

ABSTRACT
Biochemical studies of the human ribosome synthesis pathway have been hindered by technical difficulties in obtaining intact preribosomal complexes from internal regions of the nucleolus. Here we provide a detailed description of an extraction method that enables efficient detection, isolation, and characterization of nucleolar preribosomes containing large pre-rRNA species. The three-step Preribosome Sequential Extraction (PSE) protocol preserves the integrity of early preribosomal complexes and yields preparations amenable to biochemical analyses from low amounts of starting material. We validate this procedure through the detection of specific trans-acting factors and pre-rRNAs in the extracted preribosomes using affinity matrix pull-downs and sedimentation assays. In addition, we describe the application of the PSE method for monitoring cellular levels of ribosome-free 5S RNP complexes as an indicator of ribosome biogenesis stress. Our optimized experimental procedures will facilitate studies of human ribosome biogenesis in normal, mutant and stressed-cell scenarios, including the characterization of candidate ribosome biogenesis factors, preribosome interactors under specific physiological conditions or effects of drugs on ribosome maturation.
KEYWORDS: Human ribosome synthesis, ribosome assembly, preribosomes, pre-rRNA maturation
Introduction
Ribosomes are large ribonucleoprotein complexes formed by two subunits of unequal size composed of four ribosomal RNAs (18S, 5S, 5.8S and 28S in humans) and approximately 80 proteins. In eukaryotes, the synthesis of ribosomal subunits proceeds through a complex, energy-consuming route that is initiated in the nucleolus and continues in the nucleoplasm and cytoplasm (Figure 1a). The first step is the RNA polymerase I-mediated synthesis of a long polycistronic rRNA precursor (47S pre-rRNA in humans) that contains the future 18S, 5.8S and 28S rRNAs flanked by internal and external RNA segments. This common precursor enters a multi-step pathway during which the rRNA is chemically modified, the pre-rRNAs of the two subunits separated, the flanking RNA segments removed, and the ribosomal proteins assembled in an orderly manner [1–3]. The majority of those processes take place inside the nucleolus. The numerous processing, folding and assembly events of the pathway are mediated by more than 200 trans-acting ribosome biogenesis factors (RBFs) that analogous to workers at an assembly line, perform their functions at specific steps [2–6]. The precursor complexes formed during ribosome synthesis are commonly known as preribosomes or preribosomal particles.
Figure 1.
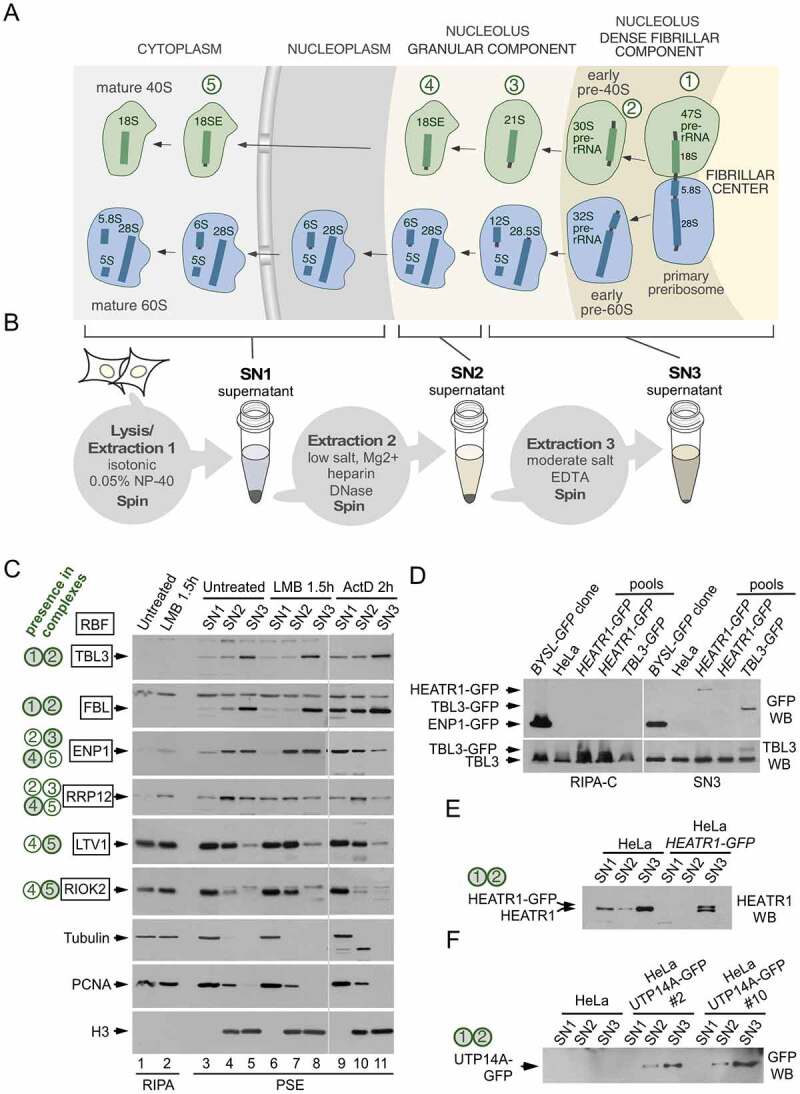
Fractionation of human preribosomes using the PSE extraction method. (a) Simplified scheme of the human ribosome biogenesis pathway showing 40S (green) and 60S (blue) preribosomal particles at different stages of maturation as they travel from the nucleolus to the cytoplasm. The names of the pre-rRNAs species at each stage are indicated. The nucleolus exhibits a tripartite organization that reflects the directionality of preribosome assembly and maturation. The sites of active RNA polymerase I transcription are at the interface between the fibrillar center and the dense fibrillar component, the early processing of the pre-rRNAs occurs in the dense fibrillar component and the late processing in the granular component. The primary transcript (47S pre-rRNA) contains the sequences of the 18S, 5.8S and 28S rRNAs. The earliest pre-40S particles, containing the 47S and 30S pre-rRNAs, are formed in the dense fibrillar component. Five pre-40S maturation stages are indicated with circled numbers. (b) Scheme of the PSE method showing the preribosome pools that are solubilized in each step. There is an additional 18S-E-containing particle (in between stages 3 and 4) that is solubilized in the SN3 fraction (not shown for simplicity). (c) Western blot analyses showing the contents of different RBFs in the SN1, SN2 and SN3 fractions obtained with the PSE method. PSE extraction was performed on HeLa cells normally growing (lanes 3 to 5), and after treatment with LMB (lanes 6 to 8) or ActD (lanes 9 to 11). Equivalent amounts of whole cell lysates prepared with RIPA buffer were analysed in parallel for comparison (lanes 1 and 2). Control proteins, unrelated to the ribosome synthesis route, include tubulin and proliferating cell nuclear antigen (extracted in the SN1 fraction) and histone H3 (extracted in the SN2 and SN3 fractions). The 40S preribosomes that contain each RBF are indicated on the left (green-filled circles are preribosome stages that mostly contain the RBF, open circles are preribosome stages in which only a subpool of particles contain the RBF). A summary of the relevant bibliography and data about the subcellular localization, loss-of-function phenotypes and hierarchy of incorporation into preribosomes of these RBFs can be found in the supplementary information of reference [24]. (d) Western blot analyses of lysates from cell pools that were subject to CRISPR editing to fuse GFP to either HEATR1 or TBL3. Panels on the left correspond to lysate samples prepared with RIPA-C buffer and panels on the right correspond to SN3 fractions obtained with the PSE method of the same pools. The presence of cells expressing the GFP-fusions in some pools is detected in the SN3 but not in the RIPA-C preparations. The parental HeLa cell line and a HeLa-derived cell line that expresses ENP1-GFP (BYSL-GFP clone, described in reference 24) were used as controls. Note that ENP1 is also known as BYSTIN (BYSL is the name of the human gene). (e, f) Western blot analyses showing the PSE fractionation profiles of HEATR1-GFP and UTP14A-GFP proteins endogenously expressed in HeLa-derived cell lines generated by CRISPR editing. Data demonstrating the functionality of the fusion proteins are shown in supplementary figures S1 and S2. HEATR1 and UTP14A are known components of 90S preribosomes in yeast (equivalent to the human particles in stages 1 and 2)
Understanding human ribosome synthesis is important not only to attain basic knowledge of a fundamental cellular process but also to inform on the mechanisms of diseases, collectively called ribosomopathies, that are caused by defects in ribosome assembly [7–10]. Furthermore, deregulation of the ribosome biogenesis pathway in cancer has fuelled the interest in the identification of drugs that could specifically block ribosome biogenesis in cancer cells [11–13]. Despite the interest, many details of human ribosome synthesis are not well-understood [14,15]. Although several nucleoplasmic and cytoplasmic pre-40S particles have been characterized [16–19], no detailed information is available at present on the composition and structure of any of the early preribosomes. This contrasts with the situation in yeast, where all major intermediates have been extensively characterized [2–4,6,20]. One reason behind the slow progress of research on human preribosomes is the technical difficulty associated with their isolation from the nucleolus.
The mammalian nucleolus exhibits an external layer of heterochromatin and a highly viscous internal subcompartment [21–23], two features that hinder the extraction of preribosomal complexes. Indeed, some common problems encountered with conventional cell lysis protocols include low yield of native preribosomes, loss of integrity of large pre-rRNA species, and inconsistent recovery of associated RBFs. All these problems impede the analyses that require good structural preservation of preribosomal complexes. For example, the association of an RBF with a specific preribosome intermediate can only be meaningfully inferred from co-precipitation and co-sedimentation experiments if preribosomes are not disrupted during extraction. Another example is the evaluation of a ribosome synthesis defect caused by a mutation in an RBF or treatments with a drug. In this case, the identification of the affected step also requires gradient sedimentation assays and compositional analyses of well-preserved preribosomes. In an effort to overcome the existing limitations to the biochemical analysis of human preribosomes, we have recently developed a procedure designated as PSE that can effectively release different classes of preribosomal complexes into a soluble form in three consecutive steps (see scheme in Figure 1b). We have shown that the PSE method preserves the integrity of human preribosomes and allows the isolation of undegraded large pre-rRNAs [24]. To further extend the utility of PSE, we demonstrate here its additional applications for studies of human ribosome biosynthesis. We provide detailed information on how to perform the analysis of the early nucleolar preribosomes by combining the PSE-based fractionation with sucrose-gradient and pull-down assays. In addition, we report a simple protocol for the detection of a small 5S RNP subcomplex important for the induction of the ribosome synthesis stress response. The accompanying experimental procedures include our in-house optimizations and technical tips to serve as a useful resource for researchers studying human ribosome biogenesis.
