


Abstract
Ribonucleases are crucial enzymes in RNA metabolism and post-transcriptional regulatory processes in bacteria. Cyanobacteria encode the two essential ribonucleases RNase E and RNase J. Cyanobacterial RNase E is shorter than homologues in other groups of bacteria and lacks both the chloroplast-specific N-terminal extension as well as the C-terminal domain typical for RNase E of enterobacteria. In order to investigate the function of RNase E in the model cyanobacterium Synechocystis sp. PCC 6803, we engineered a temperature-sensitive RNase E mutant by introducing two site-specific mutations, I65F and the spontaneously occurred V94A. This enabled us to perform RNA-seq after the transient inactivation of RNase E by a temperature shift (TIER-seq) and to map 1472 RNase-E-dependent cleavage sites. We inferred a dominating cleavage signature consisting of an adenine at the −3 and a uridine at the +2 position within a single-stranded segment of the RNA. The data identified mRNAs likely regulated jointly by RNase E and an sRNA and potential 3′ end-derived sRNAs. Our findings substantiate the pivotal role of RNase E in post-transcriptional regulation and suggest the redundant or concerted action of RNase E and RNase J in cyanobacteria.
INTRODUCTION
Ribonucleases (RNases) play central roles in bacterial RNA metabolism (1). They are crucial for RNA degradation, but also for the maturation of mRNAs, rRNAs, tRNAs and sRNAs. Furthermore, they are involved in post-transcriptional regulation and the acclimation to changing environmental conditions, e.g. by facilitating the action of regulatory sRNAs. In bacteria, RNA degradation is assumed to be an all-or-nothing event. It is initiated by a rate-limiting step, which comprises either the conversion of a 5′-triphosphate to a 5′-monophosphate or an endonucleolytic cleavage. Both result in a 5′-monophosphorylated (5′-P) RNA fragment. The first step is followed by rapid degradation by RNases with a high affinity to 5′-P RNA species, such as RNase Y, RNase J or RNase E and 3′-to-5′ exonucleases, such as PNPase or RNase II (2).
The majority of sequenced bacterial genomes encode a homologue of at least one of the three ribonucleases RNase E, RNase Y and RNase J (3). All three of these enzymes have a higher affinity to 5′-P RNA species than to 5′-triphosphorylated (5′-PPP) RNAs and are able to perform the first, rate-limiting endonucleolytic cleavage which initiates RNA degradation. This makes them key players in RNA metabolism. All three RNases share a low target specificity and can partially substitute for each other, which further highlights their functional similarity (4). Besides these common principles, different bacteria usually only encode a subset of the three mentioned RNases (2). The specific combination of these three RNases and peculiarities of the respective homologues, e.g. interaction with specific adaptor proteins or intracellular localisation, shape the RNA metabolism of an organism (3).
RNase E was intensively investigated in the gammaproteobacteria Escherichia coli (E. coli) and Salmonella enterica (Salmonella). Here, the enzyme plays a central role in rRNA (5,6), tRNA (7) and sRNA (8) maturation, the action of sRNAs and bulk RNA degradation (9,10). RNA cleavage takes place in single-stranded, adenine and uracil-rich regions (9). A uridine located two nucleotides downstream of RNase E cleavage sites was identified as an important recognition determinant in Salmonella (8). The enzyme cleaves preferentially 5′-P RNA species, which is referred to as 5′-sensing (11). Furthermore, RNase E can recognize target RNA by secondary structures in proximity to the respective cleavage site (12,13) or by the presence of several single-stranded regions within a single RNA target molecule (14). In addition to catalysing RNA cleavage, enterobacterial RNase E interacts with multiple proteins and serves as scaffold for the degradosome complex via its C-terminal ∼550 amino acids domain (15). In E. coli, RNase E interacts with specific proteins mediating the target recognition and specificity such as Hfq (16) or the adaptor protein RapZ (17). Homologues of these proteins either do not exist in Synechocystis (RapZ) or lost their RNA-binding capability in case of Hfq (18).
As the only bacteria that perform oxygenic photosynthesis, cyanobacteria are of immense ecological relevance. They are considered promising for the sustainable production of chemical feedstock and biofuels and serve as easy-to-manipulate models in synthetic biology and photosynthesis research (19). Cyanobacteria are morphologically distinct from other bacteria by the presence of thylakoids, extensive intracellular membrane systems, and carboxysomes, microcompartments specialised for the fixation of CO2. It is unknown to what extent these ultrastructural differences correlate with differences in processes such as RNA localisation and RNA metabolism. All cyanobacteria which were sequenced so far encode both RNase E as well as RNase J, but no RNase Y homologue (2). In this study, we chose Synechocystis sp. PCC 6803 (in the following Synechocystis), a unicellular model cyanobacterium. Both RNase E as well as RNase J are essential in Synechocystis (20–22). Besides cyanobacteria, only alphaproteobacteria, several actinobacteria and fibrobacteres contain both RNase E and RNase J, but lack RNase Y (2).
The Synechocystis RNase E N-terminal region is homologous to the E. coli RNase E N-terminal, catalytically active half. Cyanobacterial RNase E homologues contain only a short C-terminal non-catalytic domain (15). With usually less than 700 amino acids, they are shorter than the majority of other characterised RNase E homologues, which frequently consist of more than 900 amino acids (23). Moreover, cyanobacterial RNase E homologues lack the long N-terminal extension which is typical for RNase E homologues present in plant plastids (23,24) and certain other bacteria such as Streptomyces (25). The C-terminal region of cyanobacterial RNase E contains several conserved microdomains, of which a cyanobacterial-specific nonapeptide is binding the 3′-to-5′ exonuclease PNPase (26).
Previous studies showed that RNase E in Synechocystis triggers the degradation of the mRNAs of the two almost identical genes psbA2 and psbA3 in the dark (21), while in the light the mRNAs become protected through a unique mechanism involving the asRNAs PsbA2R and PsbA3R (27). By recruiting the sRNA PsrR1, RNase E was shown to “decapitate” the psaL mRNA after a shift from low to high light by cleaving off a fragment consisting of the 5′ UTR and the first seven nt of the coding sequence (CDS) (28). All these mRNAs encode central proteins of the photosynthetic apparatus. Therefore, Synechocystis RNase E appears to be involved in the acclimation of photosynthesis to changing light conditions, consistent with the results of a recent transcriptome analysis of an RNase E knock-down mutant (20). A surprising discovery was the identification of RNase E as the major maturation enzyme of crRNAs from the Synechocystis CRISPR3 array (29). Studies on single transcripts, such as Synechocystis psbA2, CRISPR3 or E. coli 9S RNA and RNAI pointed towards a similar cleavage specificity of Synechocystis RNase E as for the enterobacterial enzyme (15,20,21,29), consistent with the finding that Synechocystis RNase E can rescue an E. coli RNase E mutant (21).
Despite the multiple evidence for an important role of RNase E in Synechocystis, its targetome has not been determined thus far. Here, we engineered a temperature-sensitive RNase E mutant strain and applied the ‘transiently inactivation of an essential ribonuclease followed by RNA-Seq’ (TIER-seq) approach (8) for the transcriptome-wide identification of RNase-E-dependent cleavage sites. We identified 1472 RNase-E-dependent RNA processing events, a putative sequence motif for cleavage and substantiate the function of RNase E in crRNA maturation and the regulation of essential cellular functions such as photosynthesis.
MATERIALS AND METHODS
Bacterial strains and culture conditions
A motile wild-type strain of Synechocystis was used, which is capable of photoautotrophic, mixotrophic and chemoheterotrophic growth on glucose. It was originally obtained from S. Shestakov (Moscow State University, Russia) in 1993 and re-sequenced in 2012 (30). For culturing, BG-11 medium (31) substituted with 0.3% (w/v) sodium thiosulfate and 10 mM N-[Tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid (TES) buffer (pH 8.0) was used. Liquid cultures were grown in Erlenmeyer flasks under constant shaking (135 rpm) at 30°C and continuous white light (30 μmol photons m−2 s−1) in an incubator shaker (Multitron Pro, Infors AG, Switzerland). Plate cultures were grown on 0.75% bacto-agar BG-11 plates. Kanamycin (40 μg/ml) and chloramphenicol (10 μg/ml) were added to plate cultures for strain maintenance, but omitted during experiments.
Spectroscopy
Whole-cell absorption spectra were measured using a Specord 250 Plus (Analytik Jena) spectrophotometer at room temperature and normalized to absorption values at 682 and 750 nm.
Construction of mutant strains
Plasmid pUC19-3xFLAG-rne was created by assembly cloning (AQUA cloning) (32). Primer pairs P01/P02 and P03/P04 (all oligonucleotides are listed in Supplementary Table S1) were used for amplification of homologous flanks by PCR. pUC19 backbone and a kanamycin resistance cassette were amplified using primer pairs P05/P06 and P07/P08 using plasmids pUC19 and pVZ321 (33) (NCBI:AF100176.1) as templates, respectively. After assembly cloning, the resulting plasmid was introduced into wild-type Synechocystis (WT) by transformation. Chromosomal DNA of the resulting strain was used to amplify the rne-rnhB locus, i.e. the operon of rne and rnhB, encoding RNase E and RNase HII, respectively. The PCR product included an N-terminal 3xFLAG tag, promoter and terminator sequences (Supplementary Figure S1). During amplification, XhoI and SalI recognition sites were introduced using primers P09/P10. The PCR fragment was inserted into pJET 1.2 vector (CloneJET PCR Cloning Kit, Thermo Scientific™, Germany). Point mutations were introduced by site-directed mutagenesis (Q5 Site-Directed Mutagenesis Kit, New England BioLabs, Inc., USA) using primer pairs P11/P12 (G63S), P13/P14 (I65F) and P15/P16 (G63S, I65F). The resulting sub-cloning plasmids and the conjugative plasmid pVZ321 were cleaved with restriction enzymes XhoI and SalI (Thermo Scientific™, Germany), generating compatible restriction sites, and subsequently ligated using T4 DNA ligase (New England BioLabs, Inc., USA). Insertion direction was tested by multiplexed PCR using primers P17, P18 and P19. For further strain construction, plasmids were selected for which primer pair P18/P19 yielded an amplicon of 590 bp, while P17/P18 did not give a product. For an empty-vector control, cut pVZ321 was ligated without insert, resulting in pVZΔKmR. The resulting plasmids were transferred into WT by triparental mating with E. coli DH5α harbouring the constructed plasmid and E. coli J53 (NCBI:txid1144303) with the conjugative helper plasmid RP4 (NCBI:txid2503).
