statement. None declared.
REFERENCES
- 1. Macheret M., Halazonetis T.D.. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. Mech. Dis. 2015; 10:425–448. [DOI] [PubMed] [Google Scholar]
- 2. Forrer Charlier C., Martins R.A.P.. Protective mechanisms against DNA replication stress in the nervous system. Genes (Basel). 2020; 11:730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gómez-González B., Aguilera A.. Transcription-mediated replication hindrance: a major driver of genome instability. Genes Dev. 2019; 33:1008–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hamperl S., Bocek M.J., Saldivar J.C., Swigut T., Cimprich K.A.. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 2017; 170:774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lang K.S., Hall A.N., Merrikh C.N., Ragheb M., Tabakh H., Pollock A.J., Woodward J.J., Dreifus J.E., Merrikh H.. Replication-transcription conflicts generate R-loops that orchestrate bacterial stress survival and pathogenesis. Cell. 2017; 170:787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. García-Rubio M., Aguilera P., Lafuente-Barquero J., Ruiz J.F., Simon M.-N., Geli V., Rondón A.G., Aguilera A.. Yra1-bound RNA–DNA hybrids cause orientation-independent transcription–replication collisions and telomere instability. Genes Dev. 2018; 32:965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Promonet A., Padioleau I., Liu Y., Sanz L., Biernacka A., Schmitz A.-L., Skrzypczak M., Sarrazin A., Mettling C., Rowicka M.et al.. Topoisomerase 1 prevents replication stress at R-loop-enriched transcription termination sites. Nat. Commun. 2020; 11:3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lang K.S., Merrikh H.. Topological stress is responsible for the detrimental outcomes of head-on replication-transcription conflicts. Cell Rep. 2021; 34:108797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crossley M.P., Bocek M., Cimprich K.A.. R-Loops as cellular regulators and genomic threats. Mol. Cell. 2019; 73:398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bhatia V., Barroso S.I., García-Rubio M.L., Tumini E., Herrera-Moyano E., Aguilera A.. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature. 2014; 511:362–365. [DOI] [PubMed] [Google Scholar]
- 11. Hatchi E., Skourti-Stathaki K., Ventz S., Pinello L., Yen A., Kamieniarz-Gdula K., Dimitrov S., Pathania S., McKinney K.M., Eaton M.L.et al.. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell. 2015; 57:636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang X., Chiang H.-C., Wang Y., Zhang C., Smith S., Zhao X., Nair S.J., Michalek J., Jatoi I., Lautner M.et al.. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun. 2017; 8:15908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shivji M.K.K., Renaudin X., Williams Ç.H., Venkitaraman A.R.. BRCA2 regulates transcription elongation by RNA polymerase II to prevent R-loop accumulation. Cell Rep. 2018; 22:1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herold S., Kalb J., Büchel G., Ade C.P., Baluapuri A., Xu J., Koster J., Solvie D., Carstensen A., Klotz C.et al.. Recruitment of BRCA1 limits MYCN-driven accumulation of stalled RNA polymerase. Nature. 2019; 567:545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cerritelli S.M., Crouch R.J.. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009; 276:1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Groh M., Albulescu L.O., Cristini A., Gromak N.. Senataxin: genome guardian at the interface of transcription and neurodegeneration. J. Mol. Biol. 2017; 429:3181–3195. [DOI] [PubMed] [Google Scholar]
- 17. Moreira M.-C., Klur S., Watanabe M., Németh A.H., Ber I. Le, Moniz J.-C., Tranchant C., Aubourg P., Tazir M., Schöls L.et al.. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 2004; 36:225–227. [DOI] [PubMed] [Google Scholar]
- 18. Chen Y.-Z., Bennett C.L., Huynh H.M., Blair I.P., Puls I., Irobi J., Dierick I., Abel A., Kennerson M.L., Rabin B.A.et al.. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004; 74:1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rudnik-Schöneborn S., Arning L., Epplen J.T., Zerres K.. SETX gene mutation in a family diagnosed autosomal dominant proximal spinal muscular atrophy. Neuromuscul. Disord. 2012; 22:258–262. [DOI] [PubMed] [Google Scholar]