Results
1. PSE preserves the integrity of early preribosomes
The PSE method is based on the selective release of different nucleolar components using solutions containing varying concentrations of salts and Mg2+ in combination with DNase I and heparin treatments. The basic three-step PSE procedure, schematically outlined in Figure 1b, yields three separate soluble fractions, each containing different preribosome species: SN1 containing nucleoplasmic and cytoplasmic preribosomes, SN2 composed largely of intermediate-maturation preribosomes that require removal of the nucleolus-associated chromatin layer for extraction, and SN3 containing early preribosomes that are tightly associated with the interior nucleolar regions. Figure 1c (lanes 3 to 5) shows how the PSE procedure fractionates six RBFs associated with pre-40S intermediates formed at different steps during normal subunit maturation (see Figure 1a for a scheme of the 40S maturation stages). LTV1 and RIOK2, two cytoplasmic-maturation factors that are recruited to preribosomes when exiting the nucleolus (transition from stage 4 to stage 5 and stage 5 in Figure 1a) are largely extracted in the SN1 fraction; intermediate-maturation (stages 2 to 4) factors are extracted in the SN2 (RRP12) and SN2/SN3 (ENP1) fractions; and early maturation (stages 1 to 2) factors (TBL3, FBL) are extracted in the SN3 fraction. The fractionation profiles of the RBFs are not altered after the treatment of cells with leptomycin B (LMB), an inhibitor of CRM1 that blocks preribosome export from the nucleus to the cytoplasm without grossly affecting the nucleolar structure (Figure 1c; see TBL3, FBL, ENP1 and RRP12 panels, compare lanes 3 to 5 with lanes 6 to 8). In contrast, low-dose treatments with actinomycin D (ActD), an RNA Pol I transcription inhibitor that triggers nucleolar disintegration and the release of nucleolar material into the nucleoplasm leads to a redistribution of the normally tightly bound nucleolar RBFs from the SN3 fraction to the SN1/SN2 fractions (Figure 1c; see TBL3, FBL and ENP1 panels, compare lanes 3 to 5 with lanes 9 to 11).
The use of the stepwise extraction in the PSE method tackles a commonly observed failure of generic cell lysis protocols to efficiently solubilize components of early preribosomes. For example, cell lysis with the RIPA buffer results in a poor extraction of TBL3 and FBL as compared with the SN3 fraction of PSE (Figure 1c, compare lanes 1 and 5). Likewise, a more stringent RIPA-C buffer is less efficient than PSE in solubilizing TBL3-GFP and HEATR1-GFP (Figure 1d). The consistent yield of early RBFs, as illustrated above, makes PSE suitable for accurate estimations of the levels of these factors even when low amounts of cells are available. Moreover, the early pre-40S complexes at stages 1 and 2 (Figure 1a), which are recovered in the PSE fraction SN3, appear to maintain their structural integrity well, as judged from the high sedimentation rates (70–100S) expected for these complexes and the preservation of the integrity of the 47S and 30S pre-rRNAs [24], see also Figure 3c below]. This makes the PSE procedure applicable for the analysis of RBF incorporation into early preribosomes. We have also found that the PSE fractionation profile can be an excellent predictor of the functionality of tagged RBFs. The two representative cases illustrating this property are HEATR1 and UTP14A, amenable to GFP-tagging and present at stages 1 and 2 of pre-40S maturation. In CRISPR-edited cell clones that express HEATR1-GFP or UTP14A-GFP, both GFP-fusion proteins display an enrichment in the SN3 fraction that suggests proper incorporation into early preribosomes (Figure 1e,f). Consistent with their recovery in the SN3 fraction with other early maturation RBFs, the GFP fusions appear to be fully functional, as inferred from their normal subcellular localization and redistribution upon ActD treatment (Figs. S1 and S2), the specific interaction of HEATR1-GFP with early RBFs and pre-rRNA species (see below), the normal pre-rRNA processing profiles of the edited cell lines (Figs. S1 and S2), and the normal growth of UTP14A-GFP-expressing cells in the absence of wild-type protein (Figure S2).
Figure 3.
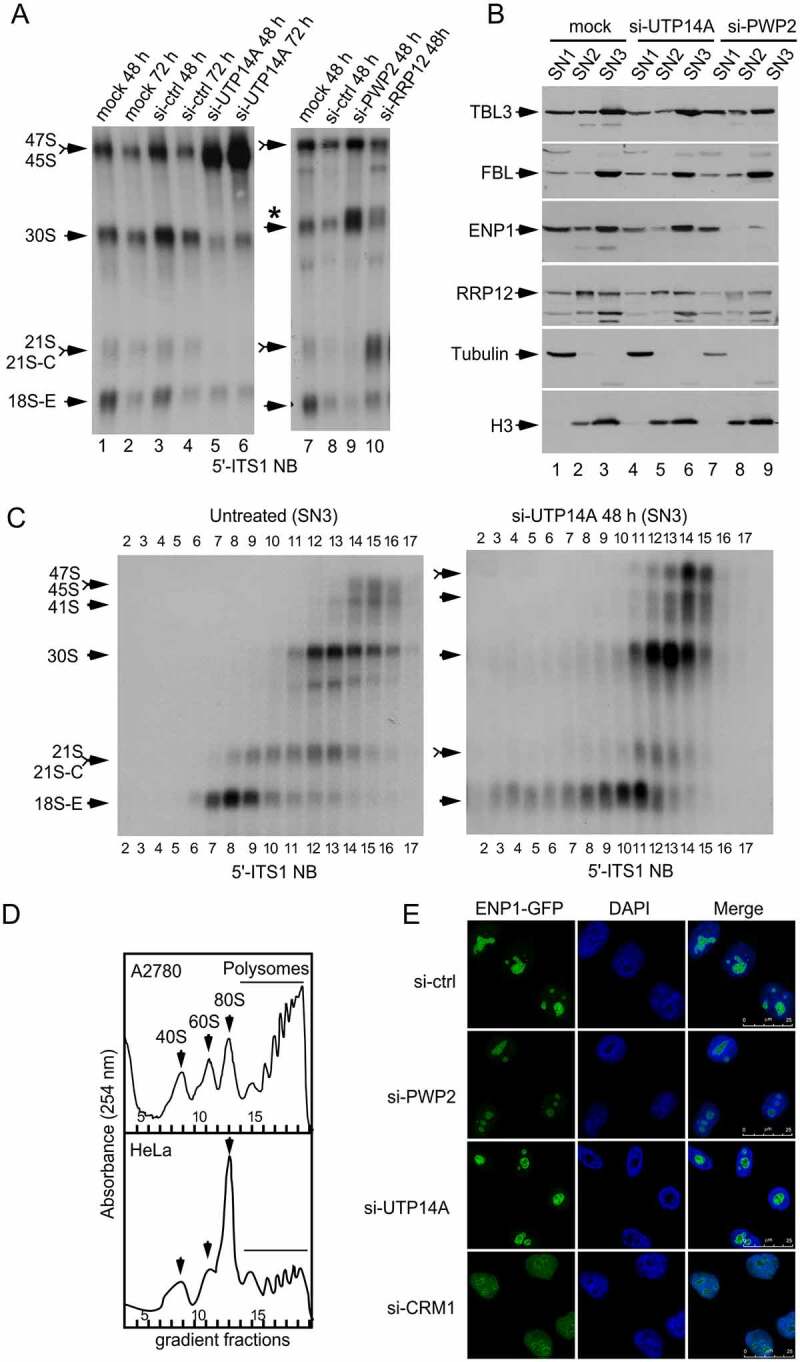
Analysis of preribosomes extracted with the PSE method in cells that lack an essential RBF. (a) Relative contents of pre-rRNA processing species in HeLa cells transfected with the indicated siRNAs and harvested at the indicated times after transfection. A probe (5′-ITS1) that recognizes most precursor species of the 18S rRNA was used for northern blot analysis of total RNAs prepared with the Trizol method. The asterisk indicates the migration position of a pre-rRNA species (34S) that is aberrantly produced in si-PWP2 and si-RRP12 KD cells. (b) Relative contents of several RBFs in the SN1, SN2, and SN3 fractions obtained with the PSE method from HeLa cells transfected with the indicated siRNAs and harvested 48 h after transfection. (c) Sedimentation profiles of pre-rRNAs extracted in the SN3 fraction of the PSE method from HeLa cells untreated or after 48 h transfection with a siRNA that depletes UTP14A. The SN3 fractions were fractionated on 7–50% sucrose gradients. The contents of pre-RNA species in each fraction of the gradients were analysed by northern blotting using a 5ʹ-ITS1 probe. (d) Polysome profiles used as reference for sucrose sedimentation analyses. Whole cell lysates (referred to as PPL lysates in the Methods section) were prepared and analysed by sucrose gradient sedimentation. The fractions in which the different ribosomal complexes sediment in these gradients are taken as references to estimate the sizes of preribosomes in the gradient analyses of SN3 fractions shown in C. Note that the conditions used to prepare and analyse PPL lysates are optimal to detect differences in global translation (A2780 cells have higher global translation than HeLa cells). (e) Microscopy analysis of HeLa-derived cells that endogenously express ENP1-GFP. Cells were transfected with the indicated siRNAs and harvested 48 h after transfection. The si-CRM1 was used as a positive control to confirm that the ENP1-GFP reporter accumulates in the nucleoplasm when nuclear export is blocked
2. Analysis of protein interactions that take place within early preribosomes
Affinity purification and co-immunoprecipitation of specific classes of preribosomes through their constituent RBFs can provide important insights into the formation and remodelling of these complexes. For these approaches to work, they must start with cell lysate preparations that contain well-preserved preribosomes, which additionally should not aggregate or bind non-specifically to affinity matrices. The protein baits through which preribosomes are purified must also: (1) Be accessible for antibody binding or amenable to tagging . (2) Maintain their interactions with preribosomes during the extraction and pull-down procedures (conversely, RBFs establishing weak or transitory associations with preribosomes are not useful). In addition, proteins chosen as baits for the pull-downs of entire preribosomes should not be components of any abundant complexes that are either pre-assembled before their incorporation into the preribosomes or released upon completion of preribosome maturation steps. The latter feature is important because the disassembly of early preribosomes involves the stepwise dislodgement of subcomplexes that are formed during particle maturation [25,26].