The plasmid used for deleting the endogenous rne-rnhB locus was generated by AQUA cloning (Supplementary Figure S1). To construct this plasmid, four PCR fragments were generated: Homologous flanks up- and downstream of the locus were amplified using primer pairs P20/P21 and P24/P25. A kanamycin resistance cassette and pUC19 plasmid backbone was amplified using primers P22/P23 and P26/P27. After assembly cloning, the resulting plasmid was transferred into the strains harbouring the plasmids containing the rne-rnhB locus. For the empty-vector control, a kanamycin resistance cassette and an N-terminal 3xFLAG tag sequence were inserted at the endogenous rne-rnhB locus using the plasmid pUC19-3xFLAG-rne. Synechocystis clones were tested for temperature sensitivity by streaking them on plates which were incubated at either 30°C or 39°C. PCR was used to verify correct construct insertion and full segregation, using primers P10/P28. The occurrence of compensatory mutations and the loss of introduced point mutations were checked by PCR and sequencing with primers P29, P30, P31, P32 and P33. A list of all strains used in this study can be found in Supplementary Table S2.
Transient inactivation of RNase E and RNA extraction
For transient inactivation of RNase E, four independent liquid cultures of WT, rne(WT) and rne(Ts) were grown at 30°C to an OD750nm of 0.7–0.8 in 50 ml culture volume. After harvesting a subsample of 20 ml culture, the temperature of the incubator shaker was set to 39°C. Further 20 ml aliquots were collected exactly 60 min after changing the set temperature. For sampling, aliquots were collected by rapid vacuum filtration through 0.8 μm polyethersulphone filter disks (Pall, Germany). Filters were immediately transferred to 1.6 ml PGTX solution (34), vortexed, frozen in liquid nitrogen and stored at −80°C. RNA was extracted according to Pinto et al. (34) with modifications as described by Wallner et al. (35). Purified RNA was quantified using a NanoDrop ND2000 spectrophotometer (Thermo Fisher Scientific, Germany).
Northern blot hybridization
Total RNA was separated either by electrophoresis on agarose/formaldehyde gels (1.3% (w/v) agarose, 1.85% (v/v) formaldehyde, 1× MOPS–EDTA–NaOAc-buffer) or on polyacrylamide (PAA)–urea gels (8% PAA, 8.3 M urea, 1× Tris–borate–EDTA buffer) and blotted onto Roti-Nylon plus membranes (Carl Roth, Germany). Hybridization of the membranes with radioactively labelled probes was carried out as described (29) with the following modifications: Incubation with washing solution I (2× SSC, 0.5% SDS) was performed at 25°C. Subsequent washes with buffer II (2× SSC, 0.5% SDS) and III (0.1× SSC, 0.1% SDS) were performed at 65°C. Signals were detected by phosphorimaging on a Typhoon FLA 9500 imaging system (GE Healthcare, USA). Oligonucleotides H01–H10 used to generate hybridisation probes are given in Supplementary Table S1. Single-stranded RNA probes were produced by in vitro transcription of PCR fragments using the Ambion T7 polymerase maxiscript kit (Thermo Fisher Scientific, Germany) in the presence of [α-32P]-UTP (Hartmann Analytics, Germany).
RNA sequencing
Biological triplicates of rne(WT) and rne(Ts) at each condition were analysed by RNA sequencing. These triplicates corresponded to three and two biologically independent clones, respectively. Residual DNA was removed from samples containing each 10 μg RNA by three subsequent incubation steps with Ambion TURBO-DNase (Thermo Fisher Scientific, Germany). For each step, 2 U TurboDNase was added, followed by 20 min incubation at 37°C. RNA was recovered using RNA Clean & Concentrator kits (Zymo Research, USA). RNA integrity was controlled on a Fragment Analyzer using the High Sensitivity RNA Analysis Kit (Agilent, USA). cDNA libraries were constructed and sequenced as a service provided by vertis Biotechnologie AG (Germany) according to the tagRNA-Seq protocol (36), including unique molecular identifiers (UMIs) (37). Ribosomal RNAs were depleted using in-house depletion probes. 5′-P RNA fragments were ligated to the 5′ Illumina TruSeq sequencing adapter carrying the sequence tag CTGAAGCT, indicating processing sites (PSS). After incubation with 5′-phosphate-dependent exoribonuclease XRN-1 (New England BioLabs, Inc., USA), samples were treated with RNA 5′ polyphosphatase (Lucigen). The 5′ Illumina TruSeq sequencing adapter carrying sequence tag TAATGCGC was ligated to newly formed 5′-P RNA ends, indicating transcriptional start sites (TSS). RNA was fragmented and an oligonucleotide adapter was ligated to the resulting 3′ ends. First-strand cDNA synthesis was performed using M-MLV reverse transcriptase and the 3′ adapter as primer. First-strand cDNA was purified and the 5′ Illumina TruSeq sequencing adapter was ligated to the 3′ end of the antisense cDNA. This was followed by PCR amplification to about 10–20 ng/μl using a high fidelity DNA polymerase. cDNA was purified using the Agencourt AMPure XP kit (Beckman Coulter Genomics). Samples were pooled in approximately equimolar amounts and the cDNA pool in the size range of 200–600 bp was eluted from a preparative agarose gel. The cDNA pool was sequenced with 75 bp read length on an Illumina NextSeq 500 system.
Bioinformatic analyses
A homology model of Synechocystis RNase E was created with iTasser (38,39) and analysed using Pymol Molecular Graphics System, (v2.4.0) (Schrödinger, LLC.). SyntTax (40) was used for synteny analyses. For RNA-Seq analysis, reads were uploaded to the usegalaxy.eu server and analysed utilizing the Galaxy web platform (41) after preliminary processing (Supplementary Methods 1). Several workflows were created to process the data further and can be accessed and reproduced at the following links: https://usegalaxy.eu/u/ute-hoffmann/w/tier-seqprocessing1, https://usegalaxy.eu/u/ute-hoffmann/w/tier-seqtranscript, https://usegalaxy.eu/u/ute-hoffmann/w/tier-seqpss-tss and https://usegalaxy.eu/u/ute-hoffmann/w/tiertranscript-coverage. For downstream analyses, reads with a mapping quality above 20 were kept. Reads with a mapping quality of exactly one were included for analyses of psbA2/psbA3 and rRNA loci. Transcript data was assigned to annotated regions using htseq-count (42). Annotation included several known sRNAs, asRNAs and small proteins (43–46). Subsequently, htseq-count files, PSS/TSS-5′ end files and transcript coverage files were downloaded and analysed using Python (v3.7.4) and R (v4.0.4) scripts available at (https://github.com/ute-hoffmann/TIER-synechocystis) (detailed description in Supplementary Methods 2). DESeq2 (v1.30.1) (47) was used for the analysis of htseq-count files of transcript data (|log2FC| > 0.8 and p.adj < 0.05), the classification of genomic positions as TSS or PSS and differential expression analysis of the resulting set of PSS and TSS positions (|log2FC| > 1.0 and p.adj < 0.05). Briefly, PSS and TSS data were filtered for a minimal count number at each position (edgeR::filterByExpr (48)). The resulting data set was used as input for DESeq2, which assessed for each position if it was significantly enriched in the TSS or PSS data set. Subsequently, DESeq2 was used to test differential expression of PSS positions in the different samples. Sequence logos were created using WebLogo (v3.7.8) (49) with a GC content of 47.4%. Minimal folding energies (ΔG) were calculated with RNAfold (v2.4.17) (50), temperature parameter set to 39°C, with a sliding window of 25 nt and a 1 nt step size.
RESULTS AND DISCUSSION
A point mutation homologous to E. coli rne-3071 conveys temperature sensitivity in the cyanobacterium Synechocystis
Synechocystis RNase E, encoded by rne, is essential (20–22) and forms an operon with rnhB (slr1130) encoding RNase HII (46), which is a widely conserved arrangement in cyanobacteria (Supplementary Figure S2). Analyses in E. coli, Salmonella, Rhodobacter sphaeroides (Rhodobacter) and Vibrio cholerae (Vibrio) demonstrated that temperature-sensitive RNase E mutant strains enable the transient inactivation of the essential RNase to perform RNA-sequencing and capture RNase-E-dependent cleavage sites on a transcriptome-wide level in vivo (8,14,51,52). Despite the low sequence identity of 36% and similarity of 55% between Synechocystis RNase E and the catalytic, N-terminal half of the E. coli enzyme (20), we aimed for a temperature-sensitive RNase E mutant strain in Synechocystis. To avoid disruption of the rne-rnhB operon and to minimize a polar effect on rnhB expression, we first introduced the rne-rnhB locus including the native promoter and 3′ UTR on a conjugative self-replicating plasmid into WT cells. This was followed by deletion of the genomic locus by homologous recombination (Supplementary Figure S1).
In E. coli, studies employing temperature-sensitive RNase E relied on one of the two point mutations ams-1 (G66S) or rne-3071 (L68F) (53). Using sequence alignments and a homology model, we identified G63S and I65F as the homologous amino acid substitutions in Synechocystis (Figure 1A, Supplementary Figure S3). The respective mutated genes were introduced into Synechocystis, following the same strategy as for the unmodified gene. Segregation between chromosomes with the deleted rne gene and those still carrying the WT rne gene was judged by PCR (Supplementary Figure S1B). Full segregation (homozygosity) was only obtained for the mutation homologous to rne-3071 (Synechocystis I65F), but not to ams-1 (Synechocystis G63S). Interestingly, all homozygous clones accumulated one of the three following second-site mutations within the RNase E gene: V94A, V297A or G281E. I65F as well as V94A and V297A are localized at the RNA-binding channel of RNase E, close to the enzyme's active site (Supplementary Figure S3B). Phenylalanine is bulkier than isoleucine. Therefore, the I65F substitution seems to destabilize the beta sheet in which it is located (E. coli L68, compare (54)). Substitution of V94 or V297 by alanine reduces this steric problem (Supplementary Figure S3E). The third mutation, G281E, is not localized within the RNA-binding channel, but in close proximity. It is part of a beta sheet which forms the RNA-binding channel, but G281E is oriented towards the protein's outer surface. How the mutation G281E might compensate for I65F is not directly apparent, since it does not seem to lower the steric issues associated with I65F. It might be part of a potential RNA-binding surface which is possibly involved in the 5′ bypass pathway (12) (Supplementary Figure S3D). Of all three detected second-site mutations, V94A seems to have the smallest effect on potential RNA-binding sites and the RNA channel, according to the homology model. Hence, for further characterization, the I65F and V94A mutations were combined in one strain, referred to as rne(Ts). The congenic control strain, which was generated in the same manner as rne(Ts) and in which the RNase E WT allele was introduced, will be referred to as rne(WT). In rne(WT), no second-site mutations within the rne gene were detected. In addition, both RNase E variants were tagged with an N-terminal 3xFLAG tag. Previous studies showed that neither the tag nor expression from a conjugative vector interfere with RNase E function (29).