- 20. Kannan A., Bhatia K., Branzei D., Gangwani L.. Combined deficiency of Senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic Acids Res. 2018; 46:8326–8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ursic D., Chinchilla K., Finkel J.S., Culbertson M.R.. Multiple protein/protein and protein/RNA interactions suggest roles for yeast DNA/RNA helicase Sen1p in transcription, transcription-coupled DNA repair and RNA processing. Nucleic Acids Res. 2004; 32:2441–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mischo H.E., Gómez-González B., Grzechnik P., Rondón A.G., Wei W., Steinmetz L., Aguilera A., Proudfoot N.J.. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol. Cell. 2011; 41:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Skourti-Stathaki K., Proudfoot N.J., Gromak N.. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell. 2011; 42:794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alzu A., Bermejo R., Begnis M., Lucca C., Piccini D., Carotenuto W., Saponaro M., Brambati A., Cocito A., Foiani M.et al.. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell. 2012; 151:835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yuce O., West S.C.. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol. Cell. Biol. 2013; 33:406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grzechnik P., Gdula M.R., Proudfoot N.J.. Pcf11 orchestrates transcription termination pathways in yeast. Genes Dev. 2015; 29:849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brustel J., Kozik Z., Gromak N., Savic V., Sweet S.M.M.. Large XPF-dependent deletions following misrepair of a DNA double strand break are prevented by the RNA:DNA helicase Senataxin. Sci. Rep. 2018; 8:3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cohen S., Puget N., Lin Y.-L., Clouaire T., Aguirrebengoa M., Rocher V., Pasero P., Canitrot Y., Legube G.. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018; 9:533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rawal C.C., Zardoni L., Di Terlizzi M., Galati E., Brambati A., Lazzaro F., Liberi G., Pellicioli A.. Senataxin ortholog Sen1 limits DNA:RNA hybrid accumulation at DNA double-strand breaks to control end resection and repair fidelity. Cell Rep. 2020; 31:107603. [DOI] [PubMed] [Google Scholar]
- 30. Grunseich C., Wang I.X., Watts J.A., Burdick J.T., Guber R.D., Zhu Z., Bruzel A., Lanman T., Chen K., Schindler A.B.et al.. Senataxin mutation reveals how R-loops promote transcription by blocking DNA methylation at gene promoters. Mol. Cell. 2018; 69:426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Achar Y.J., Adhil M., Choudhary R., Gilbert N., Foiani M.. Negative supercoil at gene boundaries modulates gene topology. Nature. 2020; 577:701–705. [DOI] [PubMed] [Google Scholar]
- 32. Chédin F. Nascent connections: R-loops and chromatin patterning. Trends Genet. 2016; 32:828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. San Martin-Alonso M., Soler-Oliva M.E., García-Rubio M., García-Muse T., Aguilera A.. Harmful R-loops are prevented via different cell cycle-specific mechanisms. Nat. Commun. 2021; 12:4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brambati A., Zardoni L., Achar Y.J., Piccini D., Galanti L., Colosio A., Foiani M., Liberi G.. Dormant origins and fork protection mechanisms rescue sister forks arrested by transcription. Nucleic Acids Res. 2018; 46:1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jensen T.H., Jacquier A., Libri D.. Dealing with pervasive transcription. Mol. Cell. 2013; 52:473–484. [DOI] [PubMed] [Google Scholar]
- 36. Wade J.T., Grainger D.C.. Pervasive transcription: illuminating the dark matter of bacterial transcriptomes. Nat. Rev. Microbiol. 2014; 12:647–653. [DOI] [PubMed] [Google Scholar]
- 37. Kuzminov A. When DNA topology turns deadly – RNA polymerases dig in their R-loops to stand their ground: new positive and negative (super)twists in the replication–transcription conflict. Trends Genet. 2018; 34:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zatreanu D., Han Z., Mitter R., Tumini E., Williams H., Gregersen L., Dirac-Svejstrup A.B., Roma S., Stewart A., Aguilera A.et al.. Elongation factor TFIIS prevents transcription stress and R-loop accumulation to maintain genome stability. Mol. Cell. 2019; 76:57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dutta D., Shatalin K., Epshtein V., Gottesman M.E., Nudler E.. Linking RNA polymerase backtracking to genome instability in E. coli. Cell. 2011; 146:533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Herrera-Moyano E., Mergui X., Garcia-Rubio M.L., Barroso S., Aguilera A.. The yeast and human FACT