

Abstract
Recent advances in 3D cell culture technology have enabled scientists to generate stem cell-derived organoids that recapitulate the structural and functional characteristics of native organs. Current organoid technologies have been striding toward identifying the essential factors for controlling the processes involved in organoid development, including physical cues and biochemical signaling. There is a growing demand for engineering dynamic niches characterized by conditions that resemble in vivo organogenesis to generate reproducible and reliable organoids for various applications. Innovative biomaterial-based and advanced engineering-based approaches have been incorporated into conventional organoid culture methods to facilitate the development of organoid research. In this review, we comprehensively summarize the recent advances in organoid engineering, including extracellular matrices and genetic modulation, to pinpoint the parameters critical for organ-specific patterning. Moreover, perspective trends in developing tunable organoids in response to exogenous and endogenous cues are discussed for next-generation developmental studies, disease modeling, and therapeutics.
Keywords: Bioengineered organoids, biomaterials, extracellular matrix, genetic engineering, disease modeling
Graphical Abstract
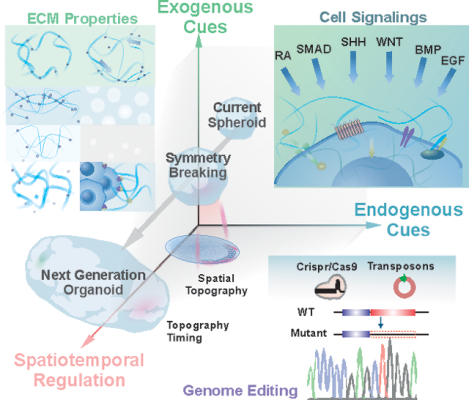
The emergence of organoid technologies, in terms of the development of 3D miniature organs in vitro, potentially can provide a revolutionized approach for understanding developmental processes and modeling disease. In this review, we discussed some recent achievements, prospects, and critical challenges in current organoid research and how to integrate Innovative biomaterial-based and advanced engineering-based approaches into conventional organoid-based culture and bioapplications.
1. INTRODUCTION
Stem cells are characterized by their unique ability to self-renew and differentiate into various cell subtypes. They have revolutionized modern biological and medical research, thereby providing a better understanding of developmental and disease progression processes.[1] As a result, stem cell-derived organoids potentially enable the study of biology and physiology at the organ level, in addition to aiding drug development and disease modeling.[2, 3]
A simple meaning of organoid is a three-dimensional (3D) multicellular tissue produced in vitro resembling in vivo organ. However, the word organoid is today limited to such constructs, which are self-organized from pluripotent stem cells or adult stem cells. Additionally, organoids should exhibit essential features, including organ-specific multiple cell types, functions of the organ, and spatially organized structures. The emergence and progression of organoid technologies have resulted from several important discoveries [Figure 1]. The formation of actual tissue-like colonies in vitro was firstly observed from a co-culture system of keratinocytes and 3T3 fibroblasts[4]. Self-organization, one of the fundamental aspects of organogenesis, was first observed via two distinct approaches, namely reaggregation and structural patterning of dissociated single cells.[5, 6] The establishment of three-dimensional (3D) culture methods for the structural organization began with the development of extracellular matrices (ECM).
Figure 1.
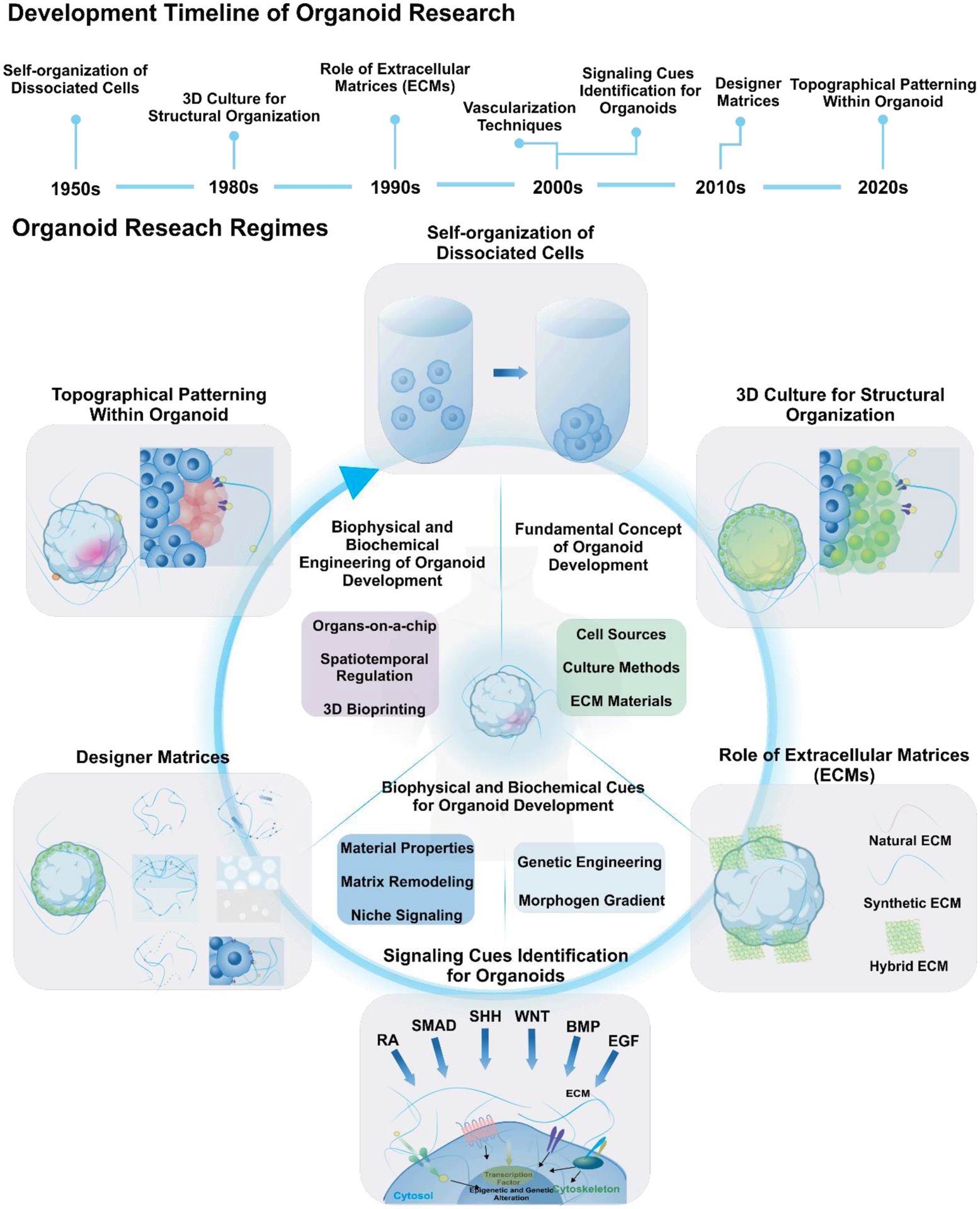
Timeline of advancement in organoid technologies, with regard to fundamental concept, biophysical and biochemical cues, and engineering for organoid development.
In the late 1980s, Bissell and colleagues observed that a laminin-rich gel could function as a basement membrane to differentiate and morphogenesis of mammary epithelial cells.[7, 8] In the 1990s, it was reported that in addition to their primary role in physical support, ECM components could regulate gene expression by interacting with integrin-based focal adhesion pathways.[9] Finally, in 2009, Hans Clevers group reported that embedding single intestinal stem cells in ECM substitute had created crypt-like structures similar to the epithelium of the native intestinal tissues, which were the first organoids.[10] Based on these recognitions, biochemical cues that include the initiation of lineage-specific genetic programs have been incorporated in 3D organoid cultures. Through exposure to morphogens, growth factors, or morphogen inhibitors, multiple research groups rapidly developed various organoid models using embryonic stem cells (ESCs) or adult stem cells (ASCs); these include intestine[10], stomach[11], liver[12], pancreas[13], prostate[14], and brain[15] organoids. At the same time, vascularization techniques were devised by several groups to embody microenvironments that are physiologically close to their actual counterparts. Microfluidic systems[16], endothelial cell-coated modules[17], and vascular endothelial growth factor delivery systems[18] have been demonstrated as in vitro vasculature systems that can facilitate oxygen or nutrients transport to the inner mass of organoids.
In the late 2010s, owing to the accumulated information on mechanisms underlying organogenesis and the remarkable advancements in the fields of biomaterial and bioengineering, the era of ‘organoid customization’ has begun. Customizable hydrogel matrices have been proposed to form intestinal organoids whose internal networks recapitulate the microenvironment of the intestinal stem cell niche.[19] These synthetic matrices could be designed and optimized to fine control critical external cues that contribute to organoid generation. In contrast, conventional ECMs, such as Matrigel, have not been fully characterized. The control of intrinsic cues within organoids became possible by taking advantage of two revolutionary technologies, patient-derived induced pluripotent stem cells (iPSCs)[20] and CRISPR/Cas-based genome editing.[21] Scientists can now generate genome-edited or mutated pluripotent stem cells (PSCs) with altered signaling cues through the generation of iPSCs from mutant-containing patients or introducing mutations to iPSCs. For example, in a recent study, brain subdivisions’ spatial topography has been recapitulated using differentially patterned PSCs exposed to signaling gradients.[22] Similar to the phenomena observed during in vivo development, Sonic Hedgehog (SHH) gradients resulted in the establishment of dorsoventral and anteroposterior axes, thereby creating polarized forebrain organoids. Genome engineering technology that modulates iPSCs via the introduction of genetic mutations has achieved accurate disease modeling by recapitulating genotypes and phenotypes of patients.[23, 24] As a result, the simultaneous use of multidisciplinary engineering methods for spatiotemporal modulation of organoids has rapidly accelerated organoid research advancements towards organ-level biology, next-generation disease modeling, and transformative regenerative medicine.
In this review, we first discuss the typical methodologies employed for in vitro organogenesis based on the defined physical and biochemical parameters that must be considered for organoid culture. Despite extensive studies, organoids from conventional methods typically lack reproducibility, reliability, and maturation. Hence, we then focus on the recent advances in engineering extracellular matrices and intrinsic cues to overcome the limitations of traditional organoid cultures. Finally, we describe the use of organoid engineering for disease modeling, and then we forecast future directions in technologies for the next generation of organoids.
2. STATE-OF-THE-ART ORGANOID RESEARCH
2.1. KEY PARAMETERS AFFECTING ORGANOID FORMATION AND DEVELOPMENT
In the last few years, increasing efforts have been dedicated to replicate the in vivo conditions for generating various organoid models. During organogenesis, biophysical and biochemical parameters that regulate stem cell niches have been shown to influence the tissues’ development, maturation, and maintenance. Two distinct parameters have been dissected to allow organoids to model the dynamic nature of mammalian tissue development [Figure 2].
Figure 2. Conventional methods to control key parameters required for organoid development.
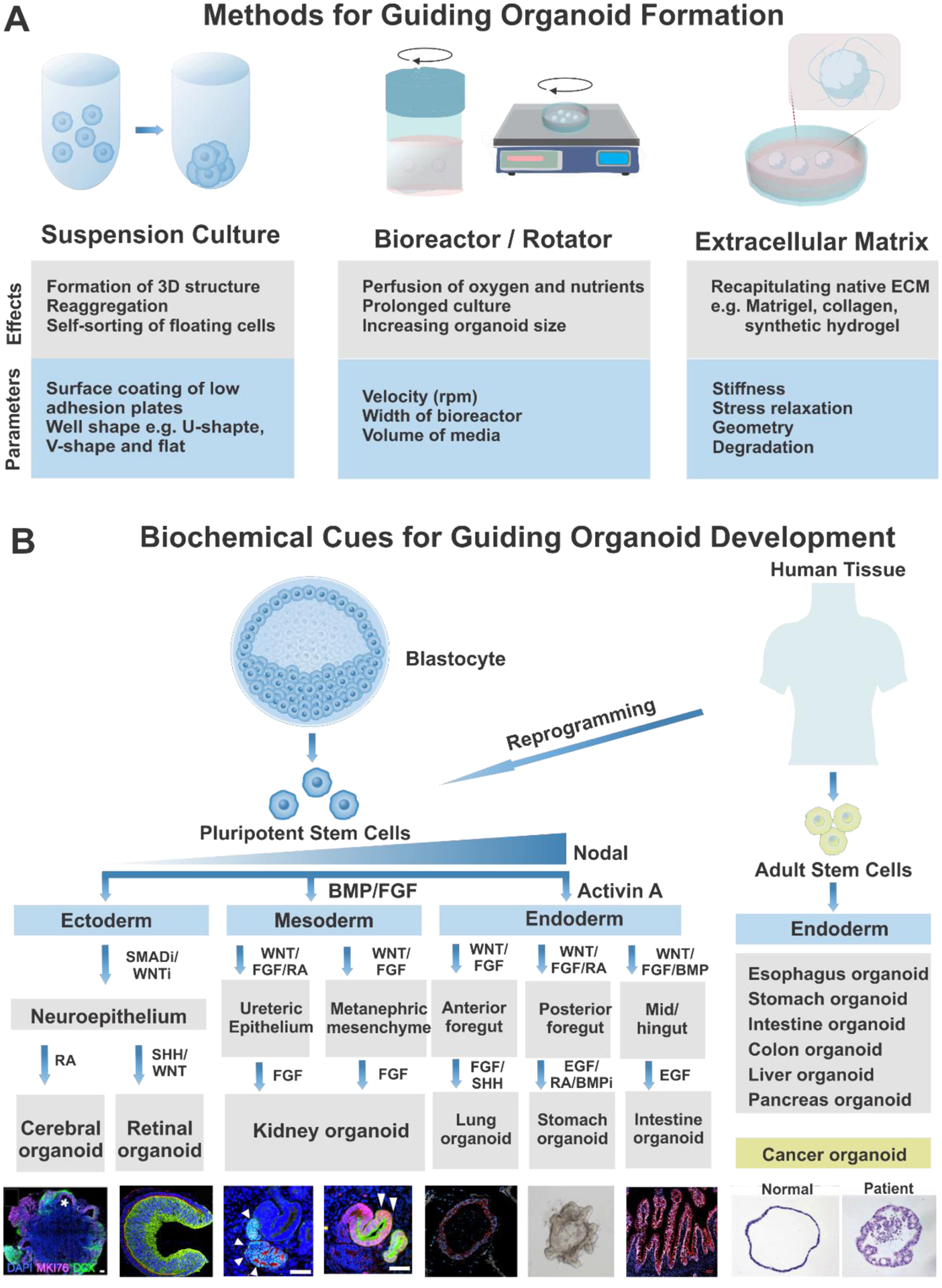
A) Methods to induce physical cues required for organoid formation. B) Biochemical cues to guide organoid development to specific lineages. ECM, extracellular matrix; BMP, bone morphogenetic protein; FGF, fibroblast growth factor; RA, retinoic acid; SHH, sonic hedgehog; EGF, epidermal growth factor. Cerebral organoid. Reproduced with permission.[62] Copyright 2015, National Academy of Sciences. Retinal organoid. Reproduced with permission.[50] Copyright 2012, Elsevier. Kidney organoid. Reproduced with permission.[45] Copyright 2014, Elsevier. Lung organoid. Reproduced with permission.[38] Copyright 2015, Dye et al. Stomach organoid. Reproduced with permission.[11] Copyright 2010, Elsevier. Intestine organoid. Reproduced with permission.[133] Copyright 2019, Elsevier. Cancer organoid. Reproduced with permission.[176] Copyright 2015, Elsevier.
First, biophysical cues by the extracellular environment significantly affect the self-organization of 3D structures and morphogenetic rearrangements of the organoids [Figure 2A].[25] During the early phases of organoid development, suspension culture conditions enable the reaggregation and self-sorting of the floating cells derived from PSCs or ASCs.[26, 27] After developing 3D structures, several physical cues such as mechanical forces and motion are employed for organotypic patterning. Spinning bioreactors or rotators can improve nutrient and oxygen perfusion levels, extending the duration of organoid culture and increasing the organoid size.[28, 29] Furthermore, to mimic the native tissues, organoids are generated using naturally-derived ECMs, such as Matrigel or collagen-based ECMs. Embedding organoids in drops of pure Matrigel provides relatively rigid ECMs.[15] However, small amounts of Matrigel are added to the culture medium to form soft epithelial structures in some instances.[26] The mechanical parameters of ECMs, including material stiffness, stress relaxation, degradation rates, and geometry, all affect cell behaviors. Hence, such parameters should be considered in the organoid generation.[30]
The other critical parameters that affect the formation and development of organoids are the intrinsic signaling pathways governing the differentiation into the specific cell lineages [Figure 2B]. As demonstrated by various in vivo studies, different organs require their distinct niche signals, which cannot be induced sufficiently and accurately by embedding in Matrigel. Therefore, to induce the lineage-specific development of organoids, culture media for organoids are usually supplemented with several ligands or compounds that can activate key patterning signaling pathways, such as TGFβ, BMP, Wnt, FGF, and SHH.[31] The three-germ layer specification from PSCs relies on the levels of TGFβ-Nodal signaling. A high level of Nodal signaling specifies endoderm differentiation, and a low level of Nodal signaling induces mesoderm differentiation, while repressed Nodal signaling leads to neuroectoderm formation.[32] These principles underlie the adoption of activin A, a molecule associated with nodal signaling, with further use of BMP to drive definitive endoderm induction during the early stages of PSC-derived endoderm organoid cultures.[33] After establishing endodermal identity, the activation of Wnt and FGF signaling promotes further patterning of mid/hindgut and posterior endoderm via the transcription factor Cdx2.[34, 35] Subsequent treatment with retinoic acid (RA) and a BMP signaling antagonist regulate foregut patterning, leading to the development of gastric organoid[36, 37], while FGF and sonic hedgehog (SHH) induce respiratory epithelium development.[38] Furthermore, EGF is required for the maintenance of the stomach[11] and intestinal identity.[35, 37] Numerous studies have demonstrated that endoderm lineages, including gastric[10, 11], liver[12, 39], and pancreas[13] niches, can also be derived from ASCs as well as PSCs. Organoids from tissue biopsies containing ASCs mimic the adult stem cell niches that support the regeneration of the tissues, while PSCs-derived organoids resemble the developmental processes of an embryo.[40] These organoids possibly can be used for autologous cell therapy by transplanted to injured organs. However, cancer organoids generated from the tumor tissues of patients may serve as a personalized drug testing tool rather than clinical transplantation.[41]
For mesoderm-derived organoids, several groups have refined the protocols for generating renal organoids. FGF and low concentrations of BMP4 direct the differentiation of PSCs into intermediate mesoderm that can subsequently differentiate into the ureteric epithelium and metanephric mesenchyme from PSCs.[42, 43] Subsequent exposure to Wnt signaling molecules, followed by FGF and RA, drives the development of the ureteric bud[42, 44], while phasic stimulation with Wnt and FGF promotes the development of metanephric mesenchyme.[45, 46] Prolonged stimulation with FGF signaling induces nephrogenesis in the kidney progenitors, namely, ureteric epithelium, and metanephric mesenchyme, in turn resulting in the production of kidney organoids.[42, 46]
Unlike endoderm and mesoderm, neuroectodermal differentiation is mediated by a ‘default pathway’ triggered by repressed extrinsic signaling cues. Hence, in vitro modeling of neuroectoderm is typically initiated by excluding morphogens or serum, instead of exposure to inhibitors of signaling molecules, such as Nodal/Activin and TGFβ/Smad.[47–49] Once the neural identity is established, subsequent patterning into organs distinct from the neuroepithelium requires the action of several biochemical factors. While retinal epithelium is developed upon stimulation by SHH and Wnt[50], the cerebral region is formed upon exposure to RA.[15, 51] During the development of cerebral organoids, as demonstrated by the Knoblich group, 3D neuroepithelial spheroids are embedded into Matrigel and cultured in spinning bioreactors to enable the development of multiple regions of the forebrain, midbrain, and hindbrain.[51] The use of modified protocols of the Sasai group[52] and Pasca group[53] can result in region-specific cortical organoids via guided differentiation.
To recapitulate the interactions among neurons in physiological and pathological circumstances, region-specific brain organoids can be assembled in vitro to form spheroids comprising at least two regions of the brain.[54] Due to the heterogeneity of the brain and other tissues derived from the endoderm and mesoderm lineages, a series of biochemical cues are required to ensure controlled organogenesis. Among the brain organoid models, whole-brain organoids primarily rely on stem cells’ intrinsic signaling and self-organization abilities for spontaneous development.[51] In contrast, region-specific organoids utilize several small molecules, which inhibit Smad or Wnt signaling.[52, 53] These approaches demonstrate that differences in signals are required for the patterning of specific regions within the brain. However, the mechanism underlying the self-patterning of multiple regions in the cerebral organoids remains still unclear.
2.2. LIMITATIONS OF THE CONVENTIONAL METHODS FOR ORGANOID PRODUCTIONS
Although the accumulated information regarding the 3D organoid models provides novel approaches that can be used for studying developmental processes and disease modeling in humans, the conventional organoid culturing methods demonstrated above have certain limitations.