Figure 1.
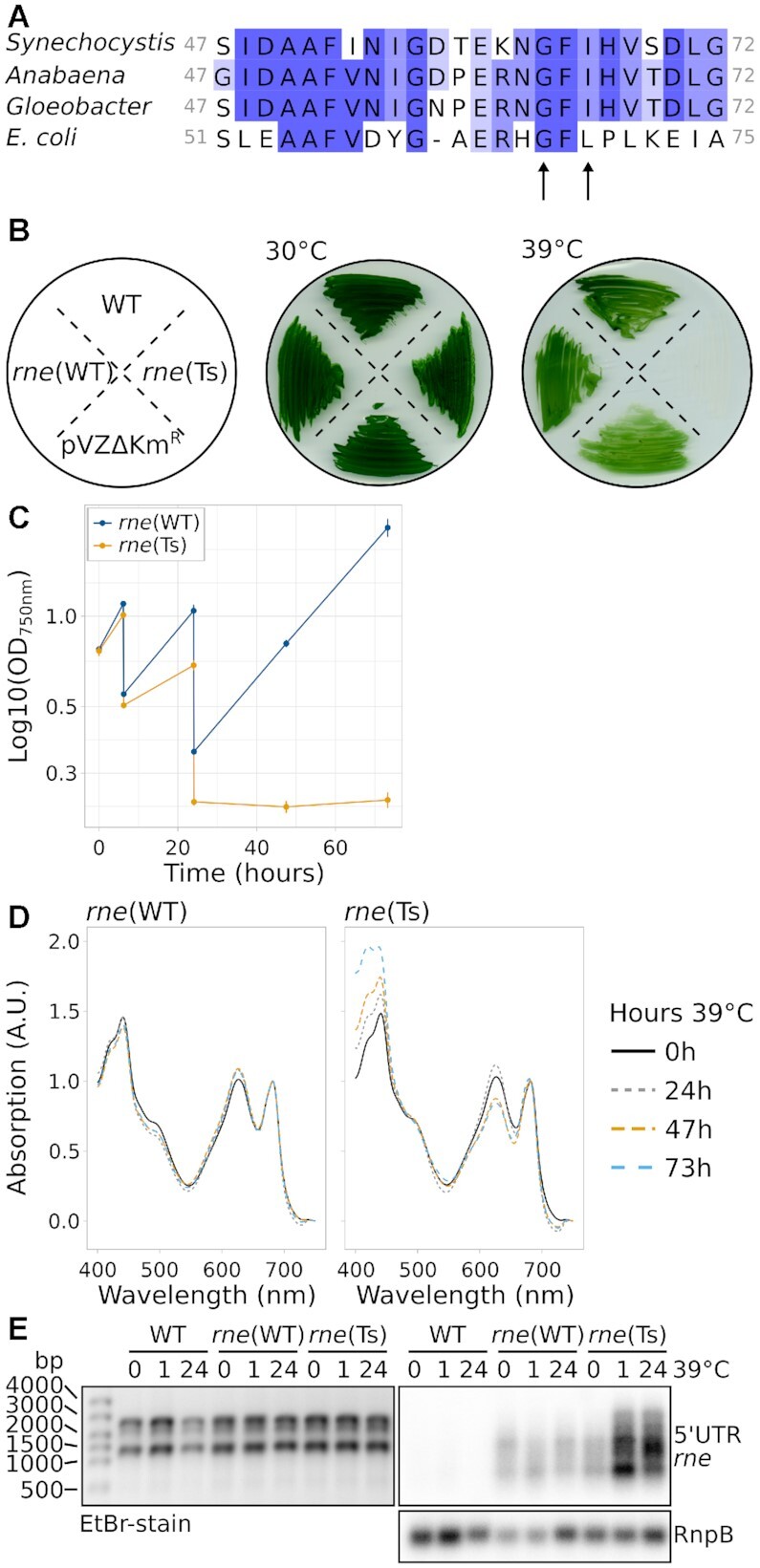
Characterisation of Synechocystis harbouring temperature-sensitive RNase E. (A) Alignment of Synechocystis RNase E residues 47–72 with the respective section in homologues from Anabaena sp. PCC 7120, Gloeobacter violaceus PCC 7421 and E. coli. The arrows point at two conserved residues which were mutated to obtain temperature sensitivity. (B) Growth of wild-type Synechocystis (WT), rne(WT), rne(Ts) and an empty-vector control strain (pVZΔKmR) at the standard temperature of 30°C and at 39°C. The scheme on the left indicates the streaking order of the strains. (C) Growth of rne(WT) and rne(Ts) at 39°C in liquid culture. Time point 0 h corresponds to the switch from 30°C to 39°C. Error bars indicate the standard deviation of biologically independent duplicates. Cultures were diluted after 6 hours and 24 hours of growth. (D) Absorption spectra of rne(WT) and rne(Ts) throughout the course of 73 h incubation at 39°C of one representative experiment (compare panel C). Spectra were normalized to absorption at 682 nm and 750 nm. (E) Northern blot analysis of the accumulation of the rne-rnhB transcript. One representative analysis is shown (n = 4). In addition to the control hybridization with RnpB (lower panel), the denaturing agarose gel which was used for blotting is shown on the left as a loading control.
Growth of rne(Ts) was not impaired in comparison to WT or rne(WT) at 30°C. We determined 39°C as non-permissive growth temperature at which, in contrast to rne(Ts), neither WT nor rne(WT) showed a severe growth deficiency as judged by growth on solid and liquid media (Figure 1B, C). Prolonged incubation of rne(Ts) liquid cultures at 39°C resulted in bleaching of the cells (Figure 1D).
The used mutation strategy led to an overexpression of the rne-rnhB transcript, which was likely caused by the higher copy number of the RSF1010-derived conjugative plasmid compared to the chromosome (Figure 1E) (33,55). In Synechocystis, the enzymatic activity of RNase E is rate-limiting for the maturation and accumulation of crRNAs from the CRISPR3 array (29). Hence, the amount of mature CRISPR3 crRNA may be used as a proxy for RNase E activity. Indeed, overexpression of RNase E led to a dramatic overaccumulation of mature CRISPR3 spacers in rne(WT) and a moderately enhanced accumulation in rne(Ts) compared to WT (Supplementary Figure S4). With increasing time at the non-permissive temperature, the level of CRISPR3 accumulation decreased in all three compared strains.
E. coli RNase E autoregulates its own transcript level (56) and there is evidence that the same holds in the cyanobacteria Synechocystis and Prochlorococcus MED4 (20,57). Here, we observed enhanced rne-rnhB transcript accumulation in rne(Ts) during prolonged incubation at 39°C (Figure 1E). This finding further supported that RNase E activity was reduced in rne(Ts) at the elevated temperature. We conclude that introduction of the I65F mutation homologous to E. coli rne-3071 in combination with the V94A substitution led to a temperature-sensitive RNase E Synechocystis mutant strain, rne(Ts).
Overview of TIER-seq experiment
We used the here generated strains rne(Ts) and rne(WT) to identify RNase-E-dependent cleavage positions on a transcriptome-wide level by performing TIER-seq (Figure 2A) (8). Inactivation of a ribonuclease should lead to increased amounts of its target RNAs. Hence, transcripts accumulating after the heat shock at a higher level in rne(Ts) compared to rne(WT) likely are direct targets of RNase E. A lowered level indicates RNA species which are normally matured by the respective RNase from an otherwise unstable precursor or may derive indirectly from the RNase E inactivation (20). The processed CRISPR3 crRNAs are an example for the former and became more abundant in rne(WT) compared to rne(Ts) and WT here (Supplementary Figure S4), consistent with previous observations (29). To further pinpoint direct targets and cleavage positions of RNase E, we analysed changes in the number of processing sites (PSS) upon RNase E inactivation. We assumed that lowered PSS counts in rne(Ts) compared to rne(WT) after transient RNase E inactivation indicated direct targets of RNase E in vivo or resulted from the combined action of RNase E and downstream processing by other RNases. More specifically, we envision a similar interaction as described by Broglia and coworkers (58) for RNase Y and PNPase as well as YhaM in Streptococcus pyogenes and assume that some observed PSS might result from cleavage by RNase E, which is followed by further processing by another RNase, e.g. RNase J. Higher PSS counts in rne(Ts) after heat inactivation relative to rne(WT) point to RNA fragments produced by the action of other RNases and which would possibly usually be further degraded by RNase E.
Figure 2.

Overview of the TIER-seq experiment and exemplary results. (A) Experimental work flow. RNA was sampled from strains encoding either wild-type (rne(WT)) or temperature-sensitive (rne(Ts)) RNase E before and after incubation at 39°C for one hour. (B) RNA was extracted and used to identify processing sites (PSS) and transcriptional start sites (TSS) after the addition of PSS- and TSS-tags, sequencing adaptors containing PSS- and TSS-specific nucleotide combinations. For the latter, 5′ pyrophosphates were removed using RNA 5′ polyphosphatase. (C) Shares in reads corresponding to PSS, TSS or unspecified transcript positions in the 12 samples. (D) TIER-seq data for the tRNA 6803t19. (E) TIER-seq data for the CDS and 3′ UTR of petD. In panels D and E, transcriptome coverage, PSS and TSS represent the average of normalised read counts of the three investigated replicates for the indicated strains. In panel E, this is compared to read counts obtained by Kopf et al. (46). CDS: coding sequences, UTR: untranslated region.
Since RNase E processing results in 5′-P RNA ends (59), we chose a library preparation protocol that distinguishes between PSS and TSS RNA ends: tagRNA-Seq (Figure 2B) (36). Briefly, 5′-P RNA species were ligated to a sequencing adaptor carrying a nucleotide combination specific for PSS: a PSS-tag. Unligated 5′-P RNA fragments were removed using the 5′-phosphate dependent exoribonuclease XRN-1. Subsequently, 5′-PPP ends (characteristic of unprocessed TSS) were converted to 5′-P ends and ligated to a TSS-tag followed by standard library preparation and sequencing. For sequencing, triplicates of RNA samples were taken before and after one hour of growth at 39°C of rne(WT) and rne(Ts). Samples were taken at an OD750nm of 0.7–0.8. In total, ∼160 million raw sequencing reads were obtained for the twelve samples. After mapping, UMI-removal and quality filtering, 48.3% of those reads remained, corresponding, on average, to 5.5 million reads (∼0.4 billion bp) per sample (Supplementary Tables S3 and S4, Supplementary Figure S5). Of those, on average, 9.5% were PSS reads, 15.7% TSS reads and 74.5% transcript reads (Figure 2C).