chromatin-reorganizing complexes solve R-loop-mediated transcription-replication conflicts. Genes Dev. 2014; 28:735–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Šviković S., Crisp A., Tan-Wong S.M., Guilliam T.A., Doherty A.J., Proudfoot N.J., Guilbaud G., Sale J.E.. R-loop formation during S phase is restricted by PrimPol-mediated repriming. EMBO J. 2019; 38:e99793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Neil A.J., Liang M.U., Khristich A.N., Shah K.A., Mirkin S.M.. RNA-DNA hybrids promote the expansion of Friedreich's ataxia (GAA)n repeats via break-induced replication. Nucleic Acids Res. 2018; 46:3487–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. García-Pichardo D., Cañas J.C., García-Rubio M.L., Gómez-González B., Rondón A.G., Aguilera A.. Histone mutants separate R loop formation from genome instability induction. Mol. Cell. 2017; 66:597–609. [DOI] [PubMed] [Google Scholar]
- 44. Groh M., Lufino M.M.P., Wade-Martins R., Gromak N.. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 2014; 10:e1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Colak D., Zaninovic N., Cohen M.S., Rosenwaks Z., Yang W.-Y., Gerhardt J., Disney M.D., Jaffrey S.R.. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 2014; 343:1002–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Salas-Armenteros I., Pérez-Calero C., Bayona-Feliu A., Tumini E., Luna R., Aguilera A.. Human THO–Sin3A interaction reveals new mechanisms to prevent R-loops that cause genome instability. EMBO J. 2017; 36:3532–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Feldman J.L., Peterson C.L.. Yeast sirtuin family members maintain transcription homeostasis to ensure genome stability. Cell Rep. 2019; 27:2978–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Almeida R., Fernández-Justel J.M., Santa-María C., Cadoret J.-C., Cano-Aroca L., Lombraña R., Herranz G., Agresti A., Gómez M.. Chromatin conformation regulates the coordination between DNA replication and transcription. Nat. Commun. 2018; 9:1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Taneja N., Zofall M., Balachandran V., Thillainadesan G., Sugiyama T., Wheeler D., Zhou M., Grewal S.I.S.. SNF2 family protein Fft3 suppresses nucleosome turnover to promote epigenetic inheritance and proper replication. Mol. Cell. 2017; 66:50–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zeller P., Padeken J., van Schendel R., Kalck V., Tijsterman M., Gasser S.M.. Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat. Genet. 2016; 48:1385–1395. [DOI] [PubMed] [Google Scholar]
- 51. Longtine M.S., Mckenzie A. III, Demarini D.J., Shah N.G., Wach A., Brachat A., Philippsen P., Pringle J.R. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998; 14:953–961. [DOI] [PubMed] [Google Scholar]
- 52. Malagon F., Kireeva M.L., Shafer B.K., Lubkowska L., Kashlev M., Strathern J.N.. Mutations in the Saccharomyces cerevisiae RPB1 gene conferring hypersensitivity to 6-azauracil. Genetics. 2006; 172:2201–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Storici F., Resnick M.A.. The Delitto Perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oin yeast. Methods in Enzymology. 2006; 329–345. [DOI] [PubMed] [Google Scholar]
- 54. Tong A.H.Y., Evangelista M., Parsons A.B., Xu H., Bader G.D., Pagé N., Robinson M., Raghibizadeh S., Hogue C.W.V., Bussey H.et al.. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001; 294:2364–2368. [DOI] [PubMed] [Google Scholar]
- 55. Zardoni L., Nardini E., Liberi G.. 2D gel electrophoresis to detect DNA replication and recombination intermediates in budding yeast. Methods in Molecular Biology. 2020; 43–59. [DOI] [PubMed] [Google Scholar]
- 56. Wahba L., Amon J.D., Koshland D., Vuica-Ross M.. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell. 2011; 44:978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dhoondia Z., Tarockoff R., Alhusini N., Medler S., Agarwal N., Ansari A.. Analysis of termination of transcription using BrUTP-strand-specific transcription run-on (TRO) approach. J. Vis. Exp. 2017; 121:55446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Roberts T.C., Hart J.R., Kaikkonen M.U., Weinberg M.S., Vogt P.K., Morris K. V.. Quantification of nascent transcription by bromouridine immunocapture nuclear run-on RT-qPCR. Nat. Protoc. 2015; 8:1198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Downs J.A., Lowndes N.F., Jackson S.P.. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 2000; 408:1001–1004. [DOI] [PubMed] [Google Scholar]
