

Abstract
The tumor microenvironment (TME), including immune cells, cancer‐associated fibroblasts, endothelial cells, adjacent normal cells, and others, plays a crucial role in influencing tumor behavior and progression. Here, we characterized the TME in 83 primary renal tumors and matched metastatic or recurrence tissue samples (n = 15) from papillary renal cell carcinoma (pRCC) types 1 (n = 20) and 2 (n = 49), collecting duct carcinomas (CDC; n = 14), and high‐grade urothelial carcinomas (HGUC; n = 5). We investigated 10 different markers of immune infiltration, vasculature, cell proliferation, and epithelial‐to‐mesenchymal transition by using machine learning image analysis in conjunction with immunohistochemistry. Marker expression was compared by Mann–Whitney and Kruskal–Wallis tests and correlations across markers using Spearman's rank correlation coefficient. Multivariable Poisson regression analysis was used to compare marker expression between histological types, while accounting for variation in tissue size. Several immune markers showed different rates of expression across histological types of renal carcinoma. Using pRCC1 as reference, the incidence rate ratio (IRR) of CD3+ T cells (IRR [95% confidence interval, CI] = 2.48 [1.53–4.01]) and CD20+ B cells (IRR [95% CI] = 4.38 [1.22–5.58]) was statistically significantly higher in CDC. In contrast, CD68+ macrophages predominated in pRCC1 (IRR [95% CI] = 2.35 [1.42–3.9]). Spatial analysis revealed CD3+ T‐cell and CD20+ B‐cell expressions in CDC to be higher at the proximal (p < 0.0001) and distal (p < 0.0001) tumor periphery than within the central tumor core. In contrast, expression of CD68+ macrophages in pRCC2 was higher in the tumor center compared to the proximal (p = 0.0451) tumor periphery and pRCC1 showed a distance‐dependent reduction, from the central tumor, in CD68+ macrophages with the lowest expression of CD68 marker at the distal tumor periphery (p = 0.004). This study provides novel insights into the TME of rare kidney cancer types, which are often understudied. Our findings of differences in marker expression and localization by histological subtype could have implications for tumor progression and response to immunotherapies or other targeted therapies.
Keywords: tumor microenvironment, rare cancer, kidney cancer, digital pathology, papillary renal cell carcinoma
Introduction
Tumor formation and progression are influenced by two main factors: genomic changes and the rearrangement of the components of the tumor microenvironment (TME) [1, 2]. The TME is tumor dependent [3] and although various immune cells may be recruited to the tumor site, their tumor killing functions are often inhibited, resulting in tumor progression. Thus, understanding the mechanisms, and cells, governing immune evasion in the TME is essential to identify novel strategies to disrupt tumor interactions with its surrounding environment and effectively treat cancer [4].
Several of the immunosuppressive components within the TME have been characterized and used in the development of novel immunotherapies, including in kidney cancer, the eighth most common malignancy in the United States [5]. However, most investigations of the TME in renal cancers have focused on clear cell renal cell carcinoma (ccRCC), which is the most frequent renal histotype in adults with a 5‐year survival rate of 68–75% [6]. ccRCCs show abundant immune cell infiltration [7, 8, 9] and there is a role for anti‐angiogenic therapies as this TME has been linked to increased angiogenic activity following Von Hippel Lindau tumor suppressor loss [10, 11]. Clinical analysis of immune checkpoint inhibitors alone [12, 13], or in combination with anti‐angiogenic therapies [14], has shown promise in relation to improved clinical outcomes for patients with ccRCC in a few studies, highlighting the relevance of the TME in facilitating therapeutic approaches and precision medicine. However, to date, little analysis of the TME has been carried out in rarer forms of renal cancers, which often present with more aggressive or metastatic disease at diagnosis [15].
Papillary renal cell carcinoma (pRCC) accounts for 10–20% of all RCC and is histologically characterized by a proliferation of papillae composed of fibrovascular cores lined by tumor cells [16]. pRCC can be further subtyped as type 1 (pRCC1) and type 2 (pRCC2), the latter being more aggressive than the former [17] with 5‐year disease‐specific survival rates of pRCC1 and pRCC2 of 94.5 and 66.4%, respectively [18]. Collecting duct carcinoma (CDC) arises from the collecting duct in the renal medulla [19]. It comprises less than 1% of all primary renal tumors and is highly aggressive, with most subjects presenting with metastatic disease [20] at the time of diagnosis and a 5‐year survival rate of just 8.8% [21]. Urothelial carcinoma arises from the urothelium of the renal pelvis and is a highly variable and aggressive disease [22]. High‐grade papillary urothelial carcinomas (HGUC) grow more quickly than low‐grade disease, and are more likely to metastasize, with a 5‐year survival rate of 6% for metastatic disease [23].
With the aim to better understand the TME in renal cell carcinomas, we characterized the TME in 103 rare kidney cancer tissue samples by applying machine learning algorithms to digitized whole slide sections stained immunohistochemically for immune, endothelial, and epithelial‐to‐mesenchymal markers.
Materials and methods
Sample collection and subject information
This study was based on archived samples collected at the Regina Elena Cancer Institute, Rome, Italy, including 103 tumor samples from 83 subjects. Specifically, we analyzed 88 primary tumor samples (20 pRCC1, 49 pRCC2, 14 CDC, 5 HGUC), 13 metastatic tissue samples, including both distant metastasis and lymph node metastasis (8 pRCC2, 4 CDC, 1 HGUC), and 2 recurrences (2 pRCC2) (supplementary material, Table S1). All tissue specimens were derived from surgical resection. Written informed consent to allow banking of biospecimens for future scientific research was obtained from each subject. This work was excluded from the NCI IRB Review per 45 CFR 46 and NIH policy for the use of specimens/data by the Office of Human Subjects Research Protections (OHSRP) of the National Institutes of Health. The data were anonymized. The pathology of all tissue samples underwent centralized review by a specialist in uropathology (SS) to confirm the diagnosis. Tumor tissue samples were formalin fixed, paraffin embedded, sectioned at 3 μm thickness, mounted on SuperFrost Plus slides (Menzel‐Gläser, Braunschweig, Germany), and stained with hematoxylin and eosin (H&E). H&E slides were inspected for the presence of neoplastic cells and the percentages of epithelial and mesenchymal regions on each slide were estimated by an expert pathologist (SS). Samples with compromised fixation/processing were excluded from the study.