First, the reproducibility of organoid formation is a frequently raised concern, which requires the establishment of robust protocols to generate organoids [Figure 3A]. Recent studies analyzed various organoids utilizing single-cell RNA sequencing, demonstrating a significant variation between different iPSC lines, protocols, and experimental batches.[55–58] In the case of brain organoids, each protocol exhibited a different degree of reproducibility, implying a trade-off between complexity and reproducibility. Patterned region-specific brain organoids showed higher consistency in shape and size with lower variations in transcriptomic profiles between individual organoids from different batches and iPSC lines than those observed in the case of self-patterned cerebral organoids.[55, 58] Whole-brain organoids exhibit the ability to generate multiple regions within the brain. However, the relatively low reproducibility is a limitation that constrains their applications in drug screening or mechanism studies. Likewise, kidney organoids produced using conventional methods are associated with other issues, such as high intrinsic variability among experimental batches when compared to the iPSC lines.[56] In contrast, other groups’ modification of protocols to enhance the differentiation efficiency and specificity of kidney organoids resulted from variability between iPSC lines, thereby reflecting the difficulty of adjusting across diverse genetic backgrounds.[46, 57] Recent efforts have developed matrix-independent culture platforms employing hydrogel-based microwells to standardize the formation of organoids with similar size and differentiation rates.[59–61] Arrayed microwells fabricated with biomimetic hydrogels such as polyethylene glycol (PEG)[59] or polycarbonate films[60] enabled size control with reduced heterogeneity.
Figure 3.
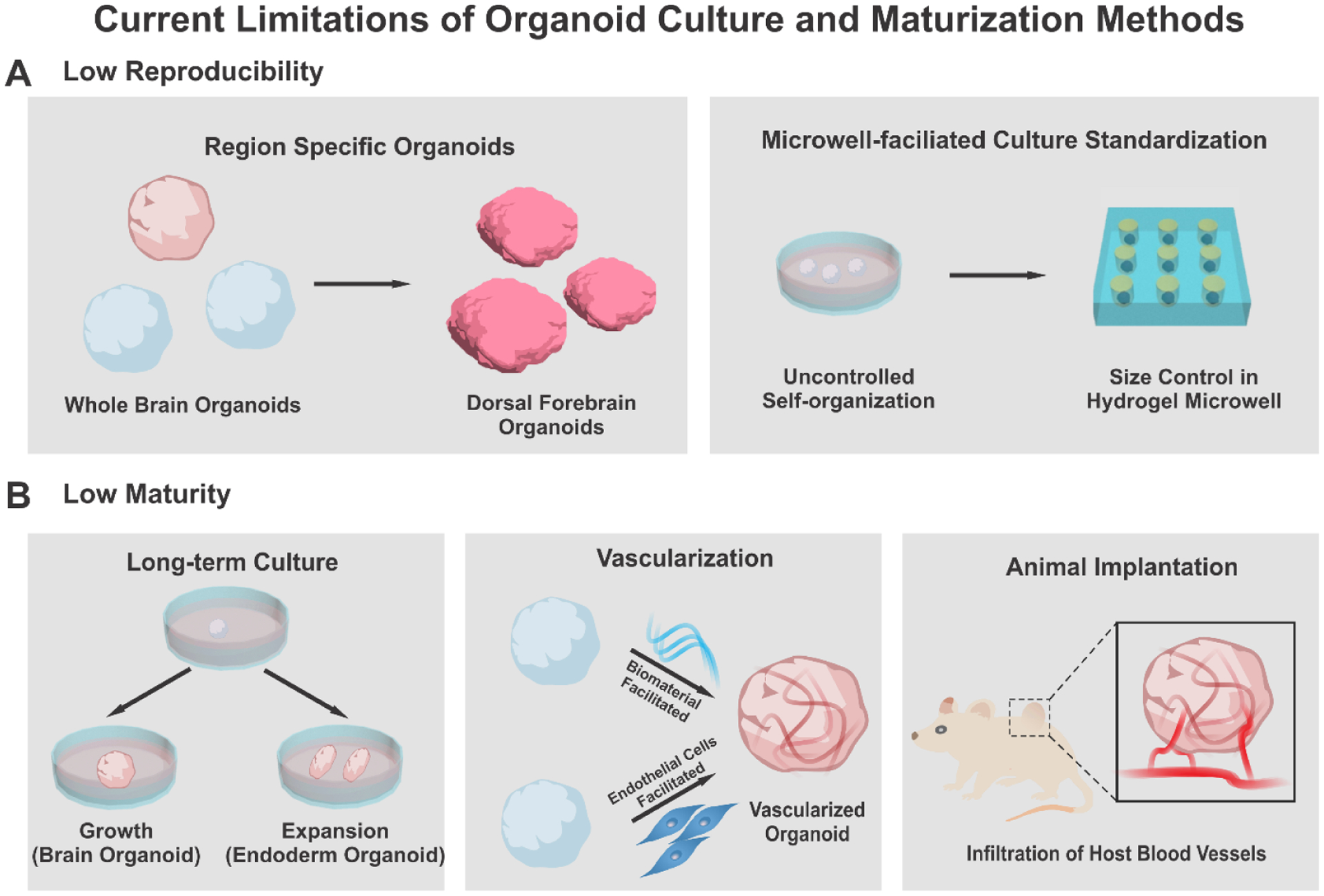
Limitations and improvement of traditional organoid culture methods. A) To improve the low reproducibility of organoids, differentiation protocols for region-specific organoids (left) and microwell-based standardization methods (right) were devised. B) To improve the low maturity of organoids, long-term culture, vascularization with microfluidics, and animal transplantation were suggested.
Another critical issue regarding organoids’ reliability is how well organoids can recapitulate the development and physiology of the actual organs. Despite a wide variety of modified protocols for organoid generation, the current organoid culture systems cannot entirely resemble all parameters of the stem cell niche in an organ-specific manner. The lack of reality in culture conditions originated from cellular stress arises from experimental conditions and the absence of vascular systems. Multiple pieces of evidence have demonstrated that PSC-derived organoids successfully mimic human organogenesis during development and reach the fetal stage but hardly resemble the adult tissue stage.[26, 35, 37, 62] Recently, a single cell-based transcriptomic analysis demonstrated that stress-related pathways activated during cortical organoid culture could impair the specification of neuronal cell types that are spatially segregated in primary human cortical cells.[63] However, several researchers have developed relatively mature organoids through long-term culture [Figure 3B].[64–68] In particular, the formation of microglia, dendritic spines, photosensitive cells, and spontaneously active neuronal circuits has been observed after extended periods of development.[66, 67] In addition to PSC-derived organoids, long-term expansion protocols of diverse ASC-derived organoids including gastric[11], colon[69], liver[70], and breast[71] organoids have been developed. These endoderm-derived organoids usually expand indefinitely and can be splitted into smaller fractions, making their long-term culture easier than indivisible organoids. However, in the case of brain or kidney organoids, which can not be splitted, prolonged culture is typically constrained by insufficient oxygen and nutrients diffusion into the central region of the organoids. To resolve this issue, the development of vascularization techniques to mimic the in vivo-like network of vasculature has been suggested.[72] Another strategy is inducing angiogenesis within organoids through animal implantation, in which host vasculatures are infiltrated into the organoids.[73, 74] Collectively, researchers are now combining techniques from multidisciplinary areas, including bioengineering, materials science, and mechanical engineering, to standardize protocols for the generation of organoids that can fulfill both reproducibility and complexity.
3. ENGINEERING EXTRACELLULAR MATRICES FOR ORGANOIDS
3.1. EXTRACELLULAR CUES FOR ORGANOID ENGINEERING
Compared to conventional 3D cell culture systems, such as spheroids and explants, organoids are derived from PSCs or ASCs, having innate self-organizing abilities to form a heterogeneous and highly organized structure. This structure mimics the morphogenetic process that occurs during development in vivo [Figure 4]. During development from PSCs, the fate, function, and plasticity of stem cells are dynamically regulated by multiple cues, including biomolecules, cell-cell interactions, and physical signals in a spatiotemporally-controlled manner.[75–78] Specifically, initial “symmetry breaking”, where one or a few cells break the initial homogenous system by changing their own identities, leading to the subsequent polarization and pattern formation.[79] Unlike PSC-derived organoids, ASC-derived organoids, including tumor organoids, lack mesenchymal lineage cells that contribute to forming the microenvironment of each tissue.[80] Thus, most ASC-derived organoids require specific biochemical factors conjugated to ECM scaffolds, providing intercellular signaling.[81] To precisely mimic natural organogenesis and biochemical support for tissue niches, it is crucial to investigate the recapitulation of various intrinsic and extracellular cues for controlling the dynamic nature of tissue niches [Figure 4A].[82]
Figure 4.
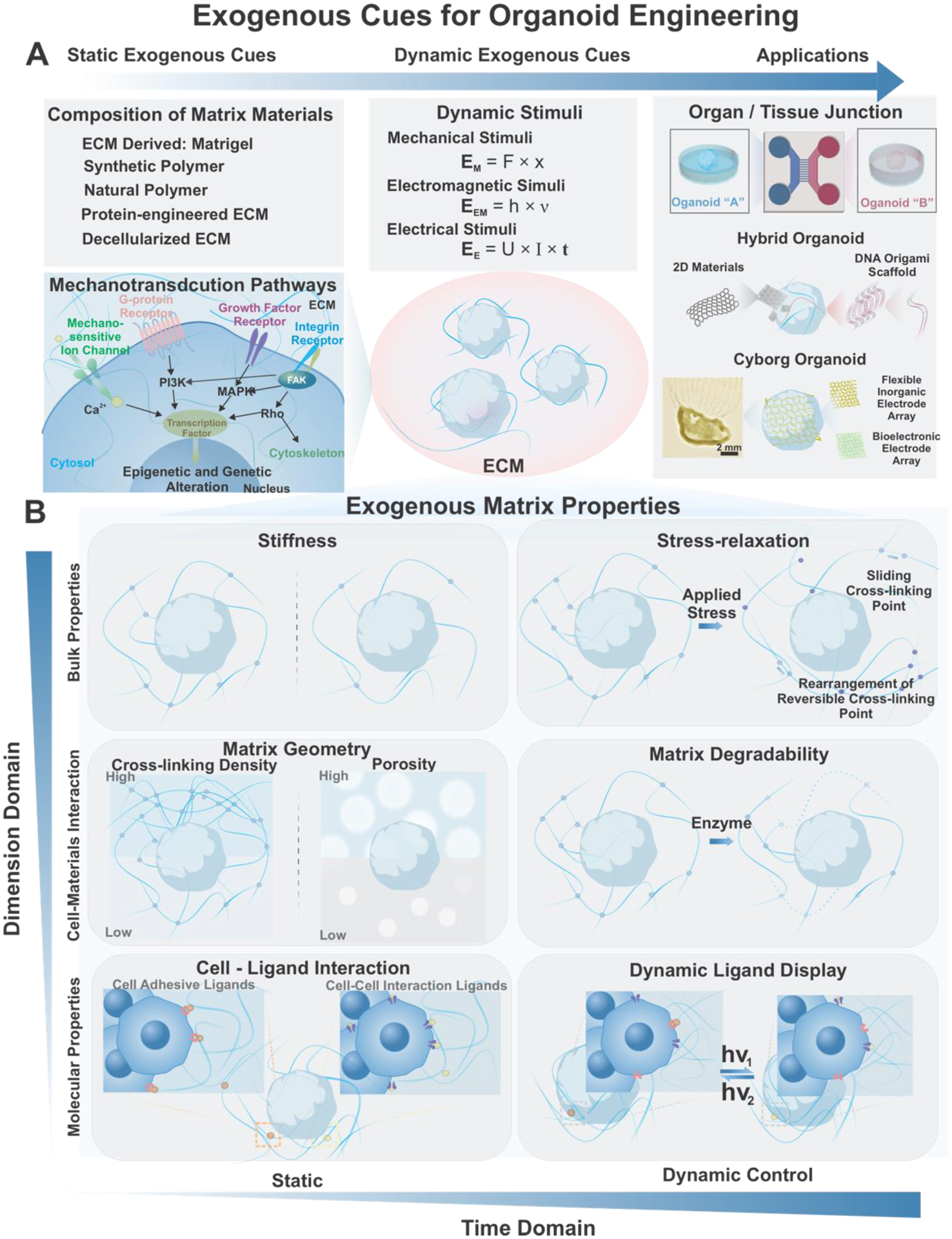
Exogenous cues for organoid engineering. A) The extracellular microenvironment can influence organoid development through matrix compositions, matrix properties, as well as systematic stimuli. Specifically, various characteristics of matrix for instance, stiffness, geometry and cell-ligand interactions have been demonstrated to have significant impact for organoid culture and development. B) Recent discoveries have indicated dynamic intriguing interactions of organoids and matrix materials as stress-relaxation, degradability, and ligand dynamics. With recent developments in biomaterials as well as bioelectronics, applications of organoid technology in organoid-on-a-chip, hybrid organoid, and cyborg organoids have become prominent. Cyborg organoid. Reproduced with permission.[144] Copyright 2019, American Chemical Society.
In terms of extracellular cues, the mechanical properties of ECMs play a significant role in regulating cell fate and the niche environment. As a result, significant research interests have been focused on the mechanotransduction signaling generated from ECM materials.[83] Still, conventional organoid culture relies heavily on scaffolds and matrices derived from animal tissues. This not only raises significant concerns about reproducibility, safety, and translatability of organoid technologies, but also makes it difficult to dissect the complex physical and biochemical organoid development ECM environment. Moreover, recent development in stem cell mechanotransduction studies have unveiled the dynamic nature of the exogenous cues. For instance, stress relaxation[84], degradability[19], and dynamic ligand display[85] have been explored to recapitulate better the features of native ECMs [Figure 4B]. It is imperative to summarize the ECM material development and categories to bridge from inceptive researches to potential applications of organoid technologies. The following sections will discuss different ECM materials and engineered ECM materials utilized for organoid engineering and studies [Table 1].
Table 1.
Extracellular materials for organoids generation and engineering
| Categories | Materials | Type of Organoid | Mechanical Properties | Applications and Findings | Ref. |
|---|---|---|---|---|---|
| Decellularized Tissue | Perfusion-decellularized Matrix | Heart | Longitudinal modulus 400 kPa; Circumferential modulus 1300 kPa (Anisotropic) | Proof-of-concept decellularized scaffold for heart | [97] |
| Decellularized liver matrix | Liver Graft | N/A | Proof-of-concept decellularized scaffold for liver graft | [222] | |
| Decellularized liver matrix | Osteochondral | N/A | Forster osteochondral differentiation | [223] | |
| Decellularized Pancreatic Matrix | Islet Organogenesis | N/A | Collagen V Regulates Islet Organogenesis | [101] | |
| Basement Membrane Extract Type 2 | Pancreas Organoid | N/A | GMP level production | [102] | |
| Natural Hydrogels | Alginate RGD Hydrogel | Breast Cancer Ductal Carcinoma | Elastic modulus: 0.04 to 2 kPa | Mechanotransduction in breast cancer progression | [224] |
| Synthetic Matrigel | Neural Tube | Stiffness: 0.5 to 12 kPa | Neurotube Morphogenesis in Synthetic ECM | [108] | |
| HA/Chitosan | Cerebral Organoids | Young’s modulus 9.8 kPa (with cell) and 10.1 kPa (without cell) | Chemical Defined Hydrogel and Defined Medium for Cerebral Oragnoid Generation | [134] | |
| HA, Fibrin | Liver Organoid, Pancreatic Organoid | Storage modulus: 0.024 to 0.492 Pa | Growth Epithelial Organoids in Defined Hydrogel | [104] | |
| Collagen | Bovine Parathyroid | N/A | Difference in 2D and 3D Cellular Behaviors | [129] | |
| Collagen | Embryonic Mesenchymal Cell | N/A | Proof-of-concept Organoid Formation | [225] | |
| Alginate Beads | Mouse limb buds differentiation | N/A | Proof-of-concept Organoid Formation | [132] | |
| Collagen Gels | Mesangial Cell | N/A | Difference in 2D and 3D Cellular Behaviors | [151] | |
| HA/Gelatin | Organoid | Storage modulus: 0.1 to 20 kPa | Proof-of-concept Organoid Formation | [142] | |
| Fibronectin | HepG2 | N/A | Difference in 2D and 3D Cellular Behaviors | [226] | |
| GAG/PEG | Mammary Epithelial | Storage modulus: 0.2 to 1.6 kPa | Modular System | [143] | |
| Agarose Gel | Cardiac Organoid | N/A | Biomimetic Development | [152] | |
| Cellulose Hydrogel | Liver Organoid | Young’s modulus: 0.255 kPa | Hepatic Differentiation | [139] | |
| Polysaccharide Hydrogel | No Cell | Young’s modulus: 3.29 to 86.73 kPa | Enzyme-based Crosslinking | [227] | |
| Protein Engineered Materials | Elastin-like Protein | Intestinal Organoid | Storage modulus: 0.18 to 1.22 kPa | Prolonged culture of primary adult intestinal organoids | [131] |
| Synthetic Hydrogels | PEG | Liver Organoid, Pancreatic Organoid | Storage modulus: 90 kPa | Growth Epithelial Organoids in Defined Hydrogel | [104] |
| PEG | Intestinal Organoid | Storage modulus: 0.3 to 1.7 kPa | PEG hydrogel for Intestinal Organoid Formation | [19] | |
| PEG | Intestinal Organoid | Storage modulus: 0.3 to 1.7 kPa | Nature Protocol For PEG-based Intestinal Organoid | [111] | |
| PEG | Human Intestinal Organoid | Storage modulus: 0.05 to 0.4 Pa Loss modulus: 0.005 to 0.02 kPa |
PEG hydrogel for intestical injury treatment | [112] | |
| Xeno Free | Retinal | N/A | Xeno-free Organoid Formation Condition | [122] | |
| PEG | Pluripotency Maintenance | Storage modulus 0.3 to 0.7 Pa | Defined ECM Boost Pluripotency | [228] | |
| PEG | Cerebral Morphogenesis | N/A | 3D Patterned NGF Guided Morphogenesis | [201] | |
| PEG | 3D Vascular Structure | Storage modulus: 0.05 to 9.4 kPa | Microfluidic Patterning | [229] |
3.2. MATRIGEL AND DECELLULARIZED MATRICES
In 3D cell culture systems, including organoid culture, scaffolds and matrices are widely used to mimic the natural ECM of tissue or cell niches.[86] Ever since the initial emergence of organoid technologies, incorporating a gelatinous protein hydrogel named Matrigel/ Geltrex/ Cultrex BME derived from Engelbreth-Holm-Swarm (EHS) mouse sarcoma cells, has enabled the culturing of various types of organoids.[87] This animal-based ECM material provides a mixture of various essential ECM components and soluble factors[88], a fostering environment with adhesion and degradation capabilities for embedded cells.[12] Many of the earlier studies producing diverse organoids, including intestinal organoids[10], brain organoids[15], retinal organoids[89], hepatocyte organoids[90], and functional liver organoids[70, 91] , have utilized Matrigel as the ECM material.
Similarly, native tissue-derived decellularized ECM (dECM) scaffolds have been developed for tissue engineering applications since 1970s.[92] As original tissue ECMs and cell niches, these dECMs provide a combination of ECM fibers, including collagen, laminin, fibronectin, and other cell-deposited ECM materials. A variety of initial studies utilizing dECM derived from other tissues, including skin[93], vasculature[94], heart valves[95], and bladders[96] have led to remarkable results for the generation of crucial tissues or organs as implants.[97, 98] By incorporating iPSCs, dECM scaffolds have been employed as exogenous platforms with patient-derived organoids, which can be applied to regenerative medicine applications.[99] In addition to their roles as basement scaffolds, dECM can provide unique combinations of ECM factors to facilitate organogenesis.[100] A recent proteomic analysis of pancreatic tissue ECM pinpoints collagen V as a key ECM material for islet organogenesis.[101] Specifically, dECM hydrogel synthesized from rat pancreas was compared to Matrigel in terms of intracellular and extracellular proteins’ composition. The pancreas-derived dECM exhibited 155 different proteins, including 63 extracellular and 92 intracellular proteins, with extracellular proteins constituting 42.3% of the total protein content. The dECM contains a certain percentage of proteins related to catalytic activity, biological regulation, and developmental processes correlated to regulatory activities in the pancreas. More importantly, collagen II, III, and V were identified in large quantities, compared to other collagen proteins in this dECM. In contrast, collagen II and III possess different α chains regulating different pathways. Collagen V was identified to be a candidate ECM material that regulates the development of pancreatic islet organoids from iPSCs via the following: (1) promoting key transcription factors, such as PDX1, NKX6.1, MAFA, MAFB, UCN3, ARX; (2) promoting the expression of a broad spectrum of genes encoding islet hormones, including insulin, glucagon, somatostatin, and pancreatic polypeptide; (3) promoting better glucose-responsive insulin secretion.
