


Abstract
G-quadruplex is an essential element in gene transcription that serves as a promising drug target. Guanine-vacancy-bearing G-quadruplex (GVBQ) is a newly identified G-quadruplex that has distinct structural features from the canonical G-quadruplex. Potential GVBQ-forming motifs are widely distributed in gene promoter regions. However, whether GVBQ can form in genomic DNA and be an effective target for manipulating gene expression is unknown. Using photo-crosslinking, dimethyl sulfate footprinting, exonuclease digestion and in vitro transcription, we demonstrated the formation of a GVBQ in the G-rich nuclease hypersensitivity element within the human PDGFR-β gene promoter region in both single-stranded and double-stranded DNA. The formation of GVBQ in dsDNA could be induced by negative supercoiling created by downstream transcription. We also found that the PDGFR-β GVBQ was specifically recognized and stabilized by a new synthetic porphyrin guanine conjugate (mPG). Targeting the PDGFR-β GVBQ in human cancer cells using the mPG could specifically alter PDGFR-β gene expression. Our work illustrates that targeting GVBQ with mPG in human cells can regulate the expression level of a specific gene, thus indicating a novel strategy for drug development.
INTRODUCTION
G-quadruplex is a four-stranded structure formed by guanine-rich (G-rich) nucleic acids. G-quadruplexes are widely distributed in genomic DNA, and enriched in regulatory regions such as gene promoters (1,2), thus acting as regulators for transcription (3,4), replication (5,6) and recombination (7,8). Targeting G-quadruplex is an emerging therapeutic approach for treating various diseases, including cancer (9–12).
The canonical G-quadruplex usually has two or more complete G-quartet planes connected by four continuous G-strands. Apart from these prefect G-quadruplexes, there exist many G-quadruplexes containing an incomplete G-quartet plane or partially folded structures, such as the guanine-vacancy-bearing G-quadruplex (GVBQ) (13,14) and G-triplex (15,16). Among them, GVBQ is a newly identified G-quadruplex that has an incomplete G-quartet plane that creates a dock for adopting a guanine from guanine derivatives (13,14,17). Sequences having the potential to form GVBQ exceed 0.46 million in human genomic DNA (2) and, notably, a number of them are enriched at the promoter region of human genes (13). The interaction between guanine derivatives and GVBQ implies that there is a connection between guanine metabolites and transcription regulation (13,18,19). However, due to a lack of detection tools and targeting drugs, the cellular function of GVBQ has not been explored and reported.
Given that filling-in by a guanine group is a unique feature of GVBQ missing in the canonical G-quadruplexes (13), a guanine-containing compounds could potentially distinguish GVBQs from canonical G- quadruplexes and specifically targeting GVBQ. In fact, we successfully developed GRPC, a compound consisting of a conjugation of a guanine and a 23-aa G4-binding domain from the RHAU protein (20), and demonstrated its ability to recognize and stabilize GVBQs with high specificity and high affinity (21). GRPC, although serving as a powerful tool for in vitro identification of GVBQ, still needs further modifications to solve the problems of intracellular delivery and improve metabolic properties (22,23) for cellular applications.
Recently, a GVBQ formed by a 19-mer G-rich DNA from the nuclease hypersensitivity element (NHE) region of the PDGFR-β gene promoter was determined using nuclear magnetic resonance (NMR) (19). PDGFR-β is a cell-surface-receptor tyrosine kinase and a therapeutic target for a wide spectrum of diseases (24–26). Previous studies indicated that targeting the G-quadruplexes of the NHE region in the PDGFR-β promoter could inhibit gene transcription (27,28), which encouraged the development of a new treatment strategy for diseases that are caused by overexpression of PDGFR-β. The newly discovered GVBQ in the PDGFR-β promoter indicated a promising new target for drugs, but a few key questions needed to be carefully addressed: first, the naturally occurring G-rich DNA from the PDGFR-β promoter contains seven continuous runs of G-tracts, from which multiple types of G-quadruplexes could form by combining four runs of G-tracts that could not coexist with each other (28,29), which may hinder the formation of a GVBQ. Second, GVBQ is generally less stable than canonical G-quadruplexes (13,14), so it is unclear whether the GVBQ of PDGFR-β is sufficiently stable to compete with other G-quadruplexes and resist double-stranded DNA pairing. Third, there are no reports of compounds that bind to GVBQ in cells; therefore, whether targeting GVBQ in the PDGFR-β gene promoter is an effective way to interfere PDGFR-β gene expression remains uncertain.
In this work, we explored GVBQ formation in the naturally occurring G-rich DNA from the PDGFR-β gene promoter and investigated the possibility of GVBQ as a drug target. Using photo-crosslinking, dimethyl sulfate (DMS) footprinting, and exonuclease digestion, we first verified the formation of a GVBQ in the wild-type single-stranded (ss) DNA including the core G-rich and additional flanking sequence from the NHE region of the PDGFR-β gene promoter. We also detected the formation of PDGFR-β GVBQ in transcribed plasmid DNA, which was induced by the negative supercoiling that was created by downstream transcription, as occured in other canonical G-quadruplexes, as reported previously (30). The formation of GVBQ was further verified by its specific interactions with GRPC and a new synthetic porphyrin guanine conjugate (mPG). mPG retains the advantages of GRPC to specifically recognize and stabilize GVBQ, and also has the enhanced ability of cellular delivery. When cells were treated with mPG, the RNA and protein level of PDGFR-β were significantly downregulated; in contrast, the expression of eight other genes with canonical G-quadruplexes in their promoters remained unchanged. Overall, our study paved a novel way to regulate gene expression by targeting GVBQ.
MATERIALS AND METHODS
Oligonucleotides, chemical ligands, peptide, DNA
All oligonucleotides were synthesized by Sangon Biotechnology (Shanghai, China) (Supplementary Tables S1 and S2). Sulfosuccinimidyl-2-[6-(biotinamido)-2-(p-azidobenzamido) hexanoamido]ethyl-1,3′-dithiopropionate (SBED)-guanosine monophosphate (GMP) was purchased from Takara Biomedical Technology (Beijing) as described (13). Guanosine monophosphate (GMP), mesoporphyrin IX dihydrochloride (MPIX) and meso-5,10,15,20-Tetrakis-(N-methyl-4-pyridyl)porphine (TMPyP4) was purchased from Sigma, GRPC was synthesized as described (21). 3,6-Bis (1-methyl-4-vinylpyridinium) carbazole diiodide (BMVC) was purchased from MedChemExpress. MPIX-(PEG)2-G (PNA)-d-Lys-d-Lys (abbreviated as mPG), (PEG)2-G (PNA)-d-Lys-d-Lys (abbreviated as OGkk), and mPG-(l-GRKKRRQRRR) (abbreviated as mPG-TAT) were purchased from Tanzhen Biotechnologies (Nanchang, China).
Preparation of plasmids
Plasmids were prepared as previously described (30), except that the PDGFR-β WT and PDGFR-β M3A sequences were inserted on the template strand between a T7 promoter and a T3 promoter. Plasmid DNA was extracted using the TIAN prep Midi Plasmid Kit (Tiangen, China).
DMS footprinting
An oligonucleotide (0.1 μM) in 50 mM LiCl or KCl was heated at 95°C for 5 min and then cooled down to room temperature at a rate of 0.1°C/s. Samples were then incubated with GMP, GRPC and mPG, respectively, at the indicated concentration for 30 min at 37°C. The mixture was then treated with 5% dimethyl sulfate (DMS) for 4 min on ice and subjected to DMS footprinting as previously described (13).
Photo-crosslinking
Photo-crosslinking was carried out as previously described (13).