Immunohistochemistry
Slides were baked at 60 °C for 1 h prior to immunostaining. Immunohistochemistry (IHC) for detection of 10 markers per tissue sample was performed on automated staining platforms using the following antibodies (supplementary material, Table S2): pan‐macrophage marker (CD68), M2‐like macrophage/tumor‐associated macrophage (TAM) marker (CD163), pan‐T‐cell markers (CD3), cytotoxic T‐cell maker (CD8), B‐cell marker (CD20), marker of immune suppression (PD‐L1), angiogenic marker (CD31), proliferation marker (Ki67), marker of tumor tissue (PanCK), and marker of epithelial‐to‐mesenchymal transition (EMT) (vimentin). In addition, staining of LAG3 (a marker of NK cells and T cells) was performed but expression levels were below detection in all samples. Staining was carried out as per the manufacturer's protocols and recommendations. After deparaffinization, rehydration, and antigen retrieval in citrate buffer (10 mm, pH 6.1), the tissue sections were stained for the marker of interest (supplementary material, Table S2). Positive control human tissues were used for all markers (supplementary material, Table S2).
Immunoreactions were revealed by Bond Polymer Refine Detection on an automated autostainer (Bond™Max, Leica Biosystems, Milan, Italy). Standard processing steps were performed according to the manufacturer's instructions. Diaminobenzidine was used as chromogenic substrate. Stained slides were rinsed in distilled water, dehydrated, cleared in xylene, and cover slipped prior to scanning.
IHC for PDL‐1 and vimentin was conducted at the Molecular Digital Pathology Laboratory of the National Cancer Institute, NIH. All other IHC slides were prepared at the Regina Elena Cancer Institute, Rome, Italy.
Image analysis
Slides were scanned using an Aperio AT2 DX scanner (Leica Biosystems, Richmond, IL, USA) at ×40 magnification. Scanned whole slide images were analyzed using the HALO image analysis platform (Indica labs, Albuquerque, NM, USA). The HALO Random Forest classifier was trained to differentiate between tissue, glass, and folds/debris and to provide quantitative data on the total tissue area on each slide. Within the tissue area, regions of interest, i.e. the tumor tissue islands, were identified by a pathologist (PL) for analysis. Image analysis settings were based on differences in protein expression and tissue localization for the different markers. For CD68, CD163, CD3, CD8, CD20, PanCK, and vimentin, which are typically localized to membranous and/or cytoplasmic compartments, we used the HALO ‘Area quantification v1.0’ algorithm, a powerful approach for analyzing tissues where clustering and aggregation inhibit reliable cell segmentation, to estimate the positive staining area (Figure 1). The percent positive marker area was calculated by dividing the positive staining area by the total tissue area and multiplying by 100. For CD31, Ki67, and PDL1, the ‘Object colocalization v1.2’ algorithm, which detects positive staining cells or objects (e.g. CD31+ endothelial cells) based on their size and shape, was used for determining the total number of positively staining objects per mm2 of tissue area (Figure 1). Algorithms to measure Ki67 and vimentin were trained on the entire tissue, considering both tumor and non‐tumor regions equally for training.
Figure 1.
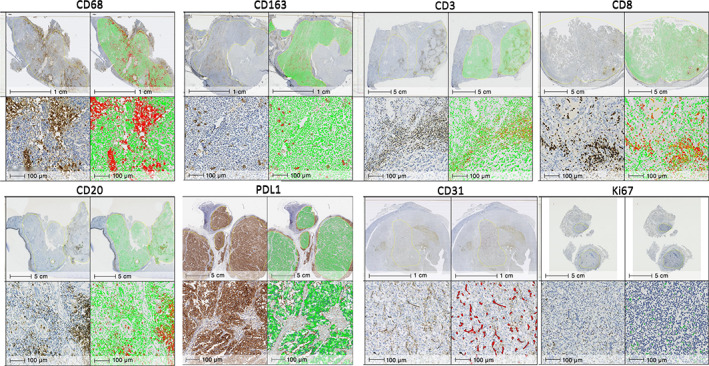
Representative images of the comparative quantification of tumor island staining by light microscopy and by digital image analysis using the HALO imagine analysis software for each marker. CD68, papillary renal cell carcinoma (pRCC) type 1; CD3, collecting duct carcinoma; Ki67, high‐grade urothelial carcinoma; all other markers, pRCC type 2.
To investigate immune infiltration patterns by histological subtype, each image was spatially characterized into tumor core, proximal tumor periphery, and distal tumor periphery, and marker expression was assessed within these compartments. In addition to careful pathology annotations, delineation of the tumor on the slide was enhanced using PanCK staining. In general, machine learning algorithms were trained to identify and annotate regions on representative sections with positive PanCK staining. For each subject, annotated PanCK regions from the representative section were digitally overlayed on serial sections from the other markers through a process of digital image registration. Accordingly, separate PanCK with CD3, CD20, or CD68 images were successfully merged by using the HALO image registration tool. For each marker, concentric rings of 140 μm in width from the tumor core to 300 μm outside the tumor region (i.e. distal tumor periphery) were analyzed using the HALO ‘Infiltration analysis’ module (supplementary material, Figure S1). Percent positive staining area and object count per mm2 were determined within each concentric region.
Statistical analyses
To compare the TME by histological types, we calculated the rate of immune markers expression (i.e. the area of tissue with positive stain divided by the total tissue area) and tested differences between individual histological types using Mann–Whitney, and across all histological types using Kruskal–Wallis tests. A single sample, chosen at random, was included from tumors with multiple samples. We examined correlations across markers using Spearman's rank correlation coefficient r.