There is a growing trend toward developing GMP-compliant dECM protocols combined with chemically defined culture media to promote the transition of dECM-based organoid research into clinical settings.[102] Coppi and his team developed a GMP-compatible dECM-based hydrogel system that enables human endoderm-derived organoid formation and development.[103] Specifically, decellularized porcine small intestinal mucosa/submucosa was processed through freeze-drying, milling, γ-irradiation treatment, and pepsin/HCl digestion to develop a clinically available ECM hydrogel. The composition of the developed dECM hydrogel was compared to that of various endodermal origin tissues, including the gut, liver, and pancreas, via proteomic analysis. Principal component analysis (PCA) revealed high similarity with 1% diversity in the identified proteins. Subsequently, human liver ductal, human fetal hepatocyte organoids, and human pediatric gastric enteroids were successfully cultured on the dECM hydrogel without significant differences compared to groups cultured on Matrigel and Cultrex BME. In vivo dECM gel organoid culture and growth were demonstrated using human fetal pancreatic duct organoids. The establishment of gelatin extraction and dECM organoid culture protocols in a GMP-compliant fashion solved the innate cons of dECM/animal-based ECM materials for human organoid development, opening new avenues for further clinical applications.
With growing demands to control organoid culture and development, uncharacterized compositions with significant batch-to-batch variation and complexity have become a significant hurdle for systematic studies, downstream characterizations, and clinical applications.[19] A recent proteomic study on Matrigel samples demonstrated that 956 different proteins, including 243 extracellular proteins and 713 proteins, were identified in Matrigel, of which 27.5% were extracellular proteins [Figure 5A].[101] A total of 1637 different proteins were observed in various Matrigel samples, demonstrating proteomic heterogeneity within and among samples. A gene ontology study on Matrigel showed a discrepancy in protein enrichment in cell organelles and nuclei. Interestingly, this study also provided a matrisome subcategory of extracellular proteins in Matrigel, containing 24% glycoproteins, including laminin, 3% collagen, 1% proteoglycans, 2% ECM-affiliated proteins, 9% ECM regulators, and 6% secretion factors. Besides, a recent combination study showed that fibrin hydrogels mixed with 10% Matrigel supported the formation of early mouse small intestinal stem cell organoids and early cyst structures, indicating that only certain Matrigel-containing signals are needed for initial organoid formation with proper material support.[104] Thus, it is imperative to develop ECM materials that support organoid culture with reproducibility and tunability, which cannot be achieved by just depending on natural features of Matrigel and dECM. As a result, engineered matrices with defined chemical and biophysical properties have been developed in recent years to achieve robust organoid development and maturation.[105]
Figure 5. Properties of extracellular materials and their effects on organoid development.
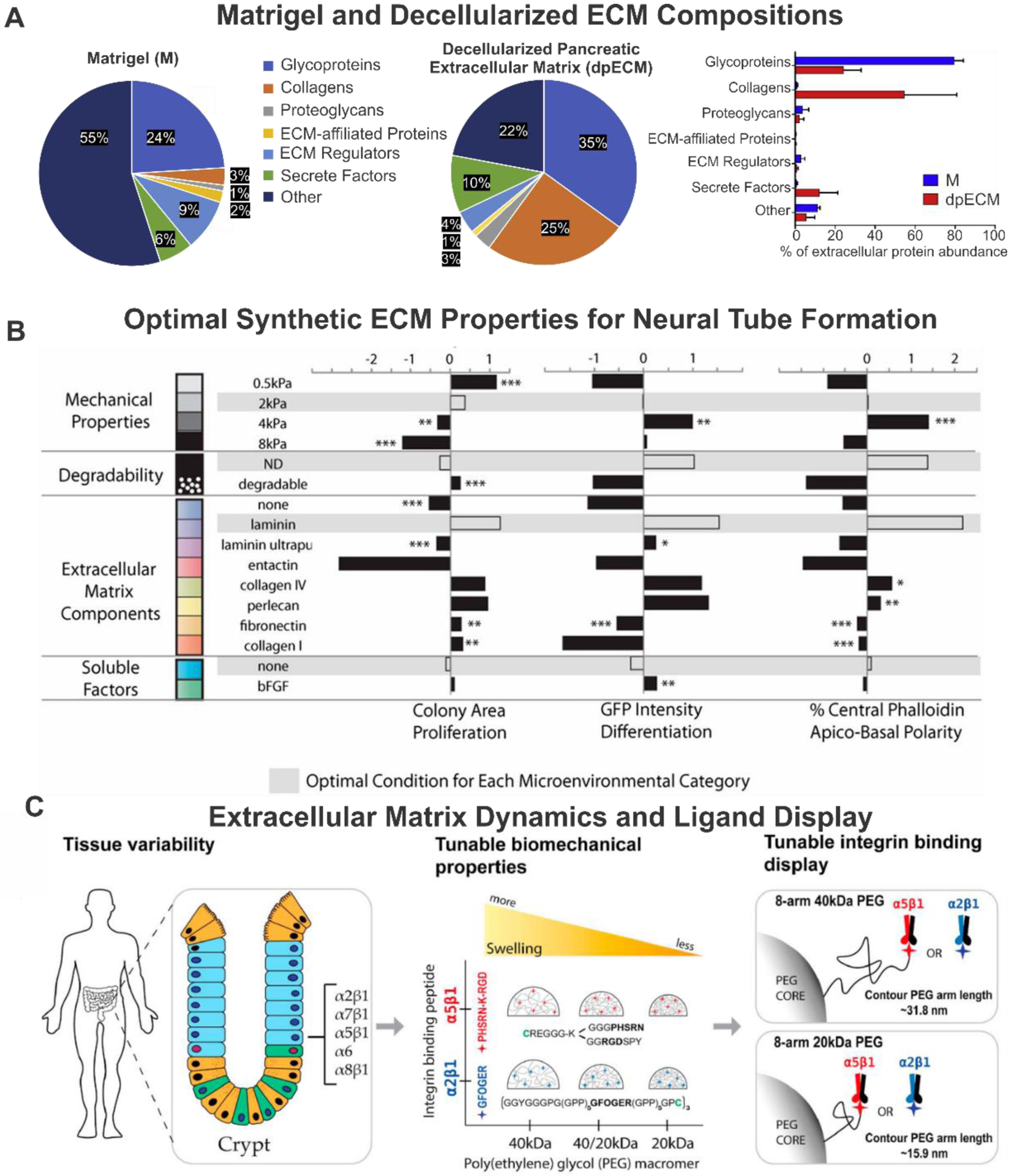
A) Matrisome subcategories of extracellular proteins in dpECM and Matrigel. Reproduced with permission.[101] Copyright 2020, Elsevier. B) A library of molecular building blocks is mixed and crosslinked in situ to form cell-containing 3D hydrogels with independently controllable mechanical and biochemical properties. Reproduced with permission.[108] Copyright 2016, National Academy of Sciences. C) Niche-inspired synthetic hydrogel network based on 8-arm PEG macromers of 20- and 40-kDa with tunable bulk biomechanical properties and tunable local integrin-binding display capability. Summary of biomechanical and its effects on enteroids emergence after encapsulation. Reproduced with permission.[116] Copyright 2020, Elsevier.
3.3. SYNTHETIC MATRICES FOR ORGANOID RESEARCH
With the recent development in the molecularly-defined synthetic matrices – i.e., polyethylene glycol (PEG) hydrogel - various biophysical cues that govern cell pluripotency[78], epigenetic states[106], and cell fate[107] have been identified. The initial research has been focused on mimicking brain organogenesis using defined ECM and media conditions. Lutolf and coworkers reported the successful generation of early cortical structures such as neural tubes in a synthetic PEG hydrogel-based ECM environment [Figure 5B].[108] Combinatorial screening of potential neurogenic modulators was performed through the modular design of this PEG hydrogel platform with parameters including degradability, mechanical stiffness (0.5 kPa – 8 kPa), soluble factor (bFGF), and various ECM components (collagen IV, collagen I, fibronectin, entactin, perlecan, laminin). This screen discovered that intermediated stiffness (2–4 kPa), non-degradable backbone, and laminin in conjunction with collagen IV and perlecan, promote apicobasal polarity and neural marker expression. More importantly, compared to those grown on Matrigel (positive control), neuroepithelial colonies cultured on PEG-based ECM showed more consistent, distinct, and polarized structures, thereby demonstrating the advantage of employing defined synthetic ECM conditions.
Moreover, as a simplified in vitro organoid model from pluripotent stem cells, epithelial organoids or intestinal organoids were also investigated using the defined ECM materials. Initial demonstration using PEG hydrogel-based ECM showed that epithelial organogenesis, cyst formation, polarization, and lumen structure formation were tightly regulated by ECM mechanical properties, adhesive ligand (RGD) density, and degradability.[109] Lutolf and colleagues utilized the chemically defined structure and the innate modularity to identify ECM parameters essential for intestinal organoid formation, expansion, and development.[19] At different stages of intestinal organoid culture, distinct ECM characteristics are required, emphasizing the need to introduce exogenous cues in a spatiotemporal manner for studying organogenesis. Incorporation of fibronectin during the initial intestinal stem cell 3D culture promotes adhesion and proliferation. A high stiffness (1.3 kPa)-mediated yes-associated protein 1 (YAP) mechano-transduction signaling[110] favors intestinal stem cell expansion. In contrast, a hydrogel with soft stiffness (~300 Pa) and laminin-111 incorporation would foster intestinal stem cell differentiation and organoid formation. Based on these findings, a well-defined protocol for human and mouse intestinal organoid culture was established[111]; hence, opening up the possibility of using modulated ECM materials for organoid culture.
García and colleagues utilized a four-armed, maleimide-terminated PEG hydrogel decorated with the adhesive peptide RGD and protease degradable peptide GPQ-W to support the growth and expansion of human intestinal organoids.[112] Variations of PEG polymer weight percentages (3.5 −6.0% w/v) revealed that lower-weight percentage density favored organoid viability. Different adhesive peptides including RGD, laminin α1 chain-derived AG73 (CGGRKRLQVQLSIRT), type I collagen-mimetic triple-helical GFOGER (GYGGGP(GPP)5 GFOGER(GPP)5GPC), and laminin α1 chain-derived IKVAV (CGGAASIKVAVSADR) were screened for intestinal organoid culture, while keeping the degradable peptide percentage and PEG polymer percentage constant. As a result, incorporation of RGD peptide resulted in significantly better viability than that observed in hydrogels containing AG73, GFOGER, and IKVAV. The difference in organoid viability can be attributed to different ECM mechanical properties (YAP-mechano-sensing pathway) instead of mesh size-mediated permeability differences. In terms of organoid differentiation outcomes, the group with organoids embedded in PEG hydrogel showed similar expression patterns of endodermal (FOXA2) and epithelial markers (ZO1, ECAD, and CLDN2) at an early stage compared to the Matrigel group. Furthermore, in vivo organoid differentiation displayed the generation of a typical mature intestinal crypt-villus structure with lamina propria, muscularis mucosa, and submucosa. This intestinal organoid was further injected into mechanically induced mucosal wounds at the distal part of the colon in immunocompromised mice, revealing a strikingly improved therapeutic effect for the colon injury.
As another step for modulating the mechanical properties of PEG-based synthetic hydrogel ECMs, property tuning and ligand display have been investigated based on the recent discovery of dynamics of cell-matrix mechanical interactions and stem cell mechano-transduction.[113] Anseth and colleagues have reported a PEG-based photodegradable hydrogel system for studying the matrix mechanical force relaxation and its effect on intestinal organoid formation, as evidenced by crypt structure formation.[114] The stiffness can be tuned through photo-degradation of the allyl sulfide-based crosslinking system, thereby rendering the modulation of organoid ECM environmental possible in a remote and in-situ fashion. Specifically, YAP/Notch signaling (a well-studied pathway in stem cell mechano-transduction) is responsible for mediating the mechanosensitivity of intestinal organoid survival, differentiation, crypt structure formation toward the ECMs.[115]
Moreover, Griffith and co-workers developed a completely synthetic ECM system with reproducible and tunable biomolecular and mechanical properties.[116] The synthetic hydrogel system was based on 8-arm PEG macromers with different combinations of adhesive peptides, ECM peptides, and matrix metalloprotease (MMP) degradable peptides. As a demonstration, human tissue-derived enteroids and organoids were encapsulated [Figure 5C]. Synthetic hydrogel with a 20 kDa stiffness containing α2β1 integrin-binding peptide (GFOGER) was shown to support organoid formation and development. In addition, intestinal enteroids were serially passaged using basolateral stimulating hydrogel systems that maintained their innate proliferative ability.
With the recent progress in microporous hydrogel engineering[117], an inverted colloidal crystal-based PEG scaffold has been fabricated by sacrificial polystyrene beads with diameters of 40, 60, 100, 140 μm corresponding to the porous size of the resultant PEG hydrogel ECM.[118] Furthermore, the porous ECM surface was functionalized with collagen I, fibronectin, or laminin 521 for promoting the attachment of iPSC-derived progenitors and inter-cluster cell-cell interactions. As a result, the 140 μm pore ECM with collagen I functionalization facilitated the liver bud formation compared to other methods, such as 3D spheroid, Matrigel, and 2D culture. This inverse colloidal crystal PEG ECM-derived liver organoid gives rise to opportunities for the recapitulation of liver organogenesis using engineered synthetic ECM.
In addition to the PEG-based ECM scaffold, other synthetic polymer-based ECM systems have been studied for culturing complex organoid structures.[119] A poly(lactic-co-glycolic acid) copolymer (PLGA) fiber microfilament-based floating scaffold has been utilized to generate elongated embryoid bodies.[120] The microfilament structure enhanced neuroectoderm formation and improved cortical development with reconstitution of the basement membrane, leading to characteristic cortical tissue architecture, including forming a polarized cortical plate and radial units. This model system could generate the distinctive radial organization of the cerebral cortex and allow for the study of neuronal migration and demonstate that combining 3D cell culture with bioengineering can increase reproducibility and improve tissue architecture. A PLGA film was shown to induce islet β-like cell organoid differentiation through dopamine and liraglutide coating. Furthermore, this PLGA-based organoid system was transplanted into a diabetic rat model, demonstrating the potential for type 1 diabetes treatment.[121] Moreover, retinal organoid differentiation was achieved by employing synthetic ECM materials, a vitronectin-mimicking oligopeptide-based scaffold, as a substitute for Matrigel.[122] By using the oligopeptide scaffold, 100% aggregation efficiency was achieved with mouse embryonic stem cells, and the size of the organoid was increased when compared to Matrigel groups. A minimal difference was observed from day 7 during retinal organoid differentiation, thereby leading to a xeno-free ECM retinal organoid culturing protocol for potential applications. Comparing to PLGA scaffold, medical-grade carbon fibers (CFs) were also investigated, showing an improved iPSC differentiation efficiency within organoids.[123] The physicochemical properties of carbon scaffolds such as porosity, microstructure, or stability in the cellular environment make them a convenient material for creating in vitro organoid models. This makes organoids formed on carbon scaffolds an improved model containing mDA neurons convenient for studying midbrain-associated neurodegenerative diseases such as Parkinson’s disease.
3.4. ENGINEERED NATURAL POLYMER MATRICES FOR ORGANOID RESEARCH
Natural polymer-derived ECMs are heavily studied to develop organoid structures due to their defined chemical structure and established the possibility for engineering based on previous research endeavors.[124, 125] These natural polymer-based ECM proteins can be divided into protein-based and polysaccharide-based categories based on their native chemical components.
Merker and coworkers utilized protein-based ECM to develop organoids, suggesting that collagen-based ECM organoid culture better promotes mesangial cell development than conventional collagen gel culture conditions.[126] Intestinal-mesenchymal 3D models were later achieved using the collagen vitrigel, incorporating fibroblast and Caco-2 cells into rigid connective tissue constructs.[127] Furthermore, the bladder mucosa organoid model was developed utilizing a type I collagen scaffold showing an anatomical and physiological resemblance to native bladder tissue.[128] Initial research efforts using collagen hydrogels to culture parathyroid organoid structure maintained innate calcium-mediated parathyroid hormone responsiveness. However, the calcium-dependent parathyroid hormone secretion is lost in primary 2D culture.[129]
Furthermore, complex organoid structures were formed using silk fibroin and collagen for disease modeling.[130] A molded cylindrical scaffold composed of silk was seeded with epithelial cells derived from human intestinal organoids and subsequently coated with intestinal myofibroblasts mimicking intestinal epithelium structure with typical epithelium markers. As a model for bacterial infection, a significant innate immune response was invoked by E. coli. treatment indicating the potential application of using this organoid system for pathogen-infected disease modeling. Heilshorn and coworkers demonstrated a protein engineering approach to generate a naturally-derived protein scaffold as the ECM for the 3D culture of primary adult intestinal organoids.[131] A similar study has demonstrated the influence of stiffness, degradability, and matrix remodeling of elastin-like protein (ELP) hydrogel as the ECM materials on maintaining the stemness of neural progenitors.[78] In this study, a recombinant engineered protein was designed with the RGD domain derived from fibronectin and elastin-based structural domains mimicking adhesive biochemical cues and elastomeric biochemical cues in the natural intestinal tissue. As a result, low mechanical stiffness (180 Pa) with increased cell adhesive domains facilitated the organoid formation.
As an example of polysaccharide-based ECM materials, De Souza and coworkers utilized alginate beads to induce chondrocyte organoid formation from mouse limb-bud-derived mesenchymal cells.[132] This discovery leads to a significant demonstration of human intestinal organoid culture in alginate hydrogel ECM without adhesive ligand modifications.[133] The cultured organoid could sustain growth under in vitro conditions for 90 days, which, in part, indicates the potential mechanical support function of ECM during the organoid growth stage. Interestingly, epithelial organoids (enteroids) cultured in this non-adhesive alginate gel showed minimal growth, while secreted laminin was discovered as a basement membrane.
Another study focused on brain organoid culture using defined polysaccharide-based ECM and media conditions.[134] Specifically, hyaluronan (HA-Na) and chitosan (CT) dry blends were infused with iPSCs and developed into cell-embedded hydrogel matrices. Interestingly, without additional neural induction processes, the organoid developed into a cerebral organoid showing typical cerebral cortex structures, such as neural rosettes and neural tubes. Moreover, an electrostatically crosslinked ECM hydrogel system from hyaluronate and chitosan was shown to sustain human brain organoid development for 10 days, showing rosette and neural-tube-like structures and functional response to glutamate or potassium treatments.[134] Within this ECM system, adrenoleukodystrophy (ALD) patient-derived iPSCs were differentiated into patient-specific cerebral organoids, showing robustness and the potential for patient-derived disease modeling using this chemically defined hydrogel system. Qin and co-workers developed a calcium-alginate fiber-based microfluidic system for brain organoid culture and development.[135] This hollow alginate gel fiber enabled the differentiation of iPSCs into brain organoids with polarized neuroepithelium and key cell heterogeneity, marking early developmental progression. This approach eased the tedious procedures for brain organoid culture and allowed for the opportunities to scale-up. Similar to the previously inverted colloidal hydrogel scaffold, a collagen-coated alginate bead-based scaffold was employed to recapitulate the void structure cultured with human lung fibroblasts and iPSC-derived mesenchymal cells.[136] The void structures mimicking the lung alveolar structures were formed successfully in the space between beads. This approach marks the potential for scaffold-based structure mimicking to guide the organogenesis process, which could be facilitated by 3D bioprinting technologies.
Pioneering work has been conducted by Kurisawa and his team utilizing gelatin-based and hyaluronic acid (HA)-based conjugates to control and mimic the native colorectal tumor organoid extracellular matrix.[137] Through the unique oxidation mediated crosslinking method, the matrix stiffness could be tuned from 2 to 34 kPa. Judged by the drug sensitivity, the gelatin-based ECMs showed retention of the colorectal tumor organoid’s susceptibility and supported Ex-vivo engraftment and tumor growth in animal models. Similarly, a Gelatin-HA-based hydrogel system was recently reported to support patient-derived colorectal cancer organoids with cancer-assisted fibroblasts (CAFs) co-culture to mimic and study the contributions of CAFs to tumor drug resistance and progression.[138]
A recent study has been reported on cellulose nanofibril-based ECM for human liver organoid development.[139] The cellulose nanofibril hydrogel showed Young’s modulus of 225 Pa, which supports hepatic differentiation and maturation while inhibiting liver organoid proliferation. Human liver organoids derived from several donors were successfully generated in the cellulose hydrogel showing enhanced metabolic functionality compared to the Matrigel group. This finding suggested that the tunability of engineered natural polymer matrices can offer potential advanced ECM materials for the further maturation of organoids.