Exonuclease digestion assay
Oligonucleotides (WT, M2A and M3A, Supplementary Table S1) labeled as 5′FAM or 3′FAM were dissolved at 0.2 μM in a buffer containing 40 mM Tris–HCl (pH 8.0), 50 mM KCl or 50 mM LiCl, heated at 95°C for 5 min, and cooled down to room temperature, then diluted to a final concentration of 20 nM in a buffer containing 40 mM Tris–HCl (pH 8.0), 50 mM KCl, or 50 mM LiCl, 8 mM MgCl2 and 2mM dithiothreitol (DTT), incubated at 37°C for 30 min in the presence of various concentrations of GRPC, mPG, or other compounds. 5′-FAM-labeled oligonucleotides were then digested with 0.25 U/μl T4 DNA Polymerase (Thermo Scientific, USA) at 37°C for 20 min, and 3′FAM labeled oligonucleotides were digested with 0.15 U/μl RecJ (NEB) at 37°C for 20 min. Reactions were stopped by adding a final concentration of 50 mM ethylenediaminetetraacetic acid and 0.2 M 2-mercaptoethanol. Samples were heated at 95°C for 5 min in 80% formamide, resolved on a 20% denaturing polyacrylamide gel and scanned on a ChemiDoc MP (Bio-Rad).
In vitro transcription and analysis of RNA transcripts
Transcription was carried out as described previously (30). When a T7 transcription was followed by a T3 transcription, the T7 transcription was stopped by adding a final concentration of 2 μM T7 inhibitor (5′-GAAATTAATACGACTCACTATA-3′) (31). Then, 0.02 mM Fluorescein-12-UTP (Roche) and 2 U/μl T3 RNA polymerase (Thermo Scientific) were added. The samples were incubated at 37°C for 60 min, followed by a treatment with 0.04 U/μl DNase I (Thermo Scientific) at 37°C for 15 min and an extraction with phenol/chloroform. RNA products were denatured in 80% formamide and resolved on an 8% denaturing polyacrylamide gel.
Fluorescence titration
5′FAM-labeled DNA (0.1 μM) was dissolved in a TE buffer (pH 7.4) containing 50 mM KCl, denatured at 95°C for 5 min and cooled down to 25°C at a rate of 0.1°C/s. Tween-20 and bovine serum albumin were added to final 0.05% (w/v) and 0.2 mg/ml each. Then, ligands of different concentrations were added to the samples and incubated at room temperature for 2 h. Samples were prepared in glass capillaries (NanoTemper), and scanned in the Monolith NT.115 to measure the fluorescence. The initial fluorescence analysis mode was selected on MO.Affinity Analysis V2.3 software. Binding of ligands to a specific DNA site is described by the equilibrium dissociation constant (Kd). Data was fitted using the single-site binding model and accounting for ligands depletion. In an equilibrium situation, the equation for fluorescence calculation can be written as:
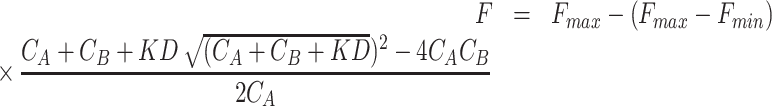 |
where F represents baseline-corrected fluorescence; 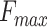 represents the maximal value of F;
represents the maximal value of F; 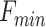 represents the minimum value of F; CA, CB (nM) represent the initial concentration of DNA and ligand, respectively.
represents the minimum value of F; CA, CB (nM) represent the initial concentration of DNA and ligand, respectively.
Fluorescence resonance energy transfer (FRET) melting
Oligonucleotides (Supplementary Table S1) labeled at the ends with a fluorescent donor FAM and an accepter TAMRA were dissolved at 0.1 μM in 10 mM lithium cacodylate buffer (pH 7.4) containing 50 mM KCl, which was denatured at 95°C for 5 min, and cooled down to 25°C at a rate of 0.1°C/s. Then mPG and other compounds were added at the indicated concentration and incubated at 37°C for 1 h. After initial equilibration at 25°C for 10 min, FRET melting was carried out as previously described (21) by monitoring the fluorescence of the FAM group on the QuantStudio 7 Flex (Thermo Scientific).
Cell culture
Cells were cultured at 37°C under 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 100 U/ml penicillin and streptomycin. U2OS cells were seeded on a six-well plate (2 × 105 cells/well) and cultured for 12 h. After incubation with mPG at final concentrations of 0, 2 and 10 μM for 48 h, cells were harvested from each well of culture plates.
RNA isolation and qRT-PCR
Total RNA was extracted from cells using an RNA extraction kit (Tiangen, China) according to the manufacturer's instructions. Then, 1.0 μg of total RNA was reverse-transcribed to cDNA using TransScript First-Strand cDNA Synthesis SuperMix (Transgen, China). Real-time PCR was performed using 2× RealStar Green Power Mixture (GenStar, China). GAPDH was used as the internal control. Primer information can be found in Supplementary Table S2. The threshold cycle (Ct) value was calculated using the QuantStudio™ Real-Time PCR Software (Applied Biosystems, USA).
Western blot
Cells were directly lysed in 2× sodium dodecyl sulfate (SDS) loading buffer and boiled for 10 min. Proteins were separated by SDS-PAGE electrophoresis and transferred to polyvinylidene fluoride membrane. The membrane was blocked by 5% fat-free milk in TBST buffer (20 mM Tris–HCl, PH 7.6, 150 mM NaCl, 0.05% (V/V) Tween 20) for 1 h at room temperature (RT). After washing for three times with TBST, the membrane was incubated with the indicated antibodies (Anti-PDGFR-β Antibody, 1:2000, (ab32570, Abcam); Alpha Tubulin Antibody, 1:5000, (66031-1-Ig, Proteintech)) for 1 h at RT. After washing three times with TBST, the membrane was incubated with 1:5000 diluted Anti-Rabbit IgG (H + L) Antibody, HRP Conjugate (5220-0336, KPL) or Anti-Mouse IgG (H + L) Antibody, HRP Conjugate (5220-0341, KPL) for 1 h at RT. After washing three more times, protein was detected using WesternBright ECL Kit (K-12045, Advansta) according to the manufacturer's instruction.
RESULTS
Detection of GVBQ in the G-rich ssDNA from the NHE region of PDGFR-β promoter
The G-rich strand of the NHE region in the PDGFR-β promoter contains seven runs of G-tracts, which have the potential to form multiple types of G-quadruplexes by using different combinations of G-tracts. Recently, a guanine-vacancy-bearing G-quadruplex (GVBQ) from a short G-rich DNA in the PDGFR-β promoter NHE region was resolved using NMR (19). However, in natural chromosomal DNA, the formation of a GVBQ in DNA can be affected by the flanking sequences, its complementary strand, and the competition of other types of G-quadruplexes. It was not clear whether the GVBQ could form in this situation.
To investigate whether a GVBQ could form in natural G-rich DNA from the PDGFR-β promoter, we first synthesized a 5′-FAM-labeled ssDNA (WT) containing the core seven runs of G-tracts with a flanking 10 nts at the 3′-end and a flanking 15 nts at the 5′-end. Using DMS footprinting (Figure 1A), we discovered that the major G-quadruplexes formed in the K+ solution containing G-tracts 2 to 6 (Figure 1B, G2–G6), which was consistent with a previous report (27). GVBQ was detected using Sulfosuccinimidyl-2-[6-(biotinamido)-2-(p-azidobenzamido) hexanoamido]-ethyl-1,3′-dithiopropionate (SBED)-guanosine monophosphate (GMP). SBED-GMP contains a guanine base that can fill in the G-vacancy of a GVBQ, and a phenyl azide group that can react with the adjacent adenine, guanine, and cytosine under UV light, thus forming a crosslinked product (13). As shown in Figure 1C, crosslinking occurred in lane 3, which led to an extra band migrating behind the original DNA, which indicated the formation of a GVBQ in the WT DNA.