To examine the associations between TME marker expression (outcome) and histological types (exposure), we fitted Poisson regression models on the log of the area of tissue with positive stain. To take into account the variability in tissue size on each slide, the log of the total tissue area per slide was included as an offset. Correlations across multiple samples from the same tumor were accommodated by using generalized estimating equations [24] with the independent working correlation to obtain variance estimates (Proc Genmod). pRCC2 was used as the reference group to ensure stable estimates, as it had the largest sample size (n = 49). To identify confounders of the association between TME markers and histological types, we verified whether marker expression (in quartiles) was associated with clinical features (listed in Table 1) using chi‐square or Fisher's exact tests (data not shown), and whether clinical features were associated with histological types using Fisher's exact tests (supplementary material, Table S3). Poisson models were first only adjusted for histological type, and then additionally adjusted for age in categories fitted with a trend (<51, 51–61, 62–70, >70), sex, tumor size (<4, 4–7, >7 cm) [25], and clinical stage (I, II, III, IV). The incidence rate ratio (IRR) estimated from the Poisson model represents the ratio of the percent positive area in a specific histological type compared to pRCC2. A Wald‐based P heterogeneity was calculated to test differences of marker expression across all four histological types. In sensitivity analyses, we additionally adjusted the Poisson model of the immune markers for the rate of immune marker expression from the same cell types (i.e. T cells or macrophages) to control for any possible residual confounding.
Table 1.
Summary of clinical characteristics of study participants.
| n (%) | ||
|---|---|---|
| Age at surgery (n = 83) | <51 | 16 (19.28) |
| 51–61 | 23 (27.71) | |
| 62–70 | 22 (26.51) | |
| >70 | 22 (26.51) | |
| Gender (n = 83) | Male | 63 (75.90) |
| Female | 20 (24.10) | |
| Histology (n = 83) | pRCC type 1 | 20 (24.10) |
| pRCC type 2 | 44 (53.00) | |
| CDC | 14 (16.90) | |
| HGUC | 5 (6.00) | |
| Clinical stage (n = 83) | I | 32 (38.60) |
| II | 7 (8.40) | |
| III | 21 (25.30) | |
| IV | 23 (27.70) | |
| Tumor size, cm (n = 83) | <4 | 16 (19.50) |
| 4–7 | 31 (37.80) | |
| >7 | 35 (42.70) | |
| Metastasis at diagnosis (n = 83) | Yes | 20 (24.10) |
| No | 63 (75.90) | |
Next, we compared the median expression of markers inside the tumor with that on the proximal (tumor border) and distal periphery using Wilcoxon matched‐pairs signed rank test. To assess marker expression by distance from the center of the tumor island region to the periphery, we fitted linear models to the log‐transformed marker values, adjusted for the log‐transformed bandwidth area. P values of <0.05 were considered significant. All statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA).
Results
As expected [26], the majority of the subjects were male (76%) (Table 1). The mean age of participants was 61 years (range 56–76 years). Over half (53%) of the samples analyzed were pRCC2. Clinical stage and tumor size showed even distribution across categories. Finally, 24% of subjects presented with metastasized tumors (Table 1).
We analyzed 103 tumor samples from 83 subjects. Variability in expression across all markers was observed when we compared multiple samples from the same primary tumor (supplementary material, Figure S2), highlighting the heterogeneity of the TME in these cancers. In contrast, we observed no significant variation between marker expression in primary tumors and matched metastatic (n ≤ 13) or recurrent (n = 2) tissue samples (supplementary material, Figure S3). However, although not statistically different, PD‐L1 showed a suggestive increase in the metastatic tissue samples (median positive cells/mm2 = 17.6 (lymph node and distant metastatic samples) versus 5.30 (primary matched samples), n = 13, p = 0.1099; nodes only, n = 4, p = 0.1250; distant metastatic samples only, n = 9, p = 0.820) (supplementary material, Figure S3 and Table S4). Moderate correlation was observed between expression of markers from the same cell types, i.e. macrophages (CD68 and CD163; r = 0.634) and T cells (CD3 and CD8; r = 0.48) (supplementary material, Table S5). Furthermore, B‐cell marker showed moderate and significant correlation with the pan‐T‐cell marker CD3 (r = 0.693) (supplementary material, Table S5), suggesting that activation of adaptive immune cells may occur concurrently.
Several immune markers showed different rates of expression across histological types (Figure 2). Kruskal–Wallis analysis showed that CD68 (p = 0.0005), CD20 (p = 0.0003), and Ki67 (p = 0.0053) expression varied the greatest between all histological types (not shown on graphs). Overall, expression of macrophage markers (i.e. CD68 and CD163) was highest in the pRCC histological types (pRCC2 versus CDC, CD68: p = 0.006; pRCC2 versus HGUC, CD68: p = 0.01, CD163; p = 0.046, Mann–Whitney test). However, lymphocyte markers (i.e. CD3, CD8, and CD20) showed a trend of higher expression in the CDC or HGUC subtypes, with CD20 expression significantly higher in CDC versus pRCC2 (p = 0.04). Moreover, pRCC1 had significantly lower expression of CD8 (p = 0.0083), CD20 (p < 0.001), PDL‐1 (p = 0.0161), CD31 (p = 0.039), and Ki67 (p = 0.0245) in comparison to the more clinically aggressive pRCC2 (Figure 2).
Figure 2.