Characterization of newly identified TSS and PSS
Following the initial data analysis, 3540 nucleotide positions were classified as TSS and 3450 as PSS (Supplementary Methods 2, Supplementary Figure S6A, supplementary file TSS_PSS_rneAnalysis.gff). Newly identified PSS were scrutinized to assure the feasibility of our approach to identify and classify TSS and PSS. We explored the sequence composition up- and downstream of the identified TSS and PSS positions (Supplementary Figure S6B). TSS positions showed an enrichment of a canonical –10 element (5′-TAnAAT-3′) and a higher frequency of purines at the +1 position matching previous observations (44). PSS positions showed a slight enrichment of T 2 nt downstream, reminiscent of the motif identified by Chao et al. for Salmonella RNase E (8).
The majority of PSS (98.7%) overlapped with transcriptional units (TUs) defined previously (46) and coding regions of Synechocystis. In total, 16.0% (656) TUs and 12.0% (737) CDS contained processing sites. We noticed a correlation between the expression level and the detection of PSS (Supplementary Results 1, Supplementary Figure S6C, Supplementary Tables S4–S8) indicating that processing sites in low abundance transcripts were possibly partially not captured.
We compared the newly obtained set of PSS to the total of 5162 TSS determined by Kopf et al. (46). In the following, those TSS will be referred to as anno-TSS, for “annotated TSS”. Only 47 of the PSS identified using tagRNA-Seq overlapped with anno-TSS. Of these, 29 (61.7%) anno-TSS were previously classified as alternative or internal TSS within another transcript (Supplementary Figure S6D), including 11 positions which correspond to the 5′ ends of matured tRNAs. Using tagRNA-Seq, both the 5′-P ends of mature tRNAs as well as corresponding TSS upstream of those positions were detected (Figure 2D). Those examples show that tagRNA-Seq efficiently discriminated PSS and TSS.
To evaluate the effect of incubating the cells at 39°C, we analysed transcriptomic differences before and after the heat treatment (Supplementary Results 2, Supplementary Figure S7, Supplementary Tables S9–S14). In both strains, the set of most strongly upregulated genes after 39°C treatment encoded heat shock proteins, while genes involved in the acclimation to inorganic carbon limitation were down-regulated.
Transient inactivation of RNase E affects a high proportion of the transcriptome
The TIER-seq data set was separately analysed for the three different data types encompassing transcript patterns, TSS and PSS (Supplementary Tables S13–S21). Principal component analyses (PCAs) indicated similar gene expression patterns and consistency between biological replicates and a stronger effect of the rne(Ts) mutation on processing rather than on transcription initiation or transcript patterns (Figure 3A). This is reflected by the number of differentially expressed transcripts, PSS and TSS positions before and after the heat treatment (Table 1, Supplementary Figure S8, p.adj < 0.05; for RNA features: |log2FC| > 0.8, for TSS/PSS: |log2FC| > 1). The 1472 PSS with significant different read counts between both strains after the heat shock mapped to 380 (9.4%) of the annotated TUs (rne(WT): 248; rne(Ts): 237) and 307 (8.4%) annotated CDS (rne(WT): 198; rne(Ts): 182) (Figure 3B, Table 2). PSS corresponding to major rRNA maturation intermediates were not affected by RNase E inactivation (Supplementary Results 3). tRNA levels were higher in rne(WT) than in rne(Ts) after RNase E inactivation (Table 3, Supplementary Figure S9A, Supplementary Tables S22). This indicates a role of Synechocystis RNase E in the maturation of tRNAs and is in line with known functions of enterobacterial RNase E (8,60). Combined with the high proportion of transcripts affected by transient RNase E inactivation (15.5% of annotated transcriptional units), these findings illustrate the central role of RNase E in cyanobacterial RNA metabolism. Gene set enrichment analyses (GSEA) and functional enrichment analyses identified mainly photosynthesis-associated KEGG and GO terms among the differentially regulated transcripts and PSS with divergent read counts (Supplementary Results 4, Supplementary Figure S9B and S9C, Supplementary Tables S22–S27).
Figure 3.

Global analysis of TIER-seq results. (A) Comparison between the triplicates for the three analysed strains using principal component analyses and differentiating between unspecified transcripts, TSS and PSS. (B) Percentages of different types of transcripts associated with PSS identified in rne(WT) and rne(Ts) after the heat treatment relative to total number of the respective transcript type in the annotation. (C) Relative lengths of different parts of the Synechocystis genome (on the left). Percentage of PSS mapped to the five major replicons comparing rne(WT) and rne(Ts) after the heat treatment (on the right). Chr: chromosome. (D) Percentages of PSS associated with different types of transcripts comparing rne(WT) and rne(Ts) after the heat treatment. (E) Distribution of PSS identified in rne(WT) and rne(Ts) after heat shock relative to start and stop codons.
Table 1.
RNA features, transcriptional units (TUs), PSS and TSS differentially expressed between rne(WT) and rne(Ts) and in rne(WT) upon heat treatment. RNA features include CDS, known sRNAs, asRNAs and small proteins (43–45) as well as 5′ UTRs and 3′ UTR based on the total of 4091 TUs defined by Kopf et al. (46). Numbers in parentheses give percentages relative to all features of respective type
| rne(WT) 1h/0h 39°C | rne(WT)/rne(Ts) (0h 39°C) | rne(WT)/rne(Ts) (1h 39°C) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Decrease | Increase | rne(WT) | rne(Ts) | rne(WT) | rne(Ts) | |||||||
| RNA features | 6117 | 360 | (5.9%) | 302 | (4.9%) | 94 | (1.5%) | 20 | (0.3%) | 443 | (7.2%) | 433 | (7.1%) |
| TUs | 4091 | 268 | (6.6%) | 227 | (5.5%) | 78 | (1.9%) | 8 | (0.2%) | 309 | (7.6%) | 327 | (8.0%) |
| PSS | 3450 | 277 | (8.0%) | 292 | (8.5%) | 133 | (3.9%) | 35 | (1.0%) | 747 | (21.7%) | 725 | (21.0%) |
| TSS | 3540 | 336 | (9.5%) | 301 | (8.5%) | 49 | (1.4%) | 14 | (0.4%) | 228 | (6.4%) | 337 | (9.5%) |
Table 2.
Numbers of different RNA features overlapping PSS. Numbers in parentheses give percentages relative to all RNA regions of respective type
| Type of RNA feature | Total number of RNA feature | rne(WT) | rne(Ts) | ||
|---|---|---|---|---|---|
| CDS | 3675 | 198 | (5.4%) | 182 | (5.0%) |
| 5UTR | 979 | 19 | (1.9%) | 22 | (2.3%) |
| 3UTR | 29 | 4 | (13.8%) | 0 | (0.0%) |
| tRNA | 42 | 0 | (0.0%) | 1 | (2.4%) |
| rRNA | 6 | 0 | (0.0%) | 0 | (0.0%) |
| ncRNA | 318 | 6 | (1.9%) | 10 | (3.1%) |
| asRNA | 1071 | 6 | (0.6%) | 11 | (1.0%) |
| CRISPR | 3 | 2 | (66.7%) | 0 | (0.0%) |
Table 3.
Number of RNA features differentially expressed between rne(WT) and rne(Ts) after heat treatment and in rne(WT) upon heat treatment. Numbers in parentheses give percentages relative to all RNA regions of respective type
| rne(WT) 1h/0h 39°C | rne(WT)/rne(Ts) (1h 39°C) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Total | Decrease | Increase | rne(WT) | rne(Ts) | |||||
| CDS | 3675 | 292 | (7.9%) | 193 | (5.3%) | 333 | (9.1%) | 226 | (6.1%) |
| 5′ UTR | 979 | 26 | (2.7%) | 25 | (2.6%) | 29 | (3.0%) | 41 | (4.2%) |
| 3′ UTR | 29 | 2 | (6.9%) | 3 | (10.3%) | 2 | (6.9%) | 1 | (3.4%) |
| tRNA | 42 | 5 | (11.9%) | 0 | (0.0%) | 17 | (40.5%) | 0 | (0.0%) |
| asRNA | 1071 | 24 | (2.2%) | 52 | (4.9%) | 31 | (2.9%) | 140 | (13.1%) |
| ncRNA | 318 | 10 | (3.1%) | 28 | (8.8%) | 28 | (8.8%) | 24 | (7.5%) |
| CRISPR | 3 | 0 | (0.0%) | 0 | (0.0%) | 3 | (100.0%) | 0 | (0.0%) |
The number of obtained RNase-E-dependent cleavage sites differ from numbers gained with a similar methodology for E. coli (6 997) (14), Salmonella (22 033) (8), Rhodobacter (41 000) (51) and Vibrio (24 962) (52). This could be interpreted as a less important role of RNase E in Synechocystis than in the mentioned species. However, these differences do more likely arise from differences between the used sequencing protocols, bioinformatic analyses and sequencing depth.
RNase E inactivation affects transcripts originating from plasmid pSYSA
In relation to the sizes of the chromosome and the seven different native plasmids, PSS located on pSYSA were overrepresented among PSS accumulating in rne(WT) compared to rne(Ts) after RNase E heat inactivation (Figure 3C). This is also reflected by the higher percentage of annotated transcripts containing RNase-E-dependent PSS and the higher number of PSS per kb (Supplementary Figure S10). We did not observe pSYSA-derived transcripts to be generally more abundant than transcripts derived from other replicons (Supplementary Table S28). However, transcripts derived from the four major plasmids accumulated differentially between rne(WT) and rne(Ts) (Supplementary Tables S29 and S30, Supplementary Figure S9D). This might indicate a role of RNase E in copy number regulation of these replicons. Such functions were described for RNase E in E. coli and RNase J1 and J2 in Staphylococcus aureus (61,62).
A large proportion of the mapped PSS correspond to processing events in the CRISPR3 array, consistent with previous observations that RNase E is the major maturation enzyme of the pSYSA-located CRISPR3 array, a type III-Bv CRISPR-Cas system (29) (Figure 4A). However, we also noticed PSS in the type I-D CRISPR1 system for which Cas6-1 was identified as the main maturation endonuclease, that cleaves 8 nt from the ends of the repeats (63). Conversely, the here identified RNase E PSS are located close to the 5′ ends of the CRISPR1 repeats suggesting possible involvement of this enzyme in the maturation or degradation of additional CRISPR RNAs. Additionally, all three CRISPR arrays were more highly expressed in rne(WT) than in rne(Ts) after the heat shock (Table 3). The large number of remaining pSYSA-located PSS might indicate further roles of RNase E associated with the multiple toxin-antitoxin systems on this plasmid (64).