Based on the data obtained in prior yeast studies, Figure 2a shows a simplified scheme of the stepwise incorporation of several conserved RBFs that are components of early pre-40S ribosomes (stages 1 and 2 in Figure 1a) [6]. Through trials with human homologs of these RBFs, we have identified HEATR1, a constituent of the UTP-A subcomplex, as a well-behaved bait protein that, after being tagged with GFP, can be efficiently used in an optimized GFP-Trap affinity enrichment protocol. Figure 2b shows a representative western blot analysis of the GFP-Trap pull-downs using HEATR1-GFP with soluble PSE fractions prepared from HeLa cells. Specific interactions of HEATR1-GFP with a component of the UTP-B subcomplex (TBL3), a component of the U3 snoRNP (FBL), and an early intermediate RBF (ENP1) are readily detected in the SN3 fractions (Figure 2b; lane 14, compare with lane 10). All these associations are no longer detected when pre-rRNA synthesis is blocked by treatment with ActD (Figure 2b, lane 16), indicating that they take place within preribosomes and not in pre-assembly or post-assembly complexes. Furthermore, the presence of early 30S pre-rRNA in the HEATR1-GFP pull-downs is detectable by northern blot analyses (Figure 2d), validating this RBF as an optimal purification bait. ENP1-GFP, a fusion protein previously used to purify 40S preribosomes [24], allows the specific enrichment of 18S-E-containing but not 30S-containing complexes. It is important to note that not all preribosome components that retain functionality after GFP tagging behave as efficient baits for GFP-Trap purifications. For example, UTP14A-GFP functionally substitutes the endogenous UTP14A protein (Figure S2) but, when bound to GFP-Trap, only pulls down subcomplexes (also containing PWP2 and DHX37) that are extracted in the SN2 fraction and accumulate upon treatment with ActD (Figure 2c, lanes 13 and 15).
Figure 2.
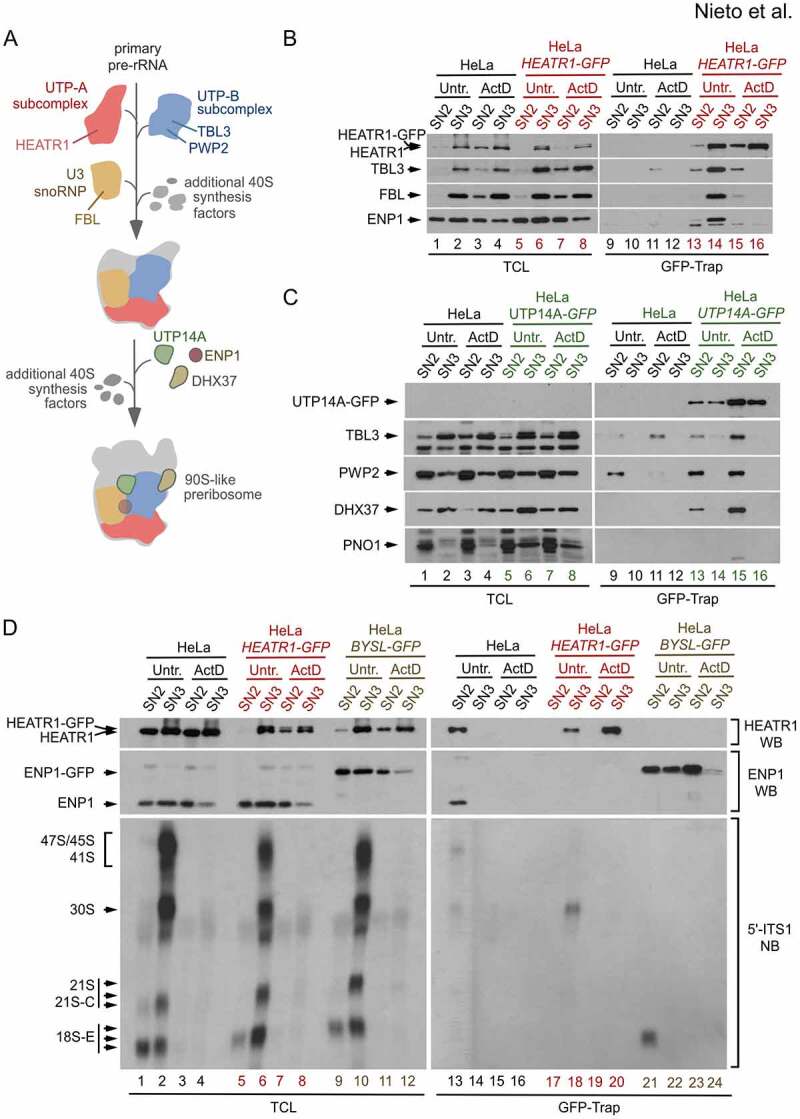
Analyses of proteins associated with early pre-40S ribosomes by GFP-Trap pull-down. (a) Possible order of incorporation of core RBFs into primary 40S preribosomes, as inferred from the steps of assembly of yeast 90S particles [6]. The human equivalents to 90S particles are the preribosomes containing the 47S pre-rRNA and 30S pre-rRNAs (stages 1 and 2 in Figure 1a). Some RBFs are recruited to the primary pre-rRNA as pre-formed subcomplexes and others are recruited individually. The scheme only specifies the RBFs analysed in panels B and C: HEATR1 (known as UTP10 in yeast, a component of the UTP-A subcomplex), TBL3 and PWP2 (TBL3 is UTP13 in yeast, both TBL3 and PWP2 are components of the UTP-B subcomplex), FBL (NOP1 in yeast, a component of the U3 snoRNP), UTP14A (UTP14 in yeast), ENP1 and DHX37 (DHR1 in yeast). (b) Interactions of several RBFs with HEATR1-GFP extracted in the SN3 fraction using the PSE method. GFP-Trap preparations from SN2 and SN3 fractions of HeLa cells and HeLa-derived cells endogenously expressing HEATR1-GFP, untreated or treated with ActD for 2 h, were analysed by western blot. A parallel western blot revealed the content of all proteins in the total fraction samples (left panels). It is detected a pre-rRNA synthesis-dependent association of HEATR1 with ENP1 and two RBFs (TBL3, PWP2) from two different subcomplexes. (c) No detection of interactions between early 40S RBFs and UTP14A-GFP extracted in the SN3 fractions with the PSE method. Parental HeLa and HeLa-derived cells endogenously expressing UTP14A-GFP were analysed as indicated in (B). UTP14A-GFP can substitute the endogenous UTP14A protein but is not an efficient bait for pulling down the 40S early particles extracted in the SN3 fraction. (b) Co-purification of early pre-rRNA species with HEATR1-GFP extracted in the SN3 fraction. GFP-Trap preparations from the SN2 and SN3 fractions of HeLa cells and HeLa-derived cells endogenously expressing HEATR1-GFP and ENP1-GFP (used as control) were analysed by northern blotting using a 5′-ITS1 probe that recognizes most precursors of the 18S rRNA, including the transient 41S, 45S and 21S-C species (right bottom panel). A parallel northern blot analysed total RNAs prepared from the same samples used for the GFP-Trap purifications (left bottom panel). Western blot analyses revealed the protein contents in the total fraction samples (left top panel) and GFP-Trap purification samples (right top panel). The 30S pre-rRNA co-precipitates with HEATR1-GFP, and the 18S-E pre-rRNA with ENP1-GFP. Both interactions are lost upon ActD treatment. HeLa-BYSL-GFP is the name of the HeLa-derived cell line that endogenously expresses ENP1-GFP (BYSL is the gene name). This cell line has been described previously [24]
Altogether, these results demonstrate that PSE extraction can be successfully combined with affinity purifications to obtain enriched preparations of early preribosomes from human cells. In the case demonstrated here, we identified a bait (HEATR1) that allows the enrichment of early (stage 2) pre-40S ribosomes, as shown by the presence of the 30S pre-rRNA and different RBFs. Our data with the HEATR1 bait indicate that early preribosomes in human cells are amenable to tagging and purification schemes similar to those previously used for the characterization of intermediate and late preribosomes [16–19,24].
3. Characterization of defects in early preribosome maturation
Defects in preribosome maturation, such as those caused by an RBF loss, usually lead to an aberrant reduction or accumulation of pre-rRNA species that can be detected by northern blot analyses. For example, the knockdown (KD) of RRP12 in HeLa cells blocks an intermediate 40S maturation step, resulting in the overaccumulation of the 21S pre-rRNA (Figure 3a, compare lanes 7 and 10; note in Figure 1a that the 21S pre-rRNA is in stage 3) [24]. The individual KDs of two other factors, UTP14A and PWP2 cause the accumulation of early pre-rRNAs (47S pre-rRNA in the UTP14A KD and 30S pre-rRNA in the PWP2 KD), concomitant with the loss of downstream species: 30S/21S/18S-E are reduced in the UTP14A KD and 21S/18S-E are reduced in the PWP2 KD (Figure 3a). These two pre-rRNA profiles are consistent with maturation defects of the initial pre-40S complexes (stages 1 and 2, Figure 1a). One potential explanation for the accumulation by the UTP14A and PWP2 KDs of different pre-rRNAs is that UTP14A is required for the maturation of preribosomes that contain the 47S pre-rRNA (stage 1), whereas PWP2 acts downstream of UTP14A at a step that matures the preribosomes that contain the 30S pre-rRNA (stage 2). However, these simple inferences might not be necessarily correct because the accumulated pre-rRNA does not always reflect the exact step of the pathway that is being blocked (see Discussion). As illustrated below, this problem can be tackled, and the RBF-depletion phenotypes characterized, by inspecting the PSE fractionation profiles of the RBFs and performing direct preribosome analyses.