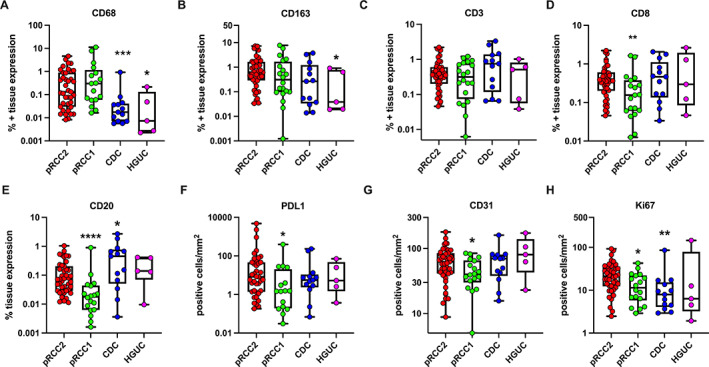
Scatter plot representation of median ± range of the percentage positive tissue area for markers measured by IHC and quantified by digital pathology using the HALO image analysis platform. Data were analyzed by unpaired Mann–Whitney test for differences between individual histological types compared. pRCC2 acted as comparison group for all analysis, as it had the largest subject size. pRCC2, papillary renal cell carcinoma type 2 (n = 44) (red); pRCC1, papillary renal cell carcinoma type 1 (n = 20) (green); CDC, collecting duct carcinoma (n = 14) (blue); HGUC, high‐grade urothelial carcinoma (n = 5) (magenta). ****p < 0.0001; ***p < 0.0005; **p < 0.005; *p < 0.05.
In multivariable Poisson models, CDC showed a higher lymphocytic infiltration in comparison to pRCC2, i.e. high T cells (CD3; IRR = 2.82, 95% confidence interval [CI] = 1.69–4.69, p < 0.001) and B cells (CD20; IRR = 4.94, 95% CI = 2.63–9.28, p < 0.001) (Table 2), but a lower infiltration of macrophages (CD68; IRR = 0.06, 95% CI = 0.00–0.95, p = 0.046). However, no variation across histological types was found for CD8, a marker of cytotoxic T cells. Additionally, PDL1 expression was higher in pRCC2 versus CDC (IRR = 0.11, 95% CI = 0.02–0.73, p = 0.02).
Table 2.
Univariable and multivariable analyses of the association between TME marker expression and kidney tumor histological types.
| Univariable model | Multivariable model* | |||||
|---|---|---|---|---|---|---|
| Marker | Histology | n | IRR (95% CI) | P value | IRR (95% CI) | P value |
| CD68 | ||||||
| pRCC type 2 | 45 | Ref. | Ref. | |||
| pRCC type 1 | 16 | 2.16 (1.09–4.29) | 0.0276 | 1.48 (0.61–3.63) | 0.3884 | |
| CDC and other | 14 | 0.06 (0.00–1.04) | 0.0532 | 0.06 (0.00–0.95) | 0.0458 | |
| HGUC | 5 | 0.02 (0.00–33.94) | 0.3160 | 0.03 (0.00–35.21) | 0.3277 | |
| P heterogeneity | 0.0662 | 0.2522 | ||||
| CD163 | ||||||
| pRCC type 2 | 49 | Ref. | Ref. | |||
| pRCC type 1 | 20 | 0.84 (0.43–1.64) | 0.6090 | 0.57 (0.26–1.27) | 0.1676 | |
| CDC and other | 14 | 0.97 (0.39–2.42) | 0.9473 | 0.96 (0.38–2.38) | 0.9239 | |
| HGUC | 5 | 0.22 (0.01–8.21) | 0.4142 | 0.31 (0.01–10.72) | 0.5170 | |
| P heterogeneity | 0.3327 | 0.571 | ||||
| CD3 | ||||||
| pRCC type 2 | 48 | Ref. | Ref. | |||
| pRCC type 1 | 20 | 0.95 (0.54–1.68) | 0.8589 | 0.79 (0.41–1.49) | 0.4632 | |
| CDC and other | 14 | 2.50 (1.56–4.02) | <0.0001 | 2.82 (1.69–4.69) | <0.0001 | |
| HGUC | 5 | 0.71 (0.24–2.12) | 0.5395 | 1.03 (0.31–3.41) | 0.9590 | |
| P heterogeneity | 0.4508 | 0.4002 | ||||
| CD8 | ||||||
| pRCC type 2 | 47 | Ref. | Ref. | |||
| pRCC type 1 | 19 | 0.65 (0.33–1.28) | 0.2145 | 0.5 (0.23–1.07) | 0.0726 | |
| CDC and other | 14 | 1.45 (0.82–2.55) | 0.1976 | 1.56 (0.86–2.83) | 0.1405 | |
| HGUC | 5 | 1.42 (0.60–3.37) | 0.4202 | 1.88 (0.72–4.91) | 0.1974 | |
| P heterogeneity | 0.364 | 0.1848 | ||||
| CD20 | ||||||
| pRCC type 2 | 48 | Ref. | Ref. | |||
| pRCC type 1 | 19 | 0.38 (0.12–1.24) | 0.1087 | 0.33 (0.09–1.24) | 0.0914 | |
| CDC and other | 13 | 4.35 (2.49–7.58) | <0.0001 | 4.94 (2.63–9.28) | <0.0001 | |
| HGUC | 5 | 1.07 (0.30–3.76) | 0.9175 | 1.29 (0.31–5.34) | 0.7210 | |
| P heterogeneity | 0.1631 | 0.3931 | ||||
| PDL1 | ||||||
| pRCC type 2 | 44 | Ref. | Ref. | |||
| pRCC type 1 | 15 | 0.11 (0.01–1.40) | 0.0886 | 0.16 (0.02–1.71) | 0.1306 | |
| CDC and other | 14 | 0.23 (0.03–2.15) | 0.1986 | 0.11 (0.02–0.73) | 0.0224 | |
| HGUC | 5 | 0.17 (0.00–16.47) | 0.4521 | 0.05 (0.00–2.39) | 0.1310 | |
| P heterogeneity | 0.4983 | 0.4304 | ||||
| CD31 | ||||||
| pRCC type 2 | 49 | Ref. | Ref. | |||
| pRCC type 1 | 18 | 0.67 (0.48–0.92) | 0.0143 | 0.76 (0.54–1.06) | 0.1043 | |
| CDC and other | 14 | 1.05 (0.78–1.41) | 0.7553 | 1.14 (0.86–1.52) | 0.3496 | |
| HGUC | 5 | 0.97 (0.52–1.83) | 0.9322 | 1.06 (0.58–1.94) | 0.8387 | |
| P heterogeneity | 0.0902 | 0.216 | ||||
| Ki67 | ||||||
| pRCC type 2 | 48 | Ref. | Ref. | |||
| pRCC type 1 | 18 | 0.51 (0.29–0.88) | 0.0162 | 0.47 (0.25–0.87) | 0.0166 | |
| CDC and other | 14 | 0.43 (0.20–0.90) | 0.0263 | 0.47 (0.22–1.00) | 0.0487 | |
| HGUC | 5 | 0.31 (0.08–1.24) | 0.0973 | 0.32 (0.08–1.31) | 0.1142 | |
| P heterogeneity | 0.0474 | 0.0282 | ||||
Data were analyzed by Poisson regression. Marker positive expression tissue area and total tissue area were quantified using HALO algorithms: ‘Area quantification v1.0’ for CD163, CD68, CD3, CD8, and CD20, and ‘Object colocalization v1.2’ for PDL‐1, CD31, and Ki67. Data were modeled as area of tissue with positive stain/positive object count as the dependent variable, and the log of the total tissue area per slide as an offset.