- 60. Hohmann S. PDC6, a weakly expressed pyruvate decarboxylase gene from yeast, is activated when fused spontaneously under the control of the PDC1 promoter. Curr. Genet. 1991; 20:373–378. [DOI] [PubMed] [Google Scholar]
- 61. DeMarini D.J., Winey M., Ursic D., Webb F., Culbertson M.R.. SEN1, a positive effector of tRNA-splicing endonuclease in Saccharomyces cerevisiae. Mol. Cell. Biol. 1992; 12:2154–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nourani A., Robert F., Winston F.. Evidence that Spt2/Sin1, an HMG-like factor, plays roles in transcription elongation, chromatin structure, and genome stability in Saccharomyces cerevisiae. Mol. Cell. Biol. 2006; 26:1496–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Soares L.M., He P.C., Chun Y., Suh H., Kim T.S., Buratowski S.. Determinants of histone H3K4 methylation patterns. Mol. Cell. 2017; 68:773–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen S., Rufiange A., Huang H., Rajashankar K.R., Nourani A., Patel D.J.. Structure–function studies of histone H3/H4 tetramer maintenance during transcription by chaperone Spt2. Genes Dev. 2015; 29:1326–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Buratowski S. Progression through the RNA Polymerase II CTD Cycle. Mol. Cell. 2009; 36:541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jimeno-González S., Haaning L.L., Malagon F., Jensen T.H.. The Yeast 5′-3′ exonuclease Rat1p functions during transcription elongation by RNA polymerase II. Mol. Cell. 2010; 37:580–587. [DOI] [PubMed] [Google Scholar]
- 67. Felipe-Abrio I., Lafuente-Barquero J., García-Rubio M.L., Aguilera A.. RNA polymerase II contributes to preventing transcription-mediated replication fork stalls. EMBO J. 2015; 34:236–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nonet M., Scafe C., Sexton J., Young R.. Eucaryotic RNA polymerase conditional mutant that rapidly ceases mRNA synthesis. Mol. Cell. Biol. 1987; 7:1602–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Han Z., Libri D., Porrua O.. Biochemical characterization of the helicase Sen1 provides new insights into the mechanisms of non-coding transcription termination. Nucleic Acids Res. 2017; 45:1355–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Martin-Tumasz S., Brow D.A.. Saccharomyces cerevisiae Sen1 helicase domain exhibits 5′- to 3′-helicase activity with a preference for translocation on DNA rather than RNA. J. Biol. Chem. 2015; 290:22880–22889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim H.-D., Choe J., Seo Y.-S.. The sen1 + gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry. 1999; 38:14697–14710. [DOI] [PubMed] [Google Scholar]
- 72. Lindstrom D.L., Squazzo S.L., Muster N., Burckin T.A., Wachter K.C., Emigh C.A., McCleery J.A., Yates J.R., Hartzog G.A.. Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol. Cell. Biol. 2003; 23:1368–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Appanah R., Lones E.C., Aiello U., Libri D., De Piccoli G.. Sen1 is recruited to replication forks via Ctf4 and Mrc1 and promotes genome stability. Cell Rep. 2020; 30:2094–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Noe Gonzalez M., Blears D., Svejstrup J.Q.. Causes and consequences of RNA polymerase II stalling during transcript elongation. Nat. Rev. Mol. Cell Biol. 2021; 22:3–21. [DOI] [PubMed] [Google Scholar]
- 75. Chappidi N., Nascakova Z., Boleslavska B., Zellweger R., Isik E., Andrs M., Menon S., Dobrovolna J., Balbo Pogliano C., Matos J.et al.. Fork cleavage-religation cycle and active transcription mediate replication restart after fork stalling at co-transcriptional R-loops. Mol. Cell. 2020; 77:528–541. [DOI] [PubMed] [Google Scholar]
- 76. De Septenville A.L., Duigou S., Boubakri H., Michel B.. Replication fork reversal after replication–transcription collision. PLoS Genet. 2012; 8:e1002622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Vos S.M., Farnung L., Boehning M., Wigge C., Linden A., Urlaub H., Cramer P.. Structure of activated transcription complex Pol II–DSIF–PAF–SPT6. Nature. 2018; 560:607–612. [DOI] [PubMed] [Google Scholar]
- 78. Mason P.B., Struhl K.. Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol. Cell. 2005; 17:831–840. [DOI] [PubMed] [Google Scholar]