In the case of the UTP14A KD, early (TBL3, FBL) and early/intermediate (ENP1) RBFs are extracted in the SN3 fraction (Figure 3b, compare lanes 1 to 3 with lanes 4 to 6), indicating that primary preribosomes are being assembled. Indeed, the complexes containing the 47S and 30S pre-rRNA are produced normally in UTP14A-depleted cells and, more importantly, they exhibit normal sedimentation behaviour (Figs. 3c, 30S pre-rRNA is in fractions 12, 13 and 14 in both the control and UTP14A KD gradients). A major defect caused by the loss of UTP14A is that the 30S-containing preribosomes do not undergo the progressive structural rearrangement and reduction in size that accompany the formation of the 18S-E pre-rRNA (Figure 3c, note that the 18S-E pre-rRNA is in fractions 7 to 9 in untreated cells, whereas in UTP14A KD cells is in fractions 10–11, see Figure 3d as reference of the locations of 40S, 60S and 80S particles in the gradients). Note that the formation of aberrant 18S-E-containing complexes is easily observed in the gradient analysis of the SN3 fraction, but this could not be inferred from the pre-rRNA pattern in the northern blot analysis (Figure 3a). The phenotype of the PWP2 KD cells is different because it alters the formation, rather than the maturation, of the primary preribosomes. Consistent with this, we can observe diminished contents of TBL3, ENP1 and RRP12 in the SN3 fraction (Figure 3b, compare lanes 1 to 3 with lanes 7 to 9) and reduced recruitment of ENP1 to the nucleolus (Figure 3e). Therefore, PWP2 acts upstream of UTP14A, and not the other way around, as could be erroneously concluded based solely on the northern blot data. These roles are fully consistent with the known roles of UTP14 and PWP2 in yeast [27,28], implying their functional conservation across eukaryotes.
4. Evaluation of ribosome biogenesis stress: analysis of ribosome-free RPL5 and RPL11
An additional application of the PSE method is the evaluation of ribosome biogenesis (also known as nucleolar) stress. Genetic defects and environmental conditions that give rise to reduced or abnormal ribosome production in cells are known to promote the accumulation of ribosome-free 5S RNP particles containing 5S rRNA, RPL5 and RPL11. Under normal conditions, the 5S RNP is associated to the large ribosomal subunit.
The ribosome-free 5S RNPs are capable of activating the tumour suppressor p53 through an inhibitory interaction with its negative regulator MDM2 (Figure 4a) [29,30]. All three components of the 5S RNPs are required for the activation of p53 by nucleolar stress, whereas p53 activation by DNA damage occurs independently of the 5S RNP-MDM2 pathway [31]. As shown in Figure 4b, siRNA-mediated depletion of RPL5 abolishes the upregulation of p53 in response to inhibition of rRNA synthesis by ActD and ribosome nuclear export by LMB. However, it only partially alleviates the p53 response to the strong DNA damage caused by doxorubicin. By comparison, p53 accumulation cannot be prevented by the depletion of RPS6, a ribosomal protein unrelated to 5S RNP complexes (Figure 4b). Because the ribosome-free RPL5 and RPL11 levels are normally very low in cells [31], detecting these proteins in a ribosome-free state can serve as a good indicator of disruptions in the nucleolar steps of ribosome production. The techniques previously used to detect free RPL5/RPL11 included sucrose gradients, multiple-step fractionations and/or TCA precipitations [31,32]. We have found that a rapid evaluation of ribosome-free RPL5 and RPL11 can be achieved by combining the PSE procedure with one additional ultracentrifugation step to pellet high-molecular-weight complexes (ribosomes and preribosomes). This leaves only low-molecular-weight complexes and free proteins in the final supernatant. As shown in Figure 4c, RPL5 and RPL11 are normally found in the SN1-SN2-derived pellets, consistent with ribosome association, and can only be partially released into a lower-molecular-weight supernatant after chelating divalent ions at the extraction step producing the SN3 fraction. In contrast, perturbation of ribosome assembly leads to a readily detectable level of RPL5 and RPL11 in the ribosome-free portion of the SN1 fraction (Figure 4c, compare lane 5 with lanes 6 to 8). Thus, performing an ultracentrifugation step with the SN1 fraction creates a simple assay for the detection of ribosome-free cellular pools of RPL5 and RPL11 indicative of ribosome biogenesis stress. This procedure could be used for performing screens of drugs and to analyse stress conditions or identify mutations that cause ribosome synthesis disruptions and p53 activation.
Figure 4.
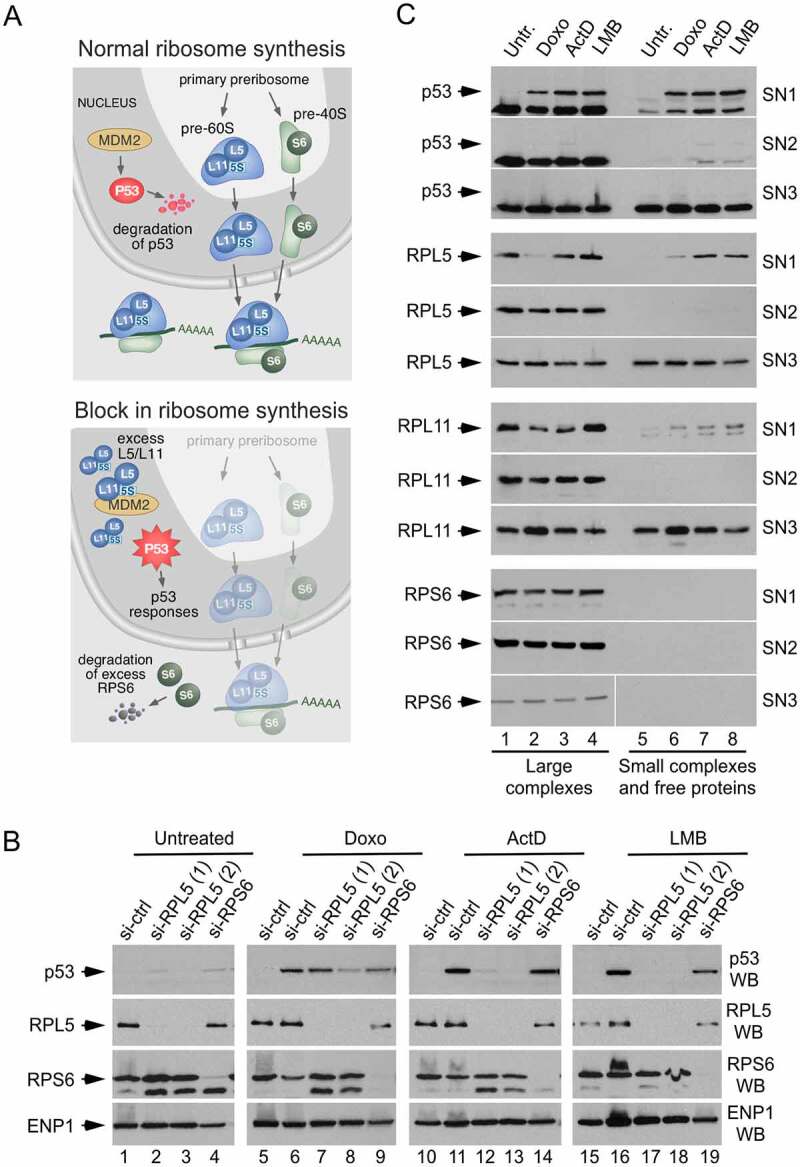
Detection of the ribosome-free RPL5/RPL11 content in cells undergoing ribosome synthesis stress. (a) Cartoon showing the 5S snRNP-mediated mechanism of p53 activation triggered by a defect in ribosome synthesis. (b) Western blot analyses of p53 levels in HCT116 cells transfected with the indicated siRNAs for 48 h and treated with doxorubicin (Doxo), ActD and LMB for 8 h. (c) Western blot analyses showing the levels of the indicated proteins in the SN1, SN2, and SN3 fractions obtained with the PSE method, after the separation of high (lanes 1–4) and low (lanes 5–8) molecular weight complexes by ultracentrifugation. There is an accumulation of free RPL5 and RPL11 in ActD- and LMB-treated cells (lanes 7 and 8). There is some increase of RPL11 in the SN1 and SN3 fractions from doxorubicin-treated cells whose significance is uncertain
Discussion
In this communication, we demonstrate a number of applications of the PSE method for the analysis of human ribosome synthesis intermediates in combinations with sucrose-gradient sedimentation and pull-down interaction assays. We also provide detailed descriptions of the protocols, materials, and technical tips for all experiments shown here to facilitate the successful implementation of these techniques by other laboratories.
RBFs and pre-rRNA species exhibit different fractionation profiles in the PSE protocol, reflecting the types of preribosomes with which they are associated. Importantly, the PSE fractionation profiles of all the RBFs we have analysed to date remain highly consistent between experiments and different cell types. This is useful when characterizing either mutant or tagged versions of the RBFs because any abnormal association with preribosomes generates altered PSE fractionation patterns that are easy to detect. Inspections of RBF fractionation patterns can also be used to detect major blockages in the early steps of ribosome synthesis. Here we show two examples, the block in pre-rRNA synthesis by ActD treatment and the impaired assembly of primary pre-40S particles by PWP2 depletion, which result in an abnormal enrichment of early RBFs in the SN2 and SN1 fractions (Figs. 1c and 3b). Another application of the PSE method illustrated here is the monitoring of ribosome synthesis stress through the detection of ribosome-free RPL5/RPL11 complexes in cells (Figure 4).
One advantageous property of the PSE procedure is that the extracted preribosomes are fully amenable to further biochemical characterization. We have recently analysed the compositions of the early/intermediate pre-40S particles extracted with this method [24]. The results presented here demonstrate that the early human preribosomes, found in the SN3 fractions, are also well-preserved and exhibit normal sedimentation behaviour in sucrose gradients. In addition, we show that these preribosome particles can be analysed for the presence of specific components by pull-downs using tagged RBFs. The preribosomes isolated by the PSE method exhibit minimal aggregation and low non-specific binding to solid matrices. For a successful pull-down, it is important, however, to use appropriate tagged RBFs as baits. HEATR1 is one such bait, since its GFP-tagged version can be used to pull down large (80S-90S) particles containing the 30S pre-rRNA associated with the UTP-A subcomplex, the UTP-B subcomplex, the U3 snoRNP and additional RBFs. These interactions are dependent on pre-rRNA synthesis, indicating that they are established upon preribosome assembly. One curious aspect of the HEATR1 pull-down SN3 preparations is the lack of primary complexes containing the 47S pre-rRNA. One possible explanation is that these complexes have low structural stability because they have both the 40S and 60S maturation machineries engaged. In yeast, purifications of 90S preribosomes with different baits are also enriched in 40S precursors that have already started the processing of the primary pre-rRNA (35S pre-rRNA) and have been separated from the 60S preribosome [25,33,34].