Figure 4.

Examples of RNase-E-dependent processing events. (A) Spacers 1, 2 and 3 (S1–S3) of the CRISPR3 array. The black arrows point at positions at which cleavage events were mapped previously by primer extension (29). (B) The 5′ UTR and begin of the psbA2 (slr1311) CDS and associated asRNA PsbA2R. Black arrows point at major (long arrows) and minor (short arrows) cleavage sites detected by in vitro cleavage assays previously (20,21,29). (C) The sRNA PmgR1 (Ncr0700). (D) The CDS of gifB (sll1515), its 5′ UTR harbouring the glutamine riboswitch and two asRNAs. The prominent PSS at position +35 of the gifB transcript is labelled by an asterisk. (E) The 5′ UTR of the rne gene (slr1129). (F) Representative northern blot analysis of PmgR1 (n = 4). A longer exposure is shown to highlight low abundance processing products. (G) Representative northern blot analysis of gifB (n = 4). The transcriptome coverage is given on top for the two indicated strains after incubation for 1 h at 39°C. Cleavage sites are displayed in the diagrams underneath by the blue and orange peaks, representing 5′-P (PSS) detected in rne(WT) and rne(Ts), respectively. 5′-PPP (TSS) may be converted to 5′-P RNA ends in vivo and also during RNA-seq library preparation. Hence, TSS show up in the PSS signal. Positions which were classified as TSS using DESeq2 are labelled with “TSS” next to the respective peaks. Transcriptome coverage and cleavage sites (PSS) represent the average of normalised read counts of the three investigated replicates. CDS: coding sequences, UTR: untranslated region.
RNase-E-dependent cleavage sites tend to be close to start and stop codons
To further analyse specific effects of RNase E, we evaluated which type of RNA features overlapped with the detected differentially accumulating PSS (Figure 3D, Table 3). This set of features included CDS, known sRNAs, asRNAs and small proteins (43–45) as well as 5′ UTRs and 3′ UTRs based on the total of 4091 TUs defined by Kopf et al. (46). The majority of PSS were mapped to CDS in both rne(WT) as well as rne(Ts). PSS mapping to 3′ UTRs were only detected in rne(WT), but not in rne(Ts) (Figure 3B and 3D). Also, we observed a tendency for 3′ UTRs to be enriched in rne(WT) compared to rne(Ts) after the heat shock (Supplementary Figure S9A, Supplementary Table S20).
The evidence towards an action of RNase E on 3′ UTRs led us to a transcriptome-wide investigation of PSS localization in start and stop codon regions (Figure 3E). Intriguingly, several PSS accumulating in rne(WT) were located exactly 1 nt upstream of start codons or 2 nt downstream of stop codons. A similar relation was previously observed for Salmonella (8), Vibrio (52) and regarding start codons for Rhodobacter (51).
RNase-E-dependent cleavage sites 2 nt downstream of stop codons were identified for psaJ (sml0008, photosystem I subunit IX), psbO (sll0427, photosystem II manganese-stabilizing polypeptide), rps14 (slr0628, 30S ribosomal protein S14) and three genes of unknown function: ssr6030, slr5110 and slr0581. Indeed, the 3′ UTRs of psaJ, psbO and rps14 were previously identified to accumulate different from their CDS under certain growth conditions (46), This also holds true for 3′ UTRs of petBD (encoding cytochrome b6 and subunit 4 of the cytochrome b6f complex; Figure 2E), ycf19 and cpcG1, which are also preceded by RNase-E-dependent PSS. Under certain growth conditions, e.g. in the stationary phase, these 3′ UTRs accumulated differently from the main transcript (46). Hence, these 3′ UTRs are top candidates to act as 3′ end-derived sRNAs, analogous to several such riboregulators identified in enterobacteria (65–67). In cyanobacteria, only a single 3′ UTR-derived sRNA is known thus far, ApcZ (68). Thus, the dataset we present provides a valuable resource for the functional characterization of further such sRNAs.
The six RNase E cleavage sites identified directly upstream of a start codon belong to the genes psbA2 (slr1311, photosystem II D1 protein), cpcB (sll1577, phycocyanin beta subunit), sigG (slr1545, sigma factor), slr0373 (hypothetical protein), rcp1 (slr0474, two-component response regulator, interacting with cyanobacterial phytochrome Cph1) and slr1563 (a fructosamine kinase family protein). Of these, the first four are the first gene within the respective transcriptional unit, whereas the latter two are the second or third and the identified cleavage sites are located exactly at the end of an intergenic region. Cutting a 5′ leader can serve as a post-transcriptional regulatory mechanism by lowering translation initiation efficiency or by initiating further degradation, as demonstrated in Synechocystis for the psbA2 (21) and psaL mRNAs (28) and hence very likely also for the here detected additional genes. RNase-E-dependent cleavage sites within intergenic regions might hint towards a wider role of Synechocystis RNase E in the process of operon discoordination, as it was shown for the rimO-crhR dicistron (69).
RNase E facilitates degradation of RNA fragments with structured 5′ ends
To identify factors determining the cleavage position of RNase E, we analysed PSS accumulating in rne(WT) or rne(Ts) regarding both their primary sequence as well as their calculated minimal folding energy (ΔG), which is indicative of the presence of structured regions (Figure 5). Analysing the minimal folding energy (ΔG), using a 25 nt sliding window approach, revealed that the regions around PSS accumulating in rne(WT) were less structured than for shuffled sequences or randomly picked positions (Figure 5A, Supplementary Figure S11). This is in concordance with the known preference of RNase E for single-stranded regions (9,29). Regions exactly downstream of PSS with a higher read count in rne(Ts) are more structured than the average minimal folding energy and there is almost no sequence preference for the respective regions. The accumulation of these RNA fragments in rne(Ts) after RNase E inactivation indicates that these PSS are products of another ribonuclease, but that RNase E activity is necessary to further degrade them (Figure 6A).
Figure 5.

Inference of a sequence signature and folding potential around cleavage sites. (A) Analysis of minimal folding energy (ΔG) around detected cleavage sites, start and stop codons and annotated starts and ends of transcriptional units. Minimal folding energy was calculated at each nucleotide position using a sliding window of 25 nt and a 1 nt step size for a segment of 150 nt up- and downstream of the respective element. (B) Sequence logos of PSS accumulating in rne(WT) or rne(Ts) after heat treatment. Sequences were aligned according to the detected cleavage site. Error bars were calculated by the WebLogo tool and represent an approximate Bayesian 95% confidence interval. (C) Nucleotide composition around detected PSS.
Figure 6.
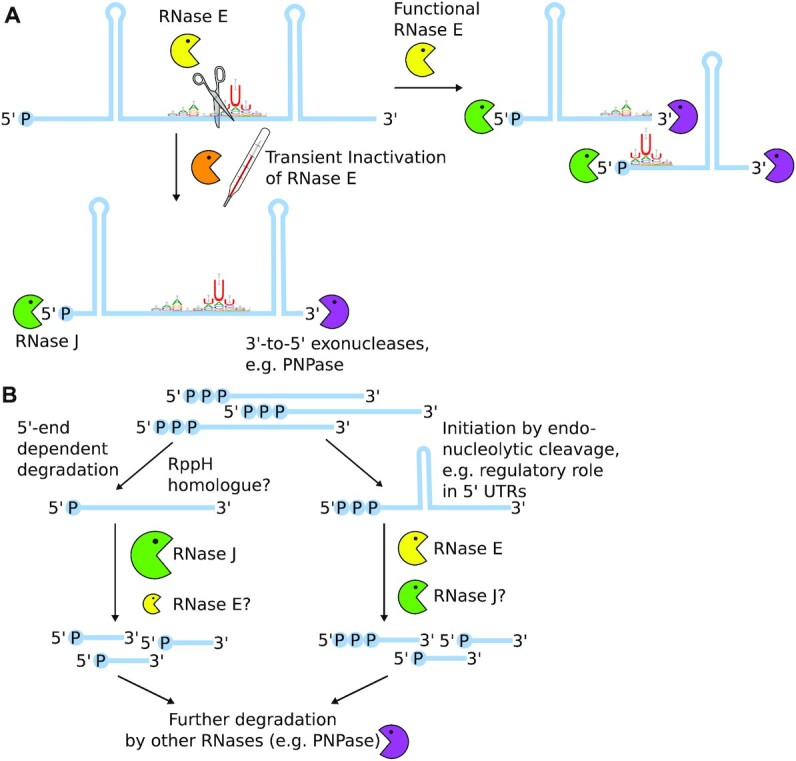
Hypothetical schemes for the coordinated action of RNase E and J in transcript turnover in Synechocystis. (A) RNase E activity is needed to make structured RNA species accessible for exonucleolytic activity of both 5′-to-3′ as well as 3′-to-5′ exonucleases. (B) Transient inactivation of RNase E did not impair 5′-end dependent RNA degradation, hinting towards an important role of RNase J therein. However, RNase E inactivation strongly affected post-transcriptional regulation via regulatory RNA elements such as 5′ UTRs.
Less pronounced secondary structure around start and stop codons might attract RNase E cleavage
We analysed the minimal folding energy of regions surrounding start and stop codons as well as starts and ends of the TUs defined by Kopf et al. (46). We observed that regions around start and stop codons as well as TU starts are less structured than other sections in the genome (Figure 5A). Shortly before the end of TUs, ΔG was slightly lowered, indicative of the presence of terminator structures. The lowered propensity for secondary structures around start and stop codons may contribute to the preference of RNase E for those positions.
RNase-E-dependent PSS reveal a role of the A-U-clamp for positioning cleavage
Sites with higher PSS counts in rne(WT) were enriched for adenine residues at positions 4 nt and 3 nt upstream of the cleavage site (Figure 5B), while the two bases exactly upstream of the cleavage site showed no base preference. The three bases following the cleavage site were enriched for uracil residues, especially at position +2 relative to the cleavage site. These findings are also reflected by the overall nucleotide content of the neighbouring positions (Figure 5C). The created A-U-clamp appears to be a low-specificity and flexible way of positioning the actual cleavage site. Although we cannot discriminate between PSS resulting from a direct endonucleolytic activity of RNase E or subsequent downstream processing by RNase J, such a sequence preference was not yet reported for RNase J. Thus, the identified A-U-clamp is likely RNase E specific and the +2 uridine ruler mechanism identified for Salmonella RNase E is conserved in cyanobacterial RNase E (8). However, gammaproteobacterial and cyanobacterial cleavage sites differ in the upstream nucleotide sequence: Salmonella and Vibrio RNase E possess a slight preference for a guanine residue at position –2 (8). The key importance of this guanine residue was shown for E. coli RNase E when acting on short RNA substrates lacking additional targeting factors such as secondary structures (70). When guanine was exchanged in these substrates for adenine, the cleavage rate of the assayed substrate was reduced by a factor of 12 (70). Another study found that the respective guanine residue is crucial for cleavage if no secondary structures are present in close distance which could guide RNase E to its cleavage site (13). According to these findings, both gammaproteobacterial and cyanobacterial RNase E prefer cleavage after purine bases and rely, to a certain extent, on a uracil residue two nucleotide downstream of the cleavage site. Interestingly, the consensus motif for RNase-E-dependent cleavage sites determined for the GC-rich bacterium Rhodobacter differs strongly from the one presented here (51). A possible reason is that the positioning of RNase E cleavage sites in a GC-rich organism relies more strongly on alternative factors such as 5′ sensing or the presence of neighbouring secondary structures.