Ref., reference category.
Adjusted for sex, age at surgery, clinical stage, and tumor size.
Bold values are statistically significant (P < 0.05).
Moreover, pRCC1 showed lower expression of Ki67, a marker of cell proliferation, in comparison to pRCC2 (IRR = 0.47, 95% CI = 0.21–0.92, p = 0.017). Adjusting for markers expressed on the same cell types did not substantially alter the findings (supplementary material, Table S6). We could not stratify analysis of marker expression by tumor size or stage because of small numbers in some categories.
By quantifying the percentage of epithelial and mesenchymal regions on each H&E slide, we investigated whether the proportions of these tissue composition metrics differed by histological type. We found the proportion of epithelium on the slide to be higher among pRCC (mean [SD] = 80% [11%] and 78% [12%] for pRCC1 and pRCC2, respectively) and CDC (mean [SD] = 72% [17%]) than HGUC (mean [SD] = 50% [29%]) histological subtypes (supplementary material, Figure S4A). In contrast, HGUC (mean [SD] = 50% [26%]) showed a higher proportion of mesenchymal regions than the other histological types (mean [SD] = 22% [11%], 20% [11%], and 28% [17%] for pRCC2, pRCC1, and CDC, respectively; supplementary material, Figure S4B). We assessed EMT by using IHC staining of vimentin as a surrogate marker, which is in keeping with previous reports [27]. No significant variation in vimentin expression was observed by histological type (supplementary material, Figure S4C).
Finally, in an effort to ascertain if immune cells remain at the tumor periphery or can penetrate the tumor tissue core, we carried out immune marker spatial analysis using the HALO 3.1 imaging platform (supplementary material, Figure S1). Notably, CD68 expression was consistently expressed both inside the tumor tissue core as well as on the periphery in the pRCC histological types. In contrast, expression of CD3 and CD20 increased at the proximal (tumor border) and distal periphery, most strongly in the CDC histological type (supplementary material, Figure S5). When we compared the median expression of markers inside the tumor island with the expression levels at the proximal or distant periphery, the highest expression levels of CD68 were inside the tumor core for pRCC2 (mean [SD] = 257.9 [270.6] cells per mm2; p = 0.045) and pRCC1 (mean [SD] = 323 [179] cells per mm2; p = 0.004) compared to the periphery (mean [SD] = 194.7 [196.7] cells per mm2; mean [SD] = 224.3 [164.5] cells per mm2, respectively) (Figure 3A).
Figure 3.
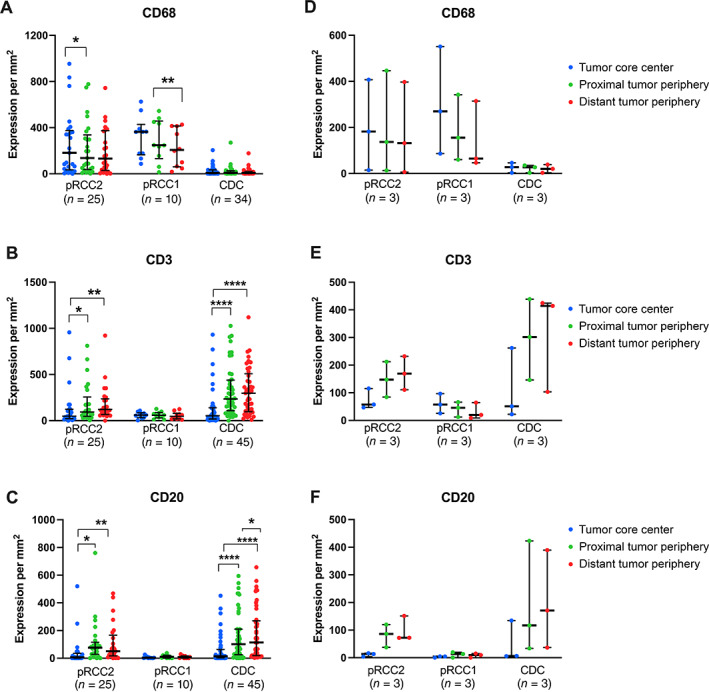
Scatter plot representation of median ± IQR expression of markers inside the tumor regions; the expression of markers across a 140‐μm band at the border between tumor and adjacent normal cells; and across a 140‐μm band outside tumor regions. Figures on the left represent the marker expression in each individual tumor island region analyzed, and the figures on the right the median expression of all tumor islands analyzed per samples (i.e. tissue slides, n = 3). Images were analyzed using the HALO 3.1 imagine analysis platform. Data were analyzed by Wilcoxon matched pairs signed rank test with ‘inside tumor’ as the comparison group unless defined by a bar. ****p < 0.0001; ***p < 0.0005; **p < 0.005; *p < 0.05.