A classic way to determine whether a protein of interest is a bona-fide RBF is to examine how its loss affects the steps of ribosome synthesis. One common approach is to examine the patterns of pre-rRNA intermediates by northern blot hybridizations. For example, hybridization-based detection of defects in pre-rRNA processing has been successfully used to identify human RBFs in large-scale siRNA-based screens [35]. However, it is not always possible to clearly define the precise ribosome maturation steps that are blocked by a deficient RBF function just by analysing the patterns of accumulated or reduced pre-rRNA species. There could be different reasons for that: (1) An accumulation of aberrant unstable pre-rRNAs may be undetected in a northern hybridization because these RNAs are rapidly degraded inside the cell or do not survive the RNA preparation procedure]. (2)Stalling the assembly pathway may trap RBFs within the aberrant complexes creating a deficiency of these RBFs at earlier steps of preribosome maturation (and skewing the picture of the pre-rRNA processing defects as a result). Another common approach is to monitor the changes in the subcellular localization of RBFs and ribosomal proteins caused by blockages in preribosome maturation in the nucleolus, nuclear export, or cytoplasm [36,37]. However, like northern blots, microscopy analyses may not always pinpoint the specific maturation steps that are being affected because they do not provide sufficient information about the content, size and composition of preribosomes. One example shown here demonstrates the importance of performing direct preribosome analysis when trying to assess the function of an RBF. We obtain crucial information about the UTP14A KD from just the sucrose-gradient sedimentation analysis of the particles extracted in the SN3 fraction. The defect observed indicates that human UTP14A, like its yeast homolog [27], is required for the dismantlement of the scaffold of RBFs that build the 30S-containing particle. The northern blot and the RBF microscopy data on UTP14A KD cells cannot reveal such a role.
Native preribosome purification schemes have been instrumental in yeast, not only to identify hundreds of RBFs but also to define how the loss of different factors affect the composition, function and localization of distinct preribosome intermediates. After two decades of continuous advances, the research in yeast has culminated with the recent cryo-EM resolution of the structures of the major preribosome intermediates displaying distinct maturation events and the RBFs performing their actions [2,4,6,20,25,26]. Getting the same degree of molecular detail on human preribosomes is a major aspiration in the field. The PSE method can contribute to reach that goal with future studies. As indicated along this manuscript, PSE-isolated preribosomes can be used to directly analyse protein-protein interactions and define preribosome modules or establish the association of conserved RBFs, non-conserved candidate RBFs, and possible regulatory proteins with early preribosomes. Importantly, the good preservation of the 40S preribosomes extracted in the PSE-SN3 fraction sets the basis to undertake multiple bait or double-tag purification schemes, similar to those used in yeast, for cryo-EM structural analyses. We have not yet attempted pull-downs on early pre-60S particles but, based on their sedimentation behaviour in sucrose gradients [24], it is anticipated that they will also be amenable to protein–protein and protein-RNA analyses after their extraction with the PSE method.
Materials and methods
1. Cell culture and treatments
HeLa and HCT116 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin/streptomycin, 2 mM L-glutamine (Gibco), and maintained under standard tissue culture conditions. Short-term treatments to block RNA polymerase I transcription and CRM1-mediated nuclear export (Figure 1) were performed with 100 ng/ml actinomycin D (Calbiochem) and 40 nM leptomycin B (Enzo) for 2 h and 1.5 h, respectively. For the analysis of p53 activation and free RPL5/RPL11 levels upon genotoxic, nucleolar and nuclear-export stress (Figure 4), cells were treated with 5 nM actinomycin D, 500 nM doxorubicin (Sigma D1515) and 20 nM leptomycin B for 8 h. To knock down the expression of specific genes, siRNA duplexes (listed in Supplementary Table 3) were purchased from Ambion (Silencer Select siRNA) and reverse transfected in cells using Lipofectamine RNAiMAX (Life Technologies) as previously described [35]. Negative controls were either untreated cells or cells transfected with a control scrambled siRNA. In the UTP14A knock-down experiments (Figure 3), cells were harvested 48 h or 72 h after transfection. The contents of the targeted mRNAs in all siRNA-mediated knock-down experiments were routinely checked by qPCR analyses. The reduction in the content of the mRNA was always higher than 80%. In the analysis of p53 activation under ribosomal protein knock-down (Figure 4b), cells were transfected with siRNAs and, after 48 h, treated for 8 h with actinomycin D, doxorubicin or leptomycin B before harvesting.
2. GFP knock-in edition
Edition of the HEATR1 (NCBI gene ID:55,127) and UTP14A (NCBI gene ID:10,813) locus involved the generation of a single plasmid to drive the expression of both the Cas9 nuclease and the scaffold/guide RNA (sgRNA), and a second plasmid to provide a DNA donor for homology-directed repair (HDR). For the generation of the first plasmid, guide sequences were chosen using open access online tools (crispr.mit.edu and benchling.com) that identify protospacer adjacent motif (PAM) sequences and inform about their on-target and off-target scores. The genomic context of sgRNA guide sequences is shown in Supplementary Figures S1 and S2. The sgRNA sequences were cloned into the plasmid pX330 using synthetic oligonucleotides that were annealed and directly ligated to BbsI-digested vector, as previously described [38]. Sequences of the sg oligonucleotides used for each locus are listed in Supplementary Table 2. For the generation of the second plasmid, HDR donor sequences were introduced into a cloning vector. The HDR plasmid for HEATR1 (pHEATR1-HDR), which contains the GFP cDNA sequence fused in-frame with HEATR1 last codon, was generated by gene synthesis (GeneArt©, Invitrogen, Life Technologies). For the construction of UTP14A HDR plasmid (pSG12), the left homology-arm fragment was cloned into pEGFP-C1 at the NheI-XmaI sites, generating a fusion of the last codon of UTP14A with the first codon of GFP. Next, the right homology-arm was introduced using the HindIII-KpnI sites. Finally, the construct containing the GFP sequence flanked by the left and right arms was excised with NheI-KpnI and cloned into SpeI-KpnI pBluescript (Stratagene). For the GFP knock-in, HeLa cells were transfected with 1–3 μg of a mixture of the Cas9/sg and HDR plasmids (1:2 molar ratio) using Lipofectamine 2000 (Life Technologies), re-transfected with the HDR plasmids 24 h after the first transfection, and sorted on the basis of GFP fluorescence intensity 4–5 days after the second transfection. One or two additional FACS separations were required to enrich the GFP positive cells. Individual cell clones were isolated, expanded, and analysed by both Western Blot and PCR to identify those carrying the knock-in modification in the targeted locus. The generation of the BYSL-GFP edited HeLa cell line has been previously described [24].
3. Preparation of whole-cell unfractionated lysates for western blot analyses
RIPA buffer (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% Triton X100, 5 mM NaF, 1 mM Na3VO4, 1 mM ß-glycerol phosphate, supplemented with cOmplete protease inhibitor cocktail) was used in the initial experiments that compared the extraction efficiency of the PSE method with that of a single-step RIPA lysis (Figure 2c). The RIPA-C buffer (25 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% NP-40, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulphate (SDS), cOmplete protease inhibitor cocktail) [39] was used in some of the analysis of contents of nucleoplasmic and cytoplasmic proteins (Figs. 2d and 4b). In both cases, the procedure and starting amounts of cells were similar. Cells from one 10 cm plate (~80% confluent) were lysed in 400 µl of lysis buffer, kept on ice for 20 min, and then the lysate was cleared by centrifugation (12,000 rpm, 10 min, 4°C). Protein concentrations in the cleared supernatants were determined with Precision Red reagent (Cytoskeleton) following the manufacturer’s directions. For western blot analyses, 50 µg of protein was used per sample. The sources of the antibodies are indicated in Supplementary Table 4.
4. Preparation of PSE fractions for western blot, northern blot, co-immunoprecipitation or gradient sedimentation analyses (detailed protocol)
We have successfully used this procedure with HeLa and HCT116 cells.
4.1. Starting material
For western and northern blot analyses of nucleolar proteins and pre-rRNAs present in PSE fractions (Figure 1c, 1e, 1f, 3b and 3c), use two 10 cm dishes of cells grown at ~80% confluency.
For GFP-Trap pull-down of GFP-tagged nucleolar proteins to analyse co-purification of proteins and RNAs (i.e. Figure 2d,e), use four 10 cm dishes or, alternatively, two 15 cm dishes of cells grown at ~80% confluency.
For sucrose sedimentation analysis of nucleolar components present in the SN3 fraction (Figure 3d), use five 10 cm dishes of cells grown at ~80% confluency.
4.2. Harvest and storage of cell pellets
Materials and equipment: liquid nitrogen, dry ice, cold 15 ml conical centrifuge tubes, cold microfuge tubes, ice-cold phosphate-buffered saline (PBS), refrigerated (4°C) benchtop centrifuge and microfuge.
Procedure:
Remove media from cell dishes with aspiration and wash twice with cold PBS.
Place the plates on a tray with ice, scrape the cells (1 ml of PBS per 10 cm-plate or 2 ml of PBS per 15 cm-plate) and combine them all into a cold 15 ml centrifuge tube.
Spin in a benchtop centrifuge (1000 rpm, 5 min, 4°C), aspirate the PBS, resuspend the cell pellet in 1 ml of ice-cold PBS, and transfer to a microfuge tube.
Spin in a microfuge (2,500 rpm, 5 min, 4°C), aspirate the PBS and start immediately the PSE procedure or flash-freeze the cell pellet in liquid nitrogen, transfer the tube to dry ice and store at −80°C. Tip: have the microfuge tubes with the lids opened when placing them in nitrogen.
4.3. Reagent set up
Prepare fresh SN1, SN2 and SN3 buffers and keep them on ice. They must be cold before use.