RNase E activity is not rate-limiting for 5′-end dependent RNA degradation
Contrasting to what would be expected after the inactivation of a 5′-P dependent RNase, the relative number of PSS reads compared to the number of TSS or transcript reads did not increase in rne(Ts) samples after the heat shock compared to the ratios in all other sequenced samples (Figure 2C). We assume that the transient inactivation of RNase E has no effect on the transformation of 5′-PPP ends to 5′-P ends, i.e. the action of a potential RNA pyrophosphohydrolase (RppH) homologue. Hence, the ratio of PSS reads to TSS reads at TSS positions is an indicator of the velocity and the extent of 5′-end-dependent degradation. We did not observe a significant difference between PSS/TSS ratios at TSS positions between different strains at the used conditions.
If RNase E activity was rate-limiting for 5′-end dependent RNA degradation, the number of 5′-P ends relative to 5′-PPP ends or transcript reads should rise after RNase E inactivation. Such an effect was observed after deletion of RNase J1 and J2 in Staphylococcus aureus (71). This implies that RNase J might be the main ribonuclease performing 5′-end dependent RNA degradation also in cyanobacteria (Figure 6B). However, a milder effect on the transcriptome was found for a knock-down of RNase J in Synechocystis than for RNase E (20). In light of this finding, we assume that both cyanobacterial RNase E and RNase J can partially substitute for each other and that their combined pool is sufficiently large to compensate for an inactivation of one of them, in regard to 5′-end dependent degradation.
Transient inactivation of RNase E leads to downregulation of sigA
Transcripts of several sigma factors, such as sigA and sigE are down-regulated in rne(Ts) compared to rne(WT) after heat treatment. The strong downregulation of sigA (log2FC = −2.0, p.adj = 1.1 × 10−41), encoding the principal sigma factor SigA, indicates that RNase E inactivation confronts the cell with a strong stress in line with previous observations that it is downregulated in response to stress conditions such as heat, high salinity or photooxidative stress (reviewed in (72)). Interestingly, no other, alternative, sigma factor was significantly upregulated compared to the control strain. In contrast, growth at 39°C appeared to be only a minor stress for the congenic control strain, judged by the negligible effect on sigA transcription (log2FC = −0.6, p.adj = 4.5 × 10−4) and moderate upregulation of the heat-responsive sigma factor sigB (log2FC = 1.2, p.adj = 1.9 × 10−19). Interestingly, an asRNA complementary to the 3′ region of sigA was strongly upregulated in rne(Ts) after the heat shock (TU3505, Supplementary Figure S12). This asRNA originates from a TSS leading to an overlap with the rRNA precursor. Also, several PSS detected in this asRNA were associated with divergent read counts between rne(WT) or rne(Ts), which is indicative of a direct action of RNase E on this transcript. Hence, this asRNA, which may impact the processing of the rRNA precursor and the level of the sigA mRNA level, is also a substrate for RNase E.
Effect of RNase E inactivation on non-coding RNAs substantiate its role in post-transcriptional regulation
The role of RNase E in post-transcriptional regulation in enterobacteria is well established, while a few examples indicated a likely important function also in Synechocystis (21,27,28). Here, we noticed several non-coding RNAs that were differentially expressed in the rne(WT) versus rne(Ts) comparison after one hour heat treatment, e.g. 16.0% of all annotated asRNAs and 16.4% of all annotated ncRNAs (Table 3). Hence, we decided to elucidate the impact of RNase E on these RNAs more closely.
A well-established target of Synechocystis RNase E is the 5′ UTR of psbA2, encoding the photosystem II D2 protein, which is degraded by RNase E during the acclimation to darkness (20,21,29). In vitro cleavage assays conducted on a 35-mer identified major cleavage positions downstream of positions 27 and 31 (20,29). Indeed, we detected PSS within the psbA2 5′ UTR which were associated with divergent read counts between rne(WT) and rne(Ts), albeit at different positions than mapped in vitro (Figure 4B). We identified such a PSS at position 17 (pos. 7, 197 on the chromosome).
PsrR1 is an sRNA upregulated in response to high light and CO2 depletion. PsrR1 regulates the expression of several photosynthesis-related proteins, e.g. PsaL, CpcA, PsbB and PsaJ on post-transcriptional level (28). For psaL mRNA, it was shown that this regulation involves RNase E. In our data set, PsrR1 was strongly downregulated after the heat shock. In rne(Ts), PsrR1 levels were further reduced than in rne(WT). PsrR1 was predicted to interact with cpcBA by base-pairing close to the start codon of both genes (28). Here, we observed PSS with enhanced read counts in rne(WT) compared to rne(Ts) after the heat shock which were located close to the start codons of cpcA and cpcB and thereby potentially removing the AUG from the respective coding region (Supplementary Figure S13). This implies that, similar to PsaL, the translation of CpcA and CpcB is also regulated by a combined action of PsrR1 and RNase E.
PmgR1 is an sRNA involved in the switch to photomixotrophic growth (73). PmgR1 was upregulated in WT, rne(WT) and rne(Ts) after the heat shock (Figure 4C, F). This upregulation was more strongly pronounced in rne(WT) than in rne(Ts). We identified several PSS in PmgR1 which accumulated in WT and rne(WT) compared to rne(Ts), indicative of a processing of PmgR1 by RNase E. When analysed via northern blot hybridisation, a shorter version of PmgR1 produced in WT and rne(WT) was missing in rne(Ts) (Figure 4F). Direct RNA targets of PmgR1 are unknown and its mechanism remains to be elucidated. Processing by RNase E might yield mature, active PmgR1 or, contrary, inactivate the sRNA, e.g. by removing a putative seed region.
The transcription and translation of gifB encoding the glutamine synthetase inactivating protein factor IF17 are controlled by the transcription factor NtcA and a glutamine riboswitch, respectively (74,75). The gifB transcript accumulates strongly in rne(Ts) after the heat shock, but not in rne(WT) or WT (Figure 4D, G). Concurrently, several PSS with higher counts for rne(Ts) than rne(WT) after the heat shock were detected in the gifB CDS and riboswitch, and in the asRNA sll1515-as2, which overlaps the gifB 5′ UTR. The single PSS within the riboswitch is located at position +35, which was experimentally demonstrated to be essential for riboswitch function (74). The exact mechanism of action has remained unknown for this riboswitch. Based on the identified PSS in a functionally relevant domain, we suggest the possibility that this riboswitch might be dual-acting, reminiscent of the lysine sensing riboswitch lysC present in E. coli, which is both controlling translation of the downstream CDS as well as exposing RNase E cleavage sites upon lysine binding (76,77). Additionally, RNase-E-dependent cleavage sites elsewhere within the mRNA and sll1515-as might affect gifB mRNA stability further.
The autoregulation of the rne transcript in E. coli is another example of 5′ UTR-mediated transcript level regulation (56). Using TIER-seq, we captured several RNase-E-dependent cleavage positions in the 5′ UTR of rne (Figure 4E). Together with the enhanced accumulation of the rne-rnhB transcript upon RNase E inactivation (Figure 1E), this provides strong evidence for the autoregulation of rne levels by RNase E also in Synechocystis.
Additional genes whose transcript level seem to be regulated via an RNase E cleavage within their 5′ UTR are sll0547, coding for an unknown protein and rbp2 (ssr1480), encoding an RNA-binding protein involved in targeting mRNAs for photosynthetic proteins to the thylakoid membrane (78). Further examples for asRNAs in which RNase-E-dependent PSS were found are slr0261-as, which is located antisense to ndhH, encoding NADH dehydrogenase subunit 7, as well as the previously studied asRNAs IsrR (79) and PsbA3R (27).
CONCLUSION
Our findings demonstrate that a substitution homologous to the rne-3071 mutation of E. coli RNase E can be successfully introduced in a cyanobacterial RNase E, if combined with one of three detected spontaneous secondary mutations. This observation opens up an important avenue for the in vivo investigation of RNase E homologues that are more distantly related to the archetypical enterobacterial model. The generated strain enabled us to identify 1472 RNase-E-dependent cleavage sites. Cleavage by an endoribonuclease results in a 5′ and a 3′ fragment, which may be rapidly further processed by other RNases. tagRNA-Seq only captures stable 3′, but not 5′ fragments, which could be added in future studies by using an RNA-Seq approach also capturing 3′ ends (58). Whereas cyanobacterial RNase E activity is not rate-limiting for 5′-end dependent RNA degradation, we showed that it is important for the degradation of RNA fragments with strong 5′ secondary structures, which hints towards one possible reason why the enzyme is essential in Synechocystis. Our data show that cyanobacterial RNase E differs slightly from gammaproteobacterial RNase E in its target affinity, by preferring adenine upstream of cleavage sites instead of guanine. Future work has to elucidate if other targeting mechanisms known for enterobacterial RNase E, e.g. 5′ sensing and 5′ bypass, are present in the compact cyanobacterial RNase E and which relevance they might have. The created mutant strain, rne(Ts), represents a promising tool for the future analysis of post-transcriptional regulation as well as the maturation and action of regulatory ncRNAs. Exploring the impact of transient RNase E inactivation under different environmental conditions will help to further understand the function of this widely conserved and versatile endoribonuclease in cyanobacteria.
DATA AVAILABILITY
Galaxy workflows used for raw read processing and several downstream analysis steps are available (https://usegalaxy.eu/u/ute-hoffmann/w/tier-seqprocessing1, https://usegalaxy.eu/u/ute-hoffmann/w/tier-seqtranscript, https://usegalaxy.eu/u/ute-hoffmann/w/tier-seqpss-tss and https://usegalaxy.eu/u/ute-hoffmann/w/tiertranscript-coverage).
All further code used for data processing and analysis are available in the GitHub and Zenodo repositories (https://github.com/ute-hoffmann/TIER-synechocystis, https://www.doi.org/10.5281/zenodo.5515447).