Figure 1.
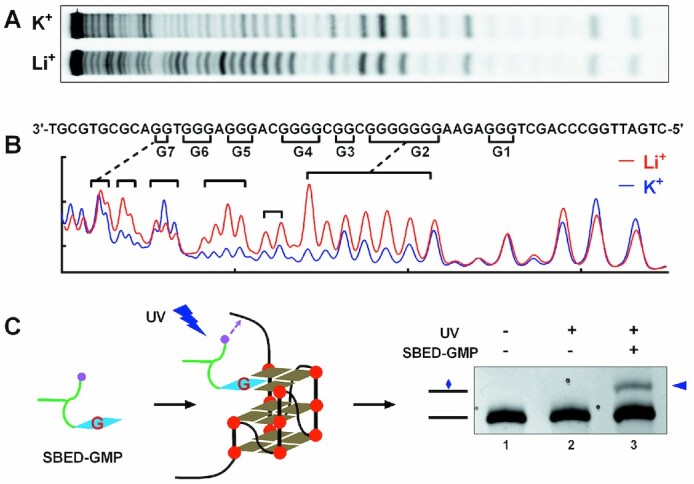
Detection of G-quadruplexes formed in the human PDGFR-β core promoter using (A and B) DMS footprinting and (C) UV crosslinking. (A) DNA cleavage fragments of PDGFR-β core promoter G-rich region (WT) resolved by denaturing gel electrophoresis. (B) Digitization of the gel in (A). (C) UV crosslinking between SBED-GMP and WT DNA detected by denaturing gel electrophoresis.
DMS footprinting detects the guanine bases involved in the formation of G-quadruplex. However, G-tracts protected from DMS in the PDGFR-β promoter G-rich region shown more than one type of G-quadruplex. To further identify the formation and motif of the GVBQ, we carried out a bi-orientational exonuclease assay with the WT and its mutant ssDNA (Figure 2A).
Figure 2.

Detection of GVBQ in the wild G-rich ssDNA from the PDGFR-β promoter NHE region by exonuclease digestion. (A) G4-forming sequence and its modifications from human PDGFR-β gene promoter. The G-tracts of wild DNA sequences (WT) are highlighted in red and mutations are highlighted in cyan. (B) Formation of G-quadruplexes protect the DNA from being hydrolyzed from the 3′ end by T4 DNA polymerase. (C) Formation of G-quadruplexes protected the DNA from being hydrolyzed from the 5′ end by RecJ exonuclease.
The 3′-end of the G-quadruplexes were first determined by the 3′ exonuclease activity of T4 DNA polymerase, which hydrolyzed nucleotides from the 3′-end until a G-quadruplex was met (32). As shown in Figure 2B (left, lane 2 versus lane 1), we observed several cleavage bands that represented at least three types of G-quadruplexes in WT DNA. To verify whether a cleavage band was caused by GVBQ, GRPC (guanine-RHAU23 peptide conjugate), a specific stabilizer of GVBQ, was added before exonuclease digestion. Cleavage in the WT ssDNA revealed that the band between G-tracts 4 and 5 was selectively enhanced by GRPC (Figure 2B, left, blue arrow), which indicated that a GVBQ formed at the 5′-end of G-tract 5. Cleavage bands before G-tract 7 were related to other types of G-quadruplexes such as the end-insertion G-quadruplexes reported previously (29), which were not sensitive to GRPC (Figure 2B, purple arrow). Hydrolysis using T4 DNA polymerase was further performed on the M2A DNA, which had two G to A mutations in G-tracts 5 and 6 compared to the WT. The result again supported the formation of a GVBQ at the 5′-end of G-tract 5 while other types of G-quadruplexes did not appear (Figure 2B, left versus right, lane 3).
The 5′-end of the GVBQ was determined by hydrolysis with the RecJ exonuclease that digested ssDNA in the 5′ to 3′ direction. GRPC was also added to highlight the locus of GVBQ. In the WT ssDNA, the formation of a G-quadruplex stopped exonuclease before G-tract 2 (Figure 2C, left), whether GRPC was added or not. This was consistent with the results of DMS footprinting shown in Figure 1A, in which G-tract 1 did not involve the formation of G-quadruplexes. We then conducted 5′hydrolysis on the M3A DNA, which had additional G to A mutations in G-tract 1 compared to that of M2A. A clear band (Figure 2C, right, lane 3) closely related to GRPC revealed that the core sequence of the PDGFR-β GVBQ was at 3′-end of G-tract 1. From the locus of the GVBQ-related cleavage band shown in Figure 2B and C, we determined that the forming sequence of the GVBQ in ssDNA was G-tracts 2, 3 and 4 (M2–3–4).
Detection of GVBQ in G-rich Double-Stranded DNA (dsDNA) from the NHE Region of the PDGFR-β promoter
The aforementioned results demonstrated that a GVBQ could form in the G-rich ssDNA from the PDGFR-β promoter. However, on chromatin DNA, G-rich sequences in the PDGFR-β promoter were paired with complementary C-rich sequences. Whether and how PDGFR-β GVBQ can form in the dsDNA was unknown. In a previous study, we found that transcription-induced negative supercoiling could induce the formation of a G-quadruplex in dsDNA at the upstream region of the transcription start site (30). To verify whether the formation of PDGFR-β GVBQ could be driven by a downstream transcription in dsDNA, we performed an in vitro transcription assay.
We first made a plasmid containing the G-core sequence of PDGFR-β NHE region. A T7 and a T3 promoters were respectively placed at the downstream and upstream regions of the G-rich sequence as previously described (30). Then we carried out the first transcription using T7 RNA polymerase. The moving T7 RNA polymerase generated dynamic negative supercoiling that transmitted upward and triggered the formation of G-quadruplexes in the G-core region. Finally, we terminated transcription of T7 RNA polymerase using a dsDNA inhibitor and started the second transcription using T3 RNA polymerase in the presence of fluorescent UTP. The formation of G-quadruplex on the template strand blocked the T3 RNA polymerase that was initiated from an upstream T3 promoter, thereby generating prematurely terminated (PT) transcripts (Figure 3A). The PT bands corresponding to marker M1–M3 (Figure 3B) appearing in K+ not in Li+ demonstrated the formation of G-quadruplexes (Figure 3C, lane 4 versus lane 3). The PT bands at M1 and M2 indicated that G-quadruplexes formed at the 3′-end region of the G-rich strand which was consistent with the exonuclease results on WT DNA in Figure 2B.
Figure 3.