SN1 buffer (prepare 1 ml per sample)
| Composition | Stock solution/storage |
|---|---|
| 20 mM HEPES-NaOH (pH 7.5) | 0.5 M/RT |
| 130 mM KCl | 3 M/RT |
| 10 mM MgCl2 | 1 M/RT |
| 0.05% Igepal CA-630 | 20%/RT |
| 600 U /ml RNasin ribonuclease inhibitor (add just before using the buffer) | RNasin (Promega N2115) 40 U/µl/-20°C |
RT: room temperature
SN1 + C buffer (prepare 500 µl per sample)
| Composition | Stock solution /storage |
|---|---|
| SN1 buffer + cOmplete protease inhibitor (1/30) | 1 tablet of cOmplete (Roche 11873580001) dissolved in 10 ml of nuclease-free H2O / -20°C |
SN2 buffer (prepare 300 µl per sample)
| Composition | Stock solution/storage |
|---|---|
| 10 mM HEPES-NaOH (pH 7.5) | 0.5 M/RT |
| 10 mM NaCl | 5 M/RT |
| 5 mM MgCl2 | 1 M/RT |
| 0.1% Igepal CA-630 | 20%/RT |
| 0.5 mg/ml heparin | 30 mg/ml heparin (Sigma H4784) in nuclease-free H2O.Filter (0.22 µm)/-20°C |
| 600 U /ml RNasin ribonuclease inhibitor (add just before using the buffer) | RNasin (Promega N2115) 40 U/µl/-20°C |
SN3 buffer (prepare 400 µl per sample)
| Composition | Stock solution/storage |
|---|---|
| 20 mM HEPES-NaOH (pH 7.5) | 0.5 M/RT |
| 200 mM NaCl | 5 M/RT |
| 4 mM EDTA | 0.5 M/RT |
| 0.1% Igepal CA-630 | 20%/RT |
| 0.04% sodium deoxycholate | 10%/RT |
| 4 mM imidazole | 1 M/RT |
| 0.1 mg/ml heparin | 30 mg/ml heparin (Sigma H4784) in nuclease-free H2O. Filter (0.22 µm)/-20°C |
| 1 mM DTT | 1 M/-20°C |
| cOmplete protease inhibitor (1/100) | 1 tablet of cOmplete dissolved in 10 ml of nuclease-free H2O/-20°C |
| 600 U /ml RNasin ribonuclease inhibitor (add just before using the buffer) | RNasin 40 U/µl −20°C |
RNase-free DNase I stock. Resuspend DNase (Qiagen 79254, 1500 U) by injecting 150 µl of 10 mM HEPES-NaOH (pH 7.5), 50% glycerol (solution prepared with nuclease-free H2O) into the DNase I vial using an RNase-free needle and syringe. Do not vortex. Transfer to an RNase-free tube and store at −20°C.
4.4. Additional materials and equipment
2x SDS-PAGE loading buffer (2x SPLB: 125 mM Tris-HCl [pH 6.8], 4% SDS, 20% glycerol, 10% β-mercaptoethanol, 0.1% bromophenol blue), dry ice/ethanol (when collecting samples for RNA extraction) and refrigerated (4°C) microfuge.
4.5. Step-by-step protocol
The protocol below is set up to process cells from two (or two and a half) 10 cm tissue culture dishes at 80% confluency. This amount of starting material is optimal for direct protein analyses of SN1, SN2 and SN3 fractions by western blot or to prepare total RNAs for northern blot (the aliquot volumes recommended for RNA preps are enough for 2–3 northern blot analyses). For GFP-Trap pull-downs, two PSE preparations are required: two cell pellets, each pellet from two 10 cm dishes (or from one 15 cm dish), are processed in parallel and then the two preparations obtained for each SN fraction are combined into the same tube (this is the sample used for the pull-down). Sucrose-gradient sedimentation analyses also require two PSE preparations: two cell pellets, each pellet from two and a half 10 cm dishes are processed in parallel and combined in step 6 to obtain 400 μl of 2x concentrated SN3 fraction.
General recommendations: have all reagents cold and maintain tubes on ice during the whole procedure.
Procedure:
Resuspend the frozen cell pellet in 500 μl SN1 + C buffer by gentle up-and-down pipetting and spin down in a microfuge (3,800 rpm, 3 min, 4°C).
The pellet will be further processed in step 3. Transfer the supernatant (SN1 fraction) to a new tube. Take a 120 µl aliquot for total RNA extraction. This aliquot has to be immediately frozen in dry ice/ethanol, and stored at −80°C until RNA is extracted (see RNA extraction section for details). If only total proteins are going to be analysed, add one volume of 2x SPLB buffer to the rest of the sample and store at −80°C (load 30 µl in gel for western blot). For protein–protein and protein-RNA co-precipitation analyses, the SN1 fraction cannot be frozen. Take a 15 µl aliquot for western blot analysis, add 15 µl of 2x SPLB solution and keep on ice. The rest of the sample is kept on ice until it is used for the GFP-Trap pull-down at the end of the PSE procedure (see details in Methods section 8).
Wash pellet with 500 µl SN1 buffer, dispersing it by vortexing, and spin it down in a microfuge (3,800 rpm, 3 min, 4°C).
Discard supernatant by aspiration. Resuspend the pellets in 300 μl of SN2 buffer, add 10 μl of DNase I (stock 10 U/µl), and incubate for 10 min at room temperature with occasional mixing (inverting the tubes gently) to digest chromatin. Spin down in microfuge (11,500 rpm, 10 min, 4°C).
The pellet will be further processed in step 6. Transfer the supernatant (SN2 fraction) to a new tube. Take a 72 µl aliquot for RNA extraction. This aliquot has to be immediately frozen in dry ice/ethanol and stored at −80°C until RNA is extracted. If only total proteins are going to be analysed, add one volume of 2xSPLB to the rest of the sample and store at −80°C (load 18 µl in gel for western blot). For protein–protein and protein-RNA pull-down assays, the SN2 fraction cannot be frozen. It is kept on ice for GFP-Trap purification at the end of the PSE procedure. Take a 9 µl aliquot for western blot analysis, add 9 µl of 2x SPLB solution and keep on ice.
Resuspend pellet/pellets* in 400 μl of SN3 buffer by up-and-down pipetting and incubate for 20 min at room temperature on a nutator to extract nucleolar RNPs. Spin down in microfuge (11,500 rpm, 10 min, 4°C). *Note that for sucrose-gradient sedimentation analysis, there are two preparations that converge at this step to obtain a 2X concentrated SN3 fraction: the pellets in the two tubes are combined by resuspending them in a total volume of 400 μl of SN3 buffer.
Transfer supernatant (SN3 fraction) to the new tube. Take 96 µl for RNA extraction. This aliquot has to be immediately frozen in dry ice/ethanol and stored at −80°C until RNA is extracted. If only total proteins are going to be analysed, add one volume of 2xSPLB to the rest of the sample and store at −80°C (load 24 µl in gel for western blot). For protein–protein and protein-RNA co-purification analyses, the SN3 fraction cannot be frozen. It must be kept on ice and be processed for GFP-Trap purification at the end of the PSE procedure. Take 12 µl for western blot analysis and add 12 µl of 2xSPLB. For sucrose-gradient sedimentation analysis, the SN3 fraction should not be frozen. Load 350–400 µl directly onto the gradient (see section 7.3 for protocol of obtention of preribosome sedimentation profiles). Store all the aliquots for western blots at −20°C.
5. Northern blot analyses of RNAs from whole cells, PSE fractions or sucrose gradient fractions
Total cellular RNAs (such as those analysed in Figure 3a) were prepared by the Trizol method using the TRI reagent (Ambion AM9738) following the manufacturer’s instructions. Quantifications were performed using a Nanodrop (VWR) spectrophotometer. For the northern blots, 4 µg of total RNA were analysed per sample. RNAs from the PSE method fractions (such as those analysed in Figure 2d, bottom left panel) and from sucrose-gradient fractions (such as those in Figure 3c) were prepared by the hot-phenol method. The procedure started with 400 µl samples containing AE buffer (50 mM sodium acetate, 10 mM EDTA [pH 5.2]). In the case of the PSE samples (see previous section), the 72 µl aliquot of freshly prepared SN2 supernatant and the 96 µl aliquot of freshly prepared SN3 supernatant were mixed with 328 µl and 304 µl of AE buffer, respectively. In the case of sucrose gradient fractions, a 120 µl aliquot of each fraction was thawed and mixed with 280 µl of AE buffer. 40 µl of 10% SDS was added to each sample and processed immediately. 530 µl of phenol (previously equilibrated with AE buffer) pre-heated to 65°C was added. The samples were incubated at 65°C in a water bath for 8 min (vortexed for 20 seconds, then rested for 20 seconds, repeating this procedure for a total of 8 min) and immediately frozen in dry ice/ethanol. Samples were centrifuged (13,000 rpm, 10 min, room temperature) and the aqueous phase (≈400 µl) was transferred to new tubes. 200 µl of pre-heated phenol and 200 µl of chloroform: isoamyl alcohol mixture (24:1) were added to and mixed by vortexing. The aqueous phase was separated again by centrifugation (13,000 rpm, 10 min, room temperature) and transferred to clean tubes. The RNA was precipitated by adding 40 µl of 3 M sodium acetate and 1 ml of −20°C-stored 100% ethanol. Tubes were kept overnight at −80°C, and the precipitated RNAs were recovered by centrifugation in a microfuge (13,000 rpm, 30 min, 4°C). The ethanol supernatant was carefully removed with a micropipette and the pellet left to dry at room temperature for 5 min before RNAs were resuspended in RNase free water by incubating them at 65°C for 10 min. The RNAs of sucrose gradient samples were resuspended in 5 µl of water and were used in total for just one northern blot analysis. The RNAs from PSE supernatants were initially resuspended in 20 µl of water, adjusted to a final concentration of 1 µg/µl and subject to northern blotting (4 µg) following the same procedures used for total cellular RNAs. The sequence of the oligonucleotide used as 5ʹ-ITS1 probe is shown in Supplementary Table 2.
6. Preparation of PPL lysates for polysome profile analysis (detailed protocol)
The following procedure is suitable for the obtention of polysome profiles by sucrose gradient sedimentation (Figure 3d). It has been successfully used with HeLa, A2780 and MDA-MB-231 cells.
6.1. Starting material
For each sample, use four 15 cm plates with cells at ~80% confluency (this prep will yield enough material for 2–3 polysome profile analyses).
6.2. Reagent set up
PPL (polysome profile lysis) buffer (1 ml per sample)
| Composition | Stock solution/storage |
|---|---|
| 15 mM Tris-HCl (pH 7.4) | 1 M/RT |
| 60 mM NaCl | 5 M/RT |
| 15 mM MgCl2 | 1 M/RT |
| 0.5% Triton X-100 | 10%/RT |
| 1 mg/ml heparin | 30 mg/ml heparin (Sigma H4784) in nuclease-free H2O. Filter (0.22 µm)/-20°C |
| 0.1 mg/ml cycloheximide | 5 mg/ml cycloheximide (Sigma C7698) in H2O. Prepare fresh (this stock is also needed to add to cells and to prepare PBS-C) |
PBS-C (120 ml per sample): cold PBS with 0.1 mg/ml cycloheximide (add cycloheximide from a fresh 5 mg/ml stock just prior to use)
80% glycerol stock
6.3. Step-by-step protocol
General recommendations : (1) have enough cycloheximide stock (freshly made) for the preparation of solutions and cell treatment, (2) benchtop centrifuge and microfuge must be refrigerated (4°C) and all tubes cold, (3) have cell plates on ice all the time and work fast during steps 2–5, (4) the PPL buffer will be needed in step 6 and also in the final step (keep it on ice all the time).