3.5. SYNTHETIC/NATURAL HYBRID MATRICES FOR ORGANOID RESEARCH
With recent advances in biomaterials engineering and synthetic ECMs, various attempts to incorporate synthetic and natural materials into organoid ECM matrices have been made to gain advantages from both types of material. Grikescheit and colleagues utilized polyglycolic acid (PG), poly-L-lactic acid, and collagen I hybrid scaffold for culturing patient and mouse colon-derived epithelium and mesenchyme cells.[140] The hybrid scaffold supports human and mouse colon organoid development showing abundant smooth muscle and neural clusters (neurons and glia). This discovery identified the enteric nervous system (ENS) development in the colon organogenesis process. Furthermore, through the incorporation of ENS progenitors, aganglionic colon organoids can be repopulated with neural clusters, making it a potential solution for the treatment of Hirschsprung disease.[141]
Interestingly, Atala and coworkers reported a HA and gelatin-based extrudable bioprinting ink, which utilizes a PEG-based crosslinker, providing tunable stiffness ranging from 100 Pa to 20 kPa.[142] This ink system also provides a customizable ECM to mimic native biochemical signals, including brain-derived neurotrophic factor (BDNF), bFGF, bone morphogenetic protein 5 (BMP-5), insulin-like growth factor binding protein 2 (IGFBP-2), TGF-β1, BMP-7, EGF, growth hormone, and neurotrophin-2 (NT-3). Primary liver spheroid printing was conducted with the system, forming liver organoids with high viability and even functional characteristics of albumin and urea productions.
Recently, a modular glycosaminoglycan (GAG)-PEG hybrid ECM material was developed, demonstrating independent tunability of biochemical and mechanical properties.[143] Specifically, GAG heparin, 4-arm PEG, and MMP-cleavable crosslinker were incorporated into the ECM formulation. Using this tunable ECM, the human mammary epithelial organoid formation was achieved, emphasizing the necessity of heparin and degradability for the organogenesis process. This hybrid ECM material provided a chemically defined culturing system showing polarized mammary epithelial acini structure and mammary epithelial cell-specific laminin 332 depositions.
Interestingly, a systematic comparison of the effects of different degradable ECM materials on epithelial organoids was conducted by Schwan and colleagues.[104] In this study, fully synthetic transglutaminase (TG) crosslinked PEG hydrogel (neutrally charged), semisynthetic TG crosslinked hyaluronan (HA) hydrogel (negatively charged), calcium crosslinked alginate hydrogel (negatively charged), and human-derived thrombin crosslinked fibrin hydrogel (zwitterionic) were investigated. Organoid formation from mouse small intestinal stem cells indicated that 10% Matrigel-fibrin mix hydrogel provided similar culture conditions to Matrigel, while other groups showed drastically lower colony formation efficiency. Hydrogel mesh size comparisons demonstrated that Matrigel possesses dense networks of pores smaller than 200 nm, while a fibrin hydrogel displays a sparse network with micron-size porosity. This observation is in accordance with the previous findings demonstrating a minimal correlation between organoid formation and mesh size.[112] Interestingly, upon the addition of soluble RGD for competitive inhibition for the binding of fibrin RGD, significantly lower colony formation was observed, corroborating the importance of RGD-mediated adhesion for organoid formation. Moreover, laminin-111/entactin combined with fibrin hydrogels were identified as suitable substitutes for the fibrin Matrigel hydrogel compared to collagen IV and heparin, the primary ECM components of Matrigel.
As a promising candidate for engineering ECM materials, hybrid active material with the capability of tissue-wide electrophysiological characterization has been demonstrated by Liu and his team[144] [Figure 6A]. In this study, a soft, stretchable mesh nano-electronic ECM composed of gold, chromium, and poly(3,4-ethylenedioxythiophene (PEDOT) was fabricated using wafer-based etching procedures.[144] A unique 2D to 3D organoid generation process was adopted to incorporate the mesh nano-electronic ECM into the human cardiac organoid system. This “cyborg” cardiac organoid presented considerable similarity in mature cardiac organoid markers troponin T (TNT), α-actinin, and actin in comparison to conventional Matrigel-based cardiac organoid culture up to 40 days. More importantly, this ECM system allows temporal observation of bursting dynamics in the whole-organoid level during the entire cardiac organogenesis process. Similarly, Dmitriev and coworkers developed a cellulose-based extracellular pH monitoring ECM for intestinal organoid culture.[145]
Figure 6.
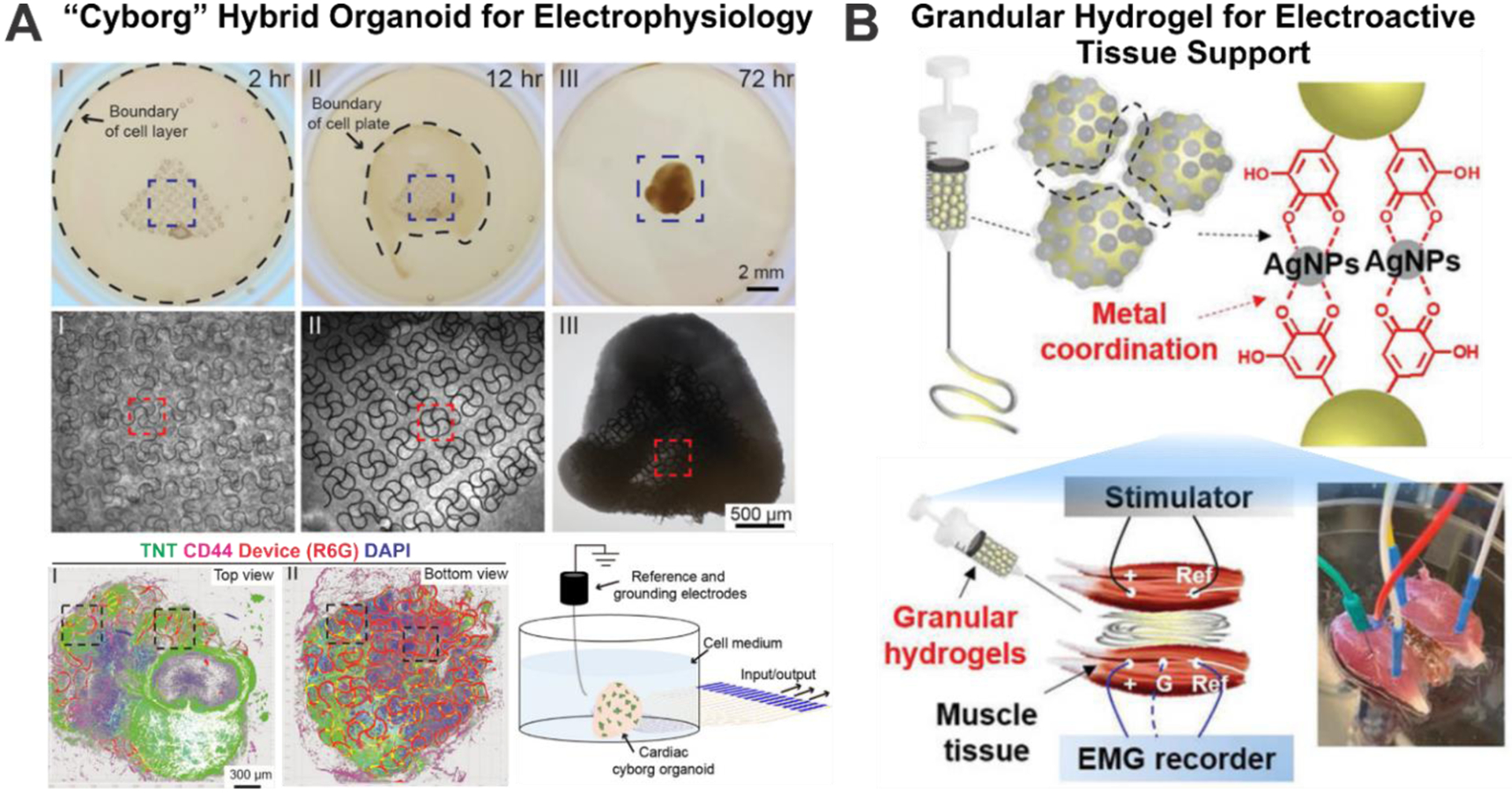
Highlights of recent developments in synthetic-natural novel materials for organoid studies. A) “Cyborg” hybrid organoid developed for in-situ tissue wide electrophysiology characterization of cardiac organoids. Reproduced with permission.[144] Copyright 2019, American Chemical Society. B) Injectable granular conductive hydrogel for electroactive tissue stimulation and electromyogram signal detection. Reproduced with permission.[146] Copyright 2019, John Wiley and Sons.
Furthermore, not only electrophysiological monitoring of organoids can be achieved, but also specific actuation capabilities could be added through incorporating functional responsive materials. For example, by combining granular hydrogel ECMs and 3D printing technologies, electroactive tissue support with the ability of muscle tissue stimulation and electromyogram monitoring has been demonstrated.[146] As shown in [Figure 6B], the researchers uniquely incorporated Ag ions inside the granular hydrogel precursor ink, which would form into silver nanoparticles (AgNPs) with an in-situ reduction reaction. This in-situ silver ion reduction not only served to generate an electroactive component in the hydrogel structure but also crosslinked the precursor onto the hydrogel structure. Interestingly, this hydrogel ECM has been applied to support muscle defects, demonstrating electro-actuation and electrophysiology characterizations.
Recent developments of bioelectronic materials with bioinspired neuron-material interfaces[147] and genetically targeted functional material assembly[148] have further explored the potential of developing novel materials at the tissue and organoid levels. Lieber and his team incorporated a biomimicry approach to develop neuron-like electronics (NeuE) with structural and mechanical similarity to native neural tissues.[147] Strong evidence suggested that structural and mechanical mismatch between conventional neural probing materials and neural tissues compromised the potential of long-term electrophysiological interrogation and modulation. However, as demonstrated in this study, long-term (90 days) tissue-level functional studies have been conducted using these novel biomimicry materials. Another breakthrough has been made by the Bao group and the Deisseroth group, demonstrating cell-type-specific chemical assembly of electroactive functional materials in living cells, tissues, and animals.[148, 149] The researchers extended genetic manipulation to local tissue structural patterns through altered local biochemical environments. Bioelectronic conductive polymers can be synthesized by genetically targeted neuron-specific expression of an engineered enzyme, ascorbate peroxidase 2 (Apex2)-mediated biocompatible polymerization reactions. Electrophysiological and behavioral characterization has been performed in freely moving animals in a cell-specific manner through this material synthesizing method. This innovative material-tissue interfacing approach opens up opportunities to create functional and active ECMs in living organoids.
Various materials have been engineered to recapitulate the in vivo organogenesis environment of different organoids. Moreover, there is an increasing trend to engineer ECM materials to recapitulate the tumor microenvironment and study the tumor organoid genesis process. These researches have been well-summarized in the following review by Kim and colleagues.[150] With the help of advanced biochemical and molecular biological characterization tools, spatiotemporal information regarding the organogenesis process will be revealed. To cope with this trend, engineered ECM materials coupled with 3D bioprinting technology have made it possible to spatially pattern biological signals in printed constructs, guiding the organogenesis process’s symmetry break.[151] On the other hand, defined scaffold methods for organoid generation could provide a comparison tool and research platform to investigate various factors for promoting organ-level development.[152] Collectively, by incorporating advanced ECM materials, cell-ECM interaction studies, and spatiotemporal signal introduction methodologies, these engineered materials could provide an innovative avenue for next-generation organoid research and applications.[153, 154]
4. GENOME ENGINEERING FOR ADVANCED ORGANOID BIOAPPLICATIONS
4.1. METHODS OF GENOME ENGINEERING
In addition to extrinsic cues such as ECM components, researchers have explored intrinsic cues such as gene editing for controlling organoid phenotype and maturation. This approach can be especially valuable, as researchers can study isogenic samples with specific mutations to reduce heterogeneity in samples. To investigate disease phenotypes or the role of essential genes during organogenesis, researchers have developed tools that can be used to edit the DNA accurately. There are two major classes of genome engineering: transient and permanent. When transient gene expression changes are necessary, methods such as adeno-associated viruses or electroporation, which allow for transient gene expression, can be used. When permanent changes are necessary, lentiviruses or transposons can be applied to add gene transfer, or CRISPR/Cas9 systems can be utilized for gene editing.
Besides the editing methods, the interval when genome engineering is performed can also play a vital role in organoid development and study. The initial cell population can be engineered and sorted to create a homogenous population of cells to form isogenic organoids. Alternatively, electroporation or other methods can be used after organoid formation to create a heterogeneous population of cells within the organoid and potentially affect organoids in a spatially controlled manner.
Lastly, viruses can be delivered after the organoids have matured, thereby mimicking gene therapy, a promising avenue for treating various debilitating diseases. Taken together, these tools aid in the advanced manipulation of endogenous signals to guide organoids towards a specific phenotype, allowing for a more advanced study of natural organ development and pathologies that affect natural organ function, as well as potential treatments of those pathologies [Figure 7]. We will discuss several genome engineering strategies for studying organogenesis and modeling various diseases with organoids in the following sections [Table 2].
Figure 7.
Genome engineering for versatile bio-applications of advanced organoid research. Methods of genome editing. Reproduced with permission.[218] Copyright 2017, Elsevier. Creation of isogenic WT or diseased iPSCs. Reproduced with permission.[219] Copyright 2020, Elsevier. Maturation of organoids. Reproduced with permission.[220] Copyright 2019, Elsevier.
Table 2.
Genome engineering for applications of organoids
| Categories | Genome Engineering Techniques | Organoid/Disease models | Results and phenotypes | Ref. |
|---|---|---|---|---|
| Fluorescence labeling | Electroporation with GFP/mCherry construct | Cerebral organoids | Live imaging of organoids | [155, 156] |
| Electroporation with GFP construct | Retinal organoids | Live imaging of organoids | [157] | |
| GFP knock-in to TUBB locus with CRISPR-HOT | Hepatocyte organoids | Visualizing subcellular structures | [222] | |
| Role of specific genes | CRISPR/Cas9-based knock-out of ODF2 and siRNA-based silencing of IFT88 in Sertoli cells | Testicular organoids | Loss of primary cilia, impaired formation of tubules | [159] |
| CRISPR/Cas9-based knock-out of RB1 in hESC | Retinal organoids | Apoptosis, reduced number of retinal cells | [160] | |
| CRISPR/Cas9-based knock-out of Wnt4 in mESC | Kidney organoids | Lack of MET, impaired nephrogenesis | [161] | |
| Modeling neurological disorders | Viral infection with mutant APPSL and PSEN1 (ΔE9) | 3D neural culture (Alzheimer’s disease) | Elevation of amyloid-β, hyperphosphorylation of tau | [230] |
| CRISPR/Cas9-based APOE4 variants in iPSCs | Cerebral organoids (Alzheimer’s disease) | Elevation of amyloid-β, hyperphosphorylation of tau | [163] | |
| Electroporation of Tau-P301S in iPSCs | Cerebral organoids (Frontotemporal dementia) | Hyperphosphorylation of tau | [164] | |
| CRISPR/Cas9-based genome editing (Δp35KI) in patient iPSCs carrying Tau-P301L | Cerebral organoids (Frontotemporal dementia) | Reduced phospho-tau and increased synaptophysin compared to patient iPSCs (Tau-P301L) | [165, 231] | |
| CRISPR/Cas9-based genome editing (LRRK2-G2019S) in iPSCs | Midbrain organoids (Parkinson’s disease) | Elevated aggregation of α-synuclein, increased expression of TXNIP | [166] | |
| Generation of patient iPSCs carrying LRRK2-G2019S | Midbrain organoids (Parkinson’s disease) | Reduced number and complexity of dopaminergic neuron, compensatory increase in FOXA2-positive progenitors | [167] | |
| Generation of iPSCs patients with idiopathic autism | Telencephalic organoids (Autism spectrum disorders) | Overproduction of inhibitory neurons, increased expression of FOXG1 | [168] | |
| CRISPR/Cas9-based dosage reduction of FOXG1 in hPSCs | MGE organoids (FOXG1 syndrome) |
Microcephaly, impaired inhibitory interneuron development | [169] | |
| Electroporation of organoids with shRNA targeting CDK5RAP2 | Cerebral organoids (Microcephaly) | Premature neuronal differentiation | [15] | |
| CRISPR/Cas9-based knock-out of GLB1 in iPSCs | Cerebral organoids (GM1 gangliosidosis) |
Accumulation of GM1 ganglioside | [170] | |
| Cancer organoids | Generation of Pdx1-Cre; Kras+/LSL-G12D (KC) and Pdx1-Cre; Kras+/LSL-G12D; Trp53+/LSL-R172H (KPC) mice | Murine pancreatic ductal organoids (pancreatic cancer) |
Neoplastic ducts, transcriptional and proteomic changes observed in pancreatic cancers | [232] |
| Lentiviral infection for gene transduction, KRASG12V and TP53R175H in pancreatic progenitor cells | Pancreatic progenitor organoids (pancreatic cancer) |
Abnormal ductal architecture, neoplastic transformation | [177] | |
| Isolation of glands from pancreatic cancer patients | Pancreatic cancer organoids | Genomic and transcriptomic alterations in patients, drug response | [176] | |
| Isolation of glands from gastric cancer patients | Gastric cancer organoids | Aneuploidy, impaired p53 pathway | [178] | |
| Isolation of tumor tissues from colorectal cancer patients | Colorectal cancer organoids | Genomic and transcriptomic alterations in patients | [179] | |
| CRISPR/Cas9-based knock-out of APC, SMAD4, and TP53; CRISPR/Cas9-based genome editing (KRASG12V and PIK3CAE545K) in organoids | Colorectal cancer organoids | Tumorigenesis | [189] | |
| Isolation of circulating tumor cells from prostate cancer patients | Prostate cancer organoids | Phenotypic diversity (AR-dependent/independent), drug response | [180] | |
| Isolation of tumor tissues from liver cancer patients | Liver cancer organoids | Histological features, expression profiles, tumorigenesis, drug response | [181] | |
| Isolation of tumor tissues from breast cancer patients | Breast cancer organoids | Histological features, copy number alterations, genomic alterations | [71, 182] | |
| Modeling of other disorders | CRISPR/Cas9-based knock-out of PKD1 or PKD2 in hESCs | Kidney organoids (Polycystic kidney) |
Formation of cyst-like structures in tubules | [190] |
| CRISPR/Cas9-based genome editing (DKC1-A386T) in iPSCs | Intestinal organoids (Dyskeratosis congenita) | Failure in crypt formation, impaired Wnt signaling, reduced telomere activity | [191] | |
| CRISPR/Cas9-based genome correction of CFTR-F508del in patient iPSCs | Intestinal organoids (Cystic fibrosis) |
Functional repair of CFTR, forskolin-mediated swelling of organoid | [192] | |
| Lentiviral infection for FUT2 overexpression in human intestinal enteroid | Norovirus-infected intestinal organoids | Susceptible to norovirus replication | [194] |
4.2. GENE EDITING FOR STUDYING ORGANOGENESIS
One of the most important focuses of organoid technologies is recapitulating organogenesis during developmental processes from embryonic or induced pluripotent stem cells, which mimic fetal cells undergoing natural development. When these cells assemble into organoids, they undergo a process similar to natural development in the presence of the right cues. However, researchers must probe these genes with knock-in or knock-out mutations to fully understand the role of specific genes in the developmental process. One standard method for studying organoids is utilizing various genome engineering techniques to label cell types for developmental tracking. Given that the organoid is too densely populated to allow the labeling of all cells or recognize cellular phenotypes, electroporation with a transposon is frequently used to label the cells sporadically. By doing so, researchers can study various complex developmental processes such as regional development of brain subsections, the dynamics of brain folding, the convoluted structure of the brain, and long-range axon growth circuit formation.[120, 155–157] In retinal organoids, using a genetically engineered stem cell line that expressed GFP when adopting specific retinal lineages was used to optimize differentiation protocols, which led to the generation of retinal organoids with enhanced biomimetic structures.[26] A new technique termed CRISPR-HOT was established for highly efficient knock-in models for the development of organoid cultures. Utilizing the CRISPR-HOT non-homologous end joining and cuvette electroporation, researchers were able to label and generate reporter lines for corresponding clonal organoid lines from human liver ductal cells. The CRISPR-HOT platform was approximately 10x as efficient as traditional homology-directed repair-based editing using CRISPR, thereby making it a handy tool for knock-in experiments with organoids.[158]
In addition to labeling cell types to study their migration, growth, and development, researchers can also use genome engineering to probe the role of specific genes during the development. Scientists can achieve organogenesis using isogenic variants with targeted genetic modifications by creating mutant cell types using CRISPR/Cas9 systems or differentially regulating genes using viral vectors or siRNAs. In one study, gene editing with the ODF2 and siRNA knockdown of IFT88 significantly reduced the number and length of cilia on cells in testicular organoids, resulting in irregular cellular assembly.[159] In retinal organoids, CRISPR/Cas9 was used to create RB1 null hESCs, which were subsequently used to form organoids. Compared to isogenic wild-type controls, RB1 null organoids showed significantly lower bipolar cells, photoreceptor cells, and ganglion cells population. Using genetically engineered organoids, these studies led to further understanding of the role of RB1 in retinal development.[160] In kidney organoids, the deletion of Wnt4 using CRISPR/Cas9 led to the failure of the organoids to undergo a mesenchymal to epithelial transition, which led to incorrect segmentation and a lack of a nephron structure compared to the isogenic controls.[161] Collectively, these studies demonstrate the ability of genome engineering, in combination with organoid technologies, to study the development of various organs and the role of specific molecular pathways. While the study of natural development can be extremely interesting for understanding how we develop as humans, we are further interested in diseases and how various genetic and environmental changes can lead to disease phenotypes.