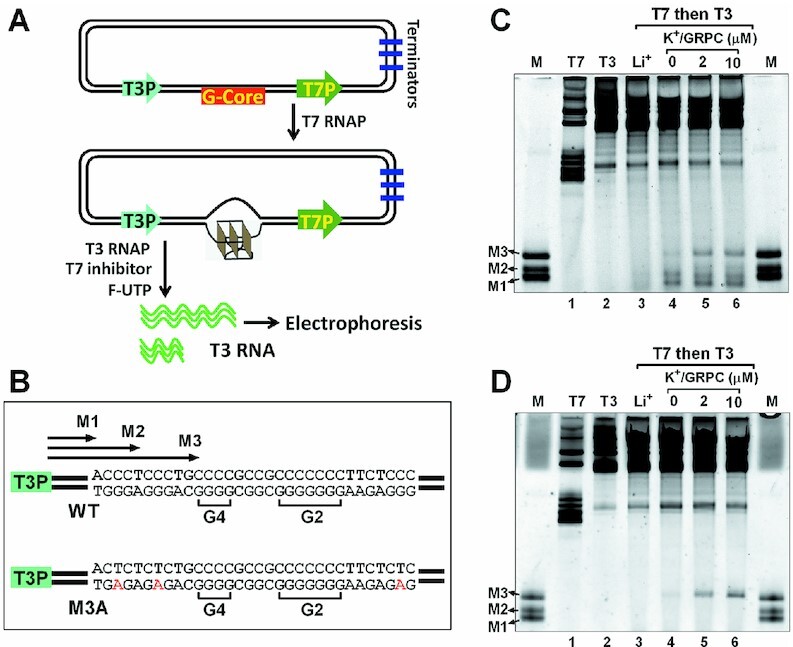
Detection of GVBQ in the wild G-rich dsDNA from the PDGFR-β promoter NHE region by RNA polymerase arrest. (A) Plasmid contained the PDGFR-β gene promoter G-core sequence, a T7 and a T3 promoter. The G-core sequence was placed at the upstream of T7 promoter and downstream of T3 promoter. The plasmid was first transcribed with T7 RNA polymerase to induce the formation of G-quadruplexes, and then T3 RNA polymerase was added together with a fluorescein-UTP and a T7 inhibitor to produce fluorescent T3 RNA transcripts. The T7 inhibitor was used to prevent the T7 RNA polymerase from further transcription. (B) Sequences of WT and mutant M3A G-core. (C) Detection of G-quadruplexes in the WT plasmid by the premature termination of T3 RNA polymerase. Marker (M) shows termination sites labeled in B. (D) Detection of a G-quadruplex in the mutant M3A plasmid by the premature termination of T3 RNA polymerase.
A weak PT band at marker M3 (Figure 3C, lane 4), which was significantly intensified by the addition of GRPC during transcription (Figure 3C, lanes 5–6), represented a formation of GVBQ in dsDNA. This was further verified by transcription, using a mutant plasmid to replace the wild G-rich sequence with M3A, which kept the G-rich sequences of G-tracts 2–4 and mutated other G-tracts. A single PT band corresponding to marker M3 again proved the formation of the GVBQ and stabilization by GRPC (Figure 3D, lanes 4–6).
Determination of the core forming sequence and structure of the GVBQ from the PDGFR-β promoter
Combined the results of exonuclease digestion and RNA polymerase arrest, we finally determined the core forming sequence of the GVBQ as G-tract 2, 3 and 4 (M 2-3-4). The structure of the GVBQ was further verified by DMS footprinting using the G-core sequence of G-tract 2–4 (M2–3–4-T), which was flanked with three Ts and labeled with a FAM group at the 5′terminal. As reported in the previous study, the guanine that was not involved in Hoogsteen hydrogen bond with its N7 site in the incomplete G-tetrad of GVBQ was poorly protected (Figure 4A and B, green and blue arrow) (13). The two cleavage bands indicated by the blue and green arrow represent two equilibrating GVBQs (Figure 4C), which were obviously reduced when a guanine from GMP or GRPC was filled in the G-vacancy (Figure 4B). Coincidentally, This same G-rich DNA of M2–3–4 had been reported to form a GVBQ by NMR spectroscopy (19).
Figure 4.
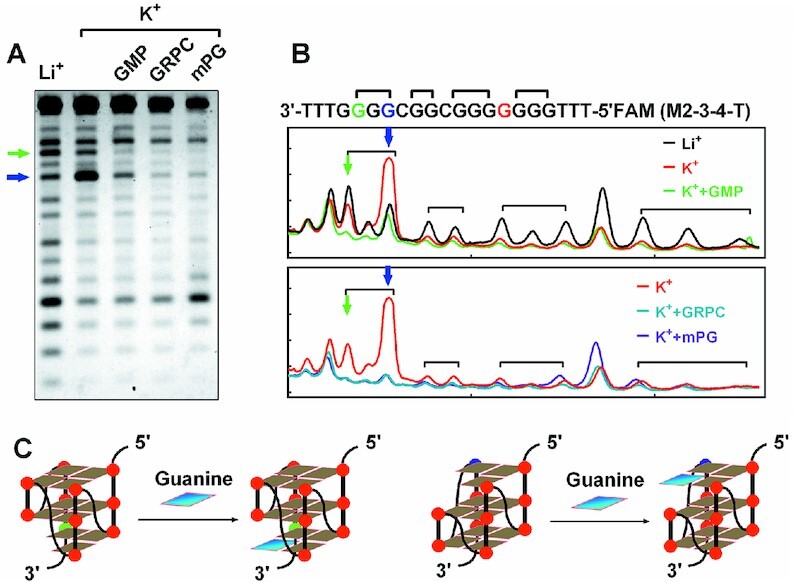
Verification of the formation of PDGFR-β GVBQ by DMS footprinting. (A) DMS footprinting of M2–3–4-T DNA in the presence of GMP, GRPC and mPG. (B) Digitization of the gel in A. (C) Possible structure of GVBQ and its completion by the G fill-in.
Targeting the PDGFR-β GVBQ with compounds
Specific ligands are key tools for studying the formation and the function of G-quadruplexes (10). Previously, we demonstrated that a guanine-RHAU23 peptide conjugate (GRPC) could selectively target and stabilize GVBQ (21). However, intracellular delivery and proteolysis limited the application of GRPC in living cells. Using the similar strategy for GRPC, we replaced the RHAU23 peptide with a porphyrin group and constructed a porphyrin-guanine conjugate (Supplementary Figures S1–S3). Porphyrins are canonical G-quadruplex-specific compounds that mostly interact with G-quadruplex at the terminal G-quartet layer (33). The porphyrin-guanine conjugate (MPIX-(PEG)2-G-d-Lys-d-Lys (mPG)) contains an MPIX that was a derivative of N-methyl mesoporphyrin IX (NMM) (34), a Guanine peptide nucleic acid (G) connected with MPIX using a (PEG)2 linker, and two d-lysine at the N-terminal to enhance solubility. mPG was expected to retain the properties of GRPC to specifically recognize and stabilize GVBQ but also overcome the shortcomings of its delivery into cells.
Interaction of mPG and PDGFR-β GVBQ was first determined by DMS footprinting. As shown in Figure 4A and B, in the presence of mPG, the two unprotected bands of GVBQ mostly disappeared, which indicated that the guanine base from mPG filled in the G-vacancy of PDGFR-β GVBQ.
Interaction of mPG and PDGFR-β GVBQ was further determined by fluorescence titration. We found that the fluorescent signal of the FAM group labeled at 5′-end of PDGFR-β GVBQ (M2–3–4-T) was greatly reduced in the presence of mPG. The cause of quenching of fluorescence is attributed to the binding of the porphyrin group to DNA, (35) which could be used to quantify the binding affinity between mPG and G-quadruplex. As shown in Figure 5A, the Kd of mPG to PDGFR-β GVBQ was calculated to be 10.2 nM, which was much lower than those of the other non-GVBQ G-quadruplexes (Figure 5B). The binding of mPG to PDGFR-β GVBQ was also stronger than to the other three GVBQs (Figure 5C), which indicated that it had a recognition preference for GVBQs. mPG did not bind non-G4 structures such as hairpin DNA, ssDNA, and C-rich ssDNA at a concentration below 1 μM (Figure 5D). The conjugation of MPIX and the guanine group conferred mPG the ability to recognize PDGFR-β GVBQ with high specificity and affinity that could not be realized by MPIX or (PEG)2-G (PNA)-d-Lys-d-Lys (OGkk) (Supplementary Figures S4 and S5) separately (Figure 5A).
Figure 5.