FastQ files from RNA sequencing have been deposited with the Gene Expression Omnibus (GEO) Data bank under accession number GSE180316.
Supplementary Material
ACKNOWLEDGEMENTS
We thank J. Behler for providing the plasmid for the chromosomal integration of a 3xFLAG tag N-terminally of rne. We thank W. Bigott and S. Oeser for technical assistance, S. Oeser and N. Schuergers for fruitful discussions and M. Treppner for biostatistical advice.
The authors acknowledge the support of the Freiburg Galaxy Team: Björn Grüning, Bioinformatics, University of Freiburg (Germany) funded by the Collaborative Research Centre 992 Medical Epigenetics (Deutsche Forschungsgemeinschaft [SFB 992/1 2012]) and Bundesministerium für Bildung und Forschung BMBF grant 031 A538A de.NBI-RBC.
Contributor Information
Ute A Hoffmann, Molecular Genetics of Prokaryotes, Institute of Biology III, University of Freiburg, 79104 Freiburg, Germany.
Florian Heyl, Bioinformatics Group, Department of Computer Science, University of Freiburg, 79110 Freiburg, Germany.
Said N Rogh, Molecular Genetics of Prokaryotes, Institute of Biology III, University of Freiburg, 79104 Freiburg, Germany.
Thomas Wallner, Molecular Genetics of Prokaryotes, Institute of Biology III, University of Freiburg, 79104 Freiburg, Germany.
Rolf Backofen, Bioinformatics Group, Department of Computer Science, University of Freiburg, 79110 Freiburg, Germany; Signalling Research Centres BIOSS and CIBSS, University of Freiburg, 79104 Freiburg, Germany.
Wolfgang R Hess, Genetics and Experimental Bioinformatics, Faculty of Biology, University of Freiburg, 79104 Freiburg, Germany.
Claudia Steglich, Genetics and Experimental Bioinformatics, Faculty of Biology, University of Freiburg, 79104 Freiburg, Germany.
Annegret Wilde, Molecular Genetics of Prokaryotes, Institute of Biology III, University of Freiburg, 79104 Freiburg, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft Research Training Group MeInBio [322977937/GRK2344 to A.W., W.R.H., R.B.]; Deutsche Forschungsgemeinschaft Grants [STE 1192/4-2 to C.S.; WI 2014/9-1 to A.W.; BA 2168/11-2 to R.B.]. The article processing charge was funded by the Baden-Wuerttemberg Ministry of Science, Research and Art and the University of Freiburg in the funding programme Open Access Publishing.
Conflict of interest statement. None declared.
REFERENCES
- 1. Durand S., Condon C.. Storz G., Papenfort K.. RNases and helicases in Gram-positive bacteria. Regulating with RNA in Bacteria and Archaea. 2018; American Society for Microbiology; 37–53. [Google Scholar]
- 2. Hui M.P., Foley P.L., Belasco J.G.. Messenger RNA degradation in bacterial cells. Annu. Rev. Genet. 2014; 48:537–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tejada-Arranz A., de Crécy-Lagard V., de Reuse H.. Bacterial RNA degradosomes: molecular machines under tight control. Trends Biochem. Sci. 2020; 45:42–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Laalami S., Cavaiuolo M., Roque S., Chagneau C., Putzer H.. Escherichia coli RNase E can efficiently replace RNase Y in Bacillus subtilis. Nucleic Acids Res. 2021; 49:4643–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ghora B.K., Apirion D.. Structural analysis and in vitro processing to p5 rRNA of a 9S RNA molecule isolated from an rne mutant of E. coli. Cell. 1978; 15:1055–1066. [DOI] [PubMed] [Google Scholar]
- 6. Li Z., Pandit S., Deutscher M.P.. RNase G (CafA protein) and RNase E are both required for the 5′ maturation of 16S ribosomal RNA. EMBO J. 1999; 18:2878–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ow M.C., Kushner S.R.. Initiation of tRNA maturation by RNase E is essential for cell viability in E. coli. Genes Dev. 2002; 16:1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chao Y., Li L., Girodat D., Förstner K.U., Said N., Corcoran C., Śmiga M., Papenfort K., Reinhardt R., Wieden H.-J.et al.. In Vivo cleavage map illuminates the central role of RNase E in coding and non-coding RNA pathways. Mol. Cell. 2017; 65:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Babitzke P., Kushner S.R.. The Ams (altered mRNA stability) protein and ribonuclease E are encoded by the same structural gene of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stead M.B., Marshburn S., Mohanty B.K., Mitra J., Castillo L.P., Ray D., van Bakel H., Hughes T.R., Kushner S.R.. Analysis of Escherichia coli RNase E and RNase III activity in vivo using tiling microarrays. Nucleic Acids Res. 2011; 39:3188–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mackie G.A. Ribonuclease E is a 5′-end-dependent endonuclease. Nature. 1998; 395:720–724. [DOI] [PubMed] [Google Scholar]
- 12. Bandyra K.J., Wandzik J.M., Luisi B.F.. Substrate recognition and autoinhibition in the central ribonuclease RNase E. Mol. Cell. 2018; 72:275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kaberdin V.R., Walsh A.P., Jakobsen T., McDowall K.J., von Gabain A.. Enhanced cleavage of RNA mediated by an interaction between substrates and the arginine-rich domain of E. coli ribonuclease E. J. Mol. Biol. 2000; 301:257–264. [DOI] [PubMed] [Google Scholar]
- 14. Clarke J.E., Kime L., Romero A.D., McDowall K.J.. Direct entry by RNase E is a major pathway for the degradation and processing of RNA in Escherichia coli. Nucleic Acids Res. 2014; 42:11733–11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaberdin V.R., Miczak A., Jakobsen J.S., Lin-Chao S., McDowall K.J., von Gabain A.. The endoribonucleolytic N-terminal half of Escherichia coli RNase E is evolutionarily conserved in Synechocystis sp. and other bacteria but not the C-terminal half, which is sufficient for degradosome assembly. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:11637–11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moll I., Afonyushkin T., Vytvytska O., Kaberdin V.R., Bläsi U.. Coincident Hfq binding and RNase E cleavage sites on mRNA and small regulatory RNAs. RNA. 2003; 9:1308–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Göpel Y., Papenfort K., Reichenbach B., Vogel J., Görke B.. Targeted decay of a regulatory small RNA by an adaptor protein for RNase E and counteraction by an anti-adaptor RNA. Genes Dev. 2013; 27:552–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schuergers N., Ruppert U., Watanabe S., Nürnberg D.J., Lochnit G., Dienst D., Mullineaux C.W., Wilde A.. Binding of the RNA chaperone Hfq to the type IV pilus base is crucial for its function in Synechocystis sp. PCC 6803. Mol. Microbiol. 2014; 92:840–852. [DOI] [PubMed] [Google Scholar]
- 19. Hagemann M., Hess W.R.. Systems and synthetic biology for the biotechnological application of cyanobacteria. Curr. Opin. Biotechnol. 2018; 49:94–99. [DOI] [PubMed] [Google Scholar]
- 20. Cavaiuolo M., Chagneau C., Laalami S., Putzer H.. Impact of RNase E and RNase J on global mRNA metabolism in the cyanobacterium Synechocystis PCC6803. Front. Microbiol. 2020; 11:1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horie Y., Ito Y., Ono M., Moriwaki N., Kato H., Hamakubo Y., Amano T., Wachi M., Shirai M., Asayama M.. Dark-induced mRNA instability involves RNase E/G-type endoribonuclease cleavage at the AU-box and SD sequences in cyanobacteria. Mol. Genet. Genomics. 2007; 278:331–346. [DOI] [PubMed] [Google Scholar]
- 22. Yao L., Shabestary K., Björk S.M., Asplund-Samuelsson J., Joensson H.N., Jahn M., Hudson E.P.. Pooled CRISPRi screening of the cyanobacterium Synechocystis sp PCC 6803 for enhanced industrial phenotypes. Nat. Commun. 2020; 11:1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aït-Bara S., Carpousis A.J.. RNA degradosomes in bacteria and chloroplasts: classification, distribution and evolution of RNase E homologs. Mol. Microbiol. 2015; 97:1021–1135. [DOI] [PubMed] [Google Scholar]
- 24. Schein A., Sheffy-Levin S., Glaser F., Schuster G.. The RNase E/G-type endoribonuclease of higher plants is located in the chloroplast and cleaves RNA similarly to the E. coli enzyme. RNA. 2008; 14:1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee K., Cohen S.N.. A Streptomyces coelicolor functional orthologue of Escherichia coli RNase E shows shuffling of catalytic and PNPase-binding domains. Mol. Microbiol. 2003; 48:349–360. [DOI] [PubMed] [Google Scholar]
- 26. Zhang J.-Y., Deng X.-M., Li F.-P., Wang L., Huang Q.-Y., Zhang C.-C., Chen W.-L.. RNase E forms a complex with polynucleotide phosphorylase in cyanobacteria via a cyanobacterial-specific nonapeptide in the noncatalytic region. RNA. 2014; 20:568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sakurai I., Stazic D., Eisenhut M., Vuorio E., Steglich C., Hess W.R., Aro E.-M.. Positive regulation of psbA gene expression by cis-encoded antisense RNAs in Synechocystis sp. PCC 6803. Plant Physiol. 2012; 160:1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Georg J., Dienst D., Schurgers N., Wallner T., Kopp D., Stazic D., Kuchmina E., Klahn S., Lokstein H., Hess W.R.et al.. The small regulatory RNA SyR1/PsrR1 controls photosynthetic functions in cyanobacteria. Plant Cell. 2014; 26:3661–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Behler J., Sharma K., Reimann V., Wilde A., Urlaub H., Hess W.R.. The host-encoded RNase E endonuclease as the crRNA maturation enzyme in a CRISPR–Cas subtype III-Bv system. Nat. Microbiol. 2018; 3:367–377. [DOI] [PubMed] [Google Scholar]
- 30. Trautmann D., Voß B., Wilde A., Al-Babili S., Hess W.R.. Microevolution in cyanobacteria: re-sequencing a motile substrain of Synechocystis sp. PCC 6803. DNA Res. 2012; 19:435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rippka R., Deruelles J., Waterbury J.B., Herdman M., Stanier R.Y.. Generic assignments, strain histories and properties of pure cultures of cyanobacteria. Microbiology. 1979; 111:1–61. [Google Scholar]
- 32. Beyer H.M., Gonschorek P., Samodelov S.L., Meier M., Weber W., Zurbriggen M.D.. AQUA cloning: a versatile and simple enzyme-free cloning approach. PLoS One. 2015; 10:e0137652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zinchenko V.V., Piven I.V., Melnik V.A., Shestakov S.V.. Vectors for the complementation analysis of cyanobacterial mutants. Russ. J. Genet. 1999; 35:228–232. [Google Scholar]
- 34. Pinto F.L., Thapper A., Sontheim W., Lindblad P.. Analysis of current and alternative phenol based RNA extraction methodologies for cyanobacteria. BMC Mol. Biol. 2009; 10:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wallner T., Pedroza L., Voigt K., Kaever V., Wilde A.. The cyanobacterial phytochrome 2 regulates the expression of motility-related genes through the second messenger cyclic di-GMP. Photochem. Photobiol. Sci. 2020; 19:631–643. [DOI] [PubMed] [Google Scholar]