4.3. MODELING NEUROLOGICAL DISORDERS USING GENETICALLY ENGINEERED ORGANOIDS
The phenotypes observed in the brain are often very complicated for the study of neurological disorders. Furthermore, owing to the lack of human tissue samples and inadequate animal models, there is a need for a better method to study the molecular mechanisms of neurodegeneration and disease development. Despite brain organoids’ ability to replicate the structures and cell types found in various brain regions, genetic engineering of organoids is needed to better understand the development of various disorders, more specifically, the genetic components of neurological disorders [Figure 8A]. In 2014, Choi and coworkers created an organoid model of Alzheimer’s disease, one of the most complex CNS disorders, by upregulating amyloid precursor protein and presenilin 1 with familial Alzheimer’s disorder (FAD) mutations using lentiviral vectors in human neural progenitor cells. The genetically modified cells formed into organoids that recapitulated the amyloid plaques, a hallmark of Alzheimer’s disease and filamentous tau proteins.[162] In another study, CRISPR/Cas9 was used to generate isogenic variants of iPSCs with APOE3 and APOE4 variants, and the iPSCs were then used to create cerebral organoids. The organoids derived from APOE4 iPSCs recapitulated the amyloid-beta and tau aggregates associated with Alzheimer’s disease in an age-dependent manner similar to those observed in sporadic Alzheimer’s disease [Figure 8A-1]. Moreover, this study also demonstrated the effects of APOE4 on microglia’s ability to degrade amyloid-beta aggregates. Compared to controls, APOE4 microglia showed longer processes and a reduced ability to break down aggregates, which is one reason for the build-up of amyloid and tau plaques.[163]
Figure 8.
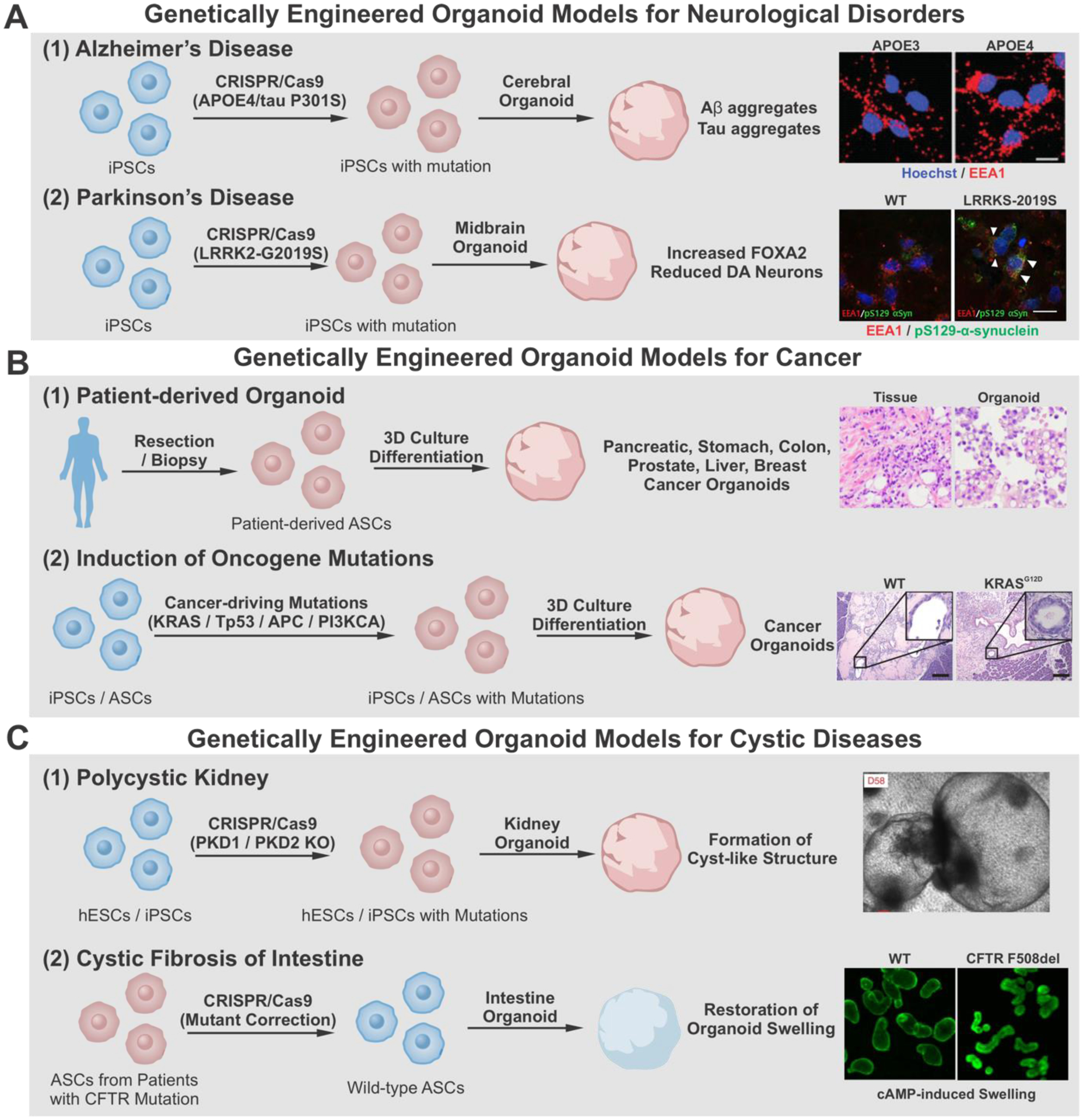
Disease modeling with genetically engineered organoids A) To model neurological diseases, introducing mutations (APOE4, Tau P301S, or LRRK2-G2019S) using CRISPR/Cas9 exhibited pathological phenotype of Alzheimer’s disease (A-1) and Parkinson’s disease (A-2) in cerebral organoid and midbrain organoid, respectively. Alzheimer’s Disease. Reproduced with permission.[164] Copyright 2018, Elsevier. Parkinson’s Disease. Reproduced with permission.[167] Copyright 2019, Elsevier. B) To generate cancer organoids, patient-derived ASCs (B-1) or iPSC/ASC with genome engineering-based oncogene mutations (B-2) are utilized. Patient-derived organoid. Reproduced with permission.[183] Copyright 2018, Elsevier. Induction of oncogene mutation. Reproduced with permission.[177] Copyright 2015, Elsevier. C) CRISPR/Cas9-based PKD deletion induces cyst-like structure in kidney organoid (C-1), whereas CRISPR/Cas9-based gene correction from patients’ cells repaired the structural phenotype of cystic fibrosis in intestine organoid (C-2). iPSCs, induced pluripotent stem cells; hESCs, human embryonic stem cells; ASCs, adult stem cells. Polycystic kidney. Reproduced with permission.[191] Copyright 2015, Springer Nature. Cystic fibrosis of intestine. Reproduced with permission.[193] Copyright 2013, Elsevier.
Like Alzheimer’s disease, iPSCs can be modified with mutant forms of tau proteins (P301S), leading to the development of hyperphosphorylated tau, thus exhibiting the canonical signs of Fronto-temporal dementia.[164] When iPSCs carrying the P301S mutation were further edited using CRISPR/Cas9 to form a non-cleavable mutant variant of p35, organoids showed reduced phosphorylation of tau proteins along with an increase in synaptophysin. This result demonstrated that the cleavage of p35 to p25 and the subsequent CDK5 signaling play a role in the phosphorylation of tau proteins. Selective inhibition of the kinase can be a potential therapeutic option for frontotemporal dementia.[165] For Parkinson’s disease, the LRRK2-G2019S mutation can be engineered into iPSCs, creating isogenic variants using CRISPR/Cas9 to study the effects of various genetic pathways [Figure 8A-2]. The study demonstrated that TXNIP was vital for the pathogenic phenotypes of LRRK2-G2019S mutation in midbrain organoids. TXNIP knockdown significantly rescued midbrain organoids from the pathological phenotype, providing a crucial link in the genetic pathway involved in sporadic Parkinson’s disease and demonstrating its potential as a therapeutic target.[166] Another study examined the FOXA2 floor plate marker and its effect on dopaminergic neurons in midbrain organoids. Compared to isogenic controls, LRRK2-G2019S mutated midbrain organoids showed an increase in FOXA2 expression and a corresponding decrease in midbrain dopaminergic neurons, suggesting a link between FOXA2 and the homeostasis of midbrain dopaminergic neurons.[167]
To study autism spectrum disorders, brain organoids developed from healthy individuals and those with severe idiopathic autism were studied. These patient samples typically showed upregulation of GABAergic inhibitory neurons, which was attributed to the overexpression of FOXG1. By engineering organoids with viruses containing siRNA to knock-down FOXG1, the balance between GABAergic and Glutamatergic neurons was restored.[168] To obtain more precise dosage control of FOXG1, researchers used the small molecule assisted shut-off (SMASh) system in which the proteins are fused to self-removing degrons to control protein concentrations in a dose-dependent manner. By incorporating a FOXG1 SMASh system into iPSCs using CRISPR/Cas9, researchers could precisely control the expression of FOXG1 in cortical organoids. When the FOXG1 expression was reduced to 60%, a reduction in GABAergic interneuron development was observed. In comparison, 30% FOXG1 expression led to a decrease in medial ganglionic eminence-derived neurons, both of which can lead to various neurological deficits such as autism, epilepsy, or seizures.[169] By applying advanced systems such as the SMASh system to organoid models, researchers can investigate the pathogenetic effects of specific genes in a dose-dependent manner.
Lastly, in addition to modeling the molecular mechanisms of disease progression, organoids can also be used to study gene therapy’s effect on various neurological disorders. In one study, researchers created a model of microencephaly by utilizing patient-derived IPSCs with a CDK5RAP2 mutation. The organoids recapitulated molecular phenotypes of microencephaly in comparison with the controls. When electroporation was used on day 12 organoids to reintroduce CDK5RAP2, a larger neuroepithelium and increased radial glial morphology were observed, thus demonstrating gene therapy’s ability in microencephaly.[15] To model GM1 gangliosidosis, iPSCs were edited using CRISPR/Cas9 with GLB1 exons 2 and 6, resulting in a deficiency of lysosomal β-galactosidase. These iPSCs and isogenic controls were grown into organoids that recapitulated the deficiencies of GM1 gangliosidosis and were treated by microinjecting AAVs expressing GLB1. Organoids receiving the gene therapy showed a significant recovery in β-galactosidase activity and a reduction in GM1 ganglioside content.[170] These studies show the ability of gene-engineered organoids as models for gene therapy.
Overall, these studies show the ability of gene-engineered organoids to demonstrate the underlying genetic and cellular mechanisms of neurological diseases and potential therapeutic avenues for treating these disorders [Figure 8A].
4.4. CANCER ORGANOIDS FROM GENETIC MODIFICATIONS
Recent developments in the field of organoids not only changed the research in neurological disorders, but also revolutionized the field of cancer studies by providing novel disease models along with new strategies for cancer therapies. Conventional 3D tumor spheroids have been utilized for validating therapeutic efficacy, thereby bridging the gap between in vitro assays and animal studies.[171] Tumor organoids possess the traits of tumor microenvironments, such as heterogeneity throughout the tumor, which are essential for preclinical models to translate cancer research into effective therapeutic avenues in human patients.[172] Cancer organoids can be generated from patient-derived primary cancer cells and adult stem cells with specific genetic modifications.[173] Cell-based organoids derived from the primary tumor of patients possess patient-specific mutations that facilitate biobank-based disease modeling, patient-specific drug screening, and personalized medicine.[174] On the other hand, cancer organoid-derived from genetically modified adult stem cells allow for understanding the mechanism and disease modeling at the molecular level [Figure 8B].[175]
Patient-derived tumor organoids have revolutionized the current understanding of tumor progression, personalized medicine, and cancer therapeutic strategies. With current 3D culture technologies, various types of cancer organoids have been generated from pancreas[176, 177], stomach[178], colon[179], prostate[180] liver[181], and breast[182] tumor tissues or metastatic biopsy tissues [Figure 8B-1]. Since these organoids harbor genetic and phenotypic characteristics from the primary tumors, a recent trend of establishing human cancer “biobank” was initiated. Moreover, through next-generation sequencing and big data analysis, characterizing various types of cancer into subtypes with specific sets of gene mutations improves established treatment outcomes and aids in discovering effective neoadjuvant treatments.[183] Rubin and coworkers showcased a precision workflow from established cancer organoid bio-banks using genetic sequencing to optimize single and combination drug screening.[184] The researchers have conducted whole-exome sequencing with a living biobank from 769 patients. Interestingly, a large portion (85.8%) of the genetic alterations are non- targetable by current FDA-approved therapeutics, indicating the great demand for such profiling initiatives. Furthermore, 56 patient tumor-derived organoids were established corresponding to uterine carcinosarcoma, pancreatic adenocarcinoma, endometrial adenocarcinoma, and colorectal cancer. Based on the genetic alteration results, different combinations of pathway inhibitors [i.e., buparisib (PI3Ki), Olaparib (HDACi), Trametinib (MEKi), and celecoxib (COX-2i)] were employed, demonstrating improved therapeutic response and outcomes with the help of patient-derived organoids as well as a patient-derived xenograft in vivo models. Similarly, drug screening with colorectal cancer organoid biobank could elucidate potential epigenetic and genetic variations that lead to drug resistance.[179] Various efforts have fortified the essence of patient-specific genetic profiling in understanding pathological progression, personalized prescription, and even disease prognosis.
Another approach indicates the introduction of oncogenic mutations in the developmental process of the organoid [Figure 8B-2].[185] It is widely accepted that cancer is a cumulative result of sequential mutations in cancer-driving genes.[186, 187] For instance, pancreatic cancer, a devastating and lethal disease, results from sequential mutations in KRAS, CDKN2A, TP53, and SMAD4.[188] Muthuswamy and coworkers utilized a lentiviral vector to induce point oncogene mutations (KRAS and TP53) in wild-type iPSCs-derived pancreatic progenitor organoids resulting in pancreatic cancer organoids. Furthermore, differentiation markers during the pancreatic organoid and pancreatic cancer organoid development processes were characterized through immune-fluorescence analysis. Interestingly, it was discovered that cytoplasm localization of SOX9 is correlated with TP53 mutation, while SOX9 maintained nuclear localization in wild-type and KRAS mutated organoids. To understand the clinical relevance of SOX9 and p53, primary patient samples were analyzed for the SOX9 distribution and the p53 expression. An impressive correlation between patient survival and SOX9 distribution was observed, indicating the potential of utilizing SOX9 as a marker for cancer malignancy. Similarly, in combination with CRISPR-Cas9 genome editing and adult stem cell-derived organoids, organ-specific cancer initiation and progression can be modeled and studied. Sato and colleagues demonstrated colorectal cancer initiation and progression from surgical resected human intestinal tissue-derived intestinal organoids.[189] A series of colorectal cancer mutations, including APC, SMAD4, TP53, KRAS, and PI3KCA, were introduced into the healthy tissue-derived intestinal organoids to understand their correlation with colorectal cancer. Interestingly, the cancer-driving mutations supported cancer organoid proliferation in vitro with no niche factors for intestinal stem cells. Moreover, after injection into mice kidney subcapsular, such organoids with KRAS activation and inactivated APC, TP53, and SMAD formed tumors and displayed metastases in response to spleen injections.
4.5. OTHER PATHOLOGIES MODELED USING GENETICALLY ENGINEERED ORGANOIDS
While neurodegenerative diseases and cancers are the most commonly studied diseases using genetically engineered organoids, several other diseases have also been modeled. Kidney organoids were used to model polycystic kidney disease by knocking out PKD1 or PKD2 [Figure 8C-1]. These mutations led to large cyst-like structures in the kidney organoids, which were not presented in isogenic controls. This mutation allows for the study of polycystic kidney disease and the genetic basis for the condition.[190] Human intestinal organoids were generated from iPSCs of patients with dyskeratosis congenita and gene-corrected isogenic controls, respectively [Figure 8C-2]. The patient-derived organoids showed downregulated Wnt pathway activity, which led to a reduction in intestinal stem cell activity. These engineered organoids also showed that the induction of telomere-capping protein TRF2, which is a Wnt target gene, can regenerate the diseased phenotype. Besides, treatment with Wnt agonists LiCl or CHIR99021 restored TRF2 function in organoids and reversed dyskeratosis congenita phenotypes. This study showed not only the ability of gene-edited organoids to replicate disease phenotypes and study the molecular mechanisms of the diseases but also the ability to lead to new treatment options and drug models.[191] Intestinal organoids replicating cystic fibrosis can be generated using patient-derived samples. In healthy organoids, increased cAMP levels led to the organoid’s swelling through cystic fibrosis transmembrane conductor receptors [Figure 8C-2]. However, this phenotype was lost in patient-derived organoids. When CRISPR/Cas9 was used to correct the patient cells’ mutation, it rescued the swelling phenotype. Although this method had to be performed before organoid formation, it still demonstrated the possibility of genetic therapies for the treatment of cystic fibrosis.[192] Another study examined the samples of intestinal organoids from a cystic fibrosis organoid biobank. Adenine base editing, a technique that can be used to change A-T base pairs to C-G base pairs, was used to correct mutations in four different organoids from the biobank and showed recovery of both genetic and phenotypic symptoms of cystic fibrosis. This study highlighted the possibility of using organoid biobanks as a source of tissue for studying diseases and the ability of adenine base editing to edit genes with little or no off-target effects for therapeutic applications.[193] Lastly, organoids can be applied to study endogenous genetic diseases and infectious diseases and their transmission. Human noroviruses are the primary cause of gastroenteritis worldwide. Secretor status and the fucosyltransferase 2 (FUT2) gene are highly linked to susceptibility to noroviruses. However, the exact role of FUT2 and whether the individual effects of this gene are enough to cause susceptibility have remained unknown. Haga et al. demonstrated that intestinal organoids from secretor patients were susceptible to Norovirus infection, but non-secretor organoids were not. CRISPR/Cas9-based knock-out and knock-in of FUT2 showed that FUT2-positive organoids were susceptible to noroviruses while FUT2-negative organoids were not, therefore, elucidating the effects of FUT2 on norovirus susceptibility.[194]
The ability to accurately control gene expression and mutation using advanced genome engineering combined with organoid technologies, allows for unprecedented control of organoid development. This allows researchers to study the molecular and cellular functions at an advanced level during natural and pathological organ development [Figure 9].
Figure 9.
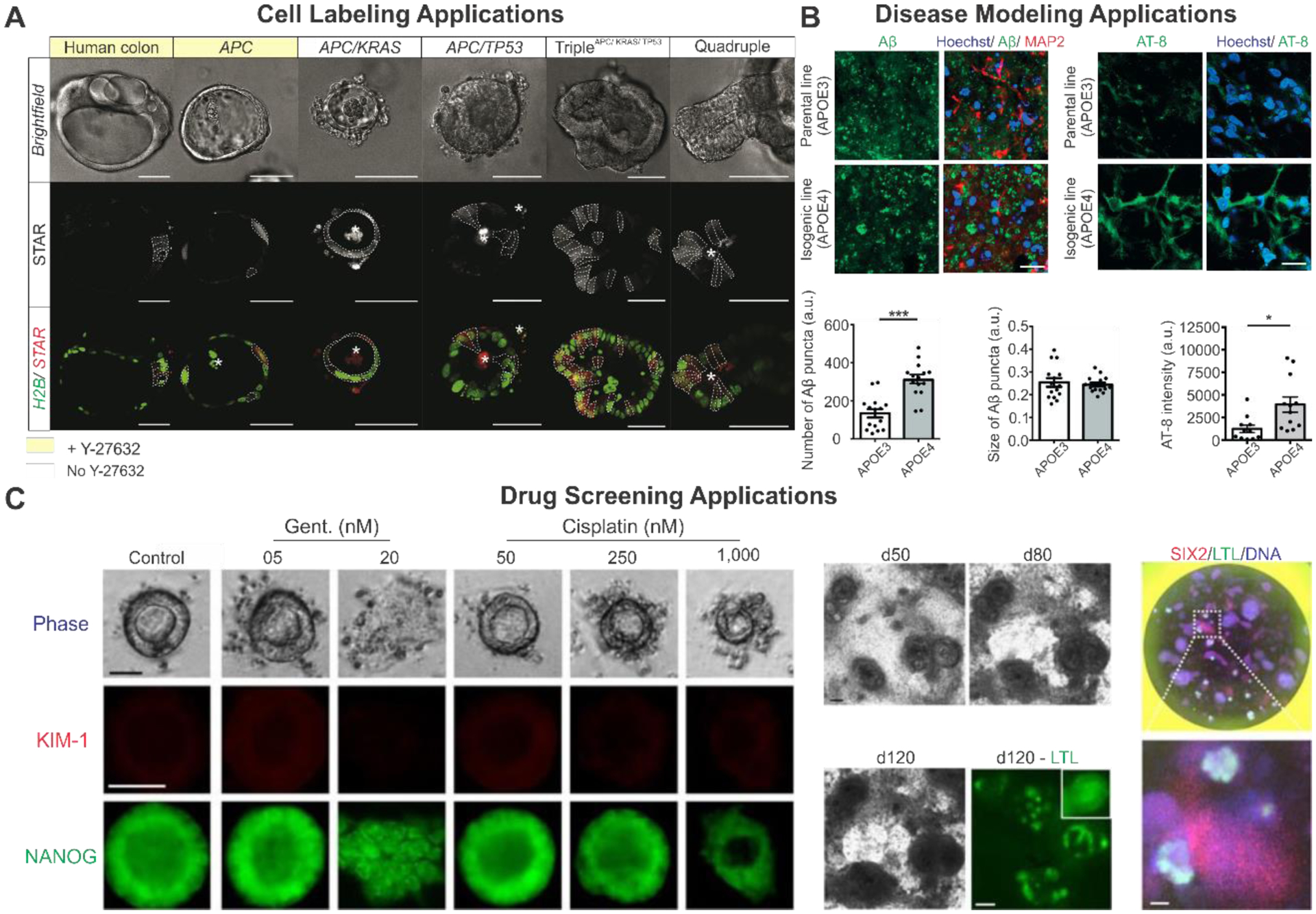
Genetically modified organoids for advanced applications. A) Example of cell labeling using STAR minigenes for labeling intestinal stem cells in tumor progression organoids. Reproduced with permission.[221] Copyright 2018, Elsevier. B) Disease modeling of Alzheimer’s disease using CRISPR to alter APOE variant found in organoids showing effect of APOE4 on disease progression. Reproduced with permission.[163] Copyright 2018, Elsevier. C) Example of drug screening using CRISPR modified kidney organoids showing dose dependent effects on nephrotoxicity. Reproduced with permission.[190] Copyright 2015, Springer Nature.
5. CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Exciting progress has been noted in organoid technologies through multidisciplinary research approaches, enabling scientists to control physical and biochemical cues. While the precedent version of organoids can simply develop organized spheroids with high cell diversity, various efforts have been undertaken to produce more complex organoids in a controlled manner using engineering-based approaches [Table 3].[195, 196] One key approach is to incorporate the signaling gradient within an organoid contributing to subdivisions along with positional identity. Recently, the self-organization of neuromuscular organoids was demonstrated, exhibiting the simultaneous generation of spinal cord neurons and skeletal muscle cells [Figure 10A].[197] Another study specified positional identity within forebrain organoids, which recapitulate the topographic organization mimicking in vivo conditions by inducing signaling gradients[22]. In addition to spatial modulation, accessory compartments such as choroid plexus-forming brain organoids[198] and hair-bearing skin organoids[199] can be developed by regulating the timing and duration of signal treatments. These recent studies opened a new horizon for dual patterning within one organoid across two organs derived from distinct lineages.
Table 3.
Technologies for spatially multi-patterned organoids
| Categories | Technologies | Results and Applications | Ref. |
|---|---|---|---|
| Intra-organoid specification | Signaling protein (SHH) gradient with genome engineering | Forebrain subdivisions that contain positional axes | [22] |
| Dual patterning from bipotent progenitors | Self-organizing neuromuscular organoids | [197] | |
| Stepwise modulation of signaling cues | Cerebrospinal fluid production of choroid plexus-forming brain organoids | [198] | |
| Stepwise modulation of signaling cues | Hair-bearing skin organoids | [199] | |
| Inter-organoid communication | Assembly of region-specific models | Mixed dorsal and ventral forebrain organoids | [54] |
| Co-culture with connective tissue | Promoted formation of alveolar organoid by addition of mesenchymal stem cells | [213] | |
| Co-culture with connective tissue/ Organ-on-a-chip | Structural arrangement in mesenchymal bodies | [214] | |
| Organ-on-a-chip | Recapitulating the connections between GI microbiome and CNS | [207] | |
| Organ-on-a-chip/Bioprinting | Multi-organ interactions upon drug administration | [208] | |
| Bioprinting | Self-patterned 3D tissue models | [209] | |
| Topographical patterning/ profiling | Light-induced small molecule release | Spatiotemporally controlled neural stem cell fate | [200] |
| Light-induced patterning | Axon guidance with NGF-patterned matrix | [201] | |
| AI-based optimization | Predicted experimental parameters for PSC self organization | [202] | |
| Micro-rheological characterization | Mechanical properties of collagen gels and cell ECM interactions | [203] | |
| Super-resolution imaging | Cellular composition of organoids with high resolution 3D imaging | [216] | |
| Spatial transcriptomics | Visualization of the distribution of mRNAs | [215] | |
| Spatial proteomics | Spatiotemporal profiling of signaling interactomes | [217] |
Figure 10.
Strategies for multi-patterned organoids. A) Human PSC-derived neuromuscular organoids producing spinal cord neurons and skeletal muscle cells simultaneously. Reproduced with permission.[197] Copyright 2020, Elsevier. B) Assembly of two or more distinct organoids to generate multi region-bearing organoids.C) A coupled tissue‐chip gut‐immune‐liver‐brain axis model. Reproduced with permission.[207] Copyright 2020, John Wiley and Sons. D) 3D bioprinting of hydrogel-based hepatic construct. Reproduced with permission.[210] Copyright 2016, Ma, X. et.al.
Topographical regulation with ECM modulation, not only cellular bioengineering, may help facilitate spatiotemporal control of organoid niches, thereby creating next-generation organoids [Figure 11]. Light-mediated release of small molecules[200] or light-mediated 3D patterning of bioactive cues[201] was developed to induce guided morphogenesis. Still, there is a critical limitation of currently used materials in that it is hard to understand which properties of each material govern the specific behavior of cells. This missing link makes it difficult to precisely customize ECM according to the purpose. To resolve this unpredictability of current system, combined technologies with artificial intelligence (AI) or physical mechanics are now utilized. AI-based computational modeling technique was recently utilized to find the relevance between material dynamics and stem cell self-organization behaviors.[202] Machine learning-based optimization of multicellular patterning enables the spatial control of organoids in a predictable manner. Moreover, the mechanical properties of ECM and organoid-matrix crosstalk can be monitored with micro-rheological characterization via optical-tweezers-based probe[203] or single-cell traction microscopy[204] in real-time. These technologies can further contribute to psychomimetic features of organoids through shape-guided morphogenesis and high-throughput setups by improving the readout.
Figure 11.
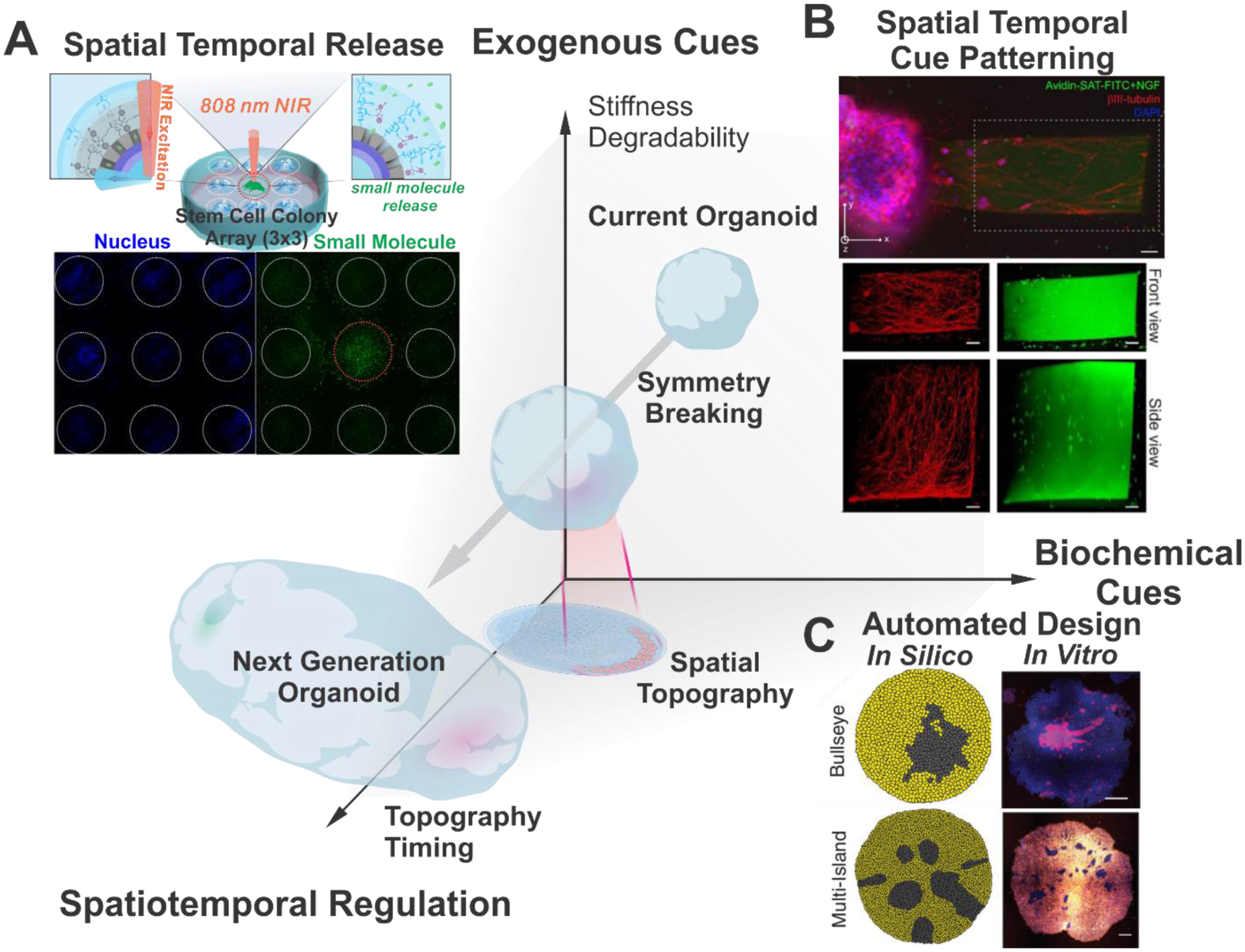
Spatiotemporal Regulation for the Development of Next-Generation Organoid. A) Example of tissue-penetrating NIR light mediated spatial temporal release. Reproduced with permission.[200] Copyright 2020, American Chemical Society. B) Light mediated spatial bioactive cue patterning. Reproduced with permission.[201] Copyright 2020, John Wiley and Sons. C) Automated design of in silico 3D cellular assembly and spontaneous patterning. Reproduced with permission.[202] Copyright 2019, Elsevier.
Besides, not only the intra-organoid specification, but also inter-organ communication should be considered to reflect integrated physiology. Interactions between two anatomically distinct tissues have emerged as essential regulators of homeostasis and disease, such as neuroimmune circuits[205], microbiome interactions in the gut-lung axis [206], or gut-liver-brain axis[207], and drug diffusion kinetics within multiple organs.[208] In addition to the assembly of multiple regions within one organ [Figure 10B][54], a multi-tissue organ-on-a-chip platform provides a circulatory perfusion system allowing for the crosstalk between several distinct organoids [Figure 10C]. This finding recapitulates the dynamically interactive environments of the human body by creating multiple organoids on a microfluidic chip.[207, 208] Bioprinting techniques has been also employed to make macro-scale architecture resembling native tissues through self-organization.[209] By controlling the geometry and cellular density, large-scale tissues containing multiple organ regions are created, potentially serving as medical transplantation [Figure 10D].[210] Still, certain technical and biological considerations are attributed to the development of human-on-a-chip models or larger bioprinted organs that reflect anatomical aspects of the actual human body. Despite the resemblance of downscaled organs, allometric scaling between organoids of distinct regions is different from that of actual organs.[211] Furthermore, organs interact with each other through different types of connective tissues derived from different origins.[212] Despite attempts to use mesenchymal cells for organoid formation[213, 214], it is poorly understood how to suppress the chaotic differentiation of mesenchymal stem cells and implement connective tissues with diverse composition and characteristics in the body.
Advancements in analytical methods, for instance, single-cell sequencing, enable researchers to decipher the transcriptional profile of cell populations that compose organoids or the connective tissues at the single-cell level. Moreover, the combination of tissue section images and gene expression data allows the visualization of spatial transcriptomics of organoids.[215] In addition to the spatial structure of RNA expression, recent innovations in optical imaging technologies, such as super-resolution confocal microscopy, multiphoton laser scanning microscopy, and light-sheet fluorescence microscopy, have enabled the high-resolution 3D visualization of an entire immunolabeled organoid at the subcellular level.[216] Furthermore, recently developed proximity proteomics enabled spatiotemporal profiling of signaling interactomes[217], which would potentially provide the analytic approach for the proteome in organoids. Collectively, deep sequencing and deep imaging systems can facilitate our understanding of the spatial distribution and dynamic interactions between multiple types of cells within organoids. The progress in organoid research will synergize with the accumulation of big data to overcome the current challenges and accelerate organoids’ clinical applications in biomedicine.
In summary, organoid technologies have been advanced by coordinating distinct research fields, including stem cell research, bioengineering, biomaterials, biophysics, and computational research. However, there are still many challenges for the clinical applications of organoids regarding biocompatibility. Designing biomaterials or bioengineering approaches with careful consideration of safety and stability issues would allow the use of organoids as organ replacement therapy in the near future.
ACKNOWLEDGEMENTS
K.-B.L. acknowledges the partial financial support from the NIH R21 (R21AR071101), NIH R01 (1R01DC016612-01, 3R01DC016612-01S1, R01DC016612-02S1, and R01DC016612-04S1), New Jersey Commission on Spinal Cord Research [CSCR17IRG010 and CSCR16ERG019], and NSF [CBET-1803517]. S.A.Y. acknowledges financial support from the National Research Foundation of Korea (2017R1A6A3A04001986). Drs. Sang Ah Yi and Yixiao Zhang contributed equally to this work.
Biographies

Sang Ah Yi received her B.S. degree from the College of Pharmacy, Sungkyunkwan University. She received her M.S. and Ph.D. in Biochemistry and Molecular Biology from the School of Pharmacy, Sungkyunkwan University. Her research has focused on epigenetic regulation of stem cell differentiation and diverse diseases, including obesity and cancer. She is now a research professor at the School of Pharmacy, Sungkyunkwan University. Her current research interest focuses on organoid engineering based on biochemical modulation and drug screening with in vitro disease modeling.

Yixiao Zhang received his B.Sc. in Chemistry from the College of Chemistry and Molecular Sciences, Wuhan University. He received his Ph.D. in Chemistry from the Department of Chemistry and Chemical Biology, Rutgers University. Under the supervision of Prof. Ki-Bum Lee, he focused on photo-responsive biomaterials for probing bio- and neuro- interfaces and biodegradable nanomaterials for cellular programming as well as cancer therapy. His current primary research interest focuses on the field of bio-nanotechnology, cell engineering, biomaterial engineering, as well as regenerative medicine.

Christopher Rathnam received his B.S. degree from Rutgers University, studying biomedical engineering. He is currently pursuing a Ph. D. in Chemistry and Chemical biology at Rutgers University in the lab of Dr. Ki-Bum Lee. His research focuses on developing novel nanomaterials to improve the efficacy and safety of cell and gene-based therapies. He is specifically interested in designing bio-inspired nanomaterials for the treatment of devastating diseases and disorders of the central nervous system, such as neurodegenerative disorders and spinal cord injury.

Thanapat Pongkulapa received his B.S. degree in Applied Chemistry from Chulalongkorn University. He is currently a Ph.D. candidate in the Department of Chemistry & Chemical Biology at Rutgers University, under the supervision of Professor Ki-Bum Lee. His research focuses on the development of novel delivery platforms for improved gene therapy in cancer and other genetic diseases. His primary research interests include genetic manipulation, genome editing, and organoid-based drug screening for precision medicine.

Ki-Bum Lee is a Professor of Chemistry and Chemical Biology at Rutgers University, where he has been a faculty member since 2008. He received his Ph.D. in Chemistry from Northwestern University (with Chad. A. Mirkin, 2004) and completed his postdoctoral training at The Scripps Research Institute (with Peter G. Schultz, 2007). The primary research interest of Prof. Lee’s group is to develop and integrate nanotechnologies and chemical functional genomics to modulate signaling pathways in cells (e.g., stem cells and cancer cells) toward specific cell lineages or behaviors.