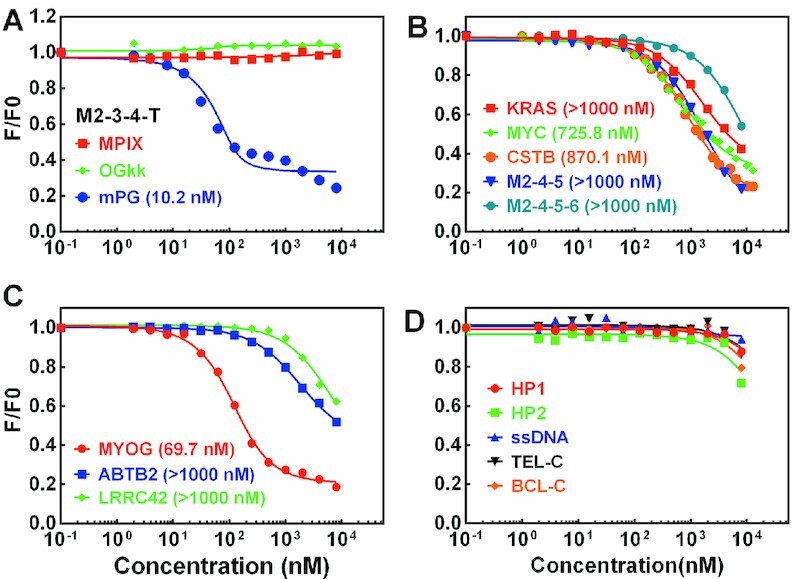
Detection of the binding ability between mPG and different DNA structures by fluorescence titration. The equilibrium dissociation constant (Kd) in parentheses was obtained by fitting the data using a single-site binding model and accounting for ligand depletion. (A) Comparison of the binding ability between PDGFR-β GVBQ (M2–3–4-T) with MPIX, OGkk and mPG. (B) Detection of the binding ability between mPG with G-quadruplexes of KRAS, MYC, CSTB, M2–4–5, and M2–4–5–6. (C) Detection of the binding ability between mPG with GVBQ of MYOG, ABTB2 and LRRC42. (D) Detection of the binding ability between mPG and non-G-quadruplex DNA.
Unlike mPG, GRPC does not quench the fluorescence of the FAM group in DNA, hence direct fluorescence titration cannot be used to determine the binding affinity. Thus, we compared the binding ability of GRPC and mPG to PDGFR-β GVBQ by using fluorescence titration in a competitive binding assay. As shown in Supplementary Figure S6A, GRPC competed with mPG and bound to PDGFR-β GVBQ at a much lower concentration than mPG. In contrast, mPG could hardly compete with GRPC at a lower concentration than GRPC (Supplementary Figure S6B). This indicates that the binding affinity of mPG to PDGFR-β GVBQ is lower than that of GRPC.
The interaction of mPG and PDGFR-β GVBQ could theoretically enhance the stability of the GVBQ. To confirm this assumption, we performed a thermal melting assay based on the FRET using PDGFR-β GVBQ (M2–3–4-F), which was labeled at the 3′-end with a FAM as a donor and at the 5′ -end with a TAMRA as an acceptor. The two functional units of mPG, MPIX and OGkk, were used as control ligands. PDGFR-β GVBQ was incubated with different concentrations of compounds, and the temperature required for the fluorescence to reach the middle value between the minimal and maximal fluorescence, which was denoted as T1/2, was used to judge the stability of the GVBQ. As shown in Figure 6A and B, mPG and GRPC increased the melting temperature of PDGFR-β GVBQ in a concentration-dependent manner. GRPC produced a greater ΔT1/2 than mPG (Figure 6C), which might be due to the stronger binding ability of GRPC to PDGFR-β GVBQ than mPG (Supplementary Figure S6). The T1/2 of PDGFR-β GVBQ increased 26.1°C in the presence of 10 μM mPG, which was much larger than in the presence of MPIX and OGkk separately (Figure 6C and Supplementary Figure S7A and B). mPG showed weaker ability to stabilize the GVBQ of MYOG or LRRC42 (Supplementary Figure S7C and D) than PDGFR-β GVBQ, which was consistent with the results in Figure 5C. Only at concentrations above 1 μM, mPG was able to show stabilization of the canonical G-quadruplex formed by M2–4–5 and M2–4–5–6 (Supplementary Figure S7E and F) derived from the G-rich sequence of the PDGFR-β gene promoter.
Figure 6.

(A, B) Stabilization of PDGFR-β GVBQ (M2–3–4-F) by mPG and GRPC. (C) Effects of ligands on the T1/2 of PDGFR-β GVBQ. T1/2 gives a temperature for the fluorescence to reach the mid-value between the minimum and maximum values.
To further explore whether mPG could specifically recognize and stabilize the GVBQ in ssDNA and dsDNA, we carried out exonuclease digestion and RNA polymerase arrest using WT and M3A DNA in the presence of mPG. As shown in Figure 7A, mPG obviously increased the protected band before GVBQ in a concentration-dependent manner. The results shown in Figure 7B showed that mPG and GRPC could effectively protect GVBQ from T4 DNA polymerase digestion, whereas TMPyP4 and BMVC, the canonical G-quadruplex binding ligands, showed little effect. We also compared the stability of PDGFR-β GVBQ in transcribed dsDNA by different ligands. The results shown in Figure 7C and D showed that mPG and GRPC could enhance the formation of GVBQ in dsDNA more effectively than the other ligands, which was in consonance with the results shown in Figure 7B.
Figure 7.
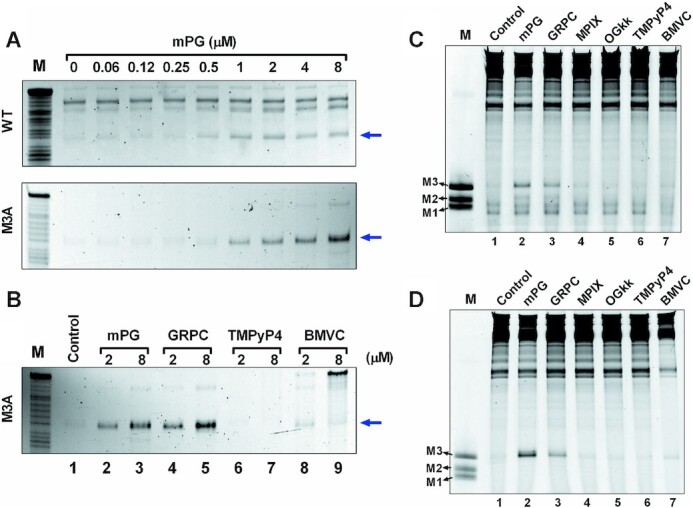
Effects of ligands on the stabilization of PDGFR-β GVBQ in the G-rich ssDNA and dsDNA. (A) Protection of WT and M3A ssDNA from being hydrolyzed by T4 DNA polymerase in the presence of mPG. The blue arrow indicates the cleavage band before GVBQ as shown in Figure 2B. (B) Compare the protection effects of mPG, GRPC, TMPyP4 and BMVC on M3A ssDNA from being hydrolyzed by T4 DNA polymerase. The blue arrow indicates the cleavage band before GVBQ as in Figure 2B. (C) Detecting the effects of mPG and other compounds (2 μM) on the stability of PDGFR-β GVBQ in transcribed WT plasmids by RNA polymerase arrest. (D) Detecting the effect of mPG and other compounds (2 μM) on the stability of PDGFR-β GVBQ in transcribed M3A plasmids by RNA polymerase arrest. Marker (M) shows termination sites as labeled in Figure 3.