In contrast, statistically significantly higher CD3+ T‐cell expression was observed at the proximal (mean [SD] = 322.7 [271.9] cells per mm2; p < 0.0001 and 186.7 [215.1] cells per mm2; p = 0.0147) and distal (mean [SD] = 317.7 [254.2] cells per mm2; p < 0.0001 and 182.8 [190.7] cells per mm2; p = 0.002) tumor periphery than within the tumor core (mean [SD] = 131.4 [204.7] and 134.6 [226.6]) for CDC and pRCC2, respectively. Similarly, for both CDC and pRCC2, CD20+ B‐cell expression was higher in the proximal (mean [SD] = 158.1 [171]; p < 0.001 and 112.0 [157.6]; p < 0.0018, respectively) and distal (mean [SD] = 177.4 [184.8]; p < 0.001 and 107.9 [137.2]; p = 0.006, respectively) tumor periphery than within the tumor core (mean [SD] = 57.94 [104.5] and 44.12 [111.7], respectively) (Figure 3C). Similar results were observed when multiple tumor islands per tissue slide were combined to represent the overall mean expression inside the tumor and at the proximal and distal tumor periphery per slide (Figure 3D–F).
To assess the statistical significance of the observed spatial patterns, we conducted linear regression analysis of the markers' expression by distance from the tumor center to the periphery. The analysis showed that CD3 expression and, to a lesser extent, that of CD20 increased with increasing distance from the tumor center (p = 0.004 and 0.01, respectively, in CDC samples). In contrast, CD68 showed no significant variation (p = 0.5471).
Discussion
The aim of this study was to evaluate the density and spatial architecture of different markers in the TME of rare histological types of RCC and other renal cancers. In particular, we focused on immune cell expression and localization in the TME. We found that the expression levels of CD68, a marker of all macrophages, was highest in the pRCC subtypes, with strong levels of infiltration into the tumor core, while higher expression of T (CD3 only) and B cells was found in CDC but localized at the proximal and distant tumor periphery.
Macrophages are innate immune cells that are primarily involved in host defense and tissue homeostasis. In response to many stimuli, macrophages are recruited to the TME and can prevent or facilitate further tumor development [28]. Macrophages can be categorized into two main subtypes based on their functions: the classical M1 phenotype, which is involved in immune response, pathogen clearance, and anti‐tumor inflammation, and the alternative M2 phenotype (i.e. CD163 expressing), which has been described as anti‐inflammatory and pro‐tumorigenic [29]. A histological hallmark found in most pRCC cases is focal accumulation of foam cell macrophages in the stroma of the papillary stalks [30]. Therefore, it is unsurprising that expression levels of macrophages were highest in pRCC types. Moreover, pRCC1 had lower expression of lymphocyte markers (i.e. CD8 and CD20) and markers of aggressiveness (i.e. CD31 and Ki67) than pRCC2, which is expected for its more ‘benign’ phenotype [17], whereas CD68 expression was higher in pRCC1 versus pRCC2. This is consistent with previously reported findings that foamy macrophages predominate in pRCC1 than pRCC2 [31]. However, little is known about the role of macrophages in pRCC biology [32]. Behnes et al [33] suggested, in a small study of 30 pRCC2 cases, that M2 TAMs may contribute to the poor prognosis of pRCC2. In our study, although not statically significant, pRCC2 expressed the highest median levels of CD163 (Figure 2) [34]. Consistent findings were seen at a transcriptomic level by Liu et al [35], based on CIBERSORT analysis, in which they found that M1 and M2 macrophages could predict pRCC2 clinical outcome. Thus, when creating a treatment plan for these cancer types, the pro‐tumorigenic presence of M2 macrophages and the low expression levels of T‐cell and B‐cell markers should be considered.
In contrast, we found higher expression of lymphocytes in the CDC histological type compared to pRCC types. CDC is an aggressive form of renal cancer, and the activation and recruitment of an adaptive immune response to the tumor site is not unexpected [36]. However, analysis of the spatial architecture of these immune cells in the TME showed that the accumulation of lymphocytes remains peritumoral. This confirms that the recruitment of these cells is occurring, but immune evasion takes place in the TME at the tumor site. Previously, a transcriptomic analysis of 17 CDC tumors showed that genes of the immune response were overexpressed, which is consistent with our finding of high T‐cell and B‐cell marker expression in this histological type [37]. That study did not analyze the localization of the overexpressed immune cells, which, based on our study, appear to remain outside the tumor island areas. The lack of infiltration of lymphocytes in the tumor mass could partly explain the aggressive phenotype of CDC. However, the opposite may also apply, i.e. that aggressive tumor phenotypes may more likely be high grade and exhibit central necrosis, which may cause the immune infiltration to be limited to the periphery. Conversely, the necrotic tumor core may be predominated by phagocytes and polymorphonuclear cells. Nonetheless, this is very unlikely to affect our results as all histological sections were carefully annotated by a pathologist to exclude regions of necrosis, artifacts, and debris. Furthermore, when we adjusted for grade in a subset of patients for whom we had these data, the results did not change (data not shown). In addition, both peripheral and intra‐tumoral regulatory T cells have been identified in ccRCC, where they were associated with poor prognosis and metastatic progression [38, 39]. As for the B lymphocytes, Sjöberg et al [40] found that B cells were independent predictors of poor prognosis in a subset of RCCs; however, their role in the TME mostly remains inconclusive and additional studies are warranted.
To the best of our knowledge, this is the first study to comprehensively characterize the TME in rare renal cell carcinomas by integrating IHC staining of 10 markers with computational pathology. Our findings provide novel insights into the density and spatial localization of immune cell subsets in pRCC and CDC, reflecting differences in TME biology between distinct subtypes of renal carcinomas. Owing to the small sample size and lack of clinical outcomes data, however, we were unable to investigate the prognostic relevance of TME in this study. Findings from recent studies suggest that, beyond immune cell densities, colocalization of immune and proliferation markers, such as CD8/Ki67, may be of prognostic relevance in pRCCs [41]. The assessment of marker colocalization can be facilitated by dual or multiplex chromogenic or immunofluorescence staining, which differ from the serial IHC staining that we performed in this analysis. Nevertheless, the HALO image registration tool enabled us to spatially characterize histological sections and to evaluate TME patterns within the tumor and its periphery. Although this approach can be extended to evaluate colocalization, the images for this study were not always from consecutive sections which precluded our ability to perform cell‐to‐cell level image registration. Future studies incorporating digital pathology with multiplex staining will allow colocalization, proximity, nearest neighbor, and infiltration analyses.