Procedure:
Incubate cells with 0.1 mg/ml cycloheximide for 5 min in the tissue culture incubator (add 500 µl of 5 mg/ml cycloheximide stock, directly in the 25 ml complete medium of each 15 cm plate).
Aspirate medium and place the four plates on ice (use a large tray).
Wash plates twice with 10 ml of cold PBS-C. Aspirate the liquid well after the second wash.
Add 2 ml of cold PBS-C to each plate, scrape the cells and pool the cells from the four plates together in one 15 ml conical tube.
Wash one plate with 2 ml of PBS-C, transfer to another plate to wash this second plate, and transfer to the 15 ml tube. Spin in benchtop centrifuge (1,000 rpm, 5 min, 4°C).
Aspirate the PBS-C, add 400 µl of PPL buffer to the pellet and lyse cells by gentle up-and-down pipetting. Transfer to a microfuge tube and spin down in a microfuge (10,000 rpm, 10 min, 4°C) to clear the lysate.
Transfer the cleared lysate to a 2 ml tube and add glycerol to a final 10%.
Take a 5 µl aliquot and read A260 absorbance. We need a total of 7–10 A260 units, in a volume not higher than 300 µl to be loaded onto each sucrose gradient (see below section Analysis of polysome and preribosome-sedimentation profiles on sucrose gradients).
Make aliquots of 200–300 µl in 2 ml tubes and store them at −80°C. Remember to prepare two 1 ml aliquots of PPL buffer with 10% glycerol and store them at −80°C together with the lysate samples. This buffer will be needed to balance the tubes before ultracentrifugation. Lysate samples can be stored at −80°C for several months.
7. Analysis of polysome and preribosome-sedimentation profiles on sucrose gradients (detailed protocol)
7.1. Preparation and long-term storage of 7%-50% sucrose gradients
General recommendation: all solutions should be either autoclaved or prepared in sterile H20 and filtered.
Materials: sucrose, buffers for sucrose solutions, container with liquid nitrogen (dry ice does not work well), 20 polypropylene 14 ml tubes (14 × 95 mm, Beckman # 331,374).
Sucrose solutions have to be prepared in different buffers depending on the experiment. For the obtention of polysome profile charts, such as those shown in Figure 3d, sucrose gradients are prepared in the following buffer: 15 mM Tris-HCl (pH 7.4); 60 mM NaCl; 15 mM MgCl2; 1 mM DTT. For sedimentation analyses of the preribosomes extracted in the PSE SN3 fraction, such as those shown in Figure 3c, sucrose gradients are prepared in 20 mM HEPES-NaOH (pH 7.5); 200 mM NaCl; 4 mM EDTA; 1 mM DTT; 0.1 mg/ml heparin (note that DTT and heparin are added once sucrose is dissolved, see below).
Procedure:
Prepare the following two solutions (120 ml each is enough for preparing 20 gradients) of sucrose in the appropriate buffer (without DTT and heparin): Solution A: 120 ml of sucrose 7%. Solution D: 120 ml of sucrose 50%. Add the corresponding DTT and heparin.
Use solutions A and D to prepare two solutions (B and C) as indicated below. This will render four solutions (60 ml each) of different sucrose concentrations: Solution A: 7% sucrose (60 ml). Solution B: 21.33% sucrose (40 ml solution A + 20 ml solution D). Solution C: 35.66% sucrose (20 ml solution A + 40 ml solution D). Solution D: 50% sucrose (60 ml)
Have a container with liquid nitrogen set up to easily introduce a rack with 20 gradient polypropylene 14 ml tubes. Choose a rack that holds well 20 tubes in a vertical still position. Have enough liquid nitrogen to submerge about two-thirds of the height of the tubes. This container is needed for steps 4 to 7.
Place 20 gradient tubes on the rack, on the bench top, and add 2.5 ml of sucrose solution D to each one of the tubes. Using long forceps or holders, introduce the rack inside the liquid nitrogen until solution D freezes down completely (it will take a short time). Take the rack out of the nitrogen container, place it on the bench top and proceed rapidly to the next step.
Add 2.5 ml of sucrose solution C to all tubes (solution C will not get mixed with solution D because this one is frozen and cold). Freeze down solution C in liquid nitrogen as indicated above and take the rack out of the nitrogen.
Add 2.5 ml of sucrose solution B to each tube and repeat the liquid-nitrogen freezing step.
Add 2.5 ml of the sucrose solution A to each tube and repeat the freezing step.
Store all the gradients at −80°C in a closed plastic container to avoid evaporation.
When needed for an experiment, take the required number of gradients out of the −80°C freezer and leave them undisturbed at 4°C overnight. The discontinuous solutions will thaw and diffuse, making a continuous 7%–50% gradient.
7.2. Obtaining polysome profiles
Materials needed in advance: (1) 7%–50% sucrose gradients (an even number) taken out of −80°C storage in advance and kept at 4°C for 12–16 hours ;(2) samples of cell extracts (for obtaining just polysome profile charts, such as those of Figure 3d, use the equivalent of 5–7 A260 units per sample; for fractionation and subsequent analysis of proteins or RNAs in the fractions, which is not shown in this article, use the equivalent of 15 A260 units per gradient) prepared in PPL buffer and stored frozen at −80°C (see above Methods section 6) ; (3) PPL-10% glycerol solution, stored at −80°C together with the PPL lysate samples.
Special equipment : (1)ultracentrifuge (Beckman) and rotor (Beckman SW40), both refrigerated at 4°C in advance ; (2)gradient reader/fractionator apparatus (we use a Brandel BR-186 Gradient Fractionator with Syringe Pump coupled to a Spectra/Chrom 280 UV Monitor and Chart Recorder).
General recommendation: always move gradient tubes very gently to avoid disturbance of the gradients.
Procedure:
Thaw cell PPL lysates and PPL-10% buffer on ice. Layer 5–7 A260 units of lysate very gently onto each gradient. It is advisable that the volume of the sample loaded onto the gradient does not exceed 300 µl. If the number of samples is not even, an ‘empty’ gradient will be needed to balance the rotor.
Balance the tubes, in pairs, using PPL-10% glycerol buffer and place them carefully inside the rotor buckets. Ultracentrifuge at 39,000 rpm for 165 min at 4°C (deceleration with the brake off).
Take the tubes out of the buckets, place them on ice and proceed to obtain the polysome profile of each gradient, one by one, in the gradient reader apparatus. In the Brandel System, the polysome profiles are obtained by continuous reading of absorbance at 254 nm from the top to the bottom of the gradient.
7.3. Sucrose gradient fractionation to obtain early preribosome sedimentation profiles
Materials prepared in advance : (1) 7%–50% sucrose gradients (an even number) that have to be taken out of the −80°C storage in advance and kept at 4°C for 12–16 hours ; (2) samples of SN3 fractions prepared fresh, the same day, by the PSE extraction protocol (see above Methods section 4) ; (3) for each gradient have three sets of 20 microfuge tubes labelled and cold: one set is to collect the gradient fractions, another set is to collect an aliquot of each fraction for RNA preparation, and the other set is to collect an aliquot for western blot analyses (each tube in this set must have 130 µl of 50% trichloroacetic acid (TCA) to precipitate the protein fraction)
Special equipment and materials : (1) ultracentrifuge (Beckman) and rotor (Beckman SW40), both refrigerated at 4°C in advance ; (2)gradient reader/fractionator apparatus (we use a Brandel BR-186 Gradient Fractionator with Syringe Pump coupled to a Spectra/Chrom 280 UV Monitor and Chart Recorder) ; (3) dry-ice/ethanol bath for freezing fraction aliquots; (4) 2x PAGE loading buffer.
General recommendations: (1) always move the gradient tubes very gently to avoid disturbance of the gradients ; (2)use sterile tubes to collect fractions and store their aliquots.
Procedure:
Layer very gently 350–400 µl of the SN3 sample onto one 7%–50% sucrose gradient.
Balance the tubes, in pairs, using the SN3 buffer and place them carefully in the rotor buckets. Ultracentrifuge at 39,000 rpm for 165 min at 4°C (deceleration with the brake off).
Take the tubes out of the rotor buckets, place them on ice and proceed to fractionate them, one by one, using the gradient fractionator apparatus. Collect twenty 0.5 ml fractions from each gradient and keep them on ice during the time needed to take aliquots for RNA and protein analysis.
Transfer a 120 µl aliquot of each fraction to the corresponding tube in the set of RNA extraction aliquots. Freeze this set of aliquots quickly in dry ice/ethanol. Store them at −80°C until RNA is prepared (see below section of Northern blot analyses). They can be stored for months.
Transfer 380 µl to the corresponding tube in the set of western blot aliquots (they contain 130 µl of 50% TCA). Mix and keep at −20°C overnight to precipitate proteins. Pellet the proteins in each sample by centrifugation (full speed, 15 min, 4°C), wash the pellets twice with 200 µl 100% acetone and dry them in a speed vac apparatus (10 min). Resuspend the pellet with 30 µl of 2x SPLB and neutralize with one-half volume (15 µl) of 2 M Tris pH 8.0. Store these samples at −20°C until western blot is performed.
8. Protein-protein and protein-RNA co-immunoprecipitation analysis of complexes extracted in PSE fractions
For HEATR1-GFP and ENP1-GFP pull-downs with GFP-Trap (such as those in Figure 2b–d), the SN2 and SN3 fractions were obtained from a starting material of either four 10 cm dishes or two 15 cm dishes (two PSE samples prepared in parallel and pooled; see above Methods section 4). After taking the corresponding aliquots for total RNA and total protein analyses, the SN2 and SN3 fractions were pre-incubated with 25 μl binding-control agarose beads (Chromotek) for 1 h at 4°C to eliminate the material that binds non-specifically to the beads. After a quick spin, each supernatant was then transferred to a new tube and incubated with 15 μl of GFP-Trap beads (Chromotek) at 4°C for 2 h. Finally, beads were washed five times with the corresponding (SN2 or SN3) ice-cold buffer. For the protein–protein interaction experiments, the whole sample of purified material was resuspended in SPLB and analysed, in parallel with total protein samples, by western blot (see Figure 2b,c, total protein samples are in lanes 1–8 and GFP-Trap pull-downs are in lanes 9–16). For the protein–RNA interaction experiments, one-fifth of the GFP-Trap beads were transferred to a new tube, and SPLB buffer was added for analysis, in parallel, with total fraction protein samples by western blot (see Figure 2d, top two panels, total protein samples are in lanes 1–12 and GFP-Trap material are in lanes 13–24). The rest of the pull-down material was resuspended in 400 μl of AE buffer (50 mM sodium acetate plus 10 mM EDTA [pH 5.2]) and processed for RNA extraction by the hot-phenol method as described above in Methods section 5. After precipitation, the recovered RNA was resuspended in formaldehyde-loading buffer and analysed by northern blot in parallel with 4 μg of the total RNA samples prepared from the aliquots that were taken prior to the GFP-Trap pull-down (see as an example bottom panels in Figure 2d, total RNA samples are in lanes 1–12 and RNAs bound to the GFP-Trapped proteins are in lanes 13–24).