Targeting PDGFR-β GVBQ in human cells for regulating gene expression
It was reported that targeting G-quadruplexes in the PDGFR-β promoter with compounds could regulate gene expression (27). To explore whether the stabilization of GVBQ could influence PDGFR-β gene expression, we treated U2OS cells with mPG and mPG-TAT. mPG-TAT is derived from mPG by connecting a classical cell-penetrating peptide (TAT: GRKKRRQRRR) at the C-terminal of mPG (Supplementary Figures S8 and S9) which will improve cellular delivery. mPG-TAT could also stabilize PDGFR-β GVBQ in transcribed plasmid as mPG did (Supplementary Figure S10). As shown in Figure 8A and B, the PDGFR-β RNA and protein levels significantly decreased in presence of 2 μM and 10 μM of each mPG and mPG-TAT. The effect of mPG and mPG-TAT was very similar, which indicated that mPG could permeate into the cells and modulate PDGFR-β gene expression efficiently. We also tested the effect of mPG and mPG-TAT on the growth of U2OS cells, and found that both mPG and mPG-TAT inhibited cell growth, and that the inhibition caused by mPG-TAT was more obvious (Supplementary Figure S11), thus indicating that mPG-TAT might have targets other than the PDGFR-β gene.
Figure 8.

Effects of mPG and mPG-TAT on the expression of PDGFR-β. (A) Detection of the mRNA level of PDGFR-β in the presence of mPG and mPG-TAT by RT-qPCR. (B) Detection of the protein level of PDGFR-β in presence of mPG and mPG-TAT by western blot. (C) Detection of the mRNA level of PDGFR-β in the presence of MPIX and TMPyP4 by RT-qPCR. (D) Detection of the mRNA level of MYC, Hif1a, VEGFA, BCL2, KRAS, BRCA1, MYB and ERBB2 genes in the presence of mPG by RT-qPCR.
In comparison with mPG, MPIX and TMPyP4, which could not alter the formation ability of PDGFR-β GVBQ (Figure 7C and D), had little effect on the expression of PDGFR-β at the same concentration of mPG (Figure 8C). We further tested the RNA level of other genes (MYC, Hif1a, VEGFA, BCL2, KRAS, BRCA1, MYB and ERBB2) that had been reported to be regulated by G-quadruplexes binding ligands (36–43). Interestingly, mPG did not influence the transcription level of these genes (Figure 8D), thus implying that its effect on PDGFR-β was specific.
DISCUSSION
Aberrant overexpression of PDGFR-β and consequent increased PDGFR-β signaling are causative factors for a variety of pathologies, including vascular disease, fibrotic disorders, and cancer (24–26). In this work, we validated the formation of a GVBQ in the natural G-rich sequence of the PDGFR-β gene promoter in both ssDNA and dsDNA. We also confirmed that the GVBQ could be specifically targeted by mPG and subsequently modulate the expression of the PDGFR-β gene (Figure 9).
Figure 9.
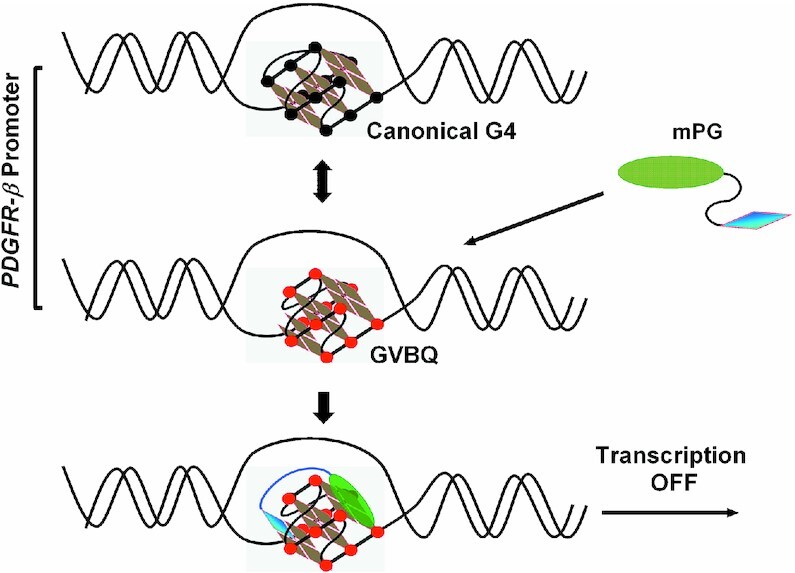
mPG selectively recognizes and stabilizes the GVBQ of the PDGFR-β gene promoter and subsequently modulates gene expression.
Previous studies reported at least four different G-quadruplexes with the potential to form from a wild G-rich sequence in the PDGFR-β promoter (27,44). These G-quadruplexes are mutually exclusive because they utilize overlapping G-runs. Due to its incomplete structure, the GVBQ was unstable and had difficulty competing with other types of G-quadruplexes (13,14). The reason for the formation of GVBQ might be the advantage of its folding speed, which requires further confirmation. Another possibility is that physiological guanine derivatives such as GTP could fill in and stabilize GVBQ, which supported its formation (13). This was verified by the fact that guanine-conjugated compounds (GRPC and mPG) could significantly improve the formation and the stability of GVBQ in ssDNA and transcribed dsDNA (Figures 6 and 7).
In a previous study, a G-quadruplex structure that formed at the 3′-end of the G-rich sequence in PDGFR-β promoter were found to be involved in transcription activation of PDGFR-β (27). Whether the formation of GVBQ can also directly affect transcriptional activity is currently unclear. However, when the GVBQ formed by the middle three G-tracts (M2–3–4, Figure 2A), the remaining G-tracts were unable to form another G-quadruplex at the 3′-end. Thus, we hypothesized that enforced stability of GVBQ by mPG in human cells will correspondingly reduce the forming probability of the 3′-end structures. This may be one of the mechanisms by which mPG causes a decrease of PDGFR-β gene expression.
The regulatory role of GVBQ in the PDGFR-β promoter indicates that GVBQ can be a new target for modulating gene expression. Compared with canonical G-quadruplexes, targeting GVBQ had several advantages. First, due to structural incompleteness, GVBQ was generally less stable than canonical G-quadruplexes (13,14). Compounds that bind GVBQ can improve its stability to a greater extent, thereby causing more significant changes in the state of DNA structures and its binding by proteins. Second, GVBQ, but not canonical standard G-quadruplexes, could be filled and stabilized by guanine derivatives (14,17). Thus, targeting GVBQ with guanine derivatives will leave the other types of G-quadruplexes unaffected (Figure 8D).
In our previous studies (13,21), the formation of most DNA GVBQs needed an additional molecular crowding reagent (PEG). For example, the GVBQ of LRRC42 and ABTB2 could hardly or partly form in the absence of PEG (13), Correspondingly, we found a weak binding between mPG with LRRC42 and ABTB2 (Figure 5C) and little stabilization of LRRC42 by mPG (Supplementary Figure S7D) in the absence of PEG. There was an exception, i.e., MYOG GVBQ, which could form in the absence and presence of PEG (13), Correspondingly, under our present experimental conditions, we detected the binding of mPG with MYOG (Kd = 69.7 nM, Figure 5C) and the stabilization of MYOG GVBQ by mPG (Supplementary Figure S7C). Compared with these GVBQs, the formation of PDGFR-β GVBQ is less dependent upon environmental conditions. Therefore, PDGFR-β GVBQ is a more robust target among GVBQs.