- 36. Innocenti N., Golumbeanu M., Fouquier d’Hérouël A., Lacoux C., Bonnin R.A., Kennedy S.P., Wessner F., Serror P., Bouloc P., Repoila F.et al.. Whole-genome mapping of 5′ RNA ends in bacteria by tagged sequencing: a comprehensive view in Enterococcus faecalis. RNA. 2015; 21:1018–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith T., Heger A., Sudbery I.. UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 2017; 27:491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roy A., Kucukural A., Zhang Y.. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010; 5:725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang J., Zhang Y.. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015; 43:W174–W181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oberto J. SyntTax: a web server linking synteny to prokaryotic taxonomy. BMC Bioinform. 2013; 14:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Čech M., Chilton J., Clements D., Coraor N., Grüning B.A.et al.. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018; 46:W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Anders S., Pyl P.T., Huber W.. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kaneko T., Sato S., Kotani H., Tanaka A., Asamizu E., Nakamura Y., Miyajima N., Hirosawa M., Sugiura M., Sasamoto S.et al.. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 1996; 3:109–136. [DOI] [PubMed] [Google Scholar]
- 44. Mitschke J., Georg J., Scholz I., Sharma C.M., Dienst D., Bantscheff J., Voß B., Steglich C., Wilde A., Vogel J.et al.. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:2124–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Baumgartner D., Kopf M., Klähn S., Steglich C., Hess W.R.. Small proteins in cyanobacteria provide a paradigm for the functional analysis of the bacterial micro-proteome. BMC Microbiol. 2016; 16:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kopf M., Klähn S., Scholz I., Matthiessen J.K.F., Hess W.R., Voß B.. Comparative analysis of the primary transcriptome of Synechocystis sp. PCC 6803. DNA Res. 2014; 21:527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Crooks G.E., Hon G., Chandonia J.-M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lorenz R., Bernhart S.H., Höner zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms for Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Förstner K.U., Reuscher C.M., Haberzettl K., Weber L., Klug G.. RNase E cleavage shapes the transcriptome of Rhodobacter sphaeroides and strongly impacts phototrophic growth. Life Sci. Alliance. 2018; 1:e201800080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hoyos M., Huber M., Förstner K.U., Papenfort K.. Gene autoregulation by 3′ UTR-derived bacterial small RNAs. eLife. 2020; 9:e58836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McDowall K.J., Hernandez R.G., Lin-Chao S., Cohen S.N.. The ams-1 and rne-3071 temperature-sensitive mutations in the ams gene are in close proximity to each other and cause substitutions within a domain that resembles a product of the Escherichia coli mre locus. J. Bacteriol. 1993; 175:4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Diwa A.A., Jiang X., Schapira M., Belasco J.G.. Two distinct regions on the surface of an RNA-binding domain are crucial for RNase E function. Mol. Microbiol. 2002; 46:959–969. [DOI] [PubMed] [Google Scholar]
- 55. Nagy C., Thiel K., Mulaku E., Mustila H., Tamagnini P., Aro E.-M., Pacheco C.C., Kallio P.. Comparison of alternative integration sites in the chromosome and the native plasmids of the cyanobacterium Synechocystis sp. PCC 6803 in respect to expression efficiency and copy number. Microb. Cell Factories. 2021; 20:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jain C., Belasco J.G.. RNase E autoregulates its synthesis by controlling the degradation rate of its own mRNA in Escherichia coli: unusual sensitivity of the rne transcript to RNase E activity. Genes Dev. 1995; 9:84–96. [DOI] [PubMed] [Google Scholar]
- 57. Stazic D., Pekarski I., Kopf M., Lindell D., Steglich C.. A novel strategy for exploitation of host RNase E activity by a marine cyanophage. Genetics. 2016; 203:1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Broglia L., Lécrivain A.-L., Renault T.T., Hahnke K., Ahmed-Begrich R., Le Rhun A., Charpentier E.. An RNA-seq based comparative approach reveals the transcriptome-wide interplay between 3′-to-5′ exoRNases and RNase Y. Nat. Commun. 2020; 11:1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Misra T.K., Apirion D.. RNase E, an RNA processing enzyme from Escherichia coli. J. Biol. Chem. 1979; 254:11154–11159. [PubMed] [Google Scholar]
- 60. Kime L., Clarke J.E., Romero A.D., Grasby J.A., McDowall K.J.. Adjacent single-stranded regions mediate processing of tRNA precursors by RNase E direct entry. Nucleic Acids Res. 2014; 42:4577–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Guimarães V.A., Le Scornet A., Khemici V., Hausmann S., Armitano J., Prados J., Jousselin A., Manzano C., Linder P., Redder P. RNase J1 and J2 are host-encoded factors for plasmid replication. Front. Microbiol. 2021; 12:966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Saramago M., Bárria C., Arraiano C.M., Domingues S.. Ribonucleases, antisense RNAs and the control of bacterial plasmids. Plasmid. 2015; 78:26–36. [DOI] [PubMed] [Google Scholar]
- 63. Scholz I., Lange S.J., Hein S., Hess W.R., Backofen R.. CRISPR-Cas systems in the cyanobacterium Synechocystis sp. PCC6803 exhibit distinct processing pathways involving at least two Cas6 and a Cmr2 protein. PLoS ONE. 2013; 8:e56470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kopfmann S., Hess W.R.. Toxin-antitoxin systems on the large defense plasmid pSYSA of Synechocystis sp. PCC 6803. J. Biol. Chem. 2013; 288:7399–7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. De Mets F., Van Melderen L., Gottesman S.. Regulation of acetate metabolism and coordination with the TCA cycle via a processed small RNA. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:1043–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang C., Chao Y., Matera G., Gao Q., Vogel J.. The conserved 3′ UTR-derived small RNA NarS mediates mRNA crossregulation during nitrate respiration. Nucleic Acids Res. 2020; 48:2126–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Miyakoshi M., Matera G., Maki K., Sone Y., Vogel J.. Functional expansion of a TCA cycle operon mRNA by a 3′ end-derived small RNA. Nucleic Acids Res. 2019; 47:2075–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhan J., Steglich C., Scholz I., Hess W.R., Kirilovsky D.. Inverse regulation of light harvesting and photoprotection is mediated by a 3′-end-derived sRNA in cyanobacteria. Plant Cell. 2021; 33:358–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rosana A.R.R., Whitford D.S., Migur A., Steglich C., Kujat-Choy S.L., Hess W.R., Owttrim G.W.. RNA helicase–regulated processing of the Synechocystis rimO–crhR operon results in differential cistron expression and accumulation of two sRNAs. J. Biol. Chem. 2020; 295:6372–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Redko Y., Tock M.R., Adams C.J., Kaberdin V.R., Grasby J.A., McDowall K.J.. Determination of the catalytic parameters of the N-terminal half of Escherichia coli ribonuclease E and the identification of critical functional groups in RNA substrates. J. Biol. Chem. 2003; 278:44001–44008. [DOI] [PubMed] [Google Scholar]
- 71. Linder P., Lemeille S., Redder P.. Transcriptome-wide analyses of 5′-ends in RNase J mutants of a Gram-positive pathogen reveal a role in RNA maturation, regulation and degradation. PLoS Genet. 2014; 10:e1004207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Srivastava A., Summers M.L., Sobotka R.. Cyanobacterial sigma factors: Current and future applications for biotechnological advances. Biotechnol. Advances. 2020; 40:107517. [DOI] [PubMed] [Google Scholar]
- 73. de Porcellinis A.J., Klähn S., Rosgaard L., Kirsch R., Gutekunst K., Georg J., Hess W.R., Sakuragi Y.. The non-coding RNA Ncr0700/PmgR1 is required for photomixotrophic growth and the regulation of glycogen accumulation in the cyanobacterium Synechocystis sp. PCC 6803. Plant Cell Physiol. 2016; 57:2091–2103. [DOI] [PubMed] [Google Scholar]
- 74. Klähn S., Bolay P., Wright P.R., Atilho R.M., Brewer K.I., Hagemann M., Breaker R.R., Hess W.R.. A glutamine riboswitch is a key element for the regulation of glutamine synthetase in cyanobacteria. Nucleic Acids Res. 2018; 46:10082–10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. García-Domínguez M., Reyes J.C., Florencio F.J.. NtcA represses transcription of gifA and gifB, genes that encode inhibitors of glutamine synthetase type I from Synechocystis sp. PCC 6803. Mol. Microbiol. 2000; 35:1192–1201. [DOI] [PubMed] [Google Scholar]
- 76. Caron M.-P., Bastet L., Lussier A., Simoneau-Roy M., Massé E., Lafontaine D.A.. Dual-acting riboswitch control of translation initiation and mRNA decay. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Richards J., Belasco J.G.. Riboswitch control of bacterial RNA stability. Mol. Microbiol. 2021; 00:361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mahbub M., Hemm L., Yang Y., Kaur R., Carmen H., Engl C., Huokko T., Riediger M., Watanabe S., Liu L.-N.et al.. mRNA localization, reaction centre biogenesis and thylakoid membrane targeting in cyanobacteria. Nat Plants. 2020; 6:1179–1191. [DOI] [PubMed] [Google Scholar]
- 79. Dühring U., Axmann I.M., Hess W.R., Wilde A.. An internal antisense RNA regulates expression of the photosynthesis gene isiA. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:7054–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Galaxy workflows used for raw read processing and several downstream analysis steps are available (https://usegalaxy.eu/u/ute-hoffmann/w/tier-seqprocessing1, https://usegalaxy.eu/u/ute-hoffmann/w/tier-seqtranscript, https://usegalaxy.eu/u/ute-hoffmann/w/tier-seqpss-tss and https://usegalaxy.eu/u/ute-hoffmann/w/tiertranscript-coverage).
All further code used for data processing and analysis are available in the GitHub and Zenodo repositories (https://github.com/ute-hoffmann/TIER-synechocystis, https://www.doi.org/10.5281/zenodo.5515447).
FastQ files from RNA sequencing have been deposited with the Gene Expression Omnibus (GEO) Data bank under accession number GSE180316.