In conclusion, this study provides novel insights into the immune TME of rare histological types of kidney cancer using a large number of markers and a spatial analysis of immune cells. Although based on a small number of samples in some analyses, distinct tumor environments were observed in different histological types, which could ultimately impact the response to immune‐based therapies or other targeted therapies. Future clinical trials of these tumors should take type‐specific immune markers and their localizations into account.
Author contributions statement
NCS, MLP and MC managed data analysis and interpretation and wrote the manuscript draft. RMP and NCS led the statistical analyses and interpretations. ML assisted in the manuscript preparation. SS, YG, PL and MDu performed pathological review of slides. SL and KM performed HALO image analyses. MA reviewed the digital pathology analysis and interpretation. GS and MG performed all surgeries. EP supervised sample collection. MLP, MC and VMF supervised the field activities and data collection. CA, MDa and MO managed slide preparation and staining. MTL led the project and reviewed all data analysis, interpretations, and manuscript drafts. All authors reviewed the manuscript.
Supporting information
Figure S1. Summary explanation of the spatial image analysis conducted using HALO 3.1 image analysis software
Figure S2. Bar chart representation of the mean expression of markers in the subjects (n = 6) with multiple tumor samples
Figure S3. Scatterplot representation of the percentage of tissue area positive for markers in primary samples and marched metastasis or recurrence samples
Figure S4. Scatterplot representations of % epithelial region or mesenchymal regions present per sample slide, as measured by H&E staining and pathologist review by eye, per whole slide
Figure S5. Scatterplot representation of markers' spatial distribution by histological subtype
Table S1. Summary table of samples from lymph nodes, distant metastases, and recurrences
Table S2. Antibodies information and test conditions
Table S3. Association between clinical features and histological type
Table S4. Summary table of % tissue expression (CD68, CD163, CD3, CD8, CD20) or cells/mm2 (PDL1, CD31, Ki67) in primary samples with matched metastatic or recurrent samples
Table S5. Spearman correlation coefficient across percentage of markers' expression
Table S6. Association between macrophage or T‐cell marker expression and renal cell carcinoma histological type. Data were analyzed using Poisson regression
Acknowledgement
This study was supported by the Intramural Program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH.
No conflicts of interest were declared.
References
- 1. Whiteside TL. The microenvironment and its role in promoting tumor growth. Oncogene 2008; 27: 5904–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arneth B. Tumor microenvironment. Medicina (Kaunas) 2020; 56: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Locy H, de Mey S, de Mey W, et al. Immunomodulation of the tumor microenvironment: turn foe into friend. Front Immunol 2018; 9: 2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang M, Zhao J, Zhang L, et al. Role of tumor microenvironment in tumorigenesis. J Cancer 2017; 8: 761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018; 68: 394–424. [DOI] [PubMed] [Google Scholar]
- 6. Feng X, Zhang L, Tu W, et al. Frequency, incidence, and survival outcomes of clear cell renal cell carcinoma in the United States from 1973 to 2014. Medicine (Baltimore) 2019; 98: e16684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thompson RH, Dong H, Lohse CM, et al. PD‐1 is expressed by tumor‐infiltrating immune cells and is associated with poor outcome for patients with renal cell carcinoma. Clin Cancer Res 2007; 13: 1757–1761. [DOI] [PubMed] [Google Scholar]
- 8. Thompson RH, Dong H, Kwon ED. Implications of B7‐H1 expression in clear cell carcinoma of the kidney for prognostication and therapy. Clin Cancer Res 2007; 13: 709s–715s. [DOI] [PubMed] [Google Scholar]
- 9. Leite KRM, Reis ST, Junior JP, et al. PD‐L1 expression in renal cell carcinoma clear cell type is related to unfavorable prognosis. Diagn Pathol 2015; 10: 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moore LE, Nickerson ML, Brennan P, et al. Von Hippel‐Lindau (VHL) inactivation in sporadic clear cell renal cancer: associations with germline VHL polymorphisms and etiologic risk factors. PLoS Genet 2011; 7: e1002312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vuong L, Kotecha RR, Voss MH, et al. Tumor microenvironment dynamics in clear cell renal cell carcinoma. Cancer Discov 2019; 9: 1349–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal‐cell carcinoma. N Engl J Med 2018; 378: 1277–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal‐cell carcinoma. N Engl J Med 2015; 373: 1803–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rini BI, Plimack ER, Stus V, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal‐cell carcinoma. N Engl J Med 2019; 380: 1116–1127. [DOI] [PubMed] [Google Scholar]
- 15. Giles RH, Choueiri TK, Heng DY, et al. Recommendations for the management of rare kidney cancers. Eur Urol 2017; 72: 974–983. [DOI] [PubMed] [Google Scholar]
- 16. Akhtar M, Al‐Bozom IA, Al Hussain T. Papillary renal cell carcinoma (PRCC): an update. Adv Anat Pathol 2019; 26: 124–132. [DOI] [PubMed] [Google Scholar]
- 17. Waldert M, Haitel A, Marberger M, et al. Comparison of type I and II papillary renal cell carcinoma (RCC) and clear cell RCC. BJU Int 2008; 102: 1381–1384. [DOI] [PubMed] [Google Scholar]
- 18. Ren W, Gao X, Zhang X, et al. Prognostic factors for the survival of patients with papillary renal cell carcinoma after surgical management. Clin Transl Oncol 2020; 22: 725–733. [DOI] [PubMed] [Google Scholar]
- 19. Petejova N, Martinek A. Renal cell carcinoma: review of etiology, pathophysiology and risk factors. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2016; 160: 183–194. [DOI] [PubMed] [Google Scholar]