Drug development by targeting GVBQ is still in its preliminary stages. We previously reported that a guanine and RHAU23 peptide conjugate (GRPC) could specifically recognize GVBQs by G-filling in the G-vacancy and binding the G-quartet plane (21). This dual-specific targeting approach had also been reported in targeting G-quadruplexes adjacent to duplex DNA (45–47). Using the same strategy as GRPC, we designed a new compound (mPG) by connecting a guanine with a porphyrin. As expected, mPG retains the ability of GRPC to specifically recognize and stabilize the GVBQ of PDGFR-β, with a binding Kd of 10.2 nM. More importantly, mPG overcame the cellular delivery shortcomings of GRPC, and successfully traveled into cells to downregulate the expression of PDGFR-β. This is an original report demonstrating that the expression level of a specific gene can be regulated by targeting GVBQ with compounds in human cells. However, the binding ability of mPG to PDGFR-β GVBQ is lower than that of GRPC, which means that mPG has room for further improvement. For example, the porphyrin group of mPG can be replaced with another molecule with higher affinity and specificity to the terminal G-quartet plane or the groove of the G-quadruplex.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Zheng Tan, Danzhou Yang and Yong Zhao for their valuable suggestions.
Contributor Information
Juan-nan Chen, School of Pharmaceutical Sciences (Shenzhen), Sun Yat-Sen University, Guangzhou 510006, P.R. China.
Yi-de He, School of Pharmaceutical Sciences (Shenzhen), Sun Yat-Sen University, Guangzhou 510006, P.R. China; School of Life Sciences, Sun Yat-Sen University, Guangzhou 510275, P.R. China.
Hui-ting Liang, School of Pharmaceutical Sciences (Shenzhen), Sun Yat-Sen University, Guangzhou 510006, P.R. China.
Ting-ting Cai, School of Pharmaceutical Sciences (Shenzhen), Sun Yat-Sen University, Guangzhou 510006, P.R. China.
Qi Chen, School of Public Health (Shenzhen), Sun Yat-Sen University, Guangzhou 510006, P.R. China.
Ke-wei Zheng, School of Pharmaceutical Sciences (Shenzhen), Sun Yat-Sen University, Guangzhou 510006, P.R. China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [21708042]; Natural Science Foundation of Shenzhen [JCYJ20190807153618850]; Guangdong Basic and Applied Basic Research Foundation [2020A1515110812, 2021A1515010611]. Funding for open access charge: Natural Science Foundation of Shenzhen.
Conflict of interest statement. None declared.
REFERENCES
- 1. Hansel-Hertsch R., Beraldi D., Lensing S.V., Marsico G., Zyner K., Parry A., Di Antonio M., Pike J., Kimura H., Narita M.et al.. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016; 48:1267–1272. [DOI] [PubMed] [Google Scholar]
- 2. Zheng K.W., Zhang J.Y., He Y.D., Gong J.Y., Wen C.J., Chen J.N., Hao Y.H., Zhao Y., Tan Z.. Detection of genomic G-quadruplexes in living cells using a small artificial protein. Nucleic Acids Res. 2020; 48:11706–11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bochman M.L., Paeschke K., Zakian V.A.. DNA secondary structures: stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012; 13:770–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Varshney D., Spiegel J., Zyner K., Tannahill D., Balasubramanian S.. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020; 21:459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kanoh Y., Matsumoto S., Fukatsu R., Kakusho N., Kono N., Renard-Guillet C., Masuda K., Iida K., Nagasawa K., Shirahige K.et al.. Rif1 binds to G quadruplexes and suppresses replication over long distances. Nat. Struct. Mol. Biol. 2015; 22:889–897. [DOI] [PubMed] [Google Scholar]
- 6. Prorok P., Artufel M., Aze A., Coulombe P., Peiffer I., Lacroix L., Guedin A., Mergny J.L., Damaschke J., Schepers A.et al.. Involvement of G-quadruplex regions in mammalian replication origin activity. Nat. Commun. 2019; 10:3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Schie J.J.M., Faramarz A., Balk J.A., Stewart G.S., Cantelli E., Oostra A.B., Rooimans M.A., Parish J.L., de Almeida Esteves C., Dumic K.et al.. Warsaw Breakage Syndrome associated DDX11 helicase resolves G-quadruplex structures to support sister chromatid cohesion. Nat. Commun. 2020; 11:4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Wietmarschen N., Merzouk S., Halsema N., Spierings D.C.J., Guryev V., Lansdorp P.M.. BLM helicase suppresses recombination at G-quadruplex motifs in transcribed genes. Nat. Commun. 2018; 9:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Balasubramanian S., Hurley L.H., Neidle S.. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy?. Nat. Rev. Drug Discov. 2011; 10:261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hansel-Hertsch R., Di Antonio M., Balasubramanian S.. DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nature reviews. Mol. Cell Biol. 2017; 18:279–284. [DOI] [PubMed] [Google Scholar]
- 11. Asamitsu S., Obata S., Yu Z., Bando T., Sugiyama H.. Recent progress of targeted G-quadruplex-preferred ligands toward cancer therapy. Molecules. 2019; 24:429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carvalho J., Mergny J.L., Salgado G.F., Queiroz J.A., Cruz C.. G-quadruplex, friend or foe: the role of the G-quartet in anticancer strategies. Trends Mol. Med. 2020; 26:848–861. [DOI] [PubMed] [Google Scholar]
- 13. Li X.M., Zheng K.W., Zhang J.Y., Liu H.H., He Y.D., Yuan B.F., Hao Y.H., Tan Z.. Guanine-vacancy-bearing G-quadruplexes responsive to guanine derivatives. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:14581–14586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heddi B., Martin-Pintado N., Serimbetov Z., Kari T.M., Phan A.T.. G-quadruplexes with (4n - 1) guanines in the G-tetrad core: formation of a G-triad.water complex and implication for small-molecule binding. Nucleic Acids Res. 2016; 44:910–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koirala D., Mashimo T., Sannohe Y., Yu Z., Mao H., Sugiyama H.. Intramolecular folding in three tandem guanine repeats of human telomeric DNA. Chem. Commun. (Camb.). 2012; 48:2006–2008. [DOI] [PubMed] [Google Scholar]
- 16. Rajendran A., Endo M., Hidaka K., Sugiyama H.. Direct and single-molecule visualization of the solution-state structures of G-hairpin and G-triplex intermediates. Angew. Chem. Int. Ed. Engl. 2014; 53:4107–4112. [DOI] [PubMed] [Google Scholar]
- 17. Li X.M., Zheng K.W., Hao Y.H., Tan Z.. Exceptionally selective and tunable sensing of guanine derivatives and analogues by structural complementation in a G-quadruplex. Angew. Chem. Int. Ed. Engl. 2016; 55:13759–13764. [DOI] [PubMed] [Google Scholar]
- 18. Winnerdy F.R., Das P., Heddi B., Phan A.T.. Solution structures of a G-quadruplex bound to linear- and cyclic-dinucleotides. J. Am. Chem. Soc. 2019; 141:18038–18047. [DOI] [PubMed] [Google Scholar]
- 19. Wang K.B., Dickerhoff J., Wu G., Yang D. PDGFR-beta promoter forms a vacancy G-quadruplex that can be filled in by dGMP: solution structure and molecular recognition of guanine metabolites and drugs. J. Am. Chem. Soc. 2020; 142:5204–5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heddi B., Cheong V.V., Martadinata H., Phan A.T.. Insights into G-quadruplex specific recognition by the DEAH-box helicase RHAU: solution structure of a peptide-quadruplex complex. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:9608–9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. He Y.D., Zheng K.W., Wen C.J., Li X.M., Gong J.Y., Hao Y.H., Zhao Y., Tan Z.. Selective targeting of guanine-vacancy-bearing G-quadruplexes by G-quartet complementation and stabilization with a guanine-peptide conjugate. J. Am. Chem. Soc. 2020; 142:11394–11403. [DOI] [PubMed] [Google Scholar]