- 79. Poli J., Gerhold C.-B., Tosi A., Hustedt N., Seeber A., Sack R., Herzog F., Pasero P., Shimada K., Hopfner K.-P.et al.. Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Genes Dev. 2016; 30:337–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hazelbaker D.Z., Marquardt S., Wlotzka W., Buratowski S.. Kinetic competition between RNA polymerase II and Sen1-dependent transcription termination. Mol. Cell. 2013; 49:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Porrua O., Libri D.. Transcription termination and the control of the transcriptome: why, where and how to stop. Nat. Rev. Mol. Cell Biol. 2015; 16:190–202. [DOI] [PubMed] [Google Scholar]
- 82. Chan Y.A., Aristizabal M.J., Lu P.Y.T., Luo Z., Hamza A., Kobor M.S., Stirling P.C., Hieter P.. Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-Chip. PLoS Genet. 2014; 10:e1004288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Costantino L., Koshland D.. Genome-wide map of R-loop-induced damage reveals how a subset of R-loops contributes to genomic instability. Mol. Cell. 2018; 71:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hawkins M., Dimude J.U., Howard J.A.L., Smith A.J., Dillingham M.S., Savery N.J., Rudolph C.J., McGlynn P.. Direct removal of RNA polymerase barriers to replication by accessory replicative helicases. Nucleic Acids Res. 2019; 47:5100–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Brüning J.-G., Marians K.J.. Replisome bypass of transcription complexes and R-loops. Nucleic Acids Res. 2020; 48:10353–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kouzminova E.A., Kuzminov A.. Ultraviolet-induced RNA:DNA hybrids interfere with chromosomal DNA synthesis. Nucleic Acids Res. 2021; 49:3888–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Saponaro M., Kantidakis T., Mitter R., Kelly G.P., Heron M., Williams H., Söding J., Stewart A., Svejstrup J.Q.. RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell. 2014; 157:1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lockhart A., Pires V.B., Bento F., Kellner V., Luke-Glaser S., Yakoub G., Ulrich H.D., Luke B.. RNase H1 and H2 are differentially regulated to process RNA-DNA hybrids. Cell Rep. 2019; 29:2890–2900. [DOI] [PubMed] [Google Scholar]
- 89. Zimmer A.D., Koshland D.. Differential roles of the RNases H in preventing chromosome instability. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:12220–12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Saveanu C., Jacquier A.. How cells kill a ‘killer’ messenger. Elife. 2016; 5:e16076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Schleich S., Strassburger K., Janiesch P.C., Koledachkina T., Miller K.K., Haneke K., Cheng Y.-S., Küchler K., Stoecklin G., Duncan K.E.et al.. DENR–MCT-1 promotes translation re-initiation downstream of uORFs to control tissue growth. Nature. 2014; 512:208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Schlissel G., Krzyzanowski M.K., Caudron F., Barral Y., Rine J.. Aggregation of the Whi3 protein, not loss of heterochromatin, causes sterility in old yeast cells. Science. 2017; 355:1184–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Nickless A., Cheruiyot A., Flanagan K.C., Piwnica-Worms D., Stewart S.A., You Z.. p38 MAPK inhibits nonsense-mediated RNA decay in response to persistent DNA damage in noncycling cells. J. Biol. Chem. 2017; 292:15266–15276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Janke R., Kong J., Braberg H., Cantin G., Yates J.R., Krogan N.J., Heyer W.-D.. Nonsense-mediated decay regulates key components of homologous recombination. Nucleic Acids Res. 2016; 44:5218–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kramer N.J., Carlomagno Y., Zhang Y.-J., Almeida S., Cook C.N., Gendron T.F., Prudencio M., Van Blitterswijk M., Belzil V., Couthouis J.et al.. Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science. 2016; 353:708–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Liu C.-R., Chang C.-R., Chern Y., Wang T.-H., Hsieh W.-C., Shen W.-C., Chang C.-Y., Chu I.-C., Deng N., Cohen S.N.et al.. Spt4 is selectively required for transcription of extended trinucleotide repeats. Cell. 2012; 148:690–701. [DOI] [PubMed] [Google Scholar]
- 97. Goodman L.D., Prudencio M., Kramer N.J., Martinez-Ramirez L.F., Srinivasan A.R., Lan M., Parisi M.J., Zhu Y., Chew J., Cook C.N.et al.. Toxic expanded GGGGCC repeat transcription is mediated by the PAF1 complex in C9orf72-associated FTD. Nat. Neurosci. 2019; 22:863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Freudenreich C.H. R-loops: targets for nuclease cleavage and repeat instability. Curr. Genet. 2018; 64:789–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.