- 20. Gupta R, Billis A, Shah RB, et al. Carcinoma of the collecting ducts of Bellini and renal medullary carcinoma: clinicopathologic analysis of 52 cases of rare aggressive subtypes of renal cell carcinoma with a focus on their interrelationship. Am J Surg Pathol 2012; 36: 1265–1278. [DOI] [PubMed] [Google Scholar]
- 21. Qian X, Wang Z, Zhang J, et al. Clinical features and prognostic outcome of renal collecting duct carcinoma: 12 cases from a single institution. Cancer Manag Res 2020; 12: 3589–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Babjuk M, Burger M, Compérat EM, et al. European Association of Urology guidelines on non‐muscle‐invasive bladder cancer (TaT1 and carcinoma in situ)—2019 update. Eur Urol 2019; 76: 639–657. [DOI] [PubMed] [Google Scholar]
- 23. Mai KT, Flood TA, Williams P, et al. Mixed low‐ and high‐grade papillary urothelial carcinoma: histopathogenetic and clinical significance. Virchows Arch 2013; 463: 575–581. [DOI] [PubMed] [Google Scholar]
- 24. Liang K‐Y, Zeger SL. Longitudinal data analysis using generalized linear models. Biometrika 1986; 73: 13–22. [Google Scholar]
- 25. Amin MB, Greene FL, Edge SB, et al. The Eighth Edition AJCC Cancer Staging Manual: continuing to build a bridge from a population‐based to a more “personalized” approach to cancer staging. CA Cancer J Clin 2017; 67: 93–99. [DOI] [PubMed] [Google Scholar]
- 26. Lucca I, Klatte T, Fajkovic H, et al. Gender differences in incidence and outcomes of urothelial and kidney cancer. Nat Rev Urol 2015; 12: 585–592. [DOI] [PubMed] [Google Scholar]
- 27. Yao JX, Chen X, Zhu YJ, et al. Prognostic value of vimentin is associated with immunosuppression in metastatic renal cell carcinoma. Front Oncol 2020; 10: 1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity 2010; 32: 593–604. [DOI] [PubMed] [Google Scholar]
- 29. Zhu J, Zhi Q, Zhou BP, et al. The role of tumor associated macrophages in the tumor microenvironment: mechanism and functions. Anticancer Agents Med Chem 2016; 16: 1133–1141. [DOI] [PubMed] [Google Scholar]
- 30. Delahunt B, Eble JN. Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol 1997; 10: 537–544. [PubMed] [Google Scholar]
- 31. Le X, Wang X‐B, Zhao H, et al. Comparison of clinicopathologic parameters and oncologic outcomes between type 1 and type 2 papillary renal cell carcinoma. BMC Urol 2020; 20: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krawczyk KM, Nilsson H, Allaoui R, et al. Papillary renal cell carcinoma‐derived chemerin, IL‐8, and CXCL16 promote monocyte recruitment and differentiation into foam‐cell macrophages. Lab Invest 2017; 97: 1296–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Behnes CL, Bremmer F, Hemmerlein B, et al. Tumor‐associated macrophages are involved in tumor progression in papillary renal cell carcinoma. Virchows Arch 2014; 464: 191–196. [DOI] [PubMed] [Google Scholar]
- 34. Hu JM, Liu K, Liu JH, et al. CD163 as a marker of M2 macrophage, contribute to predict aggressiveness and prognosis of Kazakh esophageal squamous cell carcinoma. Oncotarget 2017; 8: 21526–21538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu T, Zhang M, Sun D. Immune cell infiltration and identifying genes of prognostic value in the papillary renal cell carcinoma microenvironment by bioinformatics analysis. Biomed Res Int 2020; 2020: 5019746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gajewski TF, Schreiber H, Fu Y‐X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013; 14: 1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Malouf GG, Compérat E, Yao H, et al. Unique transcriptomic profile of collecting duct carcinomas relative to upper tract urothelial carcinomas and other kidney carcinomas. Sci Rep 2016; 6: 30988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Siddiqui SA, Frigola X, Bonne‐Annee S, et al. Tumor‐infiltrating Foxp3 − CD4 + CD25 + T cells predict poor survival in renal cell carcinoma. Clin Cancer Res 2007; 13: 2075–2081. [DOI] [PubMed] [Google Scholar]
- 39. Kang MJ, Kim KM, Bae JS, et al. Tumor‐infiltrating PD1‐positive lymphocytes and FoxP3‐positive regulatory T cells predict distant metastatic relapse and survival of clear cell renal cell carcinoma. Transl Oncol 2013; 6: 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sjöberg E, Frödin M, Lövrot J, et al. A minority‐group of renal cell cancer patients with high infiltration of CD20+B‐cells is associated with poor prognosis. Br J Cancer 2018; 119: 840–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eich M‐L, Chaux A, Rodriguez MAM, et al. Tumour immune microenvironment in primary and metastatic papillary renal cell carcinoma. Histopathology 2020; 76: 423–432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Summary explanation of the spatial image analysis conducted using HALO 3.1 image analysis software
Figure S2. Bar chart representation of the mean expression of markers in the subjects (n = 6) with multiple tumor samples
Figure S3. Scatterplot representation of the percentage of tissue area positive for markers in primary samples and marched metastasis or recurrence samples
Figure S4. Scatterplot representations of % epithelial region or mesenchymal regions present per sample slide, as measured by H&E staining and pathologist review by eye, per whole slide
Figure S5. Scatterplot representation of markers' spatial distribution by histological subtype
Table S1. Summary table of samples from lymph nodes, distant metastases, and recurrences
Table S2. Antibodies information and test conditions
Table S3. Association between clinical features and histological type
Table S4. Summary table of % tissue expression (CD68, CD163, CD3, CD8, CD20) or cells/mm2 (PDL1, CD31, Ki67) in primary samples with matched metastatic or recurrent samples
Table S5. Spearman correlation coefficient across percentage of markers' expression
Table S6. Association between macrophage or T‐cell marker expression and renal cell carcinoma histological type. Data were analyzed using Poisson regression