- 22. Fosgerau K., Hoffmann T.. Peptide therapeutics: current status and future directions. Drug Discov. Today. 2015; 20:122–128. [DOI] [PubMed] [Google Scholar]
- 23. Tesauro D., Accardo A., Diaferia C., Milano V., Guillon J., Ronga L., Rossi F.. Peptide-based drug-delivery systems in biotechnological applications: recent advances and perspectives. Molecules. 2019; 24:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He C., Medley S.C., Hu T., Hinsdale M.E., Lupu F., Virmani R., Olson L.E.. PDGFRbeta signalling regulates local inflammation and synergizes with hypercholesterolaemia to promote atherosclerosis. Nat. Commun. 2015; 6:7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ostman A., Heldin C.H.. PDGF receptors as targets in tumor treatment. Adv. Cancer. Res. 2007; 97:247–274. [DOI] [PubMed] [Google Scholar]
- 26. van Dijk F., Olinga P., Poelstra K., Beljaars L.. Targeted therapies in liver fibrosis: combining the best parts of platelet-derived growth factor BB and interferon gamma. Front. Med. (Lausanne). 2015; 2:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brown R.V., Wang T., Chappeta V.R., Wu G., Onel B., Chawla R., Quijada H., Camp S.M., Chiang E.T., Lassiter Q.R.et al.. The consequences of overlapping G-quadruplexes and i-motifs in the platelet-derived growth factor receptor beta core promoter nuclease hypersensitive element can explain the unexpected effects of mutations and provide opportunities for selective targeting of both structures by small molecules to downregulate gene expression. J. Am. Chem. Soc. 2017; 139:7456–7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen Y., Agrawal P., Brown R.V., Hatzakis E., Hurley L., Yang D. The major G-quadruplex formed in the human platelet-derived growth factor receptor beta promoter adopts a novel broken-strand structure in K+ solution. J. Am. Chem. Soc. 2012; 134:13220–13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Onel B., Carver M., Agrawal P., Hurley L.H., Yang D. The 3′-end region of the human PDGFR-beta core promoter nuclease hypersensitive element forms a mixture of two unique end-insertion G-quadruplexes. Biochim. Biophys. Acta Gen. Subj. 2018; 1862:846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zheng K.W., He Y.D., Liu H.H., Li X.M., Hao Y.H., Tan Z.. Superhelicity constrains a localized and R-loop-dependent formation of G-quadruplexes at the upstream region of transcription. ACS Chem. Biol. 2017; 12:2609–2618. [DOI] [PubMed] [Google Scholar]
- 31. Zheng K.W., Wu R.Y., He Y.D., Xiao S., Zhang J.Y., Liu J.Q., Hao Y.H., Tan Z.. A competitive formation of DNA:RNA hybrid G-quadruplex is responsible to the mitochondrial transcription termination at the DNA replication priming site. Nucleic. Acids. Res. 2014; 42:10832–10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tang J., Kan Z.Y., Yao Y., Wang Q., Hao Y.H., Tan Z.. G-quadruplex preferentially forms at the very 3′ end of vertebrate telomeric DNA. Nucleic Acids Res. 2008; 36:1200–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han H., Langley D.R., Rangan A., Hurley L.H.. Selective interactions of cationic porphyrins with G-quadruplex structures. J. Am. Chem. Soc. 2001; 123:8902–8913. [DOI] [PubMed] [Google Scholar]
- 34. Nicoludis J.M., Miller S.T., Jeffrey P.D., Barrett S.P., Rablen P.R., Lawton T.J., Yatsunyk L.A.. Optimized end-stacking provides specificity of N-methyl mesoporphyrin IX for human telomeric G-quadruplex DNA. J. Am. Chem. Soc. 2012; 134:20446–20456. [DOI] [PubMed] [Google Scholar]
- 35. Onodera M., Sueyoshi K., Umetsu M.. Fluorescence quenching by complex of a DNA aptamer and porphyrin for sensitive detection of porphyrins by capillary electrophoresis. Chem. Lett. 2021; 50:949–952. [Google Scholar]
- 36. Chaudhuri R., Bhattacharya S., Dash J., Bhattacharya S.. Recent update on targeting c-MYC G-quadruplexes by small molecules for anticancer therapeutics. J. Med. Chem. 2021; 64:42–70. [DOI] [PubMed] [Google Scholar]
- 37. Chen H., Long H., Cui X., Zhou J., Xu M., Yuan G.. Exploring the formation and recognition of an important G-quadruplex in a HIF1alpha promoter and its transcriptional inhibition by a benzo[c]phenanthridine derivative. J. Am. Chem. Soc. 2014; 136:2583–2591. [DOI] [PubMed] [Google Scholar]
- 38. Cui X., Chen H., Zhang Q., Xu M., Yuan G., Zhou J.. Exploration of the structure and recognition of a G-quadruplex in the her2 proto-oncogene promoter and its transcriptional regulation. Sci. Rep. 2019; 9:3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaiser C.E., Van Ert N.A., Agrawal P., Chawla R., Yang D., Hurley L.H.. Insight into the complexity of the i-motif and G-quadruplex DNA structures formed in the KRAS promoter and subsequent drug-induced gene repression. J. Am. Chem. Soc. 2017; 139:8522–8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moruno-Manchon J.F., Koellhoffer E.C., Gopakumar J., Hambarde S., Kim N., McCullough L.D., Tsvetkov A.S.. The G-quadruplex DNA stabilizing drug pyridostatin promotes DNA damage and downregulates transcription of Brca1 in neurons. Aging (Albany NY). 2017; 9:1957–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun D., Liu W.J., Guo K., Rusche J.J., Ebbinghaus S., Gokhale V., Hurley L.H.. The proximal promoter region of the human vascular endothelial growth factor gene has a G-quadruplex structure that can be targeted by G-quadruplex-interactive agents. Mol. Cancer Ther. 2008; 7:880–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang X.D., Ou T.M., Lu Y.J., Li Z., Xu Z., Xi C., Tan J.H., Huang S.L., An L.K., Li D.et al.. Turning off transcription of the bcl-2 gene by stabilizing the bcl-2 promoter quadruplex with quindoline derivatives. J. Med. Chem. 2010; 53:4390–4398. [DOI] [PubMed] [Google Scholar]
- 43. Zhang Q., Cui X., Lin S., Zhou J., Yuan G.. Convenient method for the synthesis of a flexible cyclic polyamide for selective targeting of c-myb G-quadruplex DNA. Org. Lett. 2012; 14:6126–6129. [DOI] [PubMed] [Google Scholar]
- 44. Qin Y., Fortin J.S., Tye D., Gleason-Guzman M., Brooks T.A., Hurley L.H.. Molecular cloning of the human platelet-derived growth factor receptor beta (PDGFR-beta) promoter and drug targeting of the G-quadruplex-forming region to repress PDGFR-beta expression. Biochemistry. 2010; 49:4208–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nguyen T.Q.N., Lim K.W., Phan A.T.. A dual-specific targeting approach based on the simultaneous recognition of duplex and quadruplex motifs. Sci. Rep. 2017; 7:11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Asamitsu S., Obata S., Phan A.T., Hashiya K., Bando T., Sugiyama H.. Simultaneous binding of hybrid molecules constructed with dual DNA-binding components to a G-quadruplex and its proximal duplex. Chemistry. 2018; 24:4428–4435. [DOI] [PubMed] [Google Scholar]
- 47. Mandal S., Kawamoto Y., Yue Z., Hashiya K., Cui Y., Bando T., Pandey S., Hoque M.E., Hossain M.A., Sugiyama H.et al.. Submolecular dissection reveals strong and specific binding of polyamide-pyridostatin conjugates to human telomere interface. Nucleic Acids Res. 2019; 47:3295–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.