9. Subfractionation of large and small complexes by ultracentrifugation
For the experiment in which PSE fractions were further fractionated into small and large molecular weight complexes (Figure 4c), samples of the fractions [200 μl of SN1, 200 μl of SN2, and 100 μl of SN3 (taken to 200 μl with SN3 buffer)] were ultracentrifuged at 155,000 xg in a TLA-100 rotor for 120 min at 4°C. Pellets were resuspended in the same original buffer and volume. Aliquots of these samples (containing the high-molecular-weight complexes) and of the corresponding ultracentrifugation supernatant (containing low-molecular weight complexes) were analysed by western blot. For SN1 and SN2 subfractions, 25 µl of those samples were loaded onto SDS polyacrylamide gels and, in the case of SN3 subfractions, 50 µl were loaded.
10. Reproducibility
All the data shown in this manuscript have been reproduced. The experiments were performed a minimum of two times and, in all cases, the obtained results were highly similar.
Supplementary Material
Funding Statement
This work was supported by the Spanish Ministry of Science and Innovation [BFU2017-88192-P to MD][RTI2018-096481-B-100 to XRB]; the National Institutes of Health [R03CA246009 to DGP]; the Castilla-León autonomous government [CSI252P18 and CSI145P2 to XRB]; the Spanish Association against Cancer [GC16173472GARC to XRB]; “la Caixa” Banking Foundation [HR20-00164 to XRB]; and the Samuel Solórzano Foundation [FS/36-2017 to MD]. BN has been supported by a predoctoral contract sponsored by the University of Salamanca and Santander Bank, SGG by a predoctoral FPU contract of the Spanish Ministry of Education, Culture and Sports, and LC by a predoctoral contract from the Spanish Association against Cancer. The Centro de Investigación del Cáncer is supported by the Programa de Apoyo a Planes Estratégicos de Investigación de Estructuras de Investigación de Excelencia of the Ministry of Education of the Castilla-León Government (CLC-2017-01). Both the Spanish and Castilla-León government-associated funding is partially supported by the European Regional Development Fund.
Disclosure statement
The authors report no competing financial interests.
Author contributions
BN and SGG contributed equally to this work. BN and SGG generated all the reagents, performed all the experiments except the polysome profiling and contributed to the design of the experiments and analysis of the data. RTS performed experiments to confirm the reproducibility of the PSE method.
LC performed the polysome profiles; XRB contributed to the analysis of data, discussion of results and to the writing of the paper; DGP developed the protocol of the PSE preribosome extraction method and contributed to the writing of the paper; MD designed the experiments, analysed the data, and wrote the paper.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Henras AK, Plisson-Chastang C, O’Donohue MF, et al. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip Rev RNA. 2015;6(2):225–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Baßler J, Hurt E.. Eukaryotic ribosome assembly. Annu Rev Biochem. 2019;88(1):281–306. [DOI] [PubMed] [Google Scholar]
- [3].Woolford JL Jr., Baserga SJ. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics. 2013;195:643–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kressler D, Hurt E, Bassler J. A puzzle of life: crafting ribosomal subunits. Trends Biochem Sci. 2017;42(8):640–654. [DOI] [PubMed] [Google Scholar]
- [5].De La Cruz J, Karbstein K, Woolford JL Jr. Functions of ribosomal proteins in assembly of eukaryotic ribosomes in vivo. Annu Rev Biochem. 2015;84(1):93129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Klinge S, Woolford JL. Ribosome assembly coming into focus. Nat Rev Mol Cell Biol. 2019;20(2):116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115(16):3196–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Farley KI, Baserga SJ. Probing the mechanisms underlying human diseases in making ribosomes. Biochem Soc Trans. 2016;44(4):1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Farley-Barnes KI, Ogawa LM, Baserga SJ. Ribosomopathies: old concepts, new controversies. Trends Genet. 2019;35(10):754–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kampen KR, Sulima SO, Vereecke S, et al. Hallmarks of ribosomopathies. Nucleic Acids Res. 2020;48(3):1013–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pelletier J, Thomas G, Volarevic S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat Rev Cancer. 2018;18(1):51–63. [DOI] [PubMed] [Google Scholar]
- [12].Bustelo XR, Dosil M. Ribosome biogenesis and cancer: basic and translational challenges. Curr Opin Genet Dev. 2018;48:22–29. [DOI] [PubMed] [Google Scholar]
- [13].Bursać S, Prodan Y, Pullen N, et al. Dysregulated ribosome biogenesis reveals therapeutic liabilities in cancer. Trends Cancer. 2021;7(1):57–76. [DOI] [PubMed] [Google Scholar]
- [14].Bohnsack KE, Bohnsack MT. Uncovering the assembly pathway of human ribosomes and its emerging links to disease. EMBO J. 2019;38(13):e100278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tomecki R, Sikorski PJ, Zakrzewska-Placzek M. Comparison of preribosomal RNA processing pathways in yeast, plant and human cells - focus on coordinated action of endo- and exoribonucleases. FEBS Lett. 2017;591:1801–1850. [DOI] [PubMed] [Google Scholar]
- [16].Ameismeier M, Cheng J, Berninghausen O, et al. Visualizing late states of human 40S ribosomal subunit maturation. Nature. 2018;558(7709):249–253. [DOI] [PubMed] [Google Scholar]
- [17].Ameismeier M, Zemp I, van den Heuvel J, et al. Structural basis for the final steps of human 40S ribosome maturation. Nature. 2020;587(7835):683–687. [DOI] [PubMed] [Google Scholar]
- [18].Larburu N, Montellese C, O’Donohue MF, et al. Structure of a human pre-40S particle points to a role for RACK1 in the final steps of 18S rRNA processing. Nucleic Acids Res. 2016;44(17):84658478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Plassart L, Shayan R, Montellese C, et al. The final step of 40S ribosomal subunit maturation is controlled by a dual key lock. Elife. 2021 Apr 28;10:e61254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Barandun J, Hunziker M, Klinge S. Assembly and structure of the SSU processome-a nucleolar precursor of the small ribosomal subunit. Curr Opin Struct Biol. 2018;49:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Farley KI, Surovtseva Y, Merkel J, et al. Determinants of mammalian nucleolar architecture. Chromosoma. 2015;124(3):323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hernandez-Verdun D, Roussel P, Thiry M, et al. The nucleolus: structure/function relationship in RNA metabolism. Wiley Interdiscip Rev RNA. 2010;1(3):415–431. [DOI] [PubMed] [Google Scholar]
- [23].Lafontaine DLJ, Riback JA, Bascetin R, et al. The nucleolus as a multiphase liquid condensate. Nat Rev Mol Cell Biol. 2021;22(3):165–182. [DOI] [PubMed] [Google Scholar]
- [24].Nieto B, Gaspar SG, Moriggi G, et al. Identification of distinct maturation steps involved in human 40S ribosomal subunit biosynthesis. Nat Commun. 2020;11(1):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cheng J, Lau B, La Venuta G, et al. 90S pre-ribosome transformation into the primordial 40S subunit. Science. 2020;369(6510):1470–1476. [DOI] [PubMed] [Google Scholar]
- [26].Du Y, An W, Zhu X, et al. Cryo-EM structure of 90. Science. 2020;369:1477–1481. [DOI] [PubMed] [Google Scholar]
- [27].Zhu J, Liu X, Anjos M, et al. Utp14 Recruits and Activates the RNA Helicase Dhr1 To Undock U3 snoRNA from the Preribosome. Mol Cell Biol. 2016;36(6):965–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dosil M, Bustelo XR. Functional characterization of Pwp2, a WD family protein essential for the assembly of the 90 S pre-ribosomal particle. J Biol Chem. 2004;279(36):37385–37397. [DOI] [PubMed] [Google Scholar]
- [29].Bursac S, Brdovcak MC, Donati G, et al. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim Biophys Acta. 2014;1842(6):817–830. [DOI] [PubMed] [Google Scholar]
- [30].Pelava A, Schneider C, Watkins NJ. The importance of ribosome production, and the 5S RNP-MDM2 pathway, in health and disease. Biochem Soc Trans. 2016;44(4):1086–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sloan KE, Bohnsack MT, Watkins NJ. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep. 2013;5(1):237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bursać S, Brdovčak MC, Pfannkuchen M, et al. Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. Proc Natl Acad Sci U S A. 2012;109(50):20467–20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Perez-Fernandez J, Martin-Marcos P, Dosil M. Elucidation of the assembly events required for the recruitment of Utp20, Imp4 and Bms1 onto nascent pre-ribosomes. Nucleic Acids Res. 2011;39(18):8105–8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Perez-Fernandez J, Roman A, De Las Rivas J, et al. The 90S preribosome is a multimodular structure that is assembled through a hierarchical mechanism. Mol Cell Biol. 2007;27(15):5414–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tafforeau L, Zorbas C, Langhendries JL, et al. The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of Pre-rRNA processing factors. Mol Cell. 2013;51(4):539–551. [DOI] [PubMed] [Google Scholar]
- [36].Wild T, Horvath P, Wyler E, et al. A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol. 2010;8:e1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Badertscher L, Wild T, Montellese C, et al. Genome-wide RNAi screening identifies protein modules required for 40s subunit synthesis in human cells. Cell Rep. 2015;13(12):2879–2891. [DOI] [PubMed] [Google Scholar]
- [38].Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Castle CD, Cassimere EK, Lee J, et al. Las1L is a nucleolar protein required for cell proliferation and ribosome biogenesis. Mol Cell Biol. 2010;30(18):4404–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.