REFERENCES
- [1].Murry CE, Keller G, Cell 2008, 132, 661. [DOI] [PubMed] [Google Scholar]
- [2].Choudhury D, Ashok A, Naing MW, Trends Mol Med 2020, 26, 245. [DOI] [PubMed] [Google Scholar]
- [3].Rossi G, Manfrin A, Lutolf MP, Nat Rev Genet 2018, 19, 671. [DOI] [PubMed] [Google Scholar]
- [4].Rheinwald JG, Green H, Cell 1975, 6, 331. [DOI] [PubMed] [Google Scholar]
- [5].Moscona A, Moscona H, J Anat 1952, 86, 287. [PMC free article] [PubMed] [Google Scholar]
- [6].Weiss P, Taylor AC, Proc Natl Acad Sci U S A 1960, 46, 1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li ML, Aggeler J, Farson DA, Hatier C, Hassell J, Bissell MJ, Proc Natl Acad Sci U S A 1987, 84, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Barcellos-Hoff MH, Aggeler J, Ram TG, Bissell MJ, Development 1989, 105, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Streuli CH, Schmidhauser C, Bailey N, Yurchenco P, Skubitz AP, Roskelley C, Bissell MJ, J Cell Biol 1995, 129, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H, Nature 2009, 459, 262. [DOI] [PubMed] [Google Scholar]
- [11].Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, Sato T, Stange DE, Begthel H, van den Born M, Danenberg E, van den Brink S, Korving J, Abo A, Peters PJ, Wright N, Poulsom R, Clevers H, Cell Stem Cell 2010, 6, 25. [DOI] [PubMed] [Google Scholar]
- [12].Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, Sato T, Hamer K, Sasaki N, Finegold MJ, Haft A, Vries RG, Grompe M, Clevers H, Nature 2013, 494, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Huch M, Bonfanti P, Boj SF, Sato T, Loomans CJ, van de Wetering M, Sojoodi M, Li VS, Schuijers J, Gracanin A, Ringnalda F, Begthel H, Hamer K, Mulder J, van Es JH, de Koning E, Vries RG, Heimberg H, Clevers H, EMBO J 2013, 32, 2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, Dowling CM, Gao D, Begthel H, Sachs N, Vries RGJ, Cuppen E, Chen Y, Sawyers CL, Clevers HC, Cell 2014, 159, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA, Nature 2013, 501, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Golden AP, Tien J, Lab Chip 2007, 7, 720. [DOI] [PubMed] [Google Scholar]
- [17].McGuigan AP, Sefton MV, Proc Natl Acad Sci U S A 2006, 103, 11461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Richardson TP, Peters MC, Ennett AB, Mooney DJ, Nat Biotechnol 2001, 19, 1029. [DOI] [PubMed] [Google Scholar]
- [19].Gjorevski N, Sachs N, Manfrin A, Giger S, Bragina ME, Ordonez-Moran P, Clevers H, Lutolf MP, Nature 2016, 539, 560. [DOI] [PubMed] [Google Scholar]
- [20].Takahashi K, Yamanaka S, Cell 2006, 126, 663. [DOI] [PubMed] [Google Scholar]
- [21].Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F, Nat Protoc 2013, 8, 2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cederquist GY, Asciolla JJ, Tchieu J, Walsh RM, Cornacchia D, Resh MD, Studer L, Nat Biotechnol 2019, 37, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Amin ND, Pasca SP, Neuron 2018, 100, 389. [DOI] [PubMed] [Google Scholar]
- [24].Qian X, Song H, Ming GL, Development 2019, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dahmann C, Oates AC, Brand M, Nat Rev Genet 2011, 12, 43. [DOI] [PubMed] [Google Scholar]
- [26].Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S, Sekiguchi K, Adachi T, Sasai Y, Nature 2011, 472, 51. [DOI] [PubMed] [Google Scholar]
- [27].Eiraku M, Watanabe K, Matsuo-Takasaki M, Kawada M, Yonemura S, Matsumura M, Wataya T, Nishiyama A, Muguruma K, Sasai Y, Cell Stem Cell 2008, 3, 519. [DOI] [PubMed] [Google Scholar]
- [28].Ovando-Roche P, West EL, Branch MJ, Sampson RD, Fernando M, Munro P, Georgiadis A, Rizzi M, Kloc M, Naeem A, Ribeiro J, Smith AJ, Gonzalez-Cordero A, Ali RR, Stem Cell Res Ther 2018, 9, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Qian X, Jacob F, Song MM, Nguyen HN, Song H, Ming GL, Nat Protoc 2018, 13, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kratochvil MJ, Seymour AJ, Li TL, Paşca SP, Kuo CJ, Heilshorn SC, Nat Rev Mater 2019, 4, 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].McCauley HA, Wells JM, Development 2017, 144, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Arnold SJ, Robertson EJ, Nat Rev Mol Cell Biol 2009, 10, 91. [DOI] [PubMed] [Google Scholar]
- [33].Huch M, Koo BK, Development 2015, 142, 3113. [DOI] [PubMed] [Google Scholar]
- [34].Zorn AM, Wells JM, Annu Rev Cell Dev Biol 2009, 25, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, Hoskins EE, Kalinichenko VV, Wells SI, Zorn AM, Shroyer NF, Wells JM, Nature 2011, 470, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fausett SR, Brunet LJ, Klingensmith J, Dev Biol 2014, 391, 111. [DOI] [PubMed] [Google Scholar]
- [37].McCracken KW, Cata EM, Crawford CM, Sinagoga KL, Schumacher M, Rockich BE, Tsai YH, Mayhew CN, Spence JR, Zavros Y, Wells JM, Nature 2014, 516, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dye BR, Hill DR, Ferguson MA, Tsai YH, Nagy MS, Dyal R, Wells JM, Mayhew CN, Nattiv R, Klein OD, White ES, Deutsch GH, Spence JR, Elife 2015, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, Zhang RR, Ueno Y, Zheng YW, Koike N, Aoyama S, Adachi Y, Taniguchi H, Nature 2013, 499, 481. [DOI] [PubMed] [Google Scholar]
- [40].Bartfeld S, Clevers H, J Mol Med 2017, 95, 729. [DOI] [PubMed] [Google Scholar]
- [41].Driehuis E, Kretzschmar K, Clevers H, Nat Protoc 2020, 15, 3380. [DOI] [PubMed] [Google Scholar]
- [42].Takasato M, Er PX, Chiu HS, Little MH, Nat Protoc 2016, 11, 1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Xia Y, Sancho-Martinez I, Nivet E, Rodriguez Esteban C, Campistol JM, Izpisua Belmonte JC, Nat Protoc 2014, 9, 2693. [DOI] [PubMed] [Google Scholar]
- [44].Takasato M, Little MH, Development 2015, 142, 1937. [DOI] [PubMed] [Google Scholar]
- [45].Taguchi A, Kaku Y, Ohmori T, Sharmin S, Ogawa M, Sasaki H, Nishinakamura R, Cell Stem Cell 2014, 14, 53. [DOI] [PubMed] [Google Scholar]
- [46].Morizane R, Bonventre JV, Nat Protoc 2017, 12, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L, Nat Biotechnol 2009, 27, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Suzuki IK, Vanderhaeghen P, Development 2015, 142, 3138. [DOI] [PubMed] [Google Scholar]
- [49].Watanabe K, Kamiya D, Nishiyama A, Katayama T, Nozaki S, Kawasaki H, Watanabe Y, Mizuseki K, Sasai Y, Nat Neurosci 2005, 8, 288. [DOI] [PubMed] [Google Scholar]
- [50].Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K, Saito K, Yonemura S, Eiraku M, Sasai Y, Cell Stem Cell 2012, 10, 771. [DOI] [PubMed] [Google Scholar]
- [51].Lancaster MA, Knoblich JA, Nat Protoc 2014, 9, 2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kadoshima T, Sakaguchi H, Nakano T, Soen M, Ando S, Eiraku M, Sasai Y, Proc Natl Acad Sci U S A 2013, 110, 20284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pasca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, Kim CH, Park JY, O’Rourke NA, Nguyen KD, Smith SJ, Huguenard JR, Geschwind DH, Barres BA, Pasca SP, Nat Methods 2015, 12, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sloan SA, Andersen J, Pasca AM, Birey F, Pasca SP, Nat Protoc 2018, 13, 2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Velasco S, Kedaigle AJ, Simmons SK, Nash A, Rocha M, Quadrato G, Paulsen B, Nguyen L, Adiconis X, Regev A, Levin JZ, Arlotta P, Nature 2019, 570, 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Phipson B, Er PX, Combes AN, Forbes TA, Howden SE, Zappia L, Yen HJ, Lawlor KT, Hale LJ, Sun J, Wolvetang E, Takasato M, Oshlack A, Little MH, Nat Methods 2019, 16, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Subramanian A, Sidhom EH, Emani M, Vernon K, Sahakian N, Zhou Y, Kost-Alimova M, Slyper M, Waldman J, Dionne D, Nguyen LT, Weins A, Marshall JL, Rosenblatt-Rosen O, Regev A, Greka A, Nat Commun 2019, 10, 5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yoon SJ, Elahi LS, Pasca AM, Marton RM, Gordon A, Revah O, Miura Y, Walczak EM, Holdgate GM, Fan HC, Huguenard JR, Geschwind DH, Pasca SP, Nat Methods 2019, 16, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Shin HS, Hong HJ, Koh WG, Lim JY, ACS Biomater Sci Eng 2018, 4, 4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kakni P, Hueber R, Knoops K, Lopez-Iglesias C, Truckenmuller R, Habibovic P, Giselbrecht S, Adv Biosyst 2020, 4, e2000126. [DOI] [PubMed] [Google Scholar]
- [61].Decembrini S, Hoehnel S, Brandenberg N, Arsenijevic Y, Lutolf MP, Sci Rep 2020, 10, 10275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-Brauninger M, Lewitus E, Sykes A, Hevers W, Lancaster M, Knoblich JA, Lachmann R, Paabo S, Huttner WB, Treutlein B, Proc Natl Acad Sci U S A 2015, 112, 15672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bhaduri A, Andrews MG, Mancia Leon W, Jung D, Shin D, Allen D, Jung D, Schmunk G, Haeussler M, Salma J, Pollen AA, Nowakowski TJ, Kriegstein AR, Nature 2020, 578, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Akbari S, Sevinc GG, Ersoy N, Basak O, Kaplan K, Sevinc K, Ozel E, Sengun B, Enustun E, Ozcimen B, Bagriyanik A, Arslan N, Onder TT, Erdal E, Stem Cell Rep 2019, 13, 627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Li G, Xie B, He L, Zhou T, Gao G, Liu S, Pan G, Ge J, Peng F, Zhong X, Stem Cells Int 2018, 2018, 4968658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ormel PR, Vieira de Sa R, van Bodegraven EJ, Karst H, Harschnitz O, Sneeboer MAM, Johansen LE, van Dijk RE, Scheefhals N, Berdenis van Berlekom A, Ribes Martinez E, Kling S, MacGillavry HD, van den Berg LH, Kahn RS, Hol EM, de Witte LD, Pasterkamp RJ, Nat Commun 2018, 9, 4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Quadrato G, Nguyen T, Macosko EZ, Sherwood JL, Min Yang S, Berger DR, Maria N, Scholvin J, Goldman M, Kinney JP, Boyden ES, Lichtman JW, Williams ZM, McCarroll SA, Arlotta P, Nature 2017, 545, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sridhar A, Hoshino A, Finkbeiner CR, Chitsazan A, Dai L, Haugan AK, Eschenbacher KM, Jackson DL, Trapnell C, Bermingham-McDonogh O, Glass I, Reh TA, Cell Rep 2020, 30, 1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, Clevers H, Gastroenterology 2011, 141, 1762. [DOI] [PubMed] [Google Scholar]
- [70].Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MM, Ellis E, van Wenum M, Fuchs SA, de Ligt J, van de Wetering M, Sasaki N, Boers SJ, Kemperman H, de Jonge J, Ijzermans JN, Nieuwenhuis EE, Hoekstra R, Strom S, Vries RR, van der Laan LJ, Cuppen E, Clevers H, Cell 2015, 160, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Dekkers JF, van Vliet EJ, Sachs N, Rosenbluth JM, Kopper O, Rebel HG, Wehrens EJ, Piani C, Visvader JE, Verissimo CS, Boj SF, Brugge JS, Clevers H, Rios AC, Nat Protoc 2021, 16, 1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Grebenyuk S, Ranga A, Front Bioeng Biotechnol 2019, 7, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Shi Y, Sun L, Wang M, Liu J, Zhong S, Li R, Li P, Guo L, Fang A, Chen R, Ge WP, Wu Q, Wang X, PLoS Biol 2020, 18, e3000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Daviaud N, Friedel RH, Zou H, eNeuro 2018, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Sasai Y, Nature 2013, 493, 318. [DOI] [PubMed] [Google Scholar]
- [76].McGuigan AP, Javaherian S, Annu Rev Biomed Eng 2016, 18, 1. [DOI] [PubMed] [Google Scholar]
- [77].Kinney MA, McDevitt TC, Trends Biotechnol 2013, 31, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Madl CM, LeSavage BL, Dewi RE, Dinh CB, Stowers RS, Khariton M, Lampe KJ, Nguyen D, Chaudhuri O, Enejder A, Heilshorn SC, Nat Mater 2017, 16, 1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ishihara K, Tanaka EM, Curr Opin Syst Biol 2018, 11, 123. [Google Scholar]
- [80].Brassard JA, Lutolf MP, Cell Stem Cell 2019, 24, 860. [DOI] [PubMed] [Google Scholar]
- [81].Thakuri PS, Liu C, Luker GD, Tavana H, Adv Healthc Mater 2018, 7, e1700980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Griffith LG, Swartz MA, Nat Rev Mol Cell Biol 2006, 7, 211. [DOI] [PubMed] [Google Scholar]
- [83].Magno V, Meinhardt A, Werner C, Adv Funct Mater 2020, 30. [Google Scholar]
- [84].Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Lee HP, Lippens E, Duda GN, Mooney DJ, Nat Mater 2016, 15, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Gawade PM, Shadish JA, Badeau BA, DeForest CA, Adv Mater 2019, 31, e1902462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Murrow LM, Weber RJ, Gartner ZJ, Development 2017, 144, 998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Lancaster MA, Knoblich JA, Science 2014, 345, 1247125. [DOI] [PubMed] [Google Scholar]
- [88].Kleinman HK, McGarvey ML, Liotta LA, Robey PG, Tryggvason K, Martin GR, Biochemistry 1982, 21, 6188. [DOI] [PubMed] [Google Scholar]
- [89].Zhong X, Gutierrez C, Xue T, Hampton C, Vergara MN, Cao LH, Peters A, Park TS, Zambidis ET, Meyer JS, Gamm DM, Yau KW, Canto-Soler MV, Nat Commun 2014, 5, 4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Hu H, Gehart H, Artegiani B, O.-I. C L, Dekkers F, Basak O, van Es J, Chuva de Sousa Lopes SM, Begthel H, Korving J, van den Born M, Zou C, Quirk C, Chiriboga L, Rice CM, Ma S, Rios A, Peters PJ, de Jong YP, Clevers H, Cell 2018, 175, 1591. [DOI] [PubMed] [Google Scholar]
- [91].Takebe T, Zhang RR, Koike H, Kimura M, Yoshizawa E, Enomura M, Koike N, Sekine K, Taniguchi H, Nat Protoc 2014, 9, 396. [DOI] [PubMed] [Google Scholar]
- [92].Badylak SF, Tullius R, Kokini K, Shelbourne KD, Klootwyk T, Voytik SL, Kraine MR, Simmons C, J Biomed Mater Res 1995, 29, 977. [DOI] [PubMed] [Google Scholar]
- [93].Chen RN, Ho HO, Tsai YT, Sheu MT, Biomaterials 2004, 25, 2679. [DOI] [PubMed] [Google Scholar]
- [94].Schmidt CE, Baier JM, Biomaterials 2000, 21, 2215. [DOI] [PubMed] [Google Scholar]
- [95].Elkins RC, Dawson PE, Goldstein S, Walsh SP, Black KS, Ann Thorac Surg 2001, 71, S428. [DOI] [PubMed] [Google Scholar]
- [96].Schultheiss D, Gabouev AI, Cebotari S, Tudorache I, Walles T, Schlote N, Wefer J, Kaufmann PM, Haverich A, Jonas U, Stief CG, Mertsching H, J Urol 2005, 173, 276. [DOI] [PubMed] [Google Scholar]
- [97].Ott HC, Matthiesen TS, Goh SK, Black LD, Kren SM, Netoff TI, Taylor DA, Nat Med 2008, 14, 213. [DOI] [PubMed] [Google Scholar]
- [98].Guyette JP, Gilpin SE, Charest JM, Tapias LF, Ren X, Ott HC, Nat Protoc 2014, 9, 1451. [DOI] [PubMed] [Google Scholar]
- [99].Garreta E, Oria R, Tarantino C, Pla-Roca M, Prado P, Fernández-Avilés F, Campistol JM, Samitier J, Montserrat N, Mater Today 2017, 20, 166. [Google Scholar]
- [100].Wang W, Jin S, Ye K, Stem Cells Dev 2017, 26, 394. [DOI] [PubMed] [Google Scholar]
- [101].Bi H, Ye K, Jin S, Biomaterials 2020, 233, 119673. [DOI] [PubMed] [Google Scholar]
- [102].Dossena M, Piras R, Cherubini A, Barilani M, Dugnani E, Salanitro F, Moreth T, Pampaloni F, Piemonti L, Lazzari L, Stem Cell Res Ther 2020, 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Giobbe GG, Crowley C, Luni C, Campinoti S, Khedr M, Kretzschmar K, De Santis MM, Zambaiti E, Michielin F, Meran L, Hu Q, van Son G, Urbani L, Manfredi A, Giomo M, Eaton S, Cacchiarelli D, Li VSW, Clevers H, Bonfanti P, Elvassore N, De Coppi P, Nat Commun 2019, 10, 5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Broguiere N, Isenmann L, Hirt C, Ringel T, Placzek S, Cavalli E, Ringnalda F, Villiger L, Zullig R, Lehmann R, Rogler G, Heim MH, Schuler J, Zenobi-Wong M, Schwank G, Adv Mater 2018, 30, e1801621. [DOI] [PubMed] [Google Scholar]
- [105].Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM, Cell 2009, 139, 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Yang C, Tibbitt MW, Basta L, Anseth KS, Nat Mater 2014, 13, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Brusatin G, Panciera T, Gandin A, Citron A, Piccolo S, Nat Mater 2018, 17, 1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Ranga A, Girgin M, Meinhardt A, Eberle D, Caiazzo M, Tanaka EM, Lutolf MP, Proc Natl Acad Sci U S A 2016, 113, E6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Enemchukwu NO, Cruz-Acuna R, Bongiorno T, Johnson CT, Garcia JR, Sulchek T, Garcia AJ, J Cell Biol 2016, 212, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Vives J, Batlle-Morera L, Stem Cell Res Ther 2020, 11, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Gjorevski N, Lutolf MP, Nat Protoc 2017, 12, 2263. [DOI] [PubMed] [Google Scholar]
- [112].Cruz-Acuna R, Quiros M, Farkas AE, Dedhia PH, Huang S, Siuda D, Garcia-Hernandez V, Miller AJ, Spence JR, Nusrat A, Garcia AJ, Nat Cell Biol 2017, 19, 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Crowder SW, Leonardo V, Whittaker T, Papathanasiou P, Stevens MM, Cell Stem Cell 2016, 18, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Hushka EA, Yavitt FM, Brown TE, Dempsey PJ, Anseth KS, Adv Healthc Mater 2020, 9, e1901214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Totaro A, Castellan M, Battilana G, Zanconato F, Azzolin L, Giulitti S, Cordenonsi M, Piccolo S, Nat Commun 2017, 8, 15206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Hernandez-Gordillo V, Kassis T, Lampejo A, Choi G, Gamboa ME, Gnecco JS, Brown A, Breault DT, Carrier R, Griffith LG, Biomaterials 2020, 254, 120125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Daly AC, Riley L, Segura T, Burdick JA, Nat Rev Mater 2019, 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Ng SS, Saeb-Parsy K, Blackford SJI, Segal JM, Serra MP, Horcas-Lopez M, No DY, Mastoridis S, Jassem W, Frank CW, Cho NJ, Nakauchi H, Glenn JS, Rashid ST, Biomaterials 2018, 182, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Borenstein JT, Barnard E, Orrick B, Cheung W, Sundback C, Vacanti JP, in Architecture and Application of Biomaterials and Biomolecular Materials, Vol. 1 (Eds: Wong JY, Plant AL, Schmidt CE, Shea L, Coury AJ, Chen CS, Barron AE, Klok HA, Saltzman WM, Chilkoti A, Luo D, Uhrich K), 2004, 9. [Google Scholar]
- [120].Lancaster MA, Corsini NS, Wolfinger S, Gustafson EH, Phillips AW, Burkard TR, Otani T, Livesey FJ, Knoblich JA, Nat Biotechnol 2017, 35, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Sun S, Cui W, Dong Y, Wang Q, Macromol Res 2019, 27, 454. [Google Scholar]
- [122].Perepelkina T, Kegeles E, Baranov P, Tissue Eng Part C Methods 2019, 25, 433. [DOI] [PubMed] [Google Scholar]
- [123].Tejchman A, Znoj A, Chlebanowska P, Fraczek-Szczypta A, Majka M, Int J Mol Sci 2020, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Lee KY, Mooney DJ, Prog Polym Sci 2012, 37, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Hughes CS, Postovit LM, Lajoie GA, Proteomics 2010, 10, 1886. [DOI] [PubMed] [Google Scholar]
- [126].Maile S, Zimmermann B, Ketteler M, Merker HJ, Histol Histopathol 2000, 15, 403. [DOI] [PubMed] [Google Scholar]
- [127].Takezawa T, Ozaki K, Nitani A, Takabayashi C, Shimo-Oka T, Cell Transplant 2004, 13, 463. [DOI] [PubMed] [Google Scholar]
- [128].Janssen DA, Geutjes PJ, Odenthal J, van Kuppevelt TH, Schalken JA, Feitz WF, Heesakkers JF, J Urol 2013, 190, 341. [DOI] [PubMed] [Google Scholar]
- [129].Ritter CS, Slatopolsky E, Santoro S, Brown AJ, J Bone Miner Res 2004, 19, 491. [DOI] [PubMed] [Google Scholar]
- [130].Chen Y, Zhou W, Roh T, Estes MK, Kaplan DL, PLoS One 2017, 12, e0187880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].DiMarco RL, Dewi RE, Bernal G, Kuo C, Heilshorn SC, Biomater Sci 2015, 3, 1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Shakibaei M, De Souza P, Cell Biol Int 1997, 21, 75. [DOI] [PubMed] [Google Scholar]
- [133].Capeling MM, Czerwinski M, Huang S, Tsai YH, Wu A, Nagy MS, Juliar B, Sundaram N, Song Y, Han WM, Takayama S, Alsberg E, Garcia AJ, Helmrath M, Putnam AJ, Spence JR, Stem Cell Rep 2019, 12, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Lindborg BA, Brekke JH, Vegoe AL, Ulrich CB, Haider KT, Subramaniam S, Venhuizen SL, Eide CR, Orchard PJ, Chen W, Wang Q, Pelaez F, Scott CM, Kokkoli E, Keirstead SA, Dutton JR, Tolar J, O’Brien TD, Stem Cells Transl Med 2016, 5, 970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Zhu Y, Wang L, Yin F, Yu Y, Wang Y, Liu H, Wang H, Sun N, Liu H, Qin J, Integr Biol 2017, 9, 774. [DOI] [PubMed] [Google Scholar]
- [136].Wilkinson DC, Alva-Ornelas JA, Sucre JM, Vijayaraj P, Durra A, Richardson W, Jonas SJ, Paul MK, Karumbayaram S, Dunn B, Gomperts BN, Stem Cells Transl Med 2017, 6, 622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Ng S, Tan WJ, Pek MMX, Tan MH, Kurisawa M, Biomaterials 2019, 219, 119400. [DOI] [PubMed] [Google Scholar]
- [138].Luo X, Fong ELS, Zhu C, Lin QXX, Xiong M, Li A, Li T, Benoukraf T, Yu H, Liu S, Acta Biomater 2020. [DOI] [PubMed] [Google Scholar]
- [139].Kruger M, Oosterhoff LA, van Wolferen ME, Schiele SA, Walther A, Geijsen N, De Laporte L, van der Laan LJW, Kock LM, Spee B, Adv Healthc Mater 2020, 9, e1901658. [DOI] [PubMed] [Google Scholar]
- [140].Wieck MM, El-Nachef WN, Hou X, Spurrier RG, Holoyda KA, Schall KA, Mojica SG, Collins MK, Trecartin A, Cheng Z, Frykman PK, Grikscheit TC, Tissue Eng Part A 2016, 22, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Suita S, Taguchi T, Ieiri S, Nakatsuji T, J Pediatr Surg 2005, 40, 197. [DOI] [PubMed] [Google Scholar]
- [142].Skardal A, Devarasetty M, Kang HW, Seol YJ, Forsythe SD, Bishop C, Shupe T, Soker S, Atala A, J Vis Exp 2016, e53606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Nowak M, Freudenberg U, Tsurkan MV, Werner C, Levental KR, Biomaterials 2017, 112, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Li Q, Nan K, Le Floch P, Lin Z, Sheng H, Blum TS, Liu J, Nano Lett 2019, 19, 5781. [DOI] [PubMed] [Google Scholar]
- [145].O’Donnell N, Okkelman IA, Timashev P, Gromovykh TI, Papkovsky DB, Dmitriev RI, Acta Biomater 2018, 80, 85. [DOI] [PubMed] [Google Scholar]
- [146].Shin M, Song KH, Burrell JC, Cullen DK, Burdick JA, Adv Sci 2019, 6, 1901229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Yang X, Zhou T, Zwang TJ, Hong G, Zhao Y, Viveros RD, Fu TM, Gao T, Lieber CM, Nat Mater 2019, 18, 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Liu J, Kim YS, Richardson CE, Tom A, Ramakrishnan C, Birey F, Katsumata T, Chen S, Wang C, Wang X, Joubert LM, Jiang Y, Wang H, Fenno LE, Tok JB, Pasca SP, Shen K, Bao Z, Deisseroth K, Science 2020, 367, 1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Otto KJ, Schmidt CE, Science 2020, 367, 1303. [DOI] [PubMed] [Google Scholar]
- [150].Lee HJ, Mun S, Pham DM, Kim P, ACS Biomater Sci Eng 2021. [DOI] [PubMed] [Google Scholar]
- [151].Zimmermann R, Hentschel C, Schron F, Moedder D, Buttner T, Atallah P, Wegener T, Gehring T, Howitz S, Freudenberg U, Werner C, Biofabrication 2019, 11, 045008. [DOI] [PubMed] [Google Scholar]
- [152].Richards DJ, Coyle RC, Tan Y, Jia J, Wong K, Toomer K, Menick DR, Mei Y, Biomaterials 2017, 142, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Sheikhi A, de Rutte J, Haghniaz R, Akouissi O, Sohrabi A, Di Carlo D, Khademhosseini A, Biomaterials 2019, 192, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Xie M, Gao Q, Zhao H, Nie J, Fu Z, Wang H, Chen L, Shao L, Fu J, Chen Z, He Y, Small 2019, 15, e1804216. [DOI] [PubMed] [Google Scholar]
- [155].Giandomenico SL, Mierau SB, Gibbons GM, Wenger LMD, Masullo L, Sit T, Sutcliffe M, Boulanger J, Tripodi M, Derivery E, Paulsen O, Lakatos A, Lancaster MA, Nat Neurosci 2019, 22, 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Karzbrun E, Kshirsagar A, Cohen SR, Hanna JH, Reiner O, Nat Phys 2018, 14, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Renner M, Lancaster MA, Bian S, Choi H, Ku T, Peer A, Chung K, Knoblich JA, EMBO J 2017, 36, 1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Artegiani B, Hendriks D, Beumer J, Kok R, Zheng X, Joore I, Chuva de Sousa Lopes S, van Zon J, Tans S, Clevers H, Nat Cell Biol 2020, 22, 321. [DOI] [PubMed] [Google Scholar]
- [159].Goldsmith TM, Sakib S, Webster D, Carlson DF, Van der Hoorn F, Dobrinski I, Cell Tissue Res 2020, 380, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Zheng C, Schneider JW, Hsieh J, Dev Biol 2020, 462, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Tan Z, Shan J, Rak-Raszewska A, Vainio SJ, Sci Rep 2018, 8, 16618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D’Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, Klee JB, Zhang C, Wainger BJ, Peitz M, Kovacs DM, Woolf CJ, Wagner SL, Tanzi RE, Kim DY, Nature 2014, 515, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, Rueda R, Kritskiy O, Abdurrob F, Peng Z, Milo B, Yu CJ, Elmsaouri S, Dey D, Ko T, Yankner BA, Tsai LH, Neuron 2018, 98, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Nassor F, Jarray R, Biard DSF, Maiza A, Papy-Garcia D, Pavoni S, Deslys JP, Yates F, Front Cell Neurosci 2020, 14, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Seo J, Kritskiy O, Watson LA, Barker SJ, Dey D, Raja WK, Lin YT, Ko T, Cho S, Penney J, Silva MC, Sheridan SD, Lucente D, Gusella JF, Dickerson BC, Haggarty SJ, Tsai LH, J Neurosci 2017, 37, 9917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Kim H, Park HJ, Choi H, Chang Y, Park H, Shin J, Kim J, Lengner CJ, Lee YK, Kim J, Stem Cell Rep 2019, 12, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Smits LM, Reinhardt L, Reinhardt P, Glatza M, Monzel AS, Stanslowsky N, Rosato-Siri MD, Zanon A, Antony PM, Bellmann J, Nicklas SM, Hemmer K, Qing X, Berger E, Kalmbach N, Ehrlich M, Bolognin S, Hicks AA, Wegner F, Sterneckert JL, Schwamborn JC, NPJ Parkinsons Dis 2019, 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, Amenduni M, Szekely A, Palejev D, Wilson M, Gerstein M, Grigorenko EL, Chawarska K, Pelphrey KA, Howe JR, Vaccarino FM, Cell 2015, 162, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Zhu W, Zhang B, Li M, Mo F, Mi T, Wu Y, Teng Z, Zhou Q, Li W, Hu B, Nat Commun 2019, 10, 928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Latour YL, Yoon R, Thomas SE, Grant C, Li C, Sena-Esteves M, Allende ML, Proia RL, Tifft CJ, Mol Genet Metab Rep 2019, 21, 100513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Rodrigues T, Kundu B, Silva-Correia J, Kundu SC, Oliveira JM, Reis RL, Correlo VM, Pharmacol Ther 2018, 184, 201. [DOI] [PubMed] [Google Scholar]
- [172].Weeber F, Ooft SN, Dijkstra KK, Voest EE, Cell Chem Biol 2017, 24, 1092. [DOI] [PubMed] [Google Scholar]
- [173].Drost J, Clevers H, Nat Rev Cancer 2018, 18, 407. [DOI] [PubMed] [Google Scholar]
- [174].Tuveson D, Clevers H, Science 2019, 364, 952. [DOI] [PubMed] [Google Scholar]
- [175].Bellin M, Marchetto MC, Gage FH, Mummery CL, Nat Rev Mol Cell Biol 2012, 13, 713. [DOI] [PubMed] [Google Scholar]
- [176].Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, Gracanin A, Oni T, Yu KH, van Boxtel R, Huch M, Rivera KD, Wilson JP, Feigin ME, Ohlund D, Handly-Santana A, Ardito-Abraham CM, Ludwig M, Elyada E, Alagesan B, Biffi G, Yordanov GN, Delcuze B, Creighton B, Wright K, Park Y, Morsink FH, Molenaar IQ, Borel Rinkes IH, Cuppen E, Hao Y, Jin Y, Nijman IJ, Iacobuzio-Donahue C, Leach SD, Pappin DJ, Hammell M, Klimstra DS, Basturk O, Hruban RH, Offerhaus GJ, Vries RG, Clevers H, Tuveson DA, Cell 2015, 160, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Huang L, Holtzinger A, Jagan I, BeGora M, Lohse I, Ngai N, Nostro C, Wang R, Muthuswamy LB, Crawford HC, Arrowsmith C, Kalloger SE, Renouf DJ, Connor AA, Cleary S, Schaeffer DF, Roehrl M, Tsao MS, Gallinger S, Keller G, Muthuswamy SK, Nat Med 2015, 21, 1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Bartfeld S, Bayram T, van de Wetering M, Huch M, Begthel H, Kujala P, Vries R, Peters PJ, Clevers H, Gastroenterology 2015, 148, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, Kester L, McLaren-Douglas A, Blokker J, Jaksani S, Bartfeld S, Volckman R, van Sluis P, Li VS, Seepo S, Sekhar Pedamallu C, Cibulskis K, Carter SL, McKenna A, Lawrence MS, Lichtenstein L, Stewart C, Koster J, Versteeg R, van Oudenaarden A, Saez-Rodriguez J, Vries RG, Getz G, Wessels L, Stratton MR, McDermott U, Meyerson M, Garnett MJ, Clevers H, Cell 2015, 161, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, Wongvipat J, Kossai M, Ramazanoglu S, Barboza LP, Di W, Cao Z, Zhang QF, Sirota I, Ran L, MacDonald TY, Beltran H, Mosquera JM, Touijer KA, Scardino PT, Laudone VP, Curtis KR, Rathkopf DE, Morris MJ, Danila DC, Slovin SF, Solomon SB, Eastham JA, Chi P, Carver B, Rubin MA, Scher HI, Clevers H, Sawyers CL, Chen Y, Cell 2014, 159, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarro LM, Bradshaw CR, Allen GE, Arnes-Benito R, Sidorova O, Gaspersz MP, Georgakopoulos N, Koo BK, Dietmann S, Davies SE, Praseedom RK, Lieshout R, JNM IJ, Wigmore SJ, Saeb-Parsy K, Garnett MJ, van der Laan LJ, Huch M, Nat Med 2017, 23, 1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, Balgobind AV, Wind K, Gracanin A, Begthel H, Korving J, van Boxtel R, Duarte AA, Lelieveld D, van Hoeck A, Ernst RF, Blokzijl F, Nijman IJ, Hoogstraat M, van de Ven M, Egan DA, Zinzalla V, Moll J, Boj SF, Voest EE, Wessels L, van Diest PJ, Rottenberg S, Vries RGJ, Cuppen E, Clevers H, Cell 2018, 172, 373. [DOI] [PubMed] [Google Scholar]
- [183].Takebe T, Wells JM, Helmrath MA, Zorn AM, Cell Stem Cell 2018, 22, 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, Froeling FEM, Burkhart RA, Denroche RE, Jang GH, Miyabayashi K, Young CM, Patel H, Ma M, LaComb JF, Palmaira RLD, Javed AA, Huynh JC, Johnson M, Arora K, Robine N, Shah M, Sanghvi R, Goetz AB, Lowder CY, Martello L, Driehuis E, LeComte N, Askan G, Iacobuzio-Donahue CA, Clevers H, Wood LD, Hruban RH, Thompson E, Aguirre AJ, Wolpin BM, Sasson A, Kim J, Wu M, Bucobo JC, Allen P, Sejpal DV, Nealon W, Sullivan JD, Winter JM, Gimotty PA, Grem JL, DiMaio DJ, Buscaglia JM, Grandgenett PM, Brody JR, Hollingsworth MA, O’Kane GM, Notta F, Kim E, Crawford JM, Devoe C, Ocean A, Wolfgang CL, Yu KH, Li E, Vakoc CR, Hubert B, Fischer SE, Wilson JM, Moffitt R, Knox J, Krasnitz A, Gallinger S, Tuveson DA, Cancer Discov 2018, 8, 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Smith RC, Tabar V, Cell Stem Cell 2019, 24, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Stratton MR, Campbell PJ, Futreal PA, Nature 2009, 458, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, Nijman IJ, Martincorena I, Mokry M, Wiegerinck CL, Middendorp S, Sato T, Schwank G, Nieuwenhuis EE, Verstegen MM, van der Laan LJ, de Jonge J, JN IJ, Vries RG, van de Wetering M, Stratton MR, Clevers H, Cuppen E, van Boxtel R, Nature 2016, 538, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, Denroche RE, Liang SB, Brown AM, Kim JC, Wang T, Simpson JT, Beck T, Borgida A, Buchner N, Chadwick D, Hafezi-Bakhtiari S, Dick JE, Heisler L, Hollingsworth MA, Ibrahimov E, Jang GH, Johns J, Jorgensen LG, Law C, Ludkovski O, Lungu I, Ng K, Pasternack D, Petersen GM, Shlush LI, Timms L, Tsao MS, Wilson JM, Yung CK, Zogopoulos G, Bartlett JM, Alexandrov LB, Real FX, Cleary SP, Roehrl MH, McPherson JD, Stein LD, Hudson TJ, Campbell PJ, Gallinger S, Nature 2016, 538, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, Watanabe T, Kanai T, Sato T, Nat Med 2015, 21, 256. [DOI] [PubMed] [Google Scholar]
- [190].Freedman BS, Brooks CR, Lam AQ, Fu H, Morizane R, Agrawal V, Saad AF, Li MK, Hughes MR, Werff RV, Peters DT, Lu J, Baccei A, Siedlecki AM, Valerius MT, Musunuru K, McNagny KM, Steinman TI, Zhou J, Lerou PH, Bonventre JV, Nat Commun 2015, 6, 8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Woo DH, Chen Q, Yang TL, Glineburg MR, Hoge C, Leu NA, Johnson FB, Lengner CJ, Cell Stem Cell 2016, 19, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, Sasaki N, Boymans S, Cuppen E, van der Ent CK, Nieuwenhuis EE, Beekman JM, Clevers H, Cell Stem Cell 2013, 13, 653. [DOI] [PubMed] [Google Scholar]
- [193].Geurts MH, de Poel E, Amatngalim GD, Oka R, Meijers FM, Kruisselbrink E, van Mourik P, Berkers G, de Winter-de Groot KM, Michel S, Muilwijk D, Aalbers BL, Mullenders J, Boj SF, Suen SWF, Brunsveld JE, Janssens HM, Mall MA, Graeber SY, van Boxtel R, van der Ent CK, Beekman JM, Clevers H, Cell Stem Cell 2020, 26, 503. [DOI] [PubMed] [Google Scholar]
- [194].Haga K, Ettayebi K, Tenge VR, Karandikar UC, Lewis MA, Lin SC, Neill FH, Ayyar BV, Zeng XL, Larson G, Ramani S, Atmar RL, Estes MK, mBio 2020, 11, e00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Takebe T, Wells JM, Science 2019, 364, 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Hofer M, Lutolf MP, Nat Rev Mater 2021, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Faustino Martins JM, Fischer C, Urzi A, Vidal R, Kunz S, Ruffault PL, Kabuss L, Hube I, Gazzerro E, Birchmeier C, Spuler S, Sauer S, Gouti M, Cell Stem Cell 2020, 26, 172. [DOI] [PubMed] [Google Scholar]
- [198].Pellegrini L, Bonfio C, Chadwick J, Begum F, Skehel M, Lancaster MA, Science 2020, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Lee J, Rabbani CC, Gao H, Steinhart MR, Woodruff BM, Pflum ZE, Kim A, Heller S, Liu Y, Shipchandler TZ, Koehler KR, Nature 2020, 582, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Zhang Y, Wiesholler LM, Rabie H, Jiang P, Lai J, Hirsch T, Lee KB, ACS Appl Mater Interfaces 2020, 12, 40031. [DOI] [PubMed] [Google Scholar]
- [201].Broguiere N, Luchtefeld I, Trachsel L, Mazunin D, Rizzo R, Bode JW, Lutolf MP, Zenobi-Wong M, Adv Mater 2020, 32, e1908299. [DOI] [PubMed] [Google Scholar]
- [202].Libby ARG, Briers D, Haghighi I, Joy DA, Conklin BR, Belta C, McDevitt TC, Cell Syst 2019, 9, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Shayegan M, Forde NR, PLoS One 2013, 8, e70590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Hall MS, Long R, Feng X, Huang Y, Hui CY, Wu M, Exp Cell Res 2013, 319, 2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Huh JR, Veiga-Fernandes H, Nat Rev Immunol 2020, 20, 217. [DOI] [PubMed] [Google Scholar]
- [206].Enaud R, Prevel R, Ciarlo E, Beaufils F, Wieers G, Guery B, Delhaes L, Front Cell Infect Microbiol 2020, 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Hawkins KG, Casolaro C, Brown JA, Edwards DA, Wikswo JP, Clin Pharmacol Ther 2020, 108, 929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Skardal A, Murphy SV, Devarasetty M, Mead I, Kang HW, Seol YJ, Shrike Zhang Y, Shin SR, Zhao L, Aleman J, Hall AR, Shupe TD, Kleensang A, Dokmeci MR, Jin Lee S, Jackson JD, Yoo JJ, Hartung T, Khademhosseini A, Soker S, Bishop CE, Atala A, Sci Rep 2017, 7, 8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Brassard JA, Nikolaev M, Hubscher T, Hofer M, Lutolf MP, Nat Mater 2021, 20, 22. [DOI] [PubMed] [Google Scholar]
- [210].Ma X, Qu X, Zhu W, Li YS, Yuan S, Zhang H, Liu J, Wang P, Lai CS, Zanella F, Feng GS, Sheikh F, Chien S, Chen S, Proc Natl Acad Sci U S A 2016, 113, 2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Magliaro C, Rinaldo A, Ahluwalia A, Sci Rep 2019, 9, 11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Nassari S, Duprez D, Fournier-Thibault C, Front Cell Dev Biol 2017, 5, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Leeman KT, Pessina P, Lee JH, Kim CF, Sci Rep 2019, 9, 6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Sart S, Tomasi RF, Barizien A, Amselem G, Cumano A, Baroud CN, Sci Adv 2020, 6, eaaw7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Stahl PL, Salmen F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, Mollbrink A, Linnarsson S, Codeluppi S, Borg A, Ponten F, Costea PI, Sahlen P, Mulder J, Bergmann O, Lundeberg J, Frisen J, Science 2016, 353, 78. [DOI] [PubMed] [Google Scholar]
- [216].Dekkers JF, Alieva M, Wellens LM, Ariese HCR, Jamieson PR, Vonk AM, Amatngalim GD, Hu H, Oost KC, Snippert HJG, Beekman JM, Wehrens EJ, Visvader JE, Clevers H, Rios AC, Nat Protoc 2019, 14, 1756. [DOI] [PubMed] [Google Scholar]
- [217].Ke M, Yuan X, He A, Yu P, Chen W, Shi Y, Hunter T, Zou P, Tian R, Nat Commun 2021, 12, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Pashos EE, Park Y, Wang X, Raghavan A, Yang W, Abbey D, Peters DT, Arbelaez J, Hernandez M, Kuperwasser N, Li W, Lian Z, Liu Y, Lv W, Lytle-Gabbin SL, Marchadier DH, Rogov P, Shi J, Slovik KJ, Stylianou IM, Wang L, Yan R, Zhang X, Kathiresan S, Duncan SA, Mikkelsen TS, Morrisey EE, Rader DJ, Brown CD, Musunuru K, Cell Stem Cell 2017, 20, 558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Michels BE, Mosa MH, Streibl BI, Zhan T, Menche C, Abou-El-Ardat K, Darvishi T, Czlonka E, Wagner S, Winter J, Medyouf H, Boutros M, Farin HF, Cell Stem Cell 2020, 26, 782. [DOI] [PubMed] [Google Scholar]
- [220].Artegiani B, van Voorthuijsen L, Lindeboom RGH, Seinstra D, Heo I, Tapia P, Lopez-Iglesias C, Postrach D, Dayton T, Oka R, Hu H, van Boxtel R, van Es JH, Offerhaus J, Peters PJ, van Rheenen J, Vermeulen M, Clevers H, Cell Stem Cell 2019, 24, 927. [DOI] [PubMed] [Google Scholar]
- [221].Oost KC, van Voorthuijsen L, Fumagalli A, Lindeboom RGH, Sprangers J, Omerzu M, Rodriguez-Colman MJ, Heinz MC, Verlaan-Klink I, Maurice MM, Burgering BMT, van Rheenen J, Vermeulen M, Snippert HJG, Cell Rep 2018, 22, 1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Uygun BE, Soto-Gutierrez A, Yagi H, Izamis ML, Guzzardi MA, Shulman C, Milwid J, Kobayashi N, Tilles A, Berthiaume F, Hertl M, Nahmias Y, Yarmush ML, Uygun K, Nat Med 2010, 16, 814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Rowland CR, Glass KA, Ettyreddy AR, Gloss CC, Matthews JRL, Huynh NPT, Guilak F, Biomaterials 2018, 177, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Lee JY, Chang JK, Dominguez AA, Lee HP, Nam S, Chang J, Varma S, Qi LS, West RB, Chaudhuri O, Nat Commun 2019, 10, 1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Mohamed-Ali H, Scholz P, Merker HJ, Cell Tissue Res 1996, 284, 509. [DOI] [PubMed] [Google Scholar]
- [226].Bhadriraju K, Hong JS, Lund SP, Reyes DR, ACS Biomater Sci Eng 2017, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Shen S, Shen J, Shen H, Wu C, Chen P, Wang Q, Front Chem 2020, 8, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Caiazzo M, Okawa Y, Ranga A, Piersigilli A, Tabata Y, Lutolf MP, Nat Mater 2016, 15, 344. [DOI] [PubMed] [Google Scholar]
- [229].Brandenberg N, Lutolf MP, Adv Mater 2016, 28, 7450. [DOI] [PubMed] [Google Scholar]
- [230].Kim YH, Choi SH, D’Avanzo C, Hebisch M, Sliwinski C, Bylykbashi E, Washicosky KJ, Klee JB, Brustle O, Tanzi RE, Kim DY, Nat Protoc 2015, 10, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Stepanichev M, Front Genome Ed 2020, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Lee JW, Komar CA, Bengsch F, Graham K, Beatty GL, Curr Protoc Pharmacol 2016, 73, 14 39 1. [DOI] [PMC free article] [PubMed] [Google